 Ling Kui1†
Ling Kui1† Baozheng Chen2†Jian Chen3
Baozheng Chen2†Jian Chen3 Rouhallah Sharifi4
Rouhallah Sharifi4 Yang Dong5,6,7Zhanjiang Zhang6*Jianhua Miao6,8*
Yang Dong5,6,7Zhanjiang Zhang6*Jianhua Miao6,8*- 1School of Pharmacy, Jiangsu University, Zhenjiang, China
- 2College of Food Science and Technology, Yunnan Agricultural University, Kunming, China
- 3International Genome Center, Jiangsu University, Zhenjiang, China
- 4Department of Plant Protection, College of Agriculture, Razi University, Kermanshah, Iran
- 5College of Biological Big Data, Yunnan Agricultural University, Kunming, China
- 6Guangxi Key Laboratory of Medicinal Resources Protection and Genetic Improvement, Guangxi Botanical Garden of Medicinal Plants, Nanning, China
- 7State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Yunnan Agricultural University, Kunming, China
- 8School of Pharmacy, Guangxi Medical University, Nanning, China
Panax notoginseng, an important Chinese medicinal herb, can be mainly cultivated in two planting patterns, cropland planting (DT) and understory planting (LX). We speculate that the rhizosphere microbiome may vary in DT and LX and may play an important role in promoting the growth and health of P. notoginseng. In the present study, culture-independent Illumina HiSeq was employed to investigate the rhizosphere bacteria and fungi under DT and LX planting patterns. Predominant phyla include Proteobacteria, Acidobacteria, Actinobacteria, Gemmatimonadetes, and Ascomycota in the two planting patterns. DT has higher alpha diversity index than LX. The predominant LX-core genera include Bradyrhizobium, Streptomyces, and Actinomadura, and the predominant DT-core genera include Sphingomonas, Variovorax, and Novosphingobium. Total relative abundance of the disease-suppression phylum (Proteobacteria, Firmicutes, and Actinobacteria) and the potential plant growth-promoting rhizobacteria (PGPR) were both significantly higher in LX than in DT. We also identified over-presented microbial functional traits mediating plant–microbe and microbe–microbe interactions, nutrition acquisition, and plant growth promotion in P. notoginseng rhizosphere. Our findings provide a valuable reference for studying beneficial microbes and pathogens of P. notoginseng planted in DT and LX.
Introduction
Panax notoginseng (P. notoginseng) is an important Chinese medicinal plant commonly known as Sanqi or Tianqi. It belongs to the family Araliaceae (Guo et al., 2010). There is a very high global demand for this species due to its antihypertensive, antithrombotic, anti-atherosclerotic, anti-tumor, anti-oxidant, and hepatoprotective activities (Zhou et al., 2009; Yang et al., 2015; Dong et al., 2019; Li et al., 2019; Liu et al., 2019; Zhang et al., 2019a; Tang et al., 2020). Recently, more than 4 million hectares of land are cultivated annually in China for this species (Shoyama et al., 1997; Deborah et al., 2005; Wei et al., 2015). After 1–3 years of cropping, root rot is prevalent in this species (Sun et al., 2015; Yang et al., 2015; Zhang et al., 2019b).
Currently, it is grown only in southern China, typically around Wenshan City of Yunnan Province and Baise Ccity of Guangxi Province (Guo et al., 2010). Besides the root rot and limited suitable cropland, the loss of genetic diversity, and degeneration of germplasm associated with P. notoginseng also hamper its sustainable supply in the global market (Zhou et al., 2005).
Studies have revealed several factors, which limit its cropping, like the alteration of its soil microbiota (Dong et al., 2016; Fan et al., 2016; Miao et al., 2016; Tan et al., 2017a), autotoxicity caused by allelochemicals (Sun et al., 2008; Yang et al., 2015), and enhanced soil salinization and acidification as well as loss of nutrients from the soil (Liu et al., 2013). To mitigate these problems, biological control, chemical control, and crop rotation have been found effective (Klose et al., 2006; Song et al., 2014; Wang et al., 2016).
Cultivation of Chinese medicinal herbs as understory plants provides a healthy microenvironment for their optimum growth (Yan et al., 2016). It also limits the costs associated with labor, fertilizer application, and shading (Zhou et al., 2016). The microbiome of the rhizosphere influences plant growth (Kwak et al., 2018; Xu J. et al., 2018). The structure and composition of the rhizosphere microbiome are, in turn, determined by host species, location, planting pattern, and growth period (Peiffer et al., 2013; Edwards et al., 2015; Jin et al., 2017; Fan et al., 2019). The advent of high-throughput sequencing (HTS) technology provides an effective platform to decipher the structures and functions of the rhizosphere microbiome (Xu J. et al., 2018; Xiao et al., 2019). The whole-genome shotgun sequencing has been applied to study the rhizobiome of multiple crops such as rice, millet, corn, wheat, sugarcane, Populus, and grapevine (Daniel, 2005; Knief et al., 2012; Edwards et al., 2015; Fan et al., 2016; Knight et al., 2018; Kwak et al., 2018; Xu J. et al., 2018).
The studies on the rhizosphere microbiome of P. notoginseng are scarce, and a comparative evaluation of the rhizobiome composition in main cropland and understory cultivation is lacking. This study was therefore carried out to decipher the microbiome variations between the understory plantations and main croplands of P. notoginseng using a high-throughput metagenomic approach in the Wenshan City of Yunnan Province and the Baise City of Guangxi Province. This study holds the promise to link the better efficiency of P. notoginseng in the understory plantations with its specific rhizosphere microbiome when compared with the main cropland. We used a metagenomic approach to reveal the rhizosphere microbiome of P. notoginseng in the Wenshan City of Yunnan Province and the Baise City of Guangxi Province.
Materials and Methods
Sample Collection
For sampling, we chose four farms where P. notoginseng was grown for the first time since 2015. These four farms were located in Wenshan City (WS) (Yunnan Province, China) and Baise City (BS) (Guangxi Province, China) (Figure 1 and Supplementary Figure 1). In order to ensure the consistency of sampling time, samples were collected from four farms through four groups at the same day. Meanwhile, sampling 1–5-year-old P. notoginseng also weakens the influence on microbial community caused by the growth period. We used the protocol developed by Bulgarelli et al. (2015) to collect the rhizosphere soil and the corresponding bulk soil samples. From each plant, three to five fine roots (approximately 0.5–1-mm diameter, 3–5-cm length) from a depth of 5–15 cm were collected. The soil closely attached to the roots was washed using PBS buffer. The washed soil from the same plant was then pooled in a 50-ml Falcon tube and constitutes the rhizosphere soil. Similarly, corresponding bulk soil was collected, approximately 10–20 cm underground and 0.5 m away from the P. notoginseng clusters, from each farm and pooled together into a 50-ml Falcon tube as one sample. The rhizosphere soil and the corresponding bulk soil were stored at 4°C until subsequent DNA extraction.
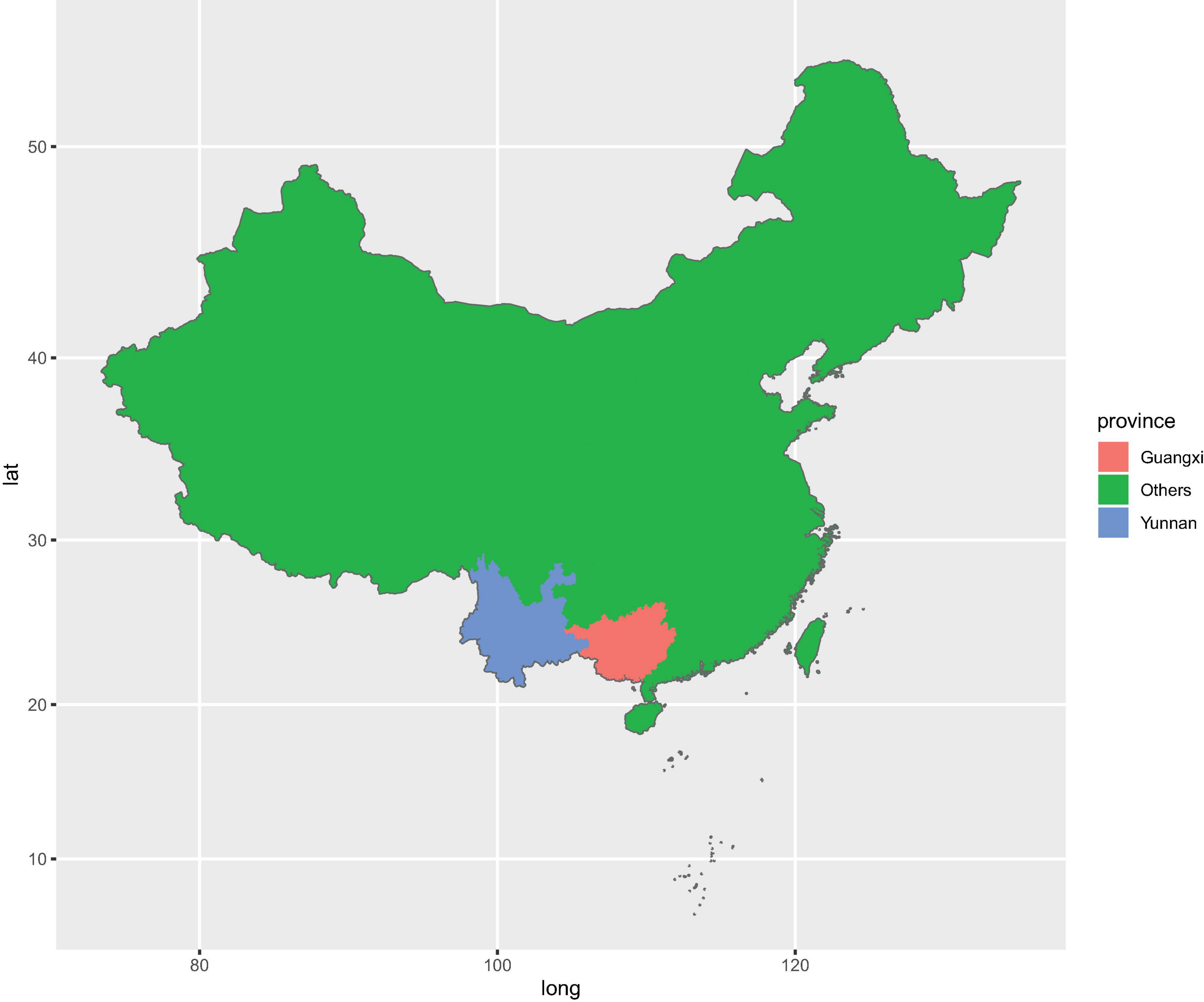
Figure 1. Geographic distribution of sampling sites across the main producing areas of Panax. notoginseng.
In total, 122 rhizosphere soil samples and 13 bulk soil samples were harvested from Wenshan City (WS) and Baise City (BS). Around 57 samples were harvested from WS, with 26 understory (LX) rhizosphere soil samples (WSLX) and three understory bulk soil samples (WSLXCK), 25 cropland (DT) rhizosphere soil samples (WSDT), and three cropland bulk soil samples (WSDTCK). Similarly, 78 samples were harvested from BS, with 44 understory (LX) rhizosphere soil samples (BSLX), three understory bulk soil samples, 27 cropland (DT) rhizosphere soil samples (BSDT), and four cropland bulk soil samples (BSDTCK). The details of the sampling sites are presented in Supplementary Table 1.
DNA Extraction and Sequencing
We used EZNA soil DNA extraction kit (OMEGA Bio-Tek, Inc., Norcross, GA, United States) for extracting DNA from the collected samples. The quality of the extracted DNA was assessed through an agarose gel (1%) electrophoresis and NanoPhotometer® spectrophotometer (IMPLEN, CA, United States). The concentration was measured through Qubit® DNA Assay kit using Qubit® 2.0 Fluorometer (Life Technologies, CA, United States). A total of 700 ng of DNA was used for library preparation. Sequencing libraries were generated using NEB ® Ultra DNA Library Prep kit for Illumina® (NEB, United States), following the manufacturer’s recommendations. The index codes were added to attribute sequences to each sample.
Briefly, DNA was purified using AMPure XP system (Beckman Coulter, Beverly, United States). Subsequent to the adenylation of 3′ ends of the DNA fragments, the NEBNext Adaptor with a hairpin loop structure was ligated. Next, electrophoresis was used to select DNA fragments of 350–400-bp size range. Then 3 μl of USER enzyme (NEB, United States) was used with size-selected and adaptor-ligated DNA fragments at 37°C for 15 min, followed by 5 min at 95°C before PCR. PCR was performed with Phusion High-Fidelity DNA Polymerase, universal PCR primers, and index (X) primer. Finally, PCR products were purified (AMPure XP system), and library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using HiSeq 4000 PE Cluster kit (Illumia), as per the manufacturer’s instructions. Totally, 135 DNA libraries were sequenced on an Illumina Hiseq 4000 platform and 1,222.690 Gb of raw data was obtained, including 51 rhizosphere soil and six bulk soil samples in WS and 71 rhizosphere soil and seven bulk soil samples in BS.
Data Analysis
The DNA from the rhizosphere and their corresponding bulk soil samples of P. notoginseng were sequenced at Novogene—Tianjing, China (Quince et al., 2017). We used SOAPnuke v2.0 to remove the adaptor sequences and low-quality reads harboring >10% N bases or < 40% bases with scores exceeding 38 (Chen et al., 2018). These cleaned reads were then aligned to the P. notoginseng reference genome released by Zhang et al. (2017) using Bowtie2 v2.3.1 (Langmead and Salzberg, 2012). The mapped reads were classified according to the farm, bulk soil, and rhizosphere soil (Supplementary Table 2).
The assemblies were generated using Megahit v1.1.3 with the default parameters (Li et al., 2016). Subsequently, the MetaGeneMark_v1_mod implemented in Prodigal v2.6.3 was used to predict the genes on the contigs (Zhu et al., 2010). All the genes of length ≥ 100 bp were retained for downstream analysis. With the assistance of the Mmseq2 software and the Linclust algorithm, all the predicted genes were combined together and then clustered into a non-redundant geneset, using the similarity threshold at 95% (Steinegger and Soding, 2017). For the taxonomic and functional information of the unigenes, their protein sequences were aligned against the NCBI non-redundant database and Kegg Orthology (KO) database, using DIAMOND at an E-value threshold of 1e–5 (Buchfink et al., 2015). Moreover, the cleaned reads were aligned against the geneset to obtain the abundance profiles (read count matrix) usingBowtie2 v2.3.1 (Langmead and Salzberg, 2012).
Comparative Analysis Across Different Planting Patterns
We used a DESeq2 v1.30.1-based negative binomial generalized linear model to statistically assess the differences in the abundance profiles of genera, phyla, and KOs across the understory (LX) and main cropland (DT) planting patterns. First, the read count matrix was normalized through the DESeqVS method (Love et al., 2014; Tang et al., 2015), followed by a comparative analysis to detect LX-enriched genera, DT-enriched genera, and enriched KOs using DESeq2. We used the criteria that if taxa or a KO is present in more than 75% of the samples, in each of the different planting patterns, they were considered as core taxa or core KOs (Xu J. et al., 2018). For visually assessing the relative abundance of the enriched taxa and the functional traits in the rhizosphere, we used the Pheatmap R package. To clearly demonstrate the differences among the core taxa and their functional traits in different planting patterns, the relative abundance was calculated based on the number of mapped reads.
Weighted Gene Co-expression Network Analysis
Weighted gene co-expression network analysis (WGCNA) was performed in prokaryotes and fungi, respectively. Classified prokaryote samples were divided into eight groups according to planting location and pattern, including WSLX, WSDT, BSLX, BSDT, WSLXCK, WSDTCK, BSLXCK, and BSDTCK. Fungi samples also follow this classification criteria. Construction of weighted co-expressed networks and identification of co-expression modules were carried out using the “WGCNA” R package with default parameters, the input file is the count table (Supplementary Table 3). In order to ensure a scale-free network, the power of β = 11 (scale-free R2 = 0.8) was selected as the soft thresholding, and MEDissThres was set as 0.25 to merge similar modules. After that, nodes were screened by the criteria MM > 0.87 for pink module in prokaryotes and MM > 0.02 for turquoise module in fungi. Then the nodes were inputted into Cytoscape to visualize the co-expression network, and the nodes and hub genera were identified (Su et al., 2014).
Results
Taxonomic Profile of Panax notoginseng Rhizosphere Microbiome
The shotgun metagenome sequencing generated approximately 30 million 150-bp paired-end reads for each sample, accounting for more than 607.7 Gbp raw sequences. After the removal of low-quality sequences (Supplementary Table 2), the de novo assembly was performed through Megahit 1.1.3. A total of 149,436,529 contigs were obtained in the final assembly, constituting a total length of 103.6 Gb. Approximately 193 million metagenes were predicted from the contigs using MetaGeneMark_v1_mod. These metagenes were then clustered into approximately 164 million non-redundant genes (unigenes). In total, 164,264,054 unigenes were generated in our study and used for downstream analysis.
Taxonomic annotations were assigned to 62.4% of the unigenes, among which 99.55% were classified into 165 prokaryotic phyla (including bacteria and archaea) and 0.43% into 31 eukaryotic phyla (Supplementary Figure 2). A negligible fraction (0.014%) of the annotated unigenes were classified as viral, indicating potential annotation biases that resulted in the underestimation of eukaryotic and viral communities (Leach et al., 2017). We focused our study on the comparative analysis of the prokaryotic and fungal rhizosphere microbiomes in the two planting patterns (DT and LX). The dominant prokaryotic (bacterial and archaeal) phyla found in the P. notoginseng rhizosphere and bulk soil included Proteobacteria (34.5%), Acidobacteria (28.6%), Actinobacteria (10.5%), Gemmatimonadetes (5.3%), and Verrucomicrobia (4.2%). These together constituted about 83.1% of the prokaryotic unigenes (Figure 2A and Supplementary Table 3-1). Fungal unigenes were primarily associated with the phyla Ascomycota (57.8%), Mucoromycota (22.1%), and Basidiomycota (17.8%), which together accounted for 97.7% of the fungal unigenes (Figure 2B and Supplementary Table 3-2).
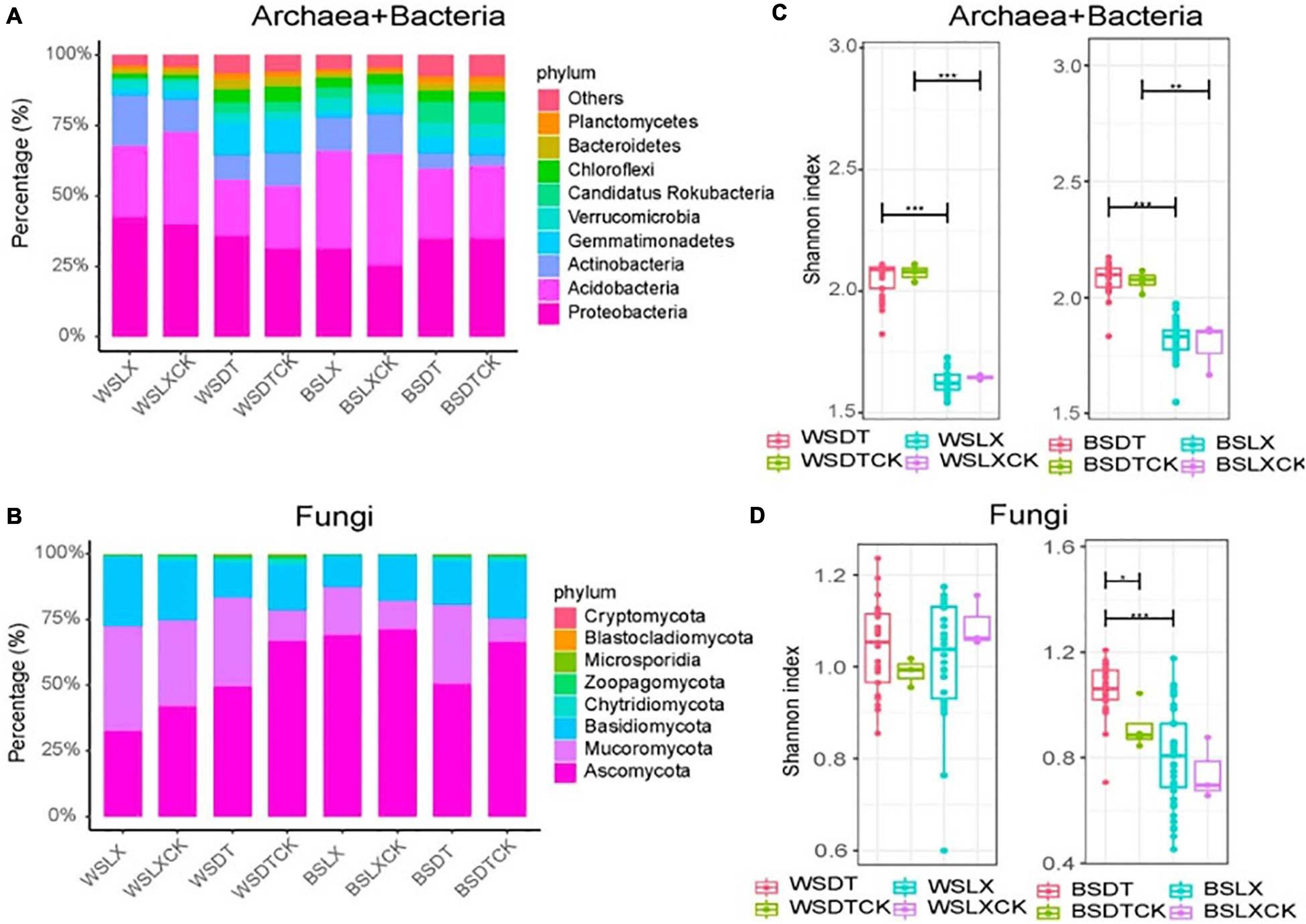
Figure 2. Taxonomic distribution and diversity comparisons in P. notoginseng rhizosphere (n = 122) and bulk soil (n = 13) microbiomes across different sites. (A,B) Phyla-level distributions of prokaryotes and fungi in the bulk soil and rhizosphere samples based on metagenomic data. (C,D) Alpha diversity comparison between the bulk soil and rhizosphere samples from each location based on the Shannon index using the metagenomic data; *P < 0.05; **P < 0.01; ***P < 0.001. One-sided t-test; center value represents the median of the Shannon index. WSDT, rhizosphere soil of cropland planting in Wenshan city; WSDTCK, bulk soil of cropland planting in Wenshan City; WSLX, rhizosphere soil of understory planting in Wenshan City; WSLXCK, bulk soil of understory planting in Wenshan City; BSDT, rhizosphere soil of cropland planting in Baise City; BSDTCK, bulk soil of cropland planting in Baise City; BSLX, rhizosphere soil of understory planting in Baise City; BSLXCK, bulk soil of understory planting in Baise City.
Estimation of the Microbial Diversity
The alpha diversity (α-diversity) analysis was estimated based on the Shannon index. We observed a significant difference in α-diversity between different planting patterns (DT and LX) in P. notoginseng rhizosphere. DT showed a higher α-diversity than LX, except for the fungal taxa in WS. A similar trend was shown by α-diversity of bulk soil microbial community, but little significant difference in α-diversity was observed between the rhizosphere and bulk soil samples, except for the fungal taxa at BSDT (Figures 2C,D).
Furthermore, the principal coordinate analysis (PCoA) based on the Bray–Curtis distance (β-diversity) revealed that the planting pattern was the principal factor in shaping the composition of the microbial communities (Figures 3A,B). Moreover, no significant variation among the microbial taxa between the rhizosphere and the bulk soil samples was revealed by the β-diversity analysis (Figures 3A,B). These results were corroborated by the permutation multivariate analysis of variance (PERMANOVA), which was according to the Bray–Curtis distance metric. PERMANOVA showed a significant variation in prokaryotic taxa (25.7%, P = 0.001) and fungal taxa (34.5%, P = 0.001) between LX and DT (Supplementary Table 4). The planting location appeared as the second-largest source to shape the microbial community difference (Supplementary Table 4).
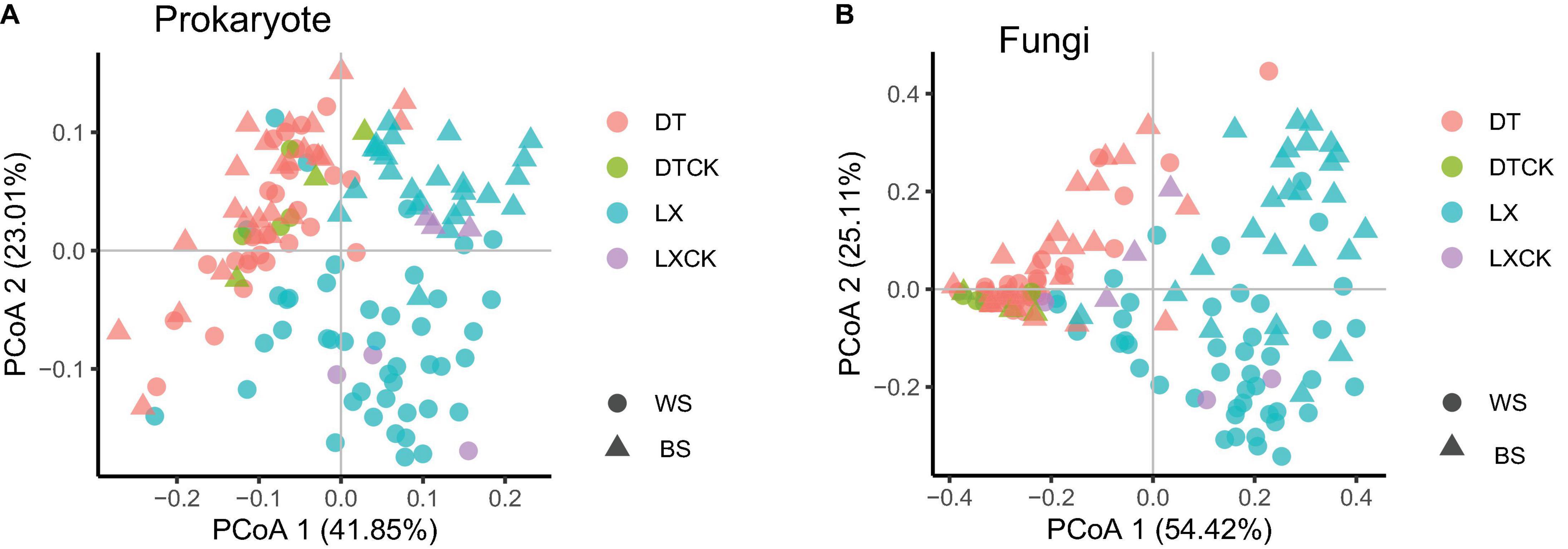
Figure 3. (A,B) Principal coordinate analysis (PCoA) of prokaryote and fungi based on the Bray–Curtis distance between the bulk soil (n = 13) and rhizosphere soil (n = 122) for each location using the metagenomic data. WS, Wenshan City; BS, Baise City; DT, cropland planting rhizosphere soil; DTCK, cropland planting bulk soil; LX, understory planting rhizosphere soil; LXCK, understory planting bulk soil.
The enrichment analysis of the microbial phyla, based on their relative abundance, revealed that 58 prokaryotic phyla and seven fungal phyla were enriched in the LX rhizosphere microbiome, whereas 73 prokaryotic phyla and one fungal phylum were enriched in the DT rhizosphere microbiome (corrected P-value < 0.05, DESeq2, Supplementary Table 5-1). Moreover, no significant variation in the relative abundance of prokaryotic or fungal phyla was observed between the rhizosphere and bulk soil microbial phyla (corrected P-value ≥ 0.05, DESeq2, Supplementary Table 5-2). Similarly, the enrichment analysis of the microbial genera revealed 902 prokaryotic genera and 251 fungal genera as enriched in the LX rhizosphere microbiome, whereas 1,087 prokaryotic genera and 27 fungal genera were enriched in the DT rhizosphere microbiome (corrected P-value < 0.05, DESeq2, Supplementary Table 6-1). These enriched genera belonged to the main prokaryotic phyla like Proteobacteria, Acidobacteria, Actinobacteria, and Verrucomicrobia and the main fungal phyla like Ascomycota and Mucoromycota (Figures 2A,B and Supplementary Tables 5, 6). In comparison with the bulk soil microbiome, the rhizosphere microbiome contained only two enriched prokaryotic genera, Nitrosopumilus and Wenzhouxiangella along with depletion of three prokaryotic genera and two fungal genera (corrected P-value < 0.05, DESeq2, Supplementary Table 6-2).
Core Genera Evaluation
Using the aforementioned criteria, the core genera (genera present in more than 75% of all samples and with enriched relative abundance) in the P. notoginseng rhizosphere of LX and DT planting patterns were evaluated. Around 875 prokaryotic and 203 fungal genera were found as core genera in LX-rhizosphere and 1,051 prokaryotic and 17 fungal genera as core genera in DT-rhizosphere of P. notoginseng (Figures 4A,B and Supplementary Table 6). The core rhizosphere genera were mainly affiliated with the prokaryotic phyla Proteobacteria, Actinobacteria, and Acidobacteria as well as with the fungal phyla Ascomycota and Mucoromycota (Figure 4C). Bradyrhizobium, Burkholderia, and Paraburkholderia have higher relative abundance (corrected P-value < 0.05, DESq2) (Figures 4C,D and Supplementary Figure 5), and were over-represented in Proteobacteria. At the same time, both LX and DT core genera contain some inhibitors and pathogenic microbiomes, which may interfere with the quality and growth of P. notoginseng.
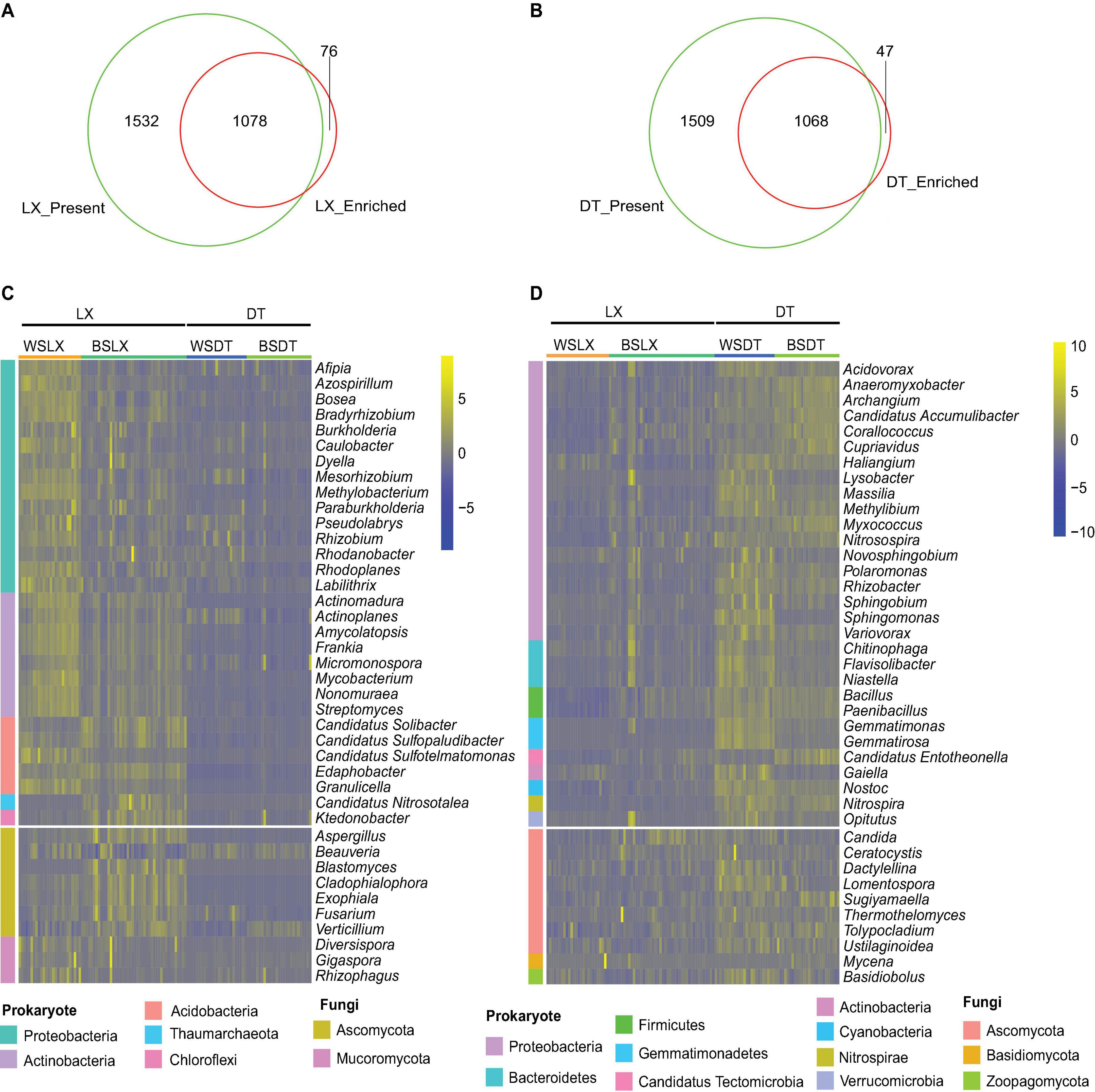
Figure 4. Characterization of the core P. notoginseng rhizosphere microbes in cropland planting (n = 52) and understory planting (n = 70) patterns. (A) Venn plot depicting the number of core rhizosphere genera based on the presence rate (>75%) in all understory planting samples and the genus have a higher relative abundance in understory planting than in cropland planting. (B) Venn plot depicting the number of core rhizosphere genera based on the presence rate (>75%) in all cropland planting samples and the genus have a higher relative abundance in cropland planting than in understory planting. (C,D) Relative abundances of the 30 prokaryotes and 10 fungi, most relatively abundant core rhizosphere genera in cropland planting and understory planting patterns based on metagenomic data. Scale, relative abundance of genus at row normalization by removing the mean (centering) and dividing by the standard deviation (scaling). The color from blue to yellow represents a relative abundance of each taxa from low to high. WS, Wenshan City; BS, Baise City; DT, cropland planting rhizosphere soil; LX, understory planting rhizosphere soil.
Characteristic Analysis of Core Rhizosphere Microbiome of Panax Notoginseng in the Different Planting Patterns
Mann–Whitney U test revealed that the total relative abundance of the core rhizosphere microbiome phyla varied significantly between LX and DT planting patterns. We observed that the multiple disease-suppression prokaryotes were highly abundant in LX than DT (P < 0.05), among which Proteobacteria, Firmicutes, and Actinobacteria were important in disease suppression (Mendes et al., 2011; Figure 5A). Furthermore, 21 potential plant growth-promoting rhizobacteria (PGPR) genera were identified, among which eight genera were related to the nitrogen cycle (Nelson et al., 2016; Supplementary Table 6), 10 genera were plant growth regulators, and 10 genera were the biological control agents (Figures 6A–C). The total relative abundance of these PGPR genera was found higher in LX than DT (Figure 6D).
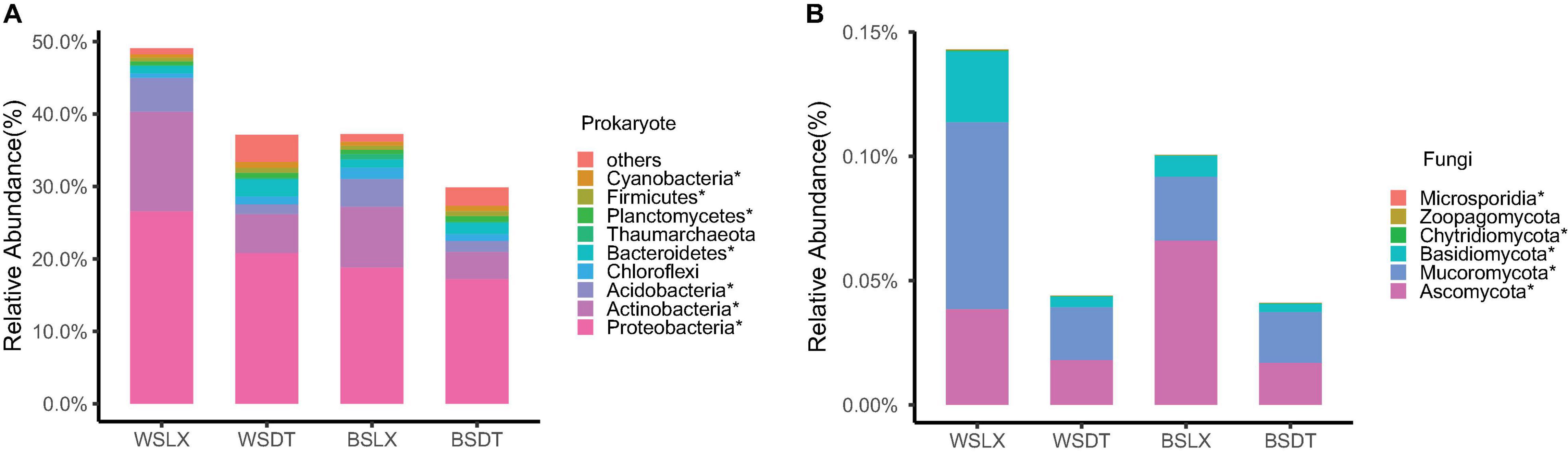
Figure 5. Phyla-level distributions of core rhizosphere microbiomes in cropland planting (n = 52) and understory planting (n = 70) patterns based on metagenomic data. (A) Detailed relative abundances of core prokaryotic rhizosphere microbiomes. (B) Detailed relative abundances of core fungal rhizosphere microbiomes. Asterisk indicates a significant difference with P < 0.05 (MW test). WSDT, rhizosphere soil of cropland planting in Wenshan City; WSLX, rhizosphere soil of understory planting in Wenshan City; BSDT, rhizosphere soil of cropland planting in Baise City; BSLX, rhizosphere soil of understory planting in Baise City.
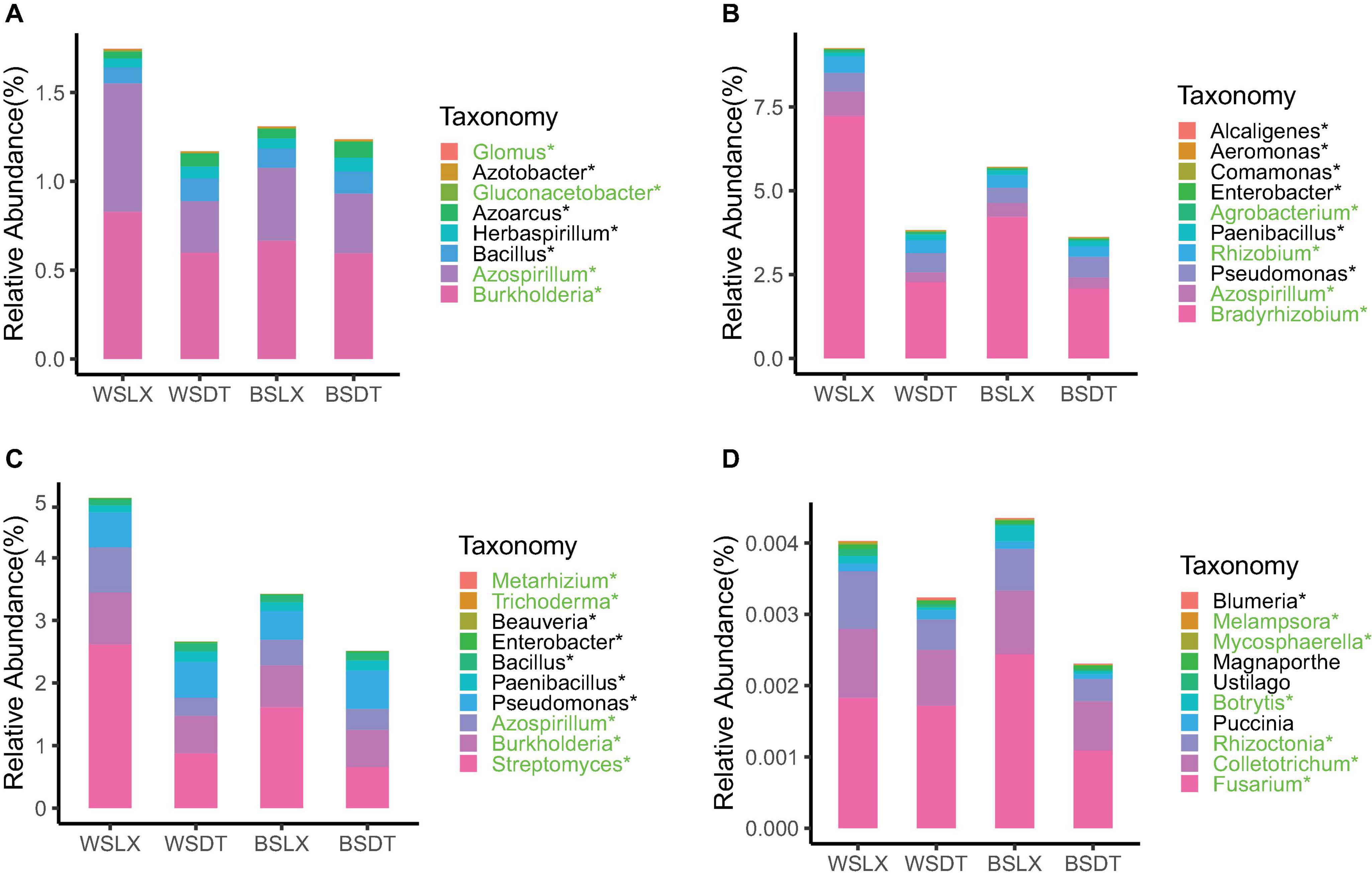
Figure 6. Core genera involved in plant growth-promoting rhizobacteria (PGPR) or plant pathogens in cropland planting (n = 52) and understory planting (n = 70) patterns across different sites based on metagenomic data. Detailed relative abundances of core rhizosphere microbiomes involved in (A) nitrogen cycle, (B) plant growth regulator, (C) biological control agent, and (D) plant pathogens. Asterisk indicates a significant difference with P < 0.05 (MW test). Green indicates the relative abundances of core rhizosphere microbiomes is significantly higher in understory planting than in cropland planting. Black indicates the relative abundances of core rhizosphere microbiomes is significantly lower in understory planting than in cropland planting. WSDT, rhizosphere soil of cropland planting in Wenshan City; WSLX, rhizosphere soil of understory planting in Wenshan City; BSDT, rhizosphere soil of cropland planting in Baise City; BSLX, rhizosphere soil of understory planting in Baise City.
However, the pathogenic fungal phyla like Basidiomycota, Mucoromycota, and Ascomycota were also significantly higher in LX than in DT (P < 0.05) (Figure 5B). The potential plant pathogen genera like Fusarium, Colletotrichum, and Rhizoctonia were higher in LX than in DT (P < 0.05, MW test) (Figure 6D; Fravel et al., 2003). Besides the pathogenic nature, the genera Fusarium has several members, which are non-pathogenic and may act as potential biocontrol agents that could control fungal pathogens (Nel et al., 2006). Similarly, Colletotrichum has also been proposed as a means to promote plant growth through hormone production and phosphorus absorption under abiotic stress (Hiruma et al., 2016; Figure 6D).
The Correlations of Microbiomes Analyzed by Weighted Gene Co-expression Network Analysis
The correlations of microbiomes under different planting patterns are shown in Figure 7. For fungi, the difference of gray and turquoise module could be seen under different planting patterns or location. However, as the number of genera is small, there are fewer modules displayed in fungi (Figure 7B), and we mainly focused on prokaryotes (Figures 7A,C,D). For the prokaryotic community, seven modules were generated; the modules represented distinct differences under different planting patterns (WSLX vs. WSDT, BSLX vs. BSDT). BSDT had a significant positive correlation with most modules: green (P = 7E-06, r = 0.38), brown (P = 4E-07, r = 0.42), pink (P = 2E-07, r = 0.43), and yellow (P = 0.02, r = 0.19) modules (Figure 7A). Comparing the rhizosphere soil and bulk soil modules, a similar correlation was shown. Besides, the modules were also varied in location (WSLXCK vs. BSLXCK, WSDTCK vs. BSDTCK). The results are consistent with the trend of PERMANOVA’s analysis, which revealed that planting pattern is the main factor to shape the structure of P. notoginseng rhizosphere microbes, and second is the location.
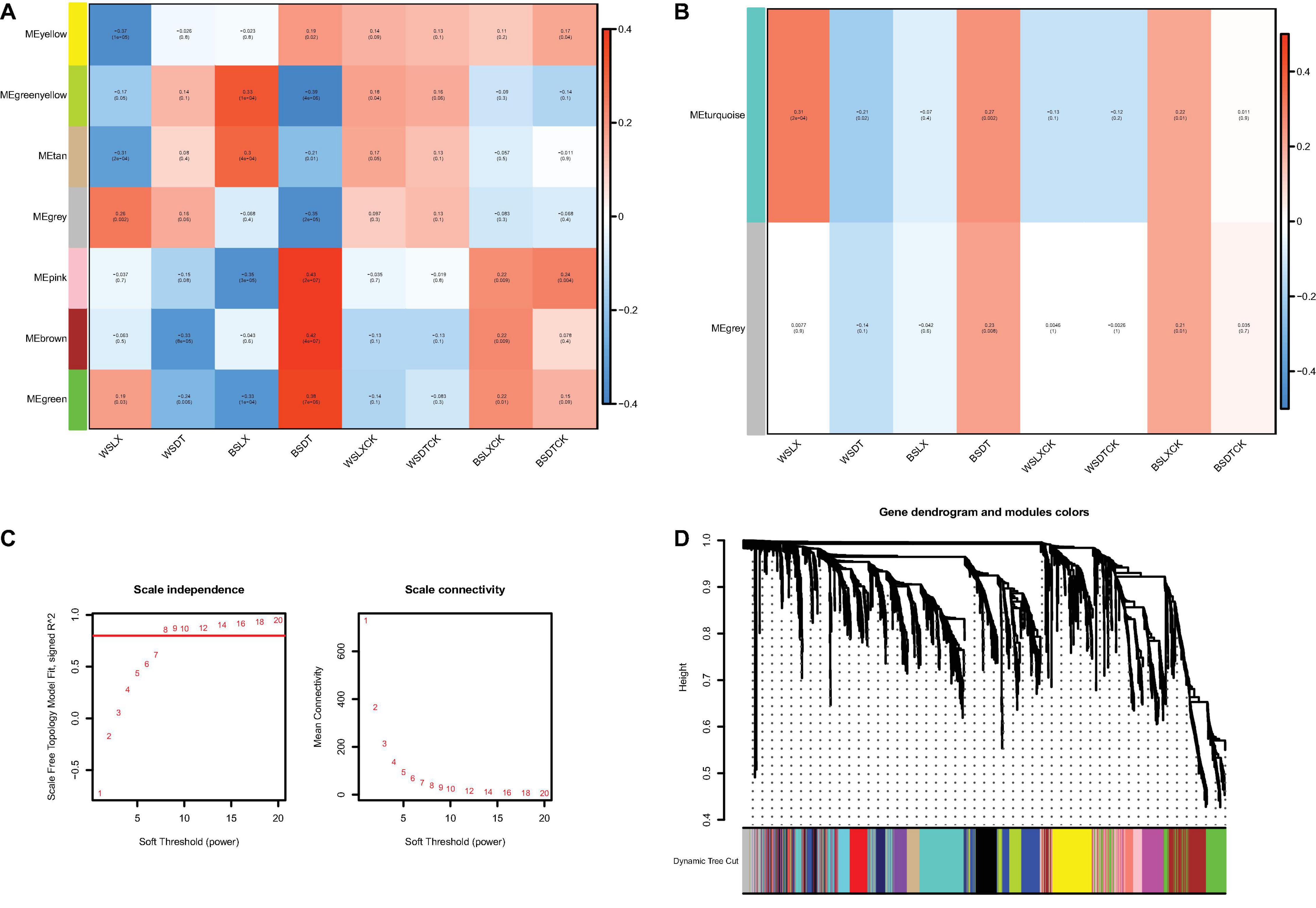
Figure 7. The correlations of microbiomes. Heat map of module-trait relationship in prokaryotes (A) and fungi (B). Each column represents the co-expression module. Red color of each box represents the positive correlation between module and trait. Blue color of each box represents the negative relationships between module and trait. The darker the module color, the more significant their relationship. (C) Estimation of soft-thresholding values in prokaryotes. Scale independence and mean connectivity of various soft-thresholding values (β), power of β = 11 (scale free R2 = 0.8). (D) Dendrogram of all prokaryotes enriched according to a dissimilarity measure (1-TOM) and the cluster module colors. The first set of modules was obtained with dynamic tree cut algorithm and the correlated modules were merged together. Each branch in the figure represents one genus and every color below represents one co-expression module.
According to the correlation results, genera from the pink module in prokaryotes and the turquoise module in fungi were extracted to generate networks (Figure 8). Nodes were screened by the criteria MM > 0.87 for the pink module and MM > 0.02 for the turquoise module. The results are shown in Figure 8. Ten hub genera were obtained in prokaryotes (Figure 8A, red color) and four hub genera in fungi (Figure 8B, red color). For prokaryotes, there are two genera, Azospirillum and Gluconacetobacter, that were highly correlated in the pink module. It is worth noting that they are also identified as PGPR’s taxa and had higher relative abundance in LX than in DT in previous analysis. Besides, two genera (Trichoderma and Metarhizium) related to biological control agent, and two genera (Fusarium and Colletotrichum) related to plant pathogen were highly correlated in the turquoise module of fungi. Similarly, these four genera had higher relative abundance in LX than in DT. It may indicate that the semi-nature planting patterns (LX) evolve a closer interaction microenvironment, which can provide us with significant reference to selectively manage microbiome in agricultural production.
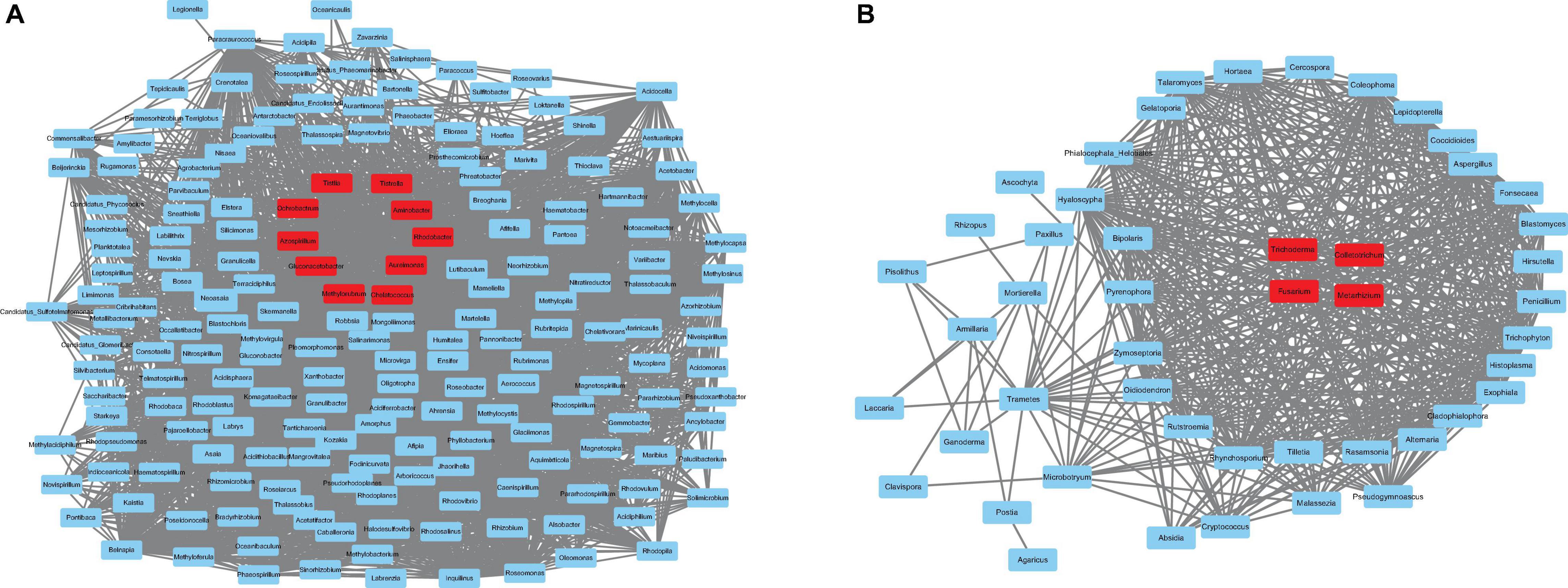
Figure 8. The co-expression network. (A) The co-expression network of the hub genera from pink module in prokaryotes, including 100 nodes. (B) The co-expression network of the hub genera from turquoise module in fungi, including 48 nodes. The hub genes are represented in red color.
Core Functional Traits in the Different Planting Patterns of Panax Notoginseng Rhizosphere
The unigene sets of the P. notoginseng rhizosphere were 4- and 18-fold larger than that of the human gut and the global Tara oceanic microbiomes, respectively (Sunagawa et al., 2015), but it had the same order as the global citrus rhizosphere microbiome (Xu J. et al., 2018). More than 35% of (58 of 164 million) the unigenes were annotated by blast against the KEGG Orthology (KO) database, and 16,396 KOs were assigned to the annotated unigenes (Supplementary Table 7-1). These KOs were mainly associated with 41 KEGG (level 2) pathways. The carbohydrate metabolism and energy metabolism had a higher relative abundance across the different planting patterns (Figure 9A). Finally, 9,585 of 15,725 (60.9%) and 9,557 of 15,757 (60.6%) KOs were identified in at least 75% of the LX-planting and DT-planting soil microbiomes, respectively. A pairwise comparative analysis using DESq2 revealed that 5,002 and 4,929 KOs were enriched in LX-planting and DT-planting soil microbiomes, respectively. The core functional characteristics of P. notoginseng rhizosphere microorganisms in different planting patterns were determined, which showed that 3,990 KOs were enriched in LX and 3,973 KOs in DT (corrected P-value < 0.05, DESeq2) (Figures 9B–D and Supplementary Tables 7-1, 7-2).
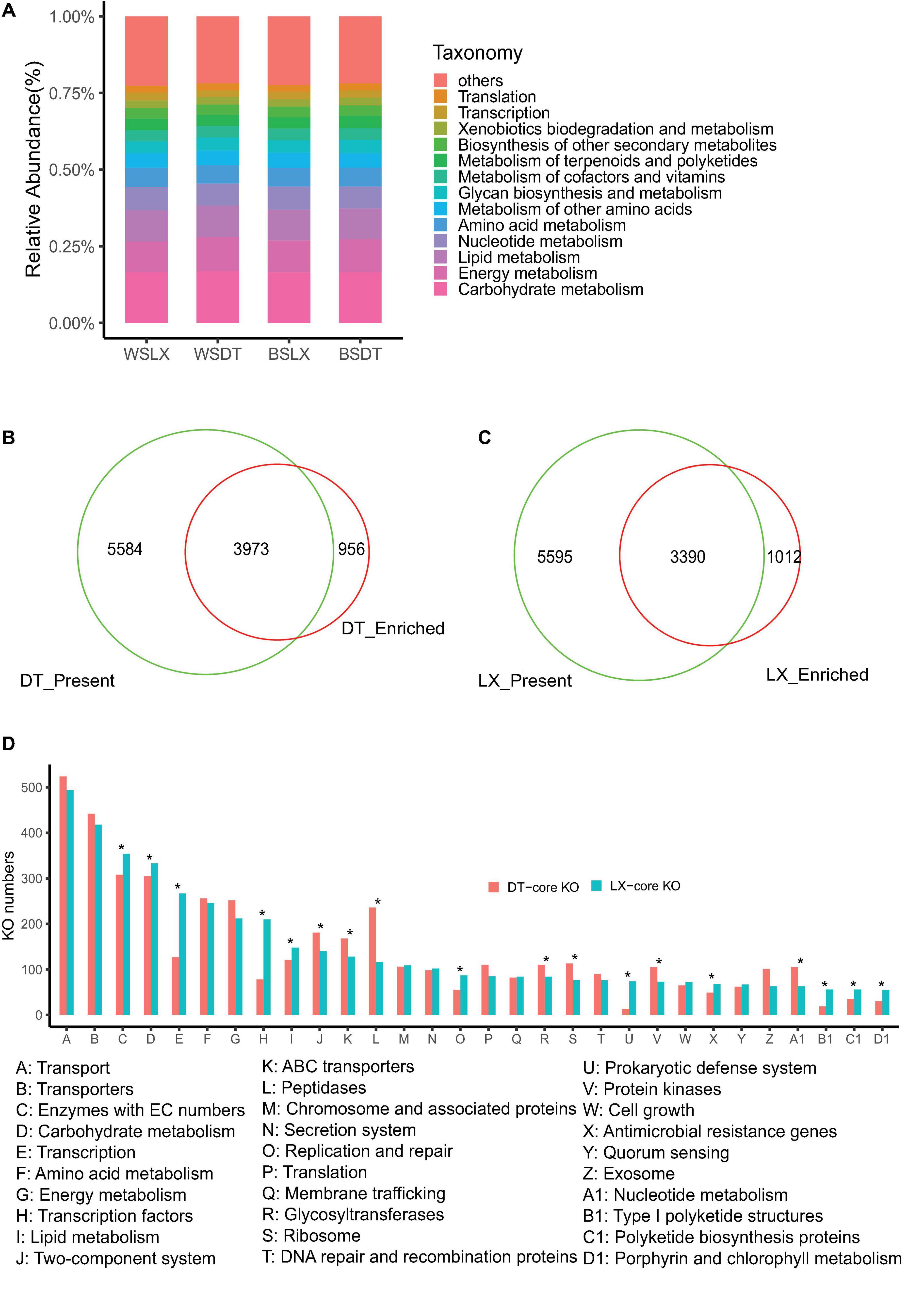
Figure 9. Characterization of the core function of the P. notoginsen rhizosphere microbiome in different planting patterns. (A) Functional KEGG level 2 pathway comparison of P. notoginsen rhizosphere microbiomes. (B) Venn plot depicting the number of core rhizosphere KOs based on the presence rate (>75%) in all understory planting samples, the KOs have a higher relative abundance in understory planting than in cropland planting. (C) Venn plot depicting the number of core rhizosphere KOs based on the presence rate (>5%) in all cropland plantin samples, the KOs have a higher relative abundance in cropland planting than in understory planting. (D) The distributions of the rhizosphere core KOs in the KEGG level 3 pathways. Asterisk indicates a significant difference with P < 0.05 (MW test). WSDT, rhizosphere soil of cropland planting in Wenshan City, WSLX, rhizosphere soil of understory planting in Wenshan City; BSDT, rhizosphere soil of cropland planting in Baise City; BSLX, rhizosphere soil of understory planting in Baise City.
The core functional characteristics involved in plant–microbe and microbe–microbe interactions were very likely to reflect the structural traits of microbiomes harboring the P. notoginseng rhizosphere. MW test to compare the core KO numbers between DT and LX revealed differences in several core functions. For example, bacterial secretion systems, bacterial chemotaxis, and flagellar assembly were over-presented in LX (Figure 10A) (P < 0.05, MW test), whereas bacterial toxins, and two-component systems were over-presented in DT (Figure 10A and Supplementary Figure 3) (P < 0.05, MW test). Quorum sensing and biofilm formation had little significance (Figure 9A) (P ≥ 0.05, MW test). The antimicrobial resistance and antibiotic synthesis were more enriched in LX than in DT (Figure 10B). For example, KOs associated with the biosynthesis of the vancomycin group of antibiotics, biosynthesis of 12-, 14-, and 16-member macrolides, biosynthesis of ansamycins, and biosynthesis of enediyne antibiotics were significantly higher in LX than in DT (P < 0.05, MW test). These observations suggested that more intimate host–microbe and microbe–microbe interactions occur in the LX-planting than in the DT-planting pattern.
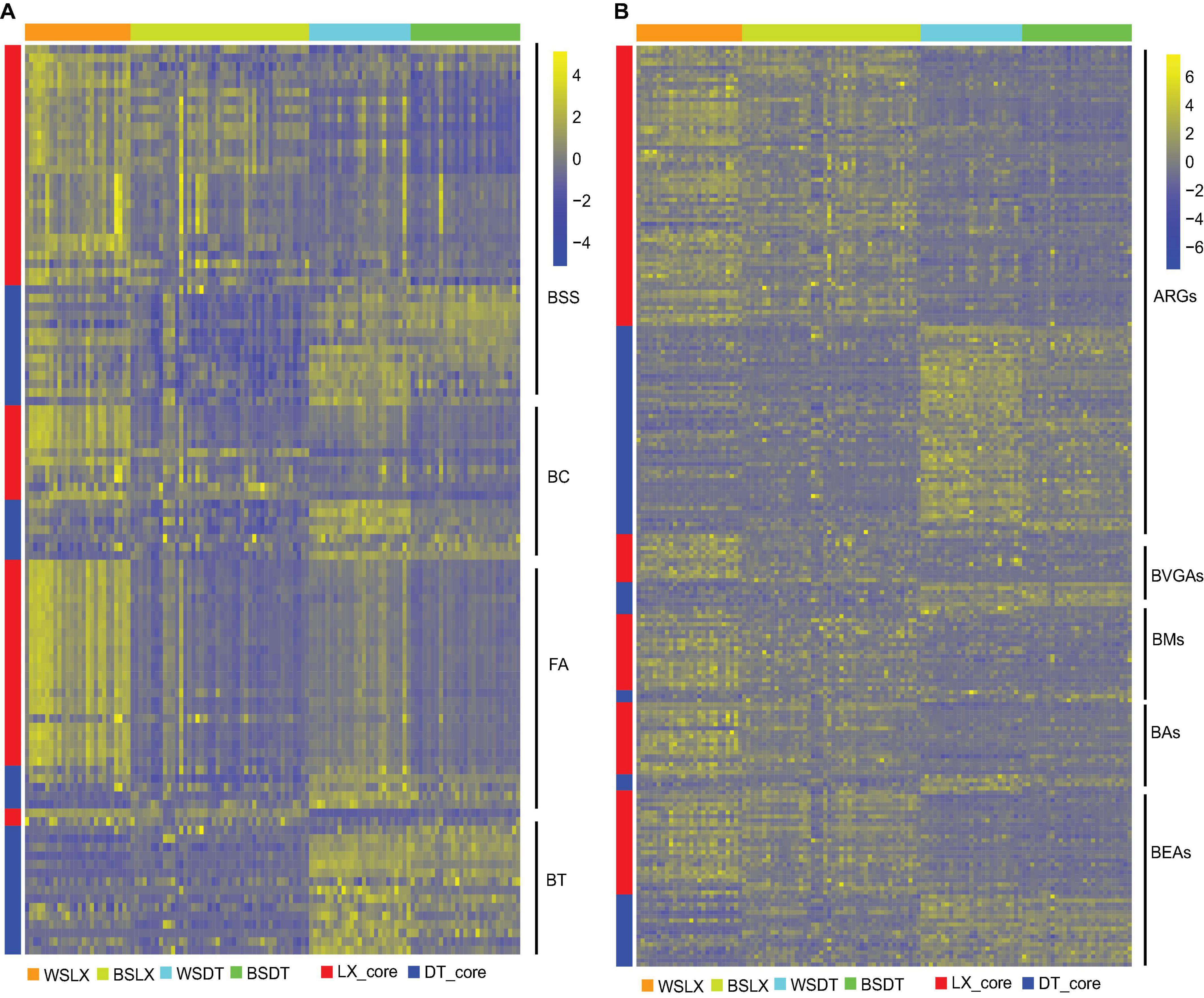
Figure 10. Relative abundances of core rhizosphere KOs in different planting patterns. (A) The relative abundance of core rhizosphere KOs involved in plant–microbe and microbe–microbe interactions. (B) The relative abundance of core rhizosphere KOs involved in antimicrobial resistant genes (ARGs), antibiotic synthesis genes (ASGs). Scale, relative abundance of KO at row normalization by removing the mean (centering) and dividing by the standard deviation (scaling). The color from blue to yellow represents a relative abundance of each KO from low to high. WSDT, rhizosphere soil of cropland planting in Wenshan City; WSLX, rhizosphere soil of understory planting in Wenshan City; BSDT, rhizosphere soil of cropland planting in Baise City; BSLX, rhizosphere soil of understory planting in Baise City; BSS, bacterial secretion systems; BC, bacterial chemotaxis; FA, flagellar assembly; BT, bacterial toxins; ARGs, antimicrobial resistant genes; BVGAs, biosynthesis of vancomycin group antibiotics; BMs. biosynthesis of 12-, 14-, and 16-membered macrolides; Bas, biosynthesis of ansamycins; BEAs, biosynthesis of enediyne antibiotics.
ABC transporters, which were mainly responsible for the transport of urea, ions, amino acids, monosaccharides, and oligosaccharides into microbial cells, were significantly more enriched in LX than in DT (Supplementary Figure 4) (P < 0.05, MW test). Metabolism of amino acids like glycine, serine, threonine, valine, leucine, isoleucine, lysine, and phenylalanine was significantly higher in LX than in DT (Supplementary Figure 5), whereas the KOs corresponding to alanine, aspartate, and glutamate metabolism were over-represented in DT (Supplementary Figure 6). The xenobiotics biodegradation-related KOs showed less difference between LX and DT (Supplementary Figure 6) (P ≥ 0.05, MW test). However, the dioxin degradation-related KOs were more enriched in DT, indicating that DT faces the problem of frequent pesticide application. The KOs corresponding to lysine, arginine, phenylalanine, tyrosine, and tryptophan biosynthesis was significantly higher in DT than in LX (P < 0.05, MW test) (Supplementary Figure 7), whereas the carbon fixation pathways in prokaryotes were significantly higher in LX than in DT (P < 0.05, MW test) (Supplementary Figure 8). A lesser number of KOs affiliated with peptidases were enriched in the LX rhizosphere microbiome (P ≥ 0.05, MW test) (Supplementary Figure 9). The transcription factors also showed a significant difference between DT and LX rhizosphere microbiomes. For example, the LuxR, AraC, GntR, MerR, TetR, AcrR, and LysR families of transcriptional regulators were more over-represented in LX than in DT (P < 0.05, MW test) (Supplementary Figure 10). Notably, such regulators were typically associated with metabolism, transport, quorum sensing, motility, stress response, and pathogenesis, which confirmed that a more intimate relationship of plant–microbe and microbe–microbe interactions appeared in LX than in DT.
Discussion
Microorganisms dwelling in the rhizosphere of plants play an important role in their growth and development. The plants and the microbes interact with each other and influence the growth of each other. P. notoginseng is an important Chinese medicinal plant with high global demand, such that its demand supersedes the production. Furthermore, several factors like root rot after few years of replanting (Sun et al., 2015), less suitable cropland, depletion of genetic diversity, and germplasm degeneration limit its production (Zhou et al., 2005). It is, therefore, imperative to evaluate the factors that could enhance the survival and growth of this species. This species is grown as an understory crop besides the main cropland species. It was observed that the understory Chinese medicinal plants experience a healthy microenvironment for their optimum growth (Yan et al., 2016). The studies on the rhizobial microbiome of P. notoginseng are scarce, and a comparative evaluation of the rhizobiome composition in the main cropland and understory cultivation is lacking. We, therefore, were interested in evaluating the microbiome variations based on the planting pattern, i.e., main cropland (DT) and understory planting (LX), in the Wenshan City of Yunnan Province and the Baise City of Guangxi Province, using a high-throughput metagenomics approach.
We found that the predominant taxa in the host rhizosphere microbiome were mainly prokaryotes, and eukaryotes accounted for a small fraction of the sequences. This finding was corroborated by Xu J. et al. (2018). The low proportion of eukaryotic taxa in the rhizosphere of P. notoginseng could be attributed to the unavailability of reference genomes for most eukaryotes (Bulgarelli et al., 2015). Approximately, 60% of the unigenes were assigned to known taxa. Taxonomic annotation at the phylum level indicates that Proteobacteria, Acidobacteria, and Actinobacteria were dominant in the rhizosphere. Similar results were obtained by several studies (Peiffer et al., 2013; Bulgarelli et al., 2015; Edwards et al., 2015; Jin et al., 2017; Xu J. et al., 2018; Xu L. et al., 2018; Fan et al., 2019). Generally, a plant will harbor specific microbial communities in its rhizosphere from the soil reservoir to facilitate growth.
The same as the citrus, little significant difference had been revealed between P. notoginseng rhizosphere and bulk samples (Xu J. et al., 2018). It may be that the threshold distance rendering the microbiomes between the rhizosphere and bulk soil distinct is species dependent upon this distance, the microbiomes of rhizosphere and bulk samples are more or less homogenous in the case of P. notoginseng. Various studies have reported that the microbial communities existing in the soil microbial reservoir can be influenced by factors such as plant genotype, location, planting pattern, and growth period (Peiffer et al., 2013; Edwards et al., 2015; Jin et al., 2017; Fan et al., 2019). Our study revealed that the planting pattern, DT or LX, was the major factor in determining the rhizosphere microbiome profile. Both alpha and beta diversities of P. notoginseng rhizosphere microbiomes in DT and LX planting patterns were significantly different. Location was the second major factor. This suggests that the planting patterns shaped the unique rhizosphere microbiomes in P. notoginseng.
The rhizosphere microbiome profile not only influences the growth of the host plants but also may be a potential bioindicator to reflect the health status of the soil. This is established by several studies concerning the microbial communities of crops such as maize, bananas, P. quinquefolius, and P. notoginseng (Fu et al., 2017; Tan et al., 2017b; Jiang et al., 2019). Dong et al. (2016) revealed that the death rate of P. notoginseng and the fungal diversity had a significantly negative correlation. In our study, LX enriches more core fungal genera than DT (203 vs. 17), and the total relative abundance of the core genera that were associated with disease suppression were significantly higher in LX than in DT, such as the genera belonging to the phyla Proteobacteria, Firmicutes, and Actinobacteria (Mendes et al., 2011). Several genera like Azospirillum, Bradyrhizobium, Burkholderia, and Streptomyces, which are potentially beneficial (Bevivino et al., 1998; Bhattacharyya and Jha, 2012), were also significantly higher in LX than in DT. Besides, although multiple genera associated with plant pathogens were also found to be significantly higher in LX than in DT such as Fusarium, Colletotrichum, and Blumeria, the non-pathogenic Fusarium may act as a potential biocontrol agent against fungal pathogens (Nel et al., 2006). Additionally, Colletotrichum has been proposed to promote plant growth through hormone production and phosphorus absorption under abiotic stress (Hiruma et al., 2016). These findings seem to evidently support that LX can create a better healthy environment, but the phenomenon still needs further and more studies to confirm.
The functional traits of the rhizosphere microbiome are closely associated with the life activities of the microorganism’s host–microbe and microbe–microbe interactions (Xu J. et al., 2018). The core functional traits involved in nutrient mobilization were over-represented in DT and LX (Figure 6D), suggesting that microbiomes need access to nutrients to form the surrounding soil, i.e., plant root exudates and humus (Peiffer et al., 2013). The rhizosphere enrichment of bacterial secretion systems, chemotaxis, flagella, assembly, antimicrobial resistance, and antibiotic synthesis genes are largely depended on the coevolution of host–microbe and microbe–microbe interactions, indicating that plants will positively harbor beneficial microbes to the rhizosphere. Interestingly, we observed more core antimicrobial resistance and antibiotic synthesis genes in LX than in DT, which indicate that the long-term use of farm chemicals in DT may cause the decrease in fungi.
Data Availability Statement
The data presented in the study are deposited in the NCBI (https://dataview.ncbi.nlm.nih.gov/objects?linked_to_id=PRJNA721004&archive=biosample) repository, accession number (PRJNA721004).
Author Contributions
JM, ZZ, and YD designed the study. LK and BC completed the experiments, completed the data analyses, and wrote the manuscript. JC, RS, and YD edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Digitalization of Biological Resource Project (No. 202002AA100007) and the Jiangsu University (No. 20JDG47).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the individuals who have helped us in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.673512/full#supplementary-material
References
Bevivino, A., Sarrocco, S., Dalmastri, C., Tabacchioni, S., Cantale, C., and Chiarini, L. (1998). Characterization of a free-living maize rhizosphere population of Burkholderia cepacia effect of seed treatment on disease suppression and growth promotion of maize. FEMS Microbiol. Ecol. 27, 225–237. doi: 10.1111/j.1574-6941.1998.tb00539.x
Bhattacharyya, P. N., and Jha, D. K. (2012). Plant growth-promoting rhizobacteria (PGPR): emergence in agriculture. World J. Microbiol. Biotechnol. 28, 1327–1350. doi: 10.1007/s11274-011-0979-9
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–63. doi: 10.1038/nmeth.3176
Bulgarelli, D., Garrido-Oter, R., Munch, P. C., Weiman, A., Droge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microb. 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6.
Deborah, Y., Hong, Q., Lau, A. J., Yeo, C. L., Liu, X. K., Yang, C. R., et al. (2005). Genetic diversity and variation of saponin contents in Panax notoginseng roots from a single farm. J. Agric. Food Chem. 53, 8460–8467. doi: 10.1021/jf051248g
Dong, L., Xu, J., Feng, G., Li, X., and Chen, S. (2016). Soil bacterial and fungal community dynamics in relation to Panax notoginseng death rate in a continuous cropping system. Sci. Rep. 6:31802.
Dong, Y., Duan, L., Chen, H. W., Liu, Y. M., Zhang, Y., and Wang, J. (2019). Network pharmacology-based prediction and verification of the targets and mechanism for Panax notoginseng saponins against coronary heart disease. Evid. Based Complement. Altern. Med. 2019:6503752.
Edwards, J., Johnson, C., Santos-Medellin, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920.
Fan, Z., Lu, S., Liu, S., Guo, H., Wang, T., Zhou, J., et al. (2019). Changes in plant rhizosphere microbial communities under different vegetation restoration patterns in karst and non-karst ecosystems. Sci. Rep. 9:8761.
Fan, Z. Y., Miao, C. P., Qiao, X. G., Zheng, Y. K., Chen, H. H., Chen, Y. W., et al. (2016). Diversity, distribution, and antagonistic activities of rhizobacteria of Panax notoginseng. J. Ginseng Res. 40, 97–104. doi: 10.1016/j.jgr.2015.05.003
Fravel, D., Olivain, C., and Alabouvette, C. (2003). Fusarium oxysporum and its biocontrol. New Phytol. 157, 493–502. doi: 10.1046/j.1469-8137.2003.00700.x
Fu, L., Penton, C. R., Ruan, Y., Shen, Z., Xue, C., Li, R., et al. (2017). Inducing the rhizosphere microbiome by biofertilizer application to suppress banana Fusarium wilt disease. Soil Biol. Biochem. 104, 39–48. doi: 10.1016/j.soilbio.2016.10.008
Guo, H. B., Cui, X. M., An, N., and Cai, G. P. (2010). Sanchi ginseng (Panax notoginseng (Burkill) F. H. Chen) in China: distribution, cultivation and variations. Genet. Resourc. Crop Evol. 57, 453–460. doi: 10.1007/s10722-010-9531-2
Hiruma, K., Gerlach, N., Sacristan, S., Nakano, R. T., Hacquard, S., Kracher, B., et al. (2016). Root Endophyte Colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent. Cell 165, 464–474. doi: 10.1016/j.cell.2016.02.028
Jiang, J., Yu, M., Hou, R., Li, L., Ren, X., Jiao, C., et al. (2019). Changes in the soil microbial community are associated with the occurrence of Panax quinquefolius L. root rot diseases. Plant Soil 438, 143–156. doi: 10.1007/s11104-018-03928-4
Jin, T., Wang, Y., Huang, Y., Xu, J., Zhang, P., Wang, N., et al. (2017). Taxonomic structure and functional association of foxtail millet root microbiome. Gigascience 6, 1–12. doi: 10.1093/gigascience/gix089
Klose, S., Acosta-Martínez, V., and Ajwa, H. A. (2006). Microbial community composition and enzyme activities in a sandy loam soil after fumigation with methyl bromide or alternative biocides. Soil Biol. Biochem. 38, 1243–1254. doi: 10.1016/j.soilbio.2005.09.025
Knief, C., Delmotte, N., Chaffron, S., Stark, M., Innerebner, G., Wassmann, R., et al. (2012). Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. 6, 1378–1390. doi: 10.1038/ismej.2011.192
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422.
Kwak, M. J., Kong, H. G., Choi, K., Kwon, S. K., Song, J. Y., Lee, J., et al. (2018). Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 36, 1100–1109. doi: 10.1038/nbt.4232
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leach, J. E., Triplett, L. R., Argueso, C. T., and Trivedi, P. (2017). Communication in the Phytobiome. Cell 169, 587–596. doi: 10.1016/j.cell.2017.04.025
Li, D., Luo, R., Liu, C. M., Leung, C. M., Ting, H. F., Sadakane, K., et al. (2016). MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Li, J., Qiu, P., Wang, S., Wu, J., He, Q., Li, K., et al. (2019). beta-N-Oxalyl-L-alpha,beta-diaminopropionic acid from Panax notoginseng plays a major role in the treatment of type 2 diabetic nephropathy. Biomed. Pharmacother. 114:108801. doi: 10.1016/j.biopha.2019.108801
Liu, L. L., Zhao, A. J., Yang, Y., Jin, H., Cui, X. M., and Liu, D. H. (2013). Comparative analysis of physical and chemical properties of Panax notoginseng replant soils in different intervals, Southwest China. J. Agric. Sci. 26, 1946–1952.
Liu, Y., Weng, W., Gao, R., and Liu, Y. (2019). New Insights for cellular and molecular mechanisms of aging and aging-related diseases: herbal medicine as potential therapeutic approach. Oxid. Med. Cell. Long. 2019:4598167.
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.
Mendes, R., Kruijt, M., de Bruijn, I., Dekkers, E., van der Voort, M., Schneider, J. H., et al. (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332, 1097–1100. doi: 10.1126/science.1203980
Miao, C. P., Mi, Q. L., Qiao, X. G., Zheng, Y. K., Chen, Y. W., Xu, L. H., et al. (2016). Rhizospheric fungi of Panax notoginseng: diversity and antagonism to host phytopathogens. J. Ginseng Res. 40, 127–134. doi: 10.1016/j.jgr.2015.06.004
Nel, B., Steinberg, C., Labuschagne, N., and Viljoen, A. (2006). The potential of nonpathogenic Fusarium oxysporum and other biological control organisms for suppressing fusarium wilt of banana. Plant Pathol. 55, 217–223. doi: 10.1111/j.1365-3059.2006.01344.x
Nelson, M. B., Martiny, A. C., and Martiny, J. B. H. (2016). Global biogeography of microbial nitrogen-cycling traits in soil. Proc. Natl. Acad. Sci. USA. 113, 8033–8040. doi: 10.1073/pnas.1601070113
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553. doi: 10.1073/pnas.1302837110
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844. doi: 10.1038/nbt.3935
Shoyama, Y., Zhu, X. X., Nakai, R., Shiraishi, S., and Kohda, H. (1997). Micropropagation of Panax notoginseng by somatic embryogenesis and RAPD analysis of regenerated plantlets. Plant Cell Rep. 16, 450–453. doi: 10.1007/s002990050258
Song, M., Yun, H. Y., and Kim, Y. H. (2014). Antagonistic Bacillus species as a biological control of ginseng root rot caused by Fusarium cf. incarnatum. J. Ginseng Res. 38, 136–145. doi: 10.1016/j.jgr.2013.11.016
Steinegger, M., and Soding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Su, G., Morris, J. H., Demchak, B., and Bader, G. D. (2014). Biological network exploration with cytoscape 3. Curr. Protoc. Bioinform. 47, 8.13.1–24.
Sun, X., Li, L., Long, G. Q., Zhang, G., Meng, Z. G., Yang, S., et al. (2015). The progress and prospect on consecutive monoculture problems of Panax notoginseng. Chin. J. Ecol. 34:885.
Sun, Y., Chen, Z., Li, G., Yang, S., Wei, M., and Cui, X. (2008). Preliminary study of allelochemicals on propagation of sanqi pathogen. Res. Pract. Chin. Med. 22, 19–21.
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Structure and function of the global ocean microbiome. Science 348, 1261351–1261359.
Tan, Y., Cui, Y., Li, H., Kuang, A., Li, X., Wei, Y., et al. (2017a). Diversity and composition of rhizospheric soil and root endogenous bacteria in Panax notoginseng during continuous cropping practices. J. Basic Microbiol. 57, 337–344. doi: 10.1002/jobm.201600464
Tan, Y., Cui, Y., Li, H., Kuang, A., Li, X., Wei, Y., et al. (2017b). Rhizospheric soil and root endogenous fungal diversity and composition in response to continuous Panax notoginseng cropping practices. Microbiol. Res. 194, 10–19. doi: 10.1016/j.micres.2016.09.009
Tang, M., Sun, J., Shimizu, K., and Kadota, K. (2015). Evaluation of methods for differential expression analysis on multi-group RNA-seq count data. BMC Bioinform. 16:361. doi: 10.1186/s12859-015-0794-7
Tang, X., Huang, M., Jiang, J., Liang, X., Li, X., Meng, R., et al. (2020). Panax notoginseng preparations as adjuvant therapy for diabetic kidney disease: a systematic review and meta-analysis. Pharm. Biol. 58, 138–145. doi: 10.1080/13880209.2020.1711782
Wang, J. S., Fan, F. F., Guo, J., Wu, A. L., Dong, E. W., Bai, W. B., et al. (2016). Effects of different crop rotations on growth of continuous cropping sorghum and its rhizosphere soil micro-environment. Ying Yong Sheng Tai Xue Bao 27, 2283–2291.
Wei, W., Wang, P., Wei, Y., Liu, Q., Yang, C., Zhao, G., et al. (2015). Characterization of Panax ginseng UDP-glycosyltransferases catalyzing protopanaxatriol and biosyntheses of bioactive ginsenosides F1 and Rh1 in metabolically engineered yeasts. Mol. Plant 8, 1412–1424. doi: 10.1016/j.molp.2015.05.010
Xiao, E., Ning, Z., Xiao, T., Sun, W., Qiu, Y., Zhang, Y., et al. (2019). Variation in rhizosphere microbiota correlates with edaphic factor in an abandoned antimony tailing dump. Environ. Pollut. 253, 141–151. doi: 10.1016/j.envpol.2019.06.097
Xu, J., Zhang, Y., Zhang, P., Trivedi, P., Riera, N., Wang, Y., et al. (2018). The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 9:4894.
Xu, L., Naylor, D., Dong, Z., Simmons, T., Pierroz, G., Hixson, K. K., et al. (2018). Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. U.S.A. 115, E4284–E4293.
Yan, Y., He, Y., Wan, G., Yin, J., and Huang, H. (2016). Preliminary study of cultivation on understory intercropping medicinal plants of Haikou forest farm in Yunnan. Mod. Chin. Med. 18, 173–177.
Yang, M., Zhang, X., Xu, Y., Mei, X., Jiang, B., Liao, J., et al. (2015). Autotoxic ginsenosides in the rhizosphere contribute to the replant failure of Panax notoginseng. PLoS One 10:e0118555. doi: 10.1371/journal.pone.0118555
Zhang, D., Li, W., Xia, E. H., Zhang, Q. J., Liu, Y., Zhang, Y., et al. (2017). The medicinal herb Panax notoginseng genome provides insights into ginsenoside biosynthesis and genome evolution. Mol. Plant 10, 903–907. doi: 10.1016/j.molp.2017.02.011
Zhang, Y., Chi, X., Wang, Z., Bi, S., Wang, Y., Shi, F., et al. (2019a). Protective effects of Panax notoginseng saponins on PME-Induced nephrotoxicity in mice. Biomed. Pharmacother. 116:108970. doi: 10.1016/j.biopha.2019.108970
Zhang, Y., Zheng, Y., Xia, P., Xun, L., and Liang, Z. (2019b). Impact of continuous Panax notoginseng plantation on soil microbial and biochemical properties. Sci. Rep. 9:13205.
Zhou, G., Xianquan, L., Jing, P., and Fanwu, G. (2016). Panax notoginseng F.H.Chen: cultivation techniques under different forests. J. Agric. 6, 41–44.
Zhou, J., Cui, X. M., Zeng, H., Yang, J., Ma, N., Zhang, W., et al. (2009). Study on chemical composition of root exudates of Panax notogineseng. Spec. Wild Econ. Anim. Plant Res. 3, 37–39.
Zhou, S., Xiong, G., Li, Z., and Wen, J. (2005). Loss of genetic diversity of domesticated Panax notoginseng F H Chen as evidenced by ITS sequence and AFLP polymorphism- a comparative study with P. stipuleanatus H Tsai et K M Feng. J. Integr. Plant Biol. 47, 107–115. doi: 10.1111/j.1744-7909.2005.00013.x
Keywords: Panax notoginseng, rhizosphere microbiome, microbial diversity, metagenome, planting patterns
Citation: Kui L, Chen B, Chen J, Sharifi R, Dong Y, Zhang Z and Miao J (2021) A Comparative Analysis on the Structure and Function of the Panax notoginseng Rhizosphere Microbiome. Front. Microbiol. 12:673512. doi: 10.3389/fmicb.2021.673512
Received: 27 February 2021; Accepted: 19 April 2021;
Published: 09 June 2021.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Ajar Nath Yadav, Eternal University, IndiaManoj Kumar Solanki, University of Silesia in Katowice, Poland
Copyright © 2021 Kui, Chen, Chen, Sharifi, Dong, Zhang and Miao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhanjiang Zhang, zzj1811@163.com; Jianhua Miao, mjh1962@163.com
†These authors have contributed equally to this work and share first authorship