 Yonggang Zhao
Yonggang Zhao Dingqiang Chen
Dingqiang Chen Boyang Ji
Boyang Ji Xingju Zhang4
Xingju Zhang4 Lars Jelsbak
Lars Jelsbak
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 14 April 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1117017
This article is part of the Research Topic Insights in Evolutionary & Genomic Microbiology: 2022 View all 11 articles
The ever-increasing prevalence of infections produced by multidrug-resistant or extensively drug-resistant Pseudomonas aeruginosa is commonly linked to a limited number of aptly-named epidemical ‘high-risk clones’ that are widespread among and within hospitals worldwide. The emergence of new potential high-risk clone strains in hospitals highlights the need to better and further understand the underlying genetic mechanisms for their emergence and success. P. aeruginosa related high-risk clones have been sporadically found in China, their genome sequences have rarely been described. Therefore, the large-scale sequencing of multidrug-resistance high-risk clone strains will help us to understand the emergence and transmission of antibiotic resistances in P. aeruginosa high-risk clones. In this study, 212 P. aeruginosa strains were isolated from 2 tertiary hospitals within 3 years (2018–2020) in Guangdong Province, China. Whole-genome sequencing, multi-locus sequence typing (MLST) and antimicrobial susceptibility testing were applied to analyze the genomic epidemiology of P. aeruginosa in this region. We found that up to 130 (61.32%) of the isolates were shown to be multidrug resistant, and 196 (92.45%) isolates were Carbapenem-Resistant Pseudomonas aeruginosa. MLST analysis demonstrated high diversity of sequence types, and 18 reported international high-risk clones were identified. Furthermore, we discovered the co-presence of exoU and exoS genes in 5 collected strains. This study enhances insight into the regional research of molecular epidemiology and antimicrobial resistance of P. aeruginosa in China. The high diversity of clone types and regional genome characteristics can serve as a theoretical reference for public health policies and help guide measures for the prevention and control of P. aeruginosa resistance.
Pseudomonas aeruginosa (P. aeruginosa) is an opportunistic pathogen and one of the major causes of nosocomial infections, particularly affecting intensive care units (ICU) and immunocompromised patients, with a very high morbidity and mortality rate (Botelho et al., 2019; Thornton and Parkins, 2023). In addition, P. aeruginosa is the most frequent chronic respiratory infection contributing to the pathogenesis of cystic fibrosis (CF) and other chronic underlying diseases (Kawalek et al., 2020). The innate and acquired antibiotics resistance mechanism of P. aeruginosa includes reduced permeability, antibiotic efflux, expression change, antibiotic modification/degradation, target protection and target modification (including the acquisition of insensitive functional target) (Pang et al., 2019).
Infections caused by multidrug-resistant (MDR) or extensively drug-resistant (XDR) P. aeruginosa are extremely difficult to treat (Kawalek et al., 2020). In Asia, the MDR and XDR rates of P. aeruginosa nosocomial pneumonia were reported to 42.8 and 4.9%, respectively (Pang et al., 2019). Over the last few decades, the occurrence of epidemic outbreaks caused by antibiotic resistance strains within hospital environments has been given more attention (Talebi Bezmin Abadi et al., 2019). Recent works have provided further evidence that a small group of MDR/XDR global clones are disseminated in many hospitals worldwide (Telling et al., 2018; Hu et al., 2021; Gravningen et al., 2022). The most prevalent of these globally disseminated clones include ST235, ST111, ST233, ST244, ST357, ST308, ST175, ST277, ST654 and ST298, and they have been denominated high-risk clones (Oliver et al., 2015; del Barrio-Tofiño et al., 2020). Furthermore, some types of P. aeruginosa strains have developed resistance to carbapenems, called Carbapenem-Resistant P. aeruginosa (CRPA). The production of carbapenemases, especially Ambler class B Metallo-β-lactamases (MBLs), is an important mechanism of carbapenem resistance in P. aeruginosa (Yoon and Jeong, 2021). Moreover, in the absence of Carbapenemase-mediated resistance, the loss of outer membrane porin OprD, together with the overexpression of efflux pumps or ampC have been shown to be the main mechanisms leading to carbapenem resistance (Botelho et al., 2019). In China, only a few reports are available on the molecular epidemiology and genome information of P. aeruginosa high-risk clones. At the same time, research on Carbapenem-Resistant P. aeruginosa (CRPA) together with high-risk clones is even more limited.
Guangdong is the most populous province with the highest domestic migration in China (Xu et al., 2022). This study aims to characterize clinical P. aeruginosa strains isolated from two hospitals in Guangdong, in terms of clone diversity, antibiotics resistance profiles, and virulence determinants. Furthermore, the relationship between the carbapenem genes and P. aeruginosa high-risk clones will be explored. The outcome of this study will fill the gap related to cultures of P. aeruginosa and their associated genome sequences in China. The genetic and phenotypic characteristics of P. aeruginosa high-risk clones would provide novel insights into the epidemic and genomic mechanism background of this pathogen in China.
The samples were collected from two-level III (tertiary care) hospitals in Guangdong Province, China, where the patients are heterogeneous. From 2018 to 2020, 212 nonduplicate P. aeruginosa isolates were obtained at The First Affiliated Hospital of Guangzhou Medical University and Zhujiang Hospital of Southern Medical University.
The Ethics Committee of the Zhujiang Hospital of Southern Medical University approved the research on May 30, 2021 (approval no.2021-KY-046-01).
Over 3 years (2018–2020), a total of 212 non-duplicate isolates were selected and distributed over different years, with 55 isolates in 2018, 104 isolates in 2019, and 53 isolates in 2020 (Supplementary Table S1). All isolates were stored in 10% glycerol-containing media and were frozen at −80°C until further use. These isolates originate from different clinical specimens processed by the hospital’s laboratory as part of the microbiological diagnostic routine.
Antibiotic susceptibility testing was performed with the Minimum Inhibitory Concentration (MIC) method according to the guidelines of the Clinical and Laboratory Standards Institute (Humphries et al., 2021). The antibiotics tested included piperacillin-tazobactam, ceftazidime, cefepime, imipenem, meropenem, aztreonam, ciprofloxacin, levofloxacin, amikacin, tobramycin and colistin. The minimum inhibitory concentrations (MICs μg/mL) were obtained for each isolate, and the isolates were classified as resistant (R), intermediate (I) and susceptible (S) according to the obtained MIC accordingly. Isolates were designated as multidrug-resistant (MDR) if they were resistant to an antibiotic from ≥3 classes and extensively drug-resistant (XDR) if they were resistant to an antibiotic from ≥6 classes tested following standardized criteria (Magiorakos et al., 2012). This study, Carbapenem-Resistance P. aeruginosa (CRPA) was defined as an isolate with imipenem and/or meropenem resistance.
Stored strains were regrown from the −80°C stocks using Nutrient agar. The isolates were grown for DNA extraction in Nutrient agar medium overnight at 37°C. DNA was extracted from pure cultures using the MagAttract HMW DNA Kit (Qiagen, Germany) according to the kit protocol. The stLFR technology uses Tn5 transposase for the co-barcoding of DNA libraries. The stLFR library was constructed following the standard protocol using the MGIEasy stLFR Library Prep kit v1.1 (PN: 1000005622) with some process improvement for better assembly of bacterial genomes according to published methods (Wang et al., 2019). The WGS was performed on MGISEQ-2000 platform (DNBSEQ™, MGI, China) according to the manufacturer’s instructions in paired-end mode (PE 100 bp).
The sequencing reads were evaluated by FastQC 0.11.3, and the reads were filtered by SOAPfilter (v2.2 version) software with the parameters set as follows: -q 33 -y -p -M 2 -f − 1 -Q 10. Athena (v1.3) was used for genome assembly (Bishara et al., 2018). The completeness and pollution of the assembly results were counted by CheckM (v1.0.13), and the SLR-superscaffolder (v0.9.0) was used for the scaffolding. After selection, an average of 5.5 Gb of sequences were kept, with an average of 6.7 million pairs of reads and mean coverage >100X according to the expected genome size (approx. 6.4 Mb). Detailed sequencing statistics and quality results are summarized (Supplementary Table S2). The quality criteria were met for all sequences.
The Comprehensive Antibiotic Resistance Database (CARD)1 was used as a reference for the antimicrobial resistance genes analysis, and Virulence Factor Database (VFDB)2 was used for the virulence factors screening (Alcock et al., 2020; Liu et al., 2022). The serotype was identified by PAst 1.0.3 The sequence types of all strains were determined from their whole genome sequence data using the MLST scheme4 sited at the University of Oxford (Jolley et al., 2018). The population snapshot of P. aeruginosa was inferred by using Phyloviz 2.0. based on the MLST allelic profiles. The minimum spanning tree (MST) analysis was implemented in BioNumerics 8.0 software (Applied-Maths, Sint Maartens-Latem, Belgium). To detect the mutations of the OprD gene, the gene sequence encoding the reference protein OprD in Pseudomonas aeruginosa strain PAO1 (accession number NC_002516) was obtained and aligned with each target strain by BLASTp script.
The MIC values together with the interpretations are shown (Table 1). 117 strains were MDR (55.19%), and 13 strains were XDR (6.13%) (Figure 1). Among these tested strains, a total of 196 strains belonging to CRPA (92.45%). The identified CRPA strains displayed reduced susceptibilities to most of the tested antimicrobials, but remained highly susceptible to colistin (98.11%) and amikacin (87.74%).
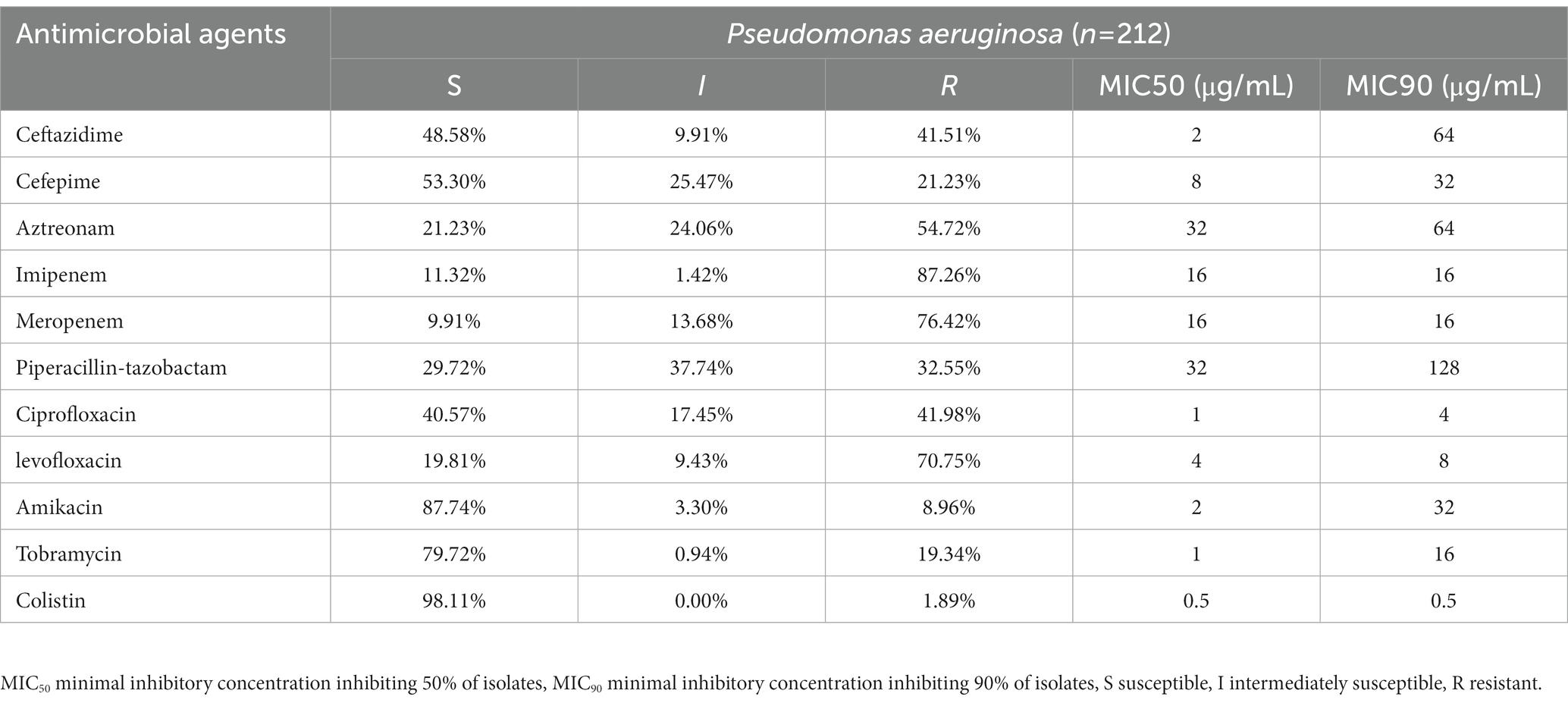
Table 1. Resistance rate (percentages) and minimum inhibitory concentrations (MICs) of P. aeruginosa isolates in Guangdong (n = 212).
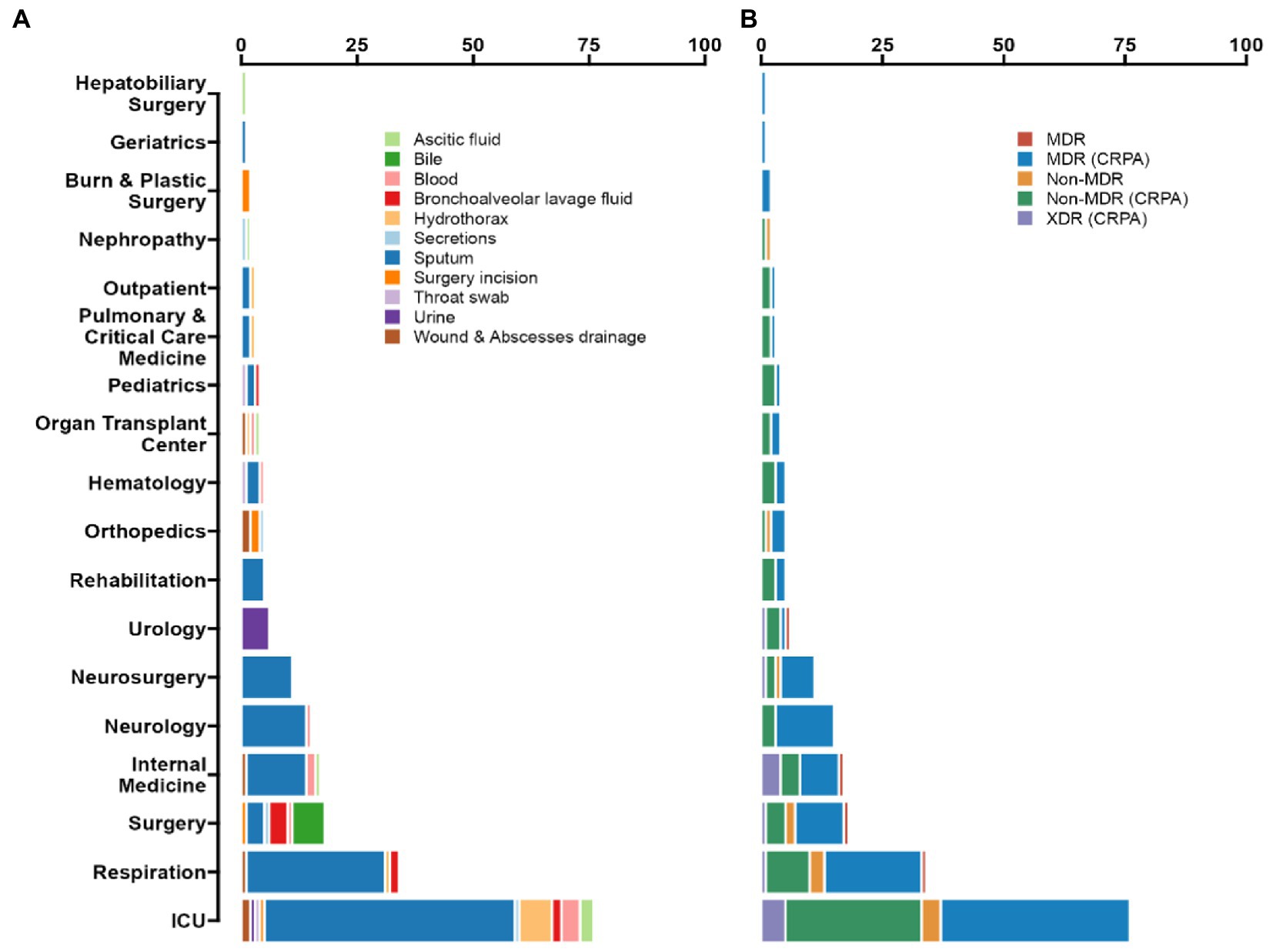
Figure 1. Prevalence of P. aeruginosa antimicrobial resistance in Guangdong province, China. (A) According to the ward distribution of the studied strains, the number of samples from ICU is 76 (35.85%), Respiratory Department samples numbered 34 (16.04%), and Neurology department samples numbered 15 (7.08%). Among all of the isolated strains, the most frequently isolated specimen was sputum (66.51%), then was hydrothorax (5.19%), blood (4.72%), and bronchoalveolar lavage fluid (BALF) (4.25%). (B) Up to 92.45% of the isolates were classified as CRPA, 55.19% as MDR (CRPA) and 6.13% as XDR (CRPA).
From 2018 to 2020, a total of 212 strains were isolated from patients in Guangdong, China. Among all the isolated strains, the most frequently clinical specimen was from sputum (n = 141, 66.51%), then hydrothorax (n = 11, 5.19%), blood (n = 10, 4.72%) and bronchoalveolar lavage fluid (BALF) (n = 9, 4.25%), as shown in Figure 1. According to the ward distribution of the studied strains, the number of samples from the intensive care unit (ICU) was 76 (35.85%), the Respiratory Department samples numbered 34 (16.04%), and the Neurology Department samples numbered 15 (7.08%) (Supplementary Table S1).
Among the 76 strains isolated from ICU, 55 multi-locus sequence types (STs) were identified. The most prevalent STs were ST244 (n = 6, 7.89%), ST274 (n = 4, 5.26%), and ST381 (n = 4, 5.26%). The 34 strains sampled from the ward at the Respiratory Department were classified into 31 STs; only ST253, ST611 and ST703 were with double strains separately. Besides the ICU samples, the intra-hospital transmission, for example, in The First Affiliated Hospital of Guangzhou Medical University, 4 ST569 MDR (CRPA) strains were isolated from the Department of Neurology, and 4 ST1971 strains were purified from the Department of Internal Medicine, which indicates that attention should also be paid to outside-ICUs-acquired-infections.
Among the isolates studied here, 122 previously reported sequence types and seven novel sequencing types were detected. Most STs are represented only by single isolates (n = 93, 43.87%). The high diversity of STs found among the isolates is also evident from the phylogenetic analysis of the strains, as shown in Supplementary Figure S1. There are no clear predominant sequence types among the collected strains, even for the identified multidrug-resistance strains. The most common STs isolated were ST244 (n = 9, 4.25%) and ST1971 (n = 7, 3.30%), followed by ST381 (n = 6, 2.83%) (Supplementary Table S1).
Six global high-risk clones (ST111, ST235, ST244, ST277, ST298 and ST357) (Oliver et al., 2015) were identified in this study (Figure 2). In addition, 12 widely disseminated clones (previously reported as potential high-risk clones) were also identified, including ST108 (Telling et al., 2018), ST179 (Moloney et al., 2020), ST253 (Fischer et al., 2020), ST260 (Martak et al., 2022), ST274 (Sewunet et al., 2022), ST348 (Dix et al., 2022), ST395 (Petitjean et al., 2017), ST446 (Pincus et al., 2019), ST463 (Hu et al., 2021), ST532 (Founou et al., 2020), ST699 (Rada et al., 2021) and ST773 (Singh et al., 2021). We observed that several MDR clones identified in this study were related to high-risk clones and were thus part of the same subclade as them (Table 2). For example, ST446 (n = 1) was part of the sub-clade with high-risk clone ST298. Additional examples include ST1338 (n = 4, ST244), ST3674 (n = 2, ST244), ST209 (n = 1, ST274), and ST2326 (n = 2, ST207) (Table 2).
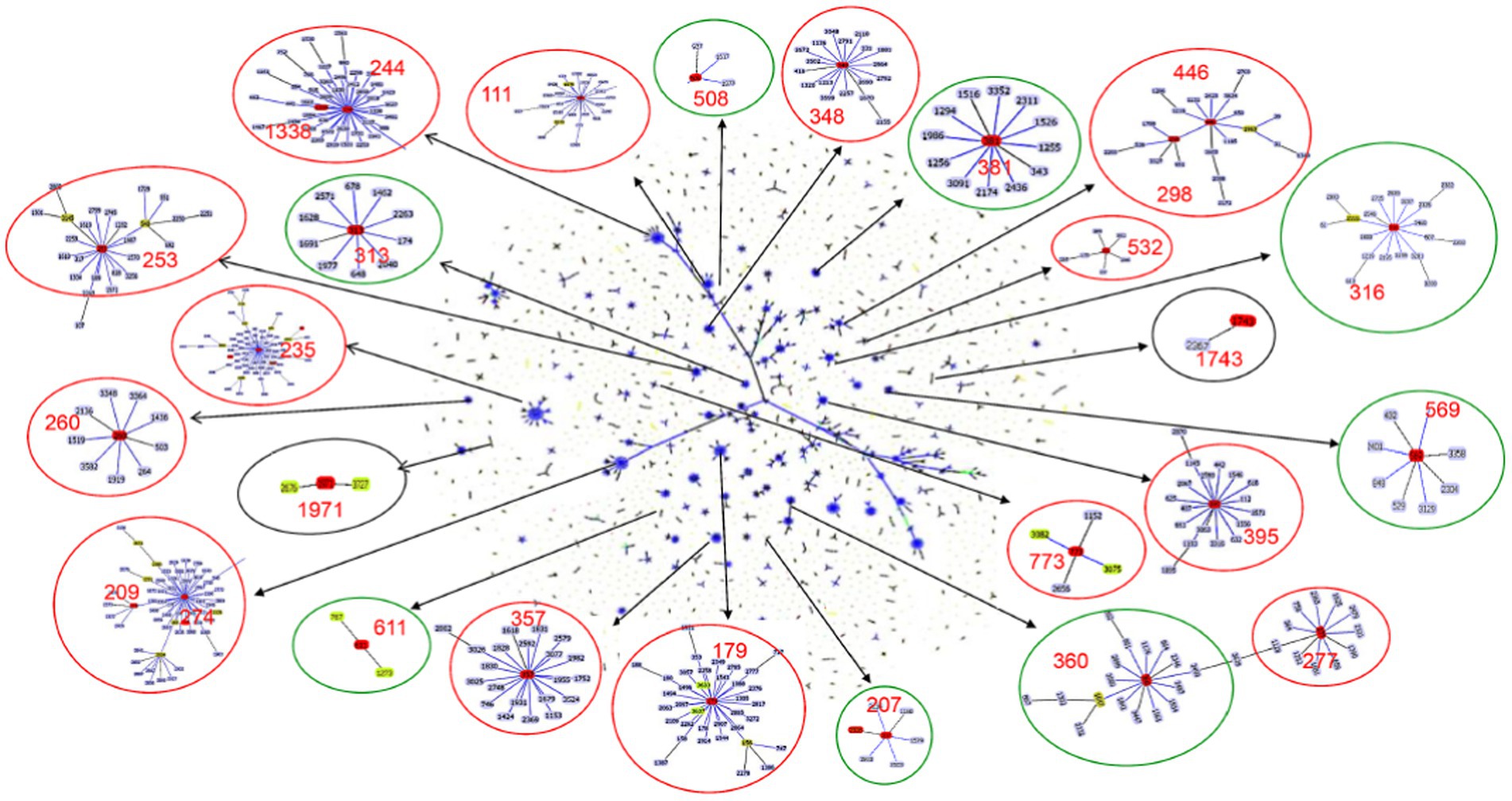
Figure 2. Population snapshot of P. aeruginosa. The 3,729 sequence types listed on the P. aeruginosa PubMLST database (http://pubmlst.org/paeruginosa, 2022/07/11) are displayed in a single eBURST diagram by setting the group definition to zero of seven shared alleles. Each dot represents a ST, and lines connect single-locus variants. The identified clones in this study were marked in red in each group of related STs. A red circle surrounded the groups reported as international high-risk clones, the groups with the new potential high-risk clones were surrounded by a green circle, and a black circle surrounded the local risk clones identified in this study.
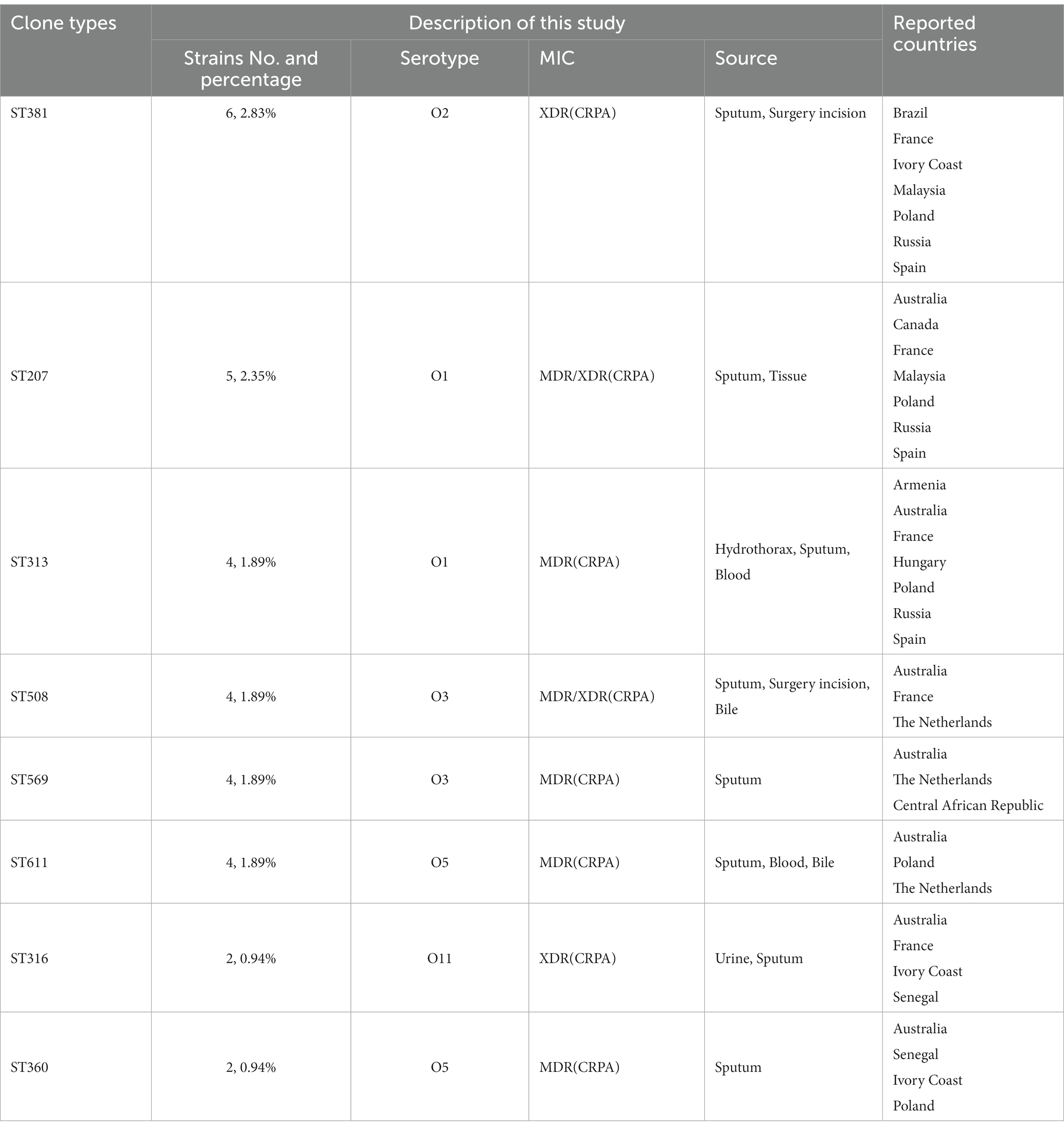
Table 2. The successful clones identified in this study with potential risks.
Besides the reported high-risk clones, we found eight groups/subgroups in the population where the founder STs were represented by multiple isolates with MDR/XDR phenotype (Green circles in Figure 2). These including ST381 (n = 6, 2.83%), ST207 (n = 5, 2.36%), ST313 (n = 4, 1.89%), ST508 (n = 4, 1.89%), ST569 (n = 4, 1.89%), and ST611 (n = 4, 1.89%). These successful emerging P. aeruginosa clones with MDR/XDR phenotype are summarized (Table 2).
In this study, 19 isolates harbored genes encoding aminoglycoside-modifying enzymes, including aminoglycoside acetyltransferase genes [aac(6')-IIa, aac(6')-Ib4, aac(6')-Ib7, aac(6')-Ib9, aac(6')-Ib10, and aac(3)-IId] and/or aminoglycoside adenylyltransferase genes (aadA, aadA2, aadA3, aadA7 and aadA13). The identified blaOXA type genes were composed of blaOXA-50-Like genes (n = 212, 100%), blaOXA-1-Like genes (n = 6, 2.83%), blaOXA-10-Like genes (n = 4, 1.89%) and blaOXA-21-Like genes (n = 1, 0.47%). In several cases, the strains were carrying multiple β-lactamases. Among them, nine strains (4.25%) simultaneously carried two different blaOXA type genes, and one ST1971 strain (0.47%) carried three different blaOXA type genes. Furthermore, one blaVIM-2 gene and two different blaOXA genes were found in each isolate of ST235 (n = 2) and ST244 (n = 1) strains. blaIMP-9/45, blaKPC-2, blaCTX-M-13 and blaCARB-1/3 co-existed with one/two blaOXA type genes were also discovered in different strains (Table 3).
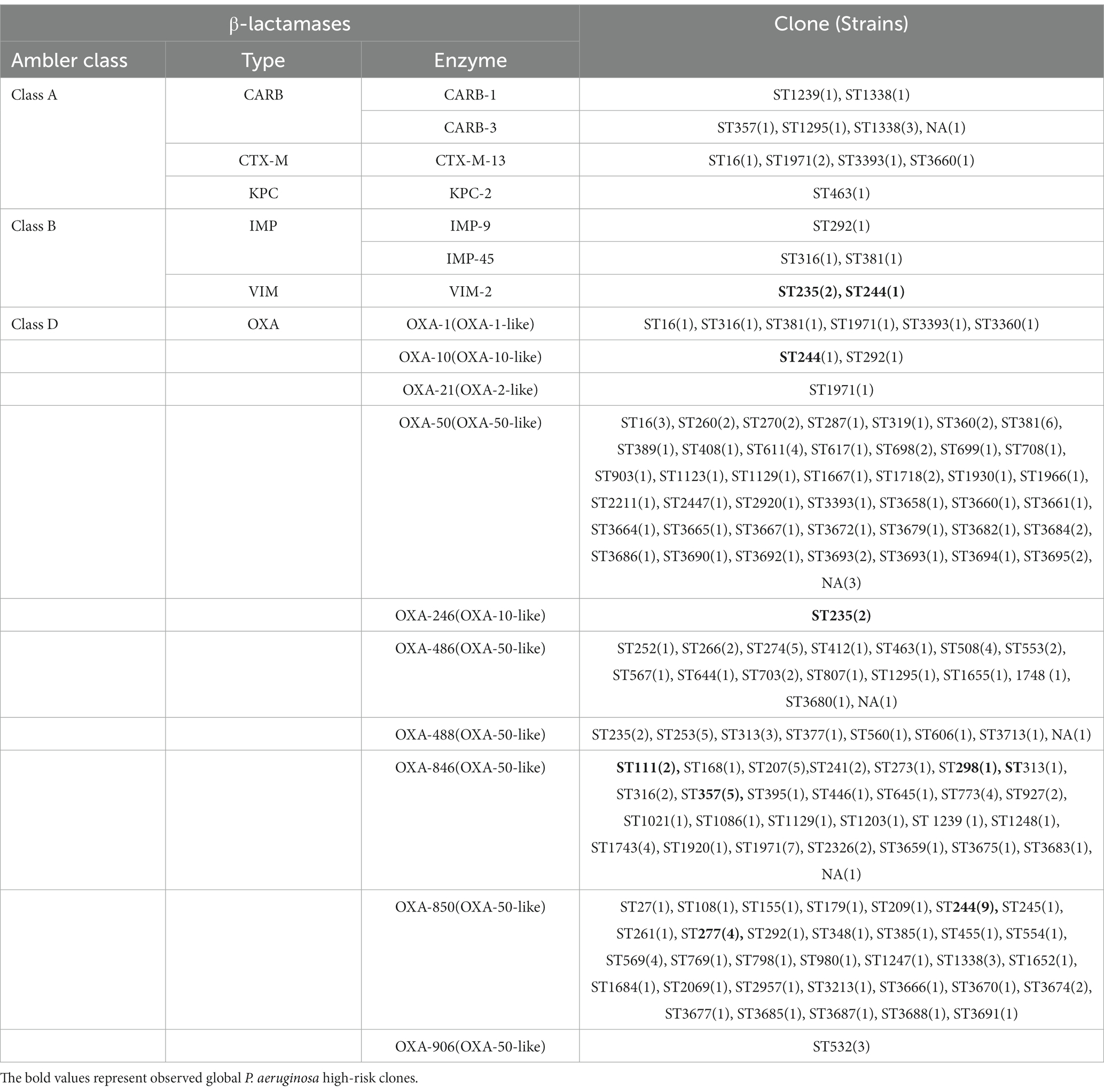
Table 3. Horizontally-acquired β-lactamases identified in collected isolates.
As with high prevalence rates of blaOXA type genes, 211 isolates harbored Pseudomonas-derived cephalosporinase (PDC) β-lactamase genes, which includes 24 blaPDC types. The dominant type was blaPDC-3 (n = 51), followed by blaPDC-5 (n = 42), which both belong to the extended-spectrum β-lactamase (ESBLs). We found blaPDC types and STs had special correspondence relationships. Among them, 7 ST244 strains were carrying blaPDC-1 and blaPDC-5, 5 ST357 strains were with blaPDC-11, and 4 ST277 strains were with blaPDC-5. Besides high-risk clones, 5 ST253 strains were with blaPDC-68, 4 ST313 strains were with blaPDC-105, 6 ST381 strains were with blaPDC-14, 5 ST207 strains were with blaPDC-337, and 7 ST1971 strains were with blaPDC-3, and so on (Supplementary Table S3).
We did not find Class A and Class B antibiotic resistance genes (ARGs) in high-risk clone ST111, ST277 and ST298, but the Klebsiella pneumoniae carbapenemase (KPC) gene blaKPC-2 was found in one ST463 strain. The Verona Integron-encoded Metallo-β-lactamase (VIM) gene was detected in one ST244 strain, and two ST235 strains carried two blaVIM-2 genes in each of the isolates (Table 3). Furthermore, two types of qnrVC variants were detected in 6 CRPA strains (2.83%) with five different STs, which included five strains carrying qnrVC1 and one strain carrying qnrVC6. The heat map showing unique resistance genes for identified P. aeruginosa high risk clone strains is illustrated (Figure 3). In order to further explore the reasons for the high proportion of CRPA in the collected strains, the OprD gene alignment was also performed. BLASTP analysis showed that the wild-type OprD gene was not detected in all collected strains (oprD mutation strains, n = 212).
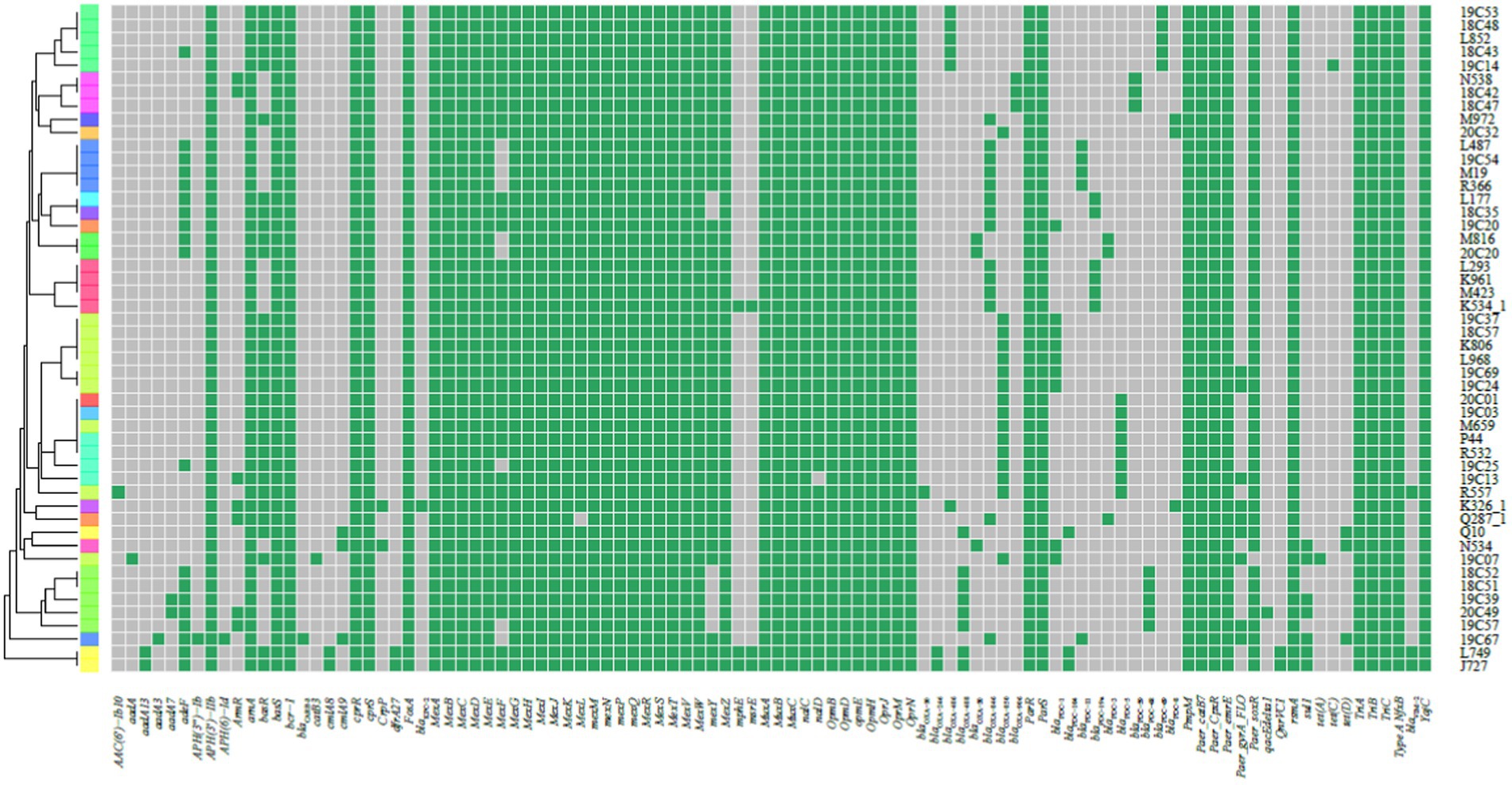
Figure 3. Heat map showing the resistance genes for the collected P. aeruginosa high-risk clone strains. The heat map was formed by performing tblastn searches using the sequences present on the Comprehensive Antibiotic Resistance Database (CARD) as queries and the genomes as subjects. Grey indicates the absence, and green indicates the presence of the resistance genes.
The exoU and exoS genes, which encoding effector proteins of type III secretion system (T3SS) in P. aeruginosa, were investigated. The more frequent distribution of exoS (n = 150, 70.75%) was observed compared with exoU (n = 57, 26.89%). More interestingly, we found the co-presence of both genes in 5 strains (ST463, n = 1; ST273, n = 1; ST3658, n = 1; ST274 n = 2).
Several class A and class B β-lactamases were identified in 22 strains (10.37%), and all these were classified as CRPA and with multidrug resistance. 8 blaCARB-3 genes carried strains were identified, and 4 of these strains belonged to ST1338 with XDR phenotype. 5 strains harboring the blaCTX-M-13 gene were identified and represented multidrug resistance. Isolates producing blaKPC type enzymes often relate to the resistance to most of β-lactams and non-β-lactams antibiotics. 1 ST463 strain was carrying blaKPC-2 gene and represented XDR (CRPA) phenotype. 1 blaIMP-9 and 2 blaIMP-45 carried isolates were distributed into three dissimilar STs, including ST292, ST316 and ST381, which were isolated from the same hospital but in different wards. The MIC profiling showed that these three strains were all representing XDR (CRPA) phenotype. Three blaVIM-2 harboring high-risk clone strains were identified which were distributed in 2 ST235 and 1 ST244 strains. The responding phenotypes were multidrug resistance. Contrary to the high proportion of CRPA, class A and B carbapenemase genes identified in collected isolates are less frequent, which means their reduced susceptibility to carbapenems resulted from various non-enzymatic mechanisms (Figure 3).
The emergence and increase of high-risk isolates, including their antibiotic resistance, has been recognized as a threat for human health (Oliver et al., 2015; del Barrio-Tofiño et al., 2020). However, our understanding of these clones’ global distribution and evolution is still incomplete due to uneven sampling in different regions of the world. Here, we investigated clinical P. aeruginosa clones isolated in Guangdong, China. The MLST analysis showed high genetic diversity among collected isolates in agreement with prior studies in China (Fan et al., 2016). Importantly, some international high-risk clones as well as new widely distributed clones (regarded as potential risk clones), were identified in our study. The observations of potential high-risk and locally distributed clones underscore the relevance of continued inspection of the population structure in clinical settings in different parts of the world.
Although previous studies had showed that high-risk clones were predominant among multidrug-resistance strains (del Barrio-Tofiño et al., 2020; Hu et al., 2021; Teo et al., 2021; Aguilar-Rodea et al., 2022), we found that 50/212 isolates (23.58%) belong to the 18 previously reported high-risk clones, and that the remaining isolates showed high diversity together with other reported STs in all strains.
Among the high-risk clones found in our study, we observed that ST111 included two MDR/non-MDR strains from different hospitals. ST235 is the most widely spread P. aeruginosa high-risk clone associated MDR/XDR isolates and carries various resistance genes (Treepong et al., 2018; Tahmasebi et al., 2022). Three ST235 MDR strains were isolated from the neurology ward, of which two were carrying the blaVIM-2 gene. The most prevalent high-risk clone in all isolated strains was ST244 (n = 9); six multidrug resistance strains were collected from the ICU, of which one harbored the blaVIM-2 gene. ST277 was reported to be highly prevalent in Brazil, and commonly associated with MBL SPM-1 (Galetti et al., 2019; Silveira et al., 2020). We found ST277 isolates composed of 4 MDR/XDR strains, but the specific MBLs genes were not identified. ST357 with blaIMP and blaVIM type MBLs was previously reported in Turkey (Çekin et al., 2021) and Czech (Papagiannitsis et al., 2017), three ST357 stains identified in our study showed MDR/XDR phenotype, and one of them harbored blaCARB-3 gene. ST298 is the subclade of the CC446 clonal complex (Pincus et al., 2019). One ST298 isolate and one ST446 isolate, both with MDR/XDR phenotype, were identified in different hospitals. In some regional research projects, the clonal diversity of P. aeruginosa is much lower among multidrug-resistant isolates (del Barrio-Tofiño et al., 2020; Hu et al., 2021; Teo et al., 2021; Aguilar-Rodea et al., 2022). This is not entirely the case among our MDR subsets from 2018–2020. We have observed 84 (39.62%) different STs in the 130 MDR/XDR strains. However, only 12.26% (n = 26) of MDR/XDR isolates are actually related to the reported international high-risk clones.
We note that our study is focused on the Guangdong Province. This region is the most populous province with the largest population inflow in China. We speculate that trade and mobility in this region have contributed to the high level of diversity observed among the isolated strains. Overall, our survey of regional P. aeruginosa high-risk clones in China may assist in a better understanding of the evolution of these important clone types.
Besides the high-risk clones, eight founder STs of clonal groups/subgroups have been identified (Figure 2), and these founder STs have been reported to be distributed in at least three different countries (Table 2). Furthermore, one special disseminated and multidrug-resistant clone, ST1971 (n = 7, 3.30%), has only been identified in China (Wang et al., 2017). This clone can be defined as a regional epidemic risk type for nosocomial healthcare. Some of the STs identified in this study have been previously described as new potential high-risk clones, including ST108 (Telling et al., 2018), ST179 (Moloney et al., 2020), ST253 (Fischer et al., 2020), ST260 (Martak et al., 2022), ST274 (Sewunet et al., 2022), ST348 (Dix et al., 2022), ST395 (Petitjean et al., 2017), ST446 (Pincus et al., 2019), ST463 (Hu et al., 2021), ST532 (Founou et al., 2020), ST699 (Rada et al., 2021) and ST773 (Singh et al., 2021).
The antibiotic profiles of all isolated P. aeruginosa strains were quantified by MIC assays. 196 strains (92.45%) were found to be CRPA, and most of them represented resistance to multiple antibiotics (126/196, 64.29%). It was previously reported that meropenem and imipenem resistance rates for P. aeruginosa range from 21.6 to 25.6% in China (Wang and Wang, 2020), and the detection rate of CRPA in Guangdong was 41.9, 17.9 and 19.2% from 2018 to 2020, respectively. The CRPA rates determined in this study were higher than the previous studies. Previous studies reported that the CRPA isolating rate of inpatients and those admitted to an ICU is higher than outpatients and those in non-ICU wards (Gill, Nicolau, and on behalf of the ERACE-PA Global Study Group, 2022). Here, 35.85% of strains were isolated from ICU patients, which might explain the high prevalence of CRPA in the collection. In future studies, collecting strains from more diverse wards and improving the randomness of the evaluation would be beneficial.
In contrast to the high rate of carbapenem resistance, the MBLs genes carrying strains were with very low proportion (n = 6, 2.83%) in all collected isolates. Only 3 blaIMP harbored strains and 3 blaVIM-2 harbored strains were found. The oprD gene has been reported as the main determinant of imipenem resistance (Quinn et al., 2022). The MIC data show that the imipenem resistance rate is 87.26%. The wild-type oprD gene was not found in any newly sequenced strains, which implied that the mutational inactivation of the oprD gene may play a role in carbapenem resistance. However, the relationship between the mutations in the oprD gene and its function cannot be fully understood only by whole genome sequencing, and future investigations should focus on mechanistic studies of these mutations.
Some variants of PDC enzymes (class C β-lactamases) are responsible for the resistance to imipenem and newer β-lactam/β-lactamase inhibitor combinations such as ceftolozane-tazobactam (Papp-Wallace et al., 2020). Furthermore, the production of class D β-lactamases (oxacillinase; OXA) can lead to third-generation cephalosporins or multiple-drug resistance (Vrancianu et al., 2020). In this study, PDC β-lactamase and OXA β-lactamase genes were prevalent in collected strains, which can induce multidrug resistance to some extent by antibiotic inactivation. Previous studies showed that the presence of qnrVC genes plays important roles in quinolone resistance (Liu et al., 2018; Saki et al., 2022), and 2.3% of collected P. aeruginosa isolates from Guangdong carried qnrVC genes (Lin et al., 2020), which is consistent with this study.
The type III secretion system is an important virulence factor of P. aeruginosa, contributing to microbial cytotoxicity and invasion (Horna and Ruiz, 2021). The exoU and exoS genes encode type III secretion virulence effector proteins. Previous studies have shown the mutual exclusion of the exoU and exoS genes (Bradbury et al., 2010; Juan et al., 2017). Through the VFDB analysis, we found the co-presence of both genes in 5 clone types strains (ST273, ST274, ST463, and ST3658). The exoS+/exoU+ strains have no significant differences in the resistance phenotype, but the strains carrying two virulence genes were all classified into XDR (CRPA).
Pseudomonas aeruginosa isolates from hospitals in this study exhibited multidrug resistance to various drugs and a high rate of resistance against carbapenem. MLST analysis further revealed the genetic diversity of these clinical isolates of P. aeruginosa. The stLFR technology was first applied for the bacteria antibiotic gene profiling, and the findings indicate that the sequence clone types are independent of carbapenem resistance or MDR/XDR phenotype. The strains harboring blaOXA and blaPDC genes mainly circulate in the hospital environments studied. The spectrum of high-risk clones is also different from previous reports and has obvious regional characteristics. This means that the control measures for the circulation and success of high-risk clones and CRPA must have a unified global standard and custom local strategies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number (s) can be found in the article/Supplementary material.
The study was approved by the Ethics Committee (full name: The Ethics Committee of the Zhujiang Hospital of Southern Medical University), the reference number 2021-KY-046-01. The samples were obtained with written informed consent and reviewed by the ethical board in accordance with the tenets of the Declaration of Helsinki.
YZ designed the experimental plan, analyzed WGS data, and finished the draft manuscript writing. DC collected strains and participated in project design. BJ prepared MLST distribution and digital chart. XZ contributed to manuscript revision. MA participated in project design. LJ contributed to conceptualization, project administration, and manuscript revision. All authors participated in the manuscript critical review and editing, and agreed with the above contribution details.
This work was supported by a research grant to LJ from the Independent Research Fund Denmark (9039-00350A).
The authors would like to thank all reviewers for their insightful comments and constructive suggestions to improve the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1117017/full#supplementary-material
3. ^https://cge.cbs.dtu.dk/services/PAst/
4. ^https://pubmlst.org/paeruginosa/; last accessed July 11, 2022.
Aguilar-Rodea, P., Estrada-Javier, E. L., Jiménez-Rojas, V., Gomez-Ramirez, U., Nolasco-Romero, C. G., Rodea, G. E., et al. (2022). New variants of Pseudomonas aeruginosa high-risk clone ST233 associated with an outbreak in a Mexican Paediatric Hospital. Microorganisms 10:1533. doi: 10.3390/microorganisms10081533
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Bishara, A., Moss, E. L., Kolmogorov, M., Parada, A. E., Weng, Z., Sidow, A., et al. (2018). High-quality genome sequences of uncultured microbes by assembly of read clouds. Nat. Biotechnol. 36, 1067–1075. doi: 10.1038/nbt.4266
Botelho, J., Grosso, F., and Peixe, L. (2019). Antibiotic resistance in Pseudomonas aeruginosa – Mechanisms, epidemiology and evolution. Drug Resist. Updat. 44:100640. doi: 10.1016/j.drup.2019.07.002
Bradbury, R. S., Roddam, L. F., Merritt, A., Reid, D. W., and Champion, A. C. (2010). Virulence gene distribution in clinical, nosocomial and environmental isolates of Pseudomonas aeruginosa. J. Med. Microbiol. 59, 881–890. doi: 10.1099/jmm.0.018283-0
Çekin, Z. K., Dabos, L., Malkoçoğlu, G., Fortineau, N., Bayraktar, B., Iorga, B. I., et al. (2021). Carbapenemase-producing Pseudomonas aeruginosa isolates from Turkey: first report of P. aeruginosa high-risk clones with VIM-5-and IMP-7-type carbapenemases in a tertiary hospital. Diagn. Microbiol. Infect. Dis. 99:115174. doi: 10.1016/j.diagmicrobio.2020.115174
del Barrio-Tofiño, E., López-Causapé, C., and Oliver, A. (2020). Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int. J. Antimicrob. Agents 56:106196. doi: 10.1016/j.ijantimicag.2020.106196
Dix, L. M. L., de Goeij, I., Manintveld, O. C., Severin, J. A., and Verkaik, N. J. (2022). Pseudomonas aeruginosa left ventricular assist device (LVAD) driveline infection acquired from the bathroom at home. Am. J. Infect. Control 50, 1392–1394. doi: 10.1016/j.ajic.2022.04.011
Fan, X., Wu, Y., Xiao, M., Xu, Z. P., Kudinha, T., Bazaj, A., et al. (2016). Diverse genetic background of multidrug-resistant Pseudomonas aeruginosa from mainland China and emergence of an extensively drug-resistant ST292 clone in Kunming. Sci. Rep. 6:26522. doi: 10.1038/srep26522
Fischer, S., Dethlefsen, S., Klockgether, J., and Tümmler, B. (2020). Phenotypic and genomic comparison of the two Most common ExoU-positive Pseudomonas aeruginosa clones, PA14 and ST235. mSystems 5, e01007–e01020. doi: 10.1128/mSystems.01007-20
Founou, R. C., Founou, L. L., Allam, M., Ismail, A., and Essack, S. Y. (2020). First report of a clinical multidrug-resistant Pseudomonas aeruginosa ST532 isolate harbouring a ciprofloxacin-modifying enzyme (CrpP) in South Africa. J. Glob. Antimicrob. Resist. 22, 145–146. doi: 10.1016/j.jgar.2020.05.012
Galetti, R., Andrade, L. N., Varani, A. M., and Darini, A. L. C. (2019). SPM-1-producing Pseudomonas aeruginosa ST277 carries a chromosomal pack of acquired resistance genes: an example of high-risk clone associated with “intrinsic resistome”. J. Glob. Antimicrob. Resist. 16, 183–186. doi: 10.1016/j.jgar.2018.12.009
Gill, C. M., and Nicolau, D. P., on behalf of the ERACE-PA Global Study Group (2022). Carbapenem-resistant Pseudomonas aeruginosa: an assessment of frequency of isolation from ICU versus non-ICU, phenotypic and genotypic profiles in a multinational population of hospitalized patients. Antimicrob. Resist Infect Control 11:146. doi: 10.1186/s13756-022-01187-8
Gravningen, K., Kacelnik, O., Lingaas, E., Pedersen, T., Iversen, B. G., Pseudomonas outbreak group, et al. (2022). Pseudomonas aeruginosa countrywide outbreak in hospitals linked to pre-moistened non-sterile washcloths, Norway, October 2021 to April 2022. Eur. Secur. 27:2200312. doi: 10.2807/1560-7917.ES.2022.27.18.2200312
Horna, G., and Ruiz, J. (2021). Type 3 secretion system of Pseudomonas aeruginosa. Microbiol. Res. 246:126719. doi: 10.1016/j.micres.2021.126719
Hu, Y., Peng, W., Wu, Y., Li, H., Wang, Q., Yi, H., et al. (2021). A potential high-risk clone of Pseudomonas aeruginosa ST463. Front. Microbiol. 12:670202. doi: 10.3389/fmicb.2021.670202
Humphries, R., Bobenchik, A. M., Hindler, J. A., and Schuetz, A. N. (2021). Overview of changes to the clinical and laboratory standards institute performance standards for antimicrobial susceptibility testing, M100, 31st edition. J. Clin. Microbiol. 59, e00213–e00221. doi: 10.1128/JCM.00213-21
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Juan, C., Peña, C., and Oliver, A. (2017). Host and pathogen biomarkers for severe Pseudomonas aeruginosa infections. J. Infect. Dis. 215, S44–S51. doi: 10.1093/infdis/jiw299
Kawalek, A., Kotecka, K., Modrzejewska, M., Gawor, J., Jagura-Burdzy, G., and Bartosik, A. A. (2020). Genome sequence of Pseudomonas aeruginosa PAO1161, a PAO1 derivative with the ICEPae1161 integrative and conjugative element. BMC Genomics 21:14. doi: 10.1186/s12864-019-6378-6
Lin, J., Chen, D. Q., Hong, J., Huang, H., and Xu, X. (2020). Prevalence of qnrVC genes in Pseudomonas aeruginosa clinical isolates from Guangdong, China. Curr. Microbiol. 77, 1532–1539. doi: 10.1007/s00284-020-01974-9
Liu, J., Yang, L., Chen, D., Peters, B. M., Li, L., Li, B., et al. (2018). Complete sequence of pBM413, a novel multidrug resistance megaplasmid carrying qnrVC6 and blaIMP-45 from Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 51, 145–150. doi: 10.1016/j.ijantimicag.2017.09.008
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Magiorakos, A.-P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Martak, D., Gbaguidi-Haore, H., Meunier, A., Valot, B., Conzelmann, N., Eib, M., et al. (2022). High prevalence of Pseudomonas aeruginosa carriage in residents of French and German long-term care facilities. Clin. Microbiol. Infect. 28, 1353–1358. doi: 10.1016/j.cmi.2022.05.004
Moloney, E. M., Deasy, E. C., Swan, J. S., Brennan, G. I., O'Donnell, M. J., and Coleman, D. C. (2020). Whole-genome sequencing identifies highly related Pseudomonas aeruginosa strains in multiple washbasin U-bends at several locations in one hospital: evidence for trafficking of potential pathogens via wastewater pipes. J. Hosp. Infect. 104, 484–491. doi: 10.1016/j.jhin.2019.11.005
Oliver, A., Mulet, X., López-Causapé, C., and Juan, C. (2015). The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 21–22, 41–59. doi: 10.1016/j.drup.2015.08.002
Pang, Z., Raudonis, R., Glick, B. R., Lin, T. J., and Cheng, Z. (2019). Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 37, 177–192. doi: 10.1016/j.biotechadv.2018.11.013
Papagiannitsis, C. C., Medvecky, M., Chudejova, K., Skalova, A., Rotova, V., Spanelova, P., et al. (2017). Molecular characterization of carbapenemase-producing Pseudomonas aeruginosa of Czech origin and evidence for clonal spread of extensively resistant sequence type 357 expressing IMP-7 metallo-β-lactamase. Antimicrob. Agents Chemother. 61, e01811–e01817. doi: 10.1128/AAC.01811-17
Papp-Wallace, K. M., Mack, A. R., Taracila, M. A., and Bonomo, R. A. (2020). Resistance to novel β-lactam–β-lactamase inhibitor combinations. Infect. Dis. Clin. N. Am. 34, 773–819. doi: 10.1016/j.idc.2020.05.001
Petitjean, M., Martak, D., Silvant, A., Bertrand, X., Valot, B., and Hocquet, D. (2017). Genomic characterization of a local epidemic Pseudomonas aeruginosa reveals specific features of the widespread clone ST395. Microb. Genom. 3:e000129. doi: 10.1099/mgen.0.000129
Pincus, N. B., Bachta, K. E. R., Ozer, E. A., Allen, J. P., Pura, O. N., Qi, C., et al. (2019). Long-term persistence of an extensively drug-resistant subclade of globally distributed Pseudomonas aeruginosa clonal complex 446 in an Academic Medical Center. Clin. Infect. Dis. 71, 1524–1531. doi: 10.1093/cid/ciz973
Quinn, A. M., Bottery, M. J., Thompson, H., and Friman, V. P. (2022). Resistance evolution can disrupt antibiotic exposure protection through competitive exclusion of the protective species. ISME J. 16, 2433–2447. doi: 10.1038/s41396-022-01285-w
Rada, A. M., de la Cadena, E., Agudelo, C. A., Pallares, C., Restrepo, E., Correa, A., et al. (2021). Genetic diversity of multidrug-resistant Pseudomonas aeruginosa isolates carrying blaVIM–2 and blaKPC–2 genes that spread on different genetic environment in Colombia. Front. Microbiol. 12:663020. doi: 10.3389/fmicb.2021.663020
Saki, M., Farajzadeh Sheikh, A., Seyed-Mohammadi, S., Asareh Zadegan Dezfuli, A., Shahin, M., Tabasi, M., et al. (2022). Occurrence of plasmid-mediated quinolone resistance genes in Pseudomonas aeruginosa strains isolated from clinical specimens in Southwest Iran: a multicentral study. Sci. Rep. 12:2296. doi: 10.1038/s41598-022-06128-4
Sewunet, T., Asrat, D., Woldeamanuel, Y., Aseffa, A., and Giske, C. G. (2022). Molecular epidemiology and antimicrobial susceptibility of Pseudomonas spp. and Acinetobacter spp. from clinical samples at Jimma medical center, Ethiopia. Front. Microbiol. 13:951857. doi: 10.3389/fmicb.2022.951857
Silveira, M. C., Rocha-de-Souza, C. M., Albano, R. M., de Oliveira Santos, I. C., and Carvalho-Assef, A. P. D.’. A. (2020). Exploring the success of Brazilian endemic clone Pseudomonas aeruginosa ST277 and its association with the CRISPR-Cas system type I-C. BMC Genomics 21:255. doi: 10.1186/s12864-020-6650-9
Singh, S., Pulusu, C. P., Pathak, A., Pradeep, B. E., and Prasad, K. N. (2021). Complete genome sequence of an extensively drug-resistant Pseudomonas aeruginosa ST773 clinical isolate from North India. J. Glob. Antimicrob. Res. 27, 244–246. doi: 10.1016/j.jgar.2021.10.010
Tahmasebi, H., Dehbashi, S., Nasaj, M., and Arabestani, M. R. (2022). Molecular epidemiology and collaboration of siderophore-based iron acquisition with surface adhesion in hypervirulent Pseudomonas aeruginosa isolates from wound infections. Sci. Rep. 12:7791. doi: 10.1038/s41598-022-11984-1
Talebi Bezmin Abadi, A., Rizvanov, A. A., Haertlé, T., and Blatt, N. L. (2019). World Health Organization report: current crisis of antibiotic resistance. Bio Nano Sci. 9, 778–788. doi: 10.1007/s12668-019-00658-4
Telling, K., Laht, M., Brauer, A., Remm, M., Kisand, V., Maimets, M., et al. (2018). Multidrug resistant Pseudomonas aeruginosa in Estonian hospitals. BMC Infect. Dis. 18:513. doi: 10.1186/s12879-018-3421-1
Teo, J. Q.-M., Tang, C. Y., Lim, J. C., Lee, S. J. Y., Tan, S. H., Koh, T. H., et al. (2021). Genomic characterization of carbapenem-non-susceptible Pseudomonas aeruginosa in Singapore. Emerg. Microbes. Infect. 10, 1706–1716. doi: 10.1080/22221751.2021.1968318
Thornton, C. S., and Parkins, M. D. (2023). Microbial epidemiology of the cystic fibrosis airways: past, present, and future. Semin. Respir. Crit. Care Med. 44, 269–286. doi: 10.1055/s-0042-1758732
Treepong, P., Kos, V. N., Guyeux, C., Blanc, D. S., Bertrand, X., Valot, B., et al. (2018). Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin. Microbiol. Infect. 24, 258–266. doi: 10.1016/j.cmi.2017.06.018
Vrancianu, C. O., Gheorghe, I., Czobor, I. B., and Chifiriuc, M. C. (2020). Antibiotic resistance profiles, molecular mechanisms and innovative treatment strategies of Acinetobacter baumannii. Microorganisms 8:935. doi: 10.3390/microorganisms8060935
Wang, K., Chen, Y. Q., Salido, M. M., Kohli, G. S., Kong, J. L., Liang, H. J., et al. (2017). The rapid in vivo evolution of Pseudomonas aeruginosa in ventilator-associated pneumonia patients leads to attenuated virulence. Open Biol. 7:170029. doi: 10.1098/rsob.170029
Wang, O., Chin, R., Cheng, X., Wu, M. K. Y., Mao, Q., Tang, J., et al. (2019). Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res. 29, 798–808. doi: 10.1101/gr.245126.118
Wang, W., and Wang, X. (2020). Prevalence of metallo-β-lactamase genes among Pseudomonas aeruginosa isolated from various clinical samples in China. J. Lab. Med. 44, 197–203. doi: 10.1515/labmed-2019-0162
Xu, J., Deng, Y., Yang, J., Huang, W., Yan, Y., Xie, Y., et al. (2022). Effect of population migration and socioeconomic factors on the COVID-19 epidemic at county level in Guangdong, China. Front. Environ. Sci. 10:841996. doi: 10.3389/fenvs.2022.841996
Keywords: Pseudomonas aeruginosa , high-risk clone, multidrug resistance, genomic epidemiology, carbapenem resistant
Citation: Zhao Y, Chen D, Ji B, Zhang X, Anbo M and Jelsbak L (2023) Whole-genome sequencing reveals high-risk clones of Pseudomonas aeruginosa in Guangdong, China. Front. Microbiol. 14:1117017. doi: 10.3389/fmicb.2023.1117017
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Carlos Henrique Camargo, Adolfo Lutz Institute, BrazilCopyright © 2023 Zhao, Chen, Ji, Zhang, Anbo and Jelsbak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Jelsbak, bGpAYmlvLmR0dS5kaw==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.