 Kelly A. Hogan1,2,3
Kelly A. Hogan1,2,3 Julianna D. Zeidler4†
Julianna D. Zeidler4† Heather K. Beasley5†
Heather K. Beasley5† Abrar I. Alsaadi1
Abrar I. Alsaadi1 Abdulkareem A. Alshaheeb1Yi-Chin Chang6
Abdulkareem A. Alshaheeb1Yi-Chin Chang6 Hua Tian3,6*
Hua Tian3,6* Antentor O. Hinton Jr5*
Antentor O. Hinton Jr5* Melanie R. McReynolds1,3*
Melanie R. McReynolds1,3*- 1Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA, United States
- 2Signal Transduction and Molecular Nutrition Laboratory, Kogod Aging Center, Department of Anesthesiology and Perioperative Medicine, Mayo Clinic College of Medicine, Rochester, MN, United States
- 3Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA, United States
- 4Instituto de Bioquímica Médica Leopoldo de Meis, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 5Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN, United States
- 6Department of Chemistry, Pennsylvania State University, University Park, PA, United States
Metabolic homeostasis balances the production and consumption of energetic molecules to maintain active, healthy cells. Cellular stress, which disrupts metabolism and leads to the loss of cellular homeostasis, is important in age-related diseases. We focus here on the role of organelle dysfunction in age-related diseases, including the roles of energy deficiencies, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, changes in metabolic flux in aging (e.g., Ca2+ and nicotinamide adenine dinucleotide), and alterations in the endoplasmic reticulum-mitochondria contact sites that regulate the trafficking of metabolites. Tools for single-cell resolution of metabolite pools and metabolic flux in animal models of aging and age-related diseases are urgently needed. High-resolution mass spectrometry imaging (MSI) provides a revolutionary approach for capturing the metabolic states of individual cells and cellular interactions without the dissociation of tissues. mass spectrometry imaging can be a powerful tool to elucidate the role of stress-induced cellular dysfunction in aging.
Introduction
Aging is the greatest risk factor for the most prevalent diseases in industrialized nations. The coordinated action of energetic molecules ensures metabolic homeostasis, normal cellular function, and increased healthspan. The cellular hallmarks of aging are epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, cellular senescence, and exhaustion of stem cells (Ziegler et al., 2021). However, dysfunctional organelles are increasingly linked to diseases of aging such as chronic metabolic disorders, neurodegenerative diseases, and shortened lifespan (Ziegler et al., 2021). Among organelles ripe for interrogation in the aging process are the mitochondria, the endoplasmic reticulum (ER), and the sites of inter-organelle communication called mitochondria-ER contact sites (MERCs). MERCs are enriched with proteins essential for mitochondrial Ca2+ flux, lipid transfer, autophagy, mitochondrial division, apoptosis, and morphology (Giacomello and Pellegrini, 2016; Csordás et al., 2018), and these contact sites may be important in understanding how metabolic fluxes are altered during the aging process. However, studies to define the role of MERCs in metabolic fluxes using single cell techniques such as mass spectrometry imaging (MSI) are lacking.
In addition to well characterized calcium fluxes between the ER and mitochondria, there are other metabolic exchanges including those associated with nicotinamide adenine dinucleotide (NAD+) and precursors to NAD+ synthesis that are strongly associated with diseases of aging (McReynolds et al., 2020; McReynolds et al., 2021; Zeidler et al., 2022). NAD+ is a cofactor for redox and non-redox enzyme reactions that maintain metabolic pathways, DNA repair, chromatin remodeling, immune cell function, and cellular senescence (Hogan et al., 2019; Covarrubias et al., 2021). A decline in cellular NAD+ levels is linked to several diseases of aging in mice including neurodegeneration and cognitive decline, cancer, metabolic disease, sarcopenia, frailty, hearing loss, stroke, cataracts, and kidney and heart disease (Hogan et al., 2019; Covarrubias et al., 2021). Senescent cells secrete pro-inflammatory factors characteristic of the senescence-associated secretory phenotype (SASP), resulting in the accumulation of CD38+ inflammatory cells (Chini et al., 2020; Covarrubias et al., 2020). As an NADase and nicotinamide mononucleotidase (NMNase), CD38 decreases cellular NAD+ and extracellular NMN, respectively, and contributes to low-grade inflammation called inflammaging (Franceschi et al., 2018). The role of NAD+ metabolism in the regulation of SASP is not well understood, but both de novo and salvage NAD+ synthesis pathways are implicated in senescence. For example, the rate-limiting NAD+ synthesis salvage pathway enzyme nicotinamide phosphoribosyltransferase (NAMPT) promotes SASP through nuclear factor kappa B (NF-κB) in IMR90 fibroblasts (Nacarelli et al., 2019). Additionally, disruption of the de novo NAD+ synthesis pathway may lead to a decline in NAD+ in macrophages and innate immune cell dysfunction observed in aging and age-related diseases (Minhas et al., 2019). Single-cell technologies that combine imaging and metabolomics to interrogate inter-organellar trafficking of molecules like NAD+ are potentially powerful tools for understanding the aging process at the cellular and subcellular levels. Defining the metabolic landscape and the variability within cells and across cell types represents a new frontier in metabolomics and an opportunity to re-envision our understanding of cellular biology.
The emerging field of single-cell “spatial omics” resolves genomics, proteomics, and transcriptomics associated with intracellular structures in tissue sections without dissociating tissues or disturbing cellular interactions (Blum et al., 2018; Gilmore et al., 2019; Peng et al., 2020; Bai et al., 2021; Stein et al., 2021; Taylor et al., 2021). However, resolving the spatial omics for metabolites important in the pathophysiology of diseases has been more challenging. For example, although lipids play a central role in signaling, regulation, inflammation, and cancer, preserving the spatiotemporal gradient of these dynamic and transient small molecules is difficult. Here, we propose that spatial omics can provide a better understanding of organellar metabolism to identify the mechanisms of cellular decline during aging. Although typical age-related processes such as autophagy and apoptosis may be regulated through age-associated MERC modifications, changes in the composition or number of MERCs may also be pro-senescent (Ziegler et al., 2021). This review focuses primarily on the structure, function, and role of mitochondria and the ER in age-related diseases, with special emphasis on MERCs as modifiers or targets of cellular aging (Janikiewicz et al., 2018; Csordás et al., 2018; Moltedo et al., 2019; Ziegler et al., 2021).
Single-cell technologies that combine imaging and metabolomics to interrogate inter-organellar trafficking of molecules, like NAD+, are potentially powerful tools for understanding the aging process at the cellular and subcellular levels. Defining the metabolic landscape and the variability within cells and across cell types represents a new frontier in metabolomics and an opportunity to re-envision our understanding of cellular biology. However, few studies have defined the role of MERCs in metabolic fluxes using single-cell techniques, such as mass spectrometry imaging (MSI). We also describe the advantages and limitations of MSI, particularly as a tool to interrogate age-related dysfunction at the single-cell level.
Spatial metabolomics/lipidomics as a tool for interrogating diseases of aging
Cell-to-cell variation is intrinsic to cell populations, organs, and tumors and includes differences in epigenetic, transcriptional, translational, and metabolic regulation; therefore, understanding these differences is important for defining age-related regulatory changes across cell and tissue types. Identifying spatially resolved age-related metabolic alterations will be key to elucidating the impact of these changes on the aging trajectory and developing effective metabolite-targeted treatments for age-related diseases. Changes in metabolite levels triggered by fluctuations in the cellular environment or altered intracellular signaling are reliable markers of cellular status. Single-cell transcriptomics provides powerful insights into differences observed in normal and pathological contexts. Likewise, studies of the metabolome in single cells are also likely to provide a comprehensive profile of individual cellular changes associated with age. Spatial and temporal snapshots of nutrient gradients, and fluxes in particular, may become the basis of discovery of new interventions for aging-related diseases.
MSI is a tool that has the potential to visualize pro-senescent MERCs because the technology has the capacity to simultaneously detect proteins, lipids, and metabolites. To date, there are several technical approaches and workflows for MSI including gas cluster ion beam secondary ion mass spectrometry (GCIB-SIMS), matrix-assisted laser desorption ionization (MALDI), desorption electrospray ionization (DESI), and liquid extraction surface analysis (LESA) (Figure 1). GCIB-SIMS is a particularly promising approach to spatially mapping the age-related metabolites in single cell or subcellular organelle level. With an innovative new desorption source, high-energy CO2 or H2O cluster ion beam, GCIB-SIMS resolves biomolecules at the lateral resolution of 1 µm without comprising signal level of low concentration biomolecules (e.g., transient metabolites). Among several advantages of the GCIB source is enhanced ionization up to ∼200 fold to facilitate detection of biomolecules; reduced ionization suppression to allow the imaging of diverse biomolecules; and extension of mass detection ranges for spatial multiomics in a single sample (Tian et al., 2014; Wucher et al., 2014; Tian et al., 2016a; Tian et al., 2016b; Tian et al., 2016c; Tian et al., 2016d; Tian et al., 2017a; Tian et al., 2019a; Tian et al., 2019b; Sheraz et al., 2019). Coupled with a unique buncher-ToF SIMS instrument (J105 SIMS) (Fletcher et al., 2008; Tian et al., 2016d), GCIB-SIMS achieves multi-omics imaging at cellular/subcelluar resolution in biological samples (Tian et al., 2019a; Tian et al., 2021a). Additionally, as one of very few MSIs capable of cryogenic analysis, GCIB-SIMS captures images of frozen-hydrated single cells and preserves the chemical microenvironments at near-nature state with minimal disruption of the tissue and cellular structures, especially for the dynamic metabolic gradients (Tian et al., 2014; Tian et al., 2016b; Tian et al., 2019a; Tian et al., 2021a). Additionally, GCIB-SIMS produces molecular species, (M + H)+ or (M-H)− for biomolecules, as well as different forms of fragments. A caveat, however, are incompatibilities in sample preparation that prevent the GCIB-SIMS cryogenic workflow from being adapted for other MSI tools, thus making them unsuitable for exploration of spatial metabolomics (Robinson and Castner, 2013; Zhang et al., 2018). As the first multiplexing pipeline to integrate multiomics in the same cells, the GCIB-SIMS multimodal imaging platform simultaneously detects metabolites and lipids with multiple protein expression markers in frozen-hydrated breast cancer tissue and mouse liver tissue at the single-cell level (Tian et al., 2021b). As shown in Figure 2 (Tian et al., 2021b), untargeted metabolites/lipids and multiple targeted proteins are imaged at 1 µm spatial resolution in human breast cancer tissue, facilitating cell segmentation and registration of multi-omics in individual cells by computational processing and discriminant analysis. The workflow permits visualization of multiple molecular coordination (>150 key species) in different types of tumor microenvironment cells including actively proliferating tumor cells and infiltrating immune cells. For example, desaturated fatty acids, phosphatidylethanolamine plasmalogen and glutathione are observed in pan cytokeratin-expressing cells (i.e., epithelial cancer cells), suggesting that cancer cells adopt antioxidant mechanism to prevent lipids oxidation-induced ferroptopic cell death under conditions of stress.
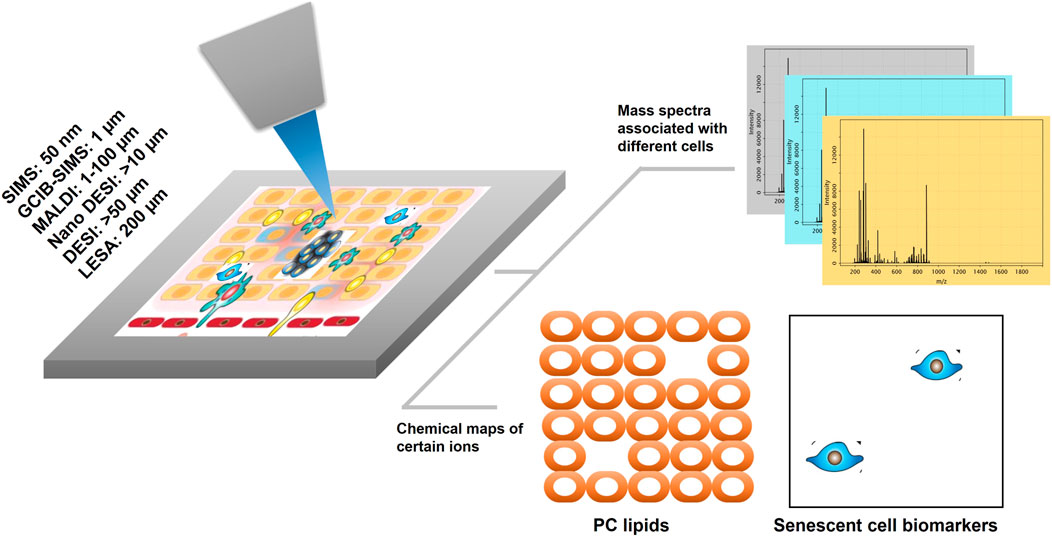
FIGURE 1. A simplified workflow of MSI for spatial omics. The focused ion desorption source scans the tissue surface (or cells in culture) pixel by pixel. Both mass spectra and x and y coordinates are generated from each pixel. With the single-cell resolution of the ion desorption source, omics information is obtained from individual cells in the tissue or cell culture without tissue dissociation. Biomolecules or biomarkers are localized to cellular structures or different cell populations. Technical approaches for MSI workflows include: SIMS, secondary ion mass spectrometry; GCIB, gas cluster ion beam; MALDI, matrix-assisted laser desorption ionization; DESI, desorption electrospray ionization; LESA, liquid extraction surface analysis.
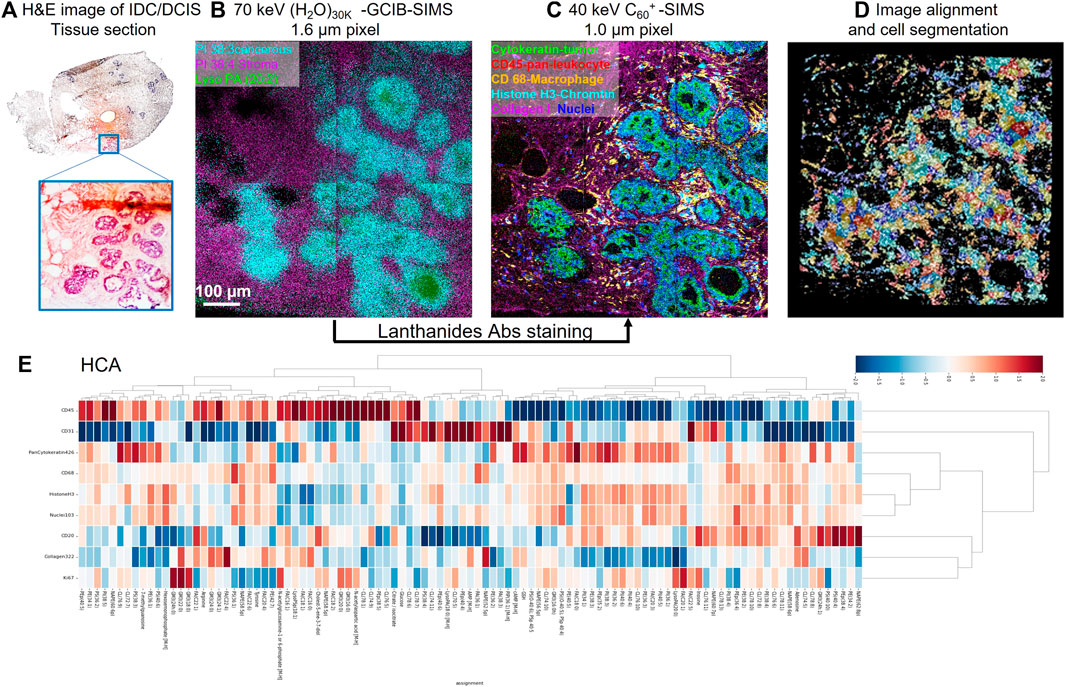
FIGURE 2. Schematic of the workflow for cell-type-specific profiling of multi-omics on IDC/DCIS tissue sections at the single-cell level with select overlays and single-ion images using (H2O)n-GCIB-SIMS and C60-SIMS. (A) H&E staining image of a semi-serial section of invasive ductal carcinoma/ductal carcinoma in situ (IDC/DCIS) tissue. The enlarged image from the region of interest highlighted in blue shows the tumor region in purple and the stromal region in pink. (B) (H2O)n-GCIB-SIMS imaging of a fresh-frozen tissue section. The cryogenic analysis at 100 K was performed on frozen-hydrated tissue sections for molecular imaging (e.g., lipids and metabolites) at a beam spot size of 1.6 µm. (C) C60-SIMS imaging of the same tissue section stained with a lanthanide-tagged cocktail of antibodies at a beam spot size of 1.0 µm. (D,E), (D) the image alignment and cell segmentation were used to integrate the omics (E) metabolites, lipids, and proteins into different cell types at single-cell resolution.
To this end, the GCIB-SIMS workflow integrates spatial omics at the single-cell level, providing a new approach for visualizing individual cells, cellular interactions, and chemical microenvironments in aging tissues for deeper insights into the mechanisms of age-related diseases. Spatially resolved approaches to elucidating metabolic activity will be particularly important for highly age-sensitive epithelial, mesenchymal, immune, senescent, and cancer cells, as well as for cells from relatively short-lived gestational tissues at the maternal-fetal interface. As demographics continue to shift toward an aging population and diseases of aging grow more prevalent, tools to interrogate compromised tissue microenvironments and disrupted cell-to-cell communication are critical for identifying druggable targets in age-related diseases. Metabolic shift in different types of cells within the tissue is vital to understanding aging-prone cell types. With high sensitivity for various biomolecules, GCIB offers a soft ionization for imaging of pathways involving molecules like NAD+ with the potential to ionize NADP+ from NADPH as distinct molecular ions. MSI, especially high-resolution GCIB-SIMS, could transform current understanding of aging tissues by: 1) mapping the variation of metabolic flux resulting from NAD+ synthesis and degradation in individual cells in tissues vulnerable to NAD+ decline; 2) characterizing cellular heterogeneity within tissues to understand mechanisms of inflammaging and the impacts of inflammation on neighboring cell types; 3) identifying metabolic imbalances and reprogramming in age-related diseases; and 4) resolving metabolic gradients in subcellular organelles, including NAD(H) and NADP(H) trafficking across MERCs.
Organelles targeted by age-associated energy deficiencies
The aging mitochondria
Mitochondria govern cellular homeostasis by coordinating the energetic needs of the cell. Mitochondrial dysfunction, a hallmark of aging, underlies several neurological disorders and disorders of metabolism such as obesity, aging, hypertension, and type 2 diabetes (Ihab Hajjar and Kotchen, 2006; Esther Phielix and Roden., 2011; Rieusset, 2018). The pathophysiology resulting from deterioration of mitochondrial function likely contributes to the age-dependent decrease in organ function driven by loss of energy production, lipid biogenesis, and activation of cell death programs (Ian Lanza and Nair, 2010; Miriam Valera-Alberni and Canto, 2018; Seungyoon Yu and Pekkurnaz, 2018). Signals necessary for maintaining cellular homeostasis are exchanged continuously between mitochondria and ER through organelle-linking contact sights known as mitochondria-ER contact sites (Csordás et al., 2018) or MERCs. Namely, the transfer of Ca2+ from ER to mitochondria is vital for maintaining mitochondrial energy metabolism. MERC distance or thickness is integral to normal Ca2+ signaling and transport (Giacomello and Pellegrini, 2016; Csordás et al., 2018). Contact sites outside of the 12–24 nm range appear to alter Ca2+ uptake and lipid dynamics, in this context (Giacomello and Pellegrini, 2016), by creating steric hindrance that disrupts Ca2+ transport machinery within MERCs (Giacomello and Pellegrini, 2016). Mitochondrial Ca2+ is required for the activation of oxidative phosphorylation (Roberto Bravo et al., 2011; Brian Glancy and Balaban, 2012; Rieusset, 2018) and dysregulation of mitochondrial Ca2+ homeostasis may lead to higher amounts of radical oxygen species (ROS) (Artyom Baev et al., 2022).
Mitochondrial ROS contributes to age-related loss of tissue homeostasis (Shigenaga et al., 1994; Carlos López-Otín et al., 2013) and changes to MERCs are likely to target the “redox triangle” which includes the mitochondria, ER, and peroxisomes. Mitochondrial ROS arises from the leakage of electrons from redox centers of respiratory complexes and associated enzymes (Hoi-Shan Wong et al., 2017). There are at least eleven sites of O2•– and H2O2 generation in mitochondria. The rates of ROS generation and the relative contribution of each site depends on substrate availability and cellular metabolic context (Hoi-Shan Wong et al., 2017). What specifically triggers increased mitochondrial ROS during aging is still under debate, but roles for the initiation of senescence by mitochondrial ROS have been established (Passos et al., 2010; Clara Correia-Melo et al., 2016). For example, excessive production of mitochondrial ROS can drive telomeric-induced senescence (Passos et al., 2007) and oncogene-induced senescence (Olga Moiseeva et al., 2009). ROS also contribute to cellular senescence by promoting DNA damage (Passos et al., 2010), which in turn activates stress-response pathways such as JNK1/2, that ultimately leads to SASP formation (Maria Grazia Vizioli et al., 2020), NLRP3 inflammasome activation (Rongbin Zhou et al., 2011), in addition to changes in mitochondrial structure, function, and dynamics (Hélène Martini and Passos, 2022). Thus, mitochondria play essential roles in establishing and maintaining the senescence phenotype with production of mitochondrial ROS as a key component.
In addition to causing DNA damage, ROS also promote lipid peroxidation, leading to decreased mitochondrial membrane fluidity and dysregulated mitochondria function (Chen and Yu, 1994; Shigenaga et al., 1994). Increased ROS can also lead to protein oxidation. A quantitative “oxi-proteome” analysis has identified oxidized proteins enriched in senescent fibroblasts by measuring 4-hydroxy-2-nonenal (HNE)-, glycoxidation (AGE)-, and carbonylation-modified proteins (Emad Ahmed et al., 2010). Consistent with increased mitochondrial ROS in senescence, half of these oxidized proteins were mitochondrial with most involved in energy metabolism and protein quality control (Emad Ahmed et al., 2010). Not surprisingly, proteins from the cytoplasm, nucleus, and ER are also vulnerable to oxidation (Emad Ahmed et al., 2010). By mapping oxidized biomolecules such as lipids and proteins, MSI could be used to determine the extent to which oxidative damage derived from mitochondrial ROS spreads throughout cells and alters metabolism in surrounding organelles or neighboring cells. This approach could help answer foundational questions concerning the role of mitochondrial ROS in aging and identify both sources and consequences of ROS in aged tissues.
The tricarboxylic acid (TCA) cycle intermediates are another link between mitochondrial metabolism and the senescence phenotype. Some TCA cycle metabolites play pleiotropic roles outside mitochondria to influence epigenetic modification, immune regulation, and cell signaling. For instance, cytosolic citrate is required to produce acetyl-CoA for histone acetylation (Wellen et al., 2009) and can act as an inflammatory signal (Vittoria Infantino et al., 2011). α-Ketoglutarate is a co-factor for 2-oxoglutarate/Fe2+-dependent dioxygenases (OGDD). These enzymes catalyze the hydroxylation of various substrates such as nucleic acids, lipids, and proteins, including histone demethylases in the nucleus (Inmaculada Martínez-Reyes and Chandel, 2020). Also, succinate accumulation can inhibit the activity of OGDDs and HIF-prolyl hydroxylases (HPH) and promote DNA hypermethylation and pseudohypoxia, respectively (Elaine MacKenzie et al., 2007; Eric Letouzé et al., 2013). Senescent cells display increased levels of TCA cycle metabolites such as citrate, α-ketoglutarate, and malate likely as a result of impaired mitochondrial function (Dörr et al., 2013; Joanna Kaplon et al., 2013; Emma et al., 2015). In vivo, α-ketoglutarate supplementation improves the lifespan and healthspan of old mice, reduces systemic inflammation, increases bone mass, and attenuates age-related bone loss (Inmaculada Martínez-Reyes and Chandel, 2020; Yuan Wang et al., 2020). In humans, citrate levels increase in the circulation during chronological aging (Cristina Menni et al., 2013; Kirsi Auro et al., 2014) and the potential implications of citrate in age-related diseases are reviewed elsewhere (Mycielska et al., 2022). Thus, the TCA cycle metabolome seems to be disrupted in cellular senescence and aging, but the consequences at the molecular level remain mostly elusive. MSI may resolve trace changes in the subcellular localization of TCA cycle metabolites in tissues during aging and provide clues to the functions those metabolites serve in age-related tissue dysfunction.
What causes age-related defects in mitochondria is still under intense investigation. Although multifactorial and context-dependent, evidence points to disrupted NAD+ metabolism as a player in mitochondrial decay. The primary cause of NAD+ decline in aging is increased NAD+ degradation without compensatory upregulation of NAD+ synthesis pathways (Camacho-Pereira et al., 2016; McReynolds et al., 2021). Upregulation of expression and increased activity of the NADase CD38 in old mice causes NAD+ levels to decline and affects the activity of Sirt3 (Camacho-Pereira et al., 2016). This mitochondria-localized sirtuin regulates mitochondrial metabolism and oxidative homeostasis. Thus, the CD38-NAD-Sirt3 axis leads to age-related mitochondrial dysfunction (Camacho-Pereira et al., 2016). In addition, NAD+ precursor supplementation improves mitochondrial function in aged mice and C. elegans (Gomes et al., 2013; Mouchiroud et al., 2013; Zhang et al., 2016), and overexpression of Nmnat3, an enzyme of the NAD+ salvage pathway, restores TCA cycle capacity and suppresses mitochondrial ROS generation in skeletal muscle of aged mice (Maryam Gulshan et al., 2018). However, only recently, SLC25A51 was identified as the mitochondrial NAD+ transporter (Girardi et al., 2020; Kory et al., 2020; Luongo et al., 2020), but little is known about the kinetics and regulation of NAD+ and NAD+ precursor influx into mitochondria in a physiological context and during aging. Flux analysis reveals incomplete equilibration of NAD+ between cytosol and mitochondria (McReynolds et al., 2021), suggesting that this organelle can be particularly sensitive to disturbances in cellular NAD+ levels. To uncover the consequences of disruptions in this dynamic during aging, MSI will likely be a powerful approach to characterizing NAD+ metabolome fluxes in and out of mitochondria.
Endoplasmic reticulum (ER)
Communication between the ER and mitochondria is tightly regulated by mitochondria associated membranes (Zhang et al., 2021). ER homeostasis drives biosynthetic pathways including lipogenesis and gluconeogenesis (Gansemer and Rutkowski, 2022). The ER is composed of a set of membranes or cisternae that are held together by the cytoskeleton. The lumen of the ER stores phospholipids which are trafficked to various organelles. The volume of the rough or smooth ER lumens are dependent on the metabolic activity of the cell. ER functions include folding and transport of proteins to the Golgi apparatus, lipid biosynthesis, and Ca2+ storage via Ca2+-dependent chaperones. In the aging cell, ER chaperones are especially sensitive to oxidative damage and vulnerable to reduced Ca2+ buffering capacity, resulting in the deposition of insoluble fibrils or plaques as a consequence of oxidative stress (Balchin et al., 2016). Deposits that collect in the liver, brain, and spleen are pathognomonic for diseases associated with aging including Alzheimer’s disease, Parkinson’s disease, and Type 2 diabetes (Tian et al., 2019b; Sheraz et al., 2019).
In addition to oxidative stress, the ER responds to various metabolic stressors by triggering the unfolded protein response (UPR). Among UPR-inducing stressors, glucose deprivation leads to ER stress which interferes with disulfide bond catalysis and N-linked protein glycosylation with implications for caspase activation (Xu et al., 2005). Additionally, aberrant Ca2+ signaling in the ER causes improper protein folding, further contributing to the UPR. The UPR is an adaptive response that permits the cell to adjust to a challenging environment and maintain normal ER function and homeostasis. Adaptation also involves transcriptional modulation to enhance protein folding and removal of misfolded proteins in the ER. The UPR is primarily a cell-signaling system to maintain normal cellular function by restoring protein homeostasis; however, chronic ER stress leads to apoptosis. During ER stress, the UPR activates rescue pathways that accelerate protein folding and upregulate molecular chaperones and enzymes to facilitate the degradation of misfolded proteins (Gorman et al., 2012; Hetz, 2012). The three branches of UPR signaling pathways (i.e., IRE1α, PERK, and ATF6) directly or indirectly influence transcriptional and translational responses to the stressor, which either alleviate the source of stress or activate cell death (Ashley Frakes and Dillin, 2017). However, when the UPR declines with age, it is often a consequence of oxidative damage sustained by UPR-regulated chaperone proteins responsible for facilitating protein folding necessary to achieve proteostasis (Rabek et al., 2003; Nuss et al., 2008). A link between protein aggregation and aging is demonstrated by inhibiting protein aggregation and extending lifespan in C. elegans suggesting an association between proteostasis and longevity (Silvestre Alavez et al., 2011). The chaperones that appear to be central players in proteome maintenance include BiP, calnexin, and protein disulfide isomerase (PDI), which are observed to decrease both centrally and peripherally with age (Gavilán et al., 2006; Nirinjini Naidoo et al., 2008; Nuss et al., 2008). UPR during senescence (Pluquet et al., 2015) occurs in a wide range of cell types activated by oncogenic Ras (Denoyelle et al., 2006; Dörr et al., 2013; Zhu et al., 2014), xenobiotics (Dörr et al., 2013; Williams et al., 2013; Matos et al., 2015), and DNA damage (Panganiban et al., 2013). It is unclear, however, whether the UPR is a cause or an effect of senescence (Pluquet et al., 2015). The role of the UPR in senescence, therefore, remains controversial, and the mechanisms linking ER and senescence are ripe for interrogation.
Mitochondria-endoplasmic reticulum contacts (MERCs)
MERCs, the sites of interactions between mitochondria and the ER, contain hundreds of proteins including enzymes, ion channels, and tethering and transport binding proteins (Barazzuol et al., 2021). Membrane fractionation and characterization of contact zones have identified components of the ER that co-purify with mitochondria (Wu et al., 2018) and comprise the biochemical fraction of MERCs known as mitochondria associated membranes (MAMs). MAMs are the sites of interaction between organelles (Vance et al., 1997; Patergnani et al., 2011) and play a role not only in metabolite trafficking and signal transduction but in membrane fusion, mitophagy, and lipid metabolism (Janikiewicz et al., 2018). MAM proteins are broadly classified as protein tethers, ion channels, or enzymes (Wang et al., 2019) and are vulnerable to age-related dysregulation that occurs as a consequence of loss or disruption of critical metabolic fluxes (Janikiewicz et al., 2018; Moltedo et al., 2019; Ziegler et al., 2021). It remains unclear whether MAM composition is implicated in the aging process (Janikiewicz et al., 2018).
Although mechanisms of MERC-mediated senescence have not been fully elucidated, MERCs are implicated in dysregulation of Ca2+ exchange and flux (Giacomello and Pellegrini, 2016), which may potentiate cellular senescence by increasing mitochondrial Ca2+ concentration. The loss of protein tethers that control Ca2+ and phospholipid fluxes disrupts mitochondrial respiration and likely contributes to the aging process (Patergnani et al., 2011). Among the MAMs across MERC sites that regulate Ca2+ flux, the tethering mitofusin proteins MFN1 and MFN2 act as conduits from the mitochondria to the ER, where flux of Ca2+ into the mitochondria is controlled by the IP3R-DJ-1-GRP75-VDAC complex (Kaul et al., 2003; Son et al., 2017; Liu et al., 2019).
Ca2+ reservoirs, gradients, and flux in the MERC space
Ca2+ is critical for mitochondrial function and energetics, and the ER acts as a reservoir for Ca2+ by maintaining a high Ca2+ gradient with the aid of the ATPase SERCA (Wang et al., 2019). Ca2+ is not only a cofactor for enzymes in cellular respiration but is also trafficked across MERCs (Szibor et al., 2020). The MERC space accommodates Ca2+ signal transfer between the ER and mitochondria via IP3 receptors (IP3R) permitting overall cell survival (Bartok et al., 2019). Proteins in the MERC space (e.g., IP3R and VDAC1) control flux of Ca2+ into mitochondria (Sander et al., 2021). In addition to Ca2+ trafficking, MERCs modulate: 1) bidirectional lipid transfer that is critical to steroid synthesis and membrane maintenance; 2) mitochondrial quality control, which employs pro-fusion proteins (e.g., MFN1 and 2, and OPA1) to drive mitophagy (e.g., PINK1 and parkin); and 3) the ER stress response and the UPR, involving PERK, among other stress-responsive proteins (Janikiewicz et al., 2018; Veeresh et al., 2019; Picca et al., 2020; Wilson and Metzakopian, 2021).
Ca2+ serves an especially important role as a first and second messenger in signal transduction and as a cofactor of enzymes in mitochondrial cellular respiration (Fink et al., 2017). Dysregulation of inter-organellar Ca2+ homeostasis impacts metabolic fluxes, results in apoptosis, and is likely a mechanism of action in diseases arising from ER stress. Proteins that are important in Ca2+ shuffling include MFN2, which is found on the outer membrane of the cell and aids in the transport of Ca2+ from the mitochondria to the ER. MFN2 is a negative regulator of MERCs; therefore, ablation of MFN2 leads to increased Ca2+ shuttling from the ER to mitochondria (Leal et al., 2016). MFN2, which is enriched in MAM fractions, regulates ER morphology by tethering the ER and mitochondria. The movement of Ca2+ from a storage organelle to an energy-producing organelle is critical to cellular metabolic homeostasis, and interrogation of this process with MSI will likely reveal mechanisms of aging and age-related diseases associated with dysregulated Ca2+ fluxes. Understanding the molecular mechanisms of diseases arising from ER stress may re-contextualize the pathophysiology of diseases of aging, including cancers (Lin et al., 2019), Parkinson’s disease (Surmeier et al., 2017), type 2 diabetes (Burgos-Morón et al., 2019), Alzheimer’s disease (Uddin et al., 2021), and muscle disorders (Gommans et al., 2002), among others.
Pyridine nucleotide fluxes
NAD(P)+/NAD(P)H and the pyridine nucleotide derivatives of NAD(P)+ (e.g., NADP and ADPR) potentiate Ca2+ efflux from the ER (Churchill and Galione, 2001; Mándi et al., 2006) and also serve as redox cofactors in enzymatic reactions that produce ATP and synthesize fatty acids, phospholipids, select amino acids, and steroids (Wang et al., 2019). NADPH reduces oxidized precursors during the synthesis of lipids, deoxyribonucleotides, and proline, and NAD+ removes electrons during the metabolism of sugars and fats, producing the NADH required to make ATP. Although the ratio of NADPH/NADP+ is high in all cell types to accommodate biosynthesis and the oxidative stress response, the NADH/NAD+ ratio depends on nutrient availability, differs across organelles, and depends upon localization of metabolic enzymes in cellular compartments (Hosios and Vander Heiden, 2018). Vesicles and membrane contact sites are responsible for the trafficking of metabolites, lipids, and proteins, thereby facilitating adaptation of cells to changing extracellular conditions (Cohen et al., 2018). Cellular homeostasis, however, requires sufficient levels of molecules such as NAD(P)+/NAD(P)H, which declines with age (McReynolds et al., 2020). How organelles affect the NAD+ levels in a cell and how aging alters this process is an area that remains open to interrogation and MSI may be useful for greater resolution of more nuanced roles of NAD+.
Visualizing cellular and subcellular metabolism in age-related disease models
Current methods for the visualization of interorganelle spaces like MERCs include electron microscopy (EM), which provides a high-resolution image of cellular or tissue ultrastructure that is ideal for study of organelles, viruses, and macromolecules. An array of EM approaches offers detailed resolution of cellular architecture. Scanning EM (SEM), for example, is a high-resolution imaging technique using a focused beam of electrons to resolve surface topography. Transmission electron microscopy (TEM) is commonly used for high-resolution 2D micrographs (Garza-Lopez et al., 2021; Lam et al., 2021); however, there are several 3D imaging approaches which permit visualization of organelles including serial block-face scanning electron microscopy (SBF-SEM), focused ion beam scanning electron microscopy (FIB-SEM), and cryo-EM among others. The main difference between SBF-SEM and FIB-SEM is that the former uses a beam of elections and latter uses a beam of gallium ions, permitting the resolution of mitochondria cristae structure or organelle-organelle interactions such as MERCs. Electron tomography, an extension of TEM that includes cryogenic electron microscopy (Cryo-EM) improves visualization of organelles by avoiding crystallization (Xu Benjin and Ling, 2020). Other techniques, including optical and fluorescence microscopy, provide visualization of the contact sites and Ca2+ signaling in MERCs using luminescent or fluorescent Ca2+ sensors. Compared to these other methods, MSI is unique in capturing individual cells, cellular interactions, and the chemical microenvironment without requiring the dissociation of tissues.
Previous techniques using 2D static images to visualize MERCs have been described (Giacomello and Pellegrini, 2016). Whereas 2D static images provide lower quality images of MERC interactions, SBF-SEM provides 3D visualization of MERC interactions but not live images of ER and mitochondrial dynamics. Similarly, whereas immunogold labeling provides a snapshot of the location of a protein in a specific area of the electron micrograph, live light microscopy in contrast shows real-time changes in cristae and MERC dynamics at high resolution. Live light microscopy, however, requires fluorophores that often produce photobleaching and poor image capture. The combination of electron and light microscopy—also called correlative light and electron microcopy (CLEM)—is powerful but is limited by the number of fluorophores and a requirement of imaging at low temperatures. The CLEM workflow for 3D reconstruction of images provides insight into spatial relationships and specific proteins. However, CLEM permits imaging of only a few proteins at one time. Imaging technologies that can capture metabolic flux across organelles are still lacking.
The tissue microenvironment comprises heterogeneous populations of cells through which metabolites flow, interact, and shape the cellular biochemical milieu, and tissue function depends on nutrient supply and a sustained flux of metabolites. Mapping the intra- and inter-cellular metabolic flux may be an approach for elucidating the aging process in parenchymal cells as well as determining how the chemical environment shapes the tissue microenvironment. Such mapping will require the application of single-cell omics to sample the spatial landscape of cells and organelles to accurately describe metabolic heterogeneity. Currently there is no in vivo method to visualize protein or metabolite trafficking in real time. However, MSI images addresses this deficit by simultaneously imaging multiple proteins and metabolites and measuring the distribution of proteins by quantifying peptide markers of the protein (Hale and Cooper, 2021). Although the complex data generated from MSI analysis of protein complexes typically requires computational information processing, use of multiple labeled isotopes at once offers improvements over traditional methods by imaging the spatial distribution of the cell’s chemical microenvironment. Taken together, whereas EM-based technologies provide insight into the cellular landscape, structures, and connections between various organelles, MSI-based technologies permit visualization of biomolecules in the sampled pixel or voxel within the cell.
MSI-based single-cell omics have been used to elucidate the biochemical variations among individual cells in the tissue microenvironment or in cell culture in order to better understand the cellular landscape of immunometabolism (Jackson et al., 2020; Manzo et al., 2020; Rovira-Clave et al., 2021) and cellular dysregulation in cancer (Zhang and Vertes, 2018; Tian et al., 2021b) and microbial resistance (Tian et al., 2017b). However, there are few applications of MSI in research on aging. Starr et al. (2016) report the use of SIMS (secondary ion mass spectrometry) to assess physiological changes in the stratum corneum of photo-aged human skin. In addition to an altered spatial distribution of sterol cholesterol sulfate, there is more lignoceric acid (C24:0) and hexacosanoic acid (C26:0) in photo-aged human skin, which have not been linked previously to aging skin. These findings provide insights not only into novel aging mechanisms, but also could inform the design of both pharmaceutical and topical cosmetic products to counter photoaging. Additionally, SIMS imaging coupled with stable isotope labeling was used to trace DNA synthesis and de novo lipogenesis to measure cell proliferation and lipid turnover. Imaging data show an age-dependent decline in new adipocyte generation and adipocyte lipid turnover likely mediated by an age-related decline in insulin-like growth factor-1 (IGF-1) (Guillermier et al., 2017).
Although studies of aging using MSI demonstrate chemical variations across individual cells, the integration of different omics in single cells for a comprehensive multilevel understanding of the molecular mechanism is challenging. Recently, improved single cell resolution has been achieved by improved MSI methods and workflows. For example, with MALDI, improved laser focus, matrix application, and molecular ionization have made the combination of metabolomics and proteomics possible in single cells (Rubakhin et al., 2003; Zavalin et al., 2015; Kompauer et al., 2017; Wang et al., 2022). Thus, MSI is an analytical tool that can image multiple untagged biomolecules to monitor metabolic pathways originating from cellular/subcellular compartments. This technology not only improves upon existing approaches but leverages substantial spatial resolution to interrogate challenging questions in the field of aging, including: 1) the intraorganellar locale of suppressed de novo lipogenesis observed during ER stress in aging tissues (Ward et al., 2022); 2) mitochondrial ATP dynamics in senescent cells (Depaoli et al., 2018); 3) metabolic fluxes through multiple single-cell types in tumor tissue (Jeong et al., 2017); and 4) the subcellular localization of lipid storage in various metabolic diseases (Kim et al., 2016). The value of MSI rests in the layers of biochemical data that can be mined with precision from individual cells in tissue without the need for tissue dissociation. MSI offers a comprehensive view of aging on the cellular and subcellular level with the potential to generate new hypotheses and identify novel therapeutic targets. The failure to achieve sufficient molecular coverage, spatial resolution, and non-cryogenic analysis has hindered the application of spatial lipidomics/metabolomics at subcellular resolution. Other challenges that all MSI tools face include metabolic isomer separation, quantification within complex biomatrices, protein imaging, low-concentration biomolecules. The development in the technology and methodology have been adapted to solve the issues, such as integration of ion mobility for isomer imaging (McLean et al., 2007; Zandkarimi et al., 2019), external standards for quantification calibration (Landgraf et al., 2011), novel desorption sources (e.g., LESA and GCIB) for visualization of protein and trace molecules (Sarsby et al., 2015; Sheraz et al., 2019; Tian et al., 2021a). MSI can also be used to understand general disease states in MERCs and how the complex metabolomic and protein makeup of MERCs is altered during pathogenesis.
In the following sections, we will consider the role of the mitochondria, the ER, MERCs, and the metabolic changes that occur in these structures in various diseases associated with aging. We propose that MSI can provide lipidomics/metabolomics information at a subcellular level to better understand the role that metabolic changes in the mitochondria, the ER, and MERCs play in the development of these diseases.
Exploring diseases of aging at the organellar level: Outstanding questions and opportunities
Neurodegenerative disorders
Diseases of aging occur from oxidative stress, mitochondrial mutations, misfolded proteins, or failure of defective organelles to turn over (Moltedo et al., 2019); therefore, the ER and ER contact sites are potentially important targets of aging. ER stress, leading to the disruption of MERCs, is observed in models of Alzheimer’s disease and Parkinson’s disease (Remondelli and Renna, 2017) and is a hallmark of other neurodegenerative disorders, including Huntington’s disease and amyotrophic lateral sclerosis (ALS). Production of the β-amyloid peptide in Alzheimer’s disease, which is processed to the amyloid precursor protein by γ-secretase, increases the physical interactions of MERC sites and augments Ca2+ shuttling between the ER and mitochondria (Zampese et al., 2011). Likewise, mutant forms of α-synuclein and parkin that are associated with the pathophysiology of Parkinson’s disease compromise Ca2+ transport across MERCs and lead to increased autophagy and diminished mitochondrial dynamics, respectively (Calì et al., 2012; Calì et al., 2013). ROS and apoptotic signals are regulated by the MERC tether PERK (Verfaillie et al., 2012), which when upregulated in Alzheimer’s disease and Parkinson’s disease likely results in increased cell death and is correlated clinically with memory loss (Ma et al., 2013). Interestingly, inhibition of PERK reduces MFN-2 contact with the ER and alleviates ER stress (Muñoz et al., 2013; Celardo et al., 2016). Visualizing the locale of proteins associated with ER stress may provide insight into mechanisms of neurodegeneration and inform therapeutic strategies for the treatment of neurodegenerative disorders.
Maintaining Ca2+ homeostasis is also a strategy to alleviate ER stress in neurons. Despite limited in vivo studies in models of neurodegenerative diseases, Drosophila studies show that targeting pro-fusion MERC proteins critical to Ca2+ homeostasis in mitochondria (e.g., PINK1) promotes the survival of neurons (Lee et al., 2018). Recent studies in dopamine neurons, which are targeted in Parkinson’s disease, suggest that ER increases its levels of NADH and NADPH in response to metabolic stress in mitochondria. This apparent crosstalk between stress-responsive organelles suggests a coordinated response involving the sensing and shuttling of redox molecules across MERCs (Tucker et al., 2016). An example of a redox-sensitive response shared across MERCs is observed in MERCs of fibroblasts derived from individuals with ALS with a complex I deficiency. In these cells, increased NADH/NAD+ ratios in mitochondria appear to activate the UPR in the ER, suggesting that metabolic remodeling of redox-sensitive molecules triggers a stress response across organelle contact sites (Straub et al., 2021). Motor neurons affected by ALS, unlike fibroblasts, may be less responsive to an adaptive stress response, given the differences in nutrient uptake between fibroblasts and neurons. Metabolic remodeling might be captured in real time through spatial resolution by MSI and provide further elucidation of the ER stress response across different cell types.
Astrocytes with a high metabolism react to the stress of inflammation or neuronal injury by upregulating Ca2+ signaling, which can be a predictor of disease severity (Shigetomi et al., 2019). Ca2+ is mobilized by the activation of the ryanodine receptor in the ER by cADPR produced by CD38-mediated breakdown of NAD+ (Yamasaki et al., 2005). Increased Ca2+ flux results in greater ATP production and mitochondrial antioxidant potential (Park et al., 2021). The role of NAD(H) and NADP(H) in MERCs is not well understood; therefore, determining the location and trafficking of these molecules across MERCs using a spatial omics approach may clarify the role of redox sensitive pyridine nucleotides in homeostasis and disease. The voltage-dependent anion channel (VDAC) interaction with IP3R and deglycase (DJ-1), which is regulated by glucose-related protein 75 (GRP-75) (Basso et al., 2020), is critical to Ca2+ homeostasis in MERCs. While the importance of the tethering of MERCs for Ca2+ homeostasis is known, the presence of DJ-1 has only recently been discovered (Liu et al., 2019; Basso et al., 2020). MSI could provide an estimation of the relative abundance of specific proteins in diseases.
Chronic metabolic disorders
Metabolic syndrome is a constellation of cardiometabolic risk factors that include obesity, insulin resistance, dyslipidemia, and hypertension (Moltedo et al., 2019). During metabolic homeostasis, the ER and mitochondria serve as nutrient sensors that modify the distance of MERC sites to adapt to changes in nutrient type and availability, also known as metabolic flexibility (Sood et al., 2014). Loss of this adaptive capacity compromises metabolic homeostasis leading to chronic metabolic diseases (Galgani et al., 2008). Dysregulation of MERCs leads to inter-organellar miscommunication resulting in mitochondrial Ca2+ overload, impaired insulin signaling by ER stress, and lipid dysregulation and may play a yet determined key role in metabolic homeostasis (Morgado-Cáceres et al., 2022). Investigating the molecular mechanisms of metabolic flexibility using a combination of imaging and omics through MSI may improve upon approaches that have been taken historically to measure physiological adaptability (Bret Goodpaster and Sparks, 2017).
Insulin resistance (IR) is perhaps the best-characterized example of miscommunication between the ER and mitochondria (Arruda et al., 2014; Rieusset et al., 2015; Theurey et al., 2016; Rieusset, 2018; Dia et al., 2022). IR or impaired insulin sensitivity results in decreased glucose uptake by cells in type II diabetes and obesity and can be attributed to mitochondrial dysfunction leading to the release of Ca2+ from the ER and ER stress (Lim et al., 2009) and dysregulation of MERCs in response to palmitate, high fat, or high sucrose (Tubbs et al., 2014; Tubbs et al., 2018a; Tubbs et al., 2018b; Beaulant et al., 2022) in the diet. Several proteins that reside in the MERCs may be involved in obesity-induced IR including MFN2 (Sebastián et al., 2012; Gan et al., 2013), IP3R1 (Arruda et al., 2014), PACS2 (Arruda et al., 2014), PDK4 (Thoudam et al., 2019) as well as many other proteins. It has been proposed by Arruda et al. (2014), that increased MAM formation drives several processes that lead to impaired insulin action and aberrant glucose metabolism. Together these finding suggest that disruption of MERCs and resident MAM proteins, are important for understanding organelle dysfunction and possibility that restoring MERCs proper function may be useful for future therapeutics (Rieusset, 2018).
In addition to regulating insulin, MERCs affect enzymes that regulate lipid metabolism (Cheng et al., 2020). Phospholipid exchange between ER and mitochondria requires intact MERCs and is required to synthesize phosphatidylserine (PS), phosphatidylethanolamine (PE), and phosphatidylcholine (PC) (Szymański et al., 2017). An increase in the ratio of PC:PE inhibits ER Ca2+ transport and induces ER stress in a murine obesity model (Fu et al., 2011). The localization of PS synthesis at contact sites and the shuttling of PS from the ER to the mitochondria by MFN2, which is the rate-limiting step in PE synthesis, provides the optimum PC:PE ratio for cell function. An altered ratio disrupts cell membrane integrity and potentiates steatohepatitis (Li et al., 2006). In mice, the accumulation of fat in the liver decreases NAD+, triggers mitochondrial dysfunction, and curtails ATP production (Gariani et al., 2016). Supplementation with the NAD+ precursor nicotinamide riboside (NR) reverses fatty liver disease in mice through SIRT1-and SIRT3-dependent UPR in mitochondria, suggesting that boosting NAD+ could reverse mitochondrial dysfunction (Gariani et al., 2016). The ability to simultaneously visualize MERC distance and measure flux of phospholipids from ER to mitochondria using MSI may enhance understanding of IR at an organellar level.
Muscle disorders
Skeletal muscle responds to a continuum of stressors that challenge metabolic homeostasis and either forge an adaptive change or result in pathogenesis. Evidence is mounting in support of a role for contact sites between mitochondria and the sarcoplasmic reticulum (SR), a specialized form of ER charged with Ca2+ storage, Ca2+ mobilization, and signaling. Disrupted transport of Ca2+ across MERCs has emerged as an important feature of muscle disorders. As a contractile tissue, skeletal muscle relies upon availability of ATP, which requires local transport of Ca2+ between SR and the mitochondria contacts. Ca2+ is required in oxidative metabolism and acts as a cofactor of TCA enzymes (e.g., isocitrate dehydrogenase, a-ketoglutarate dehydrogenase, pyruvate dehydrogenase) and synthesis of reducing equivalents (e.g., NADH, FADH2). Whereas IP3R (IP3R1, IP3R2, IP3R3) modulates release of Ca2+ stored in the ER/SR, VDACs mediates the flux of Ca2+ into mitochondria (Mikoshiba, 2015; Bartok et al., 2019; Fan et al., 2022; Schmitz et al., 2022). An additional MAM-enriched protein called SEPN1 has been identified at the ER/SR interface (Filipe et al., 2021). SEPN1 appears to enhance ER/mitochondria contacts, calcium flux, and oxidative phosphorylation and its dysregulation has been implicated in severe muscle weakness. The movement of calcium from a storage organelle to an energy producing organelle is critical to cellular metabolic homeostasis and interrogation of this process is likely to shed greater light on mechanisms of muscle disorders.
While Ca2+ flux into the mitochondria is essential for normal metabolism, an overload of Ca2+ activates the mitochondrial permeability transition pore, resulting in mitochondrial swelling and the dissemination of apoptotic signals (Zhou et al., 2022). To counter the problem of Ca2+ toxicity leading to cell death, the tethering protein MFN2, which is localized to the outer mitochondrial membrane and ER surface, protects mitochondria by creating space within MERCs (Filadi et al., 2015). de Brito and Scorrano (2008) demonstrated MFN2 to be a bonafide tether in MERCs. Conversely, this discovery was challenged (Filadi et al., 2015) by Filadi et al.; however, Naon et al. (2016), reconfirmed MFN2 as a MERC tether by providing evidence that Mfn2 ablation increases MERC distance (Sebastián et al., 2012). Additionally, Naon et al. (2016) showed that MFN2 ablation causes reduction of mitochondrial Ca2+ uptake but doesn’t affect mitochondrial Ca2+ uniporter complex, further confirming MFN2 as a critical tether important for MERC distance. Moreover, MFN2 has a critical role in glucose homeostasis and insulin signaling in skeletal muscle (Sebastián et al., 2012). This suggests that MFN2 in skeletal muscle is an important regulator of mitochondrial dynamics and MERCs. MFN2 dimerizes with itself on the outer mitochondrial membrane but also interacts with MFN1 on the inner mitochondria membrane allowing for interaction with inner membrane fusion proteins such as OPA1. OPA1 functionally requires MFN1 further confirming the distinct roles between the two proteins (Sood et al., 2014). Interestingly, ablation of OPA1 induces ER stress, which in turn upregulates MFN2, as a key MERC tethering protein (Pereira et al., 2017). This suggests that as a possible compensatory mechanism for MFN2 upregulation during loss of OPA1 in skeletal muscle. MERC ultrastructure may also contribute to the pathophysiology of certain muscle disorders. For example, MERCs decrease after endurance exercise (Merle et al., 2019) but increase in aging (Marzetti et al., 2016; Sebastián et al., 2016; Picca et al., 2017), suggesting that the ultrastructure of MERCs in muscle may respond differentially to stressors.
Although MERCs play a role in some muscle myopathies, their function is less clear in sarcopenia, a myopathy in the elderly that is accompanied by a progressive increase in body fat (Hsu et al., 2019) and risk of metabolic syndrome (Umegaki, 2015). Several muscle disorders have been linked to a failure of Ca2+ regulation by MERCs, including Duchenne muscular dystrophy (DMD) and sarcopenia. DMD results from the defective cytoskeletal protein dystrophin. In dystrophin-deficient muscle, Ca2+ homeostasis is dysregulated, which results in aberrant Ca2+ signaling and increased ROS production (Cully and Rodney, 2020). Similarly, mutations in ryanodine receptor (RyR1) lead to severe muscle myopathies, decreasing SR Ca2+ amplitudes (Lee et al., 2017). Such ryanodine receptors are important for Ca2+ leakage and mishandling, playing a major role in diseases such as DMD (Meyer et al., 2021). Ca2+ levels in dystrophin-deficient skeletal muscle are regulated by the SR membrane-bound ryanodine receptor, which promotes Ca2+ release (Meyer et al., 2021), and the SR membrane-bound Ca2+ salvaging proteins SERCA1 and SERCA2a, which in dystrophin deficiency fail to scavenge excess Ca2+ (Schneider et al., 2013; Voit et al., 2017).
Ca2+ dysregulation can lead to oxidative stress and ROS production. To counter ROS, antioxidant enzymes rely on NADPH as a source of reducing equivalents. In muscle SR, hexose-6-phosphate dehydrogenase (H6PD) reduces NADP+ to produce the NADPH needed for steroidogenesis, and H6PD-deficient skeletal muscle exhibits a myopathy characterized by abnormal SR structure (Lavery et al., 2006; Rogoff et al., 2010), activation of stress-induced UPR (Bujalska IJ et al., 2008), and dysregulation of Ca2+ metabolism (Ying, 2006). Therefore, H6PD is important not only for steroidogenesis but also for normal ER/SR redox balance. H6PD knockout mice upregulate NRK2-mediated biosynthesis of NAD+ from NR in response to dysregulated NADPH homeostasis in muscle SR and impaired metabolism in mitochondria (Doig et al., 2020). Curiously, NR supplementation of H6PD knockout mice fails to reverse the muscle myopathy phenotype, despite improvement following NR supplementation in a mouse model of mitochondrial myopathy (Khan et al., 2014). The role of H6PD modulation of NADPH in whole-cell redox and metabolism remains to be elucidated and may be visualized and quantified by MSI.
The changes in insulin signaling, loss of proteostasis, and inflammation characteristic of sarcopenia are likely mediated by mitochondrial dysfunction (Coen PM et al., 2019; Ferri et al., 2020). Mitochondrial dysfunction leads to: 1) loss of mitochondrial quality control (Joseph et al., 2013); 2) increased activity of the ryanodine Ca2+ channel receptor, resulting in Ca2+ overload and mitochondria permeability transition pore activation (Andersson et al., 2011); 3) Ca2+-induced decreased proteolysis and the formation of toxic proteins (Baehr et al., 2016); and 4) ROS leakage due to mitochondrial dysfunction (Shally and McDonagh, 2020). These findings support a role for MERCs in the pathogenesis of sarcopenia (Sebastián et al., 2016; Wiedmer et al., 2021). However, we have a very limited understanding of sarcopenia development, due to lack of appropriate animal models and the difficulty in separating “pure sarcopenia” from other age-related comorbidities (Christian CJ and Benian, 2020).
Atherosclerosis
The pathogenesis of the age-related development of atherosclerosis (Finney et al., 2017), an inflammatory disease of the arteries leading to fibrosis, clotting, and increased cardiovascular risk, can be tied to dysfunction of mitochondria-associated ER membrane formation in smooth muscle (Bennett et al., 2016; Perrotta, 2020; Wang et al., 2021) and endothelial cells (Nguyen et al., 2012). One mechanism of MERC-mediated atherosclerosis depends on the tethering protein PACS2 to transfer Ca2+ from the ER to mitochondria, resulting in Ca2+ overload, loss of mitochondrial membrane potential, increased production of ROS, the release of cytochrome c, and, eventually, apoptosis (Moulis et al., 2019; Yu et al., 2019). Oxidized low-density lipoprotein also induces inflammation in endothelial cells, thereby recruiting the NLRP3 inflammasome to contact sites with the activation of vascular remodeling (Zeisbrich et al., 2018). The NLRP3 inflammasome is also activated by ATP, ROS, ER stress, Ca2+ signaling, and mitochondrial dysfunction (Strowig et al., 2012; Chen and Chen, 2018). Ca2+ mobilization via the cell-surface type II transmembrane glycoprotein CD38 is critical for the activation of the NLRP3 inflammasome (Li et al., 2020). CD38 is a NADase that metabolizes nicotinamide nucleotides (NAD+ and NMN) to ADPR and cADPR (Hogan et al., 2019). cADPR mobilizes Ca2+ by activating the ryanodine receptor in SR (Venturi et al., 2012). Recent data in a diabetic mouse model demonstrate that CD38-mediated Ca2+ signaling leads to mitochondrial damage, the release of mitochondrial DNA, and activation of NLRP3 inflammasome in vascular smooth muscle (Li et al., 2020). The tissue specific locale of CD38-mediated ADPR and cADPR production and subsequent Ca2+ mobilization are likely candidates for spatially resolved omics available through MSI. To this end, the NAD+ consumer CD38 has potential as a therapeutic target in atherosclerosis.
Perspectives
MSI is an analytical tool that has the capability to image multiple untagged biomolecules for the purpose of monitoring metabolic processes originating from cellular/subcellular compartments. This technology not only improves upon existing approaches, but leverages its substantial spatial resolution to interrogate challenging questions in the field of aging including: the intraorganellar locale of suppressed de novo lipogenesis observed during ER stress in aging tissues (Ward et al., 2022); mitochondrial ATP dynamics in senescent cells (Depaoli et al., 2018); metabolic fluxes through multiple single cell types found in tumor tissue (Jeong et al., 2017); and the subcellular localization of lipid storage in various metabolic diseases (Kim et al., 2016). The merit of MSI lies in the layered biochemical data that can be mined with precision from individual cells in tissue without the need for tissue dissociation. MSI offers a comprehensive view of aging on the cellular and sub-cellular level with the potential to generate new hypotheses and identify novel therapeutic targets.
Author contributions
MM, KH, AH, and HT conceived the project. KH, JZ, and HB performed and analyzed literature analysis. AIA, AAA, and Y-CC, contributed to analyses and background search. KH, JZ, and HB wrote the manuscript with input from all authors. MM, HT, and AH edited the article with editing input from KH, HB, and JZ.
Acknowledgments
The authors wish to acknowledge the generous support from the Howard Hughes Medical Institute Hanna H. Gray Fellows Program Faculty Phase (Grant# GT15655 awarded to MM) and Burroughs Wellcome Fund PDEP Transition to Faculty (Grant# 1022604 awarded to MM). The UNCF/Bristol-Myers Squibb (UNCF/BMS)-E.E. Just Postgradaute Fellowship in Life Sciences Fellowship (HB) and Burroughs Wellcome Fund PDEP award (HB), the UNCF/Bristol-Myers Squibb (UNCF/BMS)-E.E. Just Faculty Fund (AH), Burroughs Wellcome Fund Career Awards at the Scientific Interface (AH), Burroughs Wellcome Fund Ad-Hoc Award (AH), NIH SRP Subaward to #5R25HL106365-12 from the NIH PRIDE Program (AH), the DRTC grant: DK020593, Vanderbilt Diabetes and Research Training Center for DRTC Alzheimer’s Disease Pilot and Feasibility Program (AH). CZI Science Diversity Leadership grant number 2022-253529 from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation (to AH).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andersson, D. C., Betzenhauser, M. J., Reiken, S., Meli, A. C., Umanskaya, A., Xie, W., et al. (2011). Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell. Metab. 14, 196–207. doi:10.1016/j.cmet.2011.05.014
Arruda, A. P., Pers, B. M., Parlakgul, G., Guney, E., Inouye, K., and Hotamisligil, G. S. (2014). Chronic enrichment of hepatic endoplasmic reticulum–mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 20, 1427–1435. doi:10.1038/nm.3735
Artyom Baev, A. Y. V., Novikova, Irina N., ViktorDremin, V., ElenaPotapova, V., Andrey, Y., and Abramov, A. Y. (2022). Interaction of mitochondrial calcium and ROS in neurodegeneration. Cells 11, 706. doi:10.3390/cells11040706
Ashley Frakes, A. D., and Dillin, A. (2017). The UPR ER: Sensor and coordinator of organismal homeostasis. Mol. Cell. 66, 761–771. doi:10.1016/j.molcel.2017.05.031
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. doi:10.1126/science.aac4354
Baehr, L. M., West, D. W. D., Marcotte, G., Marshall, A. G., De Sousa, L. G., Baar, K., et al. (2016). Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 8, 127–146. doi:10.18632/aging.100879
Bai, D., Peng, J., and Yi, C. (2021). Advances in single-cell multi-omics profiling. RSC Chem. Biol. 2, 441–449. doi:10.1039/D0CB00163E
Barazzuol, L., Giamogante, F., and Calì, T. (2021). Mitochondria associated membranes (MAMs): Architecture and physiopathological role. Cell. Calcium 94, 102343. doi:10.1016/j.ceca.2020.102343
Bartok, A., Weaver, D., Golenar, T., Nichtova, Z., Katona, M., Bansaghi, S., et al. (2019). IP(3) receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 10, 3726. doi:10.1038/s41467-019-11646-3
Basso, V., Marchesan, E., and Ziviani, E. (2020). A trio has turned into a quartet: DJ-1 interacts with the IP3R-grp75-VDAC complex to control ER-mitochondria interaction. Cell. Calcium 87, 102186. doi:10.1016/j.ceca.2020.102186
Beaulant, D. M., Pillot, B., Chauvin, M. A., Ji-Cao, J., Durand, C., Bendridi, N., et al. (2022). Endoplasmic reticulum-mitochondria miscommunication is an early and causal trigger of hepatic insulin resistance and steatosis. J. Hepatol. 77, 710–722. Epub 2022 Mar 28. PMID: 35358616 (2022). doi:10.1016/j.jhep.2022.03.017
Bennett, M. R., Sinha, S., and Owens, G. K. (2016). Vascular smooth muscle cells in atherosclerosis. Circ. Res. 118, 692–702. doi:10.1161/circresaha.115.306361
Blum, B. C., Mousavi, F., and Emili, A. (2018). Single-platform "multi-omic' profiling: Unified mass spectrometry and computational workflows for integrative proteomics-metabolomics analysis. Mol. Omics 14, 307–319. doi:10.1039/c8mo00136g
Bret Goodpaster, L. M. S., and Sparks, L. M. (2017). Metabolic flexibility in health and disease. Cell. Metab. 25, 1027–1036. doi:10.1016/j.cmet.2017.04.015
Brian Glancy, R. S. B., and Balaban, R. S. (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. doi:10.1021/bi2018909
Bujalska Ij, H. K., Hauton, D., Lavery, G. G., Tomlinson, J. W., Walker, E. A., Stewart, P. M., et al. (2008). Lack of hexose-6-phosphate dehydrogenase impairs lipid mobilization from mouse adipose tissue. Endocrinology 149, 2584–2591. doi:10.1210/en.2007-1705
Burgos-Morón, E., Abad-Jimenez, Z., Maranon, A. M. d., Iannantuoni, F., Escribano-Lopez, I., Lopez-Domenech, S., et al. (2019). Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 8, 1385. doi:10.3390/jcm8091385
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2013). Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508. doi:10.1016/j.bbadis.2013.01.004
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2012). α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 287, 17914–17929. doi:10.1074/jbc.M111.302794
Camacho-Pereira, J., Tarrago, M. G., Chini, C. C. S., Nin, V., Escande, C., Warner, G. M., et al. (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell. Metab. 23, 1127–1139. doi:10.1016/j.cmet.2016.05.006
Carlos López-Otín, M. A. B., Partridge, Ml S., Guido, K., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell. 153, 1194–1217. doi:10.1016/j.cell.2013.05.039
Celardo, I., Costa, A. C., Lehmann, S., Jones, C., Wood, N., Mencacci, N. E., et al. (2016). Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson's disease. Cell. Death Dis. 7, e2271. doi:10.1038/cddis.2016.173
Chen, J. J., and Yu, B. P. (1994). Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic. Biol. Med. 17, 411–418. doi:10.1016/0891-5849(94)90167-8
Chen, J., and Chen, Z. J. (2018). PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76. doi:10.1038/s41586-018-0761-3
Cheng, H., Gang, X., He, G., Liu, Y., Wang, Y., Zhao, X., et al. (2020). The molecular mechanisms underlying mitochondria-associated endoplasmic reticulum membrane-induced insulin resistance. Front. Endocrinol. (Lausanne) 11, 592129. doi:10.3389/fendo.2020.592129
Chini, C. C. S., Peclat, T. R., Warner, G. M., Kashyap, S., Espindola-Netto, J. M., de Oliveira, G. C., et al. (2020). CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2, 1284–1304. doi:10.1038/s42255-020-00298-z
Christian Cj, B. G., and Benian, G. M. (2020). Animal models of sarcopenia. Aging Cell. 10, e13223. doi:10.1111/acel.13223
Churchill, G. C., and Galione, A. (2001). NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. Embo J. 20, 2666–2671. doi:10.1093/emboj/20.11.2666
Clara Correia-Melo, F. D. M. M., Anderson, R., Hewitt, G., Hewitt, R., Cole, J., Carroll, B. M., et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742. doi:10.15252/embj.201592862
Coen Pm, M. R., Hinkley, J. M., and Miller, B. F. (2019). Mitochondria as a target for mitigating sarcopenia. Front. Physiol. 9, 1883. doi:10.3389/fphys.2018.01883
Cohen, S., Valm, A. M., and Lippincott-Schwartz, J. (2018). Interacting organelles. Curr. Opin. Cell. Biol. 53, 84–91. doi:10.1016/j.ceb.2018.06.003
Covarrubias, A. J., Kale, A., Perrone, R., Lopez-Dominguez, J. A., Pisco, A. O., Kasler, H. G., et al. (2020). Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2, 1265–1283. doi:10.1038/s42255-020-00305-3
Covarrubias, A. J., Perrone, R., Grozio, A., and Verdin, E. (2021). NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell. Biol. 22, 119–141. doi:10.1038/s41580-020-00313-x
Cristina Menni, G. K., Petersen, A. K., Bell, J. T., Psatha, M., Tsai, P. C., Gieger, C., et al. (2013). Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int. J. Epidemiol. 42, 1111–1119. doi:10.1093/ije/dyt094
Csordás, G., Weaver, D., and Hajnóczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: Structure and signaling functions. Trends Cell. Biol. 28, 523–540. doi:10.1016/j.tcb.2018.02.009
Cully, T. R., and Rodney, G. G. (2020). Nox4 - RyR1 - nox2: Regulators of micro-domain signaling in skeletal muscle. Redox Biol. 36, 101557. doi:10.1016/j.redox.2020.101557
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi:10.1038/nature07534
Denoyelle, C., Abou-Rjaily, G., Bezrookove, V., Verhaegen, M., Johnson, T. M., Fullen, D. R., et al. (2006). Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell. Biol. 8, 1053–1063. doi:10.1038/ncb1471
Depaoli, M. R., Karsten, F., Madreiter-Sokolowski, C. T., Klec, C., Gottschalk, B., Bischof, H., et al. (2018). Real-time imaging of mitochondrial ATP dynamics reveals the metabolic setting of single cells. Cell. Rep. 25, 501–512. doi:10.1016/j.celrep.2018.09.027
Dia, M. L. C., Chanon, S., Bendridi, N., Gomez, L., Rieusset, J., Thibault, H., et al. (2022). Effect of metformin on T2D-induced MAM Ca2+ uncoupling and contractile dysfunction in an early mouse model of diabetic HFpEF. Int. J. Mol. Sci. 23, 3569. doi:10.3390/ijms23073569
Doig, C. L., Zielinska, A. E., Fletcher, R. S., Oakey, L. A., Elhassan, Y. S., Garten, A., et al. (2020). Induction of the nicotinamide riboside kinase NAD(+) salvage pathway in a model of sarcoplasmic reticulum dysfunction. Skelet. Muscle 10, 5. doi:10.1186/s13395-019-0216-z
Dörr, J. R., Yu, Y., Milanovic, M., Beuster, G., Zasada, C., Dabritz, J. H. M., et al. (2013). Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425. doi:10.1038/nature12437
Elaine MacKenzie, M. A. S., Tennant, D. A., Payne, L. J., Crosby, S., Frederiksen, C. M., Watson, D. G., et al. (2007). Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 27, 3282–3289. doi:10.1128/MCB.01927-06
Emad Ahmed, A. R.-W., Roepstorff, P., Bulteau, A. L., and Friguet, B. (2010). Protein modification and replicative senescence of WI-38 human embryonic fibroblasts. Aging Cell. 9, 252–272. doi:10.1111/j.1474-9726.2010.00555.x
Emma, L., James, R. D. M., Pitiyage, G. N., M de Castro, A., Vignola, K., Jones, J., et al. (2015). Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 14, 1854–1871. doi:10.1021/pr501221g
Eric Letouzé, C. M., Martinelli, C., Loriot, C., Burnichon, N., Abermil, N., Ottolenghi, C., et al. (2013). SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 23, 739–752. doi:10.1016/j.ccr.2013.04.018
Esther Phielix, J. S., and Roden., M. (2011). Mitochondrial function and insulin resistance during aging: A mini-review. Gerontology 57, 387–396. doi:10.1159/000317691
Fan, G., Baker, M. R., Terry, L. E., Arige, V., Chen, M., Seryshev, A. B., et al. (2022). Conformational motions and ligand-binding underlying gating and regulation in IP3R channel. Nat. Commun. 13, 6942. doi:10.1038/s41467-022-34574-1
Ferri, E., Marzetti, E., Calvani, R., Picca, A., Cesari, M., and Arosio, B. (2020). Role of age-related mitochondrial dysfunction in sarcopenia. Int. J. Mol. Sci. 21, 5236. doi:10.3390/ijms21155236
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U. S. A. 112, E2174–E2181. doi:10.1073/pnas.1504880112
Filipe, A., Chernorudskiy, A., Arbogast, S., Varone, E., Villar-Quiles, R. N., Pozzer, D., et al. (2021). Defective endoplasmic reticulum-mitochondria contacts and bioenergetics in SEPN1-related myopathy. Cell. Death Differ. 28, 123–138. doi:10.1038/s41418-020-0587-z
Fink, B. D., Bai, F., Yu, L., and Sivitz, W. I. (2017). Regulation of ATP production: Dependence on calcium concentration and respiratory state. Am. J. Physiol. Cell. Physiol. 313, C146–c153. doi:10.1152/ajpcell.00086.2017
Finney, A. C., Stokes, K. Y., Pattillo, C. B., and Orr, A. W. (2017). Integrin signaling in atherosclerosis. Cell. Mol. Life Sci. 74, 2263–2282. doi:10.1007/s00018-017-2490-4
Fletcher, J. S., Rabbani, S., Henderson, A., Blenkinsopp, P., Thompson, S. P., Lockyer, N. P., et al. (2008). A new dynamic in mass spectral imaging of single biological cells. Anal. Chem. 80, 9058–9064. doi:10.1021/ac8015278
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C., and Santoro, A. (2018). Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590. doi:10.1038/s41574-018-0059-4
Fu, S., Yang, L., Li, P., Hofmann, O., Dicker, L., Hide, W., et al. (2011). Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531. doi:10.1038/nature09968
Galgani, J. E., Moro, C., and Ravussin, E. (2008). Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 295, E1009–E1017. doi:10.1152/ajpendo.90558.2008
Gan, K. X., Wang, C., Chen, J. H., Zhu, C. J., and Song, G. Y. (2013). Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J. Gastroenterol. 19, 1572–1581. doi:10.3748/wjg.v19.i10.1572
Gansemer, E. R., and Rutkowski, D. T. (2022). Pathways linking nicotinamide adenine dinucleotide phosphate production to endoplasmic reticulum protein oxidation and stress. Front. Mol. Biosci. 9, 858142. doi:10.3389/fmolb.2022.858142
Gariani, K., Menzies, K. J., Ryu, D., Wegner, C. J., Wang, X., Ropelle, E. R., et al. (2016). Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 63, 1190–1204. doi:10.1002/hep.28245
Garza-Lopez, E., Vue, Z., Katti, P., Neikirk, K., Biete, M., Lam, J., et al. (2021). Protocols for generating surfaces and measuring 3D organelle morphology using amira. Cells 11, 65. doi:10.3390/cells11010065
Gavilán, M. P., Castaño, A., Ramos, B., Río, J. C. d., Vitorica, J., Ruano, D., et al. (2006). Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 27, 973–982. doi:10.1016/j.neurobiolaging.2005.05.010
Giacomello, M., and Pellegrini, L. (2016). The coming of age of the mitochondria-ER contact: A matter of thickness. Cell. Death Differ. 23, 1417–1427. doi:10.1038/cdd.2016.52
Gilmore, I. S., Heiles, S., and Pieterse, C. L. (2019). Metabolic imaging at the single-cell scale: Recent advances in mass spectrometry imaging. Annu. Rev. Anal. Chem. (Palo Alto, Calif.) 12, 201–224. doi:10.1146/annurev-anchem-061318-115516
Girardi, E., Agrimi, G., Goldmann, U., Fiume, G., Lindinger, S., Sedlyarov, V., et al. (2020). Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 11, 6145. doi:10.1038/s41467-020-19871-x
Gomes, A. P., Price, N. L., Ling, A. J. Y., Moslehi, J. J., Montgomery, M. K., Rajman, L., et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 155, 1624–1638. doi:10.1016/j.cell.2013.11.037
Gommans, I. M., Vlak, M. H., de Haan, A., and van Engelen, B. G. (2002). Calcium regulation and muscle disease. J. Muscle Res. Cell. Motil. 23, 59–63. doi:10.1023/a:1019984714528
Gorman, A. M., Healy, S. J., Jäger, R., and Samali, A. (2012). Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 134, 306–316. doi:10.1016/j.pharmthera.2012.02.003
Guillermier, C., Fazeli, P. K., Kim, S., Lun, M., Zuflacht, J. P., Milian, J., et al. (2017). Imaging mass spectrometry demonstrates age-related decline in human adipose plasticity. JCI insight 2, e90349. doi:10.1172/jci.insight.90349
Hale, O. J., and Cooper, H. J. (2021). Native mass spectrometry imaging of proteins and protein complexes by nano-DESI. Anal. Chem. 93, 4619–4627. doi:10.1021/acs.analchem.0c05277
Hélène Martini, J. F. P., and Passos, J. F. (2022). Cellular senescence: All roads lead to mitochondria. FEBS J. doi:10.1111/febs.16361
Hetz, C. (2012). The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 13, 89–102. doi:10.1038/nrm3270
Hogan, K. A., Chini, C. C. S., and Chini, E. N. (2019). The multi-faceted ecto-enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol. 10, 1187. doi:10.3389/fimmu.2019.01187
Hoi-Shan Wong, P. A. D., Mezera, V., Monternier, P. A., and Brand, M. D. (2017). Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809. doi:10.1074/jbc.R117.789271
Hosios, A. M., and Vander Heiden, M. G. (2018). The redox requirements of proliferating mammalian cells. J. Biol. Chem. 293, 7490–7498. doi:10.1074/jbc.TM117.000239
Hsu, K. J., Liao, C. D., Tsai, M. W., and Chen, C. N. (2019). Effects of exercise and nutritional intervention on body composition, metabolic health, and physical performance in adults with sarcopenic obesity: A meta-analysis. Nutrients 11, 2163. doi:10.3390/nu11092163
Ian Lanza, K. S. N., and Nair, K. S. (2010). Mitochondrial function as a determinant of life span. Pflugers Arch. 459, 277–289. doi:10.1007/s00424-009-0724-5
Ihab Hajjar, J. M. K., and Kotchen, T. A. (2006). Hypertension: Trends in prevalence, incidence, and control. Annu. Rev. Public Health 27, 465–490. doi:10.1146/annurev.publhealth.27.021405.102132
Inmaculada Martínez-Reyes, N. S. C., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102. doi:10.1038/s41467-019-13668-3
Jackson, H. W., Fischer, J. R., Zanotelli, V. R. T., Ali, H. R., Mechera, R., Soysal, S. D., et al. (2020). The single-cell pathology landscape of breast cancer. Nature 578, 615–620. doi:10.1038/s41586-019-1876-x
Janikiewicz, J., Szymanski, J., Malinska, D., Patalas-Krawczyk, P., Michalska, B., Duszynski, J., et al. (2018). Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell. Death Dis. 9, 332. doi:10.1038/s41419-017-0105-5
Jeong, S., Eskandari, R., Park, S. M., Alvarrez, J., Tee, S. S., Weissleder, R., et al. (2017). Real-time quantitative analysis of metabolic flux in live cells using a hyperpolarized micromagnetic resonance spectrometer. Sci. Adv. 16, e1700341. doi:10.1126/sciadv.1700341
Joanna Kaplon, L. Z., Meissl, K., Chaneton, B., Selivanov, V. A., Mackay, G., van der Burg, S. H., et al. (2013). A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. doi:10.1038/nature12154
Joseph, A. M., Adhihetty, P. J., Wawrzyniak, N. R., Wohlgemuth, S. E., Picca, A., Kujoth, G. C., et al. (2013). Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS One 8, e69327. doi:10.1371/journal.pone.0069327
Mikoshiba, K. (2015). Role of IP3 receptor signaling in cell functions and diseases. Adv Biol Regul. 57, 217-27. doi:10.1016/j.jbior.2014.10.001
Kaul, S. C., Yaguchi, T., Taira, K., Reddel, R. R., and Wadhwa, R. (2003). Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp. Cell. Res. 286, 96–101. doi:10.1016/s0014-4827(03)00101-0
Khan, N. A., Auranen, M., Paetau, I., Pirinen, E., Euro, L., Forsstrom, S., et al. (2014). Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 6, 721–731. doi:10.1002/emmm.201403943
Kim, K., Lee, S., Yoon, J., Heo, J., Choi, C., and Park, Y. (2016). Three-dimensional label-free imaging and quantification of lipid droplets in live hepatocytes. Sci. Rep. 22, 36815. doi:10.1038/srep36815
Kirsi Auro, A. J., Fischer, K., Kettunen, J., Salo, P., Mattsson, H., Niironen, M., et al. (2014). A metabolic view on menopause and ageing. Nat. Commun. 5, 4708. doi:10.1038/ncomms5708
Kompauer, M., Heiles, S., and Spengler, B. (2017). Atmospheric pressure MALDI mass spectrometry imaging of tissues and cells at 1.4-μm lateral resolution. Nat. Methods 14, 90–96. doi:10.1038/nmeth.4071
Kory, N., de Bos, J. U., van der Rijt, S., Jankovic, N., Gura, M., Arp, N., et al. (2020). MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 6, eabe5310. doi:10.1126/sciadv.abe5310
Lam, J., Katti, P., Biete, M., Mungai, M., AshShareef, S., Neikirk, K., et al. (2021). A universal approach to analyzing transmission electron microscopy with ImageJ. Cells 10, 2177. doi:10.3390/cells10092177
Landgraf, R. R., Garrett, T. J., Conaway, M. C. P., Calcutt, N. A., Stacpoole, P. W., and Yost, R. A. (2011). Considerations for quantification of lipids in nerve tissue using matrix-assisted laser desorption/ionization mass spectrometric imaging. Rapid Commun. Mass Spectrom. 25, 3178–3184. doi:10.1002/rcm.5189
Lavery, W. E., Draper, N., Jeyasuria, P., Marcos, J., Shackleton, C. H., Parker, K. L., et al. (2006). Hexose-6-phosphate dehydrogenase knock-out mice lack 11 beta-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J. Biol. Chem. 281, 6546–6551. doi:10.1074/jbc.M512635200
Leal, N. S., Schreiner, B., Pinho, C. M., Filadi, R., Wiehager, B., Karlstrom, H., et al. (2016). Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 20, 1686–1695. doi:10.1111/jcmm.12863
Lee, C. S., Hanna, A. D., Wang, H., Dagnino-Acosta, A., Joshi, A. D., Knoblauch, M., et al. (2017). A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat. Commun. 8, 14659. doi:10.1038/ncomms14659
Lee, K. S., Huh, S., Lee, S., Wu, Z., Kim, A. K., Kang, H. Y., et al. (2018). Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. U. S. A. 115, E8844–e8853. doi:10.1073/pnas.1721136115
Li, J. P., Wei, W., Li, X. X., and Xu, M. (2020). Regulation of NLRP3 inflammasome by CD38 through cADPR-mediated Ca(2+) release in vascular smooth muscle cells in diabetic mice. Life Sci. 255, 117758. doi:10.1016/j.lfs.2020.117758
Li, Z., Agellon, L. B., Allen, T. M., Umeda, M., Jewell, L., Mason, A., et al. (2006). The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell. Metab. 3, 321–331. doi:10.1016/j.cmet.2006.03.007
Lim, J. H., Lee, H. J., Ho Jung, M., and Song, J. (2009). Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: A molecular mechanism leading to hepatic insulin resistance. Cell. Signal 21, 169–177. doi:10.1016/j.cellsig.2008.10.004
Lin, Y., Jiang, M., Chen, W., Zhao, T., and Wei, Y. (2019). Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 118, 109249. doi:10.1016/j.biopha.2019.109249
Liu, M. A., Fujioka, H., Zhu, X., Liu, J., and Chen, S. (2019). DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. PNAS 116, 25322–25328. doi:10.1073/pnas.1906565116
Luongo, T. S., Eller, J. M., Lu, M. J., Niere, M., Raith, F., Perry, C., et al. (2020). SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nat. Metab. 588, 174–179. doi:10.1038/s41586-020-2741-7
Mándi, M., Tóth, B., Timár, G., and Bak, J. (2006). Ca2+ release triggered by NAADP in hepatocyte microsomes. Biochem. J. 395, 233–238. doi:10.1042/bj20051002
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi:10.1038/nn.3486
Manzo, T., Prentice, B. M., Anderson, K. G., Raman, A., Schalck, A., Codreanu, G. S., et al. (2020). Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J. Exp. Med. 217, e20191920. doi:10.1084/jem.20191920
Maria Grazia Vizioli, T. L., Miller, K. N., Robertson, N. A., Gilroy, K., Lagnado, A. B., Perez-Garcia, A., et al. (2020). Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes. Dev. 34, 428–445. doi:10.1101/gad.331272.119
Maryam Gulshan, K. Y., Okabe, K., Mahmood, A., Sasaki, T., Yamamoto, M., Hikosaka, K., et al. (2018). Overexpression of Nmnat3 efficiently increases NAD and NGD levels and ameliorates age-associated insulin resistance. Aging Cell. 17, e12798. doi:10.1111/acel.12798
Marzetti, E., Calvani, R., Lorenzi, M., Tanganelli, F., Picca, A., Bossola, M., et al. (2016). Association between myocyte quality control signaling and sarcopenia in old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp. Gerontol. 80, 1–5. doi:10.1016/j.exger.2016.04.003
Matos, L., Gouveia, A. M., and Almeida, H. (2015). ER stress response in human cellular models of senescence. J. Gerontol. A Biol. Sci. Med. Sci. 70, 924–935. doi:10.1093/gerona/glu129
McLean, J. A., Ridenour, W. B., and Caprioli, R. M. (2007). Profiling and imaging of tissues by imaging ion mobility-mass spectrometry. J. Mass Spectrom. 42, 1099–1105. doi:10.1002/jms.1254
McReynolds, M. R., Chellappa, K., and Baur, J. A. (2020). Age-related NAD(+) decline. Exp. Gerontol. 134, 110888. doi:10.1016/j.exger.2020.110888
McReynolds, M. R., Chellappa, K., Chiles, E., Jankowski, C., Shen, Y., Chen, L., et al. (2021). NAD(+) flux is maintained in aged mice despite lower tissue concentrations. Cell. Syst. 12, 1160–1172.e4. doi:10.1016/j.cels.2021.09.001
Merle, A., Jollet, M., Britto, F. A., Goustard, B., Bendridi, N., Rieusset, J., et al. (2019). Endurance exercise decreases protein synthesis and ER-mitochondria contacts in mouse skeletal muscle. J. Appl. Physiol. 127, 1297–1306. doi:10.1152/japplphysiol.00196.2019
Meyer, N. C., Meli, A. C., Matecki, S., Hugon, G., Salvador, J., Khalil, M., et al. (2021). Skeletal ryanodine receptors are involved in impaired myogenic differentiation in Duchenne muscular dystrophy patients. Int. J. Mol. Sci. 22, 12985. doi:10.3390/ijms222312985
Minhas, P. S., Liu, L., Moon, P. K., Joshi, A. U., Dove, C., Mhatre, S., et al. (2019). Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 20, 50–63. doi:10.1038/s41590-018-0255-3
Miriam Valera-Alberni, C. C., and Canto, C. (2018). Mitochondrial stress management: A dynamic journey. Cell. Stress 2, 253–274. doi:10.15698/cst2018.10.158
Moltedo, O., Remondelli, P., and Amodio, G. (2019). The mitochondria-endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Front. Cell. Dev. Biol. 7, 172. doi:10.3389/fcell.2019.00172
Morgado-Cáceres, P. L. G., Calle, X., Briones, L., Riquelme, J. A., Bravo-Sagua, R., Parra, V., et al. (2022). The aging of ER-mitochondria communication: A journey from undifferentiated to aged cells. Front. Cell. Dev. Biol. 10, 946678. doi:10.3389/fcell.2022.946678
Mouchiroud, L. H. R., Moullan, N., Katsyuba, E., Ryu, D., Cantó, C., Mottis, A., et al. (2013). The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 154, 430–441. doi:10.1016/j.cell.2013.06.016
Moulis, M., Grousset, E., Faccini, J., Richetin, K., Thomas, G., and Vindis, C. (2019). The multifunctional sorting protein PACS-2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria-ER contact sites. Cells 8, 638. doi:10.3390/cells8060638
Muñoz, J. P., Ivanova, S., Sánchez-Wandelmer, J., Martínez-Cristóbal, P., Noguera, E., Sancho, A., et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361. doi:10.1038/emboj.2013.168
Mycielska, M. E., James, E. N., and Parkinson, E. K. (2022). Metabolic alterations in cellular senescence: The role of citrate in ageing and age-related disease. Int. J. Mol. Sci. 23, 3652. doi:10.3390/ijms23073652
Nacarelli, T., Lau, L., Fukumoto, T., Zundell, J., Fatkhutdinov, N., Wu, S., et al. (2019). NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell. Biol. 21, 397–407. doi:10.1038/s41556-019-0287-4
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. 113, 11249–11254. doi:10.1073/pnas.1606786113
Nguyen, M. T., Chen, A., Lu, W. J., Fan, W., Li, P. P., Oh, D. Y., et al. (2012). Regulation of chemokine and chemokine receptor expression by PPARγ in adipocytes and macrophages. PLoS One 7, e34976. doi:10.1371/journal.pone.0034976
Nirinjini Naidoo, M. F., Ferber, M., and Master, M. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi:10.1523/JNEUROSCI.5685-07.2008
Nuss, J. E., DeFord, J. H., and Papaconstantinou, J. (2008). Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 365, 355–361. doi:10.1016/j.bbrc.2007.10.194
Olga Moiseeva, V. B., Antoine, R., Deschênes-Simard, X., and Ferbeyre, G. (2009). Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 29, 4495–4507. doi:10.1128/MCB.01868-08
Panganiban, R. A., Mungunsukh, O., and Day, R. M. (2013). X-irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int. J. Radiat. Biol. 89, 656–667. doi:10.3109/09553002.2012.711502
Park, J. H., Lo, E. H., and Hayakawa, K. (2021). Endoplasmic reticulum interaction supports energy production and redox homeostasis in mitochondria released from astrocytes. Transl. Stroke Res. 12, 1045–1054. doi:10.1007/s12975-021-00892-7
Passos, J. F., Nelson, G., Ahmed, S., Nelson, G., Richter, T., Peters, H., et al. (2007). Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 5, e110. doi:10.1371/journal.pbio.0050110
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347. doi:10.1038/msb.2010.5
Patergnani, S., Suski, J. M., Agnoletto, C., Bononi, A., Bonora, M., De Marchi, E., et al. (2011). Calcium signaling around mitochondria associated membranes (MAMs). Cell. Commun. Signal 9, 19. doi:10.1186/1478-811x-9-19
Pereira, R. O., Tadinada, S. M., Zasadny, F. M., Oliveira, K. J., Pires, K. M. P., Olvera, A., et al. (2017). OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J. 36, 2126–2145. doi:10.15252/embj.201696179
Peng, A., Mao, X., Zhong, J., Fan, S., and Hu, Y. (2020). Single-cell multi-omics and its prospective application in cancer biology. Proteomics 20, 1900271. doi:10.1002/pmic.201900271
Perrotta, I. (2020). The microscopic anatomy of endothelial cells in human atherosclerosis: Focus on ER and mitochondria. J. Anat. 237, 1015–1025. doi:10.1111/joa.13281
Picca, A., Calvani, R., Coelho-Junior, H. J., Landi, F., Bernabei, R., and Marzetti, E. (2020). Inter-organelle membrane contact sites and mitochondrial quality control during aging: A geroscience view. Cells 9, 598. doi:10.3390/cells9030598
Picca, A., Calvani, R., Lorenzi, M., Menghi, A., Galli, M., Vitiello, R., et al. (2017). Mitochondrial dynamics signaling is shifted toward fusion in muscles of very old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp. Gerontol. 96, 63–67. doi:10.1016/j.exger.2017.06.005
Pluquet, O., Pourtier, A., and Abbadie, C. (2015). The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell. Physiol. 308, C415–C425. doi:10.1152/ajpcell.00334.2014
Rabek, J. P., Boylston, W. H., and Papaconstantinou, J. (2003). Carbonylation of ER chaperone proteins in aged mouse liver. Biochem. Biophys. Res. Commun. 305, 566–572. doi:10.1016/s0006-291x(03)00826-x
Remondelli, P., and Renna, M. (2017). The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10, 187. doi:10.3389/fnmol.2017.00187
Rieusset, F. J., Paillard, M., Belaidi, E., Tubbs, E., Chauvin, M. A., Durand, A., et al. (2015). Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 59, 614–623. doi:10.1007/s00125-015-3829-8
Rieusset, J. (2018). The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell. Death Dis. 9, 388. doi:10.1038/s41419-018-0416-1
Roberto Bravo, J. M. V., Parra, V., Troncoso, R., Munoz, J. P., Bui, M., Quiroga, C., et al. (2011). Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell. Sci. 124, 2143–2152. doi:10.1242/jcs.080762
Robinson, M. A., and Castner, D. G. (2013). Characterization of sample preparation methods of NIH/3T3 fibroblasts for ToF-SIMS analysis. Biointerphases 8, 15. doi:10.1186/1559-4106-8-15
Rogoff, D., Black, K., McMillan, D. R., and White, P. C. (2010). Contribution of hexose-6-phosphate dehydrogenase to NADPH content and redox environment in the endoplasmic reticulum. Redox Rep. 15, 64–70. doi:10.1179/174329210X12650506623249
Rongbin Zhou, A. S. Y., Yazdi, A. S., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi:10.1038/nature09663
Rovira-Clave, X., Jiang, S., Bai, Y., Zhu, B., Barlow, G., Bhate, S., et al. (2021). Subcellular localization of biomolecules and drug distribution by high-definition ion beam imaging. Nat. Commun. 12, 4628. doi:10.1038/s41467-021-24822-1
Rubakhin, S. S., Greenough, W. T., and Sweedler, J. V. (2003). Spatial profiling with MALDI MS: Distribution of neuropeptides within single neurons. Anal. Chem. 75, 5374–5380. doi:10.1021/ac034498+
Sander, P., Gudermann, T., and Schredelseker, J. (2021). A calcium guard in the outer membrane: Is VDAC a regulated gatekeeper of mitochondrial calcium uptake? Int. J. Mol. Sci. 22, 946. doi:10.3390/ijms22020946
Sarsby, J., Griffiths, R. L., Rac, A. M., Bunch, J., Randall, E. C., Creese, A. J., et al. (2015). Liquid extraction surface analysis mass spectrometry coupled with field asymmetric waveform ion mobility spectrometry for analysis of intact proteins from biological substrates. Anal. Chem. 87, 6794–6800. doi:10.1021/acs.analchem.5b01151
Schmitz, E. A., Takahashi, H., and Karakas, E. (2022). Structural basis for activation and gating of IP3 receptors. Nat. Commun. 13, 1408. doi:10.1038/s41467-022-29073-2
Schneider, J. S., Shanmugam, M., Gonzalez, J. P., Lopez, H., Gordan, R., Fraidenraich, D., et al. (2013). Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell. Motil. 34, 349–356. doi:10.1007/s10974-013-9350-0
Sebastián, D., Hernandez-Alvarez, M. I., Segales, J., Sorianello, E., Munoz, J. P., Sala, D., et al. (2012). Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. 109, 5523–5528. doi:10.1073/pnas.1108220109
Sebastián, D., Sorianello, E., Segales, J., Irazoki, A., Ruiz-Bonilla, V., Sala, D., et al. (2016). Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. Embo J. 35, 1677–1693. doi:10.15252/embj.201593084
Seungyoon Yu, G. P., and Pekkurnaz, G. (2018). Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J. Mol. Biol. 430, 3922–3941. doi:10.1016/j.jmb.2018.07.027
Shally, A., and McDonagh, B. (2020). The redox environment and mitochondrial dysfunction in age-related skeletal muscle atrophy. Biogerontology 21, 461–473. doi:10.1007/s10522-020-09879-7
Sheraz, S., Tian, H., Vickerman, J. C., Blenkinsopp, P., Winograd, N., and Cumpson, P. (2019). Enhanced ion yields using high energy water cluster beams for secondary ion mass spectrometry analysis and imaging. Anal. Chem. 91, 9058–9068. doi:10.1021/acs.analchem.9b01390
Shigenaga, M. K., Hagen, T. M., and Ames, B. N. (1994). Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. U. S. A. 91, 10771–10778. doi:10.1073/pnas.91.23.10771
Shigetomi, E., Saito, K., Sano, F., and Koizumi, S. (2019). Aberrant calcium signals in reactive astrocytes: A key process in neurological disorders. Int. J. Mol. Sci. 20, 996. doi:10.3390/ijms20040996
Silvestre Alavez, M. C. V., Zucker, D. J. S., Klang, I. M., and Lithgow, G. J. (2011). Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472, 226–229. doi:10.1038/nature09873
Son, J. M., Sarsour, E. H., Kakkerla Balaraju, A., Fussell, J., Kalen, A. L., Wagner, B. A., et al. (2017). Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell. 16, 1136–1145. doi:10.1111/acel.12649
Sood, A., Jeyaraju, D. V., Prudent, J., Caron, A., Lemieux, P., McBride, H. M., et al. (2014). A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. U. S. A. 111, 16017–16022. doi:10.1073/pnas.1408061111
Starr, N. J., Johnson, D. J., Wibawa, J., Marlow, I., Bell, M., Barrett, D. A., et al. (2016). Age-related changes to human stratum corneum lipids detected using time-of-flight secondary ion mass spectrometry following in vivo sampling. Anal. Chem. 88, 4400–4408. doi:10.1021/acs.analchem.5b04872
Stein, C. M., Weiskirchen, R., Damm, F., and Strzelecka, P. M. (2021). Single-cell omics: Overview, analysis, and application in biomedical science. J. Cell. Biochem. 122, 1571–1578. doi:10.1002/jcb.30134
Straub, I. R., Weraarpachai, W., and Shoubridge, E. A. (2021). Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 30, 687–705. doi:10.1093/hmg/ddab078
Strowig, T., Henao-Mejia, J., Elinav, E., and Flavell, R. (2012). Inflammasomes in health and disease. Nature 481, 278–286. doi:10.1038/nature10759
Surmeier, D. J., Schumacker, P. T., Guzman, J. D., Ilijic, E., Yang, B., and Zampese, E. (2017). Calcium and Parkinson's disease. Biochem. Biophys. Res. Commun. 483, 1013–1019. doi:10.1016/j.bbrc.2016.08.168
Szibor, M., Gizatullina, Z., Gainutdinov, T., Endres, T., Debska-Vielhaber, G., Kunz, M., et al. (2020). Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J. Biol. Chem. 295, 4383–4397. doi:10.1074/jbc.RA119.011902
Szymański, J. J., Michalska, B., Patalas-Krawczyk, P., Perrone, M., Ziółkowski, W., Duszyński, J., et al. (2017). Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int. J. Mol. Sci. 18, 1576. doi:10.3390/ijms18071576
Taylor, M. J., Lukowski, J. K., and Anderton, C. R. (2021). Spatially resolved mass spectrometry at the single cell: Recent innovations in proteomics and metabolomics. J. Am. Soc. Mass Spectrom. 32, 872–894. doi:10.1021/jasms.0c00439
Theurey, T. E., Vial, G., Jacquemetton, J., Bendridi, N., Chauvin, M. A., Alam, M. R., et al. (2016). Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell. Biol. 8, 129–143. doi:10.1093/jmcb/mjw004
Thoudam, T., Ha, C. M., Leem, J., Chanda, D., Park, J. S., Kim, H. J., et al. (2019). PDK4 augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 68, 571–586. doi:10.2337/db18-0363
Tian, H., Sparvero, L. J., Blenkinsopp, P., Amoscato, A. A., Watkins, S. C., Bayir, H., et al. (2019a). Secondary-ion mass spectrometry images cardiolipins and phosphatidylethanolamines at the subcellular level. Angew. Chem. Int. Ed. Engl. 131, 3156–3161. doi:10.1002/anie.201814256
Tian, H., Maciazek, D., Postawa, Z., Garrison, B. J., and Winograd, N. C-O. (2019b). C-O bond dissociation and induced chemical ionization using high energy (CO2)n+ gas cluster ion beam. J. Am. Soc. Mass Spectrom. 30, 476–481. doi:10.1007/s13361-018-2102-z
Tian, H., Sheraz Nee Rabbani, S., Vickerman, J. C., and Winograd, N. (2021a). Multiomics imaging using high-energy water gas cluster ion beam secondary ion mass spectrometry [(H2O)n-GCIB-SIMS] of frozen-hydrated cells and tissue. Anal. Chem. 93, 7808–7814. doi:10.1021/acs.analchem.0c05210
Tian, H., Six, D. A., Krucker, T., Leeds, J. A., and Winograd, N. (2017b). Subcellular chemical imaging of antibiotics in single bacteria using C60-secondary ion mass spectrometry. Anal. Chem. 89, 5050–5057. doi:10.1021/acs.analchem.7b00466
Tian, H., Sparvero, L. J., Amoscato, A. A., Bloom, A., Bayir, H., Kagan, V. E., et al. (2017a). Gas cluster ion beam time-of-flight secondary ion mass spectrometry high-resolution imaging of cardiolipin speciation in the brain: Identification of molecular losses after traumatic injury. Anal. Chem. 89, 4611–4619. doi:10.1021/acs.analchem.7b00164
Tian, H., Sparvero, L. J., Anthonymuthu, T. S., Sun, W. Y., Amoscato, A. A., He, R. R., et al. (2021b). Successive high-resolution (H2O)n-GCIB and C60-SIMS imaging integrates multi-omics in different cell types in breast cancer tissue. Anal. Chem. 93, 8143–8151. doi:10.1021/acs.analchem.0c05311
Tian, H., Wucher, A., and Winograd, N. (2014). Molecular imaging of biological tissue using gas cluster ions. Surf. Interface Anal. 46, 115–117. doi:10.1002/sia.5509
Tian, H., Wucher, A., and Winograd, N. (2016a). Dynamic reactive ionization with cluster secondary ion mass spectrometry. J. Am. Soc. Mass Spectrom. 27, 285–292. doi:10.1007/s13361-015-1283-y
Tian, H., Wucher, A., and Winograd, N. (2016b). Reduce the matrix effect in biological tissue imaging using dynamic reactive ionization and gas cluster ion beams. Biointerphases 11, 02A320. doi:10.1116/1.4941366
Tian, H., Wucher, A., and Winograd, N. (2016c). Reducing the matrix effect in organic cluster SIMS using dynamic reactive ionization. J. Am. Soc. Mass Spectrom. 27, 2014–2024. doi:10.1007/s13361-016-1492-z
Tian, H., Maciążek, D., Postawa, Z., Garrison, B. J., and Winograd, N. (2016). CO2 cluster ion beam, an alternative projectile for secondary ion mass spectrometry. J. Am. Soc. Mass Spectrom. 27, 1476–1482. doi:10.1007/s13361-016-1423-z
Tubbs, E., Vial, G., Wollheim, C. B., Rieusset, J., and Rosengren, A. H. (2018a). Sulforaphane improves disrupted ER-mitochondria interactions and suppresses exaggerated hepatic glucose production. Mol. Cell. Endocrinol. 461, 205–214. doi:10.1016/j.mce.2017.09.016
Tubbs, E., Chanon, S., Robert, M., Bendridi, N., Bidaux, G., Chauvin, M. A., et al. (2018b). Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 67, 636–650. doi:10.2337/db17-0316
Tubbs, E., Theurey, P., Vial, G., Bendridi, N., Bravard, A., Chauvin, M. A., et al. (2014). Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294. doi:10.2337/db13-1751
Tucker, K. R., Cavolo, S. L., and Levitan, E. S. (2016). Elevated mitochondria-coupled NAD(P)H in endoplasmic reticulum of dopamine neurons. Mol. Biol. Cell. 27, 3214–3220. doi:10.1091/mbc.E16-07-0479
Uddin, M. S., Yu, W. S., and Lim, L. W. (2021). Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer's disease. Ageing Res. Rev. 70, 101417. doi:10.1016/j.arr.2021.101417
Umegaki, H. (2015). Sarcopenia and diabetes: Hyperglycemia is a risk factor for age-associated muscle mass and functional reduction. J. Diabetes Investig. 6, 623–624. doi:10.1111/jdi.12365
Vance, J. E., Stone, S. J., and Faust, J. R. (1997). Abnormalities in mitochondria-associated membranes and phospholipid biosynthetic enzymes in the mnd/mnd mouse model of neuronal ceroid lipofuscinosis. Biochim. Biophys. Acta 1344, 286–299. doi:10.1016/s0005-2760(96)00153-1
Venturi, E., Pitt, S., Galfré, E., and Sitsapesan, R. (2012). From eggs to hearts: What is the link between cyclic ADP-ribose and ryanodine receptors? Cardiovasc Ther. 30, 109–116. doi:10.1111/j.1755-5922.2010.00236.x
Veeresh, P., Kaur, H., Sarmah, D., Mounica, L., Verma, G., Kotian, V., et al. (2019). Endoplasmic reticulum-mitochondria crosstalk: From junction to function across neurological disorders. Ann. N. Y. Acad. Sci. 1457, 41–60. doi:10.1111/nyas.14212
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell. Death Differ. 19, 1880–1891. doi:10.1038/cdd.2012.74
Vittoria Infantino, P. C., Cucci, L., Antonietta Panaro, M., Antonietta Di Noia, M., Calvello, R., Palmieri, F., et al. (2011). The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 438, 433–436. doi:10.1042/BJ20111275
Voit, A., Patel, V., Pachon, R., Shah, V., Bakhutma, M., Kohlbrenner, E., et al. (2017). Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 8, 1068. doi:10.1038/s41467-017-01146-7
Ying, W. Y. (2006). NAD+ and NADH in cellular functions and cell death. Front. Biosci. 11, 3129–3148. doi:10.2741/2038
Wang, L., Xing, X., Zeng, X., Jackson, S. R., TeSlaa, T., Al-Dalahmah, O., et al. (2022). Nat. Methods 19, 223–230. doi:10.1038/s41592-021-01378-y
Wang, X., Mick, G., and McCormick, K. (2019). Pyridine nucleotide regulation of hepatic endoplasmic reticulum calcium uptake. Physiol. Rep. 7, e14151. doi:10.14814/phy2.14151
Wang, Y., Zhang, X., Wen, Y., Li, S., Lu, X., Xu, R., et al. (2021). Endoplasmic reticulum-mitochondria contacts: A potential therapy target for cardiovascular remodeling-associated diseases. Front. Cell. Dev. Biol. 9, 774989. doi:10.3389/fcell.2021.774989
Ward, C. P., Peng, L., Yuen, S., Halstead, J., Palacios, H., Nyangau, E., et al. (2022). Aging alters the metabolic flux signature of the ER-unfolded protein response in vivo in mice. Aging Cell. 21, e13558. doi:10.1111/acel.13558
Wellen, K. E., Uma, M., Sachdeva, T., Bui, V., Cross, J. R., Thompson, C. B., et al. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. doi:10.1126/science.1164097
Wiedmer, P., Jung, T., Castro, J. P., Pomatto, L. C. D., Sun, P. Y., Davies, K. J. A., et al. (2021). Sarcopenia - molecular mechanisms and open questions. Ageing Res. Rev. 65, 101200. doi:10.1016/j.arr.2020.101200
Williams, C. C., Singleton, B. A., Llopis, S. D., and Skripnikova, E. V. (2013). Metformin induces a senescence-associated gene signature in breast cancer cells. J. Health Care Poor Underserved 24, 93–103. doi:10.1353/hpu.2013.0044
Wilson, E. L., and Metzakopian, E. (2021). ER-Mitochondria contact sites in neurodegeneration: Genetic screening approaches to investigate novel disease mechanisms. Cell. Death Differ. 28, 1804–1821. doi:10.1038/s41418-020-00705-8
Wu, H., Carvalho, P., and Voeltz, G. K. (2018). Here, there, and everywhere: The importance of ER membrane contact sites. Science 361, eaan5835. doi:10.1126/science.aan5835
Wucher, A., Tian, H., and Winograd, N. (2014). A mixed cluster ion beam to enhance the ionization efficiency in molecular secondary ion mass spectrometry. Rapid Commun. Mass Spectrom. 28, 396–400. doi:10.1002/rcm.6793
Xu Benjin, L. L., and Ling, L. (2020). Developments, applications, and prospects of cryo-electron microscopy. Protein Sci. 29, 872–882. doi:10.1002/pro.3805
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi:10.1172/JCI26373
Yamasaki, M., Churchill, G. C., and Galione, A. (2005). Calcium signalling by nicotinic acid adenine dinucleotide phosphate (NAADP). FEBS J. 272, 4598–4606. doi:10.1111/j.1742-4658.2005.04860.x
Yu, S., Zhang, L., Liu, C., Yang, J., Zhang, J., and Huang, L. (2019). PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca(2+) elevation. Exp. Cell. Res. 379, 191–202. doi:10.1016/j.yexcr.2019.04.002
Yuan Wang, P. D., Liu, Y., Wu, Y., Chen, Y., Guo, Y., Zhang, S., et al. (2020). Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 11, 5596. doi:10.1038/s41467-020-19360-1
Zampese, E., Fasolato, C., Kipanyula, M. J., Bortolozzi, M., Pozzan, T., and Pizzo, P. (2011). Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. U. S. A. 108, 2777–2782. doi:10.1073/pnas.1100735108
Zandkarimi, F., and Brown, L. M. (2019). in Advancements of mass spectrometry in biomedical research. Editors A G. Woods, and C C. Darie (Berlin, Germany: Springer International Publishing), 317–326.
Zavalin, A., Yang, J., Hayden, K., Vestal, M., and Caprioli, R. M. (2015). Tissue protein imaging at 1 μm laser spot diameter for high spatial resolution and high imaging speed using transmission geometry MALDI TOF MS. Anal. Bioanal. Chem. 407, 2337–2342. doi:10.1007/s00216-015-8532-6
Zeidler, J. D., Hogan, K. A., Agorrody, G., Peclat, T. R., Kashyap, S., Kanamori, K. S., et al. (2022). The CD38 glycohydrolase and the NAD sink: Implications for pathological conditions. Am. J. Physiol. Cell. Physiol. 322, C521–c545. doi:10.1152/ajpcell.00451.2021
Zeisbrich, M., Yanes, R. E., Zhang, H., Watanabe, R., Li, Y., Brosig, L., et al. (2018). Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann. Rheum. Dis. 77, 1053–1062. doi:10.1136/annrheumdis-2017-212647
Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., et al. (2016). NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. doi:10.1126/science.aaf2693
Zhang, L., and Vertes, A. (2018). Single-cell mass spectrometry approaches to explore cellular heterogeneity. Angew. Chem. Int. Ed. 57, 4466–4477. doi:10.1002/anie.201709719
Zhang, P., Konja, D., Zhang, Y., and Wang, Y. (2021). Communications between mitochondria and endoplasmic reticulum in the regulation of metabolic homeostasis. Cells 10, 2195. doi:10.3390/cells10092195
Zhang, W. F., Xia, X. P., Zhang, Y. Q., Peng, T. P., and Yang, Q. (2018). A novel sample preparation method for ultra-high vacuum (UHV) secondary ion mass spectrometry (SIMS) analysis. J. Anal. At. Spectrom. 33, 1559–1563. doi:10.1039/c8ja00087e
Zhou, Y., Jing, S., Liu, S., Shen, X., Cai, L., Zhu, C., et al. (2022). Double-activation of mitochondrial permeability transition pore opening via calcium overload and reactive oxygen species for cancer therapy. J. Nanobiotechnology 20, 188. doi:10.1186/s12951-022-01392-y
Zhu, B., Ferry, C. H., Markell, L. K., Blazanin, N., Glick, A. B., Gonzalez, F. J., et al. (2014). The nuclear receptor peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) promotes oncogene-induced cellular senescence through repression of endoplasmic reticulum stress. J. Biol. Chem. 289, 20102–20119. doi:10.1074/jbc.M114.551069
Keywords: mass spec imaging, MERCs, Golgi complex, mitocchondrial dysfunction, aging, metabolic flux, spatial omics
Citation: Hogan KA, Zeidler JD, Beasley HK, Alsaadi AI, Alshaheeb AA, Chang Y-C, Tian H, Hinton AO and McReynolds MR (2023) Using mass spectrometry imaging to visualize age-related subcellular disruption. Front. Mol. Biosci. 10:906606. doi: 10.3389/fmolb.2023.906606
Received: 28 March 2022; Accepted: 24 January 2023;
Published: 08 March 2023.
Edited by:
Sandra A. Murray, University of Pittsburgh, United StatesReviewed by:
Guillaume Thibault, Nanyang Technological University, SingaporeWilliam Griffiths, Swansea University, United Kingdom
Copyright © 2023 Hogan, Zeidler, Beasley, Alsaadi, Alshaheeb, Chang, Tian, Hinton and McReynolds. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Tian, aHV0M0Bwc3UuZWR1; Antentor O. Hinton Jr, YW50ZW50b3Iuby5oaW50b24uanJAdmFuZGVyYmlsdC5lZHU=; Melanie R. McReynolds, bWNyZXlub2xkc0Bwc3UuZWR1
†These authors have contributed equally to this work