
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 March 2024
Sec. Microbial Symbioses
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1361117
This article is part of the Research Topic Plant Microbiome: Interactions, Mechanisms of Action, and Applications, Volume III View all 20 articles
 Hongbo Guo1,2†
Hongbo Guo1,2† Weiye Liu3†Yuqi Xie1Zhenyu Wang1Chentong Huang1Jingfang Yi3Zhaoqian Yang3Jiachen Zhao3
Weiye Liu3†Yuqi Xie1Zhenyu Wang1Chentong Huang1Jingfang Yi3Zhaoqian Yang3Jiachen Zhao3 Xiaodan Yu3*
Xiaodan Yu3* Lidiya Alekseevna Sibirina2,4*
Lidiya Alekseevna Sibirina2,4*Tricholoma bakamatsutake is a delicious and nutritious ectomycorrhizal fungus. However, its cultivation is hindered owing to limited studies on its symbiotic relationships. The symbiotic relationship between T. bakamatsutake and its host is closely related to the shiro, a complex network composed of mycelium, mycorrhizal roots, and surrounding soil. To explore the symbiotic relationship between T. bakamatsutake and its host, soil samples were collected from T. bakamatsutake shiro (Tb) and corresponding Q. mongolica rhizosphere (CK) in four cities in Liaoning Province, China. The physicochemical properties of all the soil samples were then analyzed, along with the composition and function of the fungal and bacterial communities. The results revealed a significant increase in total potassium, available nitrogen, and sand in Tb soil compared to those in CK soil, while there was a significant decrease in pH, total nitrogen, total phosphorus, available phosphorus, and silt. The fungal community diversity in shiro was diminished, and T. bakamatsutake altered the community structure of its shiro by suppressing other fungi, such as Russula (ectomycorrhizal fungus) and Penicillium (phytopathogenic fungus). The bacterial community diversity in shiro increased, with the aggregation of mycorrhizal-helper bacteria, such as Paenibacillus and Bacillus, and plant growth-promoting bacteria, such as Solirubrobacter and Streptomyces, facilitated by T. bakamatsutake. Microbial functional predictions revealed a significant increase in pathways associated with sugar and fat catabolism within the fungal and bacterial communities of shiro. The relative genetic abundance of carboxylesterase and gibberellin 2-beta-dioxygenase in the fungal community was significantly increased, which suggested a potential symbiotic relationship between T. bakamatsutake and Q. mongolica. These findings elucidate the microbial community and relevant symbiotic environment to better understand the relationship between T. bakamatsutake and Q. mongolica.
Tricholoma bakamatsutake, an edible mushroom, often forms ectomycorrhizae with the roots of Fagaceae trees, such as Quercus mongolica (Deng, 2003; Ota et al., 2012). T. bakamatsutake exhibits similar morphological characteristics, aroma profiles, and nutritional composition to those of T. matsutake (Quan et al., 2005, 2007); however, it is priced lower in the market compared to T. matsutake (Kang et al., 2018). The growing demand for T. bakamatsutake necessitates its cultivation due to limited natural sources (Deng, 2019; Hou, 2020). Consequently, researchers are investigating optimal environmental conditions for the laboratory culture of T. bakamatsutake mycelia (Terashima, 1994, 1999; Wu et al., 2009; Yamanaka et al., 2019), while also attempting fruiting body production through transplantation into natural habitats (Kawai et al., 2018; Hou, 2020). However, disparities between natural and laboratory environments exist, and factors like soil physicochemical properties and microbiome composition may influence fruiting body yield in natural environments.
The underground mycelia of T. bakamatsutake form a white, sponge-like structure called shiro (Ogawa, 1977). In T. matsutake, the shiro plays an important role in the formation of fruiting bodies (Yun et al., 1997), which form at the core of the shiro. High-throughput sequencing has been widely used to study the microbiome in the shiro (Kim et al., 2014; Oh et al., 2016; Ye et al., 2018), allowing for the discovery of microbial diversity and non-culturable microorganisms by directly analyzing genetic material from shiro samples (Shokralla et al., 2012; Kim et al., 2014). Kim et al. (2014) investigated bacterial communities in T. matsutake shiro using pyrophosphate sequencing and found that environmental factors may have a greater influence on these communities than the dominance of T. matsutake. Oh et al. (2016) identified several microorganisms associated with T. matsutake, including Bacillus and Umbelopsis, which may benefit T. matsutake growth. Vaario et al. (2011) found that T. matsutake coexists with various fungal and actinobacterial species, and high enzymatic activity is involved in organic carbon degradation in the shiro. This approach can be used to study the symbiotic relationship between T. bakamatsutake and Quercus mongolica to provide a deeper understanding of T. bakamatsutake shiro and lay a foundation for future artificial cultivation. Additionally, T. bakamatsutake adapts more easily to the environment than T. matsutake, making it suitable for artificial cultivation (Deng, 2019; Hou, 2020). However, limited research on T. bakamatsutake shiro hinders its artificial cultivation and economic prospects.
Given its ectomycorrhizal (ECM) nature, the effects of T. bakamatsutake on other microorganisms, ecological milieu, and host plants necessitate careful consideration. ECM fungi colonize plant roots, modify the soil microbiome structure (Calvaruso et al., 2007; Li et al., 2019), and continuously spread their mycelia. The dominance of ECM fungi is driven by ecological niche competition, metabolite secretion, and the enrichment of mycorrhization helper bacteria (MHB) (Poole et al., 2001; Moeller and Peay, 2016). The symbiotic relationship between ECM fungi and host plants is important as the fungal mycelia closely adhere to plant roots, providing effective protection against pathogens for woody plants (Tedersoo et al., 2020). This symbiotic relationship also enhances nutrient exchange between ECM fungi and the plants; photosynthates are transported from the plants to the roots where ECM fungi reside, while minerals are transported from ECM fungi to the plants via mycorrhiza (Nehls et al., 2010; Nehls and Plassard, 2018). Additionally, ECM fungi play an important role in increasing plant resilience and improving forest environments (Song et al., 2015; Bennett et al., 2017; Yu and Yuan, 2023).
This study aimed to compare the soils of shiro and Q. mongolica rhizosphere to reveal the ecological relationship between T. bakamatsutake and Q. mongolica in natural forests. Further, the contributions of T. bakamatsutake to the host and environment were evaluated. The results herein contribute to a better understanding of the relationship between ECM fungi and their hosts in the micro-ecosystem, while providing a theoretical basis for the artificial cultivation of T. bakamatsutake.
In August 2022, 28 soil samples were collected from four cities in Liaoning Province, China, Anshan (AS), Kuandian (KD), Xinbin (XB), and Xiuyan (XY). The sampling sites were situated in a mixed forest comprising Q. mongolica and Lespedeza bicolor, with Carex callitrichos being the predominant herbaceous plant. All sampling locations were maintained at a minimum distance of 10 m from L. bicolor. These 28 samples included 14 T. bakamatsutake shiro soil (Tb) and 14 corresponding Q. mongolica rhizosphere soil (CK) samples. The Tb samples were collected from the shiro soil under the fruiting body of T. bakamatsutake. The CK soil samples were collected from the non-mycorrhizal soil of the oak rhizosphere, located 5 m away from the shiro, ensuring that both the CK and Tb samples originated from the same oak rhizosphere. Additionally, this distance not only mitigated potential impacts of shiro on the CK samples but also minimized environmental disparities between Tb and CK. All the samples were collected by first removing the surface litter layer, then collecting soil at a depth of 5–15 cm and diameter of 10 cm using a soil sampler. The samples were encoded in the format of city name followed by Tb/CK (e.g., ASTb represented the Tb soil from Anshan). Grouping and numbering details for all samples are provided in Table 1. The samples were stored at −4°C upon the removal of the gravel and roots and subsequently divided into three parts. The first was dried and passed through a 20-mesh sieve to determine the chemical properties; pH, total nitrogen (TN), total potassium (TK), total phosphorus (TP), available nitrogen (AN), available potassium (AK), available phosphorus (AP), and organic matter (OM) were measured using previous methods (Bao, 2000). The second sample was desiccated and subsequently sieved through a 100-mesh sieve to determine its mechanical composition using the Bouyoucos hydrometer method (Bouyoucos, 1936). The third was stored at −80°C for DNA extraction.
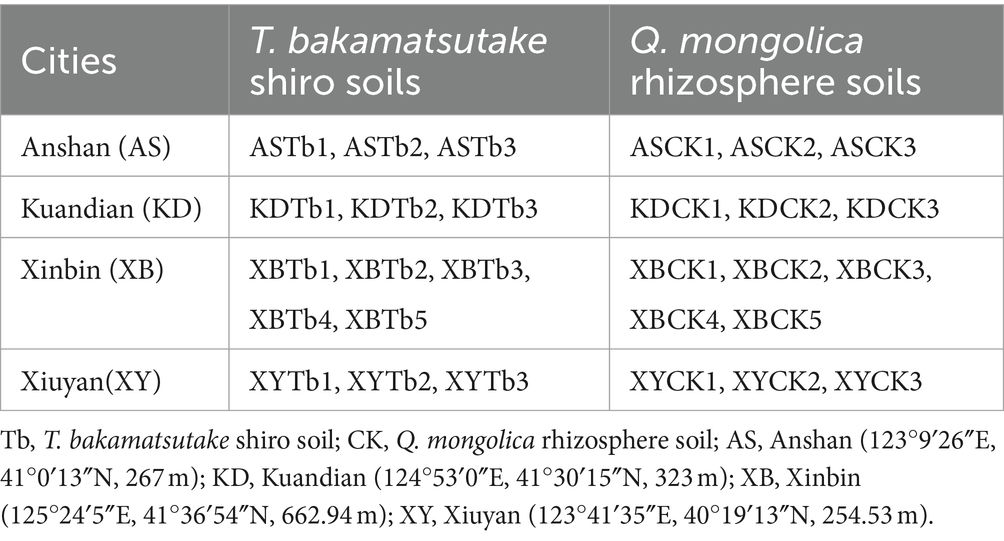
Table 1. Collection and grouping of the 28 soil samples.
Total genomic DNA of the microbiome was extracted using the E.Z.N.A.® soil DNA kit (Omega Bio-tek, Norcross, GA, United States) according to the manufacturer’s instructions. The quality of the extracted genomic DNA was determined using 1% agarose gel electrophoresis with a DYY-6C electrophoresis apparatus (Liuyi Biology, Beijing, China). The concentration and purity of the DNA were determined using a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, United States).
For bacteria, the 16S-specific primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) were used to amplify different regions of the 16S ribosomal RNA (rRNA) gene. For fungi, specific primers, including the internal transcribed spacer ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′), were used to amplify different regions of the ITS gene. The PCR mixture included 4 μL of 5 × Fast Pfu buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of Fast Pfu polymerase (TransGen Biotech, Beijing, China), 10 ng of template DNA, and ddH2O to a final volume of 20 μL. The cycling conditions were as follows: initial denaturation at 95°C for 3 min, followed by 27 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 45 s, single extension at 72°C for 10 min, and 4°C. The amplification of all the samples was performed in triplicate using the ABI GeneAmp® 9,700 instrument (ABI, CA, United States). The PCR products were extracted using a 2% agarose gel and purified with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) according to the manufacturer’s instructions. The purified PCR products were then quantified using a Quantus™ Fluorometer (Promega, Madison, WI, United States).
The purified PCR amplicons were pooled in equimolar amounts, paired-end, and sequenced on an Illumina MiSeq PE300 platform (Illumina, San Diego, CA, United States) according to the Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) protocol. Raw sequencing reads were deposited in the database of the National Center for Biotechnology Information Sequence Read Archive (BioProject: PRJNA955660).
Raw FASTQ files were de-multiplexed using an in-house perl script, quality-filtered using fastp version 0.19.6 (Chen et al., 2018), and merged using FLASH version 1.2.7 (Magoc and Salzberg, 2011). The optimized sequences were clustered into operational taxonomic units (OTUs) using UPARSE 7.1 (Stackebrandt and Goebel, 1994; Edgar, 2013), with a 97% sequence similarity level. The most abundant sequence in each OTU was selected as the representative sequence. The taxonomy of each OTU representative sequence was analyzed using RDP Classifier version 2.2 (Wang et al., 2007) against the Silva 16S rRNA and Unite ITS gene databases, with a confidence threshold of 0.7. Finally, metagenomic functions were predicted for the bacterial and fungal communities. The functions of the microbial community were predicted using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2), BugBase, FAPROTAX, and FunGuid, based on the OTU representative sequences. PICRUSt21 is a software comprising the following series of tools: HMMER is used to align OTU representative sequences with reference sequences; PA-NG and Gappa are used to place OTU representative sequences into a reference tree; castor is used to normalize gene copies; and MinPath is used to predict gene family profiles and allocate them into gene pathways. BugBase is a tool for predicting the 16S phenotypic functionality of microbiota and can be used as a web application.2 For the prediction and visualization of microbial phenotypes, OTU tables and mapping files should be inputted into the BugBase operating program. FAPROTAX is the ecological function database of prokaryotes and classifies the ecological roles of bacteria and archaea in the environment according to published literature. The collapse_table.py command in FAPROTAX can be employed to assign and summarize the functional groups of bacteria into OTU taxonomic tables (Louca et al., 2016). FunGuid3 is the ecological function database of fungi and was constructed based on existing literature. By uploading and analyzing the fungal relative OTU taxonomic table in FunGuid, predictive results can be obtained for the ecological functionality of the fungal community.
Data analyzes were performed using the Majorbio Cloud platform4 and R (v4.0.2). Mothur (Schloss et al., 2009) software5 was used to calculate the alpha diversity indices, such as the Chao and Shannon’s indices. The Wilcoxon rank-sum test was used for the inter-group variation analysis of the alpha diversity indices, and a non-metric multidimensional scaling (NMDS) analysis based on the Bray-Curtis distance algorithm was used to test the similarity between the samples’ microbial community structure. The PERMANOVA test was used to assess the percentage of variation explained by the treatment along with its statistical significance determined using the Vegan v2.5–3 package. A canonical correspondence analysis (CCA) and redundancy analysis (RDA) were used to investigate the interactions between the soil physicochemical indicators and microbial community structure. The Spearman’s correlation coefficient was used to investigate the relationship between the physicochemical properties of the soil and microbiota abundance, based on the composition distribution of the species. To detect differences between the samples and overall differences and exclude chance data, the significant differences between the two groups were analyzed using a multiple-group sample analysis (eight groups) and two-group analysis of Tb and CK. Multiple-group sample analyzes compared the mean of all samples within each group, while two-group analyzes compared the mean of all samples in the Tb groups with that of the CK groups. The means of the differences in the physicochemical properties of the samples were determined using Duncan’s multiple range test and T-tests in the SPSS Statistics software (version 17.0; IBM Inc., Armonk, NY, United States). The Kruskal–Wallis H test and Wilcoxon rank-sum test of variance were performed using the Majorbio Cloud platform.
The physicochemical properties of the samples in different groups showed significant differences after the Duncan’s multiple range test (p < 0.05) was conducted. The Tb group samples had significantly lower levels of pH, TP, TN, AP, and silt than did the CK group samples in most regions but had significantly higher levels of TK, AN, and sand (Table 2). The T-test yielded comparable findings for the overall analysis of both the CK and Tb groups (Supplementary Table S1).
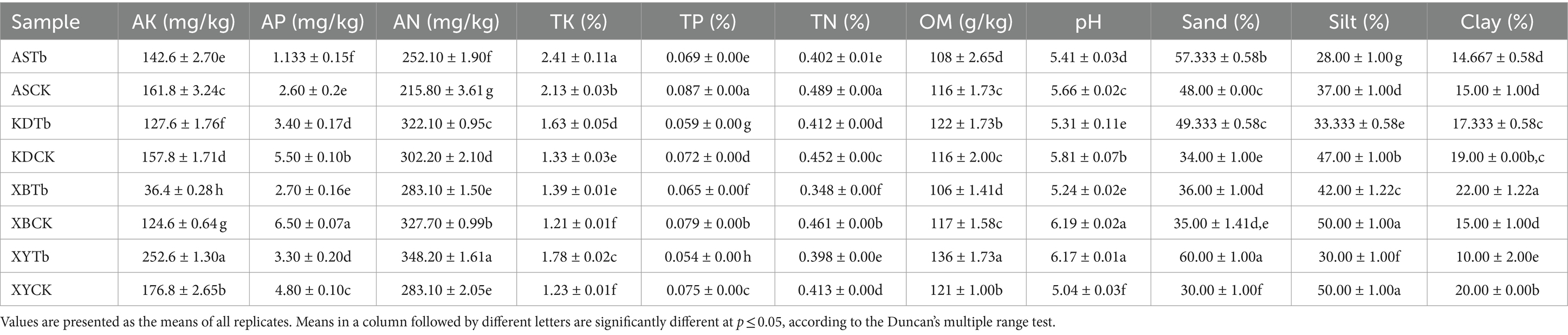
Table 2. Physicochemical properties of Tricholoma bakamatsutake shiro soils.
The raw data results from MiSeq for the fungal and bacterial communities are presented in Supplementary Tables S2, S3. Sample sequences were drawn flat according to the minimum number of sample sequences before the analysis. The fungal and bacterial coverage indices were 99.57 to 99.93% and 96.70 to 98.76%, respectively. This indicated that most of the fungal and bacterial taxa were detected in the soil samples, and the sequencing results correctly depicted the microorganisms in the samples.
For the fungal community, 47 fungal OTUs were shared between the Tb and CK groups (Supplementary Figure S1A). The CK groups had more unique OTUs than the Tb groups. The Tb and CK groups were analyzed separately (Supplementary Figure S2). The Tb groups had 63 shared OTUs, with XBTb and KDTb having the lowest (57) and highest (168) number of unique OTUs, respectively. The CK groups had 136 shared OTUs, with ASCK and XYCK having the highest (567) and lowest (92) number of unique OTUs, respectively. For the bacterial community, 663 bacterial OTUs were shared between the Tb and CK groups (Supplementary Figure S1B). The Tb groups had more unique OTUs than the CK groups. In the Tb groups, a total of 1,674 OTUs were observed (Supplementary Figure S3). XBTb had the highest number of unique OTUs (536), while KDTb had the lowest (157). In the CK groups, a total of 1,039 OTUs were observed. ASCK had the highest number of unique OTUs (816), while XYCK had the lowest (163).
The Chao and Shannon indices were used to assess the α diversity of the microbial community, with the former reflecting species richness and the latter reflecting species diversity (Supplementary Figure S4). In the fungal community, both indices of the Tb groups exhibited lower values than those of the CK groups, particularly in the Shannon index, which displayed highly significant differences across all regions (p < 0.001). Notably, the Xinbin (XB) region demonstrated both the highest (XBCK) and lowest (XBTb) values for these indices. Regarding the bacterial community, both indices of the Tb groups were higher than those of the CK groups; however, a significant difference was observed only in the Kuandian (KD) region (p < 0.01). These findings indicate a significant decrease in species richness and diversity in the fungal community of the shiro but an increasing trend in the bacterial community.
For the fungal community, Basidiomycota, Ascomycota, and Mortierellomycota were dominant in all the soil groups. Basidiomycota (96.21 to 99.77%) and Ascomycota (56.02 to 83.65%) were the dominant phyla in the Tb and CK groups, respectively. At the genus level, the heatmap and abundance bubble map (Figure 1) indicated that Tricholoma, Russula, and Mortierella were the dominant fungal genera in the different regions. Apart from Tricholoma, most genera in the Tb groups were less abundant than those in the CK groups. At the species level, T. bakamatsutake was the dominant species in the Tb groups, exhibiting 87.7, 88.0, 99.1, and 89.1% abundance in ASTb, KDTb, XBTb, and XYTb, respectively. For the bacterial community, Actinobacteria, Proteobacteria, and Acidobacteria were dominant in all the soil groups. Proteobacteria and Actinobacteria were the most abundant phyla in the CK and Tb groups, respectively. The genus-level heatmap (Figure 2A) and abundance bubble map (Figure 2B) indicated that the genera Bradyrhizobium, Acidothermus, Mycobacterium, norank_f__Xanthobacteraceae, and Burkholderia-Caballeronia-Paraburkholderia (BCP) were the dominant genera in both groups.
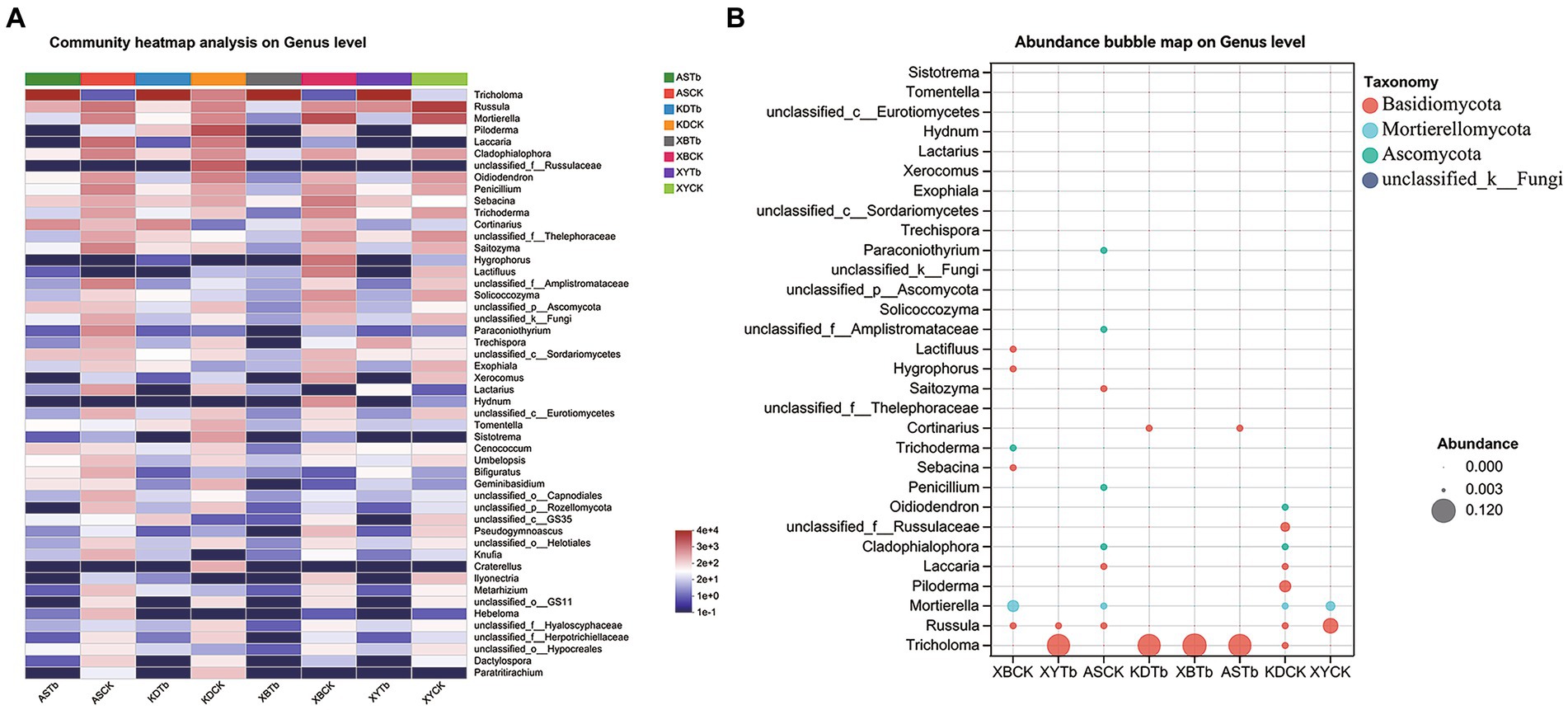
Figure 1. Composition of fungal communities at the genus level in the different groups. (A) Heatmap; (B) Abundance bubble map. Bubble size and color indicate the abundance and classification of the genus, respectively.
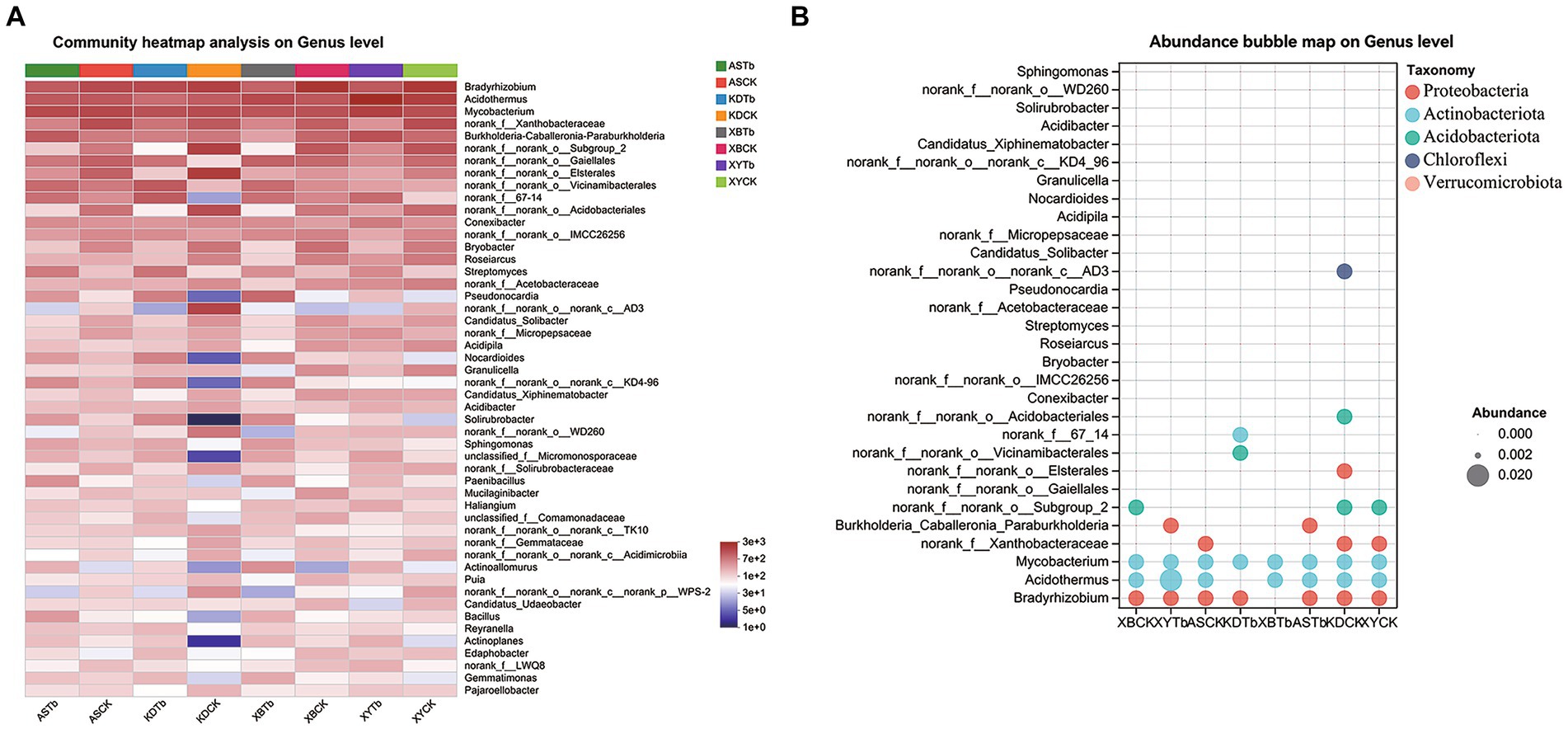
Figure 2. Composition of bacterial communities at the genus level in the different groups. (A) Heatmap; (B) Abundance bubble map. Bubble size and color indicate the abundance and classification of the genus, respectively.
The Kruskal–Wallis H test (eight groups) and Wilcoxon rank-sum test (CK and Tb) were used to analyze the differences in the abundance of microorganisms between the groups. In the fungal community (Supplementary Figure S5), T. bakamatsutake was significantly more abundant in the Tb groups than in the CK groups, whereas almost all the other fungi, such as Russula and Penicillium, were significantly lower in abundance in the Tb groups than in the CK groups. This suggests that the dominance of Tb has a broad and significant effect on other fungi. However, the dominance of one species did not result in an overwhelming change in the bacterial community. In the Tb groups (Supplementary Figure S6), there was a significant increase in Actinobacteria, such as norank_f__67–14, Streptomyces, Pseudonocardia, Bradyrhizobium, norank_f__Xanthobacteraceae, norank_f__norank_o__Elsterales, and Nocardioides. The multigroup analysis showed that BCP was significantly more abundant in ASTb and XYTb than in ASCK and XYCK but less abundant in KDTb and XBTb than in KDCK and XBCK. The abundance of common MHB (Frey-Klett et al., 2007) was further assessed in the analysis (Supplementary Figure S7). The identified MHB, which were significantly more abundant in the Tb groups than in the CK groups, were Sphingomonas, Paenibacillus, and Bacillus.
The NMDS analysis showed differences in the microbiome structures in the CK and Tb groups (Supplementary Figure S8). The fungal communities in the CK and Tb groups also differed. In the CK groups, KDCK was far from the other CK groups, indicating that the structure of the fungal communities of the oak forests in KD differed from that in other areas. The bacterial communities in both groups were generally similarly distant but different from each other.
The CCA/RDA analysis revealed significant impacts of the soil physicochemical properties on the bacterial and fungal communities (Figure 3). The AP, TP, TN, sand, and silt levels exhibited significant correlations with both the fungal and bacterial communities, indicating their pivotal roles in shaping and regulating the microbial community. The Spearman’s analysis revealed significant correlations between the microbial community and physicochemical properties of the soil (Figure 4; Supplementary Table S4). In the fungal community, T. bakamatsutake exhibited a positive correlation with the levels of clay, TK, and sand, while displaying a significant negative correlation with the levels of silt, pH, TN, TP, and AP. In the bacterial community, most of the bacteria demonstrated a significant positive correlation with the silt content as well as the TN, TP, and AP levels. Notably, several actinomycetes displayed similar patterns to T. bakamatsutake in terms of their associations with the physicochemical properties of the soil. For example, Streptomyces, Pseudonocardia, norank_f__67–14, Nocardioides, and Solirubrobacter exhibited a positive correlation with the TK and sand levels but a negative association with other physicochemical properties.
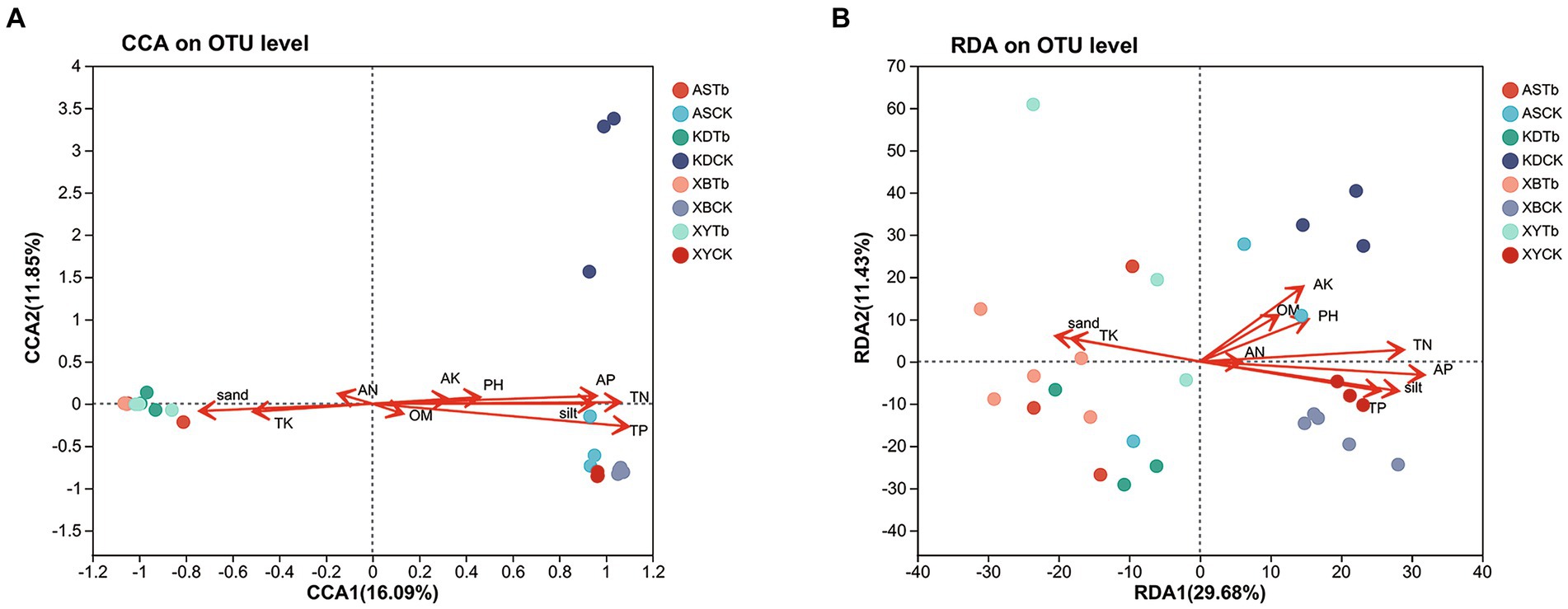
Figure 3. CCA/RDA analysis of the relationship between the microbial communities and soil physicochemical properties. (A) Fungal communities; (B) Bacterial communities. AP (r2 = 0.5386, p = 0.001), TP (r2 = 0.7456, p = 0.001), TN (r2 = 0.6587, p = 0.001), sand (r2 = 0.318, p = 0.01), and silt (r2 = 0.5227, p = 0.001) were significant factors that influenced fungal community structure. AK (r2 = 0.2546, p = 0.021), AP (r2 = 0.6052, p = 0.001), TK (r2 = 0.203, p = 0.049), TP (r2 = 0.4041, p = 0.001), TN (r2 = 0.4976, p = 0.001), sand (r2 = 0.2562, p = 0.025), and silt (r2 = 0.4882, p = 0.001) were significant factors that influenced the bacterial community structure. CCA/RDA, correspondence analysis/redundancy analysis.
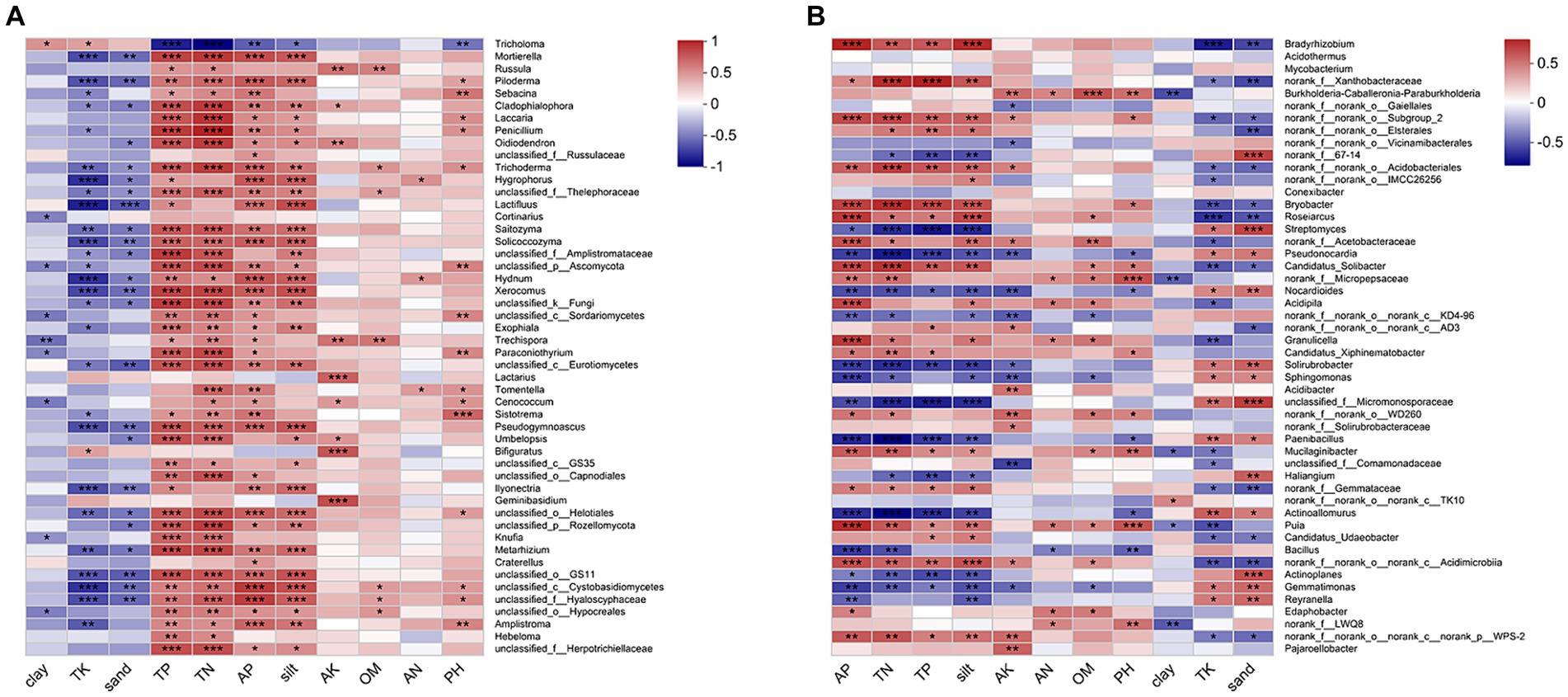
Figure 4. Heatmap of Spearman’s correlation coefficients in the microbial communities and environmental factors. (A) Fungal communities; (B) Bacterial communities. The correlation coefficient R values are shown in different colors, and the color ranges for different R values are shown in the legend on the right. The squares with a significance mark are the correlation thresholds, where |R| ≥ 0.1. The significance levels are denoted by asterisks: * for 0.01 < p ≤ 0.05, ** for 0.001 < p ≤ 0.01, and *** for p ≤ 0.001.
Functional predictions were made for the bacterial and fungal communities; the number of enzymes and pathways is explained in Supplemental Table S5. The fungal community function was predicted using PICRUSt2 and FunGuid. The results of the enzyme-level analysis facilitated by the Kyoto Encyclopedia of Genes and Genomes (KEGG) indicated that adenosine triphosphatase, glucan 1,4-alpha-glucosidase, and unspecific monooxygenase were the most abundant across the samples. The MetaCyc pathway abundance statistics showed that aerobic respiration I, aerobic respiration II, fatty acid, and beta oxidation were the most abundant metabolic pathways in each sample. The significant differences were analyzed by focusing on the top 50 data points on abundance. The abundances of enzymes (Supplementary Figures S9A,C), such as glucan 1,4-α-glucosidase and carboxylesterase, and metabolic pathways (Supplementary Figures S9B,D), such as aerobic oxidation and fatty acid degradation, were significantly higher in the Tb groups than in the CK groups. Fungal communities were classified using the fungal FunGuid function, with ECM fungi (including ECM and ECM-fungal parasites) being the most abundant in all the groups. An analysis of the significance of these differences (Supplementary Figures S10A,B) showed that ECM-fungal parasites were significantly higher in the Tb groups than in the CK groups, whereas ECM and saprotrophic parasites were significantly lower in the Tb groups than in the CK groups.
Functional predictions of the bacterial communities were performed using PICRUSt2, BugBase, and FAPROTAX. An enzyme-level analysis of the KEGG functions showed that NADH: ubiquinone reductase, DNA-directed DNA polymerase, and DNA helicase were the most abundant across the samples. The MetaCyc pathway abundance statistics showed that aerobic respiration I and pyruvate fermentation of isobutanol were the most abundant pathways in each sample. An analysis of the significant differences showed that the enzymes (Supplementary Figures S11A,C), enoyl-CoA hydratase and long-chain-fatty-acid-CoA ligase, and metabolic pathways (Supplementary Figures S11B,D), fatty acid and beta oxidation I, were significantly more abundant in the Tb groups than in the CK groups. The bacteria were phenotypically predicted using BugBase and analyzed for significant differences (Supplementary Figure S12). Seven of the nine phenotypes differed significantly. The phenotypes that were most frequent in the Tb groups were gram-positive, contained mobile elements, and exhibited stress-tolerance, whereas the phenotypes that were more frequent in the CK groups were facultative, anaerobic, potentially pathogenic, gram-negative, and aerobic. The metabolic and ecological functions of the bacteria were predicted using FAPROTAX software. The results (Supplementary Figure S13) showed that chemoheterotrophy and aerobic chemoheterotrophs were the most abundant in each sample. Chemoheterotrophy, the abundance of aerobic chemoheterotrophs, and aromatic compound degradation were higher in the Tb groups than in the CK groups, whereas nitrogen fixation was lower in the Tb groups than in the CK groups.
The pH range of all the soil samples was 5.04 to 6.19, which falls within the optimal pH range for the growth of ECM fungi, as reported by Yamanaka (2003). This pH range provided a conducive environment for the proliferation of diverse ECM-fungal species. Nitrogen (N) is the most abundant mineral nutrient required by plants (Xie et al., 2022). Due to the continuous accumulation of OM in forests, organic N dominates over inorganic N in soil (Rineau et al., 2015). However, plants cannot absorb organic N directly; thus, they often establish symbiotic relationships with microorganisms to enhance their uptake of inorganic N (Courty et al., 2015). ECM fungi represent a common example, as they convert soil organic N into inorganic forms through the secretion of extracellular enzymes and other mechanisms, thereby increasing the availability of inorganic N for plant assimilation (Rineau et al., 2015; Sebastiana et al., 2022). This may explain why the content of AN was significantly higher in most of the Tb groups than in the CK groups, while TN showed a significant decrease. Phosphorus (P) is a crucial element for plant growth; however, the content of P in all the samples analyzed in this study was found to be low. This occurrence is not uncommon, as P often limits tree productivity in forests worldwide (Plassard and Dell, 2010). Plants engage in a cooperative relationship with ECM fungi to enhance their ability to acquire P, thereby mitigating the P deficiency in the host (Cairney, 2011). This cooperation primarily manifests through ECM-mediated P solubilization and subsequent efficient mineral absorption (Cairney, 2011; Meeds et al., 2021). This could explain the significantly lower levels of TP and AP observed in the Tb groups than in the CK groups in this study. The sand content was significantly higher in the Tb groups than in the CK groups, whereas the silt content was significantly lower in the Tb groups than in the CK groups. These findings are in line with the soil structure of the T. matsutake habitat (Ji et al., 2022). Improved air and water permeability in the soil may be necessary for the survival of T. bakamatsutake. The relationship between the physicochemical properties and microorganisms was determined using subsequent correlation analyzes.
The fungal communities in the Tb groups exhibited a significant reduction in species richness and diversity, indicating that T. bakamatsutake exerted inhibitory and eliminative effects on other fungi, which is consistent with findings from previous studies on ECM fungi (Yu et al., 2021; Shen et al., 2023). Notably, XBTb displayed the lowest fungal diversity in the Tb groups. This could be attributed to the abundant presence of T. bakamatsutake fruiting bodies at the XB sampling sites, which would have resulted in a stronger effect of T. bakamatsutake on other fungi within the shiro. Conversely, XBCK exhibited the highest fungal diversity among all the samples, suggesting that the soil environment in XB was conducive to the growth of a wide range of fungi. This may also explain why T. bakamatsutake was found to be most abundant in the XB area.
For the bacterial communities, the Tb groups had higher bacterial diversity than the CK groups. ECM-fungal roots can support different types of bacteria (Liu et al., 2018), as the expanded mycelial network of ECM fungi provides a habitat for bacterial communities (Miquel Guennoc et al., 2018), and the photosynthetic products obtained from ECM fungi provide nutrition for many bacteria (Read and Perez-Moreno, 2003; van Scholl et al., 2006; Colin et al., 2017). In the present study, T. bakamatsutake received most carbohydrates from the plants, and its mycelia expanded in a denser network, attracting various types of bacteria. OM degraded or secreted by T. bakamatsutake may cause bacterial aggregation; however, the exact substances need to be confirmed.
MHB and endophytic bacteria associated with T. bakamatsutake were increased in the shiro. MHB can increase mycorrhization by inducing the rapid expansion of fungal mycelia and stimulating the formation of plant lateral roots to increase contact opportunities (Riedlinger et al., 2006; Rigamonte et al., 2010). Mycorrhizal fungi are selective regarding the type of bacteria in their rhizosphere (Izumi and Finlay, 2010), with different mycorrhizal fungi selecting different bacteria based on their requirements. In this study, Sphingomonas, Paenibacillus, and Bacillus were highly abundant and significantly different between the Tb and CK groups. These bacteria, often found in mycorrhizal environments (Bending et al., 2002; Timonen and Hurek, 2006; Nguyen and Bruns, 2015), promote the growth and colonization of ECM fungus mycorrhizae (Requena et al., 1997; Hrynkiewicz et al., 2009; Ji et al., 2022). Species of the genera Bacillus and Paenibacillus were significantly high in abundance in the T. matsutake shiro (Oh et al., 2016). Sphingomonas species are endophytic bacteria of ECM fungi, such as in T. matsutake cotyledons (Liu et al., 2021), which possess the ability to degrade aromatic compounds and exhibit metabolic activity against toxic pollutants (Yang C. et al., 2022; Yang M. et al., 2022). Thus, several of the abovementioned bacteria may contribute to the growth and development of T. bakamatsutake and its root-colonizing mycelia.
T. bakamatsutake aggregated plant growth-promoting bacteria (PGPB). Bacterial genera that have been confirmed as PGPB, such as BCP, Solirubrobacter, and Bacillus, were significantly more abundant in the Tb groups than in the CK groups. In future studies, several genera with high abundances will be explored. BCP are widely distributed in the soil and plant roots and promote plant growth, improve plant resistance, and reduce self-toxicity (Beukes et al., 2017; Luo et al., 2021). In the present study, BCP abundance was higher in both the ASTb and XYTb samples than in the corresponding CK samples. Inter-root secretions are enriched with and degraded by BCP to reduce plant auto-toxicity (Luo et al., 2021). Therefore, the possibility of Tb inter-root secretions, whether enriched with BCP or not, requires further study. In addition to being beneficial to plants, BCP aid the development of T. matsutake (Xu, 2021). Actinobacteria are considered special PGPB that promote plant growth and help plants resist pathogenic bacteria in several ways, including through the decomposition of OM, secretion of antibiotics, degradation of pollutants, and secretion of phytohormones (Bhatti et al., 2017; Olanrewaju and Babalola, 2018; Hayat et al., 2021). In the present study, the relative abundance of Actinobacteria was significantly higher in the Tb samples than in the CK samples. The significant increase in Actinobacteria was mainly concentrated in the genera Streptomyces, Pseudonocardia, norank_f__67–14, Nocardioides, and Solirubrobacter. Streptomyces, the most highly anticipated PGPB among actinomycetes, can efficiently colonize inter-root surfaces and promote plant growth through the direct production of phytohormones and decomposition of OM (Verma et al., 2011; Olanrewaju and Babalola, 2018). Norank_f__67–14 and Solirubrobacter species belong to the order Solirubrobacterales, which increases phosphorus flow and inhibits the flow of toxic aluminum and manganese from the soil to the plant, thereby promoting high crop yields (Wang et al., 2022). Nocardioides and Pseudonocardia are highly metabolically active against toxic pollutants, and their secretions exert antimicrobial activity (Carr et al., 2012; Singha and Pandey, 2021; Yang C. et al., 2022; Yang M. et al., 2022). Therefore, T. bakamatsutake aggregates Actinobacteria to degrade soil contaminants, secrete antibiotics to suppress pathogenic bacteria, and provide nutrients for itself, thereby aiding its own growth and development and the oak environment.
Microorganisms interact via metabolites. Actinomycetes in the shiro had the most significant effect on other bacteria, especially gram-negative bacteria. The abundance of norank_f__norank_o__Elsterales was lower in the Tb groups than in the CK groups. Elsterales may be associated with plant pathogenicity, soil carbon metabolism, aluminum accumulation, and soil impoverishment (Xu, 2021). This suggests that actinomycetes can suppress microorganisms that are detrimental to T. bakamatsutake and oak forests. However, actinomycetes can inhibit beneficial microorganisms, such as BCP. The analysis of the significant differences in the bacterial communities and Spearman’s analysis (Supplementary Figure S14) showed that Actinobacteria, Pseudonocardia, Solirubrobacter, and Nocardioides were significantly negatively correlated with BCP, with significant differences in the abundance of the three genera in XB and KD. Nocardicin A, a metabolite of Nocardioides, has a broad-spectrum gram-negative inhibitory effect (Aoki et al., 1976; Nishida et al., 1977; Luo et al., 2013) and is a natural product of Pseudonocardia (Carr et al., 2012). Therefore, bacteria that belong to the BCP genera may be more sensitive than other bacteria to the inhibitory effects of these three genera. This explains the low abundance of the BCP in XBTb and KDTb. Other beneficial bacteria, such as Bryobacter (Wang et al., 2022) and Roseiarcus (Yu et al., 2020), were similarly significantly affected. Despite the inhibitory effects of actinomycetes, beneficial bacteria still prevailed, some of which, including Bacillus, were not as negatively affected; therefore, this did not affect the growth and development of T. bakamatsutake.
Bacterial competition for ecological niches was considered in this study. Bradyrhizobium and norank_f__Xanthobacteraceae, which are nitrogen-fixing rhizobia (Rhizobiales) that promote the growth and root development of some plants (Yang C. et al., 2022; Yang M. et al., 2022), were highly abundant in all the samples. Therefore, Rhizobiales are symbionts of T. bakamatsutake; however, they were lower in abundance in the Tb groups than in the CK groups, which is notable. In addition to antibiotic antagonism, Rhizobiales compete with other bacteria, such as Frankia (Wall, 2000), for nitrogen fixation, ecological niches, and other aspects.
The interactions between bacterial communities are not limited to antagonistic and competitive interactions, and symbiotic interactions were observed in this study. A prime example is the manner in which actinomycetes suppress some bacteria and degrade toxic pollutants to provide nutrients and a good habitat for other bacteria and plants. These effects are not as widespread as the antagonistic effects of actinomycetes; however, they are apparent.
T. bakamatsutake dominated its shiro, with the highest relative abundance of 99.1% being in XBTb. Most fungi in the Tb groups were affected by the dominant effect of T. bakamatsutake; therefore, the fungi affected by T. bakamatsutake were divided into two main groups. The first group comprised other ECM fungi that mainly included Russula, Piloderma, Laccaria, and Sebacina. The dominant effect of T. bakamatsutake on other ECM fungi is reflected in the competition for ecological niches, such as plant root symbiosis sites and soil nutrients, whereas T. bakamatsutake metabolites may have a suppressive effect on other ECM fungi. The second group included reported plant pathogenic fungi, such as Penicillium (Li et al., 2010) and Trichoderma (Gajera et al., 2015). The dominant effect of T. bakamatsutake on pathogenic fungi is reflected in the competition for nutrients and survival space, while actinomycetes enriched by T. bakamatsutake inhibit some pathogenic fungi. The decrease in pathogenic fungi promoted the growth and development of Q. mongolica. In addition to these two groups of fungi, other common fungi in oak forests were affected by the dominance of T. bakamatsutake.
The fungal communities in the CK groups from the four areas were jointly analyzed (Figure 5). ECM fungi, such as Russula, Piloderm, and Lactifluus, shared a similar habitat with T. bakamatsutake––the oak forest. However, these ECM fungi are the main competitors of T. bakamatsutake, the most competitive being Russula, which is the most abundant among the ECM fungi (Ji et al., 2019). In addition to mycorrhizal fungi, genera, such as Mortierella, are common fungi present under oak trees and may contribute to the growth of oak forests (Guo et al., 2022) and indirectly help the formation of symbiotic relationships between oak forests and mycorrhizal fungi.
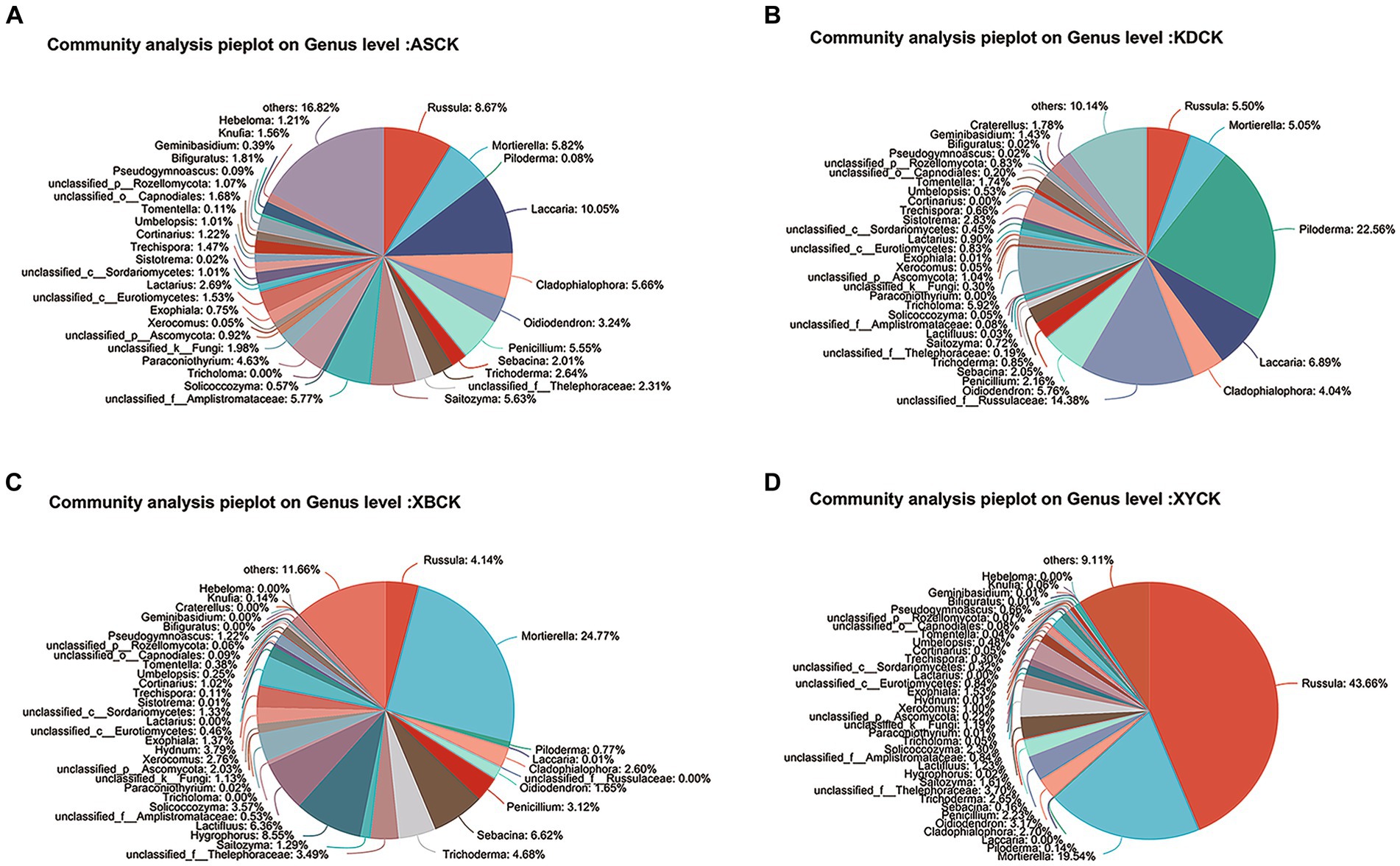
Figure 5. Pie plot of the fungal community composition at the genus level in the Quercus mongolica rhizosphere soil (CK) group. (A) ASCK; (B) KDCK; (C) XBCK; (D) XYCK.
Given the abundance of T. bakamatsutake in the Tb groups, the predictive function of the fungal community primarily reflected the functional contribution of T. bakamatsutake. The predicted results for the MetaCyc pathways showed a significantly higher abundance of sugar and lipid catabolism-related pathways, such as aerobic respiration I, tricarboxylic acid cycle II, glycolysis III, pentose phosphate pathway, and fatty acid beta oxidation, in the Tb groups compared to those in the CK groups. As a member of ECM fungi, T. bakamatsutake mainly obtains nutrients from the host plant sugars and fats for its own nutritional development (Nehls et al., 2010). Therefore, T. bakamatsutake’s function is related to the catabolism of sugars and fats. Notably, the glyoxylate cycle was significantly more abundant in the Tb groups than in the CK groups. This suggests that some sugars in T. bakamatsutake may not only come directly from the host plant but also be derived from sugars formed by acetyl CoA via the glyoxylate cycle and other pathways. Additionally, octane oxidation was significantly more abundant in the Tb groups than in the CK groups. The application of microbial degradation of aromatic hydrocarbons is of great value; however, studies on the degradation of aromatic compounds by mycorrhizal fungi are limited. Our results indicate that T. bakamatsutake may warrant further research in this regard.
The abundance of enzymes was predicted using PICRUSt2. The analysis subsequently focused on enzymes exhibiting higher abundance and greater variation. The glucan 1,4-alpha-glucosidase was the most abundant in the Tb groups and primarily acts on the 1,4-alpha glycosidic bond found in starch. The glucan 1,4-alpha-glucosidase may therefore serve as the pivotal enzyme employed by T. bakamatsutake to degrade plant photosynthetic products. Carboxylesterase was significantly more abundant in the Tb groups than in the CK groups and can hydrolyze tannins and corneum from the cell wall. The corneum is a hydroxy fatty acid mixed with wax on the outside of the cell wall to form the cuticle (Samuels et al., 2008; Arya et al., 2021). ECM fungi closely adhere to host roots and secrete cell wall-degrading enzymes to remodel the cell wall, forming a Hartig net that expands the interface for nutrient exchange with the plant (Yu and Yuan, 2023). The plant cuticle is a barrier against microbial infestation (Yeats and Rose, 2013; Ziv et al., 2018), suggesting that carboxylesterases from T. bakamatsutake may play a role in their association with host plant root cells. The abundance of chitinase was significantly higher in the Tb groups than in the CK groups and is often observed in ECM fungi, as chitinase degrades the fungal cell wall and releases spores. The abundance of gibberellin 2-beta-dioxygenase, an enzyme involved in gibberellin anabolism, was also increased in the Tb groups compared to that in the CK groups. This may play a facilitative and regulatory role for plant hosts and in the growth of T. bakamatsutake. Choline dehydrogenase can oxidize choline, which is subsequently oxidized by the enzyme betaine-aldehyde dehydrogenase to form betaine (Zhang et al., 2020); however, the latter was found to be less abundant in the Tb groups than in the CK groups. Additionally, enzymes such as tripeptidyl-peptidase I and non-specific monooxygenase were significantly more abundant in the Tb groups than in the CK groups, which could lead to interesting findings in future studies.
The bacterial community functions were predicted using PICRUSt2. The Tb groups showed a significantly higher abundance of enzymes involved in fatty acid and sugar catabolism than did the CK groups. The enzymes included enoyl-CoA hydratase, long-chain-fatty-acid-CoA ligase, pyruvate dehydrogenase, and dihydrolipoyl dehydrogenase (Martin et al., 1988; Wang et al., 2002; da Silva Coelho et al., 2010). The abundance of metabolic pathways, such as fatty acid beta oxidation I, was also significantly high in the Tb groups compared to that in the CK groups. Hence, the Tb group soils may have been enriched with bacteria related to fatty acid and sugar catabolism. Additionally, the abundance of betaine-aldehyde dehydrogenase at the enzyme level was significantly higher in the Tb groups than in the CK groups, suggesting that in the Tb groups, the bacterial community may have filled the gap in the fungal community with a lower abundance of this enzyme. Perhaps a collaborative effect between the fungi and bacteria exists for improved betaine formation.
The phenotypes of the bacterial communities were analyzed using data from BugBase. The main factor contributing to the significantly different BugBase phenotypes was the aggregation of actinomycetes in Tb. Actinomycetes had a stronger inhibitory effect on gram-negative bacteria, resulting in a higher proportion of gram-positive and lower proportion of gram-negative bacteria in the Tb groups compared to those in the CK groups (Aoki et al., 1976). The significantly high stress-tolerance phenotype in the Tb groups may have been related to antibiotic stress resistance, as fewer non-resistant bacteria were inhibited in Tb. It is possible that the bacterial communities in the Tb groups exhibited high stress tolerance. Additionally, there was a lower relative abundance of potentially pathogenic bacteria, including some plant pathogenic bacteria, in the Tb groups compared to that in the CK groups. Therefore, both plants and T. bakamatsutake itself had a low probability of being attacked by pathogenic bacteria in Tb soils.
The functional annotation of the bacterial communities was performed using FAPROTAX. Chemoheterotrophy and aerobic chemoheterotrophs were more abundant in the Tb groups compared to those in the CK groups. The carbon and energy sources of chemoheterotrophic microorganisms originate from the oxidative decomposition of OM (Zhou, 2020). Saprophytic bacteria were the most abundant among the chemoheterotrophic bacteria. In the Tb groups, T. bakamatsutake dominated and significantly reduced the abundance of saprophytic fungi. However, T. bakamatsutake had limited capacity and efficiency in decomposing OM (Shah et al., 2016); therefore, an increase in saprophytic bacteria was needed to compensate for the scope and efficiency of saprophytic functions in the Tb groups. Nitrogen fixation was significantly reduced in Tb, possibly due to antagonistic effects within the bacterial community and competition for ecological niches, resulting in a reduction in nitrogen-fixing bacteria, such as Bradyrhizobium. The degradation of aromatic compounds was higher in the Tb groups than in the CK groups. Aromatic compounds can cause serious environmental pollution (Seo et al., 2009), and the significantly high abundance of aromatic compound-degrading bacteria in the Tb groups was mainly from the several aforementioned actinomycetes (Ren, 2017). Plant pathogens were significantly less abundant in the Tb groups than in the CK groups, suggesting that some plant pathogenic bacteria were inhibited by Tb.
In this study, the diversity, microbial populations, and structure of the microbiome in the soils of the Tb and CK groups were compared. Compared to the CK groups, the Tb groups exhibited a clear dominance of T. bakamatsutake, resulting in significantly reduced species diversity and richness of other fungal species. On the contrary, the bacterial diversity increased and bacterial community structure was changed in the Tb groups compared to those of the CK groups. There were more abundant PGPB and MHB in the Tb groups than in the CK groups. Abundant MHB (Sphingomonas, Paenibacillus, and Bacillus) promotes fungal hyphal growth and mycorrhizal formation. PGPB are capable of producing plant growth regulators, promoting plant growth, and improving resistance to drought and salt tolerance. The results showed that T. bakamatsutake not only directly provided water and minerals to Q. mongolica but also promoted the growth of the host by regulating the soil microbial community structure (Figure 6). This study is important for constructing a healthy soil microbial community structure and cultivating T. bakamatsutake-Q. mongolica mycorrhizal seedlings. In future studies, the symbiotic relationship between T. bakamatsutake and Q. mongolica can be further examined at the metabolite level, using metabonomics to improve the feasibility of T. bakamatsutake artificial cultivation.
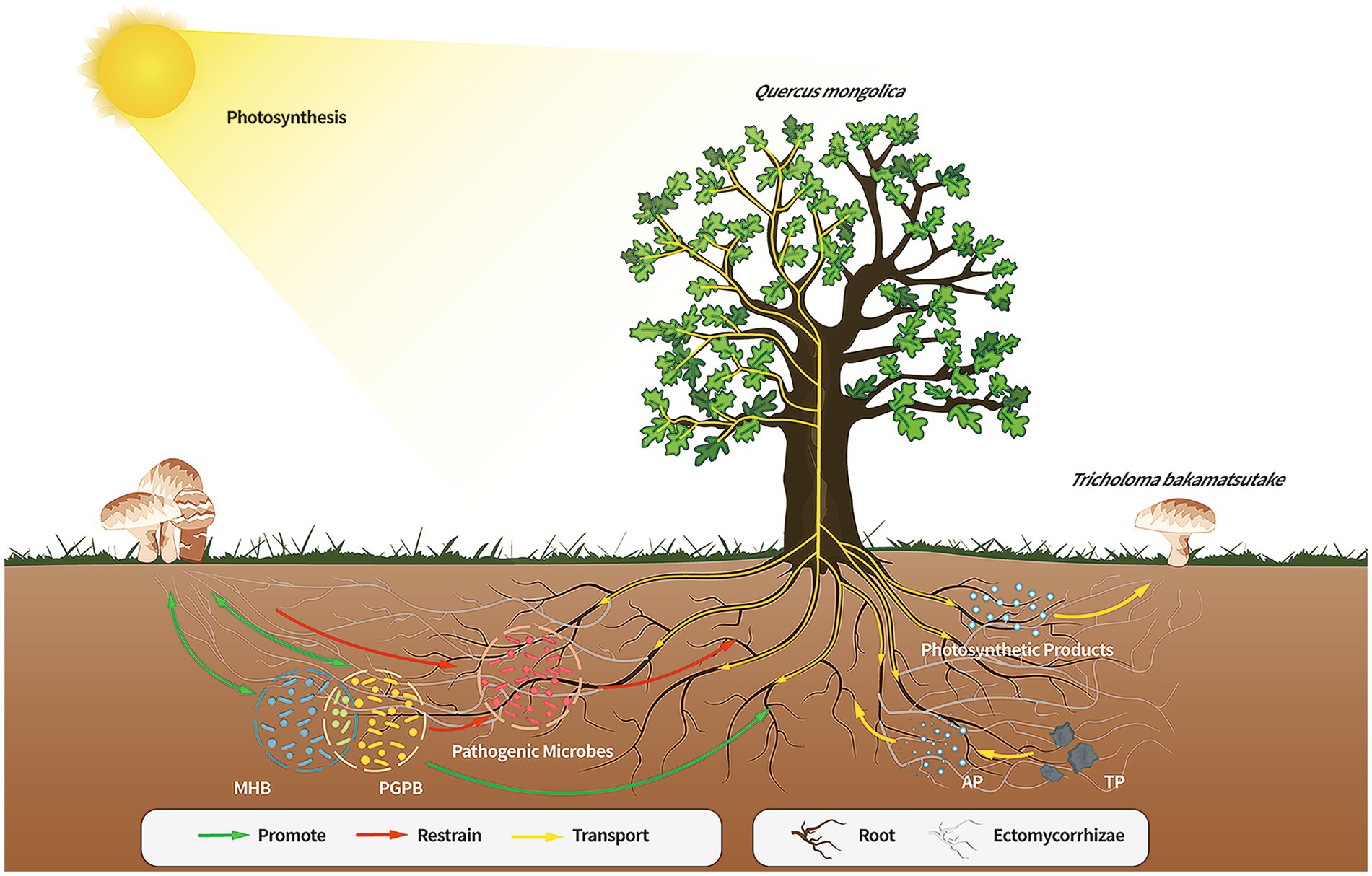
Figure 6. Direct or indirect effects of the members of the Quercus mongolica-Tricholoma bakamatsutake-associated bacteria symbiosis. The soil of the Q. mongolica forest provides a soil environment that contributes to the widespread presence of ECM fungi. However, the composition of the soil affects the colonization of different ECM species, and T. bakamatsutake may prefer to colonize environments with high sand and low silt levels owing to good aeration, permeability, and poor water storage; hence, the competition for T. bakamatsutake is low in this soil. Once T. bakamatsutake colonizes the roots of the oak tree, it receives photosynthetic products from the oak tree through its mycorrhizal roots and transports active soil material to the oak tree. As T. bakamatsutake grows, its dominance suppresses other fungi and aggregates its companion bacteria, which are beneficial to its growth and development and those of Q. mongolica, whereas Q. mongolica and T. bakamatsutake provide nutrients to these companion bacteria; ECM, ectomycorrhizal.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
HG: Writing – review & editing, Writing – original draft, Validation, Supervision, Project administration, Funding acquisition, Formal analysis, Data curation, Conceptualization. WL: Writing – review & editing, Writing – original draft, Visualization, Software, Methodology, Investigation, Formal analysis, Data curation. YX: Writing – original draft, Resources, Investigation. ZW: Writing – original draft, Resources, Investigation. CH: Writing – original draft, Resources, Investigation. JY: Writing – original draft, Validation, Resources. ZY: Writing – original draft, Resources, Investigation. JZ: Writing – original draft, Visualization, Software. XY: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Project administration, Funding acquisition, Formal analysis, Data curation, Conceptualization. LS: Writing – review & editing, Writing – original draft, Validation, Supervision, Project administration, Methodology, Conceptualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Natural Science Foundation of Liaoning Province (2022-MS-418), National Natural Science Foundation of China (32370008), and Joint Fund of Natural Science Foundation of Liaoning Province (2023-MSLH-203).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1361117/full#supplementary-material
Aoki, H., Sakai, H., Kohsaka, M., Konomi, T., and Hosoda, J. (1976). Nocardicin a, a new monocyclic beta-lactam antibiotic. I. Discovery, isolation and characterization. J. Antibiot. 29, 492–500. doi: 10.7164/antibiotics.29.492
Arya, G. C., Sarkar, S., Manasherova, E., Aharoni, A., and Cohen, H. (2021). The plant cuticle: an ancient Guardian barrier set against long-standing rivals. Front. Plant Sci. 12:663165. doi: 10.3389/fpls.2021.663165
Bending, G. D., Poole, E. J., Whipps, J. M., and Read, D. J. (2002). Characterisation of bacteria from Pinus sylvestris-Suillus luteus mycorrhizas and their effects on root-fungus interactions and plant growth. FEMS Microbiol. Ecol. 39, 219–227. doi: 10.1111/j.1574-6941.2002.tb00924.x
Bennett, J. A., Maherali, H., Reinhart, K. O., Lekberg, Y., Hart, M. M., and Klironomos, J. (2017). Plant-soil feedbacks and mycorrhizal type influence temperate forest population dynamics. Science 355, 181–184. doi: 10.1126/science.aai8212
Beukes, C. W., Palmer, M., Manyaka, P., Chan, W. K., Avontuur, J. R., Van Zyl, E., et al. (2017). Genome data provides high support for generic boundaries in Burkholderia Sensu Lato. Front. Microbiol. 8:1154. doi: 10.3389/fmicb.2017.01154
Bhatti, A. A., Haq, S., and Bhat, R. A. (2017). Actinomycetes benefaction role in soil and plant health. Microb. Pathog. 111, 458–467. doi: 10.1016/j.micpath.2017.09.036
Bouyoucos, G. (1936). Directions for making mechanical analysis of soils by the hydrometer method. Soil Sci. 42, 225–230. doi: 10.1097/00010694-193609000-00007
Cairney, J. W. G. (2011). Ectomycorrhizal fungi: the symbiotic route to the root for phosphorus in forest soils. Plant Soil 344, 51–71. doi: 10.1007/s11104-011-0731-0
Calvaruso, C., Turpault, M.-P., Leclerc, E., and Frey-Klett, P. (2007). Impact of ectomycorrhizosphere on the functional diversity of soil bacterial and fungal communities from a forest stand in relation to nutrient mobilization processes. Microb. Ecol. 54, 567–577. doi: 10.1007/s00248-007-9260-z
Carr, G., Derbyshire, E. R., Caldera, E., Currie, C. R., and Clardy, J. (2012). Antibiotic and antimalarial Quinones from fungus-growing ant-associated Pseudonocardia sp. J. Nat. Prod. 75, 1806–1809. doi: 10.1021/np300380t
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Colin, Y., Nicolitch, O., Turpault, M.-P., and Uroz, S. (2017). Mineral types and tree species determine the functional and taxonomic structures of forest soil bacterial communities. Appl. Environ. Microbiol. 83:e02684-16. doi: 10.1128/aem.02684-16
Courty, P. E., Smith, P., Koegel, S., Redecker, D., and Wipf, D. (2015). Inorganic nitrogen uptake and transport in beneficial plant root-microbe interactions. Crit. Rev. Plant Sci. 34, 4–16. doi: 10.1080/07352689.2014.897897
da Silva Coelho, I., de Queiroz, M. V., Costa, M. D., Kasuya, M. C. M., and de Araújo, E. F. (2010). Identification of differentially expressed genes of the fungus Hydnangium sp. during the pre-symbiotic phase of the ectomycorrhizal association with Eucalyptus grandis. Mycorrhiza 20, 531–540. doi: 10.1007/s00572-010-0301-y
Deng, H. (2003). Overview of research on Tricholoma bakamatsutake in China. Proceedings of the third congress of the chinese society of mycology and the sixth national symposium on mycology (Mycological Society of China).
Deng, Z. (2019). Advances in the study of Tricholoma bakamatsutake. Agri. Sci. Technol. Equip. 2:19-20+24. doi: 10.16313/j.cnki.nykjyzb.2019.02.009
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Frey-Klett, P., Garbaye, J., and Tarkka, M. (2007). The mycorrhiza helper bacteria revisited. New Phytol. 176, 22–36. doi: 10.1111/j.1469-8137.2007.02191.x
Gajera, H. P., Savaliya, D. D., Patel, S. V., and Golakiya, B. A. (2015). Trichoderma viride induces pathogenesis related defense response against rot pathogen infection in groundnut (Arachis hypogaea L.). Infect. Genet. Evol. 34, 314–325. doi: 10.1016/j.meegid.2015.07.003
Guo, P., Xing, P. J., Song, J., Wu, L. L., Li, B. Q., Si, Y. J., et al. (2022). Fungal community in roots and the root zone of Quercus mongolica and the correlations with the environmental factors. J. Fungal Res. 20, 173–182. doi: 10.13341/j.jfr.2022.1489
Hayat, S., Ashraf, A., Aslam, B., Asif, R., Muzammil, S., Asif Zahoor, M., et al. (2021). Actinobacteria: potential candidate as plant growth promoters. Plant Stress Physiol. doi: 10.5772/intechopen.93272
Hou, J. (2020). Annotation of the mitochondrial genome of Tricholoma bakamatsutake and comparison with related species. [Shanxi]: Shanxi University.
Hrynkiewicz, K., Christel, B., Niedojadło, J., and Hanna, D. (2009). Promotion of mycorrhiza formation and growth of willows by the bacterial strain Sphingomonas sp. 23L on fly ash. Biol. Fertil. Soils 45, 385–394. doi: 10.1007/s00374-008-0346-7
Izumi, H., and Finlay, R. D. (2010). Ectomycorrhizal roots select distinctive bacterial and ascomycete communities in Swedish subarctic forests. Environ. Microbiol. 13, 819–830. doi: 10.1111/j.1462-2920.2010.02393.x
Ji, R., Xing, P., Xu, Y., Li, G., Gao, T., Zhou, J., et al. (2019). Analysis of the composition of symbiotic fungal and bacterial in the roots and rhizosphere soil of Quercus mongolica. Mycosystema 38, 1894–1906. doi: 10.13346/j.mycosystema.190181
Ji, R. Q., Xu, Y., Si, Y. J., Phukhamsakda, C., Li, Y., Meng, L. P., et al. (2022). Fungal–bacterial networks in the habitat of SongRong (Tricholoma matsutake) and driving factors of their distribution rules. J.Fungi 8:575. doi: 10.3390/jof8060575
Kang, J. A., Ka, K. H., Kim, J. Y., and Kim, S. H. (2018). Mycelial growth properties of domestically collected ectomycorrhizal Tricholoma mushrooms in various culture conditions. Kor. J. Mycol. 46, 271–280. doi: 10.4489/kjm.20180037
Kawai, M., Imaji, A., Yamada, A., and Kinoshita, A. (2018). Shiro formation and fruit body flashing by inoculation of Tricholoma bakamatsutake mycelia in the forest. In abstracts of papers presented at the 62th annual meeting of the mycological society of Japan.
Kim, M., Yoon, H., Kim, Y. E., Kim, Y. J., Kong, W. S., and Kim, J. G. (2014). Comparative analysis of bacterial diversity and communities inhabiting the fairy ring of Tricholoma matsutake by barcoded pyrosequencing. J. Appl. Microbiol. 117, 699–710. doi: 10.1111/jam.12572
Li, Q., Ning, P., Zheng, L., Huang, J., Li, G., and Hsiang, T. (2010). Fumigant activity of volatiles of Streptomyces globisporus JK-1 against Penicillium italicum on Citrus microcarpa. Postharvest Biol. Technol. 58, 157–165. doi: 10.1016/j.postharvbio.2010.06.003
Li, X., Zhang, X., Yang, M., Yan, L., Kang, Z., Xiao, Y., et al. (2019). Tuber borchii shapes the Ectomycorrhizosphere microbial communities of Corylus avellana. Mycobiology 47, 180–190. doi: 10.1080/12298093.2019.1615297
Liu, D., Pérez-Moreno, J., Zhang, P., Wang, R., Chater, C., and Yu, F. (2021). Distinct compartmentalization of microbial community and potential metabolic function in the fruiting body of Tricholoma matsutake. J. Fungus 7:586. doi: 10.3390/jof7080586
Liu, Y., Sun, Q., Li, J., and Lian, B. (2018). Bacterial diversity among the fruit bodies of ectomycorrhizal and saprophytic fungi and their corresponding hyphosphere soils. Sci. Rep. 8:11672. doi: 10.1038/s41598-018-30120-6
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Luo, Q., Hiessl, S., and Steinbüchel, A. (2013). Functional diversity of Nocardia in metabolism. Environ. Microbiol. 16, 29–48. doi: 10.1111/1462-2920.12221
Luo, L., Wang, L., Deng, L., Mei, X., Liu, Y., Huang, H., et al. (2021). Enrichment of Burkholderia in the rhizosphere by autotoxic ginsenosides to alleviate negative plant-soil feedback. Microbiol. Spectr. 9:e0140021. doi: 10.1128/spectrum.01400-21
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Martin, F., Ramstedt, M., Söderhäll, K., and Canet, D. (1988). Carbohydrate and amino acid metabolism in the ectomycorrhizal ascomycete Sphaerosporella brunnea during glucose utilization. Plant Physiol. 86, 935–940. doi: 10.1104/pp.86.3.935
Meeds, J. A., Marty Kranabetter, J., Zigg, I., Dunn, D., Miros, F., Shipley, P., et al. (2021). Phosphorus deficiencies invoke optimal allocation of exoenzymes by ectomycorrhizas. ISME J. 15, 1478–1489. doi: 10.1038/s41396-020-00864-z
Miquel Guennoc, C., Rose, C., Labbé, J., and Deveau, A. (2018). Bacterial biofilm formation on the hyphae of ectomycorrhizal fungi: a widespread ability under controls? FEMS Microbiol. Ecol. 94:fiy093. doi: 10.1093/femsec/fiy093
Moeller, H. V., and Peay, K. G. (2016). Competition-function tradeoffs in ectomycorrhizal fungi. PeerJ 4:e2270. doi: 10.7717/peerj.2270
Nehls, U., Göhringer, F., Wittulsky, S., and Dietz, S. (2010). Fungal carbohydrate support in the ectomycorrhizal symbiosis: a review. Plant Biol. 12, 292–301. doi: 10.1111/j.1438-8677.2009.00312.x
Nehls, U., and Plassard, C. (2018). Nitrogen and phosphate metabolism in ectomycorrhizas. New Phytol. 220, 1047–1058. doi: 10.1111/nph.15257
Nguyen, N. H., and Bruns, T. D. (2015). The microbiome of Pinus muricata ectomycorrhizae: community assemblages, fungal species effects, and Burkholderia as important bacteria in multipartnered symbioses. Microb. Ecol. 69, 914–921. doi: 10.1007/s00248-015-0574-y
Nishida, M., Mine, Y., Nonoyama, S., and Kojo, H. (1977). Nocardicin a, a new monocyclic beta-lactam antibiotic III. In vitro evaluation. J. Antibiot. 30, 917–931. doi: 10.7164/antibiotics.30.917
Ogawa, M. (1977). Microbial ecology of 'Shiro' in Tricholoma matsutake (S. Ito & Imai) sing. And its allied species. V. Tricholoma matsutake in Tsuga sieboldii forests. Trans. Mycol. Soc. Jpn. 18, 34–46.
Oh, S., Fong, J. J., Park, M. S., and Lim, Y. W. (2016). Distinctive feature of microbial communities and bacterial functional profiles in Tricholoma matsutake dominant soil. PLoS One 11, –e0168573. doi: 10.1371/journal.pone.0168573
Olanrewaju, O. S., and Babalola, O. O. (2018). Streptomyces: implications and interactions in plant growth promotion. Appl. Microbiol. Biotechnol. 103, 1179–1188. doi: 10.1007/s00253-018-09577-y
Ota, Y., Yamanaka, T., Murata, H., Neda, H., Ohta, A., Kawai, M., et al. (2012). Phylogenetic relationship and species delimitation ofmatsutakeand allied species based on multilocus phylogeny and haplotype analyzes. Mycologia 104, 1369–1380. doi: 10.3852/12-068
Plassard, C., and Dell, B. (2010). Phosphorus nutrition of mycorrhizal trees. Tree Physiol. 30, 1129–1139. doi: 10.1093/treephys/tpq063
Poole, E. J., Bending, G. D., Whipps, J. M., and Read, D. J. (2001). Bacteria associated with Pinus sylvestris-Lactarius rufus ectomycorrhizas and their effects on mycorrhiza formation in vitro. New Phytol. 151, 743–751. doi: 10.1046/j.0028-646x.2001.00219.x
Quan, X., Fu, W., and Wu, J. (2005). Experiments on the best selected part of tissue separation of Tricholoma bakamatsutake. Edible Fungi China 5, 19–20.
Quan, X., Wang, H., Shi, T., and Zhang, M. (2007). Comparison of the nutritional composition of the seeds of Tricholoma matsutake and Tricholoma bakamatsutake. Edible Fungi 2, 54–55.
Read, D. J., and Perez-Moreno, J. (2003). Mycorrhizas and nutrient cycling in ecosystems – a journey towards relevance? New Phytol. 157, 475–492. doi: 10.1046/j.1469-8137.2003.00704.x
Ren, L. (2017). Isolation of aromatic pollutants degrading bacteria and investigation of degradation mechanism. [Beijing]: Chinese Academy of Agricultural Sciences.
Requena, N., Jimenez, I., Toro, M., and Barea, J. M. (1997). Interactions between plant-growth-promoting rhizobacteria (PGPR), arbuscular mycorrhizal fungi and Rhizobium spp. in the rhizosphere of Anthyllis cytisoides, a model legume for revegetation in mediterranean semi-arid ecosystems. New Phytol. 136, 667–677. doi: 10.1046/j.1469-8137.1997.00786.x
Riedlinger, J., Schrey, S. D., Tarkka, M. T., Hampp, R., Kapur, M., and Fiedler, H. (2006). Auxofuran, a novel metabolite that stimulates the growth of fly agaric, is produced by the mycorrhiza helper bacterium Streptomyces strain AcH 505. Appl. Environ. Microbiol. 72, 3550–3557. doi: 10.1128/aem.72.5.3550-3557.2006
Rigamonte, T. A., Pylro, V. S., and Duarte, G. F. (2010). The role of mycorrhization helper bacteria in the establishment and action of ectomycorrhizae associations. Braz. J. Microbiol. 41, 832–840. doi: 10.1590/s1517-83822010000400002
Rineau, F., Stas, J., Nguyen, N. H., Kuyper, T. W., Carleer, R., Vangronsveld, J., et al. (2015). Ectomycorrhizal fungal protein degradation ability predicted by soil organic nitrogen availability. Appl. Environ. Microbiol. 82, 1391–1400. doi: 10.1128/aem.03191-15
Samuels, L., Kunst, L., and Jetter, R. (2008). Sealing plant surfaces: Cuticular wax formation by epidermal cells. Annu. Rev. Plant Biol. 59, 683–707. doi: 10.1146/annurev.arplant.59.103006.093219
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Sebastiana, M., Serrazina, S., Monteiro, F., Wipf, D., Fromentin, J., Teixeira, R., et al. (2022). Nitrogen acquisition and transport in the ectomycorrhizal symbiosis-insights from the interaction between an oak tree and Pisolithus tinctorius. Plants (Basel, Switzerland) 12:10. doi: 10.3390/plants12010010
Seo, J., Keum, Y., and Li, Q. (2009). Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 6, 278–309. doi: 10.3390/ijerph6010278
Shah, F., Nicolás, C., Bentzer, J., Ellström, M., Smits, M., Rineau, F., et al. (2016). Ectomycorrhizal fungi decompose soil organic matter using oxidative mechanisms adapted from saprotrophic ancestors. New Phytol. 209, 1705–1719. doi: 10.1111/nph.13722
Shen, A., Shen, B., Liu, L., Tan, Y., Zeng, L., Tan, Z., et al. (2023). Diversity and network relationship construction of soil fungal communities in Lactarius hatsudake Tanaka orchard during harvest. Microorganisms 11:2279. doi: 10.3390/microorganisms11092279
Shokralla, S., Spall, J. L., Gibson, J. F., and Hajibabaei, M. (2012). Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 21, 1794–1805. doi: 10.1111/j.1365-294x.2012.05538.x
Singha, L. P., and Pandey, P. (2021). Rhizosphere assisted bioengineering approaches for the mitigation of petroleum hydrocarbons contamination in soil. Crit. Rev. Biotechnol. 41, 749–766. doi: 10.1080/07388551.2021.1888066
Song, Y. Y., Simard, S. W., Carroll, A., Mohn, W. W., and Zeng, R. S. (2015). Defoliation of interior Douglas-fir elicits carbon transfer and stress signalling to ponderosa pine neighbors through ectomycorrhizal networks. Sci. Rep. 5:8495. doi: 10.1038/srep08495
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Tedersoo, L., Bahram, M., and Zobel, M. (2020). How mycorrhizal associations drive plant population and community biology. Science 367:eaba1223. doi: 10.1126/science.aba1223
Terashima, Y. (1994). Change in medium components and colony morphology due to mycelial growth of ectomycorrhizal fungus Tricholoma bakamatsutake. Mycoscience 35, 153–159. doi: 10.1007/bf02318493
Terashima, Y. (1999). Carbon and nitrogen utilization and acid production by mycelia of the ectomycorrhizal fungus Tricholoma bakamatsutake in vitro. Mycoscience 40, 51–56. doi: 10.1007/bf02465673
Timonen, S., and Hurek, T. (2006). Characterization of culturable bacterial populations associating with Pinus sylvestris–Suillus bovinus mycorrhizospheres. Can. J. Microbiol. 52, 769–778. doi: 10.1139/w06-016
Vaario, L. M., Fritze, H., Spetz, P., Heinonsalo, J., Hanajík, P., and Pennanen, T. (2011). Tricholoma matsutake dominates diverse microbial communities in different forest soils. Appl. Environ. Microbiol. 77, 8523–8531. doi: 10.1128/aem.05839-11
van Scholl, L., Hoffland, E., and van Breemen, N. (2006). Organic anion exudation by ectomycorrhizal fungi and Pinus sylvestris in response to nutrient deficiencies. New Phytol. 170, 153–163. doi: 10.1111/j.1469-8137.2006.01649.x
Verma, V. C., Singh, S. K., and Prakash, S. (2011). Bio-control and plant growth promotion potential of siderophore producing endophytic Streptomyces from Azadirachta indica a Juss. J. Basic Microbiol. 51, 550–556. doi: 10.1002/jobm.201000155
Wall, L. G. (2000). The actinorhizal symbiosis. J. Plant Growth Regul. 19, 167–182. doi: 10.1007/s003440000027
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261. doi: 10.1128/aem.00062-07
Wang, J., Liu, K., Zhao, X., Gao, G., Wu, Y., and Shen, R. (2022). Microbial keystone taxa drive crop productivity through shifting aboveground-belowground mineral element flows. Sci. Total Environ. 811:152342. doi: 10.1016/j.scitotenv.2021.152342
Wu, Q., Liu, J., and Quan, X. (2009). Experiment on optimizing the culture medium of Tricholoma bakamatsutake parent species in Changbai Mountain area. Edible Fungi 2, 32–33.
Xie, K., Ren, Y., Chen, A., Yang, C., Zheng, Q., Chen, J., et al. (2022). Plant nitrogen nutrition: the roles of arbuscular mycorrhizal fungi. J. Plant Physiol. 269:153591. doi: 10.1016/j.jplph.2021.153591
Xu, Y. (2021). Soil microbial diversity in the habitat of Tricholoma matsutake in Northeast China. Jilin: Jilin Agricultural University.
Yamanaka, T. (2003). The effect of pH on the growth of saprotrophic and ectomycorrhizal ammonia fungi in vitro. Mycologia 95, 584–589. doi: 10.1080/15572536.2004.11833062
Yamanaka, T., Konno, M., Kawai, M., Ota, Y., Nakamura, N., and Ohta, A. (2019). Improved chlamydospore formation in Tricholoma bakamatsutake with addition of amino acids in vitro. Mycoscience 60, 319–322. doi: 10.1016/j.myc.2019.06.003
Yang, M., Qi, Y., Liu, J., Wu, Z., Gao, P., Chen, Z., et al. (2022). Dynamic changes in the endophytic bacterial community during maturation of Amorphophallus muelleri seeds. Front. Microbiol. 13:996854. doi: 10.3389/fmicb.2022.996854
Yang, C., Zeng, Z., Zhang, H., Gao, D., Wang, Y., He, G., et al. (2022). Distribution of sediment microbial communities and their relationship with surrounding environmental factors in a typical rural river, Southwest China. Environ. Sci. Pollut. Res. 29, 84206–84225. doi: 10.1007/s11356-022-21627-7
Ye, L., Fu, Y., Li, Q., Wang, Q., Zhang, B., Zou, J., et al. (2018). Bacterial community structure of Tricholoma matsutake stipe soil by high-throughput sequencing. Chin. J. Appl. Environ. Biol. 24, 583–588. doi: 10.19675/j.cnki.1006-687x.2017.08006
Yeats, T. H., and Rose, J. K. (2013). The formation and function of plant cuticles. Plant Physiol. 163, 5–20. doi: 10.1104/pp.113.222737
Yu, F., Liang, J., Song, J., Wang, S., and Lu, J. (2020). Bacterial community selection of Russula griseocarnosa mycosphere soil. Front. Microbiol. 11:347. doi: 10.3389/fmicb.2020.00347
Yu, W., Peng, M., Wang, J., Ye, W., Li, Y., Zhang, T., et al. (2021). Microbial community associated with ectomycorrhizal Russula symbiosis and dominated nature areas in southern China. FEMS Microbiol. Lett. 368:fnab028. doi: 10.1093/femsle/fnab028
Yu, J., and Yuan, H. (2023). Research progress on symbiotic interaction and host selection mechanisms of ectomycorrhizal fungi. Mycosystema 42, 86–100. doi: 10.13346/j.mycosystema.220360
Yun, W., Hall, I. R., and Evans, L. A. (1997). Ectomycorrhizal fungi with edible fruiting bodies 1.Tricholoma Matsutake and related Fungi. Econ. Bot. 51, 311–327. doi: 10.1007/bf02862101
Zhang, Y., Liao, Z., and Zhao, B. (2020). Glycine betaine: biosynthesis and biological function in halophilic microorganisms. Acta Microbiol Sin. 60, 1074–1089. doi: 10.13343/j.cnki.wsxb.20190536
Ziv, C., Zhao, Z., Gao, Y. G., and Xia, Y. (2018). Multifunctional roles of plant cuticle during plant-pathogen interactions. Front. Plant Sci. 9:1088. doi: 10.3389/fpls.2018.01088
Keywords: shiro soil, ectomycorrhizal fungi, Quercus mongolica, rhizosphere microorganism, symbiotic system, Tricholoma bakamatsutake
Citation: Guo H, Liu W, Xie Y, Wang Z, Huang C, Yi J, Yang Z, Zhao J, Yu X and Sibirina LA (2024) Soil microbiome of shiro reveals the symbiotic relationship between Tricholoma bakamatsutake and Quercus mongolica. Front. Microbiol. 15:1361117. doi: 10.3389/fmicb.2024.1361117
Edited by:
George Newcombe, University of Idaho, United StatesReviewed by:
Adnan Akhter, University of the Punjab, PakistanCopyright © 2024 Guo, Liu, Xie, Wang, Huang, Yi, Yang, Zhao, Yu and Sibirina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaodan Yu, eXV4ZDEyNkAxMjYuY29t; Lidiya Alekseevna Sibirina, c2liaXJpbmFAYmlvc29pbC5ydQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.