
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 24 January 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.805594
This article is part of the Research TopicNovel Molecular Mechanisms and Innovative Therapeutic Approaches for Age-Associated DiseasesView all 29 articles
 Wei Qin1†
Wei Qin1† Yi-Heng Li2†Jing Tong3†Jie Wu3
Yi-Heng Li2†Jing Tong3†Jie Wu3 Dong Zhao1
Dong Zhao1 Hui-Jin Li1
Hui-Jin Li1 Lu Xing1
Lu Xing1 Chun-Xia He1
Chun-Xia He1 Xin Zhou1
Xin Zhou1 Peng-Quan Li1Ge Meng3,4*Shao-Ping Wu2*
Peng-Quan Li1Ge Meng3,4*Shao-Ping Wu2* Hui-Ling Cao1,2*
Hui-Ling Cao1,2*Atrial fibrillation (AF) is the most common clinical sustained arrhythmia; clinical therapeutic drugs have low atrial selectivity and might cause more severe ventricle arrhythmias while stopping AF. As an anti-AF drug target with high selectivity on the atrial muscle cells, the undetermined crystal structure of Kv1.5 potassium channel impeded further new drug development. Herein, with the simulated 3D structure of Kv1.5 as the drug target, a series of 3-morpholine linked aromatic amino substituted 1H-indoles as novel Kv1.5 channel inhibitors were designed and synthesized based on target–ligand interaction analysis. The synthesis route was practical, starting from commercially available material, and the chemical structures of target compounds were characterized. It was indicated that compounds T16 and T5 (100 μM) exhibited favorable inhibitory activity against the Kv1.5 channel with an inhibition rate of 70.8 and 57.5% using a patch clamp technique. All compounds did not exhibit off-target effects against other drug targets, which denoted some selectivity on the Kv1.5 channel. Interestingly, twelve compounds exhibited favorable vasodilation activity on pre-contracted arterial rings in vitro using KCl or phenylephrine (PE) by a Myograph. The vasodilation rates of compounds T16 and T4 (100 μM) even reached over 90%, which would provide potential lead compounds for both anti-AF and anti-hypertension new drug development.
1. Sixteen 3-morpholine linked aromatic amino substituted 1H-indoles as novel Kv1.5 channel inhibitors were designed and synthesized targeting the simulated 3D structure of Kv1.5.
2. Compounds T16 and T5 (100 μM) exhibited favorable inhibitory activity against the Kv1.5 channel with an inhibition rate of 70.8 and 57.5% using a patch clamp technique. The vasodilation rates of compounds T16 and T4 (100 μM) reached over 90% by a Myograph.
3. The novel Kv1.5 inhibitors would provide potential lead compounds for both anti-atrial fibrillation and anti-hypertension new drug development.
As the most common clinical sustained arrhythmia, atrial fibrillation (AF) presents with a wide spectrum of symptoms and severity (Lau et al., 2019). There are now many therapeutic drugs for clinically treating AF, such as Dofetilide, Aminodarone, Sotalol, Propafenone, flecainide, etc. These drugs have low atrial selectivity while stopping AF, which could often cause severe adverse effects such as ventricle arrhythmias (e.g., life-threatening TdP) (Ang et al., 2020). Therefore, an ideal drug to treat AF should have high selectivity on the atrial muscle cells, which could terminate or postpone the occurrence of AF without affecting or prolonging the Q-T interval or the negative inotropic action to ensure the highly effective safety (Guo et al., 2016; Zimetbaum, 2017).
The cardiac IKur (Kv1.5) potassium channel is a kind of voltage-gated ion-channel which has been confirmed to be expressed only in the human atrium (Wettwer and Terlau, 2014; Du et al., 2021). Therefore, Kv1.5 is currently considered as the ideal target for designing the highly atrium-selective drugs to treat AF (Guo et al., 2012; Zhao et al., 2019). Besides, the clinical and animal research data showed that the Kv1.5 channel is the base for the atrium-reconstructing electric rhythm. The inhibitor of the Kv1.5 channel might selectively prolong the effective refractory period (ERP) of the atrium to terminate AF (Tamargo et al., 2009).
Multiple studies indicated that some compounds with the skeleton of morpholine exhibited inhibitive effects against Kv1.5 (Vaccaro et al., 2008; Guo et al., 2012), such as the compound LY294002 (Wu et al., 2009; Hong et al., 2013) (Figure 1). Some of the chemical structures with 1H-indole (Guo et al., 2013) or pyrimidine fused 1H-indole skeletons (Gasparoli et al., 2015) also showed inhibitive activity on the Kv1.5 channel (Figure 1). Sharing the common structure features, the two compounds (CD-160130 and CD-140793) contained both morpholine and 1H-indole functional groups, which were connected by several chemical bonds within a pyrimidine ring (Figure 1). The distance between morpholine and 1H-indole in the chemical structures are in a certain scope. To be specific, the distance between N on morpholine and 3-C on indole in these compounds are from 2–3 atoms to 3–4 bond length (Figure 1). The clinical drugs including Aminodarone and Dronedarone are used as the anti-arrhythmic drugs (Figure 1). Aminodarone might lengthen the monophasic action potential duration to prolong the ERP (Mujović et al., 2020). Dronedarone is a kind of multichannel-blocker with the electronic physiological and blood dynamic features similar to Aminodarone. Dronedarone could be used to prevent AF and ventricular tachycadia and restore the sinus rhythm (Kaess and Ehrlich, 2016). The common chemical structure feature of the two drugs is the linker between nitrogen and oxygen, that is, -N(CH2)nO- (n = 2–3). The chain-like linker is exactly alike to the structure feature of other kinds of Kv1.5 inhibitors (Guo et al., 2014) (Figure 1).
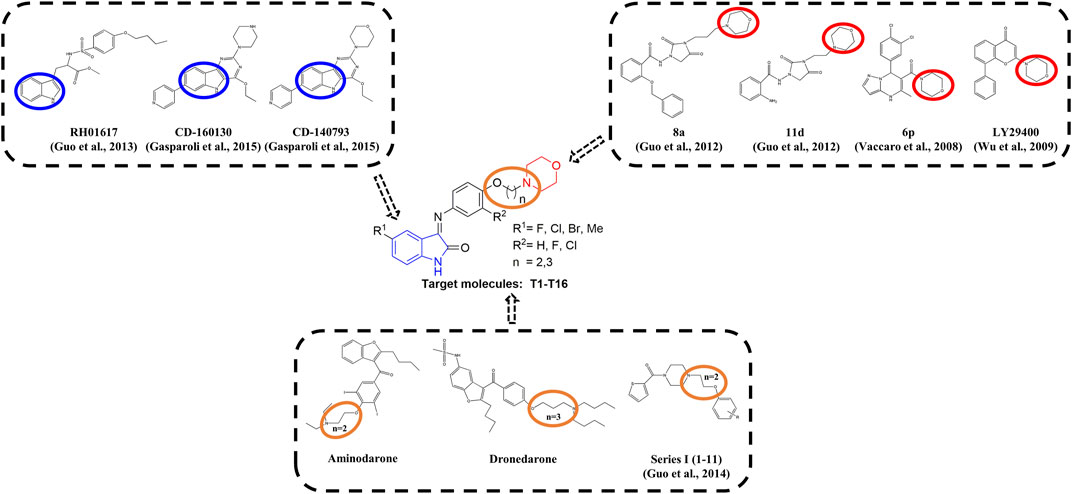
FIGURE 1. Design strategy of novel Kv1.5 inhibitors based on morpholine, 1H-indole, and the active linker (-N(CH2)nO-).
As an anti-AF drug target with high selectivity on the atrial muscle cells, the undetermined crystal structure of the Kv1.5 channel impeded further new drug development. Herein, the simulated 3D structure of the Kv1.5 channel as the drug target was built based on the crystal structure of its homolog Kv1.2 channel and its amino acid sequence. Based on target–ligand interaction and structure–activity analysis of traditional inhibitors as well as its simulated 3D structure of target Kv1.5, a series of compounds (T1–T16), containing both morpholine and 1H-indole linked with a chain-like structure (-N(CH2)nO-) in one molecule, were designed and synthesized to enhance therapeutic effects of the antiarrhythmic drugs (Figure 1).
Chloral hydrate (9.0 g, 55.0 mmol, 1.1 equiv) and water (240 ml) were added to a clean 500-ml flask under stirring, to which was added anhydrous sodium sulfate (65.0 g), substituted aniline (1a-1d, 50.0 mmol, 1.0 equiv), hydrochloride solution (4.3 ml HCl +30.0 ml water), and hydroxylamine hydrochloride solution (10.8 g + 50.0 ml water), under stirring sequentially. The reaction was heated gradually to 65°C for 2 h before heating was stopped. The hot mixture was filtered to give a clear solution, from which some pale yellow solids were precipitated while it was being cooled down to room temperature. The filtration under the reduced pressure could yield the desired product (2a-2d) (petroleum ether/ethyl acetate, 1:2, Rf = 0.2–0.4).
Concentrated sulfuric acid (24.0 ml) was added to a clean 150-ml three-neck flask. The reaction mixture was heated to 50°C, to which was added the intermediate 2a-2d (30.0 mmol, 1.0 equiv) obtained from the above procedure. The color of the reaction mixture became dark to dark-black, while the amount of the material was added increasingly. The temperature of the reaction mixture was adjusted to 80°C for 20 min. The crashed ice (100 g) was added slowly to the reaction system to allow it to turn a red-brown color. The reaction mixture was kept at room temperature overnight before being filtrated to produce the filter cake, which was washed to pH = 7 with water to yield a solid. The solid was dissolved in an aqueous solution of NaOH (10.0%, 90.0 ml) before being filtrated. The pH of the filtration was adjusted to pH = 2 with concentrated hydrochloride until there were a large amount of brick-red solids precipitated from the solution. The solid was filtered to yield the red-brown solid as the desired products (3a-3d, petroleum ether/ethyl acetate, 1:2, Rf = 0.4–0.6).
5-Fluoro-isatin (3a, 0.340 g, 2.00 mmol, 1.0 equiv) and 1-(2-(4-aminophenyloxyl)ethyl) morpholine (4a, 0.444 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to refluxing for 10 h, before being monitored by thin layer chromatography (TLC) until the reaction was completed before being cooled down to room temperature. A large amount of yellow solid precipitated, which was collected by filtering under reduced pressure to yield the yellow powder as the desired target compound (T1, 0.442 g, 60%). m. p. 155–157 °C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 1H NMR (400 MHz, DMSO-d6) δ: 10.95 (d, J = 8.4 Hz, 1H), 7.25 (t, J = 8.3 Hz, 2H), 7.08 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.95–6.82 (m, 1H), 6.34 (d, J = 7.4 Hz, 1H), 4.18–4.10 (m, 2H), 3.59 (s, 4H), 2.72 (d, J = 5.0 Hz, 2H). HRMS (ESI): m/z calculated for C20H20FN3O3 369.3895, Found 369.3895.
5-Fluoro-isatin (3a, 0.34 g, 2.0 mmol, 1.0 equiv) and 1-[2-(2-fluoro-4-amino-phenoxy) propyl] morpholine (4b, 0.508 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to refluxing for 13 h before being completed as shown by TLC analysis to ensure the reaction completion. The reaction mixture was cooled down to room temperature without any solid being precipitated. A certain amount of water was added into the reaction system to allow the large amount of red solid to be precipitated. The solids were collected via filtration under the reduced pressure and then dried under vacuo to yield the red solids as the desired target compound (T2, 0.150 g, 19%). m. p. 131–133°C (dichloromethane/methanol, 10:1, Rf = 0.5), 1H NMR (400 MHz, DMSO-d6) δ: 10.97 (d, J = 8.0 Hz, 1H), 7.28 (dd, J = 8.4, 7.5 Hz, 2H), 7.22–7.10 (m, 1H), 7.04 (dd, J = 7.0, 1.8 Hz, 1H), 6.99–6.89 (m, 1H), 6.85 (dd, J = 8.3, 6.2 Hz, 1H), 6.33–6.28 (m, 1H), 4.12 (dd, J = 5.4, 6.3 Hz, 2H), 3.58 (s, 4H), 2.48–2.36 (m, 6H), 1.92 (dd, J = 3.1, 6.6 Hz, 2H). HRMS (ESI): m/z calculated for C21H21F2N3O3 401.4065, Found 401.4066.
5-Fluoro-isatin (3a, 0.340 g, 2.00 mmol, 1.0 equiv) and 1-[2-(4-amino-phenyloxyl)propyl] morpholine (4c, 0.470 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) under stirring at room temperature. The reaction mixture was heated to reflux for 10 h, which was monitored by TLC analysis to ensure the completed reaction. The reaction system was then allowed to be cooled down to room temperature without the precipitated solid. A small amount of water was then added to this system to allow a large amount of orange-yellow solid to be precipitated. The solid was obtained by filtration to give the orange-yellow powder as the desired target compound (T3, 0.389 g, 51%). m. p. 163–165°C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 11.01 (s, 1H), 7.24 (dd, J = 8.6, 6.1, 2.7 Hz, 1H), 7.06 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 6.91 (dd, J = 8.2, 4.9 Hz, 1H), 6.34 (dd, J = 8.5, 2.3 Hz, 1H), 4.05 (dt, J = 2.1, 6.2 Hz, 2H), 3.59 (s, 4H), 2.48 (s, 1H), 2.47 (s, 1H), 2.42 (s, 4H), 1.91 (dd, J = 3.4, 6.6 Hz, 2H). HRMS (ESI): m/z calculated for C21H22FN3O3 383.4161, Found 383.4162.
5-Fluoro-isatin (3a, 0.340 g, 1.00 mmol, 1.0 equiv) and 1-[2-(2-chloro-4-aminophenoxyl) propyl] morpholine (4d, 0.540 g, 2.00 mmol, 2.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) under stirring at room temperature. The reaction system was heated to reflux for 10 h under stirring until the TLC analysis showed the reaction completion. The reaction system was then cooled down to room temperature, until there was a large amount of dark-red solid being precipitated, which was filtrated before it was dried under vacuo to produce the dark red solid as the target compound (T4, 0.428 g, 51%). m. p. 127–128°C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 10.96 (dd, J = 8.3, 9.9 Hz, 1H), 7.41–7.34 (m, 1H), 7.31–7.19 (m, 1H), 7.17–7.10 (m, 2H), 7.02 (dd, J = 1.7, 8.8, 6.5 Hz, 1H), 6.95–6.83 (m, 1H), 6.32 (dd, J = 5.2, 8.5, 2.6 Hz, 1H), 4.15 (dd, J = 5.4, 5.2 Hz, 1H), 4.13–4.01 (m, 1H), 3.60 (s, 4H), 2.53 (dd, J = 3.1, 4.5 Hz, 3H), 2.44 (s, 3H), 1.94 (dd, J = 3.9, 7.1 Hz, 2H). HRMS (ESI): m/z calculated for C21H21ClFN3O3 417.8611, Found 417.8610.
5-Chloro-isatin (3b, 0.360 g, 1.00 mmol, 1.0 equiv) and 1-[2-(4-aminophenoxyl)ethyl]morpholine (4a, 0.444 g, 2.00 mmol, 2.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 10 h until the TLC analysis showed the reaction completion. The reaction system was then allowed to be cooled down to room temperature, when there was a large amount of orange-red solids being precipitated. The solid was obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to provide the orange-red solid as the desired target compound (T5, 0.753 g, 98%). m. p. 150–152°C (dichloromethane/methanol, 10:1, Rf = 0.5) 1H NMR (400 MHz, DMSO-d6) δ: 11.07 (d, J = 8.9 Hz, 1H), 7.47–7.37 (m, 1H), 7.26 (d, J = 8.7 Hz, 1H), 7.09 (d, J = 8.7 Hz, 1H), 7.01 (d, J = 8.7 Hz, 1H), 6.90 (dd, J = 8.2, 8.3 Hz, 2H), 6.58 (d, J = 1.4 Hz, 1H), 4.19–4.08 (m, 2H), 3.59 (d, J = 4.1 Hz, 4H), 2.72 (t, J = 5.5 Hz, 2H). 13C NMR (400 MHz, DMSO-d6) HRMS (ESI): m/z calculated for C20H20ClN3O3 385.8441, Found 385.8442.
5-Chloro-isatin (3b, 0.360 g, 1.00 mmol, 1.0 equiv) and 1-[2-(2-fluoro-4-aminophenoxyl)propyl] morpholine (4b, 0.508 g, 2.00 mmol, 2.0 equiv) were added to a 50-ml flask, to which were added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was then heated to reflux for 11 h until the TLC analysis showed the reaction completion. The reaction system was then allowed to cool down to room temperature until there was a large amount of dark red solid being precipitated. The solid was obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to yield the red solid as the desired target compound (T6, 0.357 g, 64%). m. p. 148–149°C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 11.09 (d, J = 8.4 Hz, 1H), 7.46 (dd, J = 8.8, 8.4, 8 Hz, 1H), 7.31 (t, J = 8.9 Hz, 1H), 7.24–7.11 (m, 1H), 7.05 (dd, J = 8.0, 2.4 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 2.1 Hz, 1H), 4.13 (dt, J = 8.0, 6.3 Hz, 2H), 3.58 (t, J = 4.5 Hz, 4H), 2.44 (dd, J = 7.0, 6.1 Hz, 2H), 2.38 (s, 4H), 1.90 (dd, J = 3.0, 6.5 Hz, 2H). HRMS (ESI): m/z calculated for C21H21ClFN3O3 417.8611, Found 417.8612.
5-Chloro-isatin (3b, 0.360 g, 1.00 mmol, 1.0 equiv),1-[2-(4-aminophenoxyl)propyl]morpholine (4c, 0.470 g, 2.00 mmol, 2.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was then heated to reflux for 11 h until the TLC analysis showed the reaction completion. The reaction system was then allowed to cool down to room temperature until there was a large amount of orange-red solid being precipitated. The solid was obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to yield the orange-red solid as the desired target compound (T7, 0.560 g, 70%). m. p. 171–173°C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 11.13 (s, 1H), 7.43 (dd, J = 8.7, 8.4 Hz, 1H), 7.26 (d, J = 8.8 Hz, 1H), 7.07 (d, J = 8.9 Hz, 2H), 7.01 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.5 Hz, 1H), 6.90–6.85 (m, 1H), 6.59 (d, J = 2.1 Hz, 1H), 4.11–4.00 (m, 2H), 3.58 (t, J = 4.5 Hz, 4H), 2.51 (d, J = 1.6 Hz, 1H), 2.43 (dd, J = 3.4, 6.3 Hz, 2H), 2.38 (s, 4H), 1.97–1.83 (m, 2H). HRMS (ESI): m/z calculated for C21H22ClN3O3 399.8707, Found 399.8709.
5-Chloro-isatin (3b, 0.360 g, 1.00 mmol, 1.0 equiv) and 1-[2-(2-chloro-4- aminophenoxy)propyl] morpholine (4d, 0.540 g, 2.00 mmol, 2.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 11 h until the TLC analysis showed that the reaction was completed. The reaction system was then allowed to cool down to room temperature until there was a large amount of dark red solid being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to yield the dark red solid as the target compound (T8, 0.387 g, 45.0%). m. p. 134–135°C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 11.10 (dd, J = 8.3, 10.4 Hz, 1H), 7.56–7.36 (m, 1H), 7.32–7.21 (m, 1H), 7.20–7.10 (m, 1H), 7.04 (dt, J = 8.9, 8.8 Hz, 1H), 6.97–6.83 (m, 1H), 6.57 (dd, J = 8.4, 1.9 Hz, 1H), 4.21–4.00 (m, 2H), 3.60 (s, 4H), 2.55–2.49 (m, 3H), 2.46 (s, 3H), 1.95 (dd, J = 8.9, 8.7 Hz, 2H). HRMS (ESI): m/z calculated for C21H21Cl2N3O3 434.3157, Found 434.3156.
5-Methylisatin (3c, 0.319 g, 2.00 mmol, 1.0 equiv) and 1-[2-(4-amino-phenoxyl)ethyl]morpholine (4a, 0.444 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 10 h until the TLC analysis showed that the reaction was completed. The reaction system was then cooled down to room temperature until there was a small amount of dark red solid being precipitated. A small amount of water was added to the reaction mixture to allow the large amount of solids to be precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to yield the dark red solids as the desired target compound (T9, 0.327 g, 45%). m. p.127∼129°C (dichloromethane/methanol, 10:1, Rf = 0.5), 1H NMR (400 MHz, DMSO-d6) δ: 10.81 (d, J = 8.5 Hz, 1H), 7.26–7.13 (m, 2H), 7.06 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 8.9 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.49 (s, 1H), 4.13 (dt, J = 5.0, 5.8 Hz, 2H), 3.65–3.52 (m, 4H), 2.73 (s, 2H), 2.01 (s, 3H). HRMS (ESI): m/z calculated for C21H23N3O3 365.4256, Found 365.4258.
5-Methylisatin (3c, 0.319 g, 2.00 mmol, 1.0 equiv), 1-[2-(2-fluoro-4-aminophenoxyl)propyl] morpholine (4b, 0.508 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 10 h until the TLC analysis showed that the reaction was completed. The reaction system was then cooled down to room temperature until there was a large amount of orange-yellow solids being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to afford the orange-yellow solid as the target compound (T10, 0.083 g, 10%). m. p. 117–120°C (dichloromethane/methanol, 10:1, Rf = 0.4),1H NMR (400 MHz, DMSO-d6) δ: 10.83 (d, J = 8.3 Hz, 1H), 7.27 (t, J = 8.9 Hz, 1H), 7.20 (d, J = 7.9 Hz, 1H), 7.12 (dd, J = 9.9, 5.3 Hz, 1H), 7.00 (dd, J = 8.1, 1.9 Hz, 1H), 6.81 (d, J = 7.8 Hz, 1H), 6.42 (s, 1H), 4.12 (dt, J = 6.8, 6.3 Hz, 2H), 3.58 (s, 4H), 2.45 (d, J = 6.3 Hz, 2H), 2.31 (t, J = 4.7 Hz, 4H), 2.03 (s, 2H), 1.92 (dd, J = 3.8, 3.6 Hz, 2H). HRMS (ESI): m/z calculated for C22H24FN3O3 397.4427, Found 397.4425.
5-Methylisatin (3c, 0.319 g, 2.00 mmol, 1.0 equiv), 1-[2-(4-aminophenoxyl)propyl]morpholine (4c, 0.470 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 10 h until the TLC analysis showed that the reaction was completed. The reaction mixture was then cooled down to room temperature until there was a large amount of orange-red solids being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to produce the orange-red solid as the desired target compound (T11, 0.476 g, 50%). m. p. 178–180°C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 10.80 (d, J = 8.2 Hz, 1H), 7.24–7.14 (m, 2H), 7.04 (d, J = 8.9 Hz, 1H), 6.97 (d, J = 8.8 Hz, 1H), 6.92–6.71 (m, 2H), 6.50 (s, 1H), 4.10–3.98 (m, 2H), 3.61–3.53 (m, 4H), 2.51 (d, J = 1.6 Hz, 1H), 2.45 (t, J = 7.1 Hz, 2H), 2.38 (s, 4H), 2.28 (s, 1H), 2.01 (s, 3H), 1.95–1.84 (m, 2H). HRMS (ESI): m/z calculated for C22H25N3O3 379.4522, Found 379.4526.
5-Methylisatin (3c, 0.319 g, 2.00 mmol, 1.0 equiv), 1-[2-(2-chloro-4-aminophenoxyl) propylenepropyl]morpholine (4d, 0.540 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 10 h until the TLC analysis showed the completion of the reaction. The reaction system was then cooled down to room temperature until there was a large amount of orange-yellow solid being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to produce the orange-yellow solid as the desired target compound (T12, 0.282 g, 34%). m. p. 113–114°C (dichloromethane/methanol, 10:1, Rf = 0.4), 1H NMR (400 MHz, DMSO-d6) δ: 10.80 (d, J = 8.3 Hz, 1H), 7.26–7.12 (m, 1H), 7.05 (d, J = 8.6 Hz, 1H), 6.97 (d, J = 8.6 Hz, 1H), 6.92–6.71 (m, 2H), 6.50 (s, 1H), 4.10 (dt, J = 6.1, 5.8 Hz, 2H), 2.69 (t, J = 5.5 Hz, 2H), 2.51 (s, 1H), 2.46 (s, 3H), 2.29 (s, 1H), 2.01 (s, 2H), 1.53–1.47 (m, 4H), 1.39 (d, J = 4.7 Hz, 2H). HRMS (ESI): m/z calculated for C22H24ClN3O3 413.8973, Found 413.8975.
5-Bromo-isatin (3d, 0.449 g, 2.00 mmol, 1.0 equiv), 1-[2-(4-aminophenoxyl)ethyl]morpholine (4a, 0.444 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 13 h until the TLC analysis showed that the reaction was completed. The reaction system was then cooled down to room temperature until there was a large amount of orange-red solids being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to produce the orange-red solid as the target compound (T13, 0.610 g, 71%). m. p. 151–153°C (dichloromethane/methanol, 10:1, Rf = 0.5), 1H NMR (400 MHz, DMSO-d6) δ: 11.06 (d, J = 6.4 Hz, 1H), 7.56 (dd, J = 8.7, 8.3, 2.0 Hz, 1H), 7.26 (d, J = 8.8 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.95–6.80 (m, 2H), 6.72 (d, J = 1.7 Hz, 1H), 4.20–4.08 (m, 2H), 3.67–3.53 (m, 4H), 2.74 (t, J = 5.6 Hz, 2H). HRMS (ESI): m/z calculated for C20H20BrN3O3 430.2951, Found 430.2953.
5-Bromo-isatin (3d, 0.449 g, 2.00 mmol, 1.0 equiv), 1-[2-(2-fluoro-4-aminophenoxyl)propyl] morpholine (4b, 0.508 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 9 h until the TLC analysis showed that the reaction was completed. The reaction system was then cooled down to room temperature until there was a large amount of red solids being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to yield the red solid as the target compound (T14, 0.285 g, 31%). m. p. 143–144°C (dichloromethane/methanol, 10:1, Rf = 0.5), 1H NMR (400 MHz, DMSO-d6) δ: 11.09 (d, J = 8.9 Hz, 1H), 7.68–7.52 (m, 1H), 7.35–7.11 (m, 1H), 7.08–6.96 (m, 1H), 6.91–6.81 (m, 2H), 6.66 (d, J = 1.7 Hz, 1H), 4.19–4.05 (m, 2H), 3.58 (t, J = 4.4 Hz, 4H), 2.45 (dd, J = 2.9, 6.1 Hz, 2H), 2.39 (s, 4H), 1.92 (dd, J = 3.2, 6.7 Hz, 2H). HRMS (ESI): m/z calculated for C21H21BrFN3O3 462.3121, Found 462.3120.
5-Bromo-isatin (3d, 0.449 g, 2.00 mmol, 1.0 equiv) and 1-[2-(4-aminophenoxyl)propyl] morpholine (4c, 0.470 g, 4.6 mmol, 2.3 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 13 h until the TLC analysis showed that the reaction was completed. The reaction system was then cooled down to room temperature until there was a large amount of red solids being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to yield the red solid as the target compound (T15, 0.702 g, 79%). m. p. 181–182°C (dichloromethane/methanol, 10:1, Rf = 0.4). 1H NMR (400 MHz, DMSO-d6) δ: 11.06 (d, J = 8.8 Hz, 1H), 7.55 (dd, J = 9.4, 8.3, 1.9 Hz, 1H), 7.27 (d, J = 8.8 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 6.93–6.88 (m, 1H), 6.87–6.80 (m, 1H), 6.73 (d, J = 1.8 Hz, 1H), 4.10–4.00 (m, 2H), 3.58 (t, J = 4.5 Hz, 4H), 2.51 (s, 1H), 2.43 (dd, J = 3.4, 6.3 Hz, 2H), 2.37 (s, 4H), 1.96–1.83 (m, 2H). HRMS (ESI): m/z calculated for C21H22BrN3O3 444.3217, Found 444.3216.
5-Bromo-isatin (3d, 0.449 g, 2.00 mmol, 1.0 equiv) and 1-[2-(2-chloro-4-aminophenoxyl)propyl] morpholine (4d, 0.540 g, 2.00 mmol, 1.0 equiv) were added to a 50-ml flask, to which was added absolute ethanol (10.0 ml) and acetic acid (1–2 drops) at room temperature under stirring. The reaction mixture was heated to reflux for 11 h until the TLC analysis showed that the reaction was completed. The reaction system was then cooled down to room temperature until there was a large amount of red solids being precipitated. The solids were obtained via filtration under the reduced pressure. The filter cake was dried under vacuo to yield the red solid as the target compound (T16, 0.595 g, 62%). m. p. 147–149°C (dichloromethane/methanol, 10:1, Rf = 0.4). 1H NMR (400 MHz, DMSO-d6) δ: 11.08 (dd, J = 8.7, 9.4 Hz, 1H), 7.67–7.50 (m, 1H), 7.32–7.20 (m, 1H), 7.20–7.10 (m, 1H), 7.04 (dt, J = 9.0, 8.7 Hz, 2H), 6.94–6.80 (m, 1H), 6.71 (dd, J = 7.7, 1.7 Hz, 1H), 4.20–4.01 (m, 2H), 3.57 (d, J = 2.5 Hz, 4H), 2.51 (d, J = 1.5 Hz, 3H), 2.44 (s, 3H), 2.02–1.80 (m, 2H). HRMS (ESI): m/z calculated for C21H21BrClN3O3 478.7667, Found 478.7669.
For cell culture and transfection, Chinese hamster ovary (CHO) cells were cultured in Dulbecco’s modified Eagle’s medium/Ham’s F-12 (DMEM/F-12) medium supplemented with 10% fetal bovine serum and antibiotics (100 IU/ml penicillin and 100 mg/ml streptomycin) at 37°C in a humidified atmosphere with 5% CO2 and 95% air. Cells were passaged every 3–4 days with trypsin–EDTA, and a part of treated cells were seeded onto glass cover slips (5 × 3 mm2) for later transfection. The mammalian expression vector pcDNA3.1, which contained full-length cDNA encoding human Kv1.5 (hKv1.5), was kindly provided by Professor David Fedida (University of British Columbia, Canada). Wild-type hKv1.5 channel cDNA (0.5 μg) and green fluorescent protein (GFP) cDNA (0.5 μg) were transfected transiently into CHO cells by using Lipofectamine (Invitrogen Life Technologies, Carlsbad, CA, United States ). After transfecting for 48 h, the patch clamp experiments were conducted on GFP-positive cells. To prepare the materials of the drug candidates, the target compounds were dissolved in dimethyl sulphoxide (DMSO) to yield the stock solutions of 50 mM. The concentration of DMSO in the final solution was less than 0.1% (V/V), which had no effects on hKv1.5 channel current in the detective level.
The cells which could radiate the green fluorescence under UV excitation were selected for the experimental data recording. The Kv1.5 current was recorded from the cell which was green fluorescent protein (GFP)-positive 48 h after transfection at room temperature (25°C), using an EPC-8 patch-clamp amplifier (HEKA, Lambrecht, Germany), which has been recorded in the reported literature (Wu et al., 2009). Patch electrodes were fabricated from the glass capillaries (Narishige Scientific Instrument Lab, Tokyo, Japan) using a P-97 horizontal puller (Sutter Instrument Co., Novato, CA, United States ). These electrodes had a resistance of 2.0–3.0 MΩ when they were filled with the pipette solution containing 70 mM potassium aspartate, 40 mM KCl, 10 mM KH2PO4, 1 mM MgSO4, 3 mM adenosine 5-triphosphate (disodium salt; Sigma Chemical Company, St. Louis, MO, United States ), 0.1 mM dilithium salt of guanosine 5′-triphosphate (Roche Diagnostics GmbH Company, Mannheim, Germany), 5 mM EGTA, and 5 mM HEPES (pH was adjusted to 7.2 with 1 M KOH solution). Cells attached to glass cover slips were transferred to a recording chamber (0.5 ml in volume) mounted on the stage of an inverted microscope (ECLIPSE TE2000-U, Nikon, Tokyo, Japan), and perfused continuously at a flow rate of 1–2 ml/min with Tyrode solution containing 140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 0.33 mM NaH2PO4, 5.5 mM glucose, and 5.0 mM HEPES (pH was adjusted to 7.4 with 1 M NaOH). The data were then low-pass filtered at 10 kHz and sampled at 50 kHz during measuring the activation time course of hKv1.5 current, and the series resistances were usually compensated by 80%.
The hKv1.5 channel current was elicited by applying 300 ms depolarizing steps from a holding potential of −80 mV to various levels of −50 mV to +50 mV in 10 mV steps with a return potential of −40 mV. The stimulus interval of each stepwise was longer than 10 s to ensure that the channel fully recovered from the inactive state except for the frequency-dependent experiment. Apart from being specified, 30 mV was used to depolarize the voltage when the inhibition effects of drugs on the current were detected while using 300-ms bandwidth to excite the final current. The voltage-dependent activation of the hKv1.5 channel was assessed by fitting with a Boltzmann equation:
In the above equation, Itail is the tail current amplitude normalized with reference to the maximum value (as the 100%) measured at +50 mV; V1/2 is the voltage at half-maximal activation; Vm is the test potential and k is the slope factor.
The inhibition rates (IRs) of the target compounds against the Kv1.5 channel were calculated based on the value of Itail, which were recorded before and after the drug administration. The equation for calculating the IR was shown as follows.
The concentration-response curve for inhibition of hKv1.5 current by the control group was drawn by a least-squares fit of a Hill equation: %Control = 1/(1 + (IC50/[D])nH), where %Control represents the current in the presence of the drug normalized with reference to the control amplitude (expressed as a percentage). IC50 is the concentration of the control group causing a half-maximal inhibition. nH is the Hill coefficient and [D] is drug concentration. The apparent rate constants for binding (k+1) and unbinding (k-1) were obtained from fitting the equation: tD = 1/(k+1 [D] + k-1), where tD is the time constant induced by the drug, which was calculated from a single exponential fit to the traces of current decay during the depolarizing step to +30 mV. The apparent dissociation constant KD is expressed as KD = k-1/k+1. The deactivation kinetics was determined by fitting a single exponential function to the tail current trace.
All of the averaged data are presented as mean ± SEM (n = number of cells). Student’s t-test or ANOVA with Dunnett’s post hoc test was used to evaluate the statistical significance, which is considered to be statistically significant if a p value of < 0.05 was obtained.
Besides the Kv1.5 inhibition activity, the vasodilation activity of the Kv1.5 inhibitors was also tested in vitro based on our previous research work (Chen et al., 2016a; Chen et al., 2016b; Chen et al., 2016c; Chen et al., 2017; Chen et al., 2018). Briefly, the vasoconstriction model of the rat superior mesenteric artery was induced by 60 mM KCl or 10 μM PE, and target compounds were added to detect the vasodilation effects on pre-contracted vessels by using the microvascular tension measurement system Myograph (DMT620M, Danish Myo Technology A/S Company, Denmark).
The male Sprague-Dawley (SD) rats (SPF, 8 weeks old, 200–250 g) were obtained as the experimental animals from the Animal Centre of Xi’an Jiaotong University. The SD rats were euthanized using CO2. The superior mesenteric artery was gently removed and freed from adhering tissue under a dissecting microscope and then cut into 2- to 3-mm cylindrical arterial rings. The arterial rings were immersed in eight individual myograph baths (37°C) (Organ Bath Model 700MO, J.P. Trading, Aarhus, Denmark) containing 5 ml physiologic buffer solution (PSS) (pH 7.4, 119 mM NaCl, 4.6 mM KCl, 15 mM NaHCO3, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 1.5 mM CaCl2, and 5.5 mM glucose). The myograph baths were continuously aerated with 5% CO2 in O2.
The arterial segments were mounted for continuous recording of isometric tension using LabChart7 Pro software (AD Instruments, Hastings, United Kingdom). A resting tension of 2 mN was applied to each arterial ring, which was stabilized under this tension for at least 1.5–2.0 h. Then the rings were exposed to a potassium buffer solution (KPSS) (60 mM KCl in PSS). The maximum contraction induced by potassium was considered as a reference for contractile capacity. When potassium-induced reproducible responses were over 5 mN, the arterial rings could be used for experiments.
The vascular relaxation effects after the cumulative administration of tested compounds on artery rings was analyzed by the concentration-response curve for the chemicals (10–10–10−4 M) after initial potassium-induced contraction. DMSO was chosen as the control. The following formula is used to calculate the vasodilation rate of the target compound against the pre-contracted vascular rings (Equation 3).
Equation 3. Calculation equation of the vasodilation rate of the tested compounds.
The studies in vitro were approved by the Laboratory Animal Ethics Administration Committee of Xi’an Medical University and implemented according to the Guide for the Care and Use of Laboratory Animals Published by the United States National Institutes of Health (NIH Publication No. 85–23, revision in 1996).
All data were expressed as mean ± SD (standard deviation). The tested compound-induced vasodilation was presented as a percentage of response induced by KCl. One-way ANOVA with Dunnett’s post-test was applied for comparisons of more than two data sets. A t-test was applied for comparisons of two data sets. A p value of < 0.05 was considered to be significant. The analysis was performed by using GraphPad software (GraphPad Software, San Diego, United States ).
The chemical structures of all the target molecules were drawn using ChemBio Draw Ultra 12.0, which were opened in ChemBio3D Ultra 12.0. These molecules were named and minimized via the MM-semi-empirical method, which were then saved as Mol2 files one by one. The molecular descriptors were also calculated using ChemBio3D Ultra 12.0, which include the parameters of Stretch, Bend, Stretch-Bend, Torsion, Non-1,4 VDW, 1,4 VDW, Dipole/Dipole, and Total Energy.
The Mol2 file of each molecule was opened in the Sybyl workstation and was renamed again with the same name. A new database including all these 16 compounds was established based on the above information. All of the molecules were minimized in a Molecule Spread Sheet (MSS) file using the default value except for the following four settings. The force field was chosen as “Tripos.” The charge was chosen as “Gasteiger-Huckel.” “Max. Iterations” was set to be 10,000. “Gradient” was set to be 0.005. “Color option” was set to be Force. The minimization was repeated while there is still “Some columns may need to be recomputed” popped up in the column of “Console Command” of the Sybyl work-space. Minimization could also be stopped until the total energy values obtained from the last three conformation states of the molecule were the same without changing again.
Molecular modeling was carried out using the Tripos molecular modeling packages Sybyl-X2.0. All the molecules for docking were built using standard bond lengths and angles from Sybyl-X2.0/base Builder and were then optimized using the Tripos force field for 2000 generations two times or more, until the minimized conformers of the ligand were the same. The flexible docking method, called Surflex-Dock, docks the molecules automatically into the binding site of the receptor by using a protocol-based approach and an empirically derived scoring function. The protocol is a computational representation of a putative ligand that binds to the intended binding site and is a unique and essential element of the docking algorithm. The scoring function in Surflex-Dock, which contains hydrophobic, polar, repulsive, entropic, and salvation terms, was trained to estimate the dissociation constant (Kd) expressed in −log (Kd)2.
The 3D structure data of Kv1.5 was derived from its Kv1.2 homologues protein via the molecular homology method. Surflex-Dock default settings were used for other parameters, such as the number of starting conformations per molecule (set to 0), the size to expand the search grid (set to 8 Å), the maximum number of rotatable bonds per molecule (set to 100), and the maximum number of poses per ligand (set to 20). During the docking procedure, all of the single bonds in side chains of residues inside the defined binding pocket were regarded as rotatable or flexible. The ligands were allowed to rotate on all single bonds and move flexibly within the tentative binding pocket. The atomic charges were recalculated using the Kollman all-atom approach for the protein and the Gasteiger Hückel approach for the ligand. The binding interaction energy was calculated to include van der Waals, electrostatic, and torsional energy terms defined in the Tripos force field.
Based on target–ligand interaction and structure-activity analysis of traditional inhibitors and its simulated 3D structure of target Kv1.5, a series of compounds (T1–T16), containing both morpholine and 1H-indole linked with a chain-like structure (-N(CH2)nO-) in one molecule, were designed to enhance therapeutic effects of the anti-AF drugs (Figure 1).
As an anti-AF drug target with high selectivity on the atrial muscle cells, the undetermined crystal structure of the Kv1.5 channel impeded further new drug development. Herein, the simulated 3D structure of the Kv1.5 channel as the drug target was built based on the crystal structure of its homolog Kv1.2 channel and its amino acid sequence using the homology modeling technique. The crystal structure of the Kv1.2 channel (PDB ID: 2A79) was successfully determined by X-ray diffraction several years ago (Long et al., 2005). To be specific, the complete Kv1.2 channel consisted of the same four subunits and thereby formed a large tetramer. One subunit of Kv1.2 included the N terminus forming a T1 domain at the intracellular membrane surface, six transmembrane segments (S1–S6), and the C terminus. The transmembrane pore was composed by S5, pore helix, selectivity filter, and S6. Besides, the S1–S4 segments formed the voltage sensor (Long et al., 2005). The simulated 3D structure of the transmembrane region of the Kv1.5 channel was built using the homology modeling technique based on the transmembrane region of the Kv1.2 channel (PDB ID: 2A79), which reached over 70% amino acid sequence homology with the Kv1.5 channel. Unsurprisingly, the transmembrane region of Kv1.5 was similar to that of Kv1.2 and the single subunit of Kv1.5 mainly contained the pore mentioned above and the S4–S5 linker helix (Figure 2A). Furthermore, the four subunits of Kv1.5 formed a bundle crossing with a wide top and a narrow bottom; thus, the whole channel appeared like an inverted cone (Figures 2B,C).
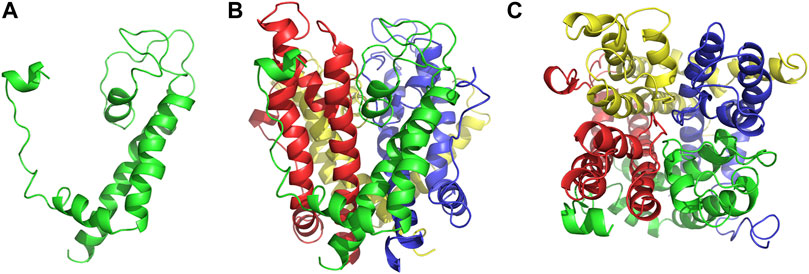
FIGURE 2. Simulated 3D structure of the transmembrane region of the Kv1.5 channel. (A) A single subunit of the transmembrane region of Kv1.5 viewed from the side. (B) Four subunits of the transmembrane region of Kv1.5 viewed from the side. (C) Four subunits of the transmembrane region of Kv1.5 viewed from the extracellular side of the pore. The subunits are represented as ribbons and colored individually.
The multiple descriptors of the target compounds via various kinds of software are summarized in Table 1 and Table 2, respectively.
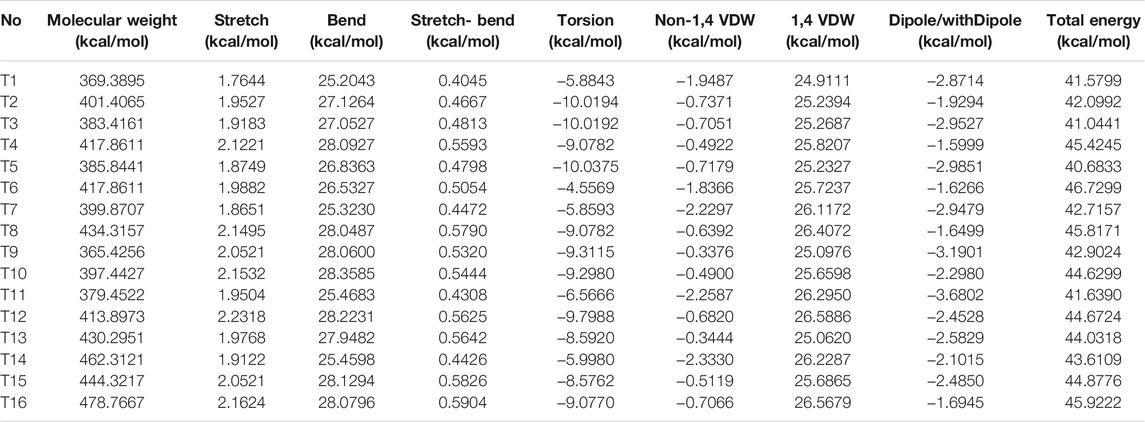
TABLE 1. Molecular descriptors of the target compounds calculated via ChemBio 3D software.
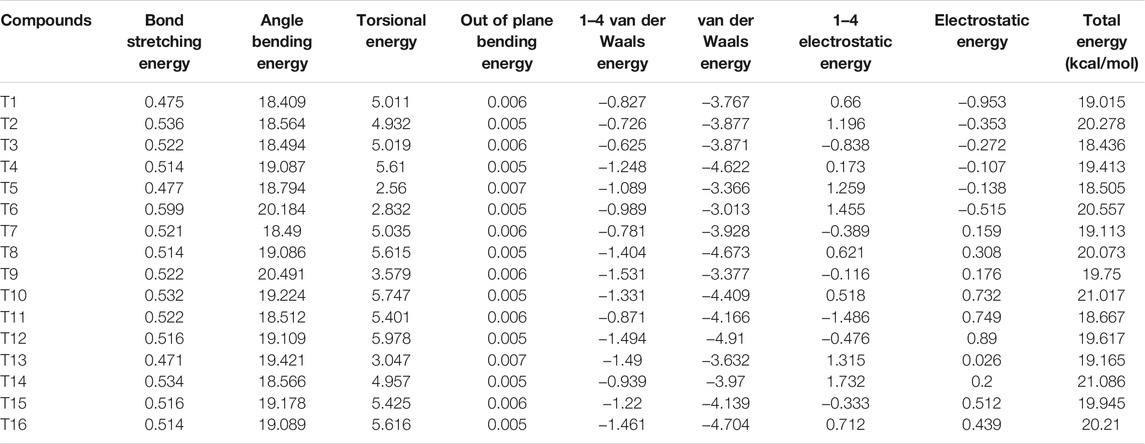
TABLE 2. Various energy data of the target compounds from energy minimization via Sybyl.
The simulated 3D structure of the Kv1.5 channel was used for docking analysis with all 16 target compounds. The docking result with the total scores (TSs) of each compound with the target protein Kv1.5 are summarized in Table 3.
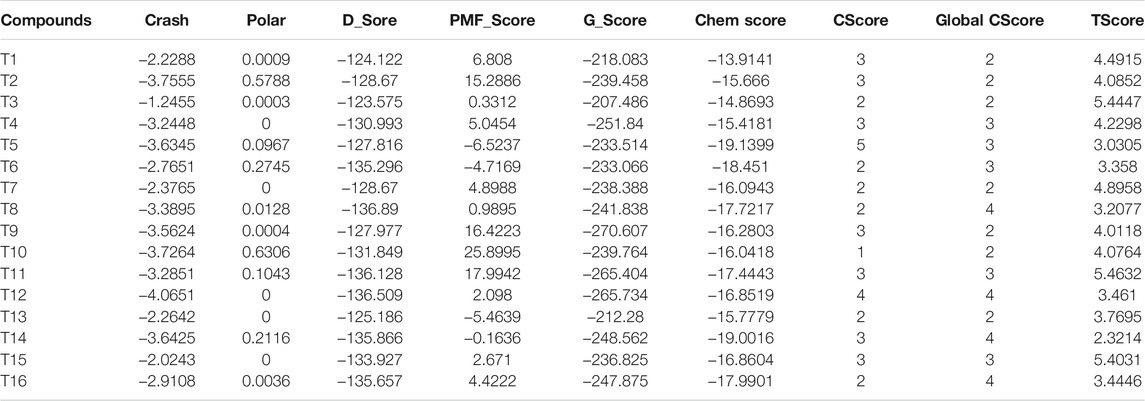
TABLE 3. Docking scores of the target compounds with Kv1.5 via Sybyl surflex software.
There are six compounds with CScore >3 and TScore >4. The docking TScore of T11 > T15 > T1 > T4 > T2 > T9 are 5.4632, 5.4031, 4.4915, 4.2298, 4.0852, and 4.0118, respectively.
Eight amino acid residues including T479, T480, R487, A501, I502, I508, V512, and V516 were identified to constitute the potential binding sites for some Kv1.5 channel blockers, such as vernakalant (Eldstrom et al., 2007), bupivacaine (Franqueza et al., 1997), anthranilic-acid derivative S0100176 (Decher et al., 2004), and arachidonic acid (Bai et al., 2015). Based on the above analysis, molecular docking of all the target compounds with the Kv1.5 channel was conducted via the Surflex dock module on the Sybyl workstation. According to the docking scores in Table 3, we choose two typical compounds (T11, CScore = 3, TScore = 5.4632; T10, CScore = 1, TScore = 4.0764) for analyzing the docking results.
The binding models of two compounds indicated that the binding site of the compounds were all in the central cavity of the Kv1.5 channel (Figure 3, Figure 4). The binding manner of T10 and T11 is similar (Figure 3, Figure 4). For both compound T11 and compound T10, hydrogen on 1H-indole ring is close to T480 on C chain (Figure 3), which forms a hydrogen bond with oxygen of the carbonyl group on the backbone of T479 on C chain (Figure 4).
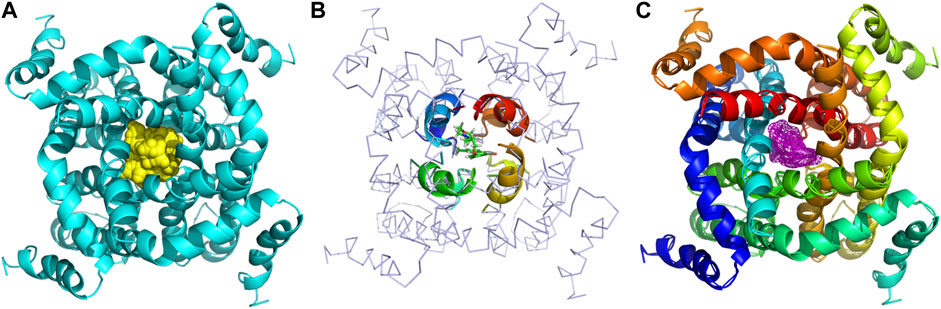
FIGURE 3. Simulated 3D complex structure of compound T11 with its target Kv1.5 channel using Surflex Docking. (A) Kv1.5 channel was shown as a tetramer colored in cyan, and the binding site in the center was shown in the surface colored in yellow. (B) Binding site of the Kv1.5 channel was shown in the rainbow cartoon. Compound T11 was shown in the green and white stick situated in the center of the channel site. (C) Kv1.5 channel was shown in the rainbow cartoon. Compound T11 in the center of the Kv1.5 channel site was shown in the magenta mesh.
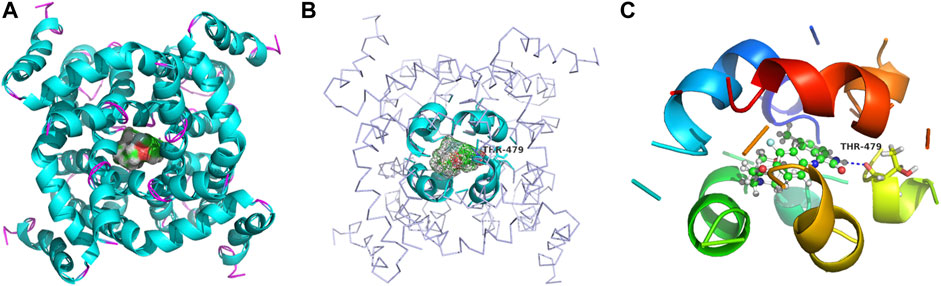
FIGURE 4. Simulated 3D complex structure of compound T10 with its target Kv1.5 channel using Surflex Docking. (A) T10 was shown in the surface while Kv1.5 was shown in the cyan cartoon. (B) Binding site around compound T10 (shown in mesh) was shown in the cyan cartoon. (C) Hydrogen bond (blue line) was formed between 1H of indole and O of the carbonyl group on the backbone of Thr479 amino acid residue.
The synthesis route of target compounds with the structure of 3-(morpholine substituted aromatic imine)-1H-indole is shown in Figure 5. The whole synthesis route contained three main reaction steps starting from the raw material of substituted aromatic amines (1a-1d). The important intermediates are 5-substituted isatin derivatives (3a-3d). Some of the main materials (4a-4d) were synthesized in the pharmaceutical chemistry laboratory of Xi’an Jiaotong University from the commercially available materials, including various substituted aromatic amines and substituted morpholines (Figure 5).
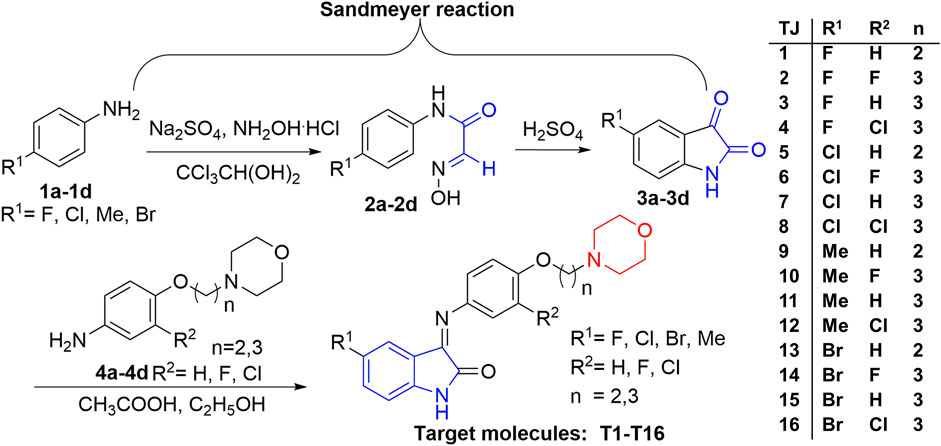
FIGURE 5. Synthesis route of the target compounds.
The yields and properties of the important intermediates including 4-substituted-N-hydroxyloxime acetyl aniline (2a-2d) and 5-substituted isatin (3a-3d) are summarized in Supplementary Table S1.
The Kv1.5 channel inhibition effects of all the target compounds were tested with the patch clamp technique. The results demonstrated that some compounds showed certain inhibition effects against the Kv1.5 channel among sixteen target compounds. Compound T16 showed the highest activity at the concentration of 100 μM with the inhibition rate of 70.8%. Compound T5 showed the relative higher activity at the concentration of 100 μM than the rest of the compounds with the inhibition rate of 57.5%. The sequence of the biological activity is listed as follows: T16 > T5 > T9 > T10 > T11 > T7. Compounds T9, T10, T11, and T7 showed the certain inhibitory effects at the low concentration (50 μM); the inhibition rates of these compounds are 20.8, 15.0, 11.3, and 9.5%, respectively (Table 4). Obviously, compounds T16 and T5 exhibited favorable inhibition effects against Kv1.5, which would provide promising lead compounds for anti-AF drug development.
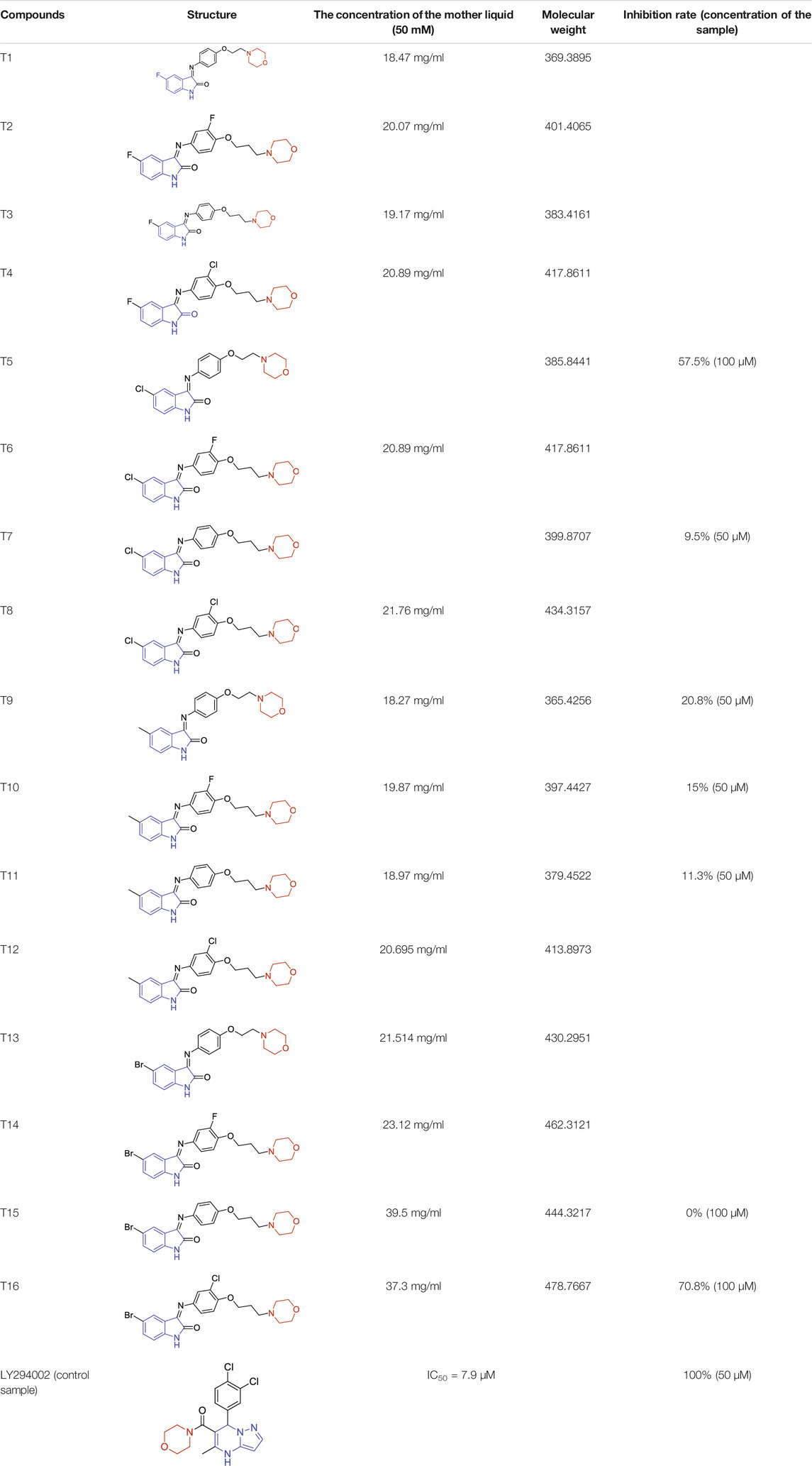
TABLE 4. Structure and the inhibitory actions on Kv1.5 of the target compounds.
There were twelve compounds (100 μM) among sixteen ones showed obvious vasodilation effects (28.96–93.39%) on superior mesenteric artery rings of SD rats in vitro pre-contracted by KCl (60 mM). To be specific, their vasodilation rates were as follows: 93.39% (T4), 91.11% (T16), 83.88% (T6), 78.12% (T15), 72.59% (T11), 68.92% (T8), 62.26% (T7), 48.30% (T5 and T9), 46.06% (T3), 43.78% (T13), and 28.96% (T1). Obviously, compounds T4 (93.39%) and T16 (91.11%) showed extraordinary vasodilation rates compared to other compounds (Figure 6A).
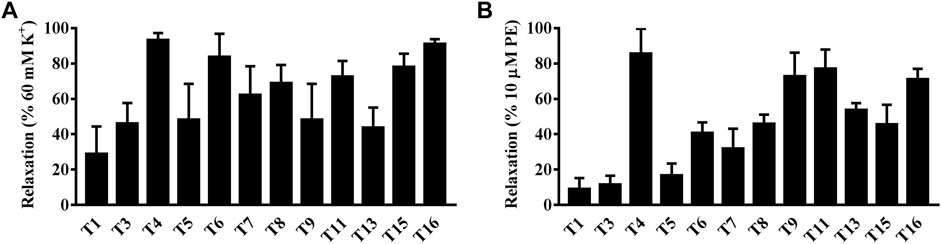
FIGURE 6. Vasodilation effects of target compounds on superior mesenteric artery rings in vitro pre-contracted by KCl (60 mM) or by phenylephrine hydrochloride (PE, 10 μM). (A) KCl. (B) PE.
There were twelve compounds (100 μM) among sixteen that showed the obvious vasodilation effects (9.12–85.63%) on the superior mesenteric artery rings of SD rats in vitro pre-contracted by PE (10 μM). To be specific, their relaxation rates were as follows: 85.63% (T4), 77.23% (T11), 72.75% (T9), 69.33% (T16), 53.88% (T13), 45.91% (T8), 45.62% (T15), 40.68% (T6), 31.96% (T7), 16.68% (T5), 11.65% (T3), and 9.12% (T1), respectively. Obviously, four samples including T4 (85.63%), T11 (77.23%), T9 (72.75%), and T16 (69.33%) showed the extraordinary relaxation rates compared to other compounds (Figure 6B).
Interestingly, both compounds T4 and T16 showed an outstanding vasodilation response on artery rings in vitro pre-contracted by KCl in a concentration-dependent manner. The vasodilation effects of compound T4 started at the concentration of 10−7 M, which could rise to 90.70% at the concentration of 10−4 M. Similarly, under the same level of concentration, the vasodilation response of compound T16 (10−4 M) was up to the highest value of 94.12% (Figure 7A). Both compounds T4 and T16 showed the remarkable vasodilation response on artery rings in vitro pre-contracted by PE in a concentration-dependent manner, which took effects at the concentration of 10−7 M. The vasodilation effects of compound T4 (10−4 M) could be up to 72.20%; compound T16 (10−4 M) was 61.00% (Figure 7B).
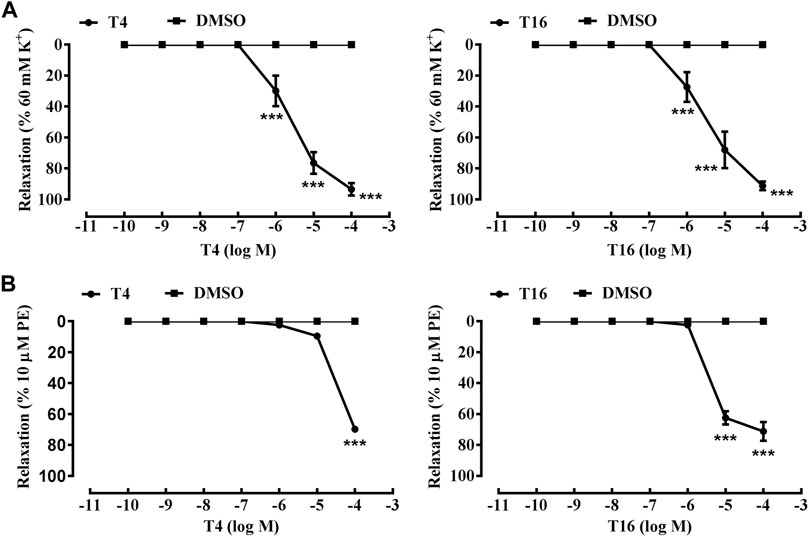
FIGURE 7. Vasodilation effects of compounds T4 and T16 on superior mesenteric artery rings in vitro pre-contracted by KCl (60 mM) or by phenylephrine hydrochloride (PE, 10 μM). (A) KCl. (B) PE. ***p < 0.05 versus a DMSO control (n = 3) indicates a significant difference compared with the control (DMSO).
In short, among sixteen target compounds, twelve compounds displayed desirable vasodilation effects on the superior mesenteric artery of SD rats in vitro pre-contracted by KCl or PE. As outstanding representatives, compounds T4 and T16 exhibited much more remarkable vasodilation effects than other compounds, which would provide reliable lead compounds for anti-hypertension drug development.
For both Kv1.5 channel inhibitory activity and vasodilation activity, T16 was the optimal one. As a result, T16 was analyzed with regard to its molecular interaction with its target Kv1.5 with the molecular docking.
The binding models of three compounds (T16, T11, and T10) indicated that the binding site of the compounds were all in the central cavity of the Kv1.5 channel (Figure 3, Figure 4, and Figure 8, Figure 9). Compounds T10 and T11 had similar binding manners (Figure 9). The most optimal compound T16 adopted a different binding manner, which is quite different from the other two compounds, T11 and T10. In compound T16, the hydrogen on indole ring is close to I508 on A chain. For both compounds T11 and T10, hydrogen on 1H-indole ring is close to T480 on C chain (Figure 3), which forms a hydrogen bond with oxygen of the carbonyl group on the backbone of T479 on C chain (Figure 3, Figure 4). Therefore, the different compounds might adopt the different manners while binding with the active site of the target Kv1.5 channel.
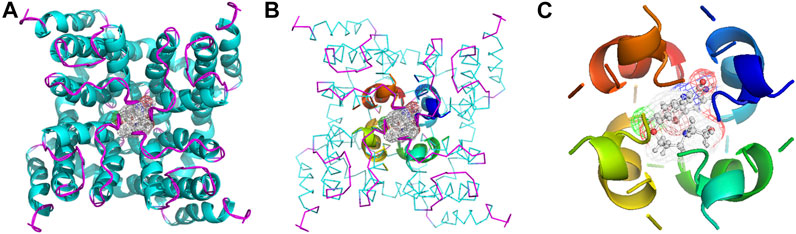
FIGURE 8. Simulated 3D complex structure of compound T16 with its target Kv1.5 channel using Surflex Docking. (A) Kv1.5 channel was shown according to its secondary structure, in which the α-helixes were shown in cyan, and β-sheets were shown in magenta. Compound T16 situated in the center of the channel was shown in mesh. (B) Binding site was shown in the rainbow cartoon. Compound T16 situated in the center of the channel was shown in mesh. Kv1.5 channel was shown in ribbon, the color of which was shown according to the secondary structure. (C) Binding site of the Kv1.5 channel was shown in the rainbow cartoon. Compound T16 in the center of the binding site was shown in the ball and stick model, which was surrounded with electronic mesh.
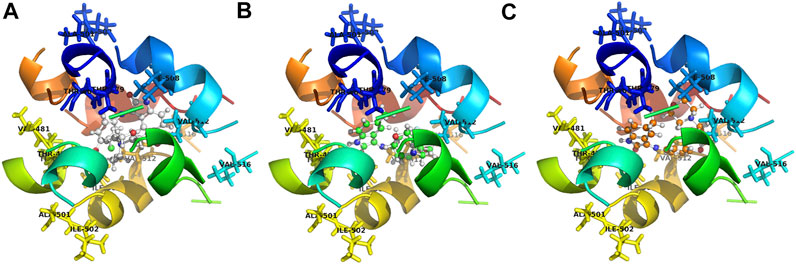
FIGURE 9. Contrasting simulated molecular interaction models of compounds T16, T11, T10 and their target Kv1.5 channel. The binding site of Kv1.5 channel is composed of the following amino acid residues: T479, T480, A501, I502, I508, V512, and V516 in the S6 domain and selectivity filter of a single subunit of the channel. The binding model of compounds T11 and T10 is similar, which is different from compound T16. (A) compound T16 (C: gray, H: gray, O: red, N: blue); (B) compound T11 (C: green, H: gray, O: red, N: blue); (C) compound T10 (C: orange, H: gray, O: red, N: blue). A chain was shown in blue, B chain was shown in green, C chain was shown in yellow, and D chain was shown in red.
It was also found that some of the amino acid residues in the binding sites on the Kv1.5 channel are crucial for binding with the compound T16 (Figure 8, Figure 9). The distances between the hydrogen atoms on I508, V512, and T480 and compound T16 were 2.8 Å, 2.5 Å, and 2.2 Å, respectively (Figure 9A). Some new target compounds in the study potently inhibited the hKv1.5 current in a voltage- and concentration-dependent manner at a lower concentration than the reported Kv1.5 inhibitors including hesperetin (Wang et al., 2016), papaverine (Choe et al., 2003), propofol (Yang et al., 2015), and cortisone (Yu et al., 2015). Therefore, acacetin showed the distances of 2.64 Å and 0.95 Å between hydrogen atoms of I508 and V512, which was set as a comparison (Wu et al., 2011). The results promoted our understanding on the molecular interaction mechanism between the target Kv1.5 and its inhibitors (Decher et al., 2004; Decher et al., 2006); however, much remains to be done.
AF is the most common clinical sustained arrhythmia; common clinical drugs have the low atrial selectivity and might cause more severe ventricle arrhythmias while stopping AF. As an anti-AF drug target with high selectivity on the atrial muscle cells, the undetermined crystal structure of the Kv1.5 channel impeded further new drug development. Herein, sixteen 3-morpholine linked aromatic amino substituted 1H-indoles as novel Kv1.5 channel inhibitors were designed and synthesized targeting to the simulated 3D structure of Kv1.5, which was built based on the crystal structure of its homolog Kv1.2 channel and its amino acid sequence using the molecular homology modeling technique. Compounds T16 and T5 (100 μM) exhibited favorable inhibitory activity against the Kv1.5 channel with an inhibition rate of 70.8 and 57.5% using a patch clamp technique. The vasodilation rates of compounds T16 and T4 (100 μM) reached over 90% using a Myograph. The novel Kv1.5 inhibitors would provide potential lead compounds for both anti-atrial fibrillation and anti-hypertension new drug development.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the Laboratory Animal Ethics Administration Committee of Xi’an Medical University.
WQ, Y-HL, and JT conducted the experiments and wrote the manuscript. Y-HL and JT synthesized the compounds. JW, C-XH, LX, and GM studied Kv1.5 inhibition activity of the target compounds. XZ and P-QL researched their vasodilation activity. WQ, DZ, and H-JL carried on the molecular docking. S-PW, GM, and H-LC designed the experiments and polished the English writing. All authors have read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (U1932130, 21871055); the Key Program of Shaanxi Provincial Science and Technology Department (2021SF-303); the Key Program of Shaanxi Provincial Education Department (20JS134), the Program of Shaanxi Administration of Traditional Chinese Medicine (2019-ZZ-ZY009); and the Key Program of Weiyang District Bureau of Science, Technology and Industry Information Technology (201928).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We appreciated Professor Minyong Li in Shandong University for his work on the molecular docking.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.805594/full#supplementary-material
Ang, Y.-S., Rajamani, S., Haldar, S. M., and Hüser, J. (2020). A New Therapeutic Framework for Atrial Fibrillation Drug Development. Circ. Res. 127 (1), 184–201. doi:10.1161/circresaha.120.316576
Bai, J.-Y., Ding, W.-G., Kojima, A., Seto, T., and Matsuura, H. (2015). Putative Binding Sites for Arachidonic Acid on the Human Cardiac Kv1.5 Channel. Br. J. Pharmacol. 172 (22), 5281–5292. doi:10.1111/bph.13314
Chen, Y., Liu, H., Wang, X., Zhang, H., Liu, E., and Su, X. (2017). Homocysteine Up-Regulates Endothelin Type A Receptor in Vascular Smooth Muscle Cells through Sirt1/ERK1/2 Signaling Pathway. Microvasc. Res. 114, 34–40. doi:10.1016/j.mvr.2017.05.010
Chen, Y., Liu, H., Zhang, H., Liu, E., Xu, C.-B., and Su, X. (2016a). The sirt1/NF-kB Signaling Pathway Is Involved in Regulation of Endothelin Type B Receptors Mediated by Homocysteine in Vascular Smooth Muscle Cells. Biomed. Pharmacother. 84, 1979–1985. doi:10.1016/j.biopha.2016.11.011
Chen, Y., Xu, C., Wei, Y., Zhang, Y., and Cao, A. (2016b). Endothelium-dependent And-independent Relaxation Induced by Resveratrol in Rat superior Mesenteric Arteries. Exp. Ther. Med. 12 (4), 2241–2246. doi:10.3892/etm.2016.3605
Chen, Y., Zhang, H., Liu, E., Xu, C.-b., and Zhang, Y. (2016c). Homocysteine Regulates Endothelin Type B Receptors in Vascular Smooth Muscle Cells. Vasc. Pharmacol. 87, 100–109. doi:10.1016/j.vph.2016.08.011
Chen, Y., Zhang, H., Liu, H., Li, K., and Su, X. (2018). Homocysteine Up-Regulates ET B Receptors via Suppression of Autophagy in Vascular Smooth Muscle Cells. Microvasc. Res. 119, 13–21. doi:10.1016/j.mvr.2018.03.010
Choe, H., Lee, Y.-K., Lee, Y.-T., Choe, H., Ko, S.-H., Joo, C.-U., et al. (2003). Papaverine Blocks hKv1.5 Channel Current and Human Atrial Ultrarapid Delayed Rectifier K+ Currents. J. Pharmacol. Exp. Ther. 304 (2), 706–712. doi:10.1124/jpet.102.042770
Decher, N., Kumar, P., Gonzalez, T., Pirard, B., and Sanguinetti, M. C. (2006). Binding Site of a Novel Kv1.5 Blocker: a "foot in the Door" against Atrial Fibrillation. Mol. Pharmacol. 70 (4), 1204–1211. doi:10.1124/mol.106.026203
Decher, N., Pirard, B., Bundis, F., Peukert, S., Baringhaus, K.-H., Busch, A. E., et al. (2004). Molecular Basis for Kv1.5 Channel Block. J. Biol. Chem. 279 (1), 394–400. doi:10.1074/jbc.M307411200
Du, Y., Wang, T., Guo, J., Li, W., Yang, T., Szendrey, M., et al. (2021). Kv1.5 Channels Are Regulated by PKC-Mediated Endocytic Degradation. J. Biol. Chem. 296, 100514. doi:10.1016/j.jbc.2021.100514
Eldstrom, J., Wang, Z., Xu, H., Pourrier, M., Ezrin, A., Gibson, K., et al. (2007). The Molecular Basis of High-Affinity Binding of the Antiarrhythmic Compound Vernakalant (RSD1235) to Kv1.5 Channels. Mol. Pharmacol. 72 (6), 1522–1534. doi:10.1124/mol.107.039388
Franqueza, L., Longobardo, M., Vicente, J., Delpón, E., Tamkun, M. M., Tamargo, J., et al. (1997). Molecular Determinants of Stereoselective Bupivacaine Block of hKv1.5 Channels. Circ. Res. 81 (6), 1053–1064. doi:10.1161/01.res.81.6.1053
Gasparoli, L., D’Amico, M., Masselli, M., Pillozzi, S., Caves, R., Khuwaileh, R., et al. (2015). New Pyrimido-Indole Compound CD-160130 Preferentially Inhibits the KV11.1B Isoform and Produces Antileukemic Effects without Cardiotoxicity. Mol. Pharmacol. 87 (2), 183–196. doi:10.1124/mol.114.094920
Guo, X., Chen, W., Sun, H., and You, Q. (2016). Kv1.5 Inhibitors for Treatment of Atrial Fibrillation: A Tradeoff between Selectivity and Non-selectivity. Ctmc 16 (16), 1843–1854. doi:10.2174/1568026616666160315142647
Guo, X., Ma, X., Yang, Q., Xu, J., Huang, L., Jia, J., et al. (2014). Discovery of 1-aryloxyethyl Piperazine Derivatives as Kv1.5 Potassium Channel Inhibitors (Part I). Eur. J. Med. Chem. 81, 89–94. doi:10.1016/j.ejmech.2014.03.075
Guo, X., Shen, Z., Yu, P., Chu, H., Tang, Y., and You, Q. (2012). Discovery of Benzamide Derivatives as Potent Kv1.5 Inhibitors. J. Chin. Pharm. Sci. 21 (06), 553–560. doi:10.5246/jcps.2012.06.070
Guo, X., Yang, Q., Xu, J., Zhang, L., Chu, H., Yu, P., et al. (2013). Design and Bio-Evaluation of Indole Derivatives as Potent Kv1.5 Inhibitors. Bioorg. Med. Chem. 21 (21), 6466–6476. doi:10.1016/j.bmc.2013.08.041
Hong, D. H., Choi, I.-W., Son, Y. K., Kim, D.-J., Na, S. H., Jung, W.-K., et al. (2013). The Effect of PI3 Kinase Inhibitor LY294002 on Voltage-dependent K+ Channels in Rabbit Coronary Arterial Smooth Muscle Cells. Life Sci. 92 (17-19), 916–922. doi:10.1016/j.lfs.2013.03.006
Kaess, B. M., and Ehrlich, J. R. (2016). Rise and Fall of Dronedarone: the Need for New Drugs. Eur. Heart J. Cardiovasc. Pharmacother. 2 (2), 95–97. doi:10.1093/ehjcvp/pvv058
Lau, D. H., Linz, D., and Sanders, P. (2019). New Findings in Atrial Fibrillation Mechanisms. Card. Electrophysiol. Clin. 11 (4), 563–571. doi:10.1016/j.ccep.2019.08.007
Long, S. B., Campbell, E. B., and Mackinnon, R. (2005). Crystal Structure of a Mammalian Voltage-dependent Shaker Family K + Channel. Science 309 (5736), 897–903. doi:10.1126/science.1116269
Moreira, D. R. M., Costa, S. P. M., Hernandes, M. Z., Rabello, M. M., de Oliveira Filho, G. B., de Melo, C. M. L., et al. (2012). Structural Investigation of Anti-trypanosoma Cruzi 2-Iminothiazolidin-4-Ones Allows the Identification of Agents with Efficacy in Infected Mice. J. Med. Chem. 55 (24), 10918–10936. doi:10.1021/jm301518v
Mujović, N., Dobrev, D., Marinković, M., Russo, V., and Potpara, T. S. (2020). The Role of Amiodarone in Contemporary Management of Complex Cardiac Arrhythmias. Pharmacol. Res. 151, 104521. doi:10.1016/j.phrs.2019.104521
Tamargo, J., Caballero, R., Gómez, R., and Delpón, E. (2009). IKur/Kv1.5 Channel Blockers for the Treatment of Atrial Fibrillation. Expert Opin. Investig. Drugs 18 (4), 399–416. doi:10.1517/13543780902762850
Vaccaro, W., Huynh, T., Lloyd, J., Atwal, K., Finlay, H. J., Levesque, P., et al. (2008). Dihydropyrazolopyrimidine Inhibitors of KV1.5 (IKur). Bioorg. Med. Chem. Lett. 18 (24), 6381–6385. doi:10.1016/j.bmcl.2008.10.099
Wang, H., Wang, H.-f., Wang, C., Chen, Y.-f., Ma, R., Xiang, J.-z., et al. (2016). Inhibitory Effects of Hesperetin on Kv1.5 Potassium Channels Stably Expressed in HEK 293 Cells and Ultra-rapid Delayed Rectifier K + Current in Human Atrial Myocytes. Eur. J. Pharmacol. 789, 98–108. doi:10.1016/j.ejphar.2016.07.015
Wettwer, E., and Terlau, H. (2014). Pharmacology of Voltage-Gated Potassium Channel Kv1.5-impact on Cardiac Excitability. Curr. Opin. Pharmacol. 15, 115–121. doi:10.1016/j.coph.2014.02.001
Wu, H.-J., Wu, W., Sun, H.-Y., Qin, G.-W., Wang, H.-B., Wang, P., et al. (2011). Acacetin Causes a Frequency- and Use-dependent Blockade of hKv1.5 Channels by Binding to the S6 Domain. J. Mol. Cell Cardiol. 51 (6), 966–973. doi:10.1016/j.yjmcc.2011.08.022
Wu, J., Ding, W.-G., Matsuura, H., Tsuji, K., Zang, W.-J., and Horie, M. (2009). Inhibitory Actions of the Phosphatidylinositol 3-kinase Inhibitor LY294002 on the Human Kv1.5 Channel. Br. J. Pharmacol. 156 (2), 377–387. doi:10.1111/j.1476-5381.2008.00017.x
Yang, L., Liu, H., Sun, H.-Y., and Li, G.-R. (2015). Intravenous Anesthetic Propofol Inhibits Multiple Human Cardiac Potassium Channels. Anesthesiology 122 (3), 571–584. doi:10.1097/aln.0000000000000495
Yu, J., Park, M.-H., and Jo, S.-H. (2015). Inhibitory Effects of Cortisone and Hydrocortisone on Human Kv1.5 Channel Currents. Eur. J. Pharmacol. 746, 158–166. doi:10.1016/j.ejphar.2014.11.007
Zhao, Z., Ruan, S., Ma, X., Feng, Q., Xie, Z., Nie, Z., et al. (2019). Challenges Faced with Small Molecular Modulators of Potassium Current Channel Isoform Kv1.5. Biomolecules 10 (1), 10. doi:10.3390/biom10010010
Keywords: Kv1.5 inhibitor, drug design, anti-atrial fibrillation, anti-hypertension, lead compound
Citation: Qin W, Li Y-H, Tong J, Wu J, Zhao D, Li H-J, Xing L, He C-X, Zhou X, Li P-Q, Meng G, Wu S-P and Cao H-L (2022) Rational Design and Synthesis of 3-Morpholine Linked Aromatic-Imino-1H-Indoles as Novel Kv1.5 Channel Inhibitors Sharing Vasodilation Effects. Front. Mol. Biosci. 8:805594. doi: 10.3389/fmolb.2021.805594
Received: 30 October 2021; Accepted: 28 December 2021;
Published: 24 January 2022.
Edited by:
Leming Sun, Northwestern Polytechnical University, ChinaReviewed by:
Zhongqun Wang, Jiangsu University, ChinaCopyright © 2022 Qin, Li, Tong, Wu, Zhao, Li, Xing, He, Zhou, Li, Meng, Wu and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ge Meng, bWVuZ2dlQG1haWwueGp0dS5lZHUuY24=; Shao-Ping Wu, d3VzaGFvcGluZ0Bud3UuZWR1LmNu; Hui-Ling Cao, Y2FvaHVpbGluZ19qenNAeGl5aS5lZHUuY24=
†The authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.