 Shunshi Ji
Shunshi Ji Zexuan Song
Zexuan Song Lijuan Luo
Lijuan Luo Yiqian Wang1
Yiqian Wang1 Changyun Ye
Changyun Ye Yan Wang
Yan Wang- 1State Key Laboratory of Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Chinese Center for Disease Control and Prevention, Beijing, China
- 2School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW, Australia
Introduction: Listeria monocytogenes is a foodborne bacterium that could persist in food and food processing environments for a long time. Understanding the population structure and genomic characterization of foodborne L. monocytogenes is essential for the prevention and control of listeriosis.
Methods: A total of 322 foodborne L. monocytogenes isolates from 13 geographical locations and four food sources in China between 2000 and 2018 were selected for whole-genome sequencing.
Results: In silico subtyping divided the 322 isolates into five serogroups, 35 sequence types (STs), 26 clonal complexes (CCs) and four lineages. Serogroup IIa was the most prevalent serogroup and ST9 was the most prevalent ST of foodborne L. monocytogenes strains isolated in China. The in-depth phylogenetic analysis on CC9 revealed that ST122 clone might be original from ST9 clone. Furthermore, 23 potentially relevant clusters were identified by pair-wised whole-genome single nucleotide polymorphism analysis, indicating that persistent- and/or cross-contamination had occurred in markets in China. ST8 and ST121 were the second and third top STs of L. monocytogenes in China, which had heterogeneity with that of L. monocytogenes isolates from other countries. The antibiotic resistance genes aacA4, tetM, tetS, dfrG carried by different mobile elements were found in L. monocytogenes strains. One lineage II strain carrying Listeria Pathogenicity Island 3 was first reported. In addition, a novel type of premature stop codon in inlA gene was identified in this study.
Discussion: These findings revealed the genomic characteristics and evolutionary relationship of foodborne L. monocytogenes in China on a scale larger than previous studies, which further confirmed that whole-genome sequencing analysis would be a helpful tool for routine surveillance and source-tracing investigation.
Introduction
Listeria monocytogenes, as an important foodborne pathogen, is the causative agent of human listeriosis with clinical symptoms ranging from self-limiting gastroenteritis to severe invasive infections (Swaminathan and Gerner-Smidt, 2007). It has a significant impact on public health around the world due to high mortality rate in hospitalized patients (Schlech III, 2019). Immunocompromised individuals, pregnant women, neonates and the elderly are often at high risk for the aggressive forms of listeriosis (Wilking et al., 2021). L. monocytogenes is widely distributed in nature and more commonly found in a variety of food products and food-associated environments (Wang et al., 2019). This bacterium can contaminate food at any time from production to consumption. It can also shed into the environment, colonize and multiply, and then cause persistent contamination and/or cross-contamination. A number of research have shown that epidemic and sporadic listeriosis were often associated with consumption of L. monocytogenes contaminated foods (McLauchlin et al., 2021a,b). Food contamination caused by L. monocytogenes poses a serious threat to food safety and public health worldwide.
L. monocytogenes has evolved into four lineages, lineage I to lineage IV. Lineage I, including serotypes 1/2b, 3b, 4b, 4d, 4e, and 7 occurs frequently among human cases of listeriosis. While lineage II including serotypes 1/2a, 3a, 1/2c, and 3c is overrepresented in food products and food-associated environments. Lineage III and IV, corresponding serotypes 4a, 4c, and 4ab, are rare and commonly isolated from animal sources (Leclercq et al., 2011). Doumith et al. developed a multiplex PCR method to classify L. monocytogenes strains into five serogroups by five marker genes (lmo0737, lmo1118, ORF2819, ORF2100 and prs), namely serogroups IIa (serotypes 1/2a and 3a), IIb (serotypes 1/2b, 3b, and 7), IIc (serotypes 1/2c and 3c), IVb (serotypes 4b, 4d, and 4e) and L (4a, 4ab, and 4c; Doumith et al., 2004, 2005). Multi-locus sequence typing (MLST) remains a practical and portable method to meet the data exchange demands of different laboratories around the world (Salcedo et al., 2003). MLST can divide L. monocytogenes isolates into large number of sequence types (STs), which can further be grouped into serval clonal complexes (CCs). A CC of L. monocytogenes is defined as a group of STs, where each ST shares six of seven identical alleles with one other ST in the group (Salcedo et al., 2003). CC1, CC2, CC4, and CC6 were hypervirulent clones and strongly associated with listeriosis outbreaks. In contrast, CC9 and CC121 were hypovirulent clones and overrepresented in food sources (Maury et al., 2019).
However, traditional molecular subtyping methods, such as MLST and pulsed-field gel electrophoresis (PFGE), have insufficient discriminative power for source-trace investigations of L. monocytogenes contamination (Moura et al., 2016). The main reason for this is the conserved genomic backbone and the limited resolution of the technologies used (Moura et al., 2016). Whole-genome sequencing (WGS) has been widely used in the investigation of listeriosis outbreaks for a decade, for example, it facilitated to identify ST6 L. monocytogenes as the causative agent of the listeriosis outbreak in South Africa and guided the traceback investigations (Thomas et al., 2020). Furthermore, WGS analysis is of great significance for both biological and epidemiological studies on L. monocytogenes, through which we can obtain more details of molecular characteristics and refine the genetic relationship among them. L. monocytogenes genome-based typing and population biology study at the national level is lacking in China, although related projects have been carried out in part of local regions. A 10-year surveillance of L. monocytogenes in Shanghai found CC87 isolates persist in different food groups (Zhang et al., 2020). Another study found L. monocytogenes isolated from pork in Wuhan differed by few SNPs indicated a common supply source contamination (Wang et al., 2021). However, there are no reports analyzed isolates from the whole country or multiple provinces in China. Here, we sequenced 322 L. monocytogenes isolates from 13 different provinces/cities across China to determine (i) the population structure and diversity of L. monocytogenes from food in China based on whole-genome sequencing, (ii) the distribution of the predominant clones in the global context of corresponding clones, and (iii) the genetic characteristics, including virulence factors, stress survival genes and antibiotic resistance genes, of food-source L. monocytogenes in China.
Materials and methods
Selection of Listeria monocytogenes isolates in this study and public genomes for comparison
A total of 322 foodborne L. monocytogenes isolates from 12 provinces and one municipality in China between 2000 and 2018 were chosen in this study. These strains were isolated from four types of food including raw meat (n = 107), raw poultry (n = 81), ready-to-eat (RTE) food (n = 44), and aquatic products (n = 31). There were also 59 isolates recorded as unknown food sources. Details of each isolate were listed in Supplementary Table S1.
To examine the relationship between the isolates of the prevalent subtypes (ST9, ST8, ST121, and ST87) in China and the isolates from other countries, a total of 310 genomes available from GenBank were selected for comparative analysis. These genomes included 79 isolates from China and 231 isolates from 17 other countries, and the detailed information was listed in Supplementary Table S2.
DNA extraction and whole-genome sequencing
All L. monocytogenes isolates were streaked on Brain Heart Infusion agar plates and incubated at 37°C for 18 h. Genomic DNA was extracted using Wizard® Genomic DNA Purification Kit (Promega, United States) according to the instructions of the manufacturer for Gram-positive bacteria. DNA quality and concentration were determined using NanoDrop Spectrophotometer (Thermo Scientific, United States). WGS was performed on Illumina HiSeq X PE150 platform by Novegene (Beijing, China), with a coverage rate of more than 100-fold. Low-quality reads, ambiguous sequences and adapter sequences were filtered using FastQC. Then, SOAP de novo v2.04 was used to assemble genome sequence. Whole genome annotation was performed using Prokka pipeline v1.14.6.
In silico subtyping and phylogenetic analysis
Serogroup identification and MLST analysis were performed in silico on BIGSdb-Lm platform.1 Snippy pipeline v4.6.0 was used to generate core genome SNPs by mapping genome sequences with the reference genome sequence EGD-e (NZ_CP023861.1). The recombination was removed by Gubbins pipeline, whole-genome phylogeny of strains was inferred by FastTree. Then the core-SNPs phylogenetic tree was visualized and edited by online software iTol2 and Phandango3 (Hadfield et al., 2018; Letunic and Bork, 2021). Roary v3.13.0 was used to define the pan genome of strains using the outputs of Prokka annotation. Pan-genome were classified according to the principles of previously reported (Page et al., 2015). The phylogenetic tree for pan-genome was visualized using online software Phandango.
Identification of genes for stress resistance, virulence factors and antibiotic resistance
According to BIGSdb-Lm database, BLASTN algorithm was used to detect genes encoding stress resistance, virulence factors and antibiotic resistance, with the minimum of 80% coverage and 80% identity (Camargo et al., 2019).
Results
Characteristics of whole genome sequencing
The high-quality reads of each isolate with an average coverage above 100X were assembled to draft genome sequences. The genome assemblies contained seven to 48 scaffolds with the sizes ranging from 2.8 Mbp to 3.2 Mbp and the GC content about 37.87%. There were 2,748 to 3,243 protein-coding sequences predicted and annotated in the genomes in this study (Supplementary Table S3).
Serogroup identification and MLST in silico
Five serogroups were identified among 322 L. monocytogenes isolates based on five genes (lmo0737, lmo1118, ORF2110, ORF2819, and prs), including serogroup IIa (n = 131, 40.68%), IIb (n = 68, 21.12%), IIc (n = 80, 24.84%), IVb (n = 24, 7.45%), and L (n = 19, 5.90%). Serogroup distributions of L. monocytogenes isolates were different in four food types (Figure 1A). Serogroups IIa, IIb, IIc and IVb accounted for a similar proportion of isolates from RTE food. Serogroup IIa and IIc isolates were predominant in raw meat with the frequency of 38.31 and 37.38%, respectively. Serogroup IIa isolates were overrepresented in raw poultry with a frequency of 56.78%. For aquatic products, serogroup L was more common with a frequency of 32.26% than other four serogroups.
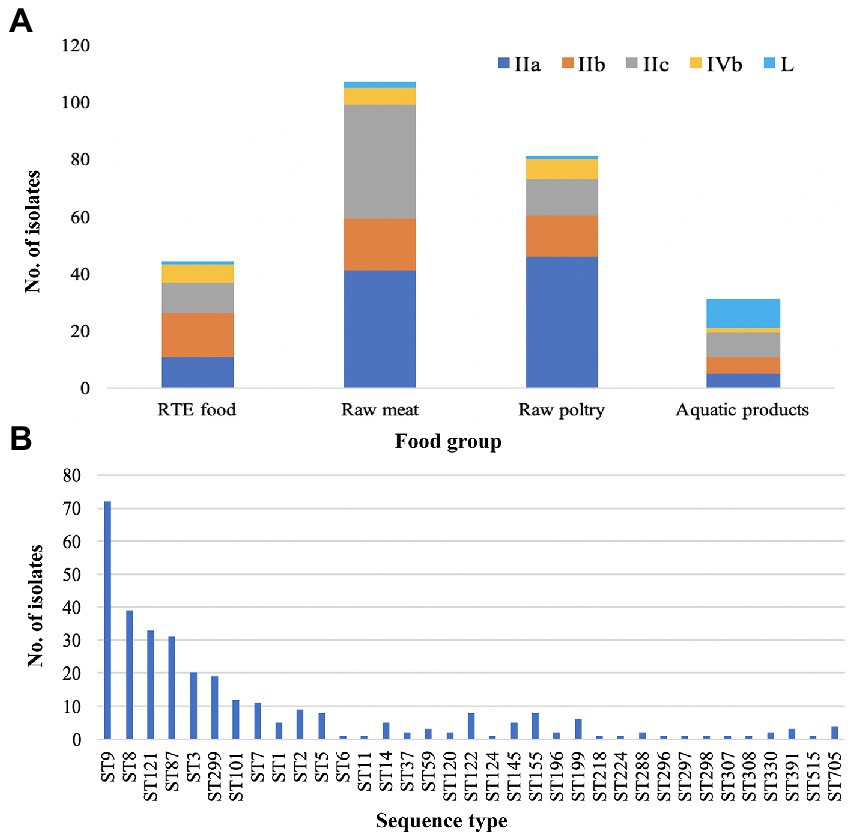
Figure 1. Distribution of Listeria monocytogenes. (A) Distribution of five serogroups (IIa, IIb, IIc, IVb and L) in four main food groups. (B) Distribution of sequence types (STs) in the 322 L. monocytogenes isolates.
Based on seven-genes MLST (Salcedo et al., 2003), all the studied isolates were classified into 35 STs which were further grouped into 26 CCs. ST9 was the predominant ST (n = 72, 22.36%), followed by ST8 (n = 39, 12.11%), ST121 (n = 33, 10.25%), ST87 (n = 31, 9.63%), ST3 (n = 20, 6.21%), ST299 (n = 19, 5.90%), ST101 (n = 12, 3.73%), and ST7 (n = 11, 3.42%). The remaining 27 STs contained less than 10 isolates for each (Figure 1B). Overall, 28.57, 65.53 and 5.90% of isolates were assigned to lineage I, lineage II and lineage III, respectively.
Core-SNP phylogenetic analysis
A maximum-likelihood phylogenetic tree was constructed based on the alignment of 16,383 core-SNPs that were identified by Snippy (Figure 2). Three clusters corresponding three lineages were displayed. In addition, the phylogenetic tree also showed that the genomes clustered based on CCs. Notably, all ST122 isolates belong to a sub-clade located within the clade of ST9, not next to the clade of ST9 (Supplementary Figure S1A). To clarify the refine relationship of ST9 and ST122, the phylogenetic analysis was performed based on the SNPs among genomes of the two STs isolates in this study and four ST122 genomes from public database (Accession numbers: GCA_002114825.1, GCA_002114845.1, GCA_002250305.1, and GCA_009664775.1; Supplementary Figure S1B). Another tree was constructed based on the binary presence and absence of accessory genes when the analysis of pan-genome was performed using Roary (Supplementary Figure S1C). Both the trees showed that a subset of ST9 isolates were phylogenetically closer to ST122 isolates than the other ST9 isolates, in other words, these ST122 isolates had a recent common ancestor with a subset of ST9 isolates.
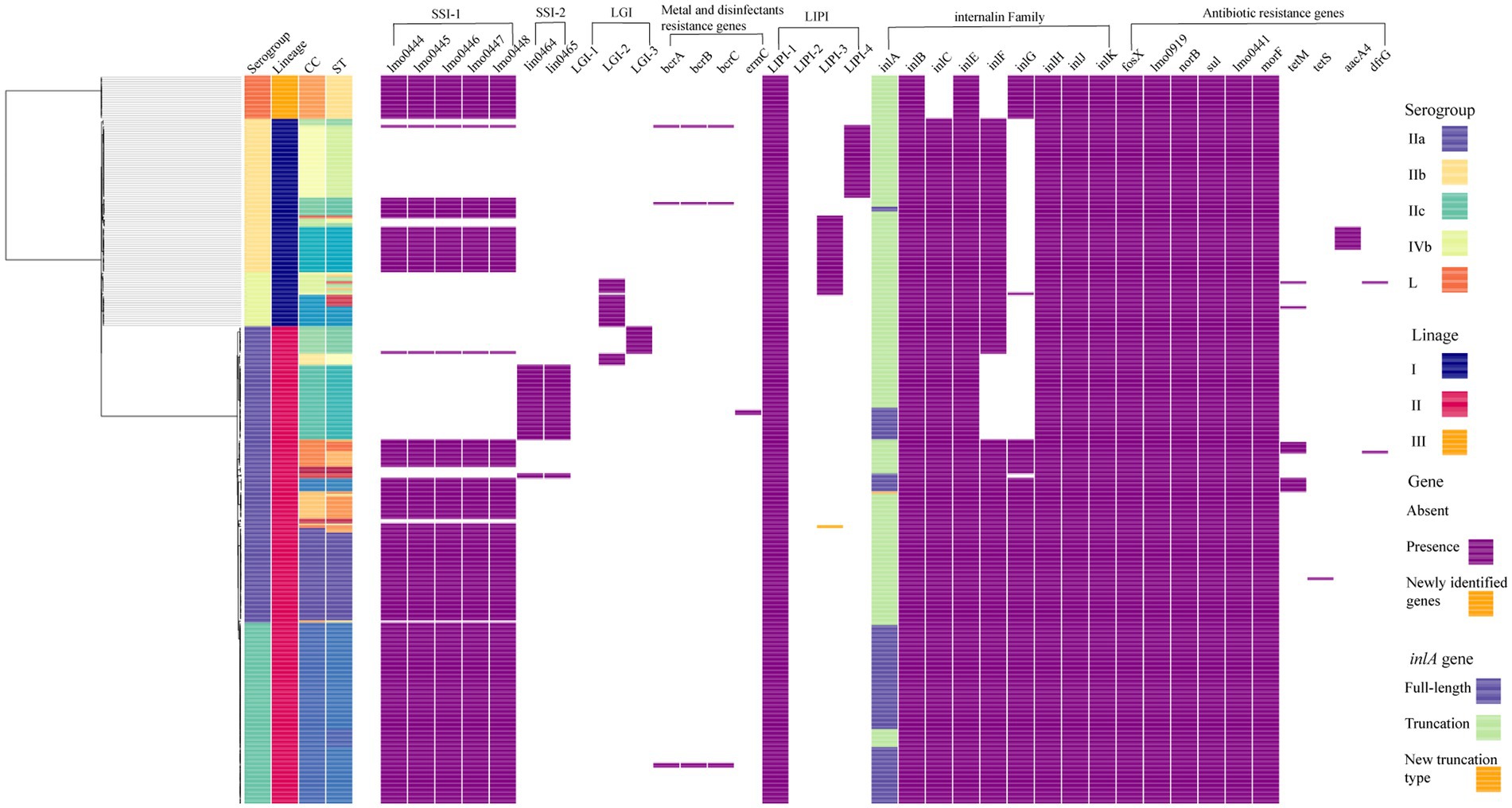
Figure 2. The core-SNP phylogenetic tree and genomic characterization of 322 foodborne L. monocytogenes isolates from China. Maximum likelihood phylogeny of 322 L. monocytogenes genomes based on 16,383 core-SNPs with EGD-e as the reference. Information of serogroup (dark purple boxes represent serogroup IIa, light orange boxes represent serogroup IIb, blue-green boxes represent serogroup IIc, light green boxes represent serogroup IVb, dark orange boxes represent serogroup L), lineage (dark blue boxes represent lineage I, rose red boxes represent lineage II, orange boxes represent lineage III), clonal complex and sequence type (they were clustered by different colors) are provided on the right. The distribution of different genes in L. monocytogenes was visualized using Phandango. The presence of a gene was marked with a purple box. The newly identified genes were marked with an orange box. For inlA gene, light green boxes represent full-length inlA gene, dark blue boxes represent truncated inlA gene.
Pair-wised wgSNP analysis of closely related isolates
Based on the above phylogenetic analysis, there were 87 core-SNP profiles represented by more than one isolates. To zoom in the differences among isolates within each profile, pair-wised SNP analysis was performed by Snippy separately. The numbers of SNP differences among isolates were minimal suggesting potential epidemiological links (Blanc et al., 2020). Twenty-three potentially relevant clusters were ≤ 12 pair-wised SNPs (Zhang et al., 2021; Table 1). These potential related isolates assigned into 10 STs with the top three STs of ST9, ST8, and ST299. Among these possible contamination events, eight clusters (Cluster 1, 3, 4, 7, 9, 10, 21, and 22) were caused by the isolates from different sources (market, farm or restaurant) in the same province in the same year, six clusters (Cluster 5, 6, 13, 17,19, and 23) were caused by the isolates from different food products from the same market in the same year, five clusters (Cluster 2, 8, 11, 12, and 14) were caused by the isolates from different sources in the same province in different years, two clusters (Cluster 15 and 20) were caused by the isolates from the same market in different years and two clusters (Cluster 16 and 18) were caused by the isolates from different provinces in different years.
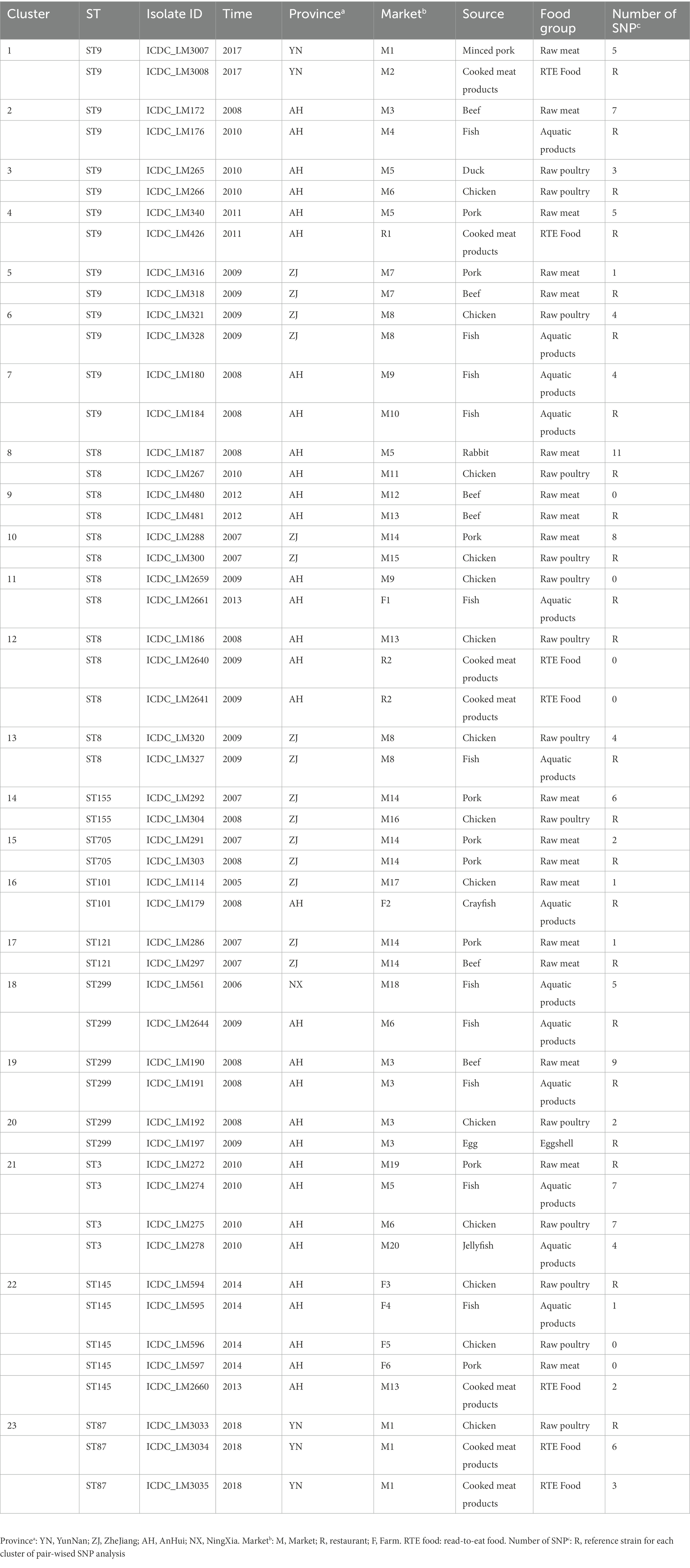
Table 1. Strains used for pair-wised wgSNP analysis.
The distribution of the prevalent subtypes of Listeria monocytogenes isolates from food in China in the context of global isolates
To determine the relationship between the top four prevalent STs from China (ST9, ST8, ST121, and ST87) and those from other countries, a total of 310 publically available genomes of L. monocytogenes were selected for comparative analysis. Here we also selected some isolates of each four ST from human cases in China to diversify the sources of isolates. For each ST, phylogenetic trees were separately constructed based on the alignment of the core-SNPs of isolates (Figure 3). The ST9 L. monocytogenes from China containing two isolates from clinical cases formed different clusters and were distributed across the phylogenetic tree of the global isolates (Figure 3A). The ST9 isolates in China were isolated from different regions and isolation years were clustered on the same branches, no epidemiological specific clustering was observed. For ST8, with exception of three outliers (one food-original and two clinical isolates), the ST8 isolates in China were grouped into two clusters (Figure 3B), and no evident temporal and spatial specificity were observed for the isolates of each cluster. For ST121, almost all isolates in China were distributed in the two out of three clusters, with exception of one food-original isolate that belonged to an international cluster. While 83.33% isolates of ST121 from other countries were grouped into the remaining cluster that was distinct from the clusters of strains isolated in China (Figure 3C). For ST87, isolates of ST87 from China and other countries were mixed with no epidemiological specific clusters observed (Figure 3D).
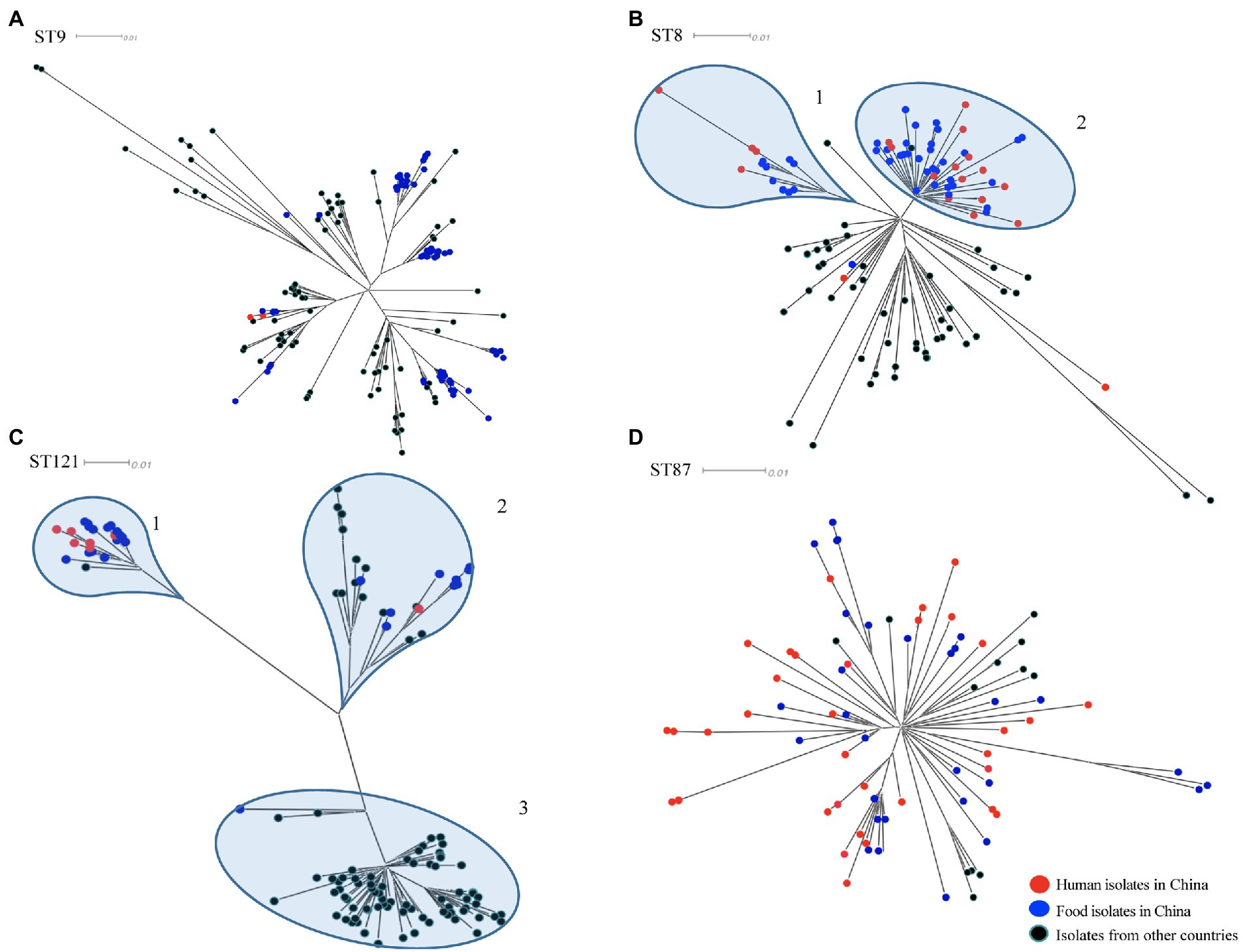
Figure 3. The core-SNP phylogenetic tree of prevalent subtypes of L. monocytogenes in China in the context of global isolates. The trees were visualized using SplitsTree 5. (A) Maximum likelihood phylogeny of 156 L. monocytogenes ST9 genomes based on 2,269 core-SNPs, annotation of SLCC2479 was used as reference. (B) Maximum likelihood phylogeny of 115 L. monocytogenes ST8 genomes based on 1,327 core-SNPs, annotation of ICDC_LM2 was used as reference. (C) Maximum likelihood phylogeny of 132 L. monocytogenes ST121 genomes based on 2,236 core-SNPs, annotation of ICDC_LM135 was used as reference. (D) Maximum likelihood phylogeny of 82 L. monocytogenes ST87 genomes based on 1,058 core-SNPs, annotation of ICDC_LM188 was used as reference. Human isolates in China are represented by red circles, food isolates in China are represented by blue circles, isolates from other countries are represented by black circles. Light blue circles represent clusters.
Stress resistance genes in the studied Listeria monocytogenes isolates
In this study, all 322 L. monocytogenes isolates were screened for the presence of stress adaptation associated genes or gene clusters, including stress survival islet 1 (SSI-1), stress survival islet 2 (SSI-2), three Listeria genomic islands (LGI1, LGI2, and LGI3), two benzalkonium chloride resistance determinants bcrABC gene cassette and the ermC gene. The SSI-1 was detected in 203 isolates (63.04%), including 32.61% (30/92) of lineage I isolates, 72.64% (154/212) of lineage II isolates and all the lineage III isolates. The SSI-2 was observed only in ST121 and ST196 isolates of lineage II (Figure 2). None of the studied isolates harbored LGI-1. There were 25 isolates harbored LGI-2 that includes genes putatively involved in arsenic and cadmium resistance. All CC2 and ST14 isolates carried LGI-2 consisting of 34 genes, which is consistent with the firstly reported strain Scott A (Kuenne et al., 2013). In addition, all the six CC1 isolates harbored a variant of LGI-2 previously reported by Gray et al. (2021), which carried an additional metC homology gene. A variant of LGI-3 was observed in all ST101 isolates in this study, lacking four genes compared to the first reported LGI-3 in ST101 isolates by Palma et al. (2020). These four genes encode the transposase (Tn3), recombinase (hin), and Cd-resistance (cadC and cadA1), the locus-tag in strain A37-02-LmUB3PA: LmUB3PA_1699 - LmUB3PA_1702. The bcrABC gene cassette was observed in two isolates (each for ST5 and ST296) of lineage I, and two ST9 isolates of lineage II. Two ST121 isolates harbored the ermC gene (Figure 2).
Virulence factors in the studied Listeria monocytogenes isolates
Three L. monocytogenes-specific pathogenicity islands (LIPI), LIPI-1, LIPI-3, and LIPI-4, were screened among the 322 foodborne isolates. LIPI-1 was highly conserved in all isolates. LIPI-3 was identified in the isolates belonging to a subset of lineage I isolates, including CC1, CC3, CC6, CC11, CC218, CC224, and CC288. Notably, one CC11/ST11 isolate ICDC_LM277 of lineage II carried the intact LIPI-3, a gene of it had 100% coverage and 96.937% identity with previously reported llsH gene in lineage I. LIPI-4 was identified in all ST87 (31/31) and ST296 (1/1) isolates belonging to lineage I.
The intact inlA gene encoding full-length InternalinA (InlA) was observed in 226 L. monocytogenes isolates, including 97.83% lineage I isolates, 55.45% lineage II isolates and 100% lineage III isolates in this study (Figure 2). The truncated inlA caused by premature stop codon (PMSC) mutations were observed in two ST5 isolates of lineage I, and 94 (44.55%) lineage II isolates belonging to ST7 (1/11), ST9 (71/72), ST121 (14/33), ST196 (2/2) and ST199 (6/6). Nine PMSC mutation types of inlA were identified, one of which was firstly reported here. The novel PMSC type existed in one ST7 isolate, a truncated InlA with a shorter peptide of 312 amino acids was caused by the deletion of cytosine in position 937 (Figure 4). For the other internalin genes, inlB, inlE, inlH, inlJ and inlK were found in all studied isolates. The inlC gene was absent in all ST299 isolates, while the inlF gene was absent in all ST14, ST121, and ST299 isolates. In addition, the inlG gene was absent in all lineage I isolates except for one ST6 isolate, and a subset of lineage II isolates which belonged to ST14, ST101, ST121, and ST196.
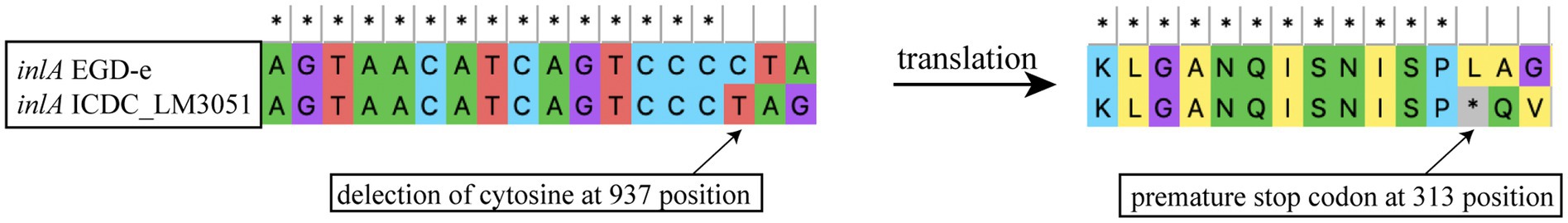
Figure 4. Newly identified inlA PMSC mutation type in L. monocytogenes as compared to full-length inlA gene from strain EGD-e.
Antibiotic resistance genes in the studied Listeria monocytogenes isolates
Ten antibiotic resistance genes were identified in this study (Figure 2). Six intrinsic antibiotic resistance genes, including fosX (resistance to fosfomycin), lmo0441 (cephalosporin), lmo0919 (lincosamides), norB (quinolones), mprF (cationic antimicrobial peptides) and sul (sulfonamides), were present in all studied isolates. Four acquired antibiotic resistance genes were identified in a small portion of the isolates. The aminoglycosides resistance gene aacA4 was identified in 50% (10/20) of ST3 isolates, locating in a 57-kb prophage that was inserted between the fosX (locus tag in EGD-e: CRH04_RS08765) and rlmD (locus tag in EGD-e: CRH04_RS08770) genes. The aacA4 gene positive isolates were found in three provinces in three different years, and they were isolated from four food sources, including RTE food, raw meat, raw poultry and aquatic products. The tetracycline resistance gene tetS was identified in one strain ICDC_LM3005 and was carried by a plasmid with a size of about 78 kb. Another tetracycline resistance gene tetM was observed in 13 isolates belonging to ST199 (n = 6), ST705 (n = 4), ST2 (n = 1), ST155 (n = 1), and ST515 (n = 1). The tetM gene from the ST199 isolates were carried by a transposon that was similar to Tn5801_B15 in Enterococcus faecalis with 100% coverage and 99.99% identity. The transposon harboring tetM gene was found in all ST199 isolates in this study, which were isolated from four provinces and three food sources. While the tetM gene from the ST705, ST2, and ST515 isolates were carried by a transposon that was similar to Tn916 in Bacillus subtilis with 100% coverage and 99.83% identity. Isolates carrying the Tn916 were distributed in three provinces and were isolated from three food sources. In addition, the tetM gene from the ST155 isolate was carried by a transposon Tn6198 that also carried the trimethoprim resistance gene dfrG. The tetM positive ST515 isolate in this study also carried dfrG, which was located downstream of a tRNA-Val gene along with two hypothetic proteins.
Discussion
As a foodborne pathogen, L. monocytogenes can survive and proliferate in different kinds of food and food-associated environments, even for a long time (Fagerlund et al., 2020; Wieczorek et al., 2020). Since 2000, the national surveillance of L. monocytogenes in different food products in China has been established. Several studies have been conducted to investigate the population structure and molecular epidemiology of L. monocytogenes in China using molecular subtyping methods, such as PFGE and MLST (Hua et al., 2018; Wang et al., 2018). Genome-wide genotyping has been used to study the biodiversity of L. monocytogenes and the genetic relationships between isolates, but is limited to local areas in China. Here, we performed whole-genome sequencing and genetic characterization of 322 L. monocytogenes isolated from food products from 13 regions between 2000 and 2018 to provide insight into the genomic diversity, molecular characteristics, and phylogenetic relationships of L. monocytogenes in China.
As previously reported, serogroup IIa was the most frequently isolated serogroup in meat products and environmental surfaces (Centorotola et al., 2021). In this study, by subtyping in silico, serogroup IIa and ST9 were the most common subtypes of food-sourced L. monocytogenes in China, which was consistent with previous studies (Wang et al., 2012; Zhang et al., 2020). ST9 was a common clone of L. monocytogenes isolated worldwide from food especially from meat products (Moura et al., 2016). The second and the third top of STs (ST8 and ST121) in this study also belonged to serogroup IIa. All the serogroup IIc isolates belonged to CC9 including ST9 and ST122, and showed limited diversity of genomes compare to serogroup IIa, which comprised of multiple CCs. ST8 was the second prevalent ST, which was proposed as the subtype of epidemic clone 5 (ECV), and was the predominant ST responsible for the human listeriosis in Canada during 1988 and 2010 (Fagerlund et al., 2016). Previous studies have shown that ST8 L. monocytogenes is commonly found in both food products and human listeriosis in China (Li et al., 2018; Zhang et al., 2021). Full-length inlA and SSI-1 were carried by all ST8 isolates, which partially supported its full pathogenic potential and increasing tolerance of stress conditions. Therefore, more attention should be paid to ST8 L. monocytogenes in the surveillance in China. ST121 was identified as the third prevalent subtype of isolates from food in China. A number of genetic determinants had been identified to involved in survival and adaptation of ST121 strains in food and food associated environments, like SSI-2, the transposon Tn6188 and a high proportion of plasmids (Schmitz-Esser et al., 2021). Consistent with our previous study, ST87 had been always identified as a concerned subtype among the L. monocytogenes from diverse sources in China (Wang et al., 2019). And the recent study had revealed that several genetic elements, such as LIPI-4 and a conserved plasmid pLM1686, may contributed to its pathogenic potential and adaptation to harsh environment (Wang et al., 2019).
So far, no listeriosis outbreak of any scale has been reported in China, partly due to the different consumption patterns and food-handling habits. However, RTE food particularly Chinese cold dishes which were reported to increase the risk of infection by 3.43-fold (Niu et al., 2022). In this study, 15 STs isolates were detected from RTE food, some STs of them were reported to have a strong link to the outbreak and sporadic listeriosis around the world, such as ST1, ST2, ST5, ST8, and ST87 (Chen et al., 2018; Maury et al., 2019; Wang et al., 2019; Zhang et al., 2021). Therefore, it is very important to strengthen hygiene monitoring and disinfection of RTE food processing and storage environment.
Pair-wised SNPs detection was performed on the indistinguishable isolates by core-SNPs analysis based on the species of L. monocytogenes. Here, we found 23 potentially associated events related to L. monocytogenes contamination. Combining with the meta-data including isolation sources (market, restaurant or farm), isolation years and isolation regions, it suggested that food safety risks exist in multiple contamination scenarios, including long-term persistent contamination over time, cross-contamination in markets, and the spread of a single source to different markets. However, caution should be exercised in inferring relationships between isolates with closely related genomes from different provinces and over time, unless clear epidemiological information is available.
Core-SNPs analysis based on the species of L. monocytogenes here apparently did not provide sufficient resolution to differentiate between isolates within a given CC/ST. ST122 was a member of CC9 with a different allele of ldh gene (the allele number of ldh is four in ST9 and 62 in ST122). Deeper phylogenetic analysis showed that the emerging ST122 clone might diverge from the ST9 clone for its most recent ancestor sharing with a subset of ST9’s. It suggested that the population of CC9 L. monocytogenes may be undergoing a diversification process.
In-depth phylogenetic study of the prevail STs in China, including ST9, ST8, ST121, and ST87, and each corresponding STs isolates from other countries, revealed the distribution of our studied isolates in the context of global isolates. ST9 L. monocytogenes in this study were grouped into several clusters throughout the phylogenetic tree of isolates from around the world, indicating that this clone from food in China was genetically diverse. ST9 was a prevalent clone isolated from foods, especially from meat products, in many countries (Martín et al., 2014; Maury et al., 2019; Wang et al., 2021). Different genetic characteristics of ST9 L. monocytogenes were spread and circulated globally with the frequent international trade of food. The majority of L. monocytogenes isolates of ST8 and ST121 from China were both grouped into two clusters, while isolates of these two STs from other countries were distributed in different clusters (Figures 3B,C). It indicated that ST8 and ST121 isolates from China were more closely related to another among each cluster, and showed heterogeneous with isolates from other countries. The possible reason is that these two STs are adapted to different food niches in China and other countries. The results of the phylogenetic analysis of ST87 L. monocytogenes in this study are consistent with that of our previous comprehensive and in-depth study of this clone in China (Wang et al., 2019). The isolates of ST87 L. monocytogenes were divided into multiple sub-clusters that were divided from the root, and minor isolates from other countries were distributed among the tree without region-clustering except for four clustered United States isolates that were from environments in the same year (2017). Overall, ST-specific core-SNP analysis well revealed the genome-level diversity of clonal groups which were prevalent in China, providing unprecedented discrimination to distinguish isolates within certain clonal groups.
Due to the use of several stress-adaptive mechanisms to withstand a wide variety of stressful conditions, L. monocytogenes isolates are able to colonize in different ecological niches. SSI-1 contributes to L. monocytogenes adapting to low pH and high salt concentration, while SSI-2 helps L. monocytogenes survival in alkaline and oxidative stress conditions (Ryan et al., 2010). In this study, 72.99% of lineage II isolates carried SSI-1, which included all ST8 and ST9 isolates. While all the ST121 isolates belonging to lineage II carried SSI-2. These findings gave further support for lineage II isolates being more prevalent in foods and food-associated environments than lineage I isolates. Interestingly, ST299 isolates that belonged to the rare lineage III and serogroup L were identified to overrepresent in aquatic products, which is similar to the results reported by Chen et al. (2018). It has been proposed that the ST299 have a specific ecological niche associated with aquatic products and the environment, while our study found that all ST299 isolates carried SSI-1. However, the contribution of SSI-1 in adapting to aquatic ecological niches in ST299 L. monocytogenes requires further functional confirmation. In addition, LGI-2 carried cadmium resistance cassette and arsenic resistance cassette which were associated with adaptation to environment, and was found a propensity with hypervirulent serogroup IVb clones, suggesting possible associations with L. monocytogenes virulence (Parsons et al., 2020). A variant of genomic island LGI3 lacking cadA1C cassette was found in all ST101/CC101 isolates in this study, which was the same with the observation of the study of Gray et al. (2021). However, Palma et al. (2020) found that the remaining 25 genes of LGI3 were significantly enriched in the persistent CC101 clone from the smoked-fish plant, indicating that this variant of LGI3 still contributes to persistent contamination of the ST101/CC101 clone.
As an important foodborne pathogen, several virulence potentials ultimately contribute to the ability of L. monocytogenes to infect humans. Except for species-specific LIPI-1, both LIPI-3, and LIPI-4 were found in a subset of lineage I L. monocytogenes isolates that were overrepresented in human infection cases. Strikingly, one ST11 strain (ICDC-LM277) of lineage II was isolated from raw meat and harbored LIPI-3. The LIPI-3 was also found in the genomes of eight ST11 L. monocytogenes isolates which caused an outbreak of listeriosis manifesting febrile gastroenteritis in Italy in 2016 (BioProject accession no.: PRJNA436467; Maurella et al., 2018). It suggested that ST11 L. monocytogenes isolates carrying LIPI-3 might have an increased risk of causing gastrointestinal infection in humans. LIPI-4, which is associated with increased neural and placental tropisms of L. monocytogenes, was found in all ST87/CC87 and ST296/CC88 isolates in this study. ST87 had been recognized as the dominant ST in clinical listeriosis in China, particularly associated with maternal-neonatal and central nervous system infections (Wang et al., 2019). Intact InlA is more favorable for L. monocytogenes to cross the intestinal barrier and establish a systemic infection, 70.19% (226/322) isolates in this study have full length of InlA, while 98.61% (71/72) ST9 isolates have truncated InlA, the under representation of the ST9 isolates in human clinical cases may be due to the frequent occurrence of PMSC mutations.
L. monocytogenes has been present sensitive to most antibiotics. For the treatment of listeriosis, ampicillin and penicillin-based agent is effective against this bacterium (Fischer et al., 2020). In present study, aminoglycosides resistance gene aacA4 was exclusively found in a subset of ST3 L. monocytogenes isolates, consistent with data obtained from Brazil and Poland (Camargo et al., 2019; Kurpas et al., 2020). The aacA4 gene was carried by a 57-kb prophage, and it is strongly associated with ST, but not with isolation location or type of food source. The standard therapy for listeriosis usually combined with gentamicin (aminoglycoside antibiotic; Fischer et al., 2020), monitoring aacA4 gene in L. monocytogenes isolates especially ST3 strains is important for listeriosis clinical treatment. Tetracycline resistance was the most common phenotype in food source L. monocytogenes in China (Yan et al., 2019), and we found that 4.3% of isolates carried the tetracycline resistance genes tetM or tetS in this study, and the tetM gene is associated with Tn916 and the tetS gene is carried by a plasmid, which was consistent with that of previous studies (Bertsch et al., 2013; Haubert et al., 2016). The high genetic identity of tetM-containing transposons between L. monocytogenes and B. subtilis or E. faecalis suggests that horizontal transfer of antibiotic genes can occur between different genera of bacteria in the same niche. In addition, the 18-kb Tn916 transposon could be found in both lineage I and lineage II isolates. It is a warning that transposon cannot be ignored for its role in the dissemination of the resistance genes across the lineages in L. monocytogenes. However, no evidence showed the harboring of resistance genes was related to isolation location, isolation year and the type of food source.
Conclusion
In this study, 322 L. monocytogenes isolates were selected and sequenced to characterize the population structure of food source isolates in China. Serogroup IIa and ST9 were identified as the most prevalent serogroup and ST, respectively. Core-SNP analysis classified all the isolates into different clusters that corresponded to the different clonal complexes which were further grouped into three lineages. In-depth core-SNP analysis based on specific CC or ST reveals the potential epidemiological relationship of isolates in China: (i) ST122 clone might diverge from ST9 clone; (ii) persistent contamination, cross-contamination, and transmission from a single source to different markets were found in markets of China; (iii) the prevalent subtypes ST8 and ST121 were heterogeneous with worldwide isolates. Genomic analysis identified LIPI-3 in lineage II (ST11/CC11) isolates and a novel PMSC type in inlA gene, which increased the understanding of the genomic diversity of L. monocytogenes.
Data availability statement
The datasets presented in this study are deposited in the China National Microbiology Data Center (NMDC), with accession number NMDC60016487 to NMDC60016827 (https://nmdc.cn/resource/genomics/genome/).
Author contributions
SJ, CY, and YaW conceived and designed the research study. SJ, YiW, LLi, and PM performed the sample collection and DNA extraction. ZS and SJ analyzed the data. SJ, LLu, CY, and YaW write and revise the manuscript. All authors have read and approved it for publication.
Funding
This work was supported by grants from National Key R&D Program of China (2018YFC1603800); National Institute for Communicable Disease Control and Prevention, China CDC (2021ZZKT003, 131031102000210003-07007); National Natural Science Foundation of China (31800004).
Acknowledgments
We sincerely thank the Centers for disease control and prevention of 11 provinces and one municipality in China for providing the L. monocytogenes isolates from food products.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1049843/full#supplementary-material
Footnotes
References
Bertsch, D., Uruty, A., Anderegg, J., Lacroix, C., Perreten, V., and Meile, L. (2013). Tn 6198, a novel transposon containing the trimethoprim resistance gene dfrG embedded into a Tn 916 element in listeria monocytogenes. J. Antimicrob. Chemother. 68, 986–991. doi: 10.1093/jac/dks531
Blanc, D. S., Magalhães, B., Koenig, I., Senn, L., and Grandbastien, B. (2020). Comparison of whole genome (wg-) and Core genome (cg-) MLST (BioNumericsTM) versus SNP variant calling for epidemiological investigation of Pseudomonas aeruginosa. Front. Microbiol. 11:1729. doi: 10.3389/fmicb.2020.01729
Camargo, A. C., Moura, A., Avillan, J., Herman, N., McFarland, A. P., Sreevatsan, S., et al. (2019). Whole-genome sequencing reveals listeria monocytogenes diversity and allows identification of long-term persistent strains in Brazil. Environ. Microbiol. 21, 4478–4487. doi: 10.1111/1462-2920.14726
Centorotola, G., Guidi, F., D’Aurizio, G., Salini, R., Di Domenico, M., Ottaviani, D., et al. (2021). Intensive environmental surveillance plan for listeria monocytogenes in food producing plants and retail stores of Central Italy: prevalence and genetic diversity. Foods 10:1944. doi: 10.3390/foods10081944
Chen, M., Cheng, J., Wu, Q., Zhang, J., Chen, Y., Xue, L., et al. (2018). Occurrence, antibiotic resistance, and population diversity of listeria monocytogenes isolated from fresh aquatic products in China. Front. Microbiol. 9:2215. doi: 10.3389/fmicb.2018.02215
Doumith, M., Buchrieser, C., Glaser, P., Jacquet, C., and Martin, P. (2004). Differentiation of the major Listeria monocytogenes Serovars by multiplex PCR. J. Clin. Microbiol. 42, 3819–3822. doi: 10.1128/JCM.42.8.3819-3822.2004
Doumith, M., Jacquet, C., Gerner-Smidt, P., Graves, L. M., Loncarevic, S., Mathisen, T., et al. (2005). Multicenter validation of a multiplex PCR assay for differentiating the major Listeria monocytogenes Serovars 1/2a, 1/2b, 1/2c, and 4b: toward an international standard. J. Food Prot. 68, 2648–2650. doi: 10.1016/j.mimet.2009.11.008
Fagerlund, A., Langsrud, S., and Mretr, T. (2020). In depth longitudinal study of Listeria monocytogenes ST9 isolates from meat processing industry: resolving diversity and transmission patterns using WGS. Appl. Environ. Microbiol. 86:e00579-20. doi: 10.1128/AEM.00579-20
Fagerlund, A., Langsrud, S., Schirmer, B. C., Møretrø, T., and Heir, E. (2016). Genome analysis of Listeria monocytogenes sequence type 8 strains persisting in salmon and poultry processing environments and comparison with related strains. PLoS One 11:e0151117. doi: 10.1371/journal.pone.0151117
Fischer, M. A., Wamp, S., Fruth, A., Allerberger, F., Flieger, A., and Halbedel, S. (2020). Population structure-guided profiling of antibiotic resistance patterns in clinical Listeria monocytogenes isolates from Germany identifies pbpB3 alleles associated with low levels of cephalosporin resistance. Emerg. Microbes Infect. 9, 1804–1813. doi: 10.1080/22221751.2020.1799722
Gray, J. A., Chandry, P. S., Kaur, M., Kocharunchitt, C., Bowman, J. P., and Fox, E. M. (2021). Characterisation of Listeria monocytogenes food-associated isolates to assess environmental fitness and virulence potential. Int. J. Food Microbiol. 350:109247. doi: 10.1016/j.ijfoodmicro.2021.109247
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2018). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Haubert, L., Mendonça, M., Lopes, G., de Itapema Cardoso, M., and Da Silva, W. (2016). Listeria monocytogenes isolates from food and food environment harbouring tetM and ermB resistance genes. Lett. Appl. Microbiol. 62, 23–29. doi: 10.1111/lam.12516
Hua, L., Pengfei, W., Ruiting, L., Lijuan, L., Xiaolong, C., Yi, W., et al. (2018). Risk factors and level of Listeria monocytogenes contamination of raw pork in retail Markets in China. Front. Microbiol. 9:1090. doi: 10.3389/fmicb.2018.01090
Kuenne, C., Billion, A., Mraheil, M. A., Strittmatter, A., Daniel, R., Goesmann, A., et al. (2013). Reassessment of the Listeria monocytogenespan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genomics 14, 1–19. doi: 10.1186/1471-2164-14-47
Kurpas, M., Osek, J., Moura, A., Leclercq, A., Lecuit, M., and Wieczorek, K. (2020). Genomic characterization of Listeria monocytogenes isolated from ready-to-eat meat and meat processing environments in Poland. Front. Microbiol. 11:1412. doi: 10.3389/fmicb.2020.01412
Leclercq, A., Chenal-Francisque, V., Dieye, H., Cantinelli, T., Drali, R., Brisse, S., et al. (2011). Characterization of the novel Listeria monocytogenes PCR serogrouping profile IVb-v1. Int. J. Food Microbiol. 147, 74–77. doi: 10.1016/j.ijfoodmicro.2011.03.010
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, W., Bai, L., Fu, P., Han, H., Liu, J., and Guo, Y. (2018). The epidemiology of Listeria monocytogenes in China. Foodborne Pathog. Dis. 15, 459–466. doi: 10.1089/fpd.2017.2409
Martín, B., Perich, A., Gómez, D., Yangüela, J., Rodríguez, A., Garriga, M., et al. (2014). Diversity and distribution of Listeria monocytogenes in meat processing plants. Food Microbiol. 44, 119–127. doi: 10.1016/j.fm.2014.05.014
Maurella, C., Gallina, S., Ru, G., Adriano, D., Bellio, A., Bianchi, D. M., et al. (2018). Outbreak of febrile gastroenteritis caused by Listeria monocytogenes 1/2a in sliced cold beef ham, Italy, may 2016. Eur. Secur. 23, 17–00155. doi: 10.2807/1560-7917.ES.2018.23.10.17-00155
Maury, M. M., Bracq-Dieye, H., Huang, L., Vales, G., Lavina, M., Thouvenot, P., et al. (2019). Hypervirulent Listeria monocytogenes clones’ adaption to mammalian gut accounts for their association with dairy products. Nat. Commun. 10, 2488–2413. doi: 10.1038/s41467-019-10380-0
McLauchlin, J., Aird, H., Amar, C., Barker, C., Dallman, T., Lai, S., et al. (2021a). An outbreak of human listeriosis associated with frozen sweet corn consumption: investigations in the UK. Int. J. Food Microbiol. 338:108994. doi: 10.1016/j.ijfoodmicro.2020.108994
McLauchlin, J., Aird, H., Amar, C., Boyd, G., Brindle, A., Dallman, T., et al. (2021b). Listeriosis associated with pre-prepared sandwich consumption in hospital in England, 2017. Epidemiol. Infect. 149:e220. doi: 10.1017/S0950268821001916
Moura, A., Criscuolo, A., Pouseele, H., Maury, M. M., Leclercq, A., Tarr, C., et al. (2016). Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2:16185. doi: 10.1038/nmicrobiol.2016.185
Niu, Y.-L., Wang, T.-Y., Zhang, X.-A., Guo, Y.-C., Zhang, Y.-W., Wang, C., et al. (2022). Risk factors for sporadic listeriosis in Beijing, China: a matched case–control study. Epidemiol. Infect. 150, 1–21. doi: 10.1017/S0950268821002673
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palma, F., Brauge, T., Radomski, N., Mallet, L., Felten, A., Mistou, M. Y., et al. (2020). Dynamics of mobile genetic elements of Listeria monocytogenes persisting in ready-to-eat seafood processing plants in France. BMC Genomics 21:130. doi: 10.1186/s12864-020-6544-x
Parsons, C., Lee, S., and Kathariou, S. (2020). Dissemination and conservation of cadmium and arsenic resistance determinants in listeria and other gram-positive bacteria. Mol. Microbiol. 113, 560–569. doi: 10.1111/mmi.14470
Ryan, S., Begley, M., Hill, C., and Gahan, C. (2010). A five-gene stress survival islet (SSI-1) that contributes to the growth of Listeria monocytogenes in suboptimal conditions. J. Appl. Microbiol. 109, 984–995. doi: 10.1111/j.1365-2672.2010.04726.x
Salcedo, C., Arreaza, L., Alcala, B., De La Fuente, L., and Vazquez, J. (2003). Development of a multilocus sequence typing method for analysis of Listeria monocytogenes clones. J. Clin. Microbiol. 41, 757–762. doi: 10.1128/JCM.41.2.757-762.2003
Schlech, W. F. III (2019). Epidemiology and clinical manifestations of Listeria monocytogenes infection. Microbiology. Spectrum 7:3. doi: 10.1128/microbiolspec.GPP3-0014-2018
Schmitz-Esser, S., Anast, J. M., and Cortes, B. W. (2021). A large-scale sequencing-based survey of plasmids in Listeria monocytogenes reveals global dissemination of plasmids. Front. Microbiol. 12:653155. doi: 10.3389/fmicb.2021.653155
Swaminathan, B., and Gerner-Smidt, P. (2007). The epidemiology of human listeriosis. Microbes Infect. 9, 1236–1243. doi: 10.1016/j.micinf.2007.05.011
Thomas, J., Govender, N., McCarthy, K. M., Erasmus, L. K., Doyle, T. J., Allam, M., et al. (2020). Outbreak of listeriosis in South Africa associated with processed meat. N. Engl. J. Med. 382, 632–643. doi: 10.1056/NEJMoa1907462
Wang, Y., Ji, Q., Li, S., and Liu, M. (2021). Prevalence and genetic diversity of Listeria monocytogenes isolated from retail pork in Wuhan. China. Front. Microbiol. 12:620482. doi: 10.3389/fmicb.2021.620482
Wang, Y., Luo, L., Li, Q., Wang, H., Wang, Y., Sun, H., et al. (2019). Genomic dissection of the most prevalent Listeria monocytogenes clone, sequence type ST87 in China. BMC Genomics 20, 1–13. doi: 10.1186/s12864-019-6399-1
Wang, H., Luo, L., Zhang, Z., Deng, J., Wang, Y., Miao, Y., et al. (2018). Prevalence and molecular characteristics of Listeria monocytogenes in cooked products and its comparison with isolates from listeriosis cases. Front. Med. 12, 104–112. doi: 10.1007/s11684-017-0593-9
Wang, Y., Zhao, A., Zhu, R., Lan, R., Jin, D., Cui, Z., et al. (2012). Genetic diversity and molecular typing of Listeria monocytogenes in China. BMC Microbiol. 12, 1–9. doi: 10.1186/1471-2180-12-119
Wieczorek, K., Bomba, A., and Osek, J. (2020). Whole-genome sequencing-based characterization of Listeria monocytogenes from fish and fish production environments in Poland. Int. J. Mol. Sci. 21:9419. doi: 10.3390/ijms21249419
Wilking, H., Lachmann, R., Holzer, A., Halbedel, S., Flieger, A., and Stark, K. (2021). Ongoing high incidence and case-fatality rates for invasive Listeriosis, Germany, 2010–2019. Emerg. Infect. Dis. 27, 2485–2488. doi: 10.3201/eid2709.210068
Yan, S., Li, M., Luque-Sastre, L., Wang, W., Hu, Y., Peng, Z., et al. (2019). Susceptibility (re)-testing of a large collection of Listeria monocytogenes from foods in China from 2012 to 2015 and WGS characterization of resistant isolates. J. Antimicrob. Chemother. 74, 1786–1794. doi: 10.1016/j.mimet.2009.11.008
Zhang, H., Chen, W., Wang, J., Xu, B., Liu, H., Dong, Q., et al. (2020). 10-year molecular surveillance of Listeria monocytogenes using whole-genome sequencing in Shanghai, China, 2009-2019. Front. Microbiol. 11:551020. doi: 10.3389/fmicb.2020.551020
Keywords: Listeria monocytogenes, foodborne, whole-genome sequencing, genomic characterization, inlA PMSCs
Citation: Ji S, Song Z, Luo L, Wang Y, Li L, Mao P, Ye C and Wang Y (2023) Whole-genome sequencing reveals genomic characterization of Listeria monocytogenes from food in China. Front. Microbiol. 13:1049843. doi: 10.3389/fmicb.2022.1049843
Edited by:
Jianmin Zhang, South China Agricultural University, ChinaReviewed by:
Moutong Chen, Guangdong Academy of Science, ChinaHongsheng Huang, Canadian Food Inspection Agency (CFIA), Canada
Copyright © 2023 Ji, Song, Luo, Wang, Li, Mao, Ye and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Wang, ✉ d2FuZ3lhbkBpY2RjLmNu; Changyun Ye, ✉ eWVjaGFuZ3l1bkBpY2RjLmNu