
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 14 April 2023
Sec. Terrestrial Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1173442
This article is part of the Research TopicSoil Microbiome Community and Functional Succession Mechanism Driven by Different Factors in Agricultural EcologyView all 43 articles
 Ruibo Sun1,2Xin Wang1,2Yousef Alhaj Hamoud3Mengxing Lu1,2Hiba Shaghaleh4Wenjie Zhang1,2Chaochun Zhang1,2,5*Chao Ma1,2*
Ruibo Sun1,2Xin Wang1,2Yousef Alhaj Hamoud3Mengxing Lu1,2Hiba Shaghaleh4Wenjie Zhang1,2Chaochun Zhang1,2,5*Chao Ma1,2*Bacteria is one of the most important drivers of straw degradation. However, the changes in bacterial community assemblage and straw-decomposing profiles during straw decomposition are not well understood. Based on cultivation-dependent and independent technologies, this study revealed that the “common species” greatly contributed to the dynamic variation of bacterial community during straw decomposition. Twenty-three functional strains involved in straw decomposition were isolated, but only seven were detected in the high-throughput sequencing data. The straw decomposers, including the isolated strains and the agents determined by functional prediction, constituted only 0.024% (on average) of the total bacterial community. The ecological network showed that most of the identified decomposers were self-existent without associations with other species. These results showed that during straw composition, community assembly might be greatly determined by the majority, but straw decomposition functions might be largely determined by the minority and emphasized the importance of the rare species in community-specific functions.
Straw is an important resource for agricultural production. It contains a high amount of nitrogen (N), phosphorus (P), potassium (K), and micronutrient elements. Thus, it is a potential substitute for chemical fertilizers (Sun et al., 2022a), and studies have found that straw returning efficiently improved soil structures and increased soil nutrient levels and crop yield (Sun et al., 2015; Chen et al., 2017, 2022). However, the direct straw return may also result in environmental and production risks in agricultural systems, such as stimulating CH4 emission and aggravating plant pest infestation (Li et al., 2018; Kerdraon et al., 2019). These issues are closely associated with the low rate of straw decomposition. Thus, accelerating straw decomposition is of great importance for the eco-friendly utilization of straw.
Microbes are the primary drivers for straw decomposition. Microbes decompose the components of straw by producing enzymes such as glucosidase, cellobiohydrolase, and xylanase (Reddy et al., 2013; Zhou et al., 2021). In addition, some microbes, such as some mycelial fungi, can physically damage the straw through the massive growth of hyphae. However, straw is made up of various complex organic substances. Besides cellulose, hemicellulose, and lignin, types of proteins, lipophilic compounds, starches, low-molecular carbohydrates, and other organic compounds have been identified in straws (Van Hung et al., 2020; Rosado et al., 2022). Thus, straw decomposition is associated with diverse microbes targeting the different components, and the microbial communities vary greatly during degradation. For example, Orlova et al. (2020) found that bacterial diversity increased during the process of oats straw decomposition, and Proteobacteria, Firmicutes, and Bacteroidetes dominated in the early stage (3rd day), while Actinobacteria dominated in the late stage (161st day). A study in a paddy field found that bacterial diversity showed a bell-shaped curve during the 2 years. Straw decomposition was dominated by copiotrophic phylotypes, such as Bacilli and Flavobacteriia, in the early stages and evolved to be dominated by oligotrophic phylotypes, such as Acidobacteria, Anaerolineae, Deltaproteobacteria, Saccharibacteria, and Sphingobacteriia, in the later stages (Wang et al., 2022).
As cellulose, hemicellulose, and lignin are straw’s main components, accounting for about 75% of rice straw’s biomass, and these biopolymers are normally decomposed slowly, the microbes targeting these components have attracted more attention than other functional types. These cellulose/lignin-decomposing microbes primarily decompose straw by producing degrading enzymes, including cellulases, hemicellulases, and lignin-degrading enzymes (Bayer et al., 2006; Floudas, 2021). Enhancing the function of these microbes is the primary pathway to accelerate straw decomposition. Many approaches have been developed to enhance microbial activities to accelerate the speed of straw decomposition, such as adding trace elements (Jin et al., 2022) or nanomaterials (Qiu et al., 2019) and improving planting strategies (Zhou et al., 2021). In addition, the construction of microbial consortiums with high straw degradation capacity is also considered a potential straw treatment method (Gong et al., 2020; Sarma et al., 2022). However, most of the present studies were performed in controlled systems; regulating microflora to enhance straw decomposition is still a great challenge in practical production. The microbial community is a complex consortium; the members are closely associated with each other (Liu et al., 2019), so it is difficult to regulate the functional groups involved in straw decomposition without considering other groups. Although many straw-decomposing strains have been isolated, and they show high capacity in straw decomposition in controlled experiments (Gupta et al., 2012; Lestari et al., 2020; Sun S. et al., 2020), how to make them perform the functions in practical systems is still a difficult problem that had not been solved, as the isolated straw-decomposing strains are difficult to colonize in the natural environments due to the great effects of environment filter and the strong competition from the indigenous residents (Sun R. et al., 2020).
Straw-decomposing microbes are widely distributed in types of ecosystems and habitats, especially in agricultural soils, but they are not alone; their growth, multiply, and functions are greatly impacted by other species (Bayer et al., 2006). Thus, understanding the associations between straw-decomposing agents and other species is crucial for regulating microbial community functions. Although many studies have revealed the variation of microbial communities during straw decomposition, we still know little about the dynamic changes of functional agents in the whole community and the relationships between the functional agents and other microbial types. This study explores the dynamic variation of bacterial community assemblage and straw-decomposing agents based on cultivation-dependent and independent techniques and the correlations between functional agents and other microbial groups. The results from this study will give new information about the microbial mechanisms of straw degradation.
A field experiment was established in June 2019 in the National Innovative and High Technology Agricultural Park of Anhui Agricultural University, Anhui Province, China (N 31°93′, E 117°20′). The soil type is Luvisols.
The field is under rice-wheat rotation. The basic properties of the studies soil were as follows, pH 6.15, soil organic matter (SOM) 11.40 g/kg, total nitrogen (TN) 0.48 g/kg, alkali-hydrolyzable nitrogen (AHN) 45.80 g/kg, available phosphorus (AP) 6.28 mg/kg, and available potassium (AK) 0.16 g/kg.
After desiccating by a dryer, wheat straw was cut into pieces (1–3 cm) and then put into nylon bags (15 cm × 20 cm, mesh size was 0.075 mm). Each bag contained 20 g wheat straw. Before rice transplanting in June 2019, the nylon bags were embedded into soils with a depth of 5–20 cm. The experiment contained three replicate plots (12 m2), and each plot contained 12 nylon bags. After treatment, rice was transplanted, and the field was managed as the local strategy.
Three nylon bags were removed from each plot at 7, 15, 60, and 120 days. Each sample was divided into two parts. One was stored at 4°C to isolate cellulose- and lignin-decomposing bacteria and the other was stored at −80°C for DNA extraction.
An enrichment culture was performed before the strain isolation. A 0.5 g sample was added to a 100 mL enrichment medium. The medium for cellulose-decomposing bacteria enrichment contained sodium carboxymethylcellulose (CMC-Na) 20 g/L, microcrystalline cellulose 5 g/L, cellulose powder 5 g/L, K2HPO4 1 g/L, KNO3 1 g/L, MgSO4·7H2O 0.2 g/L, CuCl2·2H2O 0.1 g/L, and FeCl3 0.02 g/L. The medium for lignin-decomposing bacteria enrichment contained sodium lignin sulfonate 3 g/L, NH4SO4 2 g/L, K2HPO4 1 g/L, MgSO4·7H2O 0.2 g/L, CaCl2 0.1 g/L, MnSO4 0.02 g/L, KH2PO4 1 g, FeSO4 0.05 g/L. The cellulose- and lignin-decomposing bacteria enrichment cultures were incubated at 36°C (180 rpm) and 28°C (200 rpm) for 3 days, respectively.
The functional strains were isolated using a dilution plate method (Fathallh Eida et al., 2012). 10 mL of enrichment culture was added into 90 mL sterile distilled water, and the mixture was then shaken at 150 rpm for 30 min at room temperature. Ten-fold serial dilutions were then prepared in sterilized distilled water. One-hundred microliters from the 10−6, 10−7, and 10−8 dilutions were spread on the isolation medium. The medium for cellulose-decomposing bacteria isolation contained CMC-Na 15 g/L, NH4NO3 1 g/L, yeast extract 1 g/L, MgSO4·7H2O 0.5 g/L, KH2PO4 1 g/L, and agar 15 g/L, and that for lignin-decomposing bacteria isolation contained sodium lignosulphonate 3 g/L, NH4SO4 1 g/L, KH2PO4 1 g/L, MgSO4·7H2O 0.2 g/L, CaCl2 0.1 g/L, MnSO4 0.02 g/L, MnSO4 0.02 g/L, KH2PO4 1 g, FeSO4 0.05 g/L, and agar 15 g/L. The cellulose- and lignin-decomposing bacteria isolation plates were incubated at 36°C and 28°C for 5 days, respectively.
After incubation, the cellulose- and lignin-decomposing colonies were isolated and transferred to CMC-Na Congo red medium (CMC-Na 1.88 g/L, K2HPO4 0.5 g/L, MgSO4 0.25 g/L, gelatin 2 g/L, Congo red 0.2 g/L, agar 15 g/L) and aniline blue medium (yeast extract powder 10 g/L, glucose 10 g/L, aniline blue 0.1 g/L, agar 15 g/L) respectively to confirm the decomposition capacity. All the colonies containing decomposition capacity were transferred to the LB plate to obtain a pure culture.
Colony PCR was used for the amplification of bacterial 16S rRNA using the primer sets of 27F (5′- GAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′). The amplification was performed in a 50-μL system with the thermal cycle of the initial denaturation step at 94°C for 2 min, 30 cycles with denaturation at 98°C for 10 s, annealing at 51°C for 30 s, and extension at 72°C for the 90 s, then a final extension at 72°C for 5 min. The 50-μL PCR system contained 25 μL PCR premix (Ex TaqTM; Takara, Shiga, Japan), 1 μL (10 μM) of the forward and reverse primers, and 23 μL sterile double-distilled water. After being purified using a QIAquick PCR Purification Kit (Qiagen, Hilden, Germany), the PCR products were sequenced using a 3730xl DNA analyzer (Applied Biosystems).
The bacterial community was detected as described in previous studies (Sun et al., 2022b,c). In short, total DNA was extracted from a 0.5 g sample using a FastDNA SPIN Kit (MP Biomedicals, Santa Ana, CA, United States). Before DNA extraction, the samples were frozen using liquid nitrogen and then grinded thoroughly. Primer sets 515F/907R targeting the V4-V5 region of 16S rRNA gene were used for PCR amplification, which was performed in a 25-μL system containing 12.5 μL PCR premix (Ex TaqTM; Takara, Shiga, Japan), 0.5 μL (10 μM) of each primer, 0.5 μL DNA template (20 ng), and 11 μL sterile double-distilled water with the thermocycler of an initial denaturation at 94°C for 10 min, 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C/56°C (16S rRNA/ITS) for 45 s, and extension at 72°C for 1 min, followed by a final extension at 72°C for 10 min. After purification, the PCR products were sequenced using the Illumina HiSeq2000 platform (Illumina, San Diego, CA, United States).
Bioinformatic analysis was performed primally using the VSEARCH package (version 2.21.1) (Rognes et al., 2016) according to the pipelines described in a previous study (Sun et al., 2023). The adept and primer sequences were cut using Cutadapt (version 4.2) (Martin, 2011). Then the paired-end reads were merged, and low-quality reads (length < 150 bp, expected errors per base >0.001) were filtered. After removing chimeric reads using the UCHIME algorithm (Edgar et al., 2011), the clean reads were denoised using the UNOISE algorithm (version 3), and zero-radius operational taxonomic units (zOTUs) were generated. Taxonomic assignment of each zOTU was performed using SINTAX (Edgar, 2016) based on the Silva database (version 138). The unidentified zOUTs were removed, and the zOTU table was rarified to 19,000 reads per sample for downstream analysis.
The V4-V5 region of the 16S rRNA sequences of the isolated strains was extracted using Cutadapt (version 4.2) (Martin, 2011). Then the trimmed sequences were clustered with the sequence of each zOTU at the similarity of 100% to find which zOTU they matched.
FAPROTAX (Louca et al., 2016) was used for the functional prediction of the bacterial community, and the function categories involved in straw decomposition were selected for further analysis.
R (version 4.0.2) was used for statistical analysis using the relevant libraries (Sun et al., 2019, 2021). The Kruskal–Wallis rank sum test was used to check the significance of the difference between treatments using the “dplyr” library. Principal coordinate analysis (PCoA) based on Bray–Curtis distance was performed using the “vegan” library to show the variation of bacterial community among treatments. The abundance-based beta-null model was used to determine the role of niche and neutral processes in shaping bacterial communities during straw decomposition (Tucker et al., 2016). The bacterial ecological network was inferred using SPIEC-EASI (Sparse InversE Covariance Estimation for Ecological Association Inference) (Kurtz et al., 2015). Only the zOTUs detected in more than 75% of the samples were incorporated in the construction of the network to strengthen its reliability of the network (Sun R. et al., 2020). The characteristics of the network were calculated using the “igraph” library. The beta-null deviation was calculated to infer the changes in deterministic and stochastic processes in shaping bacterial community assembly (Tucker et al., 2016).
The bacterial community in the studied samples was dominant by Bacteroidota, Firmicutes, Proteobacteria, Spirochaetota, Acidobacteriota, Actinobacteria, Chloroflexi, and Desulfobacterota, accounting for 95.89% of the total reads (Figure 1A). The composition of the bacterial community varied obviously during rice straw degradation. The dominance of Bacteroidota and Firmicutes decreased during the experiment, while the relative abundance of Proteobacteria and Acidobacteriota was significantly higher in the late stage (60 and 120 days) than in the early stage (7 and 15 days) (Figure 1A). The dynamic variation of the bacterial community was illustrated clearly in the 2D PCoA plot (Figure 1B), in which the bacterial community from different stages separated from each other. The bacterial community from D60 and D120 was far separated from that from D7 and D15, showing the great variation of the bacterial community after 15 days (Figure 1B).
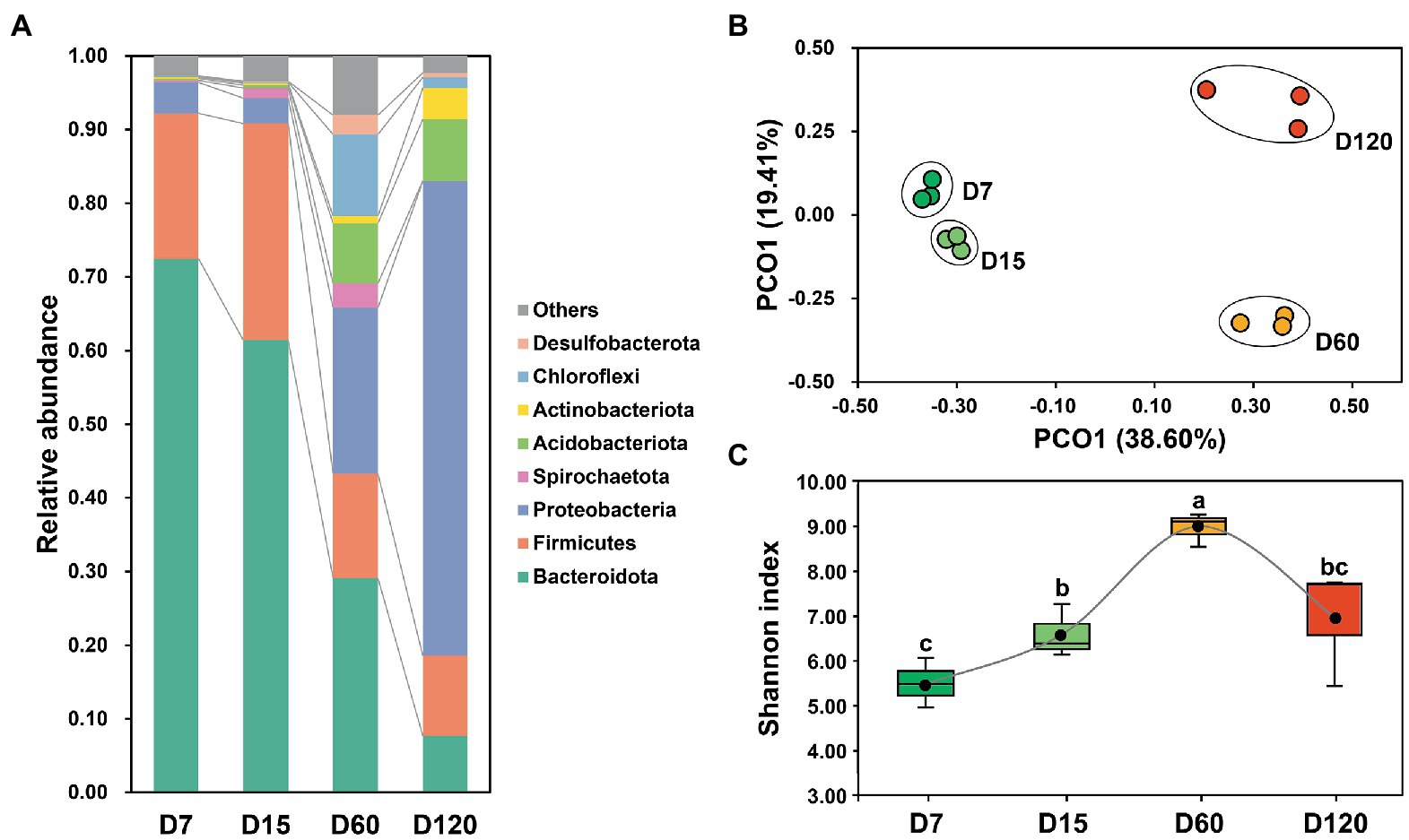
Figure 1. Variation of bacterial community composition during experimental session (A). A PCoA plot of the bacterial community in different stage (B). Dynamic variation of bacterial diversity during experimental session (C). Different letters indicate significant differences as checked by the Kruskal-Wallis rank sum test (p < 0.05).
The Shannon index was calculated to explore the variation of bacterial diversity during the experiment. During the 120 days, bacterial diversity showed a bell-shaped curve (Figure 1C). Bacterial diversity increased from D7 to D60, then decreased from D60 to D120.
In total, 3,314 zOTUs were detected in all the samples, and 782 zOTUs were shared in all samples. The number of unique zOTUs in D7, D15, D60, and D120 was 30, 19, 267, and 214, respectively (Figure 2). The unique zOTUs in each sample were rare species in the whole community; the total relative abundance of unique zOTUs in D7, D15, D60, and D120 was 0.37, 0.24, 8.74, and 8.26%, respectively. Differently, the community was dominated by the shared zOTUs, which accounted for 88.32, 85.01, 47.11, and 60.17% of the community in relative abundance for D7, D15, D60, and D120, respectively. Most of the shared OTUs were assigned as Bacteroidota, Firmicutes, Proteobacteria, Cyanobacteria, Actinobacteriota, and Acidobacteriota (Figure 2), but the taxonomic composition of the bacterial community varied greatly during the experiment. The relative abundance of shared Bacteroidota and Cyanobacteria decreased with time, while that of shared Proteobacteria, Actinobacteriota, and Acidobacteriota increased with time.
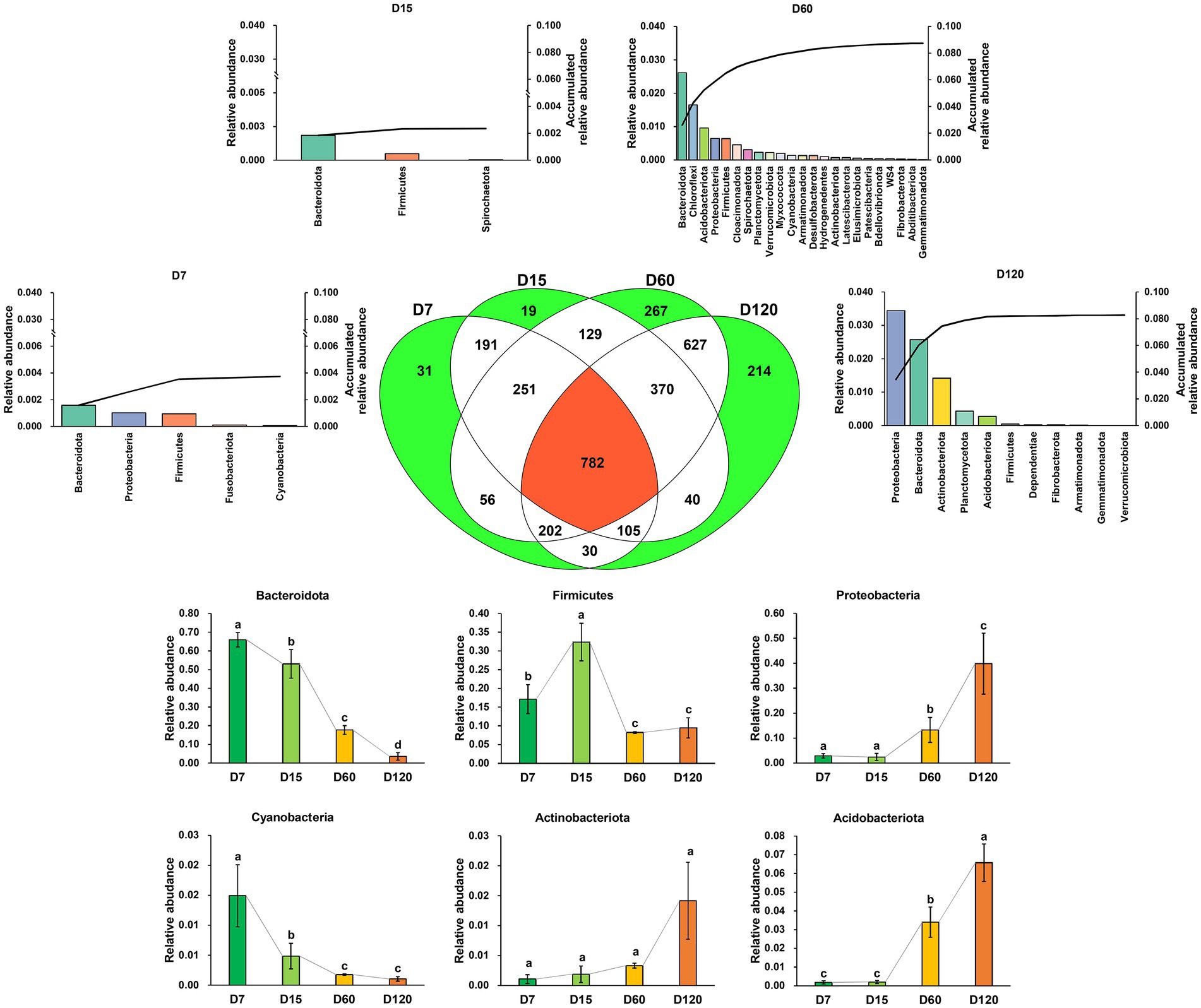
Figure 2. A Venn diagram showing the zOTU distribution in different stages. The bar charts denoted with stage name indicate the taxonomic composition and their accumulated relative abundance of the unique zOTUs in the stage. The bar charts at the bottom of the figure showed the variation of the dominant phyla of the shared zOTUs. Different letters indicate significant differences as checked by the Kruskal-Wallis rank sum test (p < 0.05).
By calculating the beta-null deviation of the bacterial community in each stage, the contribution of niche and neutral processes in shaping the bacterial community was inferred. The beta-null deviations in all the samples were positive, showing the bacterial community as less similar than expected by chance and reflecting communities structured by non-neutral assembly mechanisms. Overall, the beta-null deviation of the bacterial community was increased during the experimental session (Figure 3). Beta-null deviations in D7 and D15 were significantly lower than in D60 and D120, and the biggest beta-null deviation was observed in D120. This result showed that the contribution of niche processes increased in structuring bacterial community during straw decomposition.
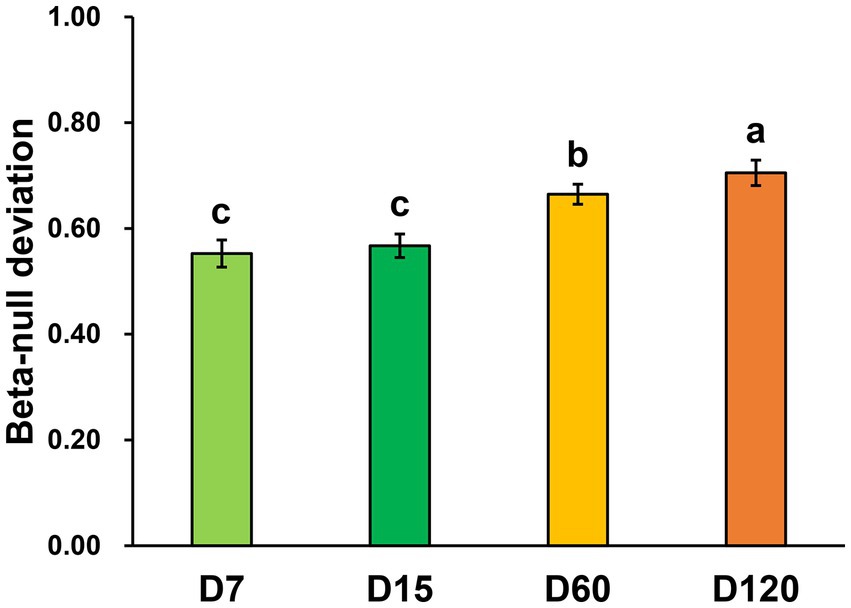
Figure 3. Abundance-based beta-null deviation for bacterial communities based on the Bray–Curtis distance. Different letters indicate significant differences as checked by the Kruskal-Wallis rank sum test (p < 0.05).
Three functional categories involved in straw decomposition were detected through functional prediction, including cellulolysis, ligninolysis, and aromatic compound degradation. The dynamic variation of the relative abundance of zOTUs involved in cellulolysis showed an inverted bell curve during the experiment, which was significantly higher in D7 and D120 than in D15 and D60 (Figure 4A). The species involved in ligninolysis were only detected in the samples from D15 and D120, and their relative abundance was quite low (Figure 4B). Differently, the species with aromatic compound degradation capacity increased with experiment time (Figure 4C).
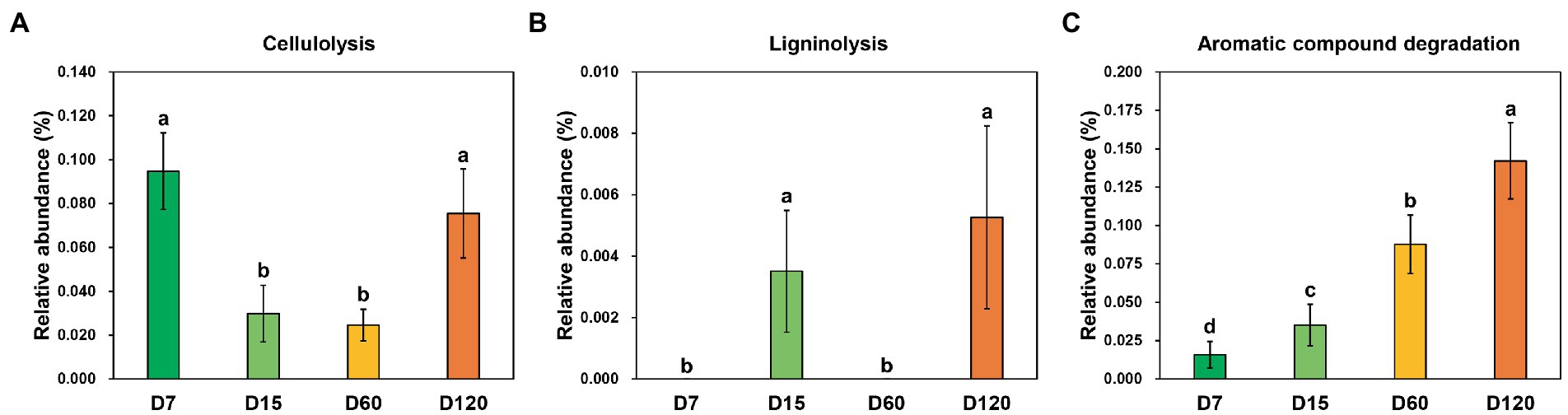
Figure 4. Variation of the functional profiles involved in straw decomposition determined by functional prediction. (A) Cellulolysis function. (B) Ligninolysis function. (C) Aromatic compound degradation. Different letters indicate significant differences as checked by the Kruskal-Wallis rank sum test (p < 0.05).
Twenty-three strains with the potential of straw decomposition were isolated, including 15 cellulose-decomposing strains and 8 lignin-decomposing strains. By comparing the sequence information of 16S rRNA, seven strains were detected in the community determined by high-throughput sequencing, which matched 5 zOTUs in the community (Table 1). The relative abundance of these functional strains was low in the whole bacterial community, and they varied during the experimental session (Table 1). The relative abundance of cellulose-decomposing zOTU increased with experimental time, while the changes of zOTUs involved in lignin decomposition were various (Table 1).
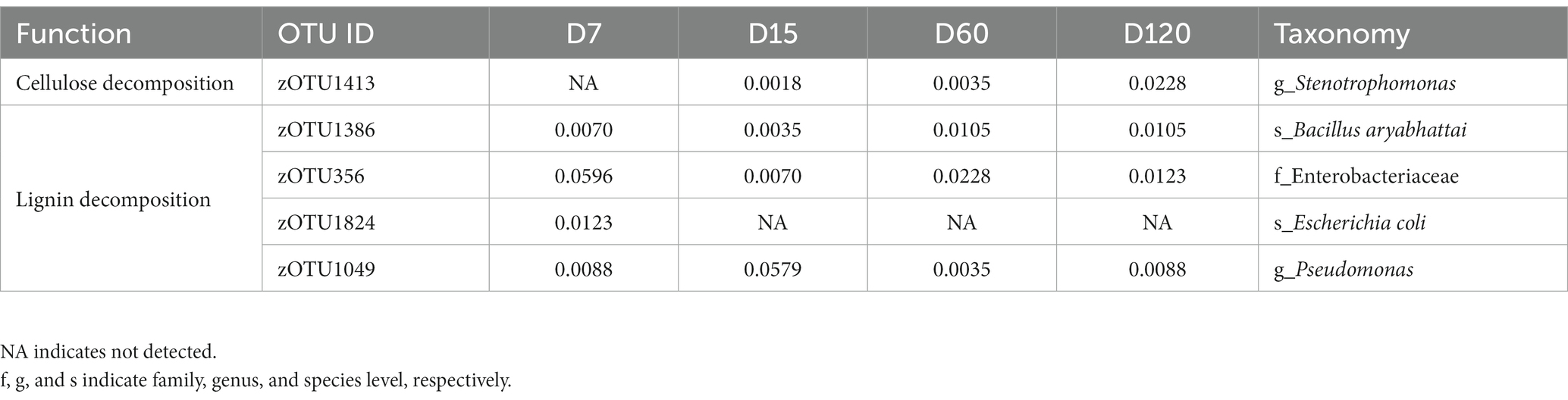
Table 1. Changes in relative abundance (%) of cellulose and lignin degrading bacteria during straw degradation.
An ecological network with 161 zOTUs (nodes) and 140 edges was constructed to infer the associations between the community members (Figure 4). Eighteen zOTUs with the capacity for cellulose, lignin, and aromatic compound degradation were incorporated into the ecological network. Only 3 zOTUs were associated with other zOTUs, and the other 15 zOTUs had no association with other zOTUs (Figure 5).
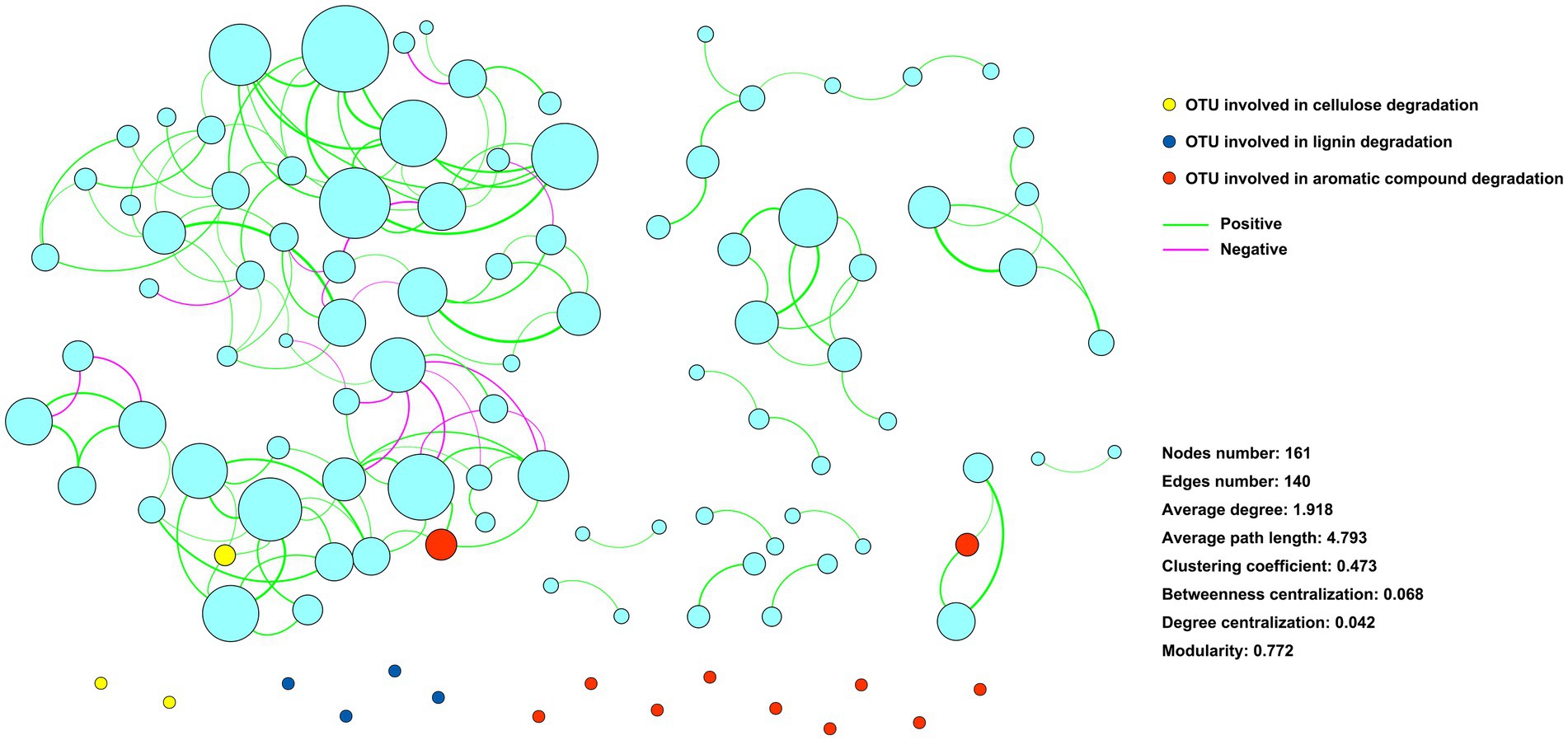
Figure 5. Ecological network showing the associations among community components. Nodes represent zOTUs, and the size of the node indicates the relative abundance of the zOTU. Nodes colored with yellow, blue, and red indicate zOTUs involved in cellulose, lignin, and aromatic compound degradation, respectively. Edges indicate associates between zOTUs.
Straw is a carbon-rich resource containing valuable nutrients for plant growth. Thus, the effective utilization of straw is of great importance for agricultural production and environmental protection, especially for large agricultural countries such as China (Chen et al., 2022). However, the low speed of decomposition strongly restricts the effects of the direct return of straw on agricultural production. As the key driver for straw decomposition, microbes are the potential solution for this issue. Understanding the mechanisms of microbial community assembly and function dynamics is the premise for microbial community regulation. The variation of microbial composition and diversity during straw decomposition has been widely reported (Orlova et al., 2020; Zhong et al., 2020; Zhang et al., 2021). In this study, through the beta-null model, we further demonstrated that the contribution of the niche process was increased during the decomposition (Figure 3). This may be partly associated with the changes in competitive intensity between community members. In the early stage, all the microbes have almost equal opportunity (without regard to their difference in population) to decompose straw. In addition, the competition between community members is weak due to the high content of readily decomposable carbon fractions in the early stage (Preston et al., 2009). Thus, the community in the early stage was more likely randomly assembled. As decomposition goes on, recalcitrant carbon fractions, such as cellulose, lignin, waxes, and tannins, gradually accumulate (Wang et al., 2022). As a result, the effect of substrate limitation becomes stronger and stronger, and the microbes targeting these substrate gains a competitive advantage and show an increase in relative abundance (Orlova et al., 2020). We also observed the gradual enrichment of aromatic compound-decomposing bacteria during the experiment (Figure 4C). Thus, the contribution of the niche processes in shaping bacterial communities increased.
The role of abundant and rare species in community assembly, diversity, and ecological functions is a key topic in microbial ecology research (Magurran and Henderson, 2003; Zhang et al., 2020, 2022). The contribution of the species with different abundance in community variation and straw decomposition is little known. The results from the present study showed that the variation of the bacterial community was primarily associated with the “common species” (Figure 2), which was the species that existed during the whole process of straw decomposition and was termed as “core microbiome” too. The core microbiome is important for community function and microbial associations (Neu et al., 2021). However, in the present study, within the core bacterial community, the species directly associated with straw decomposition, including cellulolysis and aromatic compound degradation, was not dominant in relative abundance. The average relative abundance of zOTUs involved in cellulolysis and aromatic compound degradation was 0.056 and 0.048%, respectively (Supplementary Table S1), indicating that most common species were not directly associated with straw decomposition. Even with the respect of the whole community, the species directly associated with straw decomposition were also a minority (Figure 3). These results indicate that the decomposition of straw may largely depend on minority species. Due to the high functional redundancy of microbial communities (Allison and Martiny, 2008), microbial communities’ whole potential functional profiles were more stable than the taxonomic profiles. For example, Bao et al. (2020) found that the functional composition of the microbial community associated with straw decomposition was more stable than taxonomic composition. However, this conclusion was obtained based on the broad functional profiles of the microbial community. For some specific functions, the conclusion may be different. By growing many bacterial communities under the same environmental conditions, Rivett and Bell (2018) revealed the negative statistical interactions among abundant phylotypes drive variation in broad functional measures (respiration, metabolic potential, cell yield), whereas positive interactions between rare phylotypes influence narrow functional measures (the capacity of the communities to degrade specific substrates).
In contrast, rare phylotypes positively interacted with narrow functional measures (the capacity to degrade specific substrates), demonstrating that unique components of complex communities are associated with different ecosystem functioning. In general, unique species are rare in the community. Thus, the specific function of the community may be largely determined by the rare species, which also indicates the low redundancy of the specific functions.
The low abundance of species directly involved in straw decomposition may be one important reason for the low speed of straw decomposition. However, low abundance does not mean low activity. Studies have found that low-abundance species display disproportionately high activity for specific functions (Pascoal et al., 2020). For example, the high activity of sulfate reduction was driven by the rare biosphere microorganism in a peatland (Pester et al., 2010). The rare biosphere bacteria played a predominant role in phenanthrene degradation (Sauret et al., 2014), as some low-abundance microorganisms can be highly transcriptionally active without growth (Hausmann et al., 2019). Thus, how to increase the activity of the rare species involved in straw decomposition is a significant issue for the acceleration of straw decomposition.
The interactions between microbial community components affect microbial functions (Liu et al., 2019). Our previous study revealed that the organic phosphorus-mineralizing microbial taxa were regulated by the keystone taxa of the microbial community (Chen et al., 2020). The present study showed that few of the identified straw-decomposing species were associated with other species. In contrast, most of the functional species were independent in the ecological network (Figure 5), showing the diversiform survival types of the functional species. This may be important for sustaining microbial straw-decomposing capacity in complex and dynamic environments. Understanding the role of these functional species in straw decomposition would provide crucial information for regulating microbial straw-decomposing functions.
Although many achievements in microbial community assembly and functions have been obtained during the past years, especially from devising high-throughput sequencing technologies, we may still be far from the truth, as the sequencing technologies contain large biases, and most of the microorganisms are still unculturable. In the present study, 23 functional strains were isolated. Still, only seven were detected in the high-throughput sequencing data, indicating that many species had been omitted in the sequencing data. The low coverage of the primers may be the primary issue, and using PCR-bias-free technologies, such as metagenomic sequencing, may provide more accurate results (Sun et al., 2021). In addition, due to the limitations of microbial isolation and culture technologies, types of functional strains are still unculturable. As a result, many functional microbes are omitted and the role of the functional microbes in community assemblage and ecological functions may be underestimated. However, the great challenge of strain isolation and culture may be the stumbling block lasting for a long time. What’s more, straw decomposition is associated with types of microbes, especially fungi (Krishna and Mohan, 2017; Bani et al., 2018), thus understanding the associations between different microbial types is also important.
Understanding the mechanisms of microbial community assembly and functional profiles is vital for managing straw decomposition by regulating microbial communities. This study revealed the dynamic variations of bacterial community assembly and straw-decomposing function during straw decomposition. Results showed that the variations of bacterial community greatly resulted from the changes of “common species,” and the contribution of niche process in structuring bacterial community increased during the decomposition. The relative abundance of the functional bacterial agents involved in straw decomposition was low in the whole bacterial community, and only a few of them were associated with other species, indicating that the rare species may greatly determine the functional profiles involved in straw decomposition and most of the functional agents were self-existent.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, PRJEB60739.
RS, CZ, and CM designed the experiment and wrote the manuscript. XW and ML provided experimental materials and directed the experiment. XW, ML, and WZ performed the laboratorial measurements. RS performed the data analysis. YH and HS performed language edition and correction. All authors contributed to the article and approved the submitted version.
This work was supported by the Key Science and Technology Project of Anhui Province (202103a06020012), the National Natural Science Foundation of China (32071628), and the Colleges and Universities Science Foundation of Anhui Province (KJ2021ZD0009).
We thank the staff at the experimental station for the management of the experimental field. RS appreciates Jessy for giving him the sweetest time in the past.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1173442/full#supplementary-material
Allison, S. D., and Martiny, J. B. H. (2008). Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. U. S. A. 105, 11512–11519. doi: 10.1073/pnas.0801925105
Bani, A., Pioli, S., Ventura, M., Panzacchi, P., Borruso, L., Tognetti, R., et al. (2018). The role of microbial community in the decomposition of leaf litter and deadwood. Appl. Soil Ecol. 126, 75–84. doi: 10.1016/j.apsoil.2018.02.017
Bao, Y. Y., Guo, Z. Y., Chen, R. R., Wu, M., Li, Z. P., Lin, X. G., et al. (2020). Functional community composition has less environmental variability than taxonomic composition in straw-degrading bacteria. Biol. Fertil. Soils 56, 869–874. doi: 10.1007/s00374-020-01455-y
Bayer, E. A., Shoham, Y., and Lamed, R. (2006). “Cellulose-decomposing bacteria and their enzyme systems” in The prokaryotes: Volume 2: Ecophysiology and biochemistry. eds. M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (Springer New York: New York, NY), 578–617.
Chen, Y., Sun, R. B., Sun, T. T., Chen, P., Yu, Z. Y., Ding, L. Y., et al. (2020). Evidence for involvement of keystone fungal taxa in organic phosphorus mineralization in subtropical soil and the impact of labile carbon. Soil Biol. Biochem. 148:107900. doi: 10.1016/j.soilbio.2020.107900
Chen, L. M., Sun, S. L., Yao, B., Peng, Y. T., Gao, C. F., Qin, T., et al. (2022). Effects of straw return and straw biochar on soil properties and crop growth: A review. Front. Plant Sci. 13:986763. doi: 10.3389/fpls.2022.986763
Chen, J., Zheng, M.-J., Pang, D.-W., Yin, Y.-P., Han, M.-M., Li, Y.-X., et al. (2017). Straw return and appropriate tillage method improve grain yield and nitrogen efficiency of winter wheat. J. Integr. Agric. 16, 1708–1719. doi: 10.1016/S2095-3119(16)61589-7
Edgar, R. C. (2016). SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv. doi: 10.1101/074161
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fathallh Eida, M., Nagaoka, T., Wasaki, J., and Kouno, K. (2012). Isolation and characterization of cellulose-decomposing bacteria inhabiting sawdust and coffee residue composts. Microbes Environ. 27, 226–233. doi: 10.1264/jsme2.ME11299
Floudas, D. (2021). “Chapter two – Evolution of lignin decomposition systems in fungi” in Advances in botanical research. eds. M. Morel-Rouhier and R. Sormani (London: Academic Press), 37–76.
Gong, X., Zou, H., Qian, C., Yu, Y., Hao, Y., Li, L., et al. (2020). Construction of in situ degradation bacteria of corn straw and analysis of its degradation efficiency. Ann. Microbiol. 70:62. doi: 10.1186/s13213-020-01601-9
Gupta, P., Samant, K., and Sahu, A. (2012). Isolation of cellulose-degrading bacteria and determination of their cellulolytic potential. Int. J. Microbiol. 2012:578925. doi: 10.1155/2012/578925
Hausmann, B., Pelikan, C., Rattei, T., Loy, A., and Pester, M. (2019). Long-term transcriptional activity at zero growth of a cosmopolitan rare biosphere member. mBio 10:30755506. doi: 10.1128/mBio.02189-18
Jin, M., Chen, X., Gao, M., Sun, R., Tian, D., Xiong, Q., et al. (2022). Manganese promoted wheat straw decomposition by regulating microbial communities and enzyme activities. J. Appl. Microbiol. 132, 1079–1090. doi: 10.1111/jam.15266
Kerdraon, L., Laval, V., and Suffert, F. (2019). Microbiomes and pathogen survival in crop residues, an ecotone between plant and soil. Phytobiomes J. 3, 246–255. doi: 10.1094/PBIOMES-02-19-0010-RVW
Krishna, M. P., and Mohan, M. (2017). Litter decomposition in forest ecosystems: A review. Energy Ecol. Environ. 2, 236–249. doi: 10.1007/s40974-017-0064-9
Kurtz, Z. D., Müller, C. L., Miraldi, E. R., Littman, D. R., Blaser, M. J., and Bonneau, R. A. (2015). Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 11:e1004226. doi: 10.1371/journal.pcbi.1004226
Lestari, Y., Maftu’ah, E., and Annisa, W. (2020). Rice straw composting by cellulolytic bacteria isolate and its application on rice in acid sulfate soils. BIO Web Conf. 20:01006. doi: 10.1051/bioconf/20202001006
Li, H., Dai, M., Dai, S., and Dong, X. (2018). Current status and environment impact of direct straw return in China's cropland - A review. Ecotoxicol. Environ. Saf. 159, 293–300. doi: 10.1016/j.ecoenv.2018.05.014
Liu, J., Meng, Z., Liu, X., and Zhang, X.-H. (2019). Microbial assembly, interaction, functioning, activity and diversification: A review derived from community compositional data. Mar. Life Sci. Tech. 1, 112–128. doi: 10.1007/s42995-019-00004-3
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Magurran, A. E., and Henderson, P. A. (2003). Explaining the excess of rare species in natural species abundance distributions. Nature 422, 714–716. doi: 10.1038/nature01547
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Neu, A. T., Allen, E. E., and Roy, K. (2021). Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. U. S. A. 118:e2104429118. doi: 10.1073/pnas.2104429118
Orlova, O. V., Kichko, A. A., Pershina, E. V., Pinaev, A. G., and Andronov, E. E. (2020). Succession of bacterial communities in the decomposition of oats straw in two soils with contrasting properties. Eurasian Soil Sci. 53, 1620–1628. doi: 10.1134/S1064229320090112
Pascoal, F., Costa, R., and Magalhães, C. (2020). The microbial rare biosphere: Current concepts, methods and ecological principles. FEMS Microbiol. Ecol. 97:fiaa227. doi: 10.1093/femsec/fiaa227
Pester, M., Bittner, N., Deevong, P., Wagner, M., and Loy, A. (2010). A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 4, 1591–1602. doi: 10.1038/ismej.2010.75
Preston, C. M., Nault, J. R., and Trofymow, J. A. (2009). Chemical changes during 6 years of decomposition of 11 litters in some Canadian forest sites. Part 2. 13C abundance, solid-state 13C NMR spectroscopy and the meaning of “lignin”. Ecosystems 12, 1078–1102. doi: 10.1007/s10021-009-9267-z
Qiu, C., Feng, Y., Wu, M., Zhang, J., Chen, X., and Li, Z. (2019). NanoFe3O4 accelerates methanogenic straw degradation in paddy soil enrichments. Appl. Soil Ecol. 144, 155–164. doi: 10.1016/j.apsoil.2019.07.015
Reddy, A. P., Simmons, C. W., D’haeseleer, P., Khudyakov, J., Burd, H., Hadi, M., et al. (2013). Discovery of microorganisms and enzymes involved in high-solids decomposition of rice straw using metagenomic analyses. PLoS One 8:e77985. doi: 10.1371/journal.pone.0077985
Rivett, D. W., and Bell, T. (2018). Abundance determines the functional role of bacterial phylotypes in complex communities. Nat. Microbiol. 3, 767–772. doi: 10.1038/s41564-018-0180-0
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahe, F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rosado, M. J., Marques, G., Rencoret, J., Gutiérrez, A., and Del Río, J. C. (2022). Chemical composition of lipophilic compounds from rice (Oryza sativa) straw: An attractive feedstock for obtaining valuable phytochemicals. Front. Plant Sci. 13:868319. doi: 10.3389/fpls.2022.868319
Sarma, S., Patel, N., Patel, A., Desai, C., Sharma, S., Dedania, S., et al. (2022). Rapid decomposition of rice straw by application of a novel microbial consortium and study its microbial community dynamics. World J. Microbiol. Biotechnol. 38:212. doi: 10.1007/s11274-022-03399-x
Sauret, C., Séverin, T., Vétion, G., Guigue, C., Goutx, M., Pujo-Pay, M., et al. (2014). ‘Rare biosphere’ bacteria as key phenanthrene degraders in coastal seawaters. Environ. Pollut. 194, 246–253. doi: 10.1016/j.envpol.2014.07.024
Sun, R., Chen, Y., Han, W. X., Dong, W. X., Zhang, Y. M., Hu, C. S., et al. (2020). Different contribution of species sorting and exogenous species immigration from manure to soil fungal diversity and community assemblage under long-term fertilization. Soil Biol. Biochem. 151:108049. doi: 10.1016/j.soilbio.2020.108049
Sun, R., Ding, J., Li, H., Wang, X., Li, W., Li, K., et al. (2023). Mitigating nitrate leaching in cropland by enhancing microbial nitrate transformation through the addition of liquid biogas slurry. Agric. Ecosyst. Environ. 345:108324. doi: 10.1016/j.agee.2022.108324
Sun, R. B., Li, W. Y., Hu, C. S., and Liu, B. B. (2019). Long-term urea fertilization alters the composition and increases the abundance of soil ureolytic bacterial communities in an upland soil. FEMS Microbiol. Ecol. 95:f iz044. doi: 10.1093/femsec/fiz044
Sun, R., Niu, J., Luo, B., Wang, X., Li, W., Zhang, W., et al. (2022a). Substitution of manure for mineral P fertilizers increases P availability by enhancing microbial potential for organic P mineralization in greenhouse soil. Front. Bioeng. Biotechnol. 10:1078626. doi: 10.3389/fbioe.2022.1078626
Sun, R. B., Wang, F. H., Hu, C. S., and Liu, B. B. (2021). Metagenomics reveals taxon-specific responses of the nitrogen-cycling microbial community to long-term nitrogen fertilization. Soil Biol. Biochem. 156:108214. doi: 10.1016/j.soilbio.2021.108214
Sun, R., Wang, X. G., Tian, Y. P., Guo, K., Feng, X. H., Sun, H. Y., et al. (2022c). Long-term amelioration practices reshape the soil microbiome in a coastal saline soil and alter the richness and vertical distribution differently among bacterial, archaeal, and fungal communities. Front. Microbiol. 12:768203. doi: 10.3389/fmicb.2021.768203
Sun, S., Weng, Y. T., Di, X. Y., Liu, Z. H., and Yang, G. (2020). Screening of cellulose-degrading fungi in forest litter and fungal effects on litter decomposition. Bioresources 15, 2937–2946. doi: 10.15376/biores.15.2.2937-2946
Sun, R. B., Zhang, X. X., Guo, X. S., Wang, D. Z., and Chu, H. Y. (2015). Bacterial diversity in soils subjected to long-term chemical fertilization can be more stably maintained with the addition of livestock manure than wheat straw. Soil Biol. Biochem. 88, 9–18. doi: 10.1016/j.soilbio.2015.05.007
Sun, R., Zhang, W., Liu, Y., Yun, W., Luo, B., Chai, R., et al. (2022b). Changes in phosphorus mobilization and community assembly of bacterial and fungal communities in rice rhizosphere under phosphate deficiency. Front. Microbiol. 13:953340. doi: 10.3389/fmicb.2022.953340
Tucker, C. M., Shoemaker, L. G., Davies, K. F., Nemergut, D. R., and Melbourne, B. A. (2016). Differentiating between niche and neutral assembly in metacommunities using null models of beta-diversity. Oikos 125, 778–789. doi: 10.1111/oik.02803
Van Hung, N., Maguyon-Detras, M. C., Migo, M. V., Quilloy, R., Balingbing, C., Chivenge, P., et al. (2020). “Rice straw overview: Availability, properties, and management practices” in Sustainable rice straw management. eds. M. Gummert, N. V. Hung, P. Chivenge, and B. Douthwaite (Cham: Springer International Publishing), 1–13.
Wang, X., He, P., Xu, X., Qiu, S., and Zhao, S. (2022). Characteristics of rice straw decomposition and bacterial community succession for 2 consecutive years in a paddy field in southeastern China. Sci. Rep. 12:20893. doi: 10.1038/s41598-022-25229-8
Zhang, H., Cao, Y., and Lyu, J. (2021). Decomposition of different crop straws and variation in straw-associated microbial communities in a peach orchard, China. J. Arid Land. 13, 152–164. doi: 10.1007/s40333-021-0001-9
Zhang, H., Hou, F., Xie, W., Wang, K., Zhou, X., Zhang, D., et al. (2020). Interaction and assembly processes of abundant and rare microbial communities during a diatom bloom process. Environ. Microbiol. 22, 1707–1719. doi: 10.1111/1462-2920.14820
Zhang, Z., Lu, Y., Wei, G., and Jiao, S. (2022). Rare species-driven diversity-ecosystem multifunctionality relationships are promoted by stochastic community assembly. mBio 13:e0044922. doi: 10.1128/mbio.00449-22
Zhong, Y. Q. W., Liu, J., Jia, X. Y., Shangguan, Z. P., Wang, R. W., and Yan, W. M. (2020). Microbial community assembly and metabolic function during wheat straw decomposition under different nitrogen fertilization treatments. Biol. Fertil. Soils 56, 697–710. doi: 10.1007/s00374-020-01438-z
Keywords: straw degradation, bacterial community assembly, lignocellulosic decomposing bacteria, cellulose-decomposing bacteria, microbial interaction, straw returning
Citation: Sun R, Wang X, Alhaj Hamoud Y, Lu M, Shaghaleh H, Zhang W, Zhang C and Ma C (2023) Dynamic variation of bacterial community assemblage and functional profiles during rice straw degradation. Front. Microbiol. 14:1173442. doi: 10.3389/fmicb.2023.1173442
Edited by:
Bin Huang, Tobacco Research Institute (CAAS), ChinaReviewed by:
Meng Wu, Institute of Soil Science (CAS), ChinaCopyright © 2023 Sun, Wang, Alhaj Hamoud, Lu, Shaghaleh, Zhang, Zhang and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaochun Zhang, zhangcc@cau.edu.cn; Chao Ma, chaoma@ahau.edu.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.