
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 29 October 2021
Sec. Aquatic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.701155
This article is part of the Research TopicResponses of Marine Microbes to Multiple Environmental Drivers of Global Change: the Interplay of Abiotic and Biotic FactorsView all 23 articles
 Vincent Hervé1,2†
Vincent Hervé1,2† Josie Lambourdière3
Josie Lambourdière3 Malika René-Trouillefou3Damien Alain Devault3‡
Malika René-Trouillefou3Damien Alain Devault3‡ Pascal Jean Lopez3*†
Pascal Jean Lopez3*†Rafts of drifting pelagic Sargassum that are circulating across the Atlantic Ocean are complex ecosystems composed of a large number of associated species. Upon massive stranding, they lead to various socio-environmental issues including the inflow of contaminants and human health concerns. In this study, we used metabarcoding approaches to examine the differences in both the eukaryotic- and prokaryotic-associated communities from Sargassum present in two islands of the Lesser Antilles, namely Guadeloupe and Martinique. We detected significant differences in microbial community structure and composition between landing Sargassum, the surrounding seawater, and Sargassum from inland storage sites. In total we identified 22,214 prokaryotic and 17,679 eukaryotic OTUs. Among them, functional prediction analyses revealed a number of prokaryotes that might contribute to organic matter decomposition, nitrogen cycling and gas production, including sulfate-reducing bacteria at coastal landing sites, and methanogenic archaea at inland storage sites. We also found that Metazoan was the most abundant group in Sargassum samples, with nematode clades that presented exclusive or specific richness and abundance patterns depending on their Sargassum substrate. Together, these molecular inventories of the micro- and meiofauna communities provide baseline information for further characterization of trophic interactions, algal organic matter decomposition and nutrient transfers at coastal and inland storage sites.
Numerous seaweeds, including benthic and drifting pelagic Sargassum species, are becoming invasive in various regions of the world, creating potential threats to native species and local resources, and causing economic and health concerns (Hu and Fraser, 2016). Over the last decade, the whole Caribbean region and the west coast of Africa have been faced with massive tides of Sargassum (Wang et al., 2019). These holopelagic macroalgae shoals contains several morphotypes, with the most common one being S. fluitans and S. natans (Parr, 1939; Schell et al., 2015; Amaral-Zettler et al., 2017; Martin et al., 2021). The large amounts of seaweed biomass washed up along coastlines has direct and indirect consequences on beaches and on the functioning of nearshore ecosystems (Van Tussenbroek et al., 2017; Cabanillas-Teran et al., 2019; Rodriguez-Martinez et al., 2019), and a massive influence on the tourist industry and local economies (i.e., the cost of removing and disposing of piled-up Sargassum, the effect on housing markets, etc.). Sargassum tides can also have serious health impacts, not only because of the toxicity of the associated heavy metals and pollutants, but also through their decomposition which can lead to the production of hydrogen sulfide (H2S) and ammonia (NH3), which have been shown to cause a real threat to human health (Resiere et al., 2018).
Pelagic Sargassum in the Atlantic Ocean constitutes a floating ecosystem serving as a habitat for marine species like sea turtles, seabirds, fish or invertebrates. Additionally, a number of micro- and macro-epiphytes have been described (Parr, 1939; Weis, 1968; Fine, 1970; Casazza and Ross, 1972; Ryland, 1974; Haney, 1986; Jobe and Brooks, 2009; Ballard and Rakocinski, 2012; Witherington et al., 2012; Jacobucci and Leite, 2014; Susilowati et al., 2015; Monroy-Velazquez et al., 2019), and a few of them are endemic species. Endophytic and epiphytic communities of small eukaryotes are likely to be involved in the growth of Sargassum, and might contribute to nutrient uptake, macroalgae spore release and germination, the defense against bactericidal pathogens and other competing organisms, reproduction and settlement (Egan et al., 2013; Florez et al., 2017; Van Der Loos et al., 2019). In addition, exchanges between the Sargassum holobiont and the surrounding environment might have reciprocal impacts, and micro-organisms initially associated with algae growth might also contribute to their degradation processes (Florez et al., 2017). Indeed, microorganisms are likely to contribute to algae raft formation and maintenance, algae sinking and biodegradation upon beaching (De Fouw et al., 2016). In addition, the microbial communities that are associated with the seaweeds can thrive in new habitats, as postulated for the export of surface-dwelling fauna associated with Sargassum from the surface down to the seafloor (Baker et al., 2018).
At the molecular level, bacterial diversity has been investigated for floating Sargassum from various locations and for several species. Serebryakova et al. (2018) investigated the 16S rRNA gene sequences associated with the invasive benthic S. muticum from various locations in Portugal, and showed that the epibionts were dominated by Proteobacteria, Bacteroidetes, and Actinobacteria. The structure of the bacterial community has also been analyzed for drifting S. horneri from the Yellow Sea (Mei et al., 2019). As with other macroalgae studies, Mei et al. (2019) demonstrated differences at the genus level in the bacterial composition between the surrounding water and the drifting algae, and between drifting and nearshore algae Sea. Torralba et al. (2017) found that Sargassum complexes of two main surface drifting species (S. natans and S. fluitans), collected from the Gulf of Mexico, were associated with microbial communities dominated by Rhodobacteraceae and Saprospiraceae. Another molecular study on the bacterial community of drifting Sargassum has shown that the microbial profile can vary depending on location, abundance of zooplankton and nutrient levels (Michotey et al., 2020). Surprisingly, the composition of protists associated with Sargassum have been largely investigated using taxonomic and/or morphological analyses (Maples, 1984; Huffard et al., 2014; Damare, 2015; Coelho-Souza et al., 2017; Baker et al., 2018; Kim H. M. et al., 2019), but to our knowledge, only one study have described the molecular diversity of the eukaryotic plankton and epifauna associated with floating S. horneri (Kim H. M. et al., 2019).
It was expected that the physicochemical conditions encountered along the coasts, associated to different microbial communities in both the water column and beaches could induce large modification of the micro- and macrofauna associated with Sargassum. Moreover, the significant accumulation (ca., corresponding to a new food source) and decay of seaweed biological materials at landing sites are further factors that could largely influence the local biodiversity and the microbiota associated to Sargassum. The objective of the present study was to perform the first investigations of the microbial communities associated with Sargassum from coastal landing sites (i.e., surf zone and deposited on beaches), and from inland storage sites. We used metabarcoding approaches to describe both the prokaryotic and eukaryotic diversities from two islands of the Lesser Antilles, Guadeloupe and Martinique. By comparing molecular data from the seaweed-associated fractions, we hoped to identify microorganisms that are more specifically associated with stranding or more dried seaweeds. We also aimed to gain novel information on the diversity of the species that are involved in Sargassum aging and biodegradation.
Various samples were taken along the Atlantic coasts of Martinique and Guadeloupe during the summer of 2018 (Figure 1). At coastal sites, for each sample, about 500 g of Sargassum seaweed were collected and the excess of seawater was gently drained off using a salad spinner, and then about 30 g were put into 50 ml Falcon tubes. At the same coastal landing sites, the surrounding seawater was also collected using sterile 50 ml Falcon tubes, at distances between 1 and 20 m from the shoreline and at about 0.5 m below the surface. In Martinique, we took samples from four different inland sites that were used by the local authorities to store the seaweeds collected on the beaches. For most of the tide sampling sites we collected at least one seawater marine sample and two Sargassum samples, and three samples for the inland storage sites. During the campaigns we placed all sample on ice, and then, upon arrival at the laboratory they frozen and maintained at –20°C until the nucleic acids were extracted.
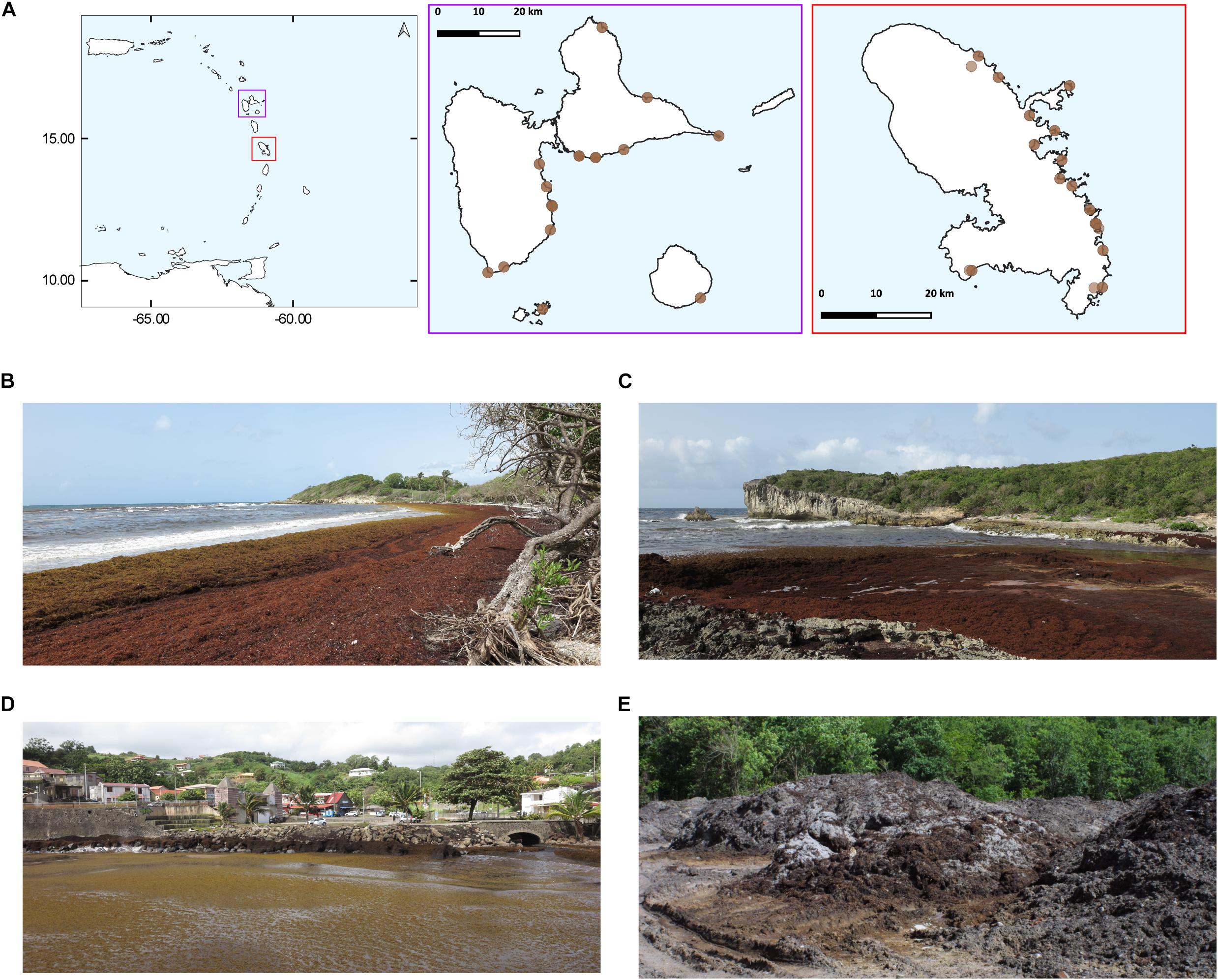
Figure 1. Maps of sampled Sargassum tide sites. The year 2018 corresponded to the biggest tide events ever recorded in the Caribbean region. (A) From left to right, the Caribbean Basin, the Guadeloupe archipelago (violet), and the island of Martinique (red). Due to the proximity of the sampled sites, some circles may overlap. (B–E) Photographs of sampling sites. Sargassum accumulated along the shorelines of Guadeloupe at Saint Félix (B), and La Porte d’Enfer (C). In Martinique, Sargassum in the small harbor of Le Marigot city (D), Sargassum pileup at an inland storage site located in the township of Le Diamant (E).
The Sargassum samples were thawed at 4°C, suspended in 50 ml RNase-free water, vortexed and then mechanically sonicated in a water bath (35 kHz) for 3 min. The latter procedure was repeated three times. Then the microbial fractions present in the supernatant and extracted from the seaweed were filtered through a 0.2 μm GTTP Biofilm Type Filter kit (Millipore). About 50 ml of marine water samples were filtered using the same filter type that was mentioned above. The environmental DNA was extracted from the filters using the DNeasy PowerBiofilm Kit (Qiagen), in accordance with the manufacturer’s recommendations. Total DNA was then quantified using Quant-it PicoGreen (Invitrogen, Thermo Fischer Scientific, Carlsbad, CA, United States) and a CFX96 Real-Time System, C1000 Touch Thermal Cycler (Biorad, California, United States).
The DNA libraries were prepared using universal primers to target both prokaryotes and eukaryotes. To amplify the V4-V5 of the 16S rRNA, we used the 16S-515F-Y (5′-GTGYCAGCMGCCGCGGTAA-3′), and 16S-926R (5′-CCGYCAATTYMTTTRAGTTT-3′) primers that cover both bacteria and archaea (Walters et al., 2016). To amplify the V4 region of the 18S rRNA gene, we used the primer pair TAReuk454FWD1 (5′-CCAGCASCYGCGGTAATTCC-3′) and TAReukREV3 (5′-ACTTTCGTTCTTGATYRA-3′) (Stoeck et al., 2010). For each sample, we duplicated the initial DNA amplification in a final volume of 25 μl following the Taq Q5 Hot Start High Fidelity DNA Polymerase recommendations (New England Biolabs). For the 16S rRNA amplification, we also added to the amplification reactions 0.4 μl of a 10 μM of peptide nucleic acid (PNA) clamps (Fitzpatrick et al., 2018), that we designed to match 16S RNA genes from Sargassum plastid (pPNA: 5′-AGCTCAACTTCAAAACT-3′) and mitochondria (mPNA: 5′- GGCTAGCCTTATTCGTC-3′). For each sample, the PCR products were checked on agarose gels, purified using Agencourt AMPure XP beads (Beckman Coulter), and quantified using a Qubit dsDNA HS assay kit (Thermo Fisher Scientific). We then quantified and pooled the technical replicates per sample. To create unique library per amplicon type (16S or 18S), we pooled same quantity of all samples. These libraries were prepared using 1 μg pooled DNA and using the TruSeq Nano Library Preparation Kit (Illumina). The supplier’s protocol was followed, with the exception of the use of a modified End-Repair mix to avoid production of chimeric constructs, and no PCR cycle was done to finalize the libraries. The resulting libraries (16S or 18S) were quantified by qPCR and the paired-end (2 × 300 bp) sequencing were done using a HiSeq 2500 by Fasteris SA (Plan-les-Ouates, Switzerland).
Amplicons of the 16S and 18S rRNA gene sequences were analyzed independently with the mothur software, version 1.41.3 (Schloss et al., 2009), following a standard operating procedure for Illumina MiSeq data (Kozich et al., 2013). First, contigs were assembled between the read pairs. Then the barcodes, primer sequences and low-quality sequences were removed (minimum length of 350 bp and maximum length of 420 bp, removing any sequences with ambiguous bases and any sequences with homopolymers longer than 8 bp). The sequences were then aligned with the SILVA reference database release 132 (Quast et al., 2013) and preclustered (pre.cluster, diffs = 1). Singletons were excluded, and chimeras were removed with vsearch (Rognes et al., 2016) implemented in mothur. The sequences were then classified using the k-nearest neighbor (knn) algorithm implemented in mothur and the BLASTN search method (cut-off of 80%) with the SILVA reference database, release 132, and the PR2 database, v4.11.1 (Guillou et al., 2013) for the 16S and 18S rRNA gene amplicons, respectively. After classification, non-prokaryotic, chloroplast, mitochondria (for the 16S rRNA gene dataset), non-eukaryotic, Sargassum (for the 18S rRNA gene dataset) and unknown (for both 16S and 18S rRNA amplicons) sequences were excluded. To account for differences in sampling efforts, 28,491 and 53,263 sequences from the 16S and 18S rRNA gene datasets, respectively, were then randomly subsampled from each sample using mothur. Finally, operational taxonomic units (OTUs) were generated using the vsearch distance-based greedy clustering algorithm, with an OTU being defined at the 97 and 99% sequence similarity level for the 16S and 18S rRNA gene reads, respectively. The raw datasets generated and analyzed during the current study were submitted to the NCBI Sequence Read Archive (SRA) under the BioProjects PRJNA630532 (16S rRNA genes) and PRJNA630533 (18S rRNA genes).
Alpha diversity indices (observed OTU richness, Shannon and inverse of Simpson indexes) as well as Good’s coverage estimates and rarefaction curves were computed with mothur. All the statistical analyses were computed using R software version 3.6.3. To compare the alpha diversity indices between compartments, we used Kruskal-Wallis tests and corrected our P-values for multiple comparisons with the Benjamini-Hochberg method. Principal coordinates analyses (PCoA) and constrained analysis of principal coordinates (CAP) were computed based on Bray-Curtis dissimilarity matrices with the phyloseq package (McMurdie and Holmes, 2013). The effect of compartments on prokaryotic and eukaryotic community composition was tested by non-parametric permutational multivariate analysis of variance (PERMANOVA) on Bray-Curtis dissimilarity matrices, as implemented in the vegan function adonis (permutations = 9,999), after checking for homogeneity of group dispersions with the vegan function betadisper (Oksanen et al., 2015). We also used permutation tests for CAP, as implemented in the vegan function anova.cca (permutations = 9,999). To test the relationship between the prokaryotic and eukaryotic community matrices, a Mantel test was performed using the ecodist package (Goslee and Urban, 2007), with Pearson correlation coefficient and 106 random permutations. To identify OTUs with differential abundances between compartments, we used the ALDEx2 algorithm on the prokaryotic and eukaryotic community matrices after filtering out the OTUs with less than 0.01% abundance (Fernandes et al., 2014). In this analysis, statistical significance was assessed based on the Wilcoxon rank test with Benjamini and Hochberg’s correction to maintain a 5% false discovery rate. Potential function among microbiota was predicted by using Functional Annotation of Prokaryotic Taxa (FAPROTAX v1.1), which was initially designed for marine samples (Louca et al., 2016). The predicted functional roles were visualized with the pheatmap R package, using Euclidean distances, and the weighted pair group method centroid (WPGMC) as a clustering method.
Phylogenetic relationships between the abundant OTUs (>1% relative abundance) of Nematoda were investigated. We first collected nearly full-length 18S rRNA gene sequences from recent studies (Meldal et al., 2007) as reference sequences. These sequences were aligned with the Silva SSU database using SINA v1.2.11 (Pruesse et al., 2012) and the subsequent alignment was filtered using trimAl v1.2rev59 with the gappyout method (Capella-Gutierrez et al., 2009). OTU sequences were added to the reference alignment using the –addfragments option of MAFFT v7.310 (Katoh and Standley, 2013). The alignment was trimmed and Smart Model Selection (Lefort et al., 2017) was used to determine the best-fit evolutionary model based on the Akaike Information criterion. Subsequently, a maximum-likelihood phylogenetic tree was built with PhyML 3.0 (Guindon et al., 2010). Branch supports were calculated using a Chi2-based parametric approximate likelihood-ratio test (aLRT) (Anisimova and Gascuel, 2006). Finally, the tree was visualized and edited with iTOL (Letunic and Bork, 2019).
In the present study, we investigated both the prokaryotic and eukaryotic communities associated with Sargassum at landing sites along the Atlantic coastlines of the Guadeloupe archipelago and the island of Martinique (Figure 1). We analyzed seawater surrounding Sargassum from different tide sites (n = 52; 34 sites). Marine Sargassum samples were collected either in water (30 samples; 27 sites) or stranded on beaches (9 samples; 6 sites). Unfortunately, we could not obtain any specific information on the arrival dates of the Sargassum and/or on landing duration. Such data are in fact very difficult to obtain also because the local parameters (i.e., currents, wind, tide, etc.) and sinking processes can vary from place to place. Nevertheless, measurements on 87% of the collected samples revealed that the dry matter content was 16.2% ± 1.8%, showing that all samples had a similar hydration level (not shown). In Martinique, we also collected samples from inland storage sites (9 samples; 4 sites). Since there was no available information on the deposition period, we took samples from the middle and the top of the piles; partial measurements indicated that the Sargassum dry matter (2 sites) was 49.9% (not shown). As mentioned above because we analyzed seaweed samples that were fresh, partially or dried we did not and often could not separate the morphotypes that composed Sargassum samples. Hereafter, the three types of delineated biotic entities, hereafter named compartments, correspond to seawater from tide sites (TS-sw), Sargassum from tide sites (TS-sarg), and Sargassum from inland storage sites (ISS-sarg).
After sequencing of the 100 samples and following bioinformatic procedures, we obtained a total of 22,214 prokaryotic and 17,679 eukaryotic operational taxonomic units (OTUs). For both datasets, the rarefaction curves (Supplementary Figure 1) as well as the high Good’s coverage estimates (median value 0.977 for the prokaryotes, Supplementary Figure 2; median value 0.997 for the eukaryotes Supplementary Figure 3) indicated that the sequencing depths were sufficient to estimate and compare the microbial diversity of our samples.
Regarding the alpha diversity of the prokaryotes, we found no significant differences in OTU richness nor diversity between the three compartments (P > 0.05) (Supplementary Figure 2). For the eukaryotes, we found significantly more OTUs in the seawater (TS-sw) than in the terrestrial and marine Sargassum samples (P < 0.001) (Supplementary Figure 3). For both the Shannon index and the inverse of Simpson index, eukaryotic diversity was significantly higher in TS-sw than in TS-sarg (P < 0.001) but no differences were found between TS-sarg and ISS-sarg (P > 0.05) (Supplementary Figure 3).
Beta diversity of the Sargassum microbiota was first visualized using unconstrained ordinations, namely principal coordinates analyses (PCoA) (Supplementary Figure 4) focusing on the three compartments (TS-sw, TS-sarg, and ISS-sarg). PERMANOVAs indicated that both the prokaryotic (R2 = 0.08, P < 0.001) and the eukaryotic (R2 = 0.09, P < 0.001) communities were significantly different across the three compartments TS-sw, TS-sarg, and ISS-sarg. Differences between compartments were further investigated using constrained ordinations, namely canonical analysis of principal coordinates (CAP) (Figure 2), and confirmed by permutation tests for CAP (P < 0.001). For both the prokaryotic and the eukaryotic communities, the first axis discriminated between TS-sarg and TS-sw, while the second axis discriminated between ISS-sarg and both the TS-sarg and TS-sw samples (Figure 2). Among the TS-sarg samples, we found no differences between samples collected in the water or on the beaches (ANOSIM, R = –0.07, P = 0.78 and R = 0.01, P = 0.42, for the prokaryotic and eukaryotic communities, respectively). We also examined the relationship between the prokaryotic and eukaryotic dissimilarity matrices. A Mantel test revealed that the community structures of prokaryotes and eukaryotes were strongly correlated (rM = 0.43, P < 0.0001), indicating potential biotic associations between microbiota.
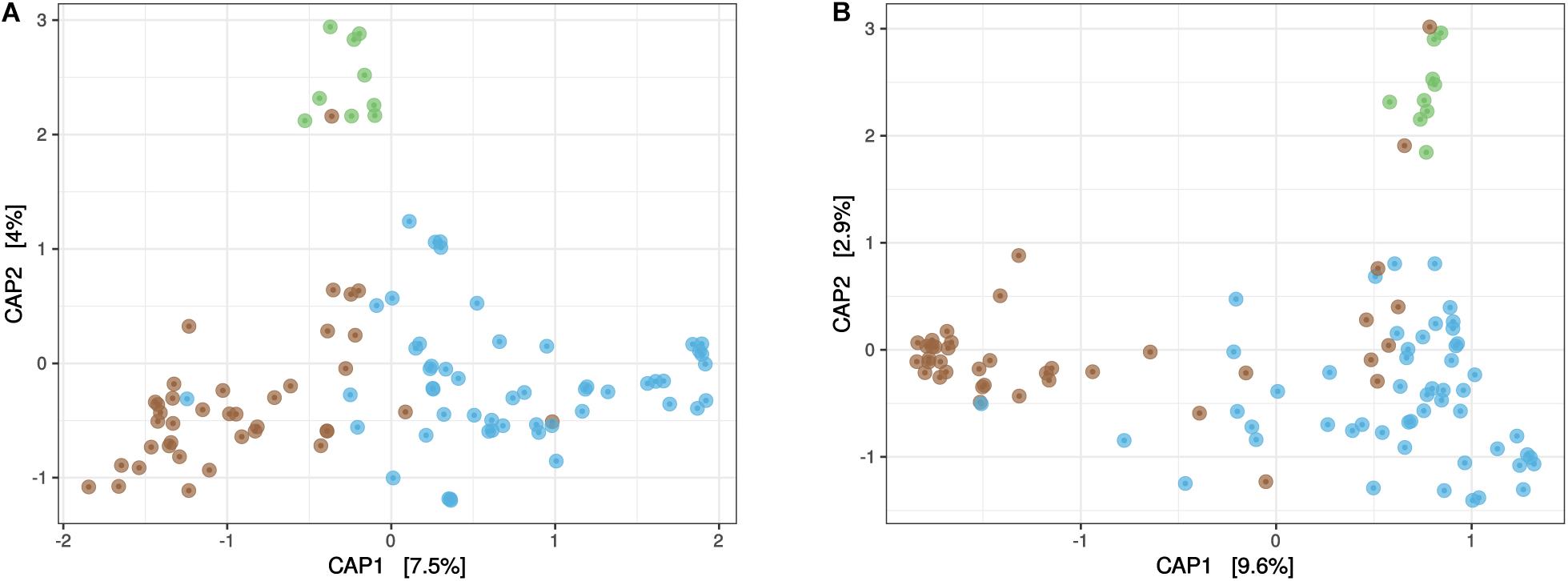
Figure 2. Differences in community composition. Both the prokaryotic community (A) and eukaryotic communities (B) presented significant differences (permutation tests for CAP, P < 0.001) between seawater at tide sites (TS-sw, in blue), Sargassum at marine tide sites (TS-sarg, in brown), and Sargassum at inland storage sites (ISS-sarg, in green).
The overall prokaryotic diversity corresponded to 658 bacterial and archaeal families distributed across 350 orders, and 57 phyla. For the TS-sarg samples we found that the most diverse phyla were Proteobacteria (34.3% of the richness in this compartment), Bacteroidetes (22.9%), and Planctomycetes (9.0%) (Figure 3A, also see Supplementary Figure 5A). At the phylum taxonomic rank, the diversity pattern present similarities with the TS-sw compartment, but differences were observed for the ISS-sarg samples (Figure 3A, and Supplementary Figure 5). Differences in diversity pattern of the three compartments were visible when analyzed at lower taxonomic ranks, with differences in the ten most rich orders (Supplementary Figure 6) or families (Supplementary Figure 7).
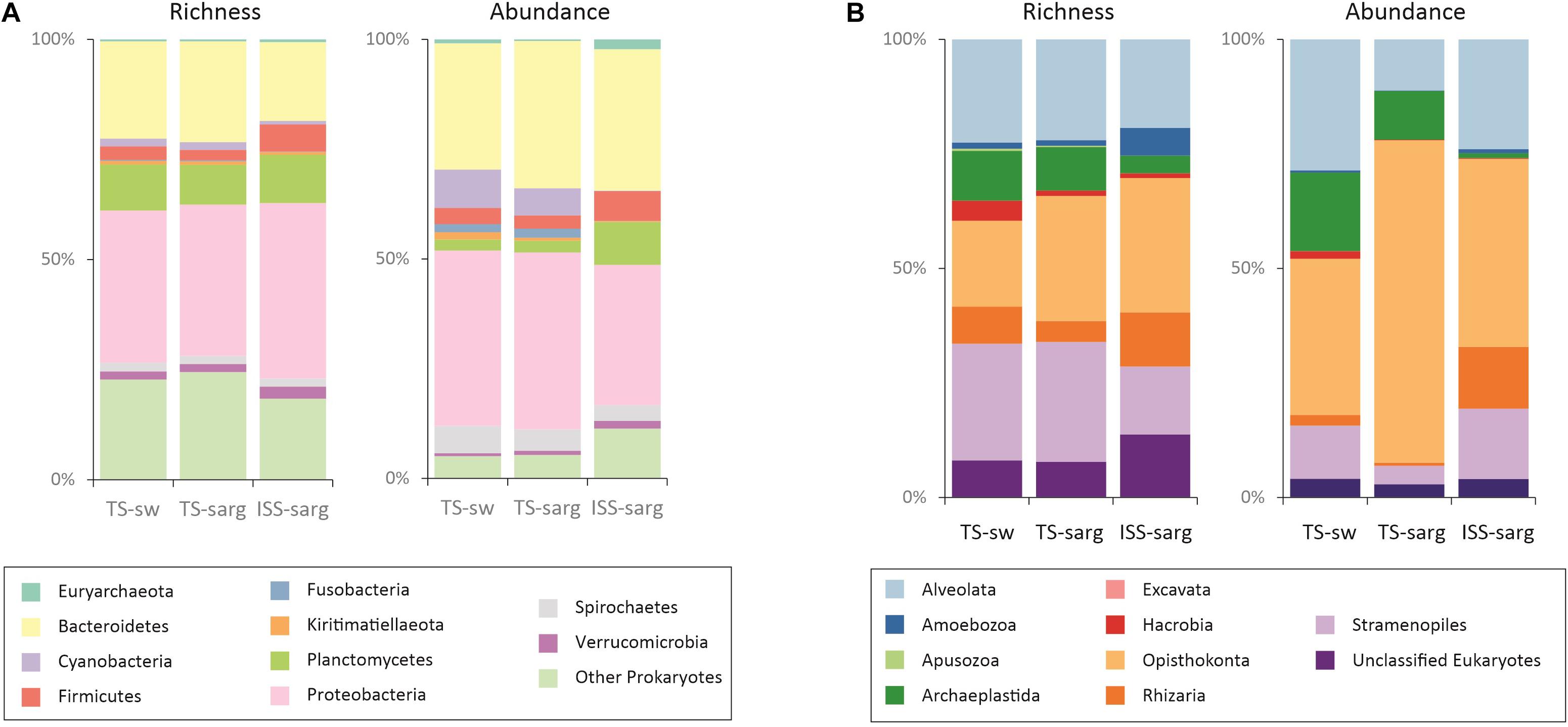
Figure 3. Molecular inventory of the biodiversity associated with Sargassum tides from 100 sampled sites. Differences in the observed OTU richness and relative abundance of seawater at tide sites (TS-sw), Sargassum at tide sites (TS-sarg) and inland Sargassum from storage sites (ISS-sarg). (A) Prokaryotic diversity corresponds to 22,214 OTUs obtained from 2,849,100 reads. (B) Eukaryotic diversity corresponds to 17,679 OTUs obtained from 5,326,300 reads.
In the samples from coastal tide sites (TS-sw and TS-sarg), the most abundant phyla were the Proteobacteria, followed by Bacteroidetes, Cyanobacteria, Spirochaetes and Firmicutes. For the inland storage sites (ISS-sarg), the most abundant phyla corresponded to Bacteroidetes, Proteobacteria, Planctomycetes, Firmicutes and Acidobacteria (Figure 3, also see Supplementary Figure 5B). In fact, differences in relative abundances between the three compartments and specially the terrestrial one, were discernible at all taxonomic levels analyzed, as shown at the order (Supplementary Figure 6) and family (Supplementary Figure 7) levels.
Even though there were differences in the number of samples per compartment, we analyzed the number of OTUs shared between them. The Venn diagram showed that 9.3% of the richness was shared between the three compartments (Supplementary Figure 8), corresponding to members of 4 archaeal and 41 bacterial phyla.
The eukaryotic diversity spread across 278 orders, with an overall dominance of Opisthokonta in terms of both richness (24.9%) and read abundance (48.9%), followed by the Alveolata (22.3% of richness and 21.4% of relative abundance), and the Stramenopiles (20.7% of richness and 9% of relative abundance). Independent analyses of the three compartments revealed differences in both OTU richness and relative abundance (Figure 3B, and Supplementary Figure 5). We found that 2.2% of the eukaryotic OTUs were present in all three compartments, a result which was lower than the one obtained for the prokaryotes (Supplementary Figure 8).
Specially in TS-sw samples we found that the phototrophs were among the most abundant organisms with key groups corresponding to Bacilliophyta, Florideophyceae, Ulvophyceae and Dinophyceae (Supplementary Figures 9, 10). In fact, in both kind of tide site samples (TS-sw and TS-sarg), the phylum that presented the highest richness were the Bacilliophyta (Supplementary Figure 9), with highest abundance of polar centric and raphid pennate diatoms in TS-sw and TS-sarg, respectively (Supplementary Figure 10). Compared to the tide site samples, differences in both richness and relative abundance were visible at all taxonomic ranks for the ISS-sarg ones (Figure 3B and Supplementary Figures 5–10).
Focusing on the Metazoa, we found strong differences between the compartments, with the Bryozoa dominating the TS-sarg samples (Supplementary Figure 11). A large abundance of Bryozoans associated with Sargassum (42% of average relative abundance for the TS-sarg samples) was expected because of the large number of them visible on axe, blade, receptacle and vesicle surfaces (see Supplementary Figure 12). Because the Nematoda were the most dominant group in terrestrial samples (Supplementary Figure 11), the taxonomy of the most abundant OTUs was refined by phylogenetic analysis and their putative membership to trophic groups were inferred following Meldal et al.’s (2007) classification (Figure 4). We identified 24 OTUs belonging to five orders, namely Enoplida, Chromadorida, Monhysterida, Tylenchida, and Rhabditida. Among the Monhysterida, ten OTUs including seven potentially bacterivorous were particularly abundant in TS-sarg. Conversely, seven Tylenchida OTUs, all assigned to the genus Halicephalobus, and the four Rhabditida OTUs were mainly present in the ISS-sarg samples. Besides bacterivores, we identified nematodes that are related to species known to be algivores-omnivores-predators, entomopathogens and vertebrate parasites (Figure 4).
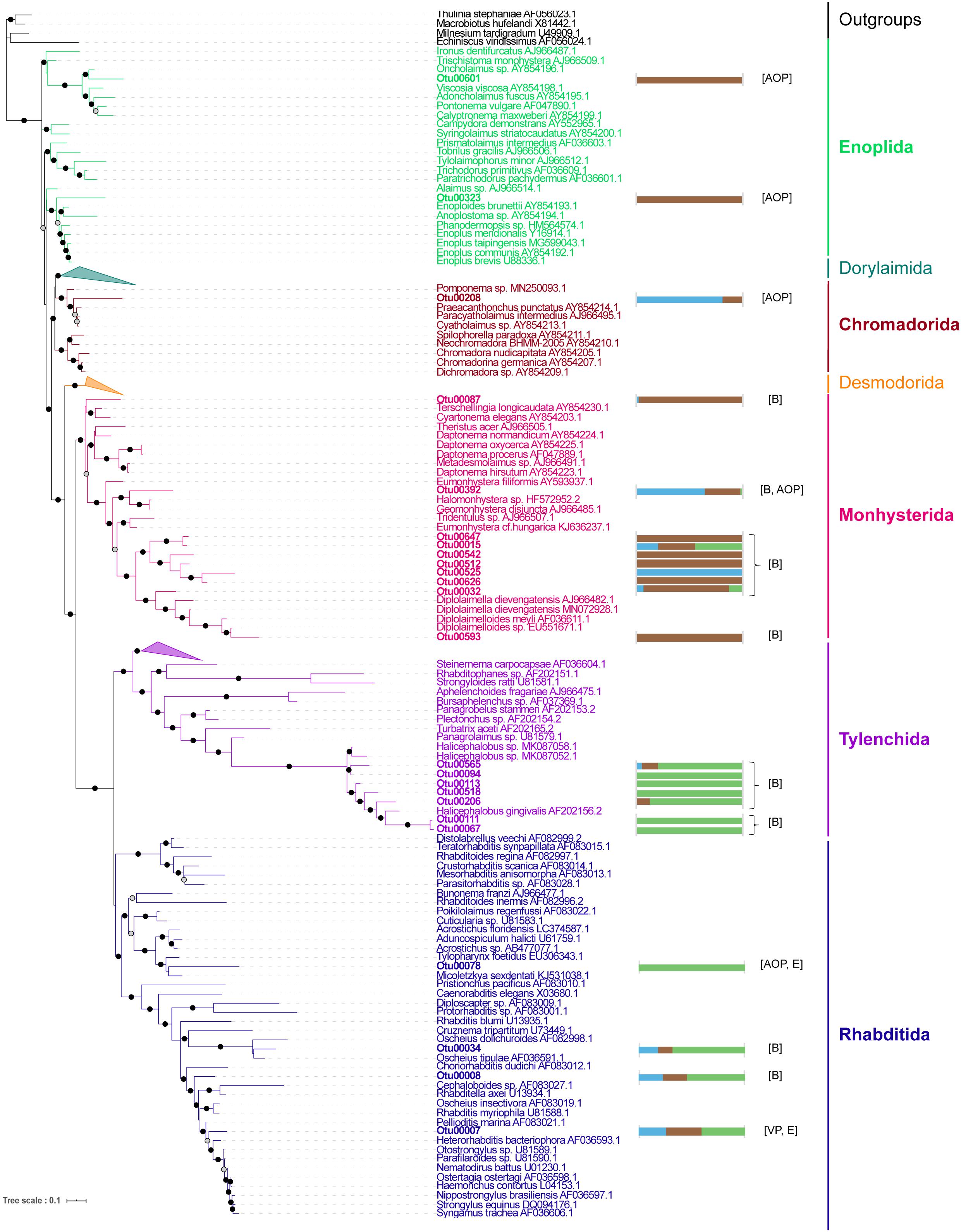
Figure 4. Phylogenetic relationships between the most abundant nematode OTUs associated with Sargassum. The maximum likelihood phylogenetic tree was constructed with PhyML v3.0 using the GTR + G + I model. Branch supports were calculated using a Chi2-based parametric approximate likelihood-ratio test and are represented by black circles for values above 0.90 and gray circles for values between 0.7 and 0.89. OTU sequences are in bold. The horizontal bar plots represent the normalized total abundance of each OTU in the different compartments: TS-sw in blue, TS-sarg in brown, and ISS-sarg in green. Putative trophic groups were assigned following recommendations by Meldal et al. (2007): algivore-omnivore-predator (AOP), bacterivore (B), entomopathogen (E), and vertebrate parasite (VP).
Because we demonstrated significant differences in community structure between the three compartment (Figure 2), we then investigated the OTUs that present differential abundance using ALDEx2 algorithm. We first made comparisons between the surrounding seawater and Sargassum at tide sites. We identified 68 prokaryotic and 30 eukaryotic OTUs with differential abundance (Figures 5A,B and Supplementary Table 2). For the prokaryotes, the TS-sw samples presented 46 OTUs that were significantly more abundant than in the TS-sarg samples (Figure 5A, blue bars). On the other hand, we found 22 OTUs preferentially associated with the TS-sarg samples compared to the seawater (Figure 5A, brown bars). In the case of the eukaryotes, the 20 OTUs were significantly more abundant with the TS-sw seawater compartment, and 10 OTUs for the TS-sarg one (Figure 5B).
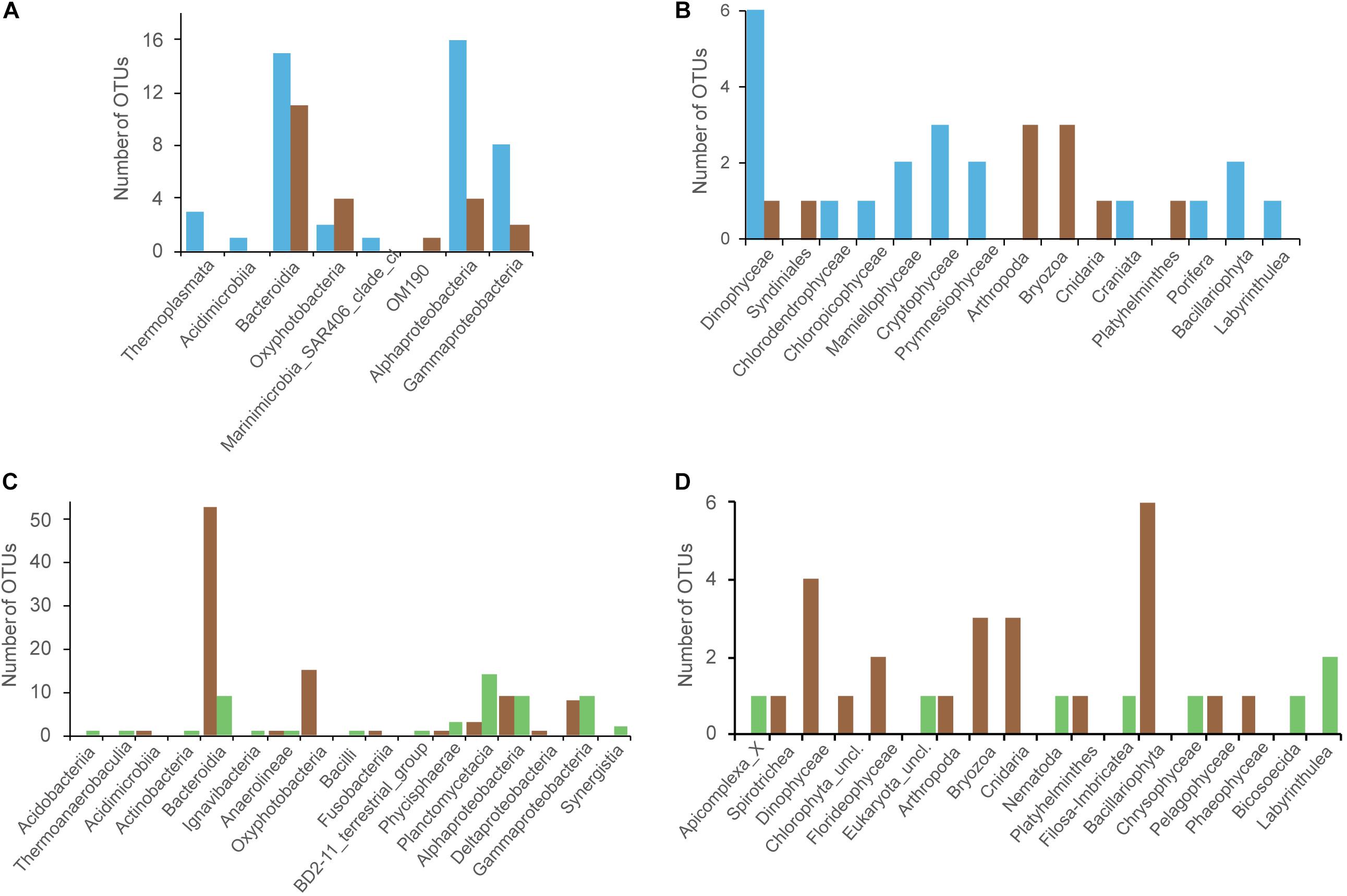
Figure 5. Identification of the OTUs presenting significant differential abundance for the different Sargassum-associated samples, via the ANOVA-like differential expression (ALDEx2) analysis. Comparison between tide sites OTUs that are differentially represented between seawater (TS-sw, blue) or Sargassum (TS-sarg, brown): (A) Prokaryotic, and (B) Eukaryotic. Analyses of the Sargassum-associated OTUs that are differentially represented between marine Sargassum (TS-sarg, brown) or terrestrial Sargassum (ISS-sarg, green): (C) Prokaryotic, and (D) Eukaryotic.
When comparing the communities associated to Sargassum collected at tide sites compared to Sargassum from inland storage sites, we identified 146 prokaryotic and 32 eukaryotic OTUs with differential abundance (Figures 5C,D and Supplementary Table 2). Coastal Sargassum samples showed a significant enrichment of 93 OTUs prokaryotic OTUs, including members of the Saprospiraceae, Flavobacteriaceae, Cyanobacteria, Alphaproteobacteria, and Gammaproteobacteria (Figure 5C). The prokaryotes found to be enriched in terrestrial samples represented 53 OTUs distributed over 13 classes (Figure 5C). For the eukaryotes, the marine Sargassum showed a significant enrichment of 24 OTUs, among them we found Dynophyceae, Bryozoans, Hydrozoans, or Bacillariophyceae (Figure 5D). The Sargassum samples from storage sites showed a significant enrichment of 8 OTUs, including one nematode (Figure 5D, green bars).
To investigate the putative roles of the prokaryotic taxa associated with the Sargassum samples, we used FAPROTAX and obtained putative functional assignment for 2,831 prokaryotic OTUs (12.7% of the global dataset) distributed into a minimum set of 22 functional groups (Figure 6 and Supplementary Table 3). The five most abundant ones corresponded to chemoheterotrophy (1,260 OTUs; 23.19% of the relative abundance), fermentation (314 OTUs; 13.04% of the relative abundance), nitrate reduction (71 OTUs; 9.32% of the relative abundance), phototrophy (388 OTUs; 6.98% of the relative abundance), and respiration of sulfur compounds (404 OTUs; 3.91% of the relative abundance) (Figure 6). Intracellular parasites, predators or exoparasites, and animal parasites or symbionts, corresponded to a relatively large number of organisms with 491, 263 and 189 OTUs, respectively. Interestingly, other functional groups specifically abundant in terrestrial samples corresponded to methanogenesis (32 OTUs; 0.21%), hydrocarbon degradation (21 OTUs; 0.31%), and dark oxidation of sulfur compounds (20 OTUs; 0.09%).
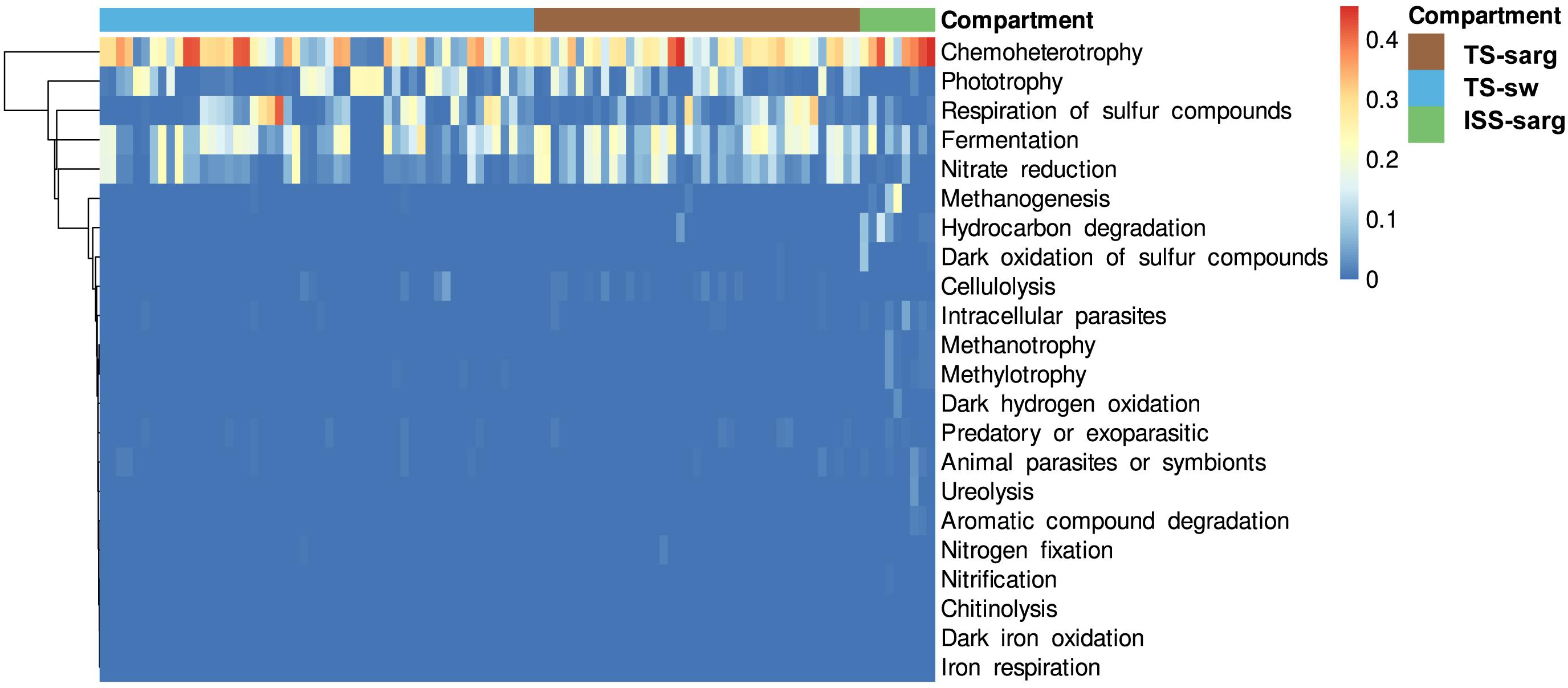
Figure 6. Heatmap of predicted functions based on the FAPROTAX database. The 22 categories presented here correspond to 2,831 prokaryotic OTUs. Functions were clustered with the weighted pair group method centroid (WPGMC) algorithm based on Euclidean distances. The color scale represents the proportion of each predicted function within a sample.
Sargassum washout in the coast of the Caribbean region, including the Gulf of Mexico, and in the coasts of Africa are causing many environmental and health concerns. Part of these concerns are probably the results of the load of new microbes associated with the seaweed, and the putative transient or persistent modifications of the microbial communities at tide sites. Here we investigated samples from three different compartments, namely from seawater and Sargassum from tide sites (TS-sw and TS-sarg samples, respectively) and Sargassum from inland storage sites (ISS-sarg samples). We found that the alpha diversity was similar for prokaryotes in the three compartments, and that the richness was higher for eukaryotes in seawater compared to the Sargassum-related samples. Beta diversity analyses revealed clear distinctions between the three compartments and proved a correlation between eukaryotic and prokaryotic communities. When analyzed at the high taxonomic ranks, compositional analyses revealed some similarities between the Sargassum and the surrounding water, even if differences exist at lower taxonomic ranks, also see below. This result somehow contrasted with previous reports on Sargassum (Torralba et al., 2017; Michotey et al., 2020), but those corresponded to analyses of Sargassum that were collected in offshore regions. In fact, beside the well documented technical biases when comparing samples across studies (DNA extraction, primer choice, PCR program, sequencing platform, etc.), we believe that differences between published data on the Sargassum microbiota could be due to a number ecological reasons including spatial and temporal variability in Sargassum-associated communities (Huffard et al., 2014; Torralba et al., 2017; Serebryakova et al., 2018; Mei et al., 2019; Michotey et al., 2020), biogeochemical factors at tide sites compared to offshore, or aging and drowning of the seaweeds (Baker et al., 2018; Sanders-Smith et al., 2020).
The most abundant prokaryotic family associated with the Sargassum samples from tide sites (TS-sarg) was the Vibrionaceae (Proteobacteria) (ca. 18% of the relative abundance, see Supplementary Figure 7), a family of ubiquitous marine bacteria. The second most abundant family was the Flavobacteriaceae (Bacteroidetes). Interestingly, utilization of macromolecules such as polysaccharides is a common feature of many members of this family (McBride, 2014; Thomas et al., 2021), and more generally of the Bacteroidetes (McKee et al., 2021). In fact, several genera of the Flavobacteriaceae that were found to possess conserved polysaccharide utilization loci, as defined by Kruger et al. (2019), were abundant at tide sites, with the NS5_marine_group, Polaribacter spp., Formosa spp., Dokdonia spp., Gramella spp., Cellulophaga spp., Lutibacter spp., Winogradskyella spp., Cryomorphaceae, Crocinitomicaceae, and Cyclobacteriaceae together making up to 12.9 and 4.3% of the relative abundance in TS-sw and TS-sarg samples, respectively. These results suggest that these bacteria might be involved in the degradation of polysaccharides either exudate or release by the seaweeds and the associated epifauna, and probably contribute to Sargassum degradation. The third most abundant family was the Saprospiraceae (Bacteroidetes), another bacterial clade harboring species capable of degrading complex macromolecules produced by seaweeds and their associated algae (Mcilroy and Nielsen, 2014). The Flavobacteriaceae (Bacteroidetes) family was the most abundant for terrestrial storage sites (Supplementary Figure 7), again suggesting a possible contribution of some of these bacteria to organic matter decay. Further studies should address the shift in microbial communities in the water column of coastal areas, depending on the Sargassum morphotypes, algal biomass and aging.
Using FAPROTAX, we obtained functional prediction for a number of prokaryotic OTUs. We found that the most abundant ones corresponded to chemoheterotrophy, nitrate reduction, phototrophy, and respiration of sulfur compounds. Interestingly, putative intracellular parasites, predators or exoparasites, and animal parasites or symbionts, corresponded to a relatively large number of organisms. Within terrestrial samples, these functional predictions identified abundant OTUs (2.0% of the relative abundance in ISS-sarg) potentially involved in methanogenesis, and corresponding to euryarchaeota assigned to Methanomicrobiaceae, Methanosarcinaceae, and Methanococcaceae. Members of these families have been reported to be involved in methane production from algal biomass (Bapteste et al., 2005; Adam et al., 2017; Klassen et al., 2017). Our results suggest that at inland storage sites, a portion of the carbohydrates from the seaweed alga biomass (for Sargassum see Devault et al., 2021, and references therein) is probably degraded anaerobically by these archaea, and used as potential energy source (Smetacek and Zingone, 2013; Helbert, 2017; Milledge et al., 2020).
When washed ashore, Sargassum decomposition processes can lead to hydrogen sulfide (H2S) and ammonia (NH3) gas emissions, which can result in significant environmental issues (Van Tussenbroek et al., 2017; Cabanillas-Teran et al., 2019; Rodriguez-Martinez et al., 2019) and health concerns (Resiere et al., 2018, 2020). As mentioned above, functional prediction identified OTUs involved in the respiration of sulfur compounds (Figure 6), which represent 3.8, 4.7, and 0.9% of the relative abundance in TS-sarg, TS-sarg and ISS-sarg compartment, respectively. These OTUs were assigned to several orders including Desulfobacterales (257 OTUs), Desulfovibrionales (87 OTUs), Clostridiales (51 OTUs), Desulfarculales (35 OTUs), Desulfuromonadales (3 OTUs), Syntrophobacterales (3 OTUs), and Thermococcales (1 OTU). Member of these orders, except the Thermococcales were present in the three compartments. In addition to the FAPROTAX predictions, we found in the three compartments additional OTUs assigned to Sulfurovum and Sulfurimonas (Campylobacterales) which are genera known to be involved in sulfur oxidation, and thus probably also contribute to the sulfur cycle in our samples (Han and Perner, 2015). A deeper understanding of the diversity of sulfate-reducing and sulfur-oxidizing microorganisms associated with Sargassum decomposition processes will require specific investigations, such as the presence of functional sulfur cycling-related genes (Pelikan et al., 2016). Furthermore, it will be important to further link the sulfur cycle to several others such as carbon, metals and metalloids such as arsenic (As) (Fisher et al., 2008; Edwardson and Hollibaugh, 2017). This issue seems particularly relevant to us given the large amount of arsenic that bioaccumulates in various Sargassum species (see Rodriguez-Martinez et al., 2020; Devault et al., 2021, and our unpublished data for Guadeloupe and Martinique).
Nitrate reduction is another predicted functional role that might be played by prokaryotes. The 71 OTUs functionally predicted to contribute to nitrate reduction belong to 16 bacterial families (Supplementary Table 3) with the most abundant OTUs assigned to the genus Vibrio (4.6 and 17.4% of the relative abundance in TS-sw and TS-sarg). This genus, was found to present variable and eventually very high abundance in pelagic Sargassum samples (Michotey et al., 2020), could contribute to nitrogen cycling (Urdaci et al., 1988; Herbert, 1999) and potentially to the host nitrogen metabolism. Vibrios have been shown to also contribute to the marine organic carbon cycle (Martin et al., 2015; Zhang et al., 2018), and to potential sanitary risks because many of them are pathogenic to animals or humans (Baker-Austin et al., 2017). Regarding the nitrogen cycle, we also found 12 prokaryotic OTUs potentially involved in nitrogen fixation as well as 19 OTUs potentially involved in nitrification. Recent analyses have shown that %N has increased over the years in Sargassum and that this percentage is higher in Sargassum from coastal waters (see Lapointe et al., 2021 and references therein) raising the importance to further study N metabolism of landing Sargassum.
The eukaryotic community associated to Sargassum has been rarely investigated by molecular approaches (Kim H. M. et al., 2019). Here we demonstrate that the eukaryotic communities of Sargassum samples is dominated by Metazoa, with 24.8% of diversity and 69.9% of the relative abundance from tide sites, and 20.3% of diversity and 38.0% of the relative abundance for the inland storage sites. We expected that some of them correspond to the non-motile and motile epifauna species (Stoner and Greening, 1984; Martinsmith, 1994; Gestoso et al., 2012; Martin et al., 2021). Indeed, crustacea were the most abundant clade in the TS-sw and the second most abundant in the TS-sarg samples, and corresponded essentially to the class Maxillopoda. Copepods (Maxillopoda) have already been described as an abundant invertebrate fauna associated with floating Sargassum (Ólafsson et al., 2001; Abé et al., 2013). Among the 555 Maxillopoda OTUs found from the tide site samples, 16.4% were shared between the seawater (TS-sw) and Sargassum (TS-sarg) samples, and 36.9% were exclusive to TS-sarg. The most abundant copepod that was found to be shared between these two compartments was assigned to the genus Zaus (best blast hit), a genus already found to be associated with macroalgae (Kangtia et al., 2015). We hypothesized that colonization of Sargassum by copepods could have occurred during rafting, explaining why different communities could be found between Sargassum and seawater at tide sites. Alternatively, some of the copepods identified might correspond to genuine coastal species, and that for some of them the arrival of Sargassum and macroalgal detritus could have correspond to new ecological niches to colonize.
Association between nematodes and floating Sargassum in the Atlantic region was already noted a century ago (Micoletzky, 1922). The colonization of such usually benthic meiofauna could be the result of accidental encounters and dispersal when clumps of algae are carried by wind or currents. Indeed, benthic nematode species were previously found attached to the surface of pelagic or benthic Sargassum spp. (Gerlach, 1977; Kito, 1982; Vranken et al., 1986; Kim H. G. et al., 2019). Within our dataset, we specifically investigated the most abundant nematode-related OTUs and, interestingly, found specific patterns depending on the substrate (Figure 4). The nematodes that were found to be associated with Sargassum at tide sites belong to the Enoplia and Monhysterida clades that have been described in marine habitats. The trophic behaviors of species from these clades have been shown to correspond to bacterivore and algivore-omnivore-predator (Meldal et al., 2007; Smythe, 2015). Nematodes were largely dominant in terrestrial samples, with 92% of the metazoan’s read abundance. OTUs assigned to Tylenchina, which corresponds mostly to plants, invertebrate and vertebrate parasites, and to Rhabditina, which are generally terrestrial free-living nematode species (Meldal et al., 2007), showed exclusive or specific patterns with inland Sargassum samples (Figure 4). Regarding the OTUs related to Halicephalobus, the best blast hit for Otu00067 and Otu00111 was Halicephalobus sp. (accession number GQ918144), a bacterivore nematode that has been found in the terrestrial deep subsurface in hypoxic conditions (Borgonie et al., 2011). The hypoxic conditions within the piles of Sargassum at storage sites are likely to be one of the important factors explaining differences in the distribution of nematode species within compartments.
In the present study we also identified OTUs that presented differential abundance at tide sites, or between coastal and terrestrial Sargassum samples. In total, we found 199 prokaryotic and 56 eukaryotic OTUs with differential abundance in our compartments (Supplementary Table 2). At tide sites, the eukaryotic OTUs significantly more abundant in the surrounding water corresponded to Dinophyceae, unclassified Maxillipoda, Bryozoa [probably to the genus Membranipora, a hydrozoan of the genus Zanclea that was previously described as being common on a hydroid assemblage on holopelagic Sargassum from the Sargasso Sea (Calder, 1995)], and one OTU of flatworm of the Rhabdocoela order. Some of these later species are known to live in association with Sargassum (Willems et al., 2004). Among the 24 OTUs that were significantly associated with the TS-sarg samples, we found one OTU assigned to a toxic dinoflagellate of the Amphidinium carterae species, another to Ulvella that are endophytic microalgae, one related to the raphid diatom genus Aneumastus, one OTU with the best BLASTN results as Sargassococcus simulans, an epiphyte on floating Sargassum thallus, isolated in the Sargasso Sea, one to the copepod genus Zaus, which are organisms living in the phytal zone, and five OTUs belonging to Hydrozoans, which are also frequently encountered as Sargassum epibionts (Huffard et al., 2014). The Sargassum samples from storage sites showed significant enrichment of 8 OTUs, including a flagellated protist of the Colpodellidae order, a nematode assigned to the Monhysterida order, and a filose amoeba.
Among the prokaryotes differentially abundant in Sargassum from tide sites, we identified several Oxyphotobacteria and Saprospiraceae which have often been reported as associated with macroalgae surface (Florez et al., 2017). Members of these family were also differentially abundant when comparing Sargassum samples between tide sites and inland storage sites. In particular, we found at tide sites 3 Rubinisphaeraceae OTUs preferentially associated with inland storage sites, and this family of Planctomycetes was also recently found in wastewater treatment plants (Al Ali et al., 2020), a putatively new member of the Rivularia-like cyanobacterium, and members of Rhizobiales Altogether, our data revealed a number of potential new molecular markers associated with Sargassum racks from the Caribbean. Even if further researches will have to address more specifically the intra and inter-sites variability as well as the variation of the communities depending on Sargassum morphotypes, we have identified here a number of potentially new or poorly described species, with some of these taxa performing selective functions for the host and/or involved in Sargassum degradation processes.
Our study revealed specific micro- and meiofauna depending on the Sargassum samples. We also identified prokaryotic (archaea and bacteria) and eukaryotic biomarkers for marine waters at tide sites, near-shore and beach stranded wracks, and at inland storage sites. The description of this biodiversity provides a grounding for further investigations of the functions played by the epiphytes for the host, algal organic matter decomposition or nutrient transfers between trophic levels at both sea to land sites. Among other things, comparative and targeted investigations on biogeography and Sargassum holobionte composition, as well as the role of the degradation processes are required to further elucidate the environmental changes induced by Sargassum tides and their local consequences on biodiversity.
The datasets presented in this study can be found in online repositories. The names of the repository and accession numbers can be found in the article/Supplementary Material.
VH and PJL: conceptualization and investigations. VH, JL, MR-T, DAD, and PJL: sampling. VH, JL, and PJL: methodology and writing—original draft. PJL: funding acquisition. All authors contributed to the article and approved the submitted version.
This work was supported by the Labex DRIIHM, the French “Investissements d’Avenir” program (ANR-11-LABX-0010), managed by the ANR and the Human-Environment Observatory Littoral Caraïbe. We also acknowledge the financial support of the Agence de l’Environnement et de la Maîtrise de l’Energie (ADEME).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.701155/full#supplementary-material
Abé, H., Komatsu, T., Kokubu, Y., Natheer, A., Rothäusler, E. A., Shishido, H., et al. (2013). Invertebrate fauna associated with floating Sargassum horneri (Fucales: Sargassaceae) in the east China Sea. Species Divers. 18, 75–85. doi: 10.12782/sd.18.1.075
Adam, P. S., Borrel, G., Brochier-Armanet, C., and Gribaldo, S. (2017). The growing tree of Archaea: new perspectives on their diversity, evolution and ecology. ISME J. 11, 2407–2425. doi: 10.1038/ismej.2017.122
Al Ali, A. A., Naddeo, V., Hasan, S. W., and Yousef, A. F. (2020). Correlation between bacterial community structure and performance efficiency of a full-scale wastewater treatment plant. J. Water Process Enginee. 37:101472. doi: 10.1016/j.jwpe.2020.101472
Amaral-Zettler, L. A., Dragone, N. B., Schell, J., Slikas, B., Murphy, L. G., Morrall, C. E., et al. (2017). Comparative mitochondrial and chloroplast genomics of a genetically distinct form of Sargassum contributing to recent “Golden Tides” in the Western Atlantic. Ecol. Evolut. 7, 516–525. doi: 10.1002/ece3.2630
Anisimova, M., and Gascuel, O. (2006). Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552. doi: 10.1080/10635150600755453
Baker, P., Minzlaff, U., Schoenle, A., Schwabe, E., Hohlfeld, M., Jeuck, A., et al. (2018). Potential contribution of surface-dwelling Sargassum algae to deep-sea ecosystems in the southern North Atlantic. Deep Sea Res. Part II Topic. Stud. Oceanogr. 148, 21–34. doi: 10.1016/j.dsr2.2017.10.002
Baker-Austin, C., Trinanes, J., Gonzalez-Escalona, N., and Martinez-Urtaza, J. (2017). Non-Cholera Vibrios: The Microbial Barometer of Climate Change. Trends Microbiol. 25, 76–84. doi: 10.1016/j.tim.2016.09.008
Ballard, S. E., and Rakocinski, C. F. (2012). Flexible feeding strategies of juveline gray triggerfish (Balistes capriscus) and planehead filefish (Stephanolepis huspidus) with Sargassum habitat. Gulf Caribb. Res. 24, 31–40. doi: 10.18785/gcr.2401.05
Bapteste, E., Brochier, C., and Boucher, Y. (2005). Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea 1, 353–363. doi: 10.1155/2005/859728
Borgonie, G., Garcia-Moyano, A., Litthauer, D., Bert, W., Bester, A., Van Heerden, E., et al. (2011). Nematoda from the terrestrial deep subsurface of South Africa. Nature 474, 79–82. doi: 10.1038/nature09974
Cabanillas-Teran, N., Hernandez-Arana, H. A., Ruiz-Zarate, M. A., Vega-Zepeda, A., and Sanchez-Gonzalez, A. (2019). Sargassum blooms in the Caribbean alter the trophic structure of the sea urchin Diadema antillarum. PeerJ 7:e7589. doi: 10.7717/peerj.7589
Calder, D. R. (1995). Hydroid assemblages on holopelagic Sargassum from the Sargasso Sea at Bermuda. Bull. Mar. Sci. 56, 537–546.
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Casazza, T. L., and Ross, S. (1972). Fishes associated with the pelagic Sargassum complex with a discussion of Sargassum community. Fishery Bull. 16:1.
Coelho-Souza, S. A., Jenkins, S. R., Casarin, A., Baeta-Neves, M. H., Salgado, L. T., Guimaraes, J. R. D., et al. (2017). The effect of light on bacterial activity in a seaweed holobiont. Microb. Ecol. 74, 868–876. doi: 10.1007/s00248-017-0995-x
Damare, V. S. (2015). Diversity of thraustochytrid protists isolated from brown alga, Sargassum cinereum using 18S rDNA sequencing and their morphological response to heavy metals. J. Mar. Biol. Associat. U K. 95, 265–276. doi: 10.1017/S0025315414001696
De Fouw, J., Govers, L. L., Van De Koppel, J., Van Belzen, J., Dorigo, W., Sidi Cheikh, M. A., et al. (2016). Drought, mutualism breakdown, and landscape-scale degradation of seagrass beds. Curr. Biol. 26, 1051–1056. doi: 10.1016/j.cub.2016.02.023
Devault, D. A., Pierre, R., Marfaing, H., Dolique, F., and Lopez, P. J. (2021). Sargassum contamination and consequences for downstream uses: a review. J. Appl. Phycol. 33, 567–602. doi: 10.1007/s10811-020-02250-w
Edwardson, C. F., and Hollibaugh, J. T. (2017). Metatranscriptomic analysis of prokaryotic communities active in sulfur and arsenic cycling in Mono Lake, California, USA. ISME J. 11, 2195–2208. doi: 10.1038/ismej.2017.80
Egan, S., Harder, T., Burke, C., Steinberg, P., Kjelleberg, S., and Thomas, T. (2013). The seaweed holobiont: understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 37, 462–476. doi: 10.1111/1574-6976.12011
Fernandes, A. D., Reid, J. N. S., Macklaim, J. M., Mcmurrough, T. A., Edgell, D. R., and Gloor, G. B. (2014). Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2:15.
Fine, M. L. (1970). Faunal variation on pelagic Sargassum. Mar. Biol. 7, 112–122. doi: 10.1007/BF00354914
Fisher, J. C., Wallschlager, D., Planer-Friedrich, B., and Hollibaugh, J. T. (2008). A new role for sulfur in arsenic cycling. Environ. Sci. Technol. 42, 81–85. doi: 10.1021/es0713936
Fitzpatrick, C. R., Lu-Irving, P., Copeland, J., Guttman, D. S., Wang, P. W., Baltrus, D. A., et al. (2018). Chloroplast sequence variation and the efficacy of peptide nucleic acids for blocking host amplification in plant microbiome studies. Microbiome 6:144. doi: 10.1186/s40168-018-0534-0
Florez, J. Z., Camus, C., Hengst, M. B., and Buschmann, A. H. (2017). A functional perspective analysis of macroalgae and epiphytic bacterial community interaction. Front. Microbiol. 8:2561. doi: 10.3389/fmicb.2017.02561
Gerlach, S. A. (1977). Attraction of decaying organisms as a possible cause for pachy distribution of nematodes in a Bermuda beach. Ophelia 16, 151–165. doi: 10.1080/00785326.1977.10425467
Gestoso, I., Olabarria, C., and Troncoso, J. S. (2012). Effects of macroalgal identity on epifaunal assemblages: native species versus the invasive species Sargassum muticum. Helgoland Mar. Res. 66, 159–166. doi: 10.1007/s10152-011-0257-0
Goslee, S. C., and Urban, D. L. (2007). The ecodist package for dissimilarity-based analysis of ecological data. J. Statist. Softw. 22, 1–19. doi: 10.18637/jss.v022.i07
Guillou, L., Bachar, D., Audic, S., Bass, D., Berney, C., Bittner, L., et al. (2013). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/nar/gks1160
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Han, Y., and Perner, M. (2015). The globally widespread genus Sulfurimonas: versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 6:989. doi: 10.3389/fmicb.2015.00989
Haney, J. C. (1986). Seabird patchiness in tropical oceanic waters - the influence of Sargassum “reefs”. Auk 103, 141–151. doi: 10.1093/auk/103.1.141
Helbert, W. (2017). Marine Polysaccharide Sulfatases. Front. Mar. Sci. 4:00006. doi: 10.3389/fmars.2017.00006
Herbert, R. A. (1999). Nitrogen cycling in coastal marine ecosystems. FEMS Microbiol. Rev. 23, 563–590. doi: 10.1111/j.1574-6976.1999.tb00414.x
Hu, Z.-M., and Fraser, C. (2016). Seaweed phylogeography. Adaptation and evolution of seaweeds under environmental change. Berlin: Springer Netherlands. doi: 10.1007/978-94-017-7534-2
Huffard, C. L., Von Thun, S., Sherman, A. D., Sealey, K., and Smith, K. L. (2014). Pelagic Sargassum community change over a 40-year period: temporal and spatial variability. Mar. Biol. 161, 2735–2751. doi: 10.1007/s00227-014-2539-y
Jacobucci, G., and Leite, F. (2014). The role of epiphytic algae and different species of Sargassum in the distribution and feeding of herbivorous amphipods. Latin Am. J. Aquat. Res. 42, 353–363. doi: 10.3856/vol42-issue2-fulltext-6
Jobe, C. F., and Brooks, W. R. (2009). Habitat selection and host location by symbiotic shrimps associated with Sargassum communities: The role of chemical and visual cues. Symbiosis 49, 77–85. doi: 10.1007/s13199-009-0017-y
Kangtia, P., Dahms, H. U., Song, S. J., Myoung, J. G., Park, J., and Khim, J. S. (2015). On the occurrence of a new species of benthic copepod, Zaus wonchoelleei (Harpacticoida, Harpacticidae), in a macroalgal habitat from Tongyong, Korea. Proc. Biol. Soc. Washington 127, 585–602. doi: 10.2988/0006-324X-127.4.585
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evolut. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, H. G., Hawkins, L. E., Godbold, J. A., Oh, C. W., Rho, H. S., and Hawkins, S. J. (2019). Comparison of nematode assemblages associated with Sargassum muticum in its native range in South Korea and as an invasive species in the English Channel. Mar. Ecol. Prog. Ser. 611, 95–110. doi: 10.3354/meps12846
Kim, H. M., Jo, J., Park, C., Choi, B. J., Lee, H. G., and Kim, K. Y. (2019). Epibionts associated with floating Sargassum horneri in the Korea Strait. Algae 34, 303–313. doi: 10.4490/algae.2019.34.12.10
Kito, K. (1982). Phytal marine nematode assemblage on Sargassum confusum Agardh, with reference to the structure and seasonal fluctuations. J. Facul. Sci. Hokkaido Univers. 23, 143–161.
Klassen, V., Blifernez-Klassen, O., Wibberg, D., Winkler, A., Kalinowski, J., Posten, C., et al. (2017). Highly efficient methane generation from untreated microalgae biomass. Biotechnol. Biofuels 10:186. doi: 10.1186/s13068-017-0871-4
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Kruger, K., Chafee, M., Ben Francis, T., Del Rio, T. G., Becher, D., Schweder, T., et al. (2019). In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. ISME J. 13, 2800–2816. doi: 10.1038/s41396-019-0476-y
Lapointe, B. E., Brewton, R. A., Herren, L. W., Wang, M., Hu, C., Mcgillicuddy, D. J., et al. (2021). Nutrient content and stoichiometry of pelagic Sargassum reflects increasing nitrogen availability in the Atlantic Basin. Nat. Communicat. 12:3060. doi: 10.1038/s41467-021-23135-7
Lefort, V., Longueville, J. E., and Gascuel, O. (2017). SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Maples, R. S. (1984). The epiphytic diatom flora of two Sargassum species. Gulf Res. Rep. 7, 373–375. doi: 10.18785/grr.0704.09
Martin, L. M., Taylor, M., Huston, G., Goodwin, D. S., Schell, J. M., and Siuda, A. N. S. (2021). Pelagic Sargassum morphotypes support different rafting motile epifauna communities. Mar. Biol. 168, 3910–3912. doi: 10.1007/s00227-021-03910-2
Martin, M., Barbeyron, T., Martin, R., Portetelle, D., Michel, G., and Vandenbol, M. (2015). The cultivable surface microbiota of the brown alga Ascophyllum nodosum is Enriched in Macroalgal-Polysaccharide-Degrading Bacteria. Front. Microbiol. 6:1487. doi: 10.3389/fmicb.2015.01487
Martinsmith, K. M. (1994). Short-term dynamics of tropical macroalgal epifauna: patterns and processes in recolonization of Sargassum fissifolium. Mar. Ecol. Prog. Ser. 110, 177–185. doi: 10.3354/meps110177
McBride, M. J. (2014). “The Family Flavobacteriaceae,” in The Prokaryotes, eds E. Rosenberg, E. F. Delong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer).
Mcilroy, S. J., and Nielsen, P. H. (2014). “The Family Saprospiraceae,” in The Prokaryotes, eds E. Rosenberg, E. F. Delong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 863–889. doi: 10.1007/978-3-642-38954-2_138
McKee, L. S., La Rosa, S. L., Westereng, B., Eijsink, V. G., Pope, P. B., and Larsbrink, J. (2021). Polysaccharide degradation by the Bacteroidetes: mechanisms and nomenclature. Environ. Microbiol. Rep. 13, 559–581. doi: 10.1111/1758-2229.12980
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Mei, X., Wu, C., Zhao, J., Yan, T., and Jiang, P. (2019). Community structure of bacteria sssociated with drifting Sargassum horneri, the causative species of golden tide in the Yellow Sea. Front. Microbiol. 10:1192. doi: 10.3389/fmicb.2019.01192
Meldal, B. H., Debenham, N. J., De Ley, P., De Ley, I. T., Vanfleteren, J. R., Vierstraete, A. R., et al. (2007). An improved molecular phylogeny of the Nematoda with special emphasis on marine taxa. Mol. Phylogenet. Evol. 42, 622–636. doi: 10.1016/j.ympev.2006.08.025
Michotey, V., Blanfune, A., Chevalier, C., Garel, M., Diaz, F., Berline, L., et al. (2020). In situ observations and modelling revealed environmental factors favouring occurrence of Vibrio in microbiome of the pelagic Sargassum responsible for strandings. Sci. Total Environ. 748:141216. doi: 10.1016/j.scitotenv.2020.141216
Micoletzky, H. (1922). Freilebende Nematoden aus den treibenden Tangen der Sargassosee. Mitteilungen Hamburger Zoologischen Museums Instit. 39, 1–11.
Milledge, J. J., Maneein, S., Lopez, E. A., and Bartlett, D. (2020). Sargassum inundations in turks and caicos: methane potential and proximate, ultimate, lipid, amino acid, metal and metalloid analyses. Energies 13:1523. doi: 10.3390/en13061523
Monroy-Velazquez, L. V., Rodriguez-Martinez, R. E., Van Tussenbroek, B. I., Aguiar, T., Solis-Weiss, V., and Briones-Fourzan, P. (2019). Motile macrofauna associated with pelagic Sargassum in a Mexican reef lagoon. J. Environ. Manage. 252:109650. doi: 10.1016/j.jenvman.2019.109650
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R. B., et al. (2015). vegan: community ecology package. R package version 2.3-2. Vienna: R Core Team.
Ólafsson, E., Ingólfsson, A., and Steinarsdóttir, M. B. (2001). Harpacticoid copepod communities of floating seaweed: Controlling factors and implications for dispersal. Hydrobiologia 45, 189–200. doi: 10.1023/A:1013196724039
Parr, A. E. (1939). Quantitative observations on the pelagic Sargassum vegetation of the Western North Atlantic. New Haven: Yale University.
Pelikan, C., Herbold, C. W., Hausmann, B., Muller, A. L., Pester, M., and Loy, A. (2016). Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrA and dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics. Environ. Microbiol. 18, 2994–3009. doi: 10.1111/1462-2920.13139
Pruesse, E., Peplies, J., and Glockner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Resiere, D., Valentino, R., Banydeen, R., Megarbane, B., Neviere, R., and Mehdaoui, H. (2020). Long-term exposure to Sargassum-seaweed pollution in the French Caribbean Islands: clinical consequences and outcome. Clin. Toxicol. 58, 550–550.
Resiere, D., Valentino, R., Neviere, R., Banydeen, R., Gueye, P., Florentin, J., et al. (2018). Sargassum seaweed on Caribbean islands: an international public health concern. Lancet 392, 2691–2691. doi: 10.1016/S0140-6736(18)32777-6
Rodriguez-Martinez, R. E., Medina-Valmaseda, A. E., Blanchon, P., Monroy-Velazquez, L. V., Almazan-Becerril, A., Delgado-Pech, B., et al. (2019). Faunal mortality associated with massive beaching and decomposition of pelagic Sargassum. Mar. Pollut. Bull. 146, 201–205. doi: 10.1016/j.marpolbul.2019.06.015
Rodriguez-Martinez, R. E., Roy, P. D., Torrescano-Valle, N., Cabanillas-Teran, N., Carrillo-Dominguez, S., Collado-Vides, L., et al. (2020). Element concentrations in pelagic Sargassum along the Mexican Caribbean coast in 2018-2019. PeerJ 8:e8667. doi: 10.7717/peerj.8667
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahe, F. (2016). VSEARCH: a versatile open source tool for metagenomics. Peerj 4:e2584.
Ryland, J. S. (1974). Observations on some epibionts of gulf-weed, Sargassum natans (L) Meyen. J. Exp. Mar. Biol. Ecol. 14, 17–25. doi: 10.1016/0022-0981(74)90034-3
Sanders-Smith, R., Segovia, B. T., Forbes, C., Hessing-Lewis, M., Morien, E., Lemay, M. A., et al. (2020). Host-specificity and core taxa of seagrass leaf microbiome identified across tissue age and geographical regions. Front. Ecol. Evolut. 8:605304. doi: 10.3389/fevo.2020.605304
Schell, J. M., Goodwin, D. S., and Siuda, A. N. S. (2015). Recent Sargassum inundation events in the Caribbean shipboard observations reveal dominance of a previously rare form. Oceanography 28, 8–10. doi: 10.5670/oceanog.2015.70
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541.
Serebryakova, A., Aires, T., Viard, F., Serrao, E. A., and Engelen, A. H. (2018). Summer shifts of bacterial communities associated with the invasive brown seaweed Sargassum muticum are location and tissue dependent. PLoS One 13:e0206734. doi: 10.1371/journal.pone.0206734
Smetacek, V., and Zingone, A. (2013). Green and golden seaweed tides on the rise. Nature 504, 84–88. doi: 10.1038/nature12860
Smythe, A. B. (2015). Evolution of feeding structures in the marine nematode order Enoplida. Integr. Comp. Biol. 55, 228–240. doi: 10.1093/icb/icv043
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D., Breiner, H. W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19(Suppl. 1), 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Stoner, A. W., and Greening, H. S. (1984). Geographic-variation in the macrofaunal associates of pelagic Sargassum and some biogeographic implications. Mar. Ecol. Prog. Ser. 20, 185–192. doi: 10.3354/meps020185
Susilowati, R., Sabdono, A., and Widowati, I. (2015). Isolation and characterization of bacteria associated with brown algae Sargassum spp. from Panjang Island and their antibacterial activities. Basic Res. Trop. Coast. Reg. Eco Dev. 23, 240–246. doi: 10.1016/j.proenv.2015.01.036
Thomas, F., Le Duff, N., Wu, T. D., Cebron, A., Uroz, S., Riera, P., et al. (2021). Isotopic tracing reveals single-cell assimilation of a macroalgal polysaccharide by a few marine Flavobacteria and Gammaproteobacteria. ISME J. 15, 3062–3075. doi: 10.1038/s41396-021-00987-x
Torralba, M. G., Franks, J. S., Gomez, A., Yooseph, S., Nelson, K. E., and Grimes, D. J. (2017). Effect of Macondo prospect 252 oil on microbiota associated with pelagic Sargassum in the northern Gulf of Mexico. Microb. Ecol. 73, 91–100. doi: 10.1007/s00248-016-0857-y
Urdaci, M. C., Stal, L. J., and Marchand, M. (1988). Occurrence of nitrogen-fixation among vibrio spp. Arch. Microbiol. 150, 224–229. doi: 10.1007/BF00407784
Van Der Loos, L. M., Eriksson, B. K., and Falcao Salles, J. (2019). The macroalgal holobiont in a changing sea. Trends Microbiol. 27, 635–650. doi: 10.1016/j.tim.2019.03.002
Van Tussenbroek, B. I., Arana, H. A. H., Rodriguez-Martinez, R. E., Espinoza-Avalos, J., Canizales-Flores, H. M., Gonzalez-Godoy, C. E., et al. (2017). Severe impacts of brown tides caused by Sargassum spp. on near-shore Caribbean seagrass communities. Mar. Pollut. Bullet. 122, 272–281. doi: 10.1016/j.marpolbul.2017.06.057
Vranken, G., Herman, P. M. J., Vincx, M., and Heip, C. (1986). A re-evaluation of marine nematode productivity. Hydrobiologia 135, 193–196. doi: 10.1007/BF00006530
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., Humphrey, G., Parada, A., et al. (2016). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, e00009–e00015. doi: 10.1128/mSystems.00009-15
Wang, M. Q., Hu, C. M., Barnes, B. B., Mitchum, G., Lapointe, B., and Montoya, J. P. (2019). The great Atlantic Sargassum belt. Science 365, 83–87. doi: 10.1126/science.aaw7912
Weis, J. S. (1968). Fauna associated with pelagic Sargassum in the Gulf Stream. Am. Natural. 80, 554–558. doi: 10.2307/2423550
Willems, W. R., Artois, T., Vermin, W., and Chockaert, E. (2004). Revision of Trigonostomum Schmidt, 1852 (Platyhelminthes, Typhloplanoida, Trigonostomidae) with the description of seven new species. Zool. J. Linnean Soc. 141, 271–296. doi: 10.1111/j.1096-3642.2004.00124.x
Witherington, B., Hirama, S., and Hardy, R. (2012). Young sea turtles of the pelagic Sargassum-dominated drift community: habitat use, population density, and threats. Mar. Ecol. Prog. Ser. 463, 1–22. doi: 10.3354/meps09970
Keywords: macroalgae, methanogenic archaea (MA), sulfate-reducing bacteria (SRB), epibiont, microbial communities, protists, nematodes, Sargassum
Citation: Hervé V, Lambourdière J, René-Trouillefou M, Devault DA and Lopez PJ (2021) Sargassum Differentially Shapes the Microbiota Composition and Diversity at Coastal Tide Sites and Inland Storage Sites on Caribbean Islands. Front. Microbiol. 12:701155. doi: 10.3389/fmicb.2021.701155
Received: 27 April 2021; Accepted: 01 October 2021;
Published: 29 October 2021.
Edited by:
Alison Buchan, The University of Tennessee, Knoxville, United StatesReviewed by:
Valerie Michotey, Aix-Marseille Université, FranceCopyright © 2021 Hervé, Lambourdière, René-Trouillefou, Devault and Lopez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascal Jean Lopez, cGFzY2FsLWplYW4ubG9wZXpAbW5obi5mcg==
†ORCID: Vincent Hervé, orcid.org/0000-0002-3495-561X; Pascal Jean Lopez, orcid.org/0000-0002-9914-4252
‡Present address: Damien Alain Devault, CUFR, Dembeni, Mayotte
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.