 Miaomiao Luo1
Miaomiao Luo1 Yiying She1
Yiying She1 Yixiang Jiang2Li Xie3
Yixiang Jiang2Li Xie3 Chao Yang4Yaqun Qiu2Rui Cai2Yinghui Li2
Chao Yang4Yaqun Qiu2Rui Cai2Yinghui Li2 Liangcai Xu5Lulu Hu2
Liangcai Xu5Lulu Hu2 Lei Wang2Shuang Wu2Qiongcheng Chen2Xiaolu Shi2
Lei Wang2Shuang Wu2Qiongcheng Chen2Xiaolu Shi2 Min Jiang2*
Min Jiang2* Qinghua Hu1,2*
Qinghua Hu1,2*- 1School of Public Health, Shanxi Medical University, Taiyuan, China
- 2Shenzhen Center for Disease Control and Prevention, Shenzhen, China
- 3School of Public Health, University of South China, Hengyang, China
- 4The Center for Microbes, Development and Health, CAS Key Laboratory of Molecular Virology and Immunology, Institute Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, China
- 5Shenzhen Futian District Center for Disease Control and Prevention, Shenzhen, China
Salmonella enterica subsp. enterica serovar Derby (S. Derby) is one of the most common serotypes responsible for salmonellosis in humans and animals. The two main sequence types (ST) observed in China are ST40 and ST71, with ST40 presently being the most common in Shenzhen. Recent years have seen an increasing number of cases of salmonella caused by ST40 S. Derby, but the epidemiology is not clear. We gathered 314 ST40 S. Derby isolates from food and patient samples for 11 years in Shenzhen; 76 globally prevalent representative strains were also collected. Whole-genome sequencing (WGS) combined with drug resistance phenotyping was used to examine population structural changes, inter-host associations, drug resistance characteristics, and the food-transmission risks of ST40 S. Derby in Shenzhen over this period. The S. enterica evolutionary tree is divided into five clades, and the strains isolated in Shenzhen were primarily concentrated in Clades 2, 4, and 5, and thus more closely related to strains from Asian (Thailand and Vietnam) than European countries. Our 11-year surveillance of S. Derby in Shenzhen showed that Clades 2, 4, and 5 are now the dominant epidemic branches, and branches 2 and 5 are heavily multi-drug resistant. The main resistance pattern is ampicillin-tetracycline-ciprofloxacin-chloramphenicol-nalidixic acid-streptomycin-sulfamethoxazole/trimethoprim. This may lead to a trend of increasing resistance to ST40 S. Derby in Shenzhen. Using a segmentation of ≤3 SNP among clone clusters, we discovered that Clades 2 and 4 contained multiple clonal clusters of both human- and food-derived strains. The food-derived strains were mainly isolated from pig liver, suggesting this food has a high risk of causing disease outbreaks in Shenzhen.
Introduction
Non-typhoidal Salmonella (NTS) is a significant zoonotic food-borne pathogen and one of the most serious public health issues worldwide (Majowicz et al., 2010). NTS can result in a range of clinical presentations, most frequently appearing as very minor gastrointestinal symptoms; however, the infection can occasionally become life-threatening, especially in young children and older adult individuals (Ruiz et al., 2004). The global burden of NTS gastroenteritis is estimated to be 93.8 million cases and 155,000 deaths per year (Balasubramanian et al., 2019). Food of animal origin is a primary vector of human Salmonella infection and has been linked to outbreaks of human Salmonella enterica subsp. enterica serovar Derby (S. Derby), a serotype that primarily affects high-risk people and was first isolated by Peckham in 1923 from pork patties that caused food poisoning (Zheng et al., 2017). In Europe, S. Derby is the most common serotype isolated from pork. It accounts for 22.9% of all isolates, followed by S. 4,[5],12:i:- (i.e., the monophasic variant of S. typhimurium) (22.3%) and S. typhimurium (20.6%) (EFSA, 2016). In the United States, S. Derby is the fourth most common isolate of non-human origin (Nguyen et al., 2015), while in China, it is the most common isolate from the sera of slaughtered pigs and the third most common from sera of clinical cases (Deng et al., 2012). Hence, S. Derby is the most common Salmonella serotype found in many countries, including countries in Europe, America, and Asia, and has been linked to several food-borne disease outbreaks in recent years.
In 1946, S. Derby caused an epidemic in Australia that affected 68 infants and resulted in the deaths of 10 babies (Mushin, 1948). In the United States, an outbreak in 1963 associated with contaminated eggs involved 822 cases in 53 hospitals (Sanders et al., 1963). In Germany, an outbreak between late 2013 and early 2014 associated with pork contaminated with S. Derby involved 145 patients, the majority of whom were elderly (Simon et al., 2018). In addition to these well-known foodborne outbreaks, S. Derby is frequently implicated in causing widespread human cases not associated with any specific food. S. Derby is the fifth most common serovar isolated from humans in Europe and caused 612 confirmed cases in 2017. Cai et al. investigated the contamination of pork samples from slaughterhouses and farmers’ markets in Jiangsu Province, and S. Derby was the serovar most frequently isolated (Cai et al., 2016). In China, S. Derby is the third most commonly reported serovar in clinical cases (Ran et al., 2011) and the most frequent serovar in infants and toddlers (Cui et al., 2009).
Antibiotic resistance to Salmonella is one of the most important public health problems worldwide and has increased significantly over recent years due to the long-term use of antibiotics in animal production practices (Barza, 2002). Multidrug-resistant (MDR) Salmonella may pose a serious threat to humans through the food chain, potentially contributing to long-term illness, disability, and death (Valdezate et al., 2005). The U.S. Centers for Disease Control and Prevention (CDC) estimated that at least 2 million people in the United States are infected with drug-resistant bacteria each year, resulting in at least 23,000 deaths and posing a serious threat to human health (Hu et al., 2020). Salmonella antibiotic resistance has increased over the last 20 years (Zhang et al., 2006). This phenomenon is especially severe in China. Here, Salmonella isolates in the 1960s were not multi-drug resistant, but since the mid-1970s, when antibiotics in animal feed became popular, a large number of new drug-resistant strains have emerged (Lin et al., 2004).
According to the Salmonella multilocus sequence typing (MLST) database (Maiden et al., 2013), there are more than 20 different sequence types associated with S. Derby, and the prevalence of these varies among countries; for example, six different sequence types (ST39, ST40, ST71, ST678, ST682, and ST683) are associated with S. Derby in Denmark (Litrup et al., 2010), and there are five in Germany (ST39, ST40, ST71, ST682, and ST774), of which ST39 is the most prevalent (Hauser et al., 2011). In China, there are two main sequence types, ST40 and ST71, with ST40 being the most common (Li et al., 2016), ST40 is also the current most common ST in Shenzhen. ST classification is based on the number of different alleles present, but these different STs are not sufficient to describe the evolutionary relationship of affinities between different isolates. In recent years, rapid developments in technology have meant whole-genome sequencing (WGS) has become more convenient and versatile. In addition to predicting drug resistance genes, WGS data can also be used in species identification, serotype prediction, the screening of virulence genes, and the rapid tracing of disease outbreaks. WGS is gradually becoming the most important prevention and control tool for providing early warnings of infectious disease epidemics (You et al., 2022).
Salmonella infection is a major food safety concern, with S. Derby rated among the top 10 human-derived Salmonella serotypes according to data from the Food Safety Risk Surveillance of Shenzhen (FSS), infectious diarrhea pathogen spectrum sentinel surveillance (IDDS), and food poisoning outbreak surveillance (FDOS) in Shenzhen (Lin et al., 2019). Very little research on the genomics of S. Derby has been conducted in China to date, and often the source of illness remains unknown. We sequenced whole genomes of S. Derby isolates collected by IDDS, FDOS, and FSS in Shenzhen from 2011 to 2021 and compared them with those of representative global S. Derby isolates. Understanding changes in population structure, host associations, resistance characteristics, and the transmission risk of ST40 S. Derby in foods in Shenzhen can provide an important reference for subsequent salmonellosis preventive measures and infection source tracing, and the identification of high-risk foods.
Materials and methods
Strain sources
The Shenzhen CDC has established a functioning Foodborne Diseases surveillance network consisting of three systems: FDOS, IIDDS, and FSS. We sequenced all the S. Derby isolates archived between 2011 and 2021. No statistical methods were used to determine sample size, and there were no data excluded from the analyses. The experiments were not randomized (Yang et al., 2022). FSS isolated 129 Salmonella strains from food samples (mainly livestock meat and poultry meat). A sentinel surveillance by IDDS and FDOS collected 188 human samples from stool samples of outpatients with diarrhea. Because data collection is part of the infectious disease surveillance, individual informed consent was waived. All strains were isolated, purified, and cultured using a VITEK2- compact fully automatic microbial identifier (BioMérieux, France) and were identified as S. Derby serotype by glass-slide agglutination according to the White Kauffmann-Le Minor method (Grimont and Weill, 2007).
In total, 129 isolates were collected from food, including livestock meat (73.6%, 95/129), poultry meat (13.2%, 17/129), frozen food (4.7%, 6/129), aquatic products (3.9%, 5/129), ready-to-eat foods (3.1%, 4/129), and pastries (1.6%, 2/129). Salmonella-infected livestock meat mainly included pig liver (44.2%, 42/95) and pork (42.1%, 40/95), and the others were beef (7.4%, 7/95) and pig kidney (6.3%, 6/95; Supplementary Figure S1A). FSS could not continuously conduct sampling because of new coronavirus epidemic, resulting in no strains isolated in 2020, and only one strain isolated in 2021. However, human-derived strains were detected every year, these data collections are part of infectious disease surveillance and sample collection was less affected by new coronavirus outbreak (Supplementary Figure S1B).
WGS and genomic datasets
Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. WGS was performed by Tianjin Novozyme Bioinformatics (Tianjin, China). After passing the quality control assessment, a library was prepared using the NEBNextUltra DNA Library Prep Kit for Illumina (NEB, United States) with an average insert size of 350 bp for sequencing on the Illumina NovaSeq 6000 platform. The average read length was 150 bp, the minimum theoretical coverage was 100×, and an average of 1.2 Gb clean data were produced for each isolate. The data presented in the study are deposited in the National Center for Biotechnology Information (NCBI) sequence read archive (SRA) under BioProject: PRJNA883032.
A total of 390 genomes were analyzed in this study, including 314 newly sequenced S. Derby genomes from Shenzhen isolates and 76 S. Derby ST40 genomes from 12 countries worldwide obtained from the EnteroBase1 (Zhou et al., 2020). Single SRA accession numbers of all the strains and associated epidemiological data are listed in Supplementary Table S1.
Bioinformatics analysis
Genomic contig sequences were obtained by de novo sequence splicing of genomic data from each strain using Shovill (v. 1.0.4; Seemann, 2018). Raw data were subjected to quality control using Trimmomatic2 (v. 0.39; Bolger et al., 2014) to obtain valid data. Genome assembly quality was assessed using QUAST3 (Gurevich et al., 2013), and the mean N50 was 313,894 bp. Ab initio genome assembly was performed using SPAdes4 gene assembly software (v. 3.9.1; Bankevich et al., 2012). Strain 2014LSAL02547 (NCBI no. CP029486) was used as a reference strain for ST40 analysis (Sevellec et al., 2018). Mapping-based single-nucleotide polymorphism (SNP) typing was performed using Snippy5 (v. 4.3.6; Olawoye et al., 2020). Gubbins6 (Croucher et al., 2015) with default parameters was used for core genome de-recombination. The resulting SNP matrix of preserved sites was then used to construct a phylogeny tree with FastTree (v. 2.1.10; Price et al., 2009) software and the maximum likelihood method, which was embellished using ITOL7 (Letunic and Bork, 2021).
Analysis of antibiotic resistance genes, Salmonella pathogenicity islands, plasmid replicons, and multi-locus sequence typing
Resistance genes and chromosomal mutation regions in assembled contigs were identified using Resfinder (v 0.3.2; Zankari et al., 2012) with an identity threshold of 75% and coverage of 75%. Salmonella pathogenicity islands (SPIs) were detected using SPIfinder (v. 1.0; Roer et al., 2016) with default settings of coverage ≥75% and identity ≥75%. PlasmidFinder (v 0.2.0.1; Carattoli and Hasman, 2020) was applied to predict plasmid replicons with sequence identity ≥80% and coverage ≥80%.
Each MLST sequence type (MLST-ST) was obtained by scanning the sequences of seven house-keeping genes (aroC, dnaN, hemD, hisD, purE, sucA, and thrA) against PubMLST typing schemes using mlst.2 (Carattoli and Hasman, 2020).
Antibacterial drug susceptibility testing
The Gram-negative aerobic bacterial susceptibility panel (Shanghai Xingbai Biotechnology Co., Ltd.) was used according to the recommendations of Clinical and Laboratory Standards Institute document M100-S30, using Escherichia coli ATCC25922 as the quality control strain. The minimum inhibitory concentrations of 17 antibacterial drugs, ampicillin (AMP), ampicillin/sulbactam (AMS), tetracycline (TET), meropenem (MEM), polymyxin E (CT), ertapenem (ETP), ceftazidime/avibactam (CZA), tigecycline (TGC), cefotaxime (CTX), ceftazidime (CAZ), ciprofloxacin (CIP), azithromycin (AZI), chloramphenicol (CHL), nalidixic acid (NAL), streptomycin (STR), trimethoprim/sulfamethoxazole (SXT), and amikacin (AMK), were tested for S. Derby strains.
Results
Sequence type
Two different ST profiles were identified among the 317 studied genomes: ST40 [aroC (19), dnaN (20), hemD (3), hisD (20), purE (5), sucA (22), thrA (22)] and ST71 [aroC (39), dnaN (35), hemD (8), hisD (36), purE (29), sucA (9), and thrA (36)]. The most frequent profile in the collection was ST40 (n = 314 genomes), followed by ST71 (n = 3). All ST40 isolates were included in this study.
Phylogenetic analysis of Salmonella Derby
The genomes of local S. Derby ST40 samples from Shenzhen (n = 314) and 76 globally prevalent representative strains from France (n = 6), Germany (n = 4), Italy (n = 5), United Kingdom (n = 6), Poland (n = 17), Vietnam (n = 10), Thailand (n = 5), United States (n = 5), Australia (n = 3), Brazil (n = 1), and other Chinese provinces (n = 14) were analyzed based on SNP frequencies. A total of 4,963 core-SNP loci were detected. Chinese isolates were found to have significant diversity and could be divided into five main and several small branches, with distances between strains within each branch of <215 SNPs. Chinese isolates were mainly concentrated in Clades 2 (41.3%, 161/390), 5 (24%, 94/390), and 4 (17.9%, 70/390), with the remaining isolates concentrated in Clade 1 (0.8%, 3/390). Strains from Shenzhen, part of the Chinese inland city of Shandong, and other Asian countries (Thailand and Vietnam) were closely related. Strains from European countries were concentrated in an independent branch of Clade 3 and were distantly related to the Chinese strains (Figure 1). During the survey, Clades 2, 4, and 5 were dominant. Clade 5 appeared in 2013 and stabilized after 2016; both Clades 2 and 4 persisted during the 11 sampled years, but the proportion of the latter was variable, with multiple co-existing clonal clusters appearing in 2017 (Figure 2).
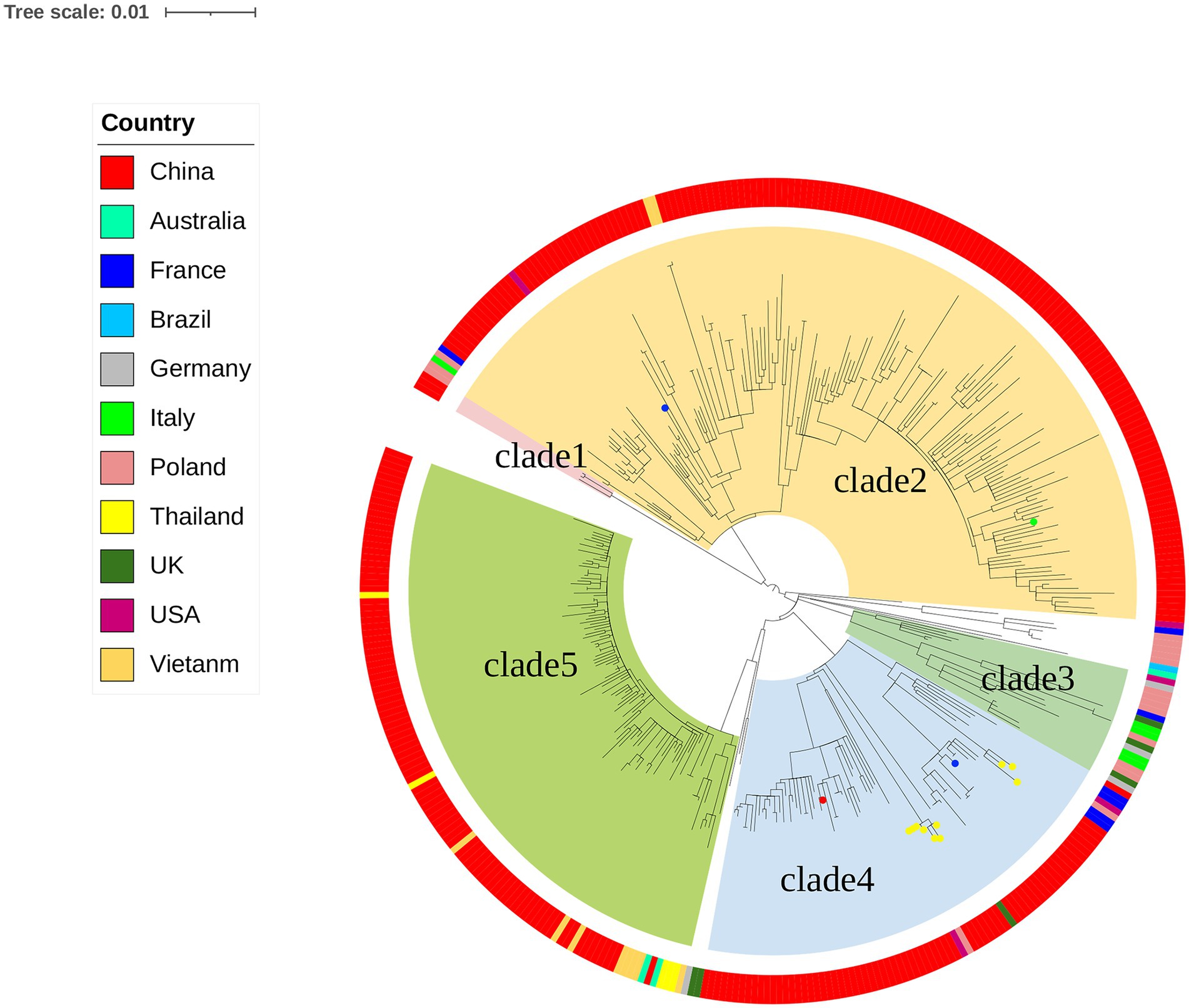
Figure 1. Maximum likelihood phylogenetic tree of 390 Salmonella strains. The background colors of the phylogenetic branches indicate the different evolutionary branches. Outer circles indicate different countries and the circles at the ends of the branches indicate other cities in China (blue, Shandong, China; green, Yangzhou, China; yellow, Taiwan, China; red, Tibet, China).
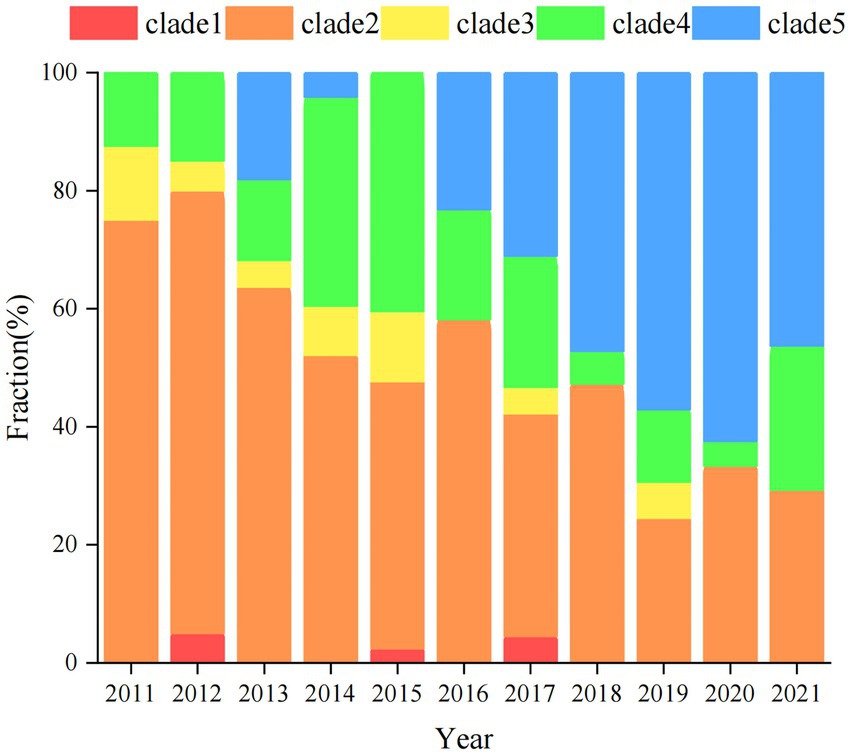
Figure 2. Distribution of S. Derby ST40 evolutionary branches globally. Different strip colors indicate different clades, clade1 (red), clade2 (orange), clade3 (yellow), clade4 (green), and clade5 (blue).
A total of 5,408 core-SNP sites were detected in the 314 strains isolated from Shenzhen. With reference to the cutoff set for S. enteritidis outbreak clonal clusters, we identified clonal clusters in the Shenzhen S. Derby ST40 maximum likelihood evolutionary tree, in which there were three SNPs across strains (Taylor et al., 2015; Jiang et al., 2020). There were 11, 4, and 2 clusters in Clades 2, 4, and 5, respectively (Figure 3). In an evolutionary tree constructed using ≤3 SNPs between two strains, 18 clusters involved 45 strains, with 2–4 isolates per cluster, differing by 0 ~ 3 SNPs; three clonal clusters included only human strains (C1, C2, and C3), three clusters had both human and food strains (C4, C7, and C9), and 12 clusters comprised only food-derived strains (C5, C6, C8, C10, C11, C12, C13, C14, C15, C16, C17, and C18; Figure 4). Among the 45 strains involved, 34 were food-derived and 11 were human-derived. Most of the 34 food-derived strains (13/34), and all food-derived strains on C4 and C7, were from pig liver. We also found that most of the strains in the same clusters came from the same market or supermarket and were isolated at similar times (Figure 4).
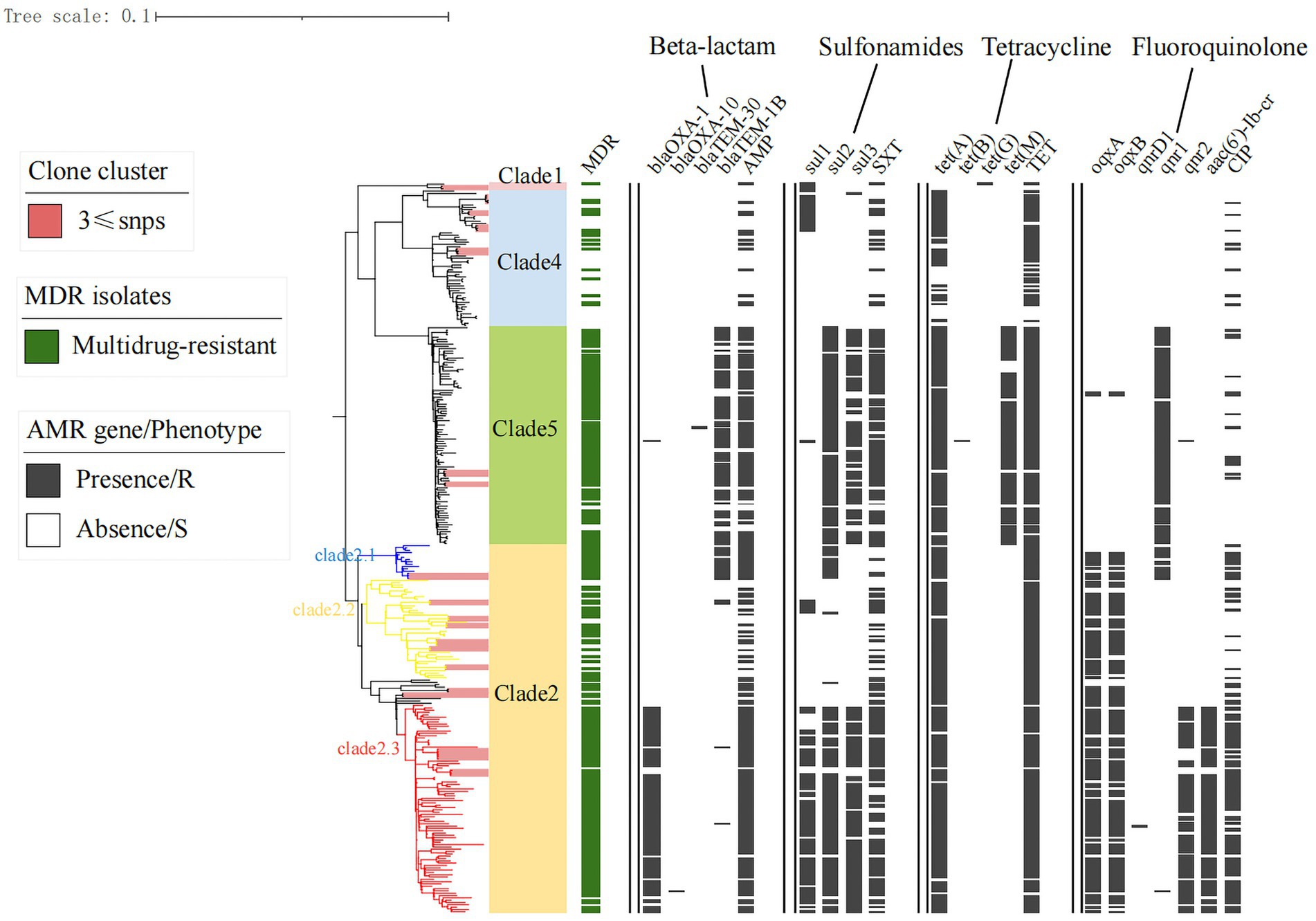
Figure 3. Antimicrobial resistance and gene distribution of Shenzhen strains. The maximum likelihood tree of 314 Shenzhen isolates is shown on the left, with the evolutionary branch of ST40 S. Derby in background color (as in Figure 1); on the right, the distribution of multidrug-resistant (MDR) isolates (green bars), representative isolates for phenotypic testing, and the presence (black bars) or absence (white bars) of antimicrobial resistance-associated mutations and genes.
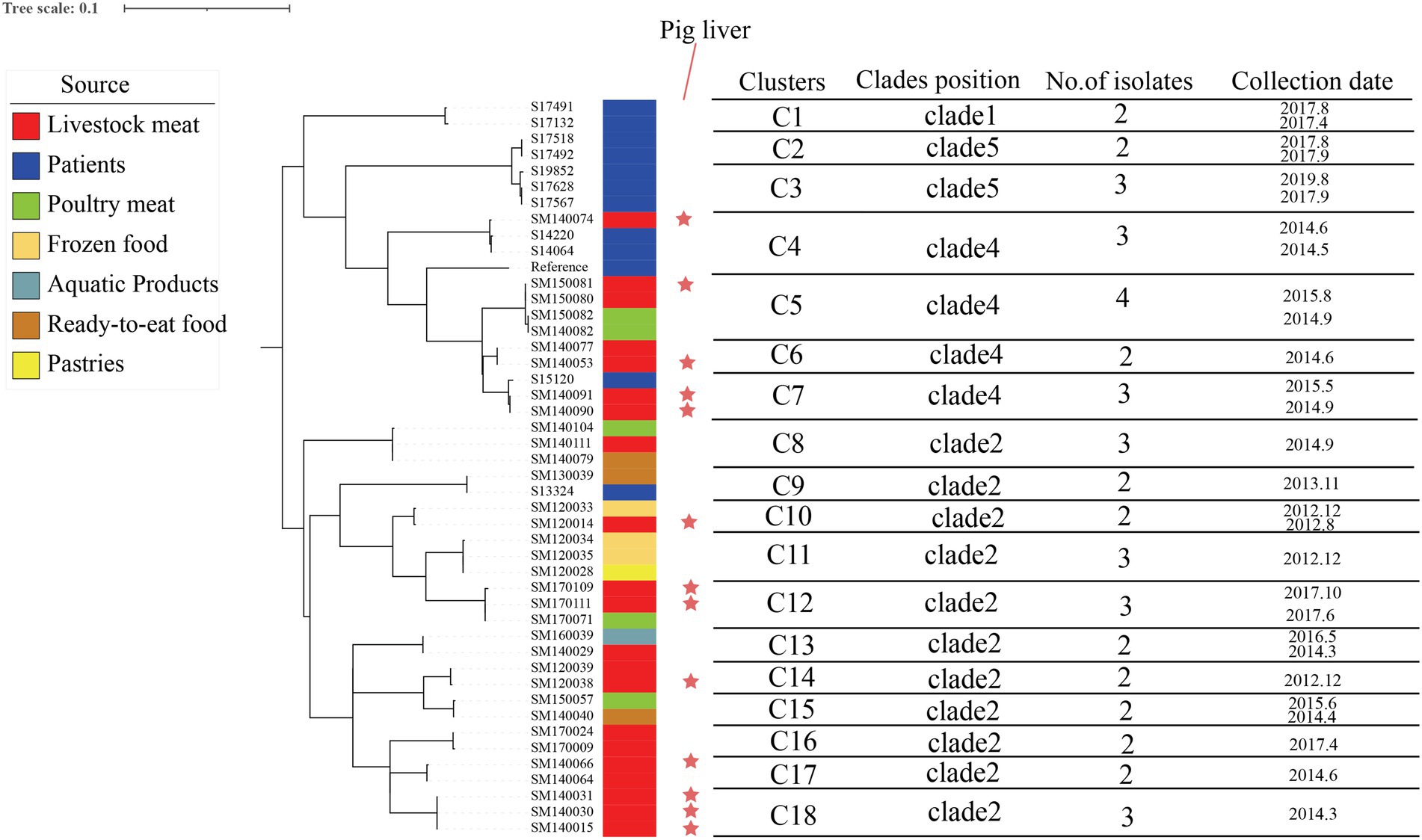
Figure 4. Maximum likelihood phylogenetic tree and strain information for 18 clonal clusters. The maximum likelihood tree of 18 clonal clusters is shown on the left; the color of the strip reports the origin of the strain: patients (blue), livestock meat (red), poultry meat (green), frozen food (earthy yellow), pastries (bright yellow), ready-to-eat food (brown), and aquatic products (lake blue); the red stars indicate that the strains were isolated from pig liver; and the contents of the line table contain the clades position, no. of isolates, and collection date.
Drug resistance genes, plasmid replicons, and virulence gene assays
To characterize the AMR profile of Shenzhen S. Derby isolates, we first screened the genome sequence data to identify AMR-associated mutations and genes. A total of 45 different AMR mutations/genes were detected in 11 classes, including those involved in resistance to aminoglycosides (13 genes), β-lactams (7 genes), sulfonamides (3 genes), TET (4 genes), fluoroquinolones (3 mutations and 6 genes), CHL (4 genes), methicillin (3 genes), macrolides (1 gene), polymyxins (1 gene), fosfomycin (1 gene), and rifampicin (2 genes). Aminoglycoside aac(6′)-Iaa and fosfomycin fosA7-resistance genes were detected in all strains. The quinolone-resistance genes mainly comprised oqxA/B (42%), aac(6′)-Ib-cr (25.6%), and qnrD1/S1/S2 (55.5%). The plasmid gene qnr was dominated by qnrS1. One and eight strains were detected carrying the gyrA p.S83F and p.D87N mutations, respectively. The blaTEM-1B allele was the most common β-lactam-resistance gene (28.7%), followed by blaOXA-1 (25.9%), which was also plasmid-mediated. Sulfonamide-, chloramphenicol-, and rifampicin-resistance genes were dominated by sul2 (58.4%), floR (53%), and arr-3 (25.2%), respectively.
Fifty-eight strains carrying 17 incompatibility group (Inc) plasmid replicons were detected among the 314 S. Derby ST40 strains; the three most common were IncI1_1 (8.28%, 26/314), IncHI2_1 (7.32%, 23/314), and IncHI2A_1 (7.00%, 22/314), with each of the 58 strains carrying 1–5 plasmid replicons. All strains carried six pathogenicity islands (SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, and SPI-9) simultaneously, and one strain carried both SPI-13 and SPI-14 (Table 1).
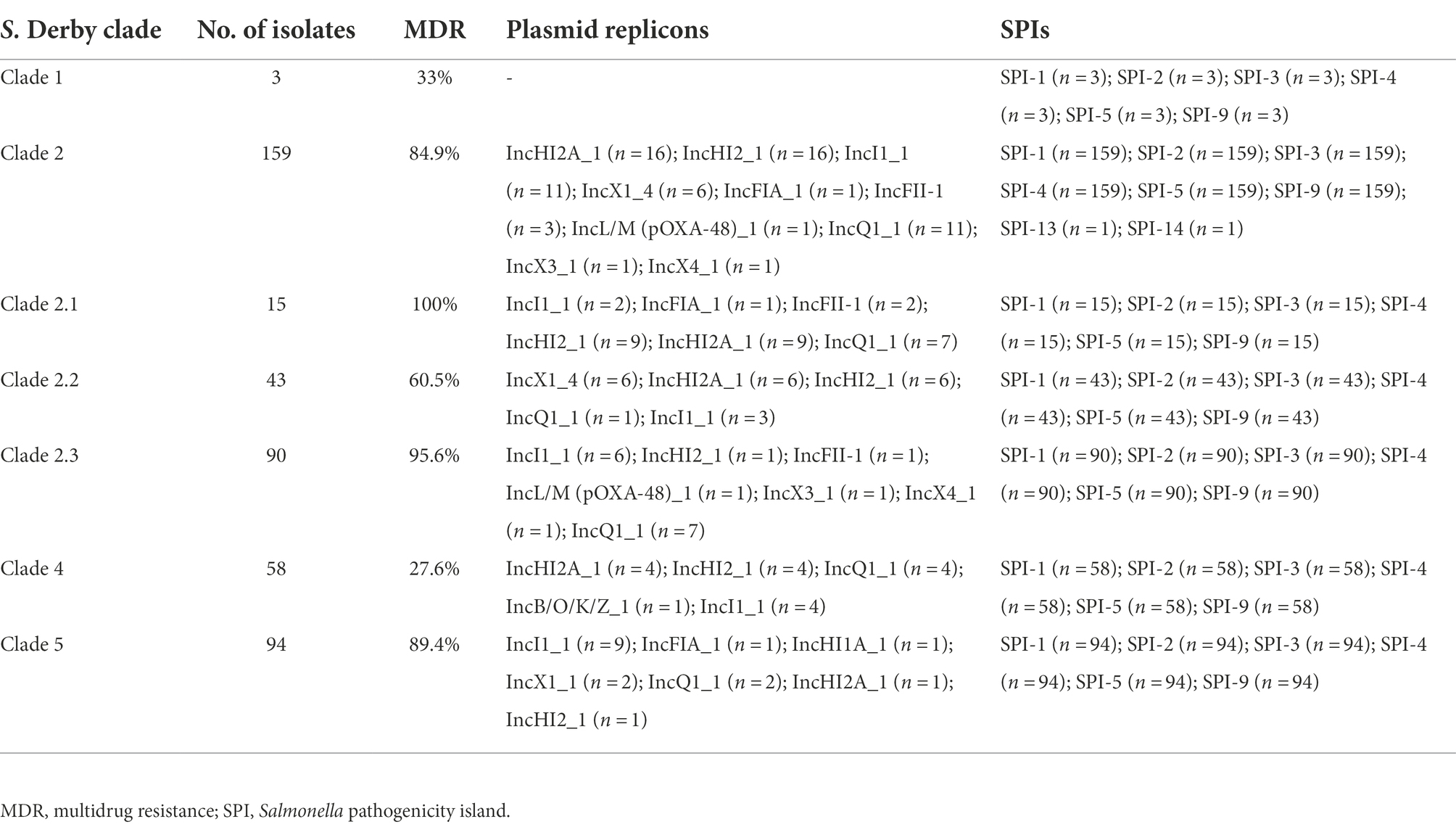
Table 1. Results of plasmid replicon and virulence gene analysis of Salmonella Derby ST40 strains (n = 314).
Antimicrobial susceptibility
The resistance genes found in Salmonella in food and patient samples from the 314 strains isolated in Shenzhen were generally concordant with phenotype testing results. The 314 strains of S. Derby ST40 were resistant to 16 antibiotics, not including TGC, to varying degrees (0.3–90.54%; Table 2). TET resistance was closely related to the presence of tet(A), with 274 (95.5%) of 287 TET-resistant strains encoding tet(A); while resistance to TGC, another antibiotic in the tetracycline class, was not detected. Resistance to β-lactams, including AMP (68.5%, 215/314), AMS (14%, 44/314), CZA (0.3%, 1/314), CAZ (1.3%, 4/314), and CTX (1.6%, 5/314), was detected, of which 79.5% (171/215) encoded at least one type of β-lactamase-associated gene, with blaOXA-1 (38.1%, 82/215) and blaTEM-1B (42.3%, 91/215) being the most common. In addition, resistance to the sulfonamide antibiotics SXT (62.4%, 196/314) and CHL (75.4%, 237/314) was detected; 83.2% (163/196) of strains resistant to sulfonamide antibiotics encoded at least one sulfonamide-resistance gene, and 73.4% of chloramphenicol-resistant strains contained floR (70.5%, 167/237) and cmlA1 (60.8%, 144/237) genes. Of the two azithromycin-resistant strains, one carried the mph(A) resistance gene. Six strains were resistant to the quinolone antibiotic NAL (42%, 132/314) and carried the gyrA p.S83F or p.D87N mutations, while four CIP-resistant strains (40%, 126/314) carried the gyrA p.D87N mutation. Resistance to CTX, AMK, CZA, ertapenem, and meropenem was detected in one strain each. By comparison, we found that both food- and human-derived strains were severely resistant to AMP, TET, and AZI, while food-derived strains were more resistant to CTX, CZA, and CHL than human-derived strains (Supplementary Table S2).
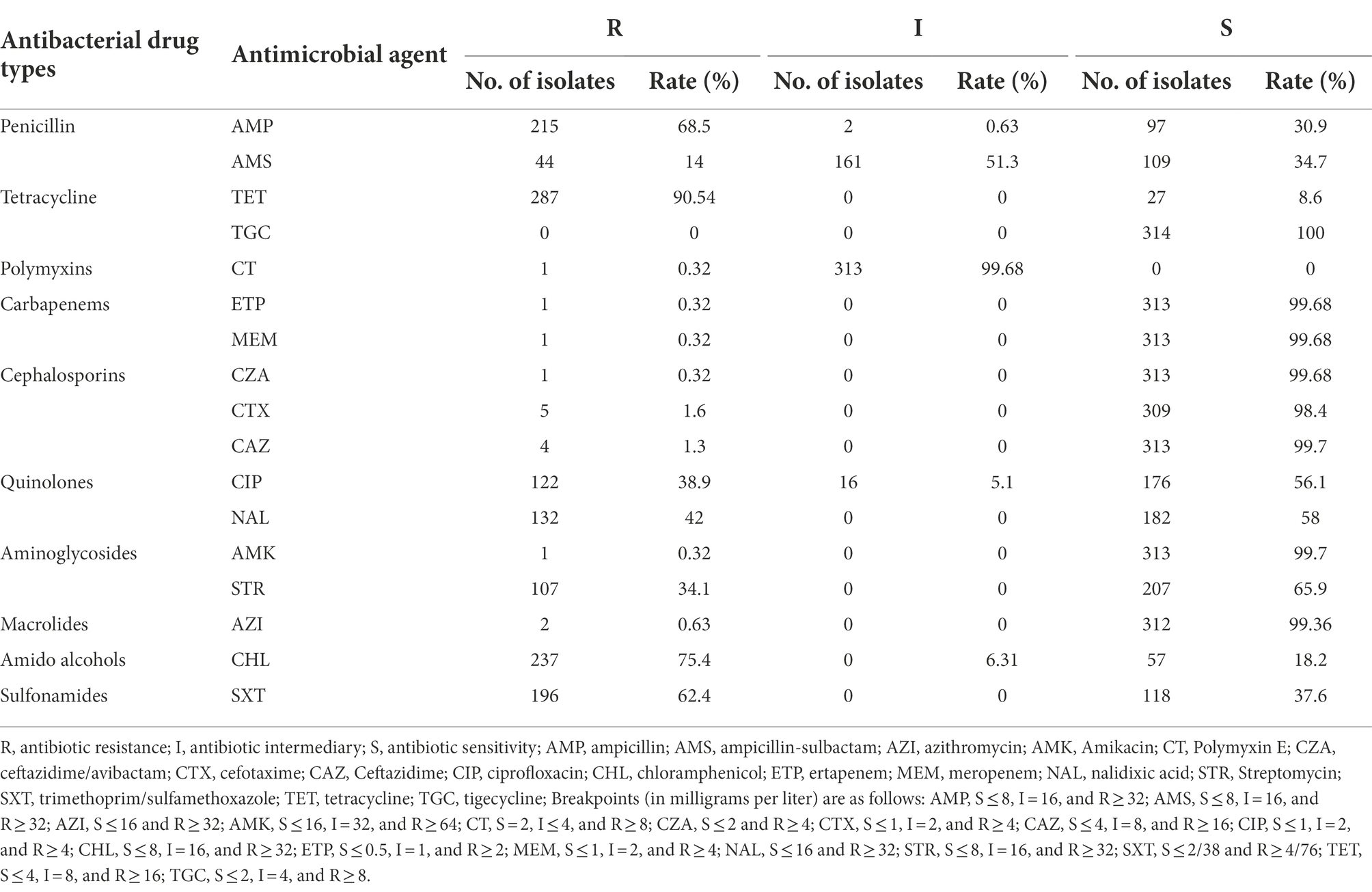
Table 2. Drug-resistant phenotypes of S. Derby ST40 strains (n = 314).
Notably, unlike the plasmids and virulence factors, the drug-resistance genes/phenotypes were associated with evolutionary branches to some extent. There were higher rates of β-lactam, sulfonamide, tetracycline, and quinolone-related resistance genes/phenotypes in Clade 2.1, 2.3, and 5 strains than those of other evolutionary branches (Figure 3). MDR strains were defined as those resistant to three or more antibiotics; 6% (18/314) of strains were susceptible to all 17 antimicrobial drugs, and almost 75% (236/314) exhibited MDR, with the most common MDR pattern being AMP-TET-CIP-CHL-NAL-STR-SXT (Supplementary Table S3).
Discussion
Our WGS-based reconstruction of the population structure of Shenzhen S. Derby ST40 isolates in the context of the global epidemic revealed the genomic diversity of the bacteria and the associations among various hosts in the Shenzhen region. This is the first study of the genomic epidemiology and drug resistance characteristics of S. Derby ST40 in China, and it was conducted over an extended period of time. Here, we traced the high-risk food and pig liver, and identified two evolutionary branches. Some of those strains clustered closely together indicating a potential foodborne outbreak with pig liver as vehicle. Isolates from Shenzhen showed simultaneous susceptibility to cephalosporins while being severely resistant to TET, CHL, and AMP. S. Derby is a significant serotype, but little is known about its genetic diversity in China. Our data expand on the publicly available sequence data and information on the genetic diversity of S. Derby in Shenzhen. At the same time, this study had some limitations. Because of the impact of the new coronavirus epidemic, FSS sampling was not continuous, resulting in a discontinuous collection of food-derived strains in the last 2 years.
Shenzhen is a developed metropolis in the south of China with a population of over 20 million. It has little arable land and no farms for raising poultry or livestock; therefore, the majority of food is imported from other Chinese cities and other countries. According to the global phylogenetic evolutionary tree constructed in this study, Shenzhen S. Derby strains are closely related to those from the inland Chinese city of Shandong as well as other Asian nations, including Thailand and Vietnam, while strains in European nations are primarily concentrated in an independent branch of Clade 3 and are more distantly related to Chinese strains. However, fewer prevalent representative strains from other regions have been uploaded to the public database, and more representative strains need to be collected to construct a global evolutionary tree for a more comprehensive picture of their prevalence. The ubiquity of S. Derby ST40 in Shenzhen for 11 years has resulted in significant diversity, and strains in all four evolutionary branches were present, indicating that the serotype has undergone microevolution over the 11-year epidemic. In recent years, outbreaks in the area have been dominated by Clades 2, 4, and 5, which may be connected to their higher levels of adaptability and MDR. The MDR rate of Clade 2 (especially Clades 2.1 and 2.3) and Clade 5 was 80% or more (Table 1).
Pigs have the potential to spread Salmonella infection during group feeding. S. Derby can also be found in the area where the pig carcasses are divided. According to U.S. CDC data, S. Derby is most frequently isolated from pig production units. S. Derby can cause long-term infections in pigs and can remain in several organs for a long time, which explains why Salmonella is easily isolated from pig organ parts, and this leads to contaminated pork at slaughter (Cevallos-Almeida et al., 2019). In our study, we identified 18 clonal clusters based on a threshold of three SNPs for the definition of S. enteritidis outbreaks (Taylor et al., 2015; Jiang et al., 2020), and 15 of the 34 strains in food were isolated from pig liver. Three clonal clusters (C4, C7, and C9) were detected in both patients and food, and the food-derived strains of two of the clonal clusters (C4 and C7) were isolated from pig liver and were in Clade 4 (Figure 4). We surmised that pig liver poses a high risk for foodborne illness outbreaks. We also discovered that the majority of foods containing bacteria in the same clonal clusters originated from the same market or supermarket and that there were various food types with comparable collection times in the same clusters. Furthermore, our market research found that different meats are sold on the same counter at supermarkets or markets and that the same cutting board and knives are used for most meats, indicating that there may be cross-contamination during food processing and selling. Our results suggest that WGS and clonal cluster analysis can be used for the identification of high-risk food types, pointing to new avenues for subsequent outbreak prevention and control strategies. These actions are also consistent with a positive One Health vision: pathogens supplied by various stakeholders from many sources (including human clinical samples, animal, food, and environmental samples) can be pooled and studied for various purposes across various analytical platforms (Timme et al., 2020), which provides a large amount of data that can be used by public health agencies for outbreak detection and tracking (Marc et al., 2016).
Plasmids are circular DNA molecules that can replicate independently from the bacterial chromosome and carry genetic material such as virulence and drug-resistance genes. Through horizontal gene transfer, mobile plasmids can spread drug resistance and virulence rapidly among bacteria of the same or other species, increasing the challenge of treating clinical infectious illnesses (Liu et al., 2009). The propagation of β-lactamase- and quinolone-resistance genes is directly related to the detection of both IncI1- and IncHI2-type plasmids (Sukmawinata et al., 2020). SPIs are located on chromosomes and code virulence-associated proteins that help Salmonella to invade, reproduce, and spread within its complex environment propagation. There are 23 known SPIs, of which SPI-1 to SPI-5 are shared by all S. enterica serovars, while the others are scattered among other serotypes. In our analysis, all strains had six pathogenicity islands (SPI1 to SPI5 and SPI9), while SPI13 and SPI14 were recognized in only one strain. SPI-9 is strongly related to biofilm development (Latasa et al., 2005), as it encodes proteins that share sequence similarity with members of the Bap family. In Staphylococcus aureus, Bap is a cell wall protein that strongly encourages biofilm development and has the potential to stimulate the emergence of drug-resistant strains (Cucarella et al., 2001). Prior research found SPI-13 and SPI-14 only in S. typhimurium and S. enteritidis (Shah et al., 2005), and the current study was the first to identify these islands in S. Derby. However, their mechanisms of action are still unclear and need further study.
Clarification of the antibiotic susceptibility profile of S. Derby will inform researchers on how to develop more effective clinical treatments. Unsurprisingly, strains from food sources had higher levels of resistance to antibiotics, such as TET, AMP, CHL, sulfonamides, and NAL, than those from human sources, as these drugs are frequently included in animal feed to treat illness or boost growth. Because of worries over the emergence of antibiotic resistance and the transmission of antibiotic-resistance genes from animals to people, EU countries began to outlaw the use of antibiotics as growth promoters in 2006 (Castanon, 2007). Recently, numerous other nations have documented significant S. Derby drug resistances to these widely used medications. In Sichuan and Guangzhou, China, TET is frequently used in feed for poultry livestock, and the food-derived strains identified in this study were primarily isolated from livestock meat. Because people in China consume a lot of pork, there is a chance that the increasing antibiotic resistance in farmed animals will spread to humans through the food chain and result in the failure of clinical antibiotics. NAL is a first-generation quinolone that bacteria have quickly developed resistance to. We discovered that the rate of NAL resistance of bacteria in food (67.4%) in China was significantly greater than that in Europe (10%; Jong et al., 2009). We concluded that to prevent the further emergence and spread of antibiotic resistance and to ensure food safety in China, intervention measures must be developed to manage food sources and restrict the use of antibiotics in animal husbandry.
Finally, this study reconstructed the WGS-based population structure of S. Derby ST40 in Shenzhen in the context of globally endemic representative strains. The kinship evolutionary tree formed five evolutionary branches, revealing that S. Derby ST40 strains in this region have closer affinities with endemic strains in other Asian countries than those in European countries. By constructing an evolutionary tree of relatedness, we discovered multiple epidemic clonal turnovers in the region over the 11 years of sampling. Notably, this study identified three currently prevalent evolutionary branches with high-resistance and high-transmission risk, Clades 2, 4, and 5, and we identified a high-risk food, pig liver. This information has important implications for salmonellosis prevention, source tracing, and risk-factor analysis, as well as laying the groundwork for future S. Derby studies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
ML, MJ, and QH conceived and designed the study. YQ, CY, LH, LW, SW, and XS performed article data collection. YS, ML, and LX experiments and data analysis were conducted. ML wrote the original draft. CY, LX, and YJ conducted raw letter analysis guidance. ML, CY, and QH reviewed and revised the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Sanming Project of Medicine in Shenzhen under grant (no. SZSM201811071); Shenzhen Key Medical Discipline Construction Fund (SZXK064); Key scientific and technological project of Shenzhen Science and Technology Innovation Committee (KCXFZ202002011006190); and Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2020-PT330-006).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1065672/full#supplementary-material
Footnotes
1. ^http://enterobase.warwick.ac.uk
2. ^https://github.com/timflutre/trimmomatic
3. ^https://github.com/ablab/quast
4. ^https://github.com/ablab/spades
5. ^https://github.com/ablab/snippy
References
Balasubramanian, R., Im, J., Lee, J. S., Jeon, H. J., Mogeni, O. D., Kim, J. H., et al. (2019). The global burden and epidemiology of invasive non-typhoidal Salmonella infections. Hum. Vaccin. Immunother. 15, 1421–1426. doi: 10.1080/21645515.2018.1504717
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barza, M. (2002). Potential mechanisms of increased disease in humans from antimicrobial resistance in food animals. Clin. Infect. Dis. 34, S123–S125. doi: 10.1086/340249
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cai, Y., Tao, J., Jiao, Y., Fei, X., Zhou, L., Wang, Y., et al. (2016). Phenotypic characteristics and genotypic correlation between Salmonella isolates from a slaughterhouse and retail markets in Yangzhou, China. Int. J. Food Microbiol. 222, 56–64. doi: 10.1016/j.ijfoodmicro.2016.01.020
Carattoli, A., and Hasman, H. (2020). PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). Methods Mol. Biol. 2075, 285–294. doi: 10.1007/978-1-4939-9877-7_20
Castanon, J. I. (2007). History of the use of antibiotic as growth promoters in European poultry feeds. Poult. Sci. 86, 2466–2471. doi: 10.3382/ps.2007-00249
Cevallos-Almeida, M., Martin, L., Houdayer, C., Rose, V., Guionnet, J. M., Paboeuf, F., et al. (2019). Experimental infection of pigs by Salmonella derby, S. typhimurium and monophasic variant of S. typhimurium: comparison of colonization and serology. Vet. Microbiol. 231, 147–153. doi: 10.1016/j.vetmic.2019.03.003
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Cucarella, C., Solano, C., Valle, J., Amorena, B., Lasa, I., and Penades, J. R. (2001). Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J. Bacteriol. 183, 2888–2896. doi: 10.1128/JB.183.9.2888-2896.2001
Cui, S., Li, J., Sun, Z., Hu, C., Jin, S., Li, F., et al. (2009). Characterization of Salmonella enterica isolates from infants and toddlers in Wuhan, China. J. Antimicrob. Chemother. 63, 87–94. doi: 10.1093/jac/dkn452
Deng, X., Ran, L., Wu, S., Ke, B., He, D., Yang, X., et al. (2012). Laboratory-based surveillance of non-typhoidal Salmonella infections in Guangdong Province, China. Foodborne Pathog. Dis. 9, 305–312. doi: 10.1089/fpd.2011.1008
EFSA (2016). The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2016 15, 5077. doi: 10.2903/j.efsa.2017.5077
Grimont, P., and Weill, F. X. (2007). Antigenic Formulae of the Salmonella Serovars. 9th Edn. Paris: WHO Collaborating Center for Reference and Research on Salmonella, Institut Pasteur
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hauser, E., Hebner, F., Tietze, E., Helmuth, R., Junker, E., Prager, R., et al. (2011). Diversity of Salmonella enterica serovar Derby isolated from pig, pork and humans in Germany. Int. J. Food Microbiol. 151, 141–149. doi: 10.1016/j.ijfoodmicro.2011.08.020
Hu, L., Cao, G., Brown, E. W., Allard, M. W., Ma, L. M., Khan, A. A., et al. (2020). Antimicrobial resistance and related gene analysis of Salmonella from egg and chicken sources by whole-genome sequencing. Poult. Sci. 99, 7076–7083. doi: 10.1016/j.psj.2020.10.011
Jiang, M., Zhu, F., Yang, C., Deng, Y., Kwan, P. S. L., Li, Y., et al. (2020). Whole-genome analysis of Salmonella enterica Serovar Enteritidis isolates in outbreak linked to online food delivery, Shenzhen, China, 2018. Emerg. Infect. Dis. 26, 789–792. doi: 10.3201/eid2604.191446
Jong, D. A., Bywater, R., Butty, P., Deroover, E., Godinho, K., Klein, U., et al. (2009). A pan-European survey of antimicrobial susceptibility towards human-use antimicrobial drugs among zoonotic and commensal enteric bacteria isolated from healthy food-producing animals. J. Antimicrob. Chemother. 63, 733–744. doi: 10.1093/jac/dkp012
Latasa, C., Roux, A., Toledo-Arana, A., Ghigo, J. M., Gamazo, C., Penadés, J. R., et al. (2005). Bap a, a large secreted protein required for biofilm formation and host colonization of Salmonella enterica serovar Enteritidis. Mol. Microbiol. 58, 1322–1339. doi: 10.1111/j.1365-2958.2005.04907.x
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, Y. C., Cai, Y. Q., Tao, J., Kang, X. L., Jiao, Y., Guo, R. X., et al. (2016). Salmonella isolated from the slaughterhouses and correlation with pork contamination in free market. Food Control 59, 591–600. doi: 10.1016/j.foodcont.2015.06.040
Lin, H. S., Cheng, H. C., Chishih, C., and Jonathan, T. O. (2004). Antimicrobial resistance in nontyphodial Salmonella serotypes: a global challenge. Clin. Infect. Dis. 39, 546–551. doi: 10.1086/422726
Lin, A. H., Xia, J. J., Liang, C. N., He, L. H., and Chen, M. L. (2019). Serotyping and drug resistance analysis of foodborne Salmonella in Shenzhen from 2011-2017. Pract. Prev. Med. 26, 495–496. doi: 10.3969/j.issn.1006-3110.2019.04.032
Litrup, E., Torpdahl, M., Malorny, B., Huehn, S., Christensen, H., and Nielsen, E. M. (2010). Association between phylogeny, virulence potential and serovars of Salmonella enterica. Infect. Genet. Evol. 10, 1132–1139. doi: 10.1016/j.meegid.2010.07.015
Liu, W., Chen, L., Li, H., Duan, H., Zhang, Y., Liang, X., et al. (2009). Novel CTX-M {beta}-lactamase genotype distribution and spread into multiple species of Enterobacteriaceae in Changsha, southern China. J. Antimicrob. Chemother. 63, 895–900. doi: 10.1093/jac/dkp068
Maiden, M. C., Jansen van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Majowicz, S. E., Musto, J., Scallan, E., Angulo, F. J., Kirk, M., Obrien, S. J., et al. (2010). The global burden of nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. 50, 882–889. doi: 10.1086/650733
Marc, W. A., Errol, S., David, M., Kelly, B., Steven, M. M., Eric, W. B., et al. (2016). Practical value of food pathogen traceability through building a whole-genome sequencing network and database. J. Clin. Microbiol. 54, 1975–1983. doi: 10.1128/JCM.00081-16
Mushin, R. (1948). An outbreak of gastro-enteritis due to Salmonella derby. J Hyg (Lond) 46, 151–157. doi: 10.1017/s0022172400036238
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Olawoye, I. B., Frost, S. D. W., and Happi, C. T. (2020). The bacteria genome pipeline (BAGEP): an automated, scalable workflow for bacteria genomes with Snakemake. PeerJ 8:e10121. doi: 10.7717/peerj.10121
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Ran, L., Wu, S., Gao, Y., Zhang, X., Feng, Z., Wang, Z., et al. (2011). Laboratory-based surveillance of nontyphoidal Salmonella infections in China. Foodborne Pathog. Dis. 8, 921–927. doi: 10.1089/fpd.2010.0827
Roer, L., Hendriksen, R. S., Leekitcharoenphon, P., Lukjancenko, O., Kaas, R. S., Hasman, H., et al. (2016). Is the evolution of Salmonella enterica subsp. enterica linked to restriction-modification systems? mSystems 1:e00009-16. doi: 10.1128/mSystems.00009-16
Ruiz, M., Rodríguez, J. C., Escribano, I., and Royo, G. (2004). Available options in the management of non-typhi Salmonella. Expert. Opin. Pharmacother. 5, 1737–1743. doi: 10.1517/14656566.5.8.1737
Sanders, E., Sweeney, F. J., Friedman, E. A., Boring, J. R., Randall, E. L., and Polk, L. D. (1963). An outbreak of hospital-associated infections due to Salmonella Derby. JAMA 186, 984–986. doi: 10.1001/jama.1963.03710110036007
Seemann, T. (2018). Shovill GitHub. Assemble Bacterial Isolate Genomes from Illumina Paired-End Reads. Available at: https://github.com/tseemann/shovill
Sevellec, Y., Granier, S. A., Radomski, N., Felten, A., Le Hello, S., Feurer, C., et al. (2018). Complete genome sequence of Salmonella enterica subsp. enterica serotype Derby, associated with the pork sector in France. Microbiol Resour Announc 7:e01027–18. doi: 10.1128/MRA.01027-18
Shah, D. H., Lee, M. J., Park, J. H., Lee, J. H., Eo, S. K., Kwon, J. T., et al. (2005). Identification of Salmonella gallinarum virulence genes in a chicken infection model using PCR-based signature-tagged mutagenesis. Microbiology (Reading) 151, 3957–3968. doi: 10.1099/mic.0.28126-0
Simon, S., Trost, E., Bender, J., Fuchs, S., Malorny, B., Rabsch, W., et al. (2018). Evaluation of WGS based approaches for investigating a food-borne outbreak caused by Salmonella enterica serovar Derby in Germany. Food Microbiol. 71, 46–54. doi: 10.1016/j.fm.2017.08.017
Sukmawinata, E., Uemura, R., Sato, W., Mitoma, S., Kanda, T., and Sueyoshi, M. (2020). IncI1 plasmid associated with blaCTX-M-2 transmission in ESBL-producing Escherichia coli isolated from healthy thoroughbred racehorse, Japan. Antibiotics (Basel) 9:70. doi: 10.3390/antibiotics9020070
Taylor, A. J., Lappi, V., Wolfgang, W. J., Lapierre, P., Palumbo, M. J., Medus, C., et al. (2015). Characterization of foodborne outbreaks of Salmonella enterica serovar enteritidis with whole-genome sequencing single nucleotide polymorphism-based analysis for surveillance and outbreak detection. J. Clin. Microbiol. 53, 3334–3340. doi: 10.1128/JCM.01280-15
Timme, R. E., Wolfgang, W. J., Balkey, M., Venkata, S. L. G., Randolph, R., Allard, M., et al. (2020). Optimizing open data to support one health: best practices to ensure interoperability of genomic data from bacterial pathogens. One Health Outlook 2:20. doi: 10.1186/s42522-020-00026-3
Valdezate, S., Vidal, A., Herrera- León, S., Pozo, J., Rubio, P., Usera, M. A., et al. (2005). Salmonella Derby clonal spread from pork. Emerg. Infect. Dis. 11, 694–698. doi: 10.3201/eid1105.041042
Yang, C., Li, Y., Jiang, M., Wang, L., Jiang, Y., Hu, L., et al. (2022). Outbreak dynamics of foodborne pathogen Vibrio parahaemolyticus over a seventeen year period implies hidden reservoirs. Nat. Microbiol. 7, 1221–1229. doi: 10.1038/s41564-022-01182-0
You, X. Y., Peng, S. L., Houde, Z., Yang, L., Daofeng, L., Hu, K., et al. (2022). Genome-wide sequencing analysis of clinical isolates of non-typhoid Salmonella resistance studies in Jiangxi Province in 2018 32, 5.
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, R., Eggleston, K., Rotimi, V., and Zeckhauser, R. J. (2006). Antibiotic resistance as a global threat: evidence from China, Kuwait and the United States. Glob. Health 2:6. doi: 10.1186/1744-8603-2-6
Zheng, H., Hu, Y., Li, Q., Tao, J., Cai, Y., Wang, Y., et al. (2017). Subtyping Salmonella enterica serovar Derby with multilocus sequence typing (MLST) and clustered regularly interspaced short palindromic repeats (CRISPRs). Food Control 73, 474–484. doi: 10.1016/j.foodcont.2016.08.051
Keywords: Salmonella Derby, phylogenetic analysis, foodborne illness, virulence factor, whole-genome sequencing, antimicrobial resistance
Citation: Luo M, She Y, Jiang Y, Xie L, Yang C, Qiu Y, Cai R, Li Y, Xu L, Hu L, Wang L, Wu S, Chen Q, Shi X, Jiang M and Hu Q (2022) Population dynamics and antimicrobial resistance of Salmonella Derby ST40 from Shenzhen, China. Front. Microbiol. 13:1065672. doi: 10.3389/fmicb.2022.1065672
Edited by:
Frank T. Robb, University of Maryland, United StatesReviewed by:
Abdelaziz Ed-Dra, Sultan Moulay Slimane University, MoroccoBurkhard Malorny, Bundesanstalt für Risikobewertung (BfR), Germany
Copyright © 2022 Luo, She, Jiang, Xie, Yang, Qiu, Cai, Li, Xu, Hu, Wang, Wu, Chen, Shi, Jiang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinghua Hu, aHVxaW5naHVhMDNAMTYzLmNvbQ==; Min Jiang, amlhbmdtaW5fMTE1QDE2My5jb20=