 Thanyaporn Srimahaeak1,2‡
Thanyaporn Srimahaeak1,2‡ Narumon Thongdee1†‡
Narumon Thongdee1†‡ Jurairat Chittrakanwong1Sopapan Atichartpongkul3Juthamas Jaroensuk1†Kamonwan Phatinuwat1
Jurairat Chittrakanwong1Sopapan Atichartpongkul3Juthamas Jaroensuk1†Kamonwan Phatinuwat1 Narumon Phaonakrop4Janthima Jaresitthikunchai4
Narumon Phaonakrop4Janthima Jaresitthikunchai4 Sittiruk Roytrakul4
Sittiruk Roytrakul4 Skorn Mongkolsuk1,3,5
Skorn Mongkolsuk1,3,5 Mayuree Fuangthong1,3,5*
Mayuree Fuangthong1,3,5*- 1Program in Applied Biological Sciences, Chulabhorn Graduate Institute, Bangkok, Thailand
- 2Department of Biotechnology, Faculty of Engineering and Industrial Technology, Silpakorn University, Sanamchandra Palace Campus, Nakhon Pathom, Thailand
- 3Laboratory of Biotechnology, Chulabhorn Research Institute, Bangkok, Thailand
- 4Functional Ingredients and Food Innovation Research Group, National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency, Pathum Thani, Thailand
- 5Center of Excellence on Environmental Health and Toxicology (EHT), OPS, MHESI, Bangkok, Thailand
Pseudomonas aeruginosa gidA, which encodes a putative tRNA-modifying enzyme, is associated with a variety of virulence phenotypes. Here, we demonstrated that P. aeruginosa gidA is responsible for the modifications of uridine in tRNAs in vivo. Loss of gidA was found to have no impact on the mRNA levels of katA and katB, but it decreased KatA and KatB protein levels, resulting in decreased total catalase activity and a hydrogen peroxide-sensitive phenotype. Furthermore, gidA was found to affect flagella-mediated motility and biofilm formation; and it was required for the full virulence of P. aeruginosa in both Caenorhabditis elegans and macrophage models. Together, these observations reveal the posttranscriptional impact of gidA on the oxidative stress response, highlight the complexity of catalase gene expression regulation, and further support the involvement of gidA in the virulence of P. aeruginosa.
1. Introduction
Pseudomonas aeruginosa is a Gram-negative bacterium and one of the major causes of nosocomial infection in immunocompromised and weak patients (Nathwani et al., 2014; Moradali et al., 2017; Ciofu and Tolker-Nielsen, 2019). During P. aeruginosa infection, the first line of host defense involves the production of reactive oxygen and nitrogen species to kill the invading pathogen (Lavoie et al., 2011). Therefore, patients with deficient immune responses may not be able to cope with the infection leading to high mortality and morbidity rates (Lavoie et al., 2011). In 2017, the World Health Organization listed P. aeruginosa as a high priority organism for which new antibiotics are needed, as it poses a serious threat to human health (World Health Organization, 2017). P. aeruginosa infections are considered to have acute and chronic phases. A number of virulence factors are involved in the acute phase, including motility activity. Swimming motility is required for the initial step of the infection, wherein bacteria adhere to the host surface and expand their degree of infections. Meanwhile, in the beginning of a chronic infection, most activities related to virulence factors are reduced. Biofilm, one such virulence factor, plays a key role in developing multidrug resistance during this phase and thus leads to high mortality rates, especially among weak patients (Shi et al., 2019).
tRNA modifications are crucial for gene expression, particularly for the accuracy and efficacy of protein synthesis (de Crecy-Lagard and Jaroch, 2021). In addition to their global impact on tRNAs functions, many studies have demonstrated the regulatory role of tRNA modifications in controlling the translations of certain codon-biased mRNAs under specific conditions (Chan et al., 2018). In Pseudomonas, several tRNA-modifying genes have been shown to be associated with virulence gene expression or oxidative stress response (Kinscherf and Willis, 2002; Ahn et al., 2004; Gupta et al., 2009; Jaroensuk et al., 2016; Thongdee et al., 2019). GidA (also known as MnmE) and TrmE (also known as MnmG) together catalyze the incorporation of carboxymethylaminomethyl (cmnm) or aminomethyl (nm) into uridine (U) or 2-thio uridine (s2U) at the position 34 in the anticodons of tRNAs (Yim et al., 2006). Modifications formed by the GidA/TrmE complex are important for translation fidelity (Brégeon et al., 2001). The deletion of gidA affects a variety of phenotypes in several bacteria (Shippy and Fadl, 2014). In Pseudomonas syringe, gidA contributes to swarming motility and the production of lipodepsipeptide antibiotic and pyoverdine (Kinscherf and Willis, 2002). gidA also impacts the levels of LasA protease, rhamnolipid, and pyocyanin through the posttranscriptional control of the transcriptional regulator RhlR in the quorum sensing system of P. aeruginosa (Gupta et al., 2009). Furthermore, studies in other pathogenic bacteria have shown that a lack of gidA or trmE decreases the infection ability of Salmonella enterica serovar Typhimurium (Shippy et al., 2011), Aeromonas hydrophila (Sha et al., 2004), Streptococcus pyogenes (Cho and Caparon, 2008), and Streptococcus suis (Gao et al., 2020), suggesting GidA is a potential candidate for future vaccine development and a target for antibiotic development.
Although, the previous studies on gidA in P. syringe and P. aeruginosa have indicated the pleiotropic phenotype of gidA deletion, whether gidA influences Pseudomonas infection and how gidA controls gene expression remain unknown. Here, gidA was characterized in vivo as being required for the biosynthesis of cmcm5U and mnm5U tRNA modifications, revealing a novel role of gidA in the cellular response to hydrogen peroxide (H2O2) in P. aeruginosa. The impact of gidA deletion on biofilm formation that has not yet been linked with Pseudomonas gidA was examined in this study. In addition, a lack of GidA was found to attenuate P. aeruginosa virulence both in vitro and in vivo.
2. Materials and methods
2.1. Bacterial strains, plasmids, and cell lines
Pseudomonas aeruginosa strain UCBPP-PA14 (PA14) was used as the wild-type strain. All Escherichia coli and Pseudomonas strains were grown at 37°C in lysogeny broth (LB) or lysogeny agar (Lennox) unless otherwise indicated in the method. The following concentrations of antibiotic(s) were added: carbenicillin (Cb) at 200 μg/ml and gentamicin (Gm) at 75 μg/ml for P. aeruginosa; gentamicin (Gm) at 15 μg/ml and ampicillin (Ap) at 100 μg/ml for E. coli. All bacterial strains and plasmids used in this study are listed in Table 1.
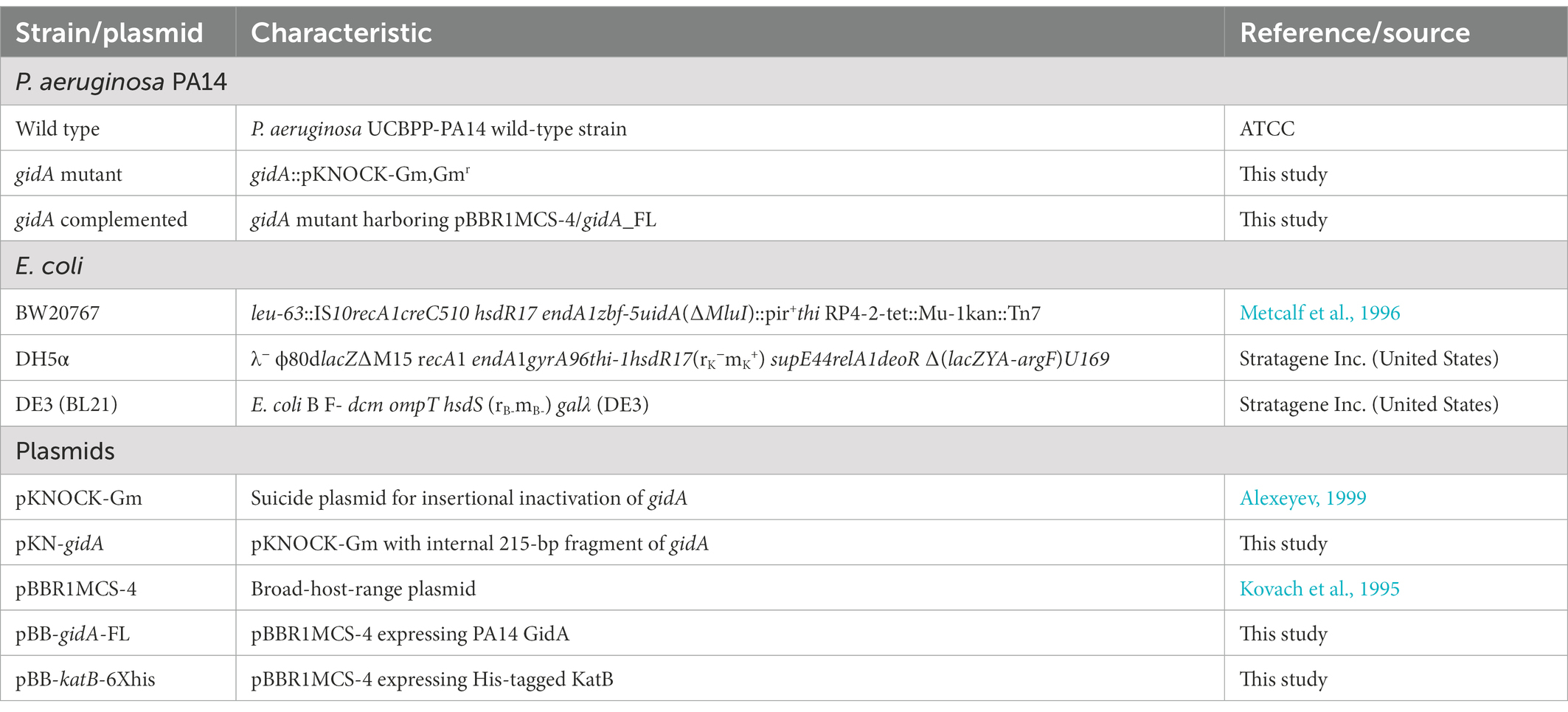
Table 1. Strains and plasmids used in this study.
2.2. Construction of the gidA mutant strain and gidA complemented strain
A gidA mutant strain was constructed using the pKNOCK suicide vector (Alexeyev, 1999). A 200 bp gidA fragment was amplified from the genomic DNA of P. aeruginosa PA14 using polymerase chain reaction (PCR) with the primer pair BT4409 (5′-CACATCGGCCTGGAGAAC-3′) and BT4410 (5′-GAACCGAGGAAGGACATCAC-3′). The amplified fragment was ligated into the SmaI site of the pKNOCK vector, which carried a GmR cassette (pKNOCK-GmR). The ligation reaction was transformed into E. coli strain BW20767, which was screened for the gentamicin resistance phenotype (Metcalf et al., 1996). The resulting plasmid was named pKN-gidA and transferred to the recipient P. aeruginosa PA14 by biparental mating using E. coli BW20767 as the donor strain. The gidA mutant was verified using colony PCR and confirmed using Southern blot analysis.
A full-length DNA fragment of the gidA gene was amplified from PA14 using primers BT5113 (5′-CCGAGGTGCGTGGTGGATT-3′) and BT5114 (5′-TGGGTTACCGCAGACATCAAG-3′). The gidA full-length DNA fragment was ligated into the SmaI restriction site of the pBBR1MCS-4 vector (Kovach et al., 1995), which contains an AmpR cassette, to produce a pBB-gidA plasmid. To obtain the gidA complemented strain, the verified pBB-gidA was transformed into a gidA mutant by electroporation. The gidA complemented strain was then screened on LB agar plates containing carbenicillin and gentamicin and confirmed using colony PCR analysis.
2.3. Ribonucleoside analysis
Total tRNA in vivo was purified as previously described (Jaroensuk et al., 2016; Thongdee et al., 2019). Total RNA was purified from logarithmic phase cultures using Trizol reagent (Invitrogen). Large RNA species were separated from small RNA species using 35% ethanol precipitation. tRNA was purified from the small RNA pool using size-exclusion high performance liquid chromatography (SEC-HPLC) with an Agilent SEC3 300 Å, 7.8 × 300 mm column. The SEC-HPLC was operated under isocratic elution mode using 100 mM ammonium acetate as the mobile phase. The concentration and quality of total tRNA were assessed using a bioanalyzer (Agilent Technologies). To produce ribonucleoside products, the purified tRNA was treated with benzonase nuclease (Sigma), bacterial alkaline phosphatase (Invitrogen), and phosphodiesterase in the presence of deaminase inhibitors (0.5 μg/ml coformycin and 5 μg/ml tetrahydrouridine) and antioxidants (50 μM desferrioxamine and 50 μM butylated hydroxytoluene) at 37°C overnight. The ribonucleosides were fractionated using reverse phase chromatography (Thermo Hypersil Gold a Q column, 100 × 2.1 mm, 1.9 μm particle size) with a gradient of 0.1% (v/v) formic acid in water (solvent A) and 0.1% (v/v) formic acid in acetonitrile (solvent B). The HPLC column was directly connected to a triple quadrupole mass spectrometer (Agilent LC/QQQ 6490) operated in the positive ion mode. Ribonucleosides were identified by comparing their m/z, CID fragmentation patterns, fragmentor voltages, collision energies, and HPLC retention times with chemical synthetic standards.
2.4. Minimum inhibitory concentration (MIC) assay
MIC assays were performed to determine the minimum inhibitory concentration of H2O2 - that is, the lowest concentration that inhibits bacterial growth (Wiegand et al., 2008). Briefly, an overnight cell culture was diluted in Mueller Hinton Broth (MHB) and grown to a turbidity of 0.5 McFarland standard. The culture was then diluted by a factor of 1:20 with 0.9% sterile normal saline. The diluted culture was used to inoculate each well of a 96-well plate containing different concentrations of a test chemical solution prepared in MHB. MHB without cells were used as a negative control, while MHB with cells inoculated but without any test chemical was used as a positive control. The 96-well plate was incubated at 37°C. After 16–20 h, the turbidity level in each well was measured using a microplate reader.
2.5. Catalase activity assay
Exponential phase cultures were treated with or without H2O2 at 5 mM or 10 mM for 25 min at 37°C with shaking. The cell pellets were washed three times with 50 mM phosphate buffer at pH 7.4 before being subjected to sonication. Crude protein concentration was then measured using the Bradford assay. Specific catalase activity (U/mg protein) was spectrophotometrically measured (A420, light path = 1 cm). U was defined as one unit of catalase decomposing 1.0 μmole of H2O2 per minute at pH 7.0 at 25°C. 10 mM H2O2 in 50 mM phosphate buffer (pH 7.0) was used as the substrate.
2.6. Western blot analysis
Exponential phase cultures were treated with or without 5 mM H2O2 for 25 min at 37°C with shaking. 50 μg of crude protein was separated using 12.5% SDS-PAGE. The separated proteins were transferred onto the polyvinylidene difluoride (PVDF) membranes (GE Healthcare). The membranes were blocked with 5% skim milk in TBST buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, and 0.05% Tween 20) for at least 1 h before incubating with antibody solution (horse radish peroxidase-conjugated anti-His tag antibody diluted in 3% skim milk and 1% BSA in TBST) for 1 h. The membranes were washed three times for 30 min in TBST buffer. The antibody reaction was observed using a chemiluminescent (ECL) detection reagent kit (GE Healthcare) according to the company’s recommendation. KatA and KatB were expressed from the pBB-katA-6xHis plasmid and the pBB-katB-6xHis plasmid, respectively. The pBB-katB 6XHis plasmid was previously described (Thongdee et al., 2019). pBB-katA 6XHis plasmid was constructed in a similar fashion to the pBB-katB 6XHis plasmid. The full-length katA gene was amplified from PA14 chromosomal DNA using PCR with the primer pair BT5117 (5′-GTTCTCCGTGGTCGCCC-3′) and BT6458 (5′-TTAGTGGTGGTGGTGGTGGTGGTCCAGCTTCAGGCCGAGG-3′), which contains hexahistidine (6XHis) tag. The katA-6XHis fragment was ligated into the BamHI and EcoICRI restriction sites of the pBBR1MCS-4 vector (Kovach et al., 1995) resulting in pBB-katA 6XHis plasmid for gene expression in PA14. The verified pBB-katA 6XHis plasmid was introduced into the wild-type and gidA mutant strains for Western blot analysis.
2.7. Semiquantitative real-time PCR
Exponential phase cultures grown in LB medium and treated with or without 5 mM H2O2 for 10 min at 37°C were used to determine the level of gene expression. A hot acid phenol extraction protocol was used for total RNA extraction (Chomczynski and Sacchi, 1987). Following DNase treatment, cDNA was synthesized using random hexamer primers and a RevertAid™ M-MuLV Reverse Transcriptase Kit (Thermo Fisher Scientific). Semiquantitative real-time PCR was performed using KAPA SYBR FAST reagent (KAPA Biosystems) and a StepOnePlus™ real-time PCR system (Thermo Fisher Scientific) according to the manufacturers’ instructions. The specific primers for the genes of interest were as follows: BT5637 (5′-TCTCCATGCGTTTCTACACC-3′) and BT5638 (5′-CGCATTGATGAAGCTGAAGG-3′) for katA; BT5639 (5′-CGACGCTTCGATTTCTTCTC-3′) and BT5640 (5′-TTCGGATCGAGGTTCTTCTG-3′) for katB; BT5641 (5′-CTACGGCGAGTTCCAGAAAG-3′) and BT5642 (5′-AGTGGATCTCGACGGTCTTG-3′) for ahpC; BT8259 (5′-CGCGAAGAAATACGACGCCG-3′) and BT8260 (5′-GTCGCTGAGGATGCCGTAGTA-3′) for fliA; BT8261 (5′-AAGGTCGAGGTCAGCGATGAC-3′) and BT8262 (5′-TCACCACGGTCTGTTCGTTGAT-3′) for fliD; BT8263 (5′-AACCCGCACCGTCTGATCC-3′) and BT8264 (5′-GCCTCGACCAGACGAGCGA-3′) for fliS; BT8253 (5′-TGGACAAGCAGACCGGCGAC-3′) and BT8254 (5′-TACATCCTTGGGCAGGCAGG-3′) for algD; BT8255 (5′-GGTGCCGCTCATCGTGCTCTA-3′) and BT8256 (5′-CGCGTTGCCCTGCATCTGGTA-3′) for algK; BT8257 (5′-CTACCCTGCCGAAGCTGGAT-3′) and BT8258 (5′-TCGCTGGACGAGGAGTTGGT-3′) for mucA; and BT2781 (5′-GCCCGCACAAGCGGTGGAG-3′) and BT2782 (5′-ACGTCATCCCCACCTTCCT-3′) for 16 s RNA gene. The 16S ribosomal RNA gene was used as the internal control to normalize gene expression.
2.8. Motility assays
Motility was examined on BM2-swimming plates (0.3% bacto agar, 62 mM potassium phosphate buffer pH 7, 7 mM (NH4)2SO4, 2 mM MgSO4, 10 μM FeSO4, 0.4% glucose) and BM2-swarming plates (0.5% bacto agar, 62 mM potassium phosphate buffer pH 7, 0.1% casamino acid, 2 mM MgSO4, 10 μM FeSO4, 0.4% glucose; Overhage et al., 2007, 2008; Filloux and Ramos, 2014). Homogenized colonies were stabbed into the center of the BM2-swimming plate while 5 μl of the overnight cultures was inoculated on the surface of each BM2-swarming plate. The swimming and swarming plates were incubated overnight at 37°C. The relative motility of each strain was determined by comparing the diameters of the swim and swarm colonies.
2.9. Biofilm formation assay
Two methods were used to observe the biofilm formation, i.e., dye straining and scanning electron microscope (SEM). For the dye staining method, 100 μl of a diluted overnight culture (OD600nm ~ 0.2) was added to a well of a 96-well plate and incubated overnight at 37°C. After overnight incubation, the plate was gently rinsed with distilled water before staining with 150 μl of 1% crystal violet for 10 min. Then, the crystal violet solution was removed. The plate was rinsed with distilled water again and allowed to air dry for at least 15 min. 100 μl of absolute ethanol was added into the well to dissolve the remaining crystal violet. Then, a spectrophotometer measurement was conducted at an optical density of 600 nm. The SEM method was performed using the method of Jin et al. with some adaptations (Jin et al., 2005). The biofilm of wild-type, gidA mutant, and complemented strains were developed on 1 × 1 cm plastic slide. The plastic slides were pretreated with concentrated sulfuric acid and 95% ethanol. 100 μl of cell suspensions (107 CFU/ml) in PBS were dropped on the slide and were incubated for 90 min at 37°C to promote the bacterial cell adhesion. The plastic slides were washed twice with 200 μl PBS to remove nonadherent cells and filled with 100 μl Brain Heart Infusion (BHI) culture medium. The bacterial cultures were incubated at 37°C for 36 h and the BHI culture medium was replenished daily. The biofilm samples were fixed with 2.5% glutaraldehyde for 2 h and serially dehydrated with ethanol (70%, 95%, and absolute ethanol). The samples were kept in absolute ethanol until analysis. Before observing under the SEM, all samples were dried using critical-point drying in an EMITECH K850 dryer (Quorum Technologies Ltd.) at 39.1°C and 1,000 psi. The dried samples were then coated with gold, and the images were obtained using a TESCAN MIRA3 FEG-SEM (Field Emission Scanning Electron Microscope).
2.10. Alginate measurement
Alginate measurement was adapted from methods of Knutson and Jeanes (1968) and Zheng et al. (2016). Wild type, gidA mutant and the complement strain were spread onto BHI agar to promote alginate production. The cells were incubated at 37°C for 40–48 h, harvested and resuspended in 5 ml PBS (pH 7.4). Approximately 107 cells were centrifugation at 12,000 g for 20 min to remove the cell pellet. Alginate was precipitated by adding equal volume of 2% cetylpyimidine to the cell free medium followed by centrifugation at 12,000 g for 15 min. The pellets were resuspended in 5 ml of 1 M NaCl. Then, 5 ml ice-cold isopropanol (−20°C) was added before centrifugation at 12,000 g for 15 min to collect the alginate pellet. Completely air-dried alginate pellets were then resuspended in 1 ml 0.9% NaCl. Measurement of alginate in the solutions using the standard alginic acid (0.01–1.0 mg/ml in 0.9% NaCl) was performed in 1.5 ml microtubes. 70 μl of alginate suspension was mixed using vortex with freshly prepared ice-cold 0.1 M H3BO3 in concentrated H2SO4. Then, 20 μl of 0.1% carbazole in ethanol was added. The solution was mixed and incubated at 55°C for 30 min. The absorbance at 530 nm was measured. The amount of alginate in each sample was calculated against alginate standards and normalized with OD600 of the bacterial cell suspension.
2.11. In vitro phagocytosis assay
The phagocytosis assay was modified from previously described protocols (Kuang et al., 2011; Charoenlap et al., 2012; Gao et al., 2016). Macrophage cells (RAW264.7) were activated with mouse IFN-gamma. Activated macrophage cells (RAW264.7) were seeded (2 × 106 cells/well) in six-well tissue culture plates overnight. The cells were infected with mid-log phase P. aeruginosa cultures (MOI 1:10). The live and dead macrophage cells were counted at 0, 2, 4, and 8 h.
2.12. Caenorhabditis elegans killing assay
The virulence of P. aeruginosa was tested using Caenorhabditis elegans as the animal model (Tan et al., 1999). P. aeruginosa strains were grown in 5 ml of King’s broth at 37°C overnight with shaking. 10 μl of each overnight culture was spread onto high osmotic strength PGS agar (peptone-glucose-sorbital; 1% Bacto-peptone, 1% NaCl, 1% glucose, 0.15 M orbital, 1.7% Bacto agar) in a 60 × 15 mm plate. The plates were incubated overnight at 37°C and then subsequently transferred to 22°C for 8–12 h. Each bacterial lawn plate was seeded with 20–200 L4 stage larvae of C. elegans (Bristol-N2 wild-type strain from BP. Braeckman’s lab), incubated at room temperature, and scored for live and dead worms at time intervals over 3 days.
2.13. Total shotgun proteome analysis
The exponential phase cells were snap-frozen in liquid nitrogen. Total protein was isolated using 0.5% SDS solution. Protein content was measured using the Lowry assay with bovine serum albumin as a standard (Lowry et al., 1951). 5 μg of protein sample was subjected to in-solution digestion. All samples were completely dissolved in 10 mM ammonium bicarbonate (AMBIC). Disulfide bonds were reduced using 5 mM dithiothreitol (DTT) in 10 mM AMBIC at 60°C for 1 h. Alkylation of sulfhydryl groups was done by incubating them with 15 mM iodoacetamide (IAA) in 10 mM AMBIC at room temperature for 45 min in the dark. Then, the samples were mixed with 50 ng/μL of sequencing grade trypsin (1:20 ratio; Promega, United States) and incubated at 37°C overnight. The digested samples were dried and redissolved with 0.1% formic acid before injection into an Ultimate3000 Nano/Capillary LC System (Thermo Scientific, United Kingdom) coupled to an HCTUltra LC–MS system (Bruker Daltonics Ltd.; Hamburg, Germany) equipped with a nano-captive spray ion source. 5 μl of peptide digest was enriched on a μ-Precolumn 300 μm i.d. × 5 mm C18 Pepmap 100, 5 μm, 100 A (Thermo Scientific, United Kingdom), separated on a 75 μm i.d. × 15 cm and packed with Acclaim PepMap RSLC C18, 2 μm, 100 Å, nanoViper (Thermo Scientific, United Kingdom). The C18 column was enclosed in a thermostatted column oven set at 60°C. Solvents A and B containing 0.1% formic acid in water and 0.1% formic acid in 80% acetonitrile, respectively, were used as mobile phases. A gradient of 5–55% solvent B was used to elute the peptides at a constant flow rate of 300 nl/min for 30 min. Electrospray ionization was carried out at 1.6 kV using CaptiveSpray. Nitrogen was used as a drying gas at a flow rate of about 50 l/h. Collision-induced dissociation (CID) product ion mass spectra were obtained using nitrogen gas as the collision gas. Mass spectra (MS) and MS/MS spectra were obtained in the positive-ion mode at 2 Hz over a range of 150–2,200 m/z. The collision energy was adjusted to 10 eV as a function of the m/z value. Liquid chromatography-mass spectrometry (LC–MS) analysis of each sample was done in triplicate. The raw MS/MS spectra data are available in ProteomeXchange: JPST001884 and PXD037219.1
2.14. Bioinformatics and shotgun proteome data analysis
For protein quantitation, DeCyder MS Differential Analysis software (DeCyderMS, GE Healthcare) was used (Johansson et al., 2006). The analyzed MS/MS data from DeCyderMS were submitted for database searches using Mascot software (Matrix Science, London, United Kingdom). The data were searched against the NCBI database for protein identification. Database interrogation was performed for taxonomy (Pseudomonas aeruginosa); enzyme (trypsin); variable modifications (carbamidomethyl, oxidation of methionine residues); mass values (monoisotopic); protein mass (unrestricted); peptide mass tolerance (1.2 Da); fragment mass tolerance (±0.6 Da); peptide charge state (1+, 2+ and 3+); and missed cleavages. The level of protein in each sample was expressed as a log2 value. Gene ontology annotation was performed using Panther (Mi et al., 2019).
3. Results
3.1. Pseudomonas aeruginosa GidA is a tRNA-modifying enzyme in the mnm5(s2)U biosynthetic pathway
P. aeruginosa GidA displays 70% identity to the E. coli MnmE protein (Yim et al., 2006). To assess whether P. aeruginosa GidA is responsible for the formation of cmnm5(s2)U or nm5(s2)U in the mnm5(s2)U biosynthetic pathway (Figure 1A), tRNA modification profiles of the wild type, a gidA insertional inactivation mutant, and a complemented strain were analyzed using LC–MS/MS. The result of this study showed that the inactivation of gidA specifically altered the level of tRNA modifications linked to the mnm5(s2)U biosynthetic pathway (Figure 1B). Among the 24 modifications detected in P. aeruginosa’s tRNAs, only the s2U, cmnm5U, and mnm5U levels were found to change due to the loss of gidA (Table 2; Figure 1B). The s2U levels increased around fourfold, while the cmnm5U and mnm5U levels significantly reduced in the gidA mutant compared to those in the wild-type and complemented strains (Figure 1B; Table 2). These findings denote the crucial function of gidA in the biosynthesis of 5–carboxymethylaminomethyl uridine (cmnm5U) and 5-methylaminomethyl uridine (mnm5U) tRNA modifications in P. aeruginosa. In addition, hypomodifications of cmnm5U and mnm5U also resulted from the disruption of P. aeruginosa trmE (Supplementary Figure S1), indicating that the formation of cmnm5U and mnm5U is dependent on gidA as well as trmE.
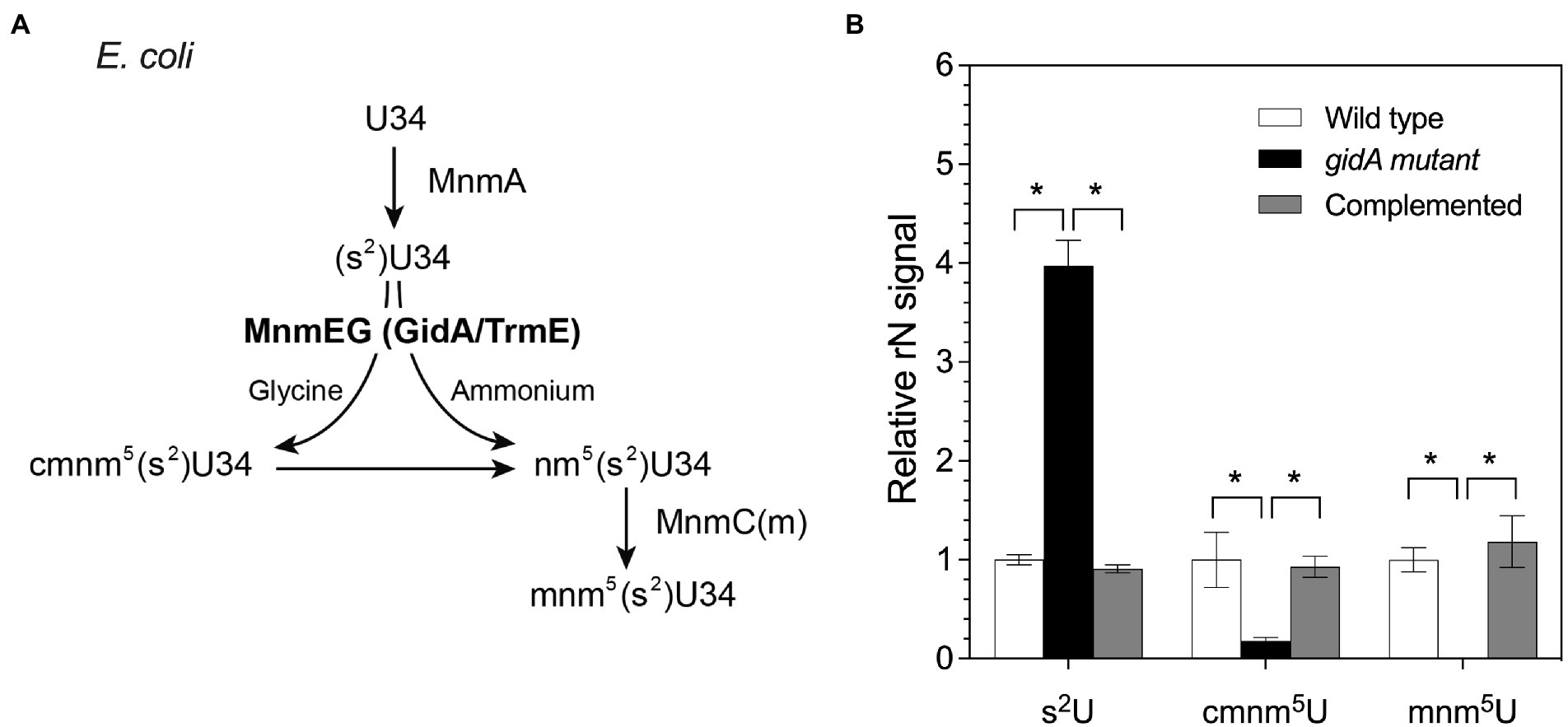
Figure 1. The amount of tRNA modifications in the mnm5(s2)U biosynthetic pathway is altered in the gidA mutant. (A) The pathway of mnm5(s2)U biosynthesis in E. coli. (B) Change in the levels of s2U, cmnm5U, and mnm5U in total tRNA isolated from wild-type, gidA mutant, and complemented strains. The wild type and gidA mutant carried the pBBR1MCS-4 plasmid as a vector control. The level of each modified ribonucleoside was quantified based on the MRM signal intensity and normalized by dividing the quantified amounts by the summed signals of adenosine, guanosine, cytidine, and uridine in the sample. The data represent the mean ± SD values of three biological replicates. The statistical analysis was performed using Graphpad Prism (GraphPad Software). Asterisks denote a significant difference in the one-way analysis of variance (ANOVA; * p≤ 0.05).
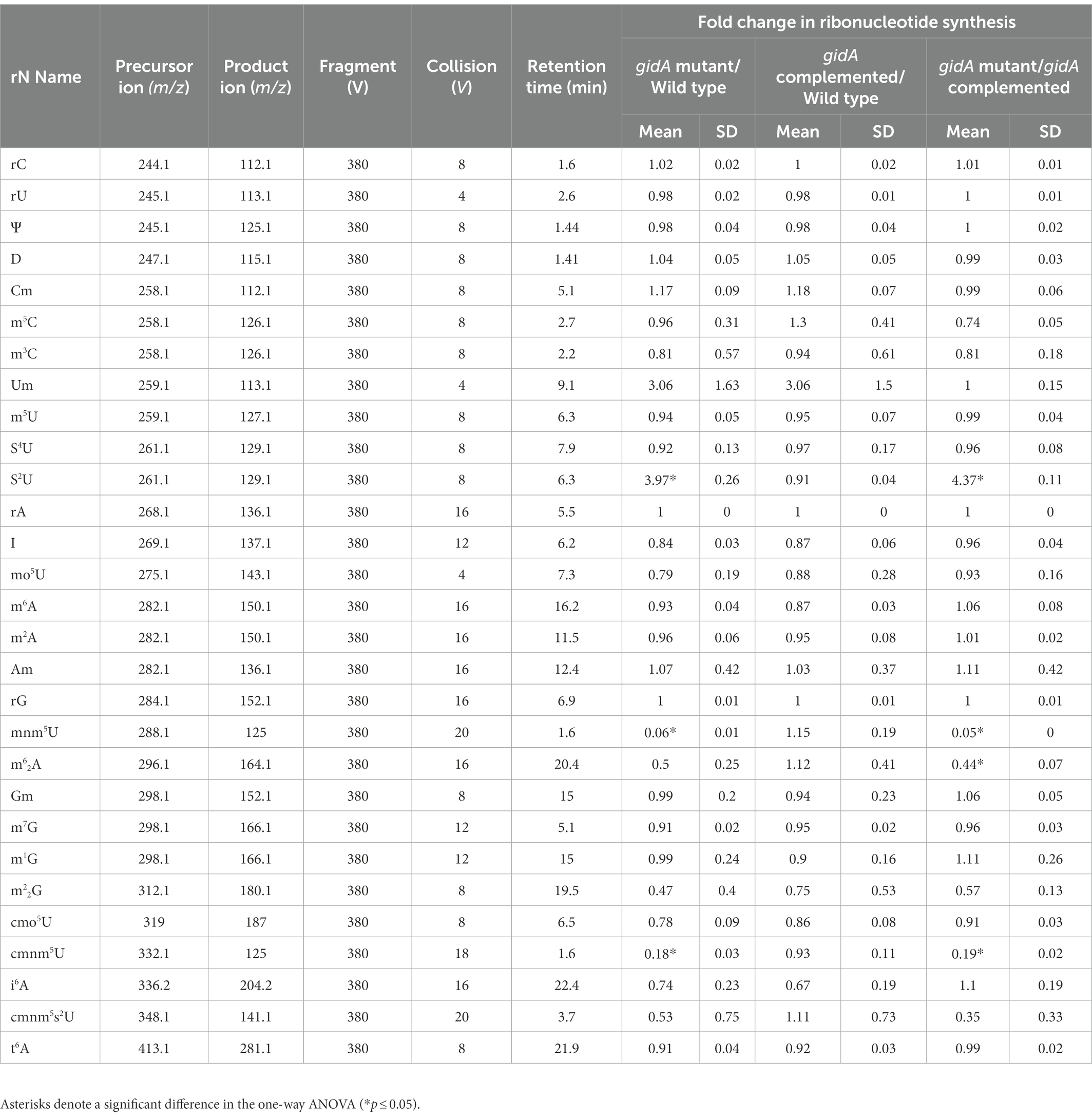
Table 2. Ribonucleotides identified by LC–MS/MS analysis of P. aeruginosa PA14 tRNA.
3.2. Loss of gidA decreases the expression of katA and katB at the posttranscriptional level leading to H2O2 susceptibility in Pseudomonas aeruginosa
Cell response to oxidative stress is an important mechanism of Pseudomonas for counteracting the toxic effects of oxidizing agents such as hydrogen peroxide, superoxide, and hypochlorous acid, which are produced by the host immune system during infection. In our H2O2 sensitive phenotype screening from the mutant library of Pseudomonas aeruginosa, gidA mutant is one of mutant strains showing such phenotype (unpublished data). To further determine the role of gidA in H2O2 stress response, the sensitivity of a gidA mutant strain to H2O2 was tested using a minimal inhibition concentration (MIC) assay. The results revealed that the gidA mutant (78 nM) was more susceptible to H2O2 than the wild-type and complemented strains (156 nM). The same result was found in the experiment involving the use of a plate sensitivity assay (data not shown). Therefore, it is possible that gidA is necessary for full resistance against H2O2.
The detoxification of H2O2 into harmless H2O and O2 molecules is primarily catalyzed by catalase enzymes such as KatA and KatB (Zamocky et al., 2010). Disruption of gidA may affect the capability of P. aeruginosa to detoxify H2O2. To test this hypothesis, the total catalase activity of the gidA mutant, the wild-type, and complemented strains were compared. Under unexposed conditions, the total catalase activities were not statistically different (143.3 ± 11.1, 121.6 ± 23.0, and 109.3 ± 5.8 units/mg protein, respectively; Figure 2A). Upon exposure to 5 mM H2O2, the total catalase activities of all strains increased from the basal level (Figure 2A). However, the catalase activity of the gidA mutant (462.9 ± 13.4 units/mg protein) was significantly lower than those of the wild-type (801.1 ± 72.1 units/mg protein) and complemented strains (690.6 ± 45.2 units/mg protein). This observation shows that the disruption of gidA has a negative effect on catalase-mediated H2O2 detoxification in P. aeruginosa. The reduced catalase activity of the gidA mutant may not be sufficient for cell survival during H2O2 treatment, resulting in a higher H2O2 sensitivity of the gidA mutant than those of the wild-type and complemented strains.
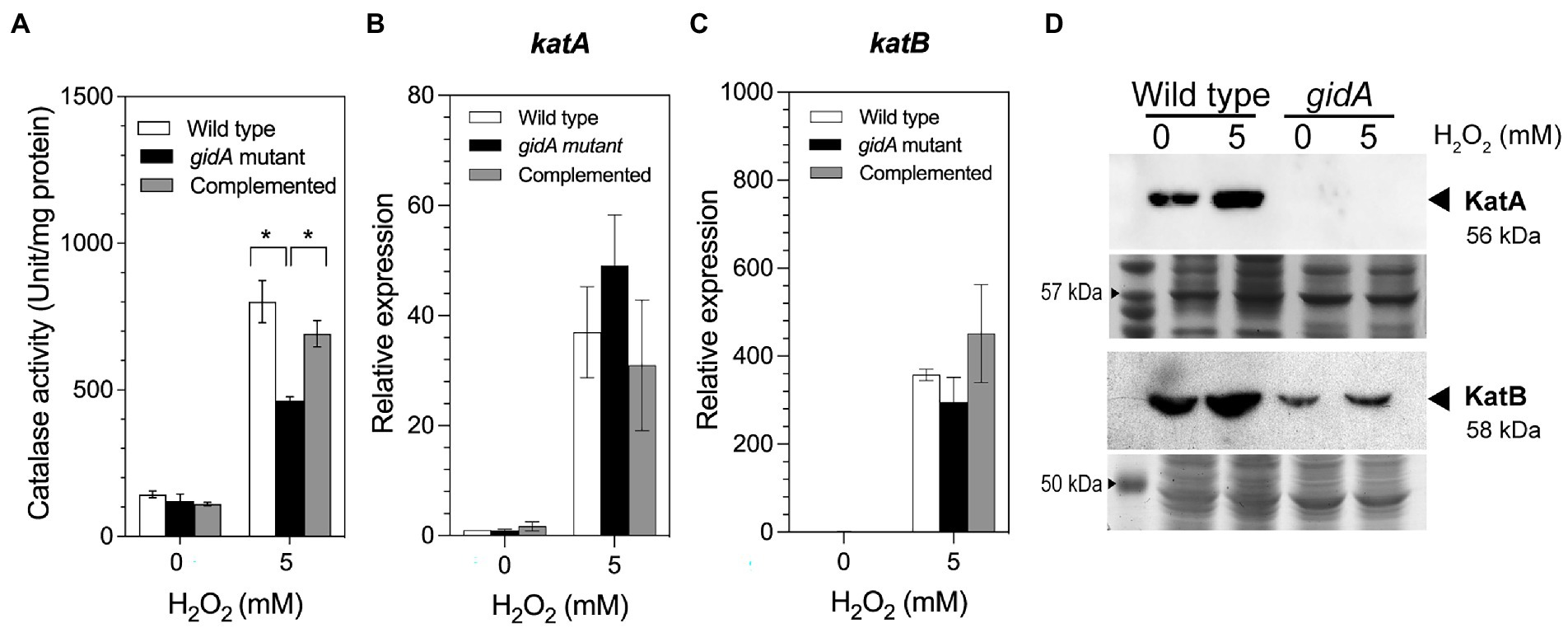
Figure 2. Impact of gidA disruption on catalase gene expression. (A) The catalase-specific activity of the wild-type, gidA inactivation mutant, and complemented strains. (B) Relative mRNA expression levels of katA, and (C) katB, in the wild-type, gidA mutant, and complemented strains upon H2O2 exposure. The wild-type and gidA mutant carried the pBBR1MCS-4 plasmid as a vector control. The data represent the mean ± SD values of three biological replicates for (A–C). The statistical analysis was performed using Graphpad Prism (GraphPad Software). Asterisks denote a significant difference in the one-way ANOVA (* p ≤ 0.05). (D) Western blot analysis of the KatA and KatB proteins. Anti-KatA antibody was used to assess KatA level. The amounts of KatA and KatB proteins overexpressed from the pBB-katA-6xHis plasmid and pBB-katB-6xHis plasmids, respectively, were assessed using the anti-His antibody. The amount of total protein loaded in each gel is shown as the loading control. The data shown are representative results of three biological replicates.
P. aeruginosa possesses three catalase enzymes; KatA, KatB, and KatE. To further explain how the loss of gidA affects catalase activity, the impact of gidA disruption on catalase gene regulation was determined. The regulation of P. aeruginosa katA and katB, which encode major catalases expressed during exponential phase growth, was investigated. The results of a semiquantitative real-time PCR assay demonstrated that the transcript levels of katA and katB increased in response to H2O2 exposure in all strains, as expected, and that the transcript levels were not statistically different. Moreover, no significant differences in transcript levels were observed between the strains in the absence of H2O2 (Figures 2B,C).
Given that total catalase activity was significantly reduced due to the absence of gidA (Figure 2A), it is thus highly possible that gidA exerts its effect on katA and katB gene expression at the posttranscriptional level. Western blot analysis was used to investigate the impact of gidA on the protein level of KatA and KatB during exposure to 5 mM H2O2. We investigated the level of KatA and KatB expressed from the plasmid copy, where gene is expressed using the lac promoter and ribosome binding site from the plasmid. The result demonstrated that KatA protein expression was not detectable in the gidA inactivation mutant compared to the wild-type strain under both normal and 5 mM H2O2 treatment conditions (Figure 2D). In addition, the level of KatB protein was markedly decreased in the gidA mutant compared with that in the wild type (Figure 2D). The decreased level of both KatA and KatB protein, but not katA and katB mRNA level, indicates that gidA exerts a posttranscriptional effect on katA and katB expression.
3.3. The gidA disruption has a negative effect on motility and biofilm formation in Pseudomonas aeruginosa
It is known that gidA is associated with virulence mechanisms and the expression of a wide range of virulence genes in bacteria, including quorum sensing and stress responses (Gupta et al., 2009; Shippy and Fadl, 2014; Zhang et al., 2014; Gao et al., 2016). In P. aeruginosa PA14, gidA is involved in the Rhl-quorum sensing system affecting the bacterium’s growth and proteolytic activities (Gupta et al., 2009). Therefore, genes associated with bacterial motility and biofilm formation assays were performed in this study. Figures 3A,B present the results for both motility activities, i.e., swimming and swarming. With respect to swimming motility (Figure 3A), the wild-type and complemented strains both presented swimming behaviors on the semisolid swimming plates by 5.7 ± 0.1 cm and 5.1 ± 0.1 cm, respectively, compared to 3.7 ± 0.1 cm in the gidA inactivation mutant indicating a decreased in the swimming ability of the gidA mutant strain. In addition, the swarming plates revealed that the loss of gidA reduced the swarming ability of P. aeruginosa PA14 in covering the semisolid plate (Figure 3B). Furthermore, the lack of gidA was also found to affect biofilm production in P. aeruginosa PA14. Biofilm formation in the gidA mutant significantly decreased in relation to biofilm formation in the wild-type and complemented strains as determined by the dye staining method (Figure 3C). Biofilm formation was also observed using scanning electron microscope. The results demonstrated that gidA mutant was unable to form the confined biofilm compared to the wild-type and complemented strains (Figure 3D). In addition, alginate concentration was measured as it is an important component of biofilm (Gardner et al., 1987; Tatnell et al., 1994; Keiski et al., 2010). Consistent with the biofilm formation data, the alginate production was lower in the gidA mutant by 21.2 ± 1.8 folds. Taken together, these observations indicate that gidA contributes to various virulence genes in P. aeruginosa PA14, alter the flagellum-mediated motility activities (swimming and swarming) and biofilm formation.
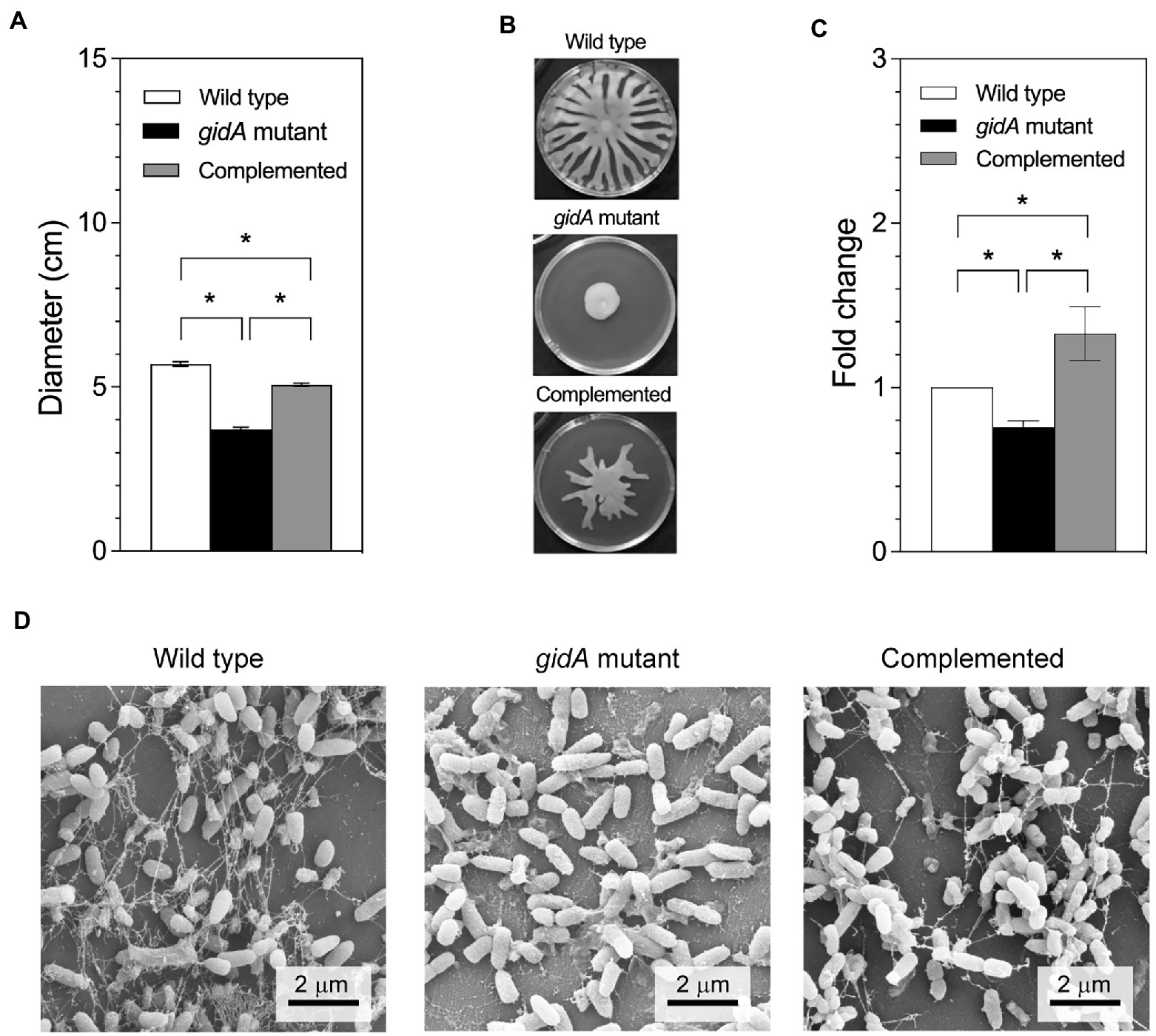
Figure 3. The gidA mutant strain shows defects in motility and biofilm production. (A) Swim colony diameter. Bacteria were stabbed into 0.3% BM2 medium agar. After incubation, the diameter of bacterial migration was measured from the stab point. (B) Images of the swarming plates. (C) The fold change in biofilm production for the wild-type, gidA mutant, and complemented strains using dye staining method. (D) Scanning electron microscope showing the biofilm formation. The images were recorded at magnification of 15,000x. The wild type and gidA mutant carried the pBBR1MCS-4 plasmid as a vector control. The data represent the mean ± SD values of three biological replicates for (A,C). The statistical analysis was performed using Graphpad Prism (GraphPad Software). Asterisks denote a significant difference in the one-way ANOVA (* p ≤ 0.05).
3.4. The expression of some motility and biofilm formation genes gets altered in the gidA mutant
To further investigate the changes in the protein profile of the gidA mutant in comparison to the wild-type strain, proteomic analysis using liquid chromatography–tandem mass spectrometry was performed. A total of 1,106 protein identities were detected, 377 of which were differentially produced between the gidA mutant and wild-type strains (Supplementary Figure S2). Among these differentially produced proteins, 267 were upregulated and 110 were downregulated in the gidA mutant relative to the wild type. The abundance levels of several proteins involved in bacterial pathogenesis were altered (Table 3). A lower abundance of some components of the flagellar system, including FliD, FliS, and FliA, was seen in the gidA mutant than in the wild-type strain (Table 3). In addition, the levels of some proteins in the alginic acid biological process, which plays a role in biofilm formation, were found to decrease in the gidA mutant when compared with the levels in the wild-type strain; these included GDP-mannose dehydrogenase (AlgD) and multiple tetratricopeptide-like (TPR-like) repeats lipoprotein (AlgK). The level of MucA, an anti-sigma factor involved in the regulation of alginic acid biosynthesis, was higher in the gidA mutant than in the wild type. These data support the defects in mobility and biofilm formation observed in the gidA mutant.
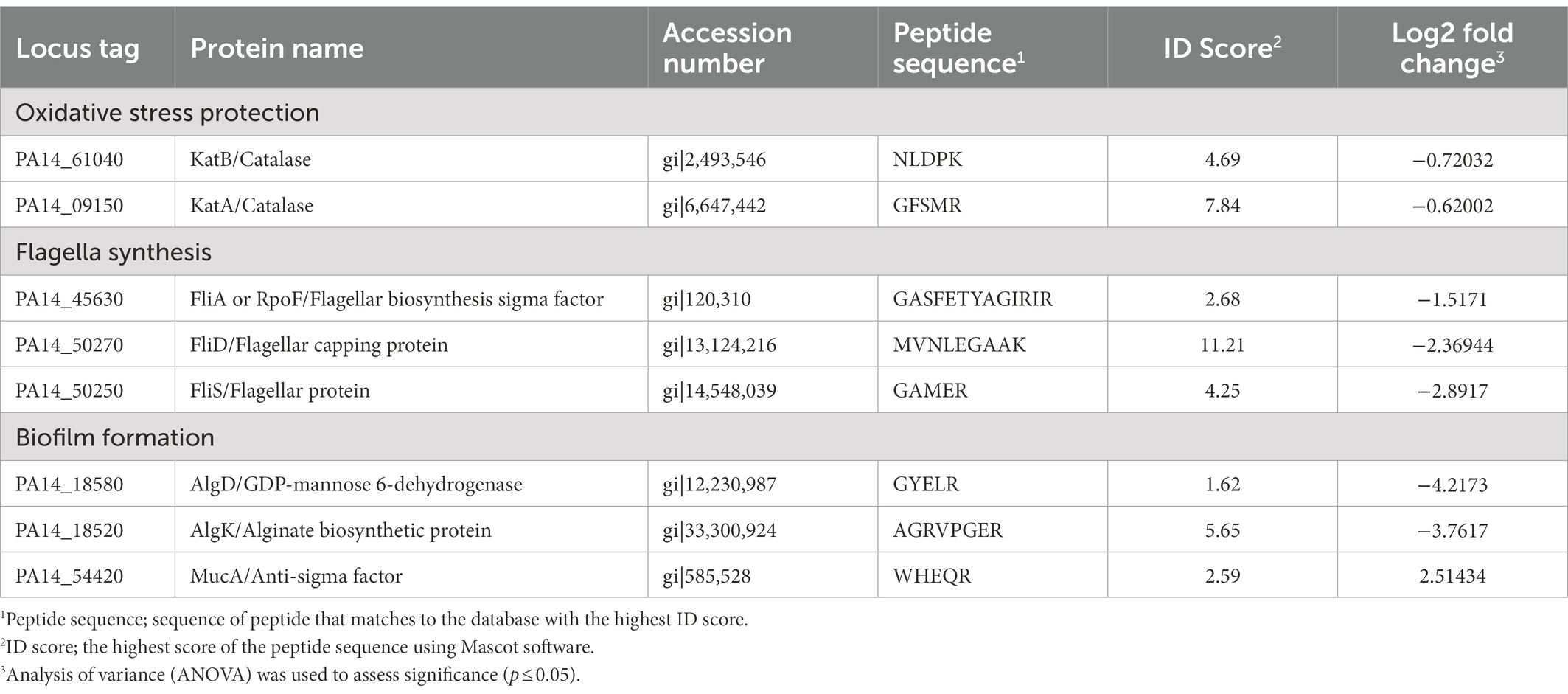
Table 3. List of differentially produced proteins in gidA mutant compared to Pseudomonas aeruginosa wild type discussed in this report.
The mRNA levels of fliA, fliD, and fliS, which are involved in flagella formation, were investigated using semiquantitative RT-PCR analysis (Figure 4). The results revealed reduced mRNA levels in the gidA mutant strain compared to the wild-type and complemented strains (Figures 4A–C). With respect to genes involved in biofilm formation, the mRNA levels of the algD and algK genes were slightly lower in the gidA mutant strain than in the wild-type strain, while the mRNA level of the mucA gene remained unchanged in the gidA mutant but not in the wild-type strain (Figures 4D–F). The mRNA levels of the algD and algK genes changed in the same direction as the corresponding protein levels, indicating that such changes might not involve regulation at the translation level of those genes. The unchanged mucA mRNA level and upregulated protein level indicate the effect of gidA on mucA expression at the posttranscriptional level.
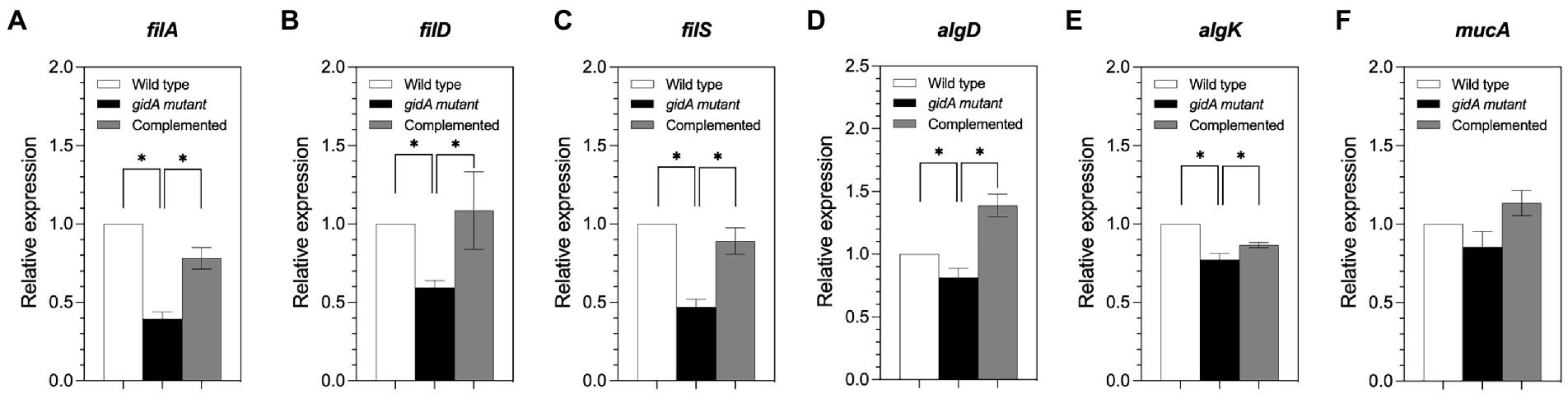
Figure 4. The gidA mutant strain shows defects in some motility and biofilm formation gene expression at the mRNA level. (A) Relative mRNA expression levels of fliA, (B) fliD, (C) fliS, (D) algD, (E) algK, and (F) mucA in the wild-type, gidA mutant, and complemented strains. The mRNA expressions of fliA, fliD, fliS (involved in motility), algD, and algK (involved in biofilm formation) genes were reduced in gidA mutant strains. The data represent the mean ± SD values of three biological replicates. The statistical analysis was performed using Graphpad Prism (GraphPad Software). Asterisks denote a significant difference in the one-way ANOVA (* p ≤ 0.05).
3.5. The gidA inactivation mutant attenuates virulence
In this study, the inactivation of gidA affected the oxidative stress response, motility, and biofilm formation capability of P. aeruginosa PA14 (Figures 2, 3), suggesting that the loss of gidA attenuates its virulence. Therefore, in vitro phagocytosis assays were performed using a mouse macrophage cell line (RAW264.7). The cells were infected with the wild-type, gidA mutant, and complemented strains of P. aeruginosa PA14 at a ratio of 10 bacterial cells per macrophage (MOI 1:10). Macrophages infected with the gidA inactivation mutant showed a significantly high survival rate compared to those infected with either the wild-type or complemented strains (Figure 5A). Further, the ability of the wild type, gidA inactivation mutant, and complemented strain to kill Caenorhabditis elegans was evaluated in vivo using a slow-killing assay. The results mirrored the macrophage assays. P. aeruginosa was clearly attenuated when gidA was disrupted. After 72 h of infection, the C. elegans fed with the gidA mutant had a significantly lower % of death (* p ≤ 0.05) than those fed with either the wild type or complemented strain (Figure 5B). Overall, these results confirmed that the inactivation of gidA attenuates the virulence of P. aeruginosa PA14.
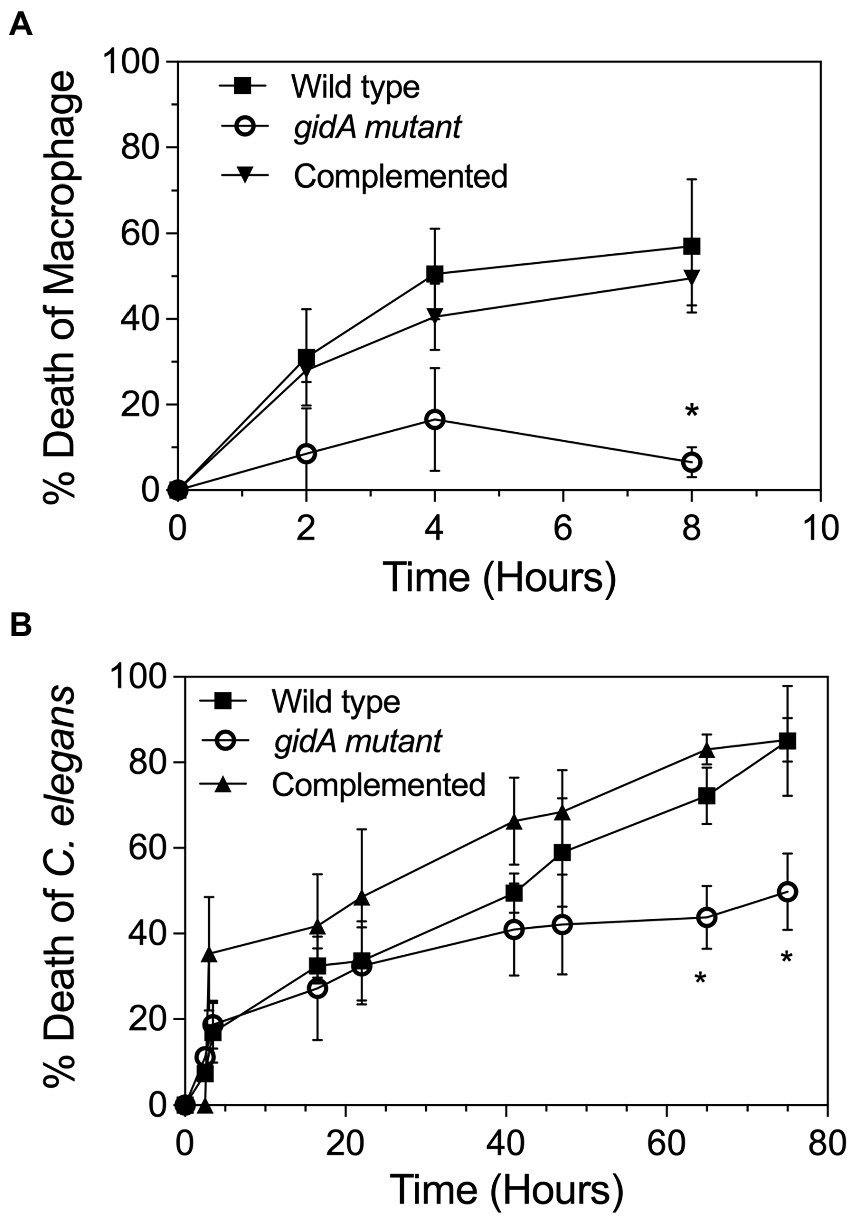
Figure 5. gidA is needed for the full virulence of P. aeruginosa. (A) Cell viability of the RAW264.7 macrophage cell line after treatment with the wild-type, gidA mutant, and complemented cells for 24 h. Cell viability at each timepoint was examined and compared with that of the untreated cells. (B) L4 stage larvae of C. elegans were added to the lawn of the wild-type, gidA mutant, and complemented cells. Live and dead worms were counted at time intervals for 3 days. The wild type and gidA mutant carried the pBBR1MCS-4 plasmid as a vector control. The data represent the mean ± SD values of three biological replicates. The statistical analysis was performed using Graphpad Prism (GraphPad Software). Asterisks denote a significant difference in the one-way ANOVA (* p ≤ 0.05).
4. Discussion
This study provided in vivo evidence that P. aeruginosa gidA functions as a tRNA-modifying enzyme involved in the biosynthetic pathway of mnm5(s2)U tRNA modification. Furthermore, the roles of gidA in the P. aeruginosa cellular response to oxidative stress, motility, biofilm production, and virulence were determined. The biosynthesis of 5-carboxymethylaminomethyl uridine (cmnm5U) and 5-methylaminomethyl uridine (mnm5U) tRNA modifications in P. aeruginosa were found to depend on both gidA and trmE (Figure 1B; Table 2; Supplementary Figure S1). The accumulation of s2U modification was detected in the total tRNA isolated from either the gidA and trmE mutant strains (Figure 1; Supplementary Figure S1), suggesting that the loss of either GidA or TrmE activity impairs the conversion of s2U to cmnm5(s2)U and/or to nm5(s2)U. According to the model for the biosynthetic pathway of mnm5(s2)U in E. coli (Moukadiri et al., 2014), the formation of cmnm5U in P. aeruginosa may be catalyzed by a GidA/TrmE complex via the glycine pathway (Figure 1A). The presence of nm5(s2)U in P. aeruginosa remains undetermined due to the unavailability of the standard. However, it is highly likely that the tRNAs of P. aeruginosa contain nm5(s2)U modifications. This is supported by the presence of the final product, mnm5U, in the wild-type tRNAs (Figure 1B) and the presence of PA14_19400, a homolog of E. coli MnmC in the genome of P. aeruginosa. MnmC is an enzyme that catalyzes the conversion of cmnm5(s2)U to nm5(s2)U.
The study results indicate that gidA contributes to H2O2 resistance in P. aeruginosa. In our previous studies, three tRNA modifying genes-trmB, ttcA, and trmJ-were shown to be associated with the H2O2 response of P. aeruginosa (Jaroensuk et al., 2016; Romsang et al., 2018; Thongdee et al., 2019). Among these three modifications, the molecular mechanism of the trmB-mediated H2O2 response and H2O2-dependent transcription regulation of ttcA have been described. The trmB gene, which encodes the tRNA m7G46 methyltransferase, modulates the H2O2 response by controlling the translation of katA and katB mRNAs via a bias codon usage mechanism (Thongdee et al., 2019). The ttcA gene, which encodes a tRNA thiolating enzyme, is directly regulated by OxyR, a transcription regulator that plays a global role in the oxidative stress response (Romsang et al., 2018). The observations in the present study suggest that the posttranscriptional regulation of katA and katB by tRNA modification is more complicated than we thought as gidA was also found to have a posttranscriptional impact on katA and katB expression (Figure 2; Table 3).
Furthermore, gidA is also involved in the posttranscriptional regulation of phlA, phlD, rsmA, and rsmE in P. fluorescens and rhlR in P. aeruginosa (Gupta et al., 2009; Zhang et al., 2014). The precise mechanism by which gidA selectively affects the translation of certain transcripts is unknown. Considering that the modification of wobble uridine by the GidA/TrmE complex ensures the discrimination of pyrimidine-ending codons from purine-ending codons and prevents translational frame shifting (Brégeon et al., 2001; Urbonavičius et al., 2001; El Yacoubi et al., 2012), the translation reduction in the gidA mutant strain may be a result of misreading problems.
In the present study, the gidA mutant showed significantly attenuated virulence in both the C. elegans model and the macrophage model (Figure 5). The disruption of gidA was found to reduce swimming, swarming, and biofilm formation, all of which contribute to bacterial virulence (Figures 3). These results are consistent with findings from previous studies that gidA deletion leads to decreased motility and biofilm formation in Lysobacter capsica, and virulence factor in S. suis and Salmonella (Rehl et al., 2013; Gao et al., 2016; Zhao et al., 2022). The proteomic study of P. aeruginosa also revealed that many proteins involved in motility and biofilm formation were downregulated in the gidA mutant compared to the wild type (Table 3). Bacterial flagella are essential for pathogenesis in P. aeruginosa, as they mediate swimming and swarming motility, bacterial adhesion, host invasion, and biofilm formation (Arora et al., 1998; Kirov, 2003). FliD, FliS and FliA, which are involved in flagellar biosynthesis, were found at lower levels in the gidA mutant (Table 3; Figure 4; Das et al., 2018; Bouteiller et al., 2021). This finding aligns with a previous report that a fliS mutant displays weak motility in Yersinia pseudotuberculosis (Xu et al., 2014). In addition, FliA, a positive regulator of cell motility, was slightly reduced in the gidA mutant. The above results imply that the depletion of fliD, fliS, and fliA genes at both mRNA and protein levels is involved in the reduced swimming and swarming motility levels in the gidA mutant of P. aeruginosa.
In the gidA mutant, the level of alginate was lower compared to wild type. The levels of AlgD, AlgK and MucA proteins were also altered (Table 3). These proteins are involved in the production of alginate, which is an important component of biofilm formation and is required for initial bacterial adhesion to a solid surface in P. aeruginosa (Gardner et al., 1987; Tatnell et al., 1994; Keiski et al., 2010). The levels of both AlgD and AlgK levels were downregulated in the gidA mutant. In contrast, MucA, an anti-AlgU sigma factor, was upregulated in the gidA mutant. Generally, MucA binds to AlgU, which is required for the transcription of the algD promoter. Thus, an increased MucA level might lead to reduced alginate biosynthesis due to AlgU down-regulation (Deretic et al., 1994). Interestingly, the mRNA level of mucA was unchanged in the gidA mutant but not in the wild-type strain indicating that mucA is subjected to post-transcription regulation by GidA.
In conclusion, the present study demonstrated that GidA is a tRNA-modifying enzyme of the mnm5U biosynthetic pathway. We revealed a novel role for gidA in the H2O2 stress response by demonstrating that gidA is required for full resistance against H2O2 and that it affects katA, katB and mucA expression at the posttranscriptional level. The results suggest that the posttranscriptional regulation of katA and katB by tRNA modification is a complex process involving multiple tRNA-modifying genes. In addition, gidA was shown to be involved in virulence of P. aeruginosa.
Data availability statement
The data presented in the study are deposited in the ProteomeXchange repository (http://www.proteomexchange.org/, accession numbers JPST001884 and PXD037219.
Author contributions
TS and NT contributed to the conception and design of the experiments, performed parts of the experiments, analyzed the results, and prepared the manuscript. JC performed the proteome data analysis and prepared the manuscript. SA performed the virulence assay. JJ, NP, and SR performed the mass spectrometry experiment. KP performed real-time PCR. SM was involved in the manuscript preparation. SA and KP performed SEM and alginate measurement. MF contributed to the conception and design of the experiments, analyzed the results, and prepared the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Chulabhorn Graduate Institute [ABS-01 to MF], by Thailand Science Research and Innovation (TSRI), Chulabhorn Research Institute (Grant no. 313/2231), and in part by the grant from Center of Excellence on Environmental Health and Toxicology (EHT), OPS, Ministry of Higher Education, Science, Research and Innovation. JC received Chulabhorn Graduate Scholarship and the Royal Golden Jubilee (RGJ) Ph.D. scholarship (Grant no. PHD/0196/2561) through the National Research Council of Thailand (NRCT), and Thailand Research Fund (TRF).
Acknowledgments
The authors are grateful to Peter C. Dedon from Massachusetts Institute of Technology, United States for kindly providing the modified ribonucleotide standards and James Dubbs for kindly reviewing the manuscript. We also thank Kisana Bhinija and Chutima Thummawat for their technical assistances.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1079710/full#supplementary-material
Footnotes
References
Ahn, K. S., Ha, U., Jia, J., Wu, D., and Jin, S. (2004). The truA gene of Pseudomonas aeruginosa is required for the expression of type III secretory genes. Microbiology (Reading) 150, 539–547. doi: 10.1099/mic.0.26652-0
Alexeyev, M. F. (1999). The pKNOCK series of broad-host-range mobilizable suicide vectors for gene knockout and targeted DNA insertion into the chromosome of gram-negative bacteria. BioTechniques 26, 824–828. doi: 10.2144/99265bm05
Arora, S. K., Ritchings, B. W., Almira, E. C., Lory, S., and Ramphal, R. (1998). The Pseudomonas aeruginosa flagellar cap protein, FliD, is responsible for mucin adhesion. Infect. Immun. 66, 1000–1007. doi: 10.1128/IAI.66.3.1000-1007.1998
Bouteiller, M., Dupont, C., Bourigault, Y., Latour, X., Barbey, C., Konto-Ghiorghi, Y., et al. (2021). Pseudomonas flagella: generalities and specificities. Int. J. Mol. Sci. 22:3337. doi: 10.3390/ijms22073337
Brégeon, D., Colot, V., Radman, M., and Taddei, F. (2001). Translational misreading: a tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev. 15, 2295–2306. doi: 10.1101/gad.207701
Chan, C., Pham, P., Dedon, P. C., and Begley, T. J. (2018). Lifestyle modifications: coordinating the tRNA epitranscriptome with codon bias to adapt translation during stress responses. Genome Biol. 19:228. doi: 10.1186/s13059-018-1611-1
Charoenlap, N., Shen, Z., McBee, M. E., Muthupalani, S., Wogan, G. N., Fox, J. G., et al. (2012). Alkyl hydroperoxide reductase is required for helicobacter cinaedi intestinal colonization and survival under oxidative stress in BALB/c and BALB/c interleukin-10−/− mice. Infect. Immun. 80, 921–928. doi: 10.1128/IAI.05477-11
Cho, K. H., and Caparon, M. G. (2008). tRNA modification by GidA/MnmE is necessary for streptococcus pyogenes virulence: a new strategy to make live attenuated strains. Infect. Immun. 76, 3176–3186. doi: 10.1128/IAI.01721-07
Chomczynski, P., and Sacchi, N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159. doi: 10.1006/abio.1987.9999
Ciofu, O., and Tolker-Nielsen, T. (2019). Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents-how P. aeruginosa can escape antibiotics. Front. Microbiol. 10:913. doi: 10.3389/fmicb.2019.00913
Das, C., Mokashi, C., Mande, S. S., and Saini, S. (2018). Dynamics and control of flagella assembly in salmonella typhimurium. Front. Cell. Infect. Microbiol. 8:36. doi: 10.3389/fcimb.2018.00036
de Crecy-Lagard, V., and Jaroch, M. (2021). Functions of bacterial tRNA modifications: from ubiquity to diversity. bTrends Microbiol 29, 41–53. doi: 10.1016/j.tim.2020.06.010
Deretic, V., Schurr, M. J., Boucher, J. C., and Martin, D. W. (1994). Conversion of Pseudomonas aeruginosa to mucoidy in cystic fibrosis: environmental stress and regulation of bacterial virulence by alternative sigma factors. J. Bacteriol. 176, 2773–2780. doi: 10.1128/jb.176.10.2773-2780.1994
El Yacoubi, B., Bailly, M., and de Crecy-Lagard, V. (2012). Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 46, 69–95. doi: 10.1146/annurev-genet-110711-155641
Filloux, A., and Ramos, J. L. (eds.). (2014). Pseudomonas methods and protocols. London: Springer New York Heidelberg Dordrecht London.
Gao, T., Tan, M., Liu, W., Zhang, C., Zhang, T., Zheng, L., et al. (2016). GidA, a tRNA modification enzyme, contributes to the growth, and virulence of Streptococcus suis serotype 2. Front. Cell. Infect. Microbiol. 6:44. doi: 10.3389/fcimb.2016.00044
Gao, T., Yuan, F., Liu, Z., Liu, W., Zhou, D., Yang, K., et al. (2020). Proteomic and metabolomic analyses povide insights into the mechanism on arginine metabolism regulated by tRNA modification enzymes GidA and MnmE of Streptococcus suis. Front. Cell. Infect. Microbiol. 10:597408. doi: 10.3389/fcimb.2020.597408
Gardner, R. M., Allison, M. J., Hartman, P. A., Reinhardt, T. A., and Horst, R. L. (1987). Factors influencing production of 5(E)-19-nor-10-keto-vitamin D3 by rumen bacteria. J. Steroid Biochem. 28, 189–192. doi: 10.1016/0022-4731(87)90376-1
Gupta, R., Gobble, T. R., and Schuster, M. (2009). GidA posttranscriptionally regulates rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 191, 5785–5792. doi: 10.1128/JB.00335-09
Jaroensuk, J., Atichartpongkul, S., Chionh, Y. H., Wong, Y. H., Liew, C. W., McBee, M. E., et al. (2016). Methylation at position 32 of tRNA catalyzed by TrmJ alters oxidative stress response in Pseudomonas aeruginosa. Nucleic Acids Res. 44, 10834–10848. doi: 10.1093/nar/gkw870
Jin, Y., Zhang, T., Samaranayake, Y. H., Fang, H. H., Yip, H. K., and Samaranayake, L. P. (2005). The use of new probes and stains for improved assessment of cell viability and extracellular polymeric substances in Candida albicans biofilms. Mycopathologia 159, 353–360. doi: 10.1007/s11046-004-6987-7
Johansson, C., Samskog, J., Sundstrom, L., Wadensten, H., Bjorkesten, L., and Flensburg, J. (2006). Differential expression analysis of Escherichia coli proteins using a novel software for relative quantitation of LC-MS/MS data. Proteomics 6, 4475–4485. doi: 10.1002/pmic.200500921
Keiski, C. L., Harwich, M., Jain, S., Neculai, A. M., Yip, P., Robinson, H., et al. (2010). AlgK is a TPR-containing protein and the periplasmic component of a novel exopolysaccharide secretin. Structure 18, 265–273. doi: 10.1016/j.str.2009.11.015
Kinscherf, T. G., and Willis, D. K. (2002). Global regulation by gidA in pseudomonas syringae. J. Bacteriol. 184, 2281–2286. doi: 10.1128/JB.184.8.2281-2286.2002
Kirov, S. M. (2003). Bacteria that express lateral flagella enable dissection of the multifunctional roles of flagella in pathogenesis. FEMS Microbiol. Lett. 224, 151–159. doi: 10.1016/S0378-1097(03)00445-2
Knutson, C. A., and Jeanes, A. (1968). A new modification of the carbazole analysis: application to heteropolysaccharides. Anal. Biochem. 24, 470–481. doi: 10.1016/0003-2697(68)90154-1
Kovach, M. E., Elzer, P. H., Hill, D. S., Robertson, G. T., Farris, M. A., Roop, R. M. 2nd, et al. (1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176. doi: 10.1016/0378-1119(95)00584-1
Kuang, Z., Hao, Y., Walling, B. E., Jeffries, J. L., Ohman, D. E., and Lau, G. W. (2011). Pseudomonas aeruginosa elastase provides an escape from phagocytosis by degrading the pulmonary surfactant protein-a. PLoS One 6:e27091. doi: 10.1371/journal.pone.0027091
Lavoie, E. G., Wangdi, T., and Kazmierczak, B. I. (2011). Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect. 13, 1133–1145. doi: 10.1016/j.micinf.2011.07.011
Lowry, O. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275. doi: 10.1016/S0021-9258(19)52451-6
Metcalf, W. W., Jiang, W., Daniels, L. L., Kim, S. K., Haldimann, A., and Wanner, B. L. (1996). Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35, 1–13. doi: 10.1006/plas.1996.0001
Mi, H., Muruganujan, A., Ebert, D., Huang, X., and Thomas, P. D. (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. doi: 10.1093/nar/gky1038
Moradali, M. F., Ghods, S., and Rehm, B. H. (2017). Pseudomonas aeruginosa lifestyle: a paradigm for adaptation, survival, and persistence. Front. Cell. Infect. Microbiol. 7:39. doi: 10.3389/fcimb.2017.00039
Moukadiri, I., Garzon, M. J., Bjork, G. R., and Armengod, M. E. (2014). The output of the tRNA modification pathways controlled by the Escherichia coli MnmEG and MnmC enzymes depends on the growth conditions and the tRNA species. Nucleic Acids Res. 42, 2602–2623. doi: 10.1093/nar/gkt1228
Nathwani, D., Raman, G., Sulham, K., Gavaghan, M., and Menon, V. (2014). Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: a systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 3:32. doi: 10.1186/2047-2994-3-32
Overhage, J., Bains, M., Brazas, M. D., and Robert, H. E. W. (2008). Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J. Bacteriol. 190, 2671–2679. doi: 10.1128/JB.01659-07
Overhage, J., Lewenza, S., Marr, A. K., and Hancock, R. E. (2007). Identification of genes involved in swarming motility using a Pseudomonas aeruginosa PAO1 mini-Tn5-lux mutant library. J. Bacteriol. 189, 2164–2169. doi: 10.1128/JB.01623-06
Rehl, J. M., Shippy, D. C., Eakley, N. M., Brevik, M. D., Sand, J. M., Cook, M. E., et al. (2013). GidA expression in salmonella is modulated under certain environmental conditions. Curr. Microbiol. 67, 279–285. doi: 10.1007/s00284-013-0361-2
Romsang, A., Duang-nkern, J., Khemsom, K., Wongsaroj, L., Saninjuk, K., Fuangthong, M., et al. (2018). Pseudomonas aeruginosa ttcA encoding tRNA-thiolating protein requires an iron-sulfur cluster to participate in hydrogen peroxide-mediated stress protection and pathogenicity. Sci. Rep. 8:11882. doi: 10.1038/s41598-018-30368-y
Sha, J., Kozlova, E., Fadl, A., Olano, J., Houston, C., Peterson, J., et al. (2004). Molecular characterization of a glucose-inhibited division gene, gidA, that regulates cytotoxic enterotoxin of Aeromonas hydrophila. Infect. Immun. 72, 1084–1095. doi: 10.1128/IAI.72.2.1084-1095.2004
Shi, N., Gao, Y., Yin, D., Song, Y., Kang, J., Li, X., et al. (2019). The effect of the sub-minimal inhibitory concentration and the concentrations within resistant mutation window of ciprofloxacin on MIC, swimming motility and biofilm formation of Pseudomonas aeruginosa. Microb. Pathog. 137:103765. doi: 10.1016/j.micpath.2019.103765
Shippy, D. C., Eakley, N. M., Bochsler, P. N., Chopra, A. K., and Fadl, A. A. (2011). Biological and virulence characteristics of salmonella enterica serovar typhimurium following deletion of glucose-inhibited division (gidA) gene. Microb. Pathog. 50, 303–313. doi: 10.1016/j.micpath.2011.02.004
Shippy, D. C., and Fadl, A. A. (2014). tRNA modification enzymes GidA and MnmE: potential role in virulence of bacterial pathogens. Int. J. Mol. Sci. 15, 18267–18280. doi: 10.3390/ijms151018267
Tan, M.-W., Mahajan-Miklos, S., and Ausubel, F. M. (1999). Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 96, 715–720. doi: 10.1073/pnas.96.5.2408
Tatnell, P. J., Russell, N. J., and Gacesa, P. (1994). GDP-mannose dehydrogenase is the key regulatory enzyme in alginate biosynthesis in Pseudomonas aeruginosa: evidence from metabolite studies. Microbiology (Reading) 140, 1745–1754. doi: 10.1099/13500872-140-7-1745
Thongdee, N., Jaroensuk, J., Atichartpongkul, S., Chittrakanwong, J., Chooyoung, K., Srimahaeak, T., et al. (2019). TrmB, a tRNA m7G46 methyltransferase, plays a role in hydrogen peroxide resistance and positively modulates the translation of katA and katB mRNAs in Pseudomonas aeruginosa. Nucleic Acids Res. 47, 9271–9281. doi: 10.1093/nar/gkz702
Urbonavičius, J., Qian, Q., Durand, J. M., Hagervall, T. G., and Björk, G. R. (2001). Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 20, 4863–4873. doi: 10.1093/emboj/20.17.4863
Wiegand, I., Hilpert, K., and Hancock, R. E. (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175. doi: 10.1038/nprot.2007.521
World Health Organization (2017). WHO publishes list of bacteria for which new antibiotics are urgently needed [online]. Available at: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (Accessed April 21, 2020).
Xu, S., Peng, Z., Cui, B., Wang, T., Song, Y., Zhang, L., et al. (2014). FliS modulates FlgM activity by acting as a non-canonical chaperone to control late flagellar gene expression, motility and biofilm formation in Yersinia pseudotuberculosis. Environ. Microbiol. 16, 1090–1104. doi: 10.1111/1462-2920.12222
Yim, L., Moukadiri, I., Björk, G. R., and Armengod, M.-E. (2006). Further insights into the tRNA modification process controlled by proteins MnmE and GidA of Escherichia coli. Nucleic Acids Res. 34, 5892–5905. doi: 10.1093/nar/gkl752
Zamocky, M., Furtmuller, P. G., and Obinger, C. (2010). Evolution of structure and function of class I peroxidases. Arch. Biochem. Biophys. 500, 45–57. doi: 10.1016/j.abb.2010.03.024
Zhang, W., Zhao, Z., Zhang, B., Wu, X. G., Ren, Z. G., and Zhang, L. Q. (2014). Posttranscriptional regulation of 2,4-diacetylphloroglucinol production by GidA and TrmE in Pseudomonas fluorescens 2P24. Appl. Environ. Microbiol. 80, 3972–3981. doi: 10.1128/AEM.00455-14
Zhao, D., Wang, H., Li, Z., Han, S., Han, C., and Liu, A. (2022). LC_glucose-inhibited division protein is required for motility, biofilm formation, and stress response in Lysobacter capsici X2-3. Front. Microbiol. 13:840792. doi: 10.3389/fmicb.2022.840792
Keywords: Pseudomonas aeruginosa, tRNA modification, oxidative stress, gidA, U34 modification, catalases, virulence
Citation: Srimahaeak T, Thongdee N, Chittrakanwong J, Atichartpongkul S, Jaroensuk J, Phatinuwat K, Phaonakrop N, Jaresitthikunchai J, Roytrakul S, Mongkolsuk S and Fuangthong M (2023) Pseudomonas aeruginosa GidA modulates the expression of catalases at the posttranscriptional level and plays a role in virulence. Front. Microbiol. 13:1079710. doi: 10.3389/fmicb.2022.1079710
Edited by:
Mitsuo Ogura, Tokai University, JapanReviewed by:
Rodolfo García-Contreras, National Autonomous University of Mexico, MexicoLaurent Aussel, Aix-Marseille Université, France
Copyright © 2023 Srimahaeak, Thongdee, Chittrakanwong, Atichartpongkul, Jaroensuk, Phatinuwat, Phaonakrop, Jaresitthikunchai, Roytrakul, Mongkolsuk and Fuangthong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mayuree Fuangthong, ✉ bWF5dXJlZUBjcmkub3IudGg=
†Present addresses: Narumon Thongdee, Division of Molecular and Cellular Biology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
Juthamas Jaroensuk, School of Biomolecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong, Thailand
‡These authors have contributed equally to this work