
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 06 July 2021
Sec. Infectious Agents and Disease
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.664386
 Priti Devi1,2†Azka Khan1†
Priti Devi1,2†Azka Khan1† Partha Chattopadhyay1,2†
Partha Chattopadhyay1,2† Priyanka Mehta1
Priyanka Mehta1 Shweta Sahni1Sachin Sharma1
Shweta Sahni1Sachin Sharma1 Rajesh Pandey1,2*
Rajesh Pandey1,2*Human host and pathogen interaction is dynamic in nature and often modulated by co-pathogens with a functional role in delineating the physiological outcome of infection. Co-infection may present either as a pre-existing pathogen which is accentuated by the introduction of a new pathogen or may appear in the form of new infection acquired secondarily due to a compromised immune system. Using diverse examples of co-infecting pathogens such as Human Immunodeficiency Virus, Mycobacterium tuberculosis and Hepatitis C Virus, we have highlighted the role of co-infections in modulating disease severity and clinical outcome. This interaction happens at multiple hierarchies, which are inclusive of stress and immunological responses and together modulate the disease severity. Already published literature provides much evidence in favor of the occurrence of co-infections during SARS-CoV-2 infection, which eventually impacts the Coronavirus disease-19 outcome. The availability of biological models like 3D organoids, mice, cell lines and mathematical models provide us with an opportunity to understand the role and mechanism of specific co-infections. Exploration of multi-omics-based interactions across co-infecting pathogens may provide deeper insights into their role in disease modulation.
Typically, an infectious disease is thought to be caused by a single pathogen. However, concomitant infections by two or more pathogens are often observed in the real-world context. Co-infections may be detrimental, insignificant or beneficial for the disease outcomes depending on the levels of interactions, such as modulation of host response, diagnostic and therapeutic interventions (McArdle et al., 2018). Broadly, the interactions may be positive or negative. Positive interaction includes when pathogens in the host system act synergistically and lead to deterioration of disease symptoms, severity and outcomes. Negative interaction occurs when one pathogen impedes the growth of another pathogen through competition, parasitism or interference (Hoffman et al., 2006). When multiple pathogens co-infect a host, one pathogen may influence the replication and disease severity caused by the other. This is mostly observed during viral infections and is known as viral interference. Viral interference may lead to early clearance of one infection and persistence of the other one (Kumar et al., 2018). This is regulated by interferons (IFNs), transacting protease, defective interfering particles, and non-specific double-stranded RNA (Salas-Benito and De Nova-Ocampo, 2015; Kumar et al., 2016). Apart from the direct interactions between co-infecting pathogens, host response also plays a pivotal role in shaping the outcome of co-infections.
Research performed in the last two decades has established that infection triggers oxidative stress by inducing the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Oxidative stress-mediated epithelial barrier dysfunction and lung tissue injury have also been reported in acute respiratory infections. As a result of the oxidative burst during acute respiratory infection, the chances of secondary infection get increased (Ivanov et al., 2017). In many cases, oxidative stress is more pronounced during co-infection compared to a single infection alone (Rajopadhye et al., 2017). The memory T cells generated after one infection may alter the degree of immune response elicited against subsequent infection or co-infection. This is known as the heterologous immune response. Many immune cells are involved in a heterologous immune response, which may induce either protective or immune-pathological reaction upon a secondary infection or co-infection. The extent of oxidative stress and immune response together regulate the disease severity. Figure 1 depicts the inter-relationship between oxidative stress, inflammatory response, immune response and disease severity during co-infection by multiple pathogens. Co-infections such as in the case of Mycobacterium tuberculosis (MTb) or Hepatitis C virus (HCV) infection with Human Immunodeficiency Virus (HIV) are synergistically associated and have multi-faceted detrimental effects on the host. Apart from the cellular effect of increased oxidative stress, studies have reported the regulation of one infection by another co-infecting pathogen. For example, Mycobacterium infection exacerbates HIV-1 infection by oxidative stress induction mediated through the exosomes released from the infected macrophages (Tyagi et al., 2020). This review attempts to highlight the often ignored aspect of co-infections in infectious disease biology, albeit with limited specific examples, including its relevance in the context of the current Coronavirus disease-19 (COVID-19) pandemic. Finally, studying co-infections enables a better understanding of the effectors and outcomes of co-infections in terms of the role of oxidative stress and immune response.
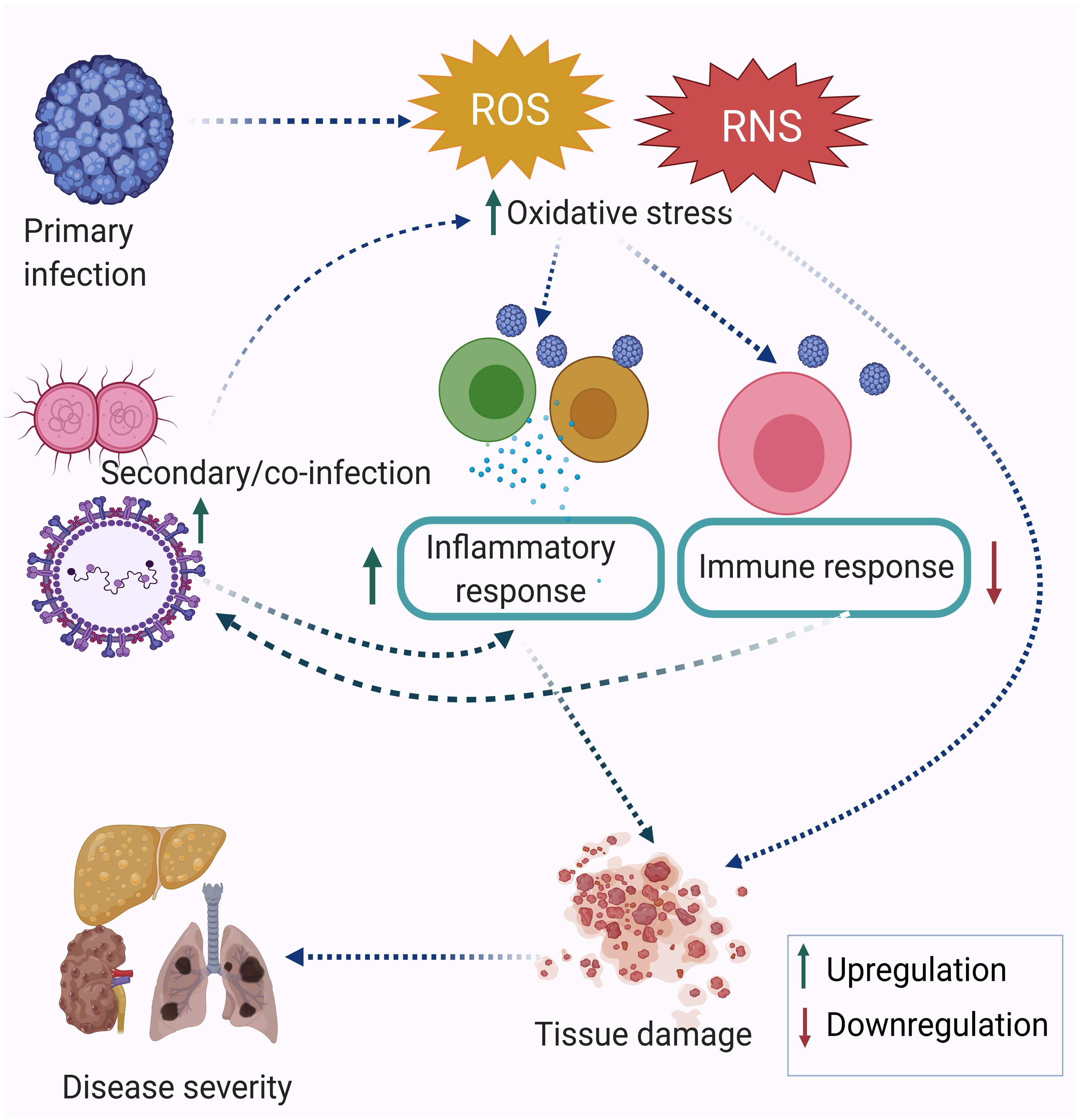
Figure 1. Co-infection modulating oxidative stress, immune response, and disease severity. The primary infection leads to an increase in oxidative stress, which is further enhanced by the secondary infection and eventually leads to dysregulation of the immune response. Taken together, this leads to tissue damage and acute disease outcomes.
Reactive oxygen species are highly reactive oxygen ions due to the presence of unpaired electrons in their outermost shell. Examples of ROS include superoxide (O2•−), hydroxyl radical (OH•), nitric oxide (NO•), hydrogen peroxide (H2O2), singlet oxygen (1O2), and ozone/trioxygen (O3). Mitochondria and peroxisomes are the major sites for production of ROS (Liou and Storz, 2010). It plays many important roles in physiological processes, including signaling, apoptosis and maintenance of cellular homeostasis. ROS is scavenged by antioxidant enzymes such as catalase, superoxide dismutase (SOD), and glutathione (GSH). An imbalance between the production and scavenging of ROS disrupts the redox homeostasis, leading to a state of oxidative stress with many detrimental effects on the cells. Elevated levels of ROS have been reported in many diseases, including infections (Bolukbas et al., 2005; Ivanov et al., 2017).
The immunological consequences of HIV infection open the door for opportunistic co-infecting pathogens to invade. One such co-infecting pathogen in HIV patients is MTb. Awodele and group reported reduced levels of antioxidant enzymes (GSH, SOD, and catalase) in HIV–MTb co-infected patients compared with patients having HIV infection only. The same group also reported an elevated level of malondialdehyde (MDA), a marker of lipid peroxidation, in HIV–MTb co-infection compared to HIV infection only (Awodele et al., 2011). MTb infection induces both mononuclear and polynuclear phagocytes to produce the ROS. Increased levels of ROS promote tissue injury and inflammatory response, which in turn, leads to immune-suppression (Jack et al., 1994; Rajopadhye et al., 2017). The situation gets further complicated in people with compromised immune systems, like patients with HIV infection, exacerbating the disease complications and possible morbidity. Reduced levels of antioxidant markers such as Vitamin A, C, E, selenium, GSH, SOD, catalase, as well as an increased level of MDA are observed in MTb patients co-infected with HIV (Rajopadhye et al., 2017; Gravier-Hernández and Gil-del Valle, 2018).
Another common example of co-infection is the concurrent infection with HIV and HCV. Multiple studies have reported a significant difference in the levels of oxidative stress markers and antioxidant stress markers between individuals with HIV or HCV co-infection and those with only one infection (Stehbens, 2004; Grønbaek et al., 2005; Baum et al., 2011). The chronic HIV infection leads to chronic oxidative stress with severe liver damage, cirrhosis, fibrosis, and carcinogenesis (De Maria et al., 1996). HCV infection leads to depleted levels of GSH, which causes immune suppression (Müller et al., 2017). The immune suppression, in turn, deteriorates the health condition of the patient in case of co-infection with HIV. Increased oxidative stress together with a compromised antioxidant defense system is one of the major causes of severe liver damage in HIV–HCV co-infection. Malondialdehyde, a marker of oxidative stress, was found to be elevated in patients with HCV or HIV infections (Paradis et al., 1997; Parola and Robino, 2001). Besides, antioxidant markers such as Vitamin A and E, zinc and selenium, are also found to be depleted in HIV patients. The combined effect results in a higher prevalence of liver fibrosis (Parola and Robino, 2001). Furthermore, in an in vitro study, it was found that treatment with antioxidants reduces the oxidative stress and disease severity induced by HIV and HCV (Price et al., 2006). A lower mitochondrial DNA copy number was observed in the HIV–HCV co-infected patients compared to either HIV or HCV infection alone, and this causes a further increase in oxidative stress (de Mendoza et al., 2005). It is presumed that the enhanced oxidative stress found in HIV–HCV co-infection may contribute to more rapid progression of liver fibrosis by stimulating HCV replication and enhanced production of ROS in hepatocytes (Farinati et al., 1995; Paradis et al., 1997; García-Mediavilla et al., 2005). On the flipside, multiple studies have evidenced ROS mediated modulation of viral replication. ROS was found to promote the HIV replication (Pace and Leaf, 1995; Ranjit et al., 2018). However, the effect of elevated ROS on HCV replication is yet to be fully understood (Medvedev et al., 2016; Anticoli et al., 2019). These pieces of evidence highlight an important role of co-infection in modulating the oxidative stress response and shaping the disease outcomes (Figure 2).
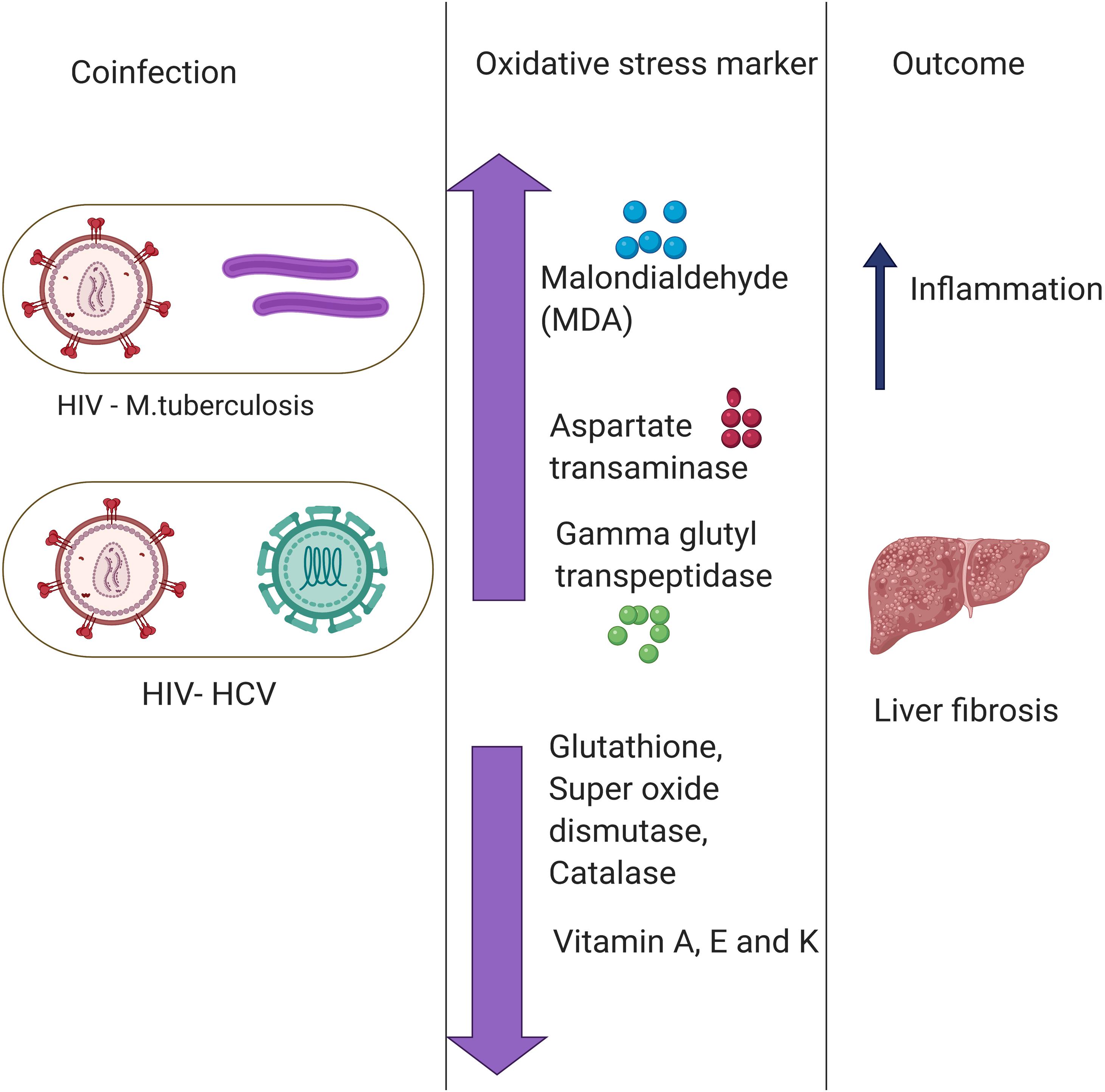
Figure 2. Role of co-infections in regulating oxidative stress. Co-infection of Hepatitis C Virus (HIV) with Mycobacterium tuberculosis (MTb) and human immunodeficiency virus (HCV) primes to a disproportion of oxidative stress and antioxidant markers leading to various disease outcomes such as liver fibrosis and inflammation. There are multiple Oxidative stress markers as highlighted above.
Within the ambit of this review, this section focuses on the immunological aspects of HIV–MTb, and HIV–HCV co-infection. We have also attempted to give an insight into HIV and MTb co-infections with SARS-CoV-2, even though research involving SARS-CoV-2 co-infections is a work in progress. Besides the already mentioned role of oxidative stress in mediating immune suppression, the former is also known to induce Unfolded Protein Response (UPR), which in turn stimulates the immune response (Grootjans et al., 2016). The oxidative stress-mediated regulation of immune response, therefore, depends on the extent of oxidative stress during the course of infection.
Several in vitro studies have shown that MTb infection stimulates the release of interleukin (IL-6) and the production of C-reactive protein (CRP)–a marker of inflammation (Bekker et al., 1998; Unsal et al., 2005; Rath et al., 2013). CRP level was found to be more pronounced in the HIV–MTb co-infection group compared to MTb infection alone (Rajopadhye et al., 2017). HIV–MTb co-infection also enhances the levels of pro-inflammatory cytokines such as Tumor Necrosis Factor-alpha (TNF-α) that further increases the replication of the virus and promotes the progression of the HIV to Acquired Immunodeficiency Syndrome (AIDS) (Kitaura et al., 2001). Figure 3 summaries the immunological outcomes of HIV–MTb and HIV–HCV co-infections.
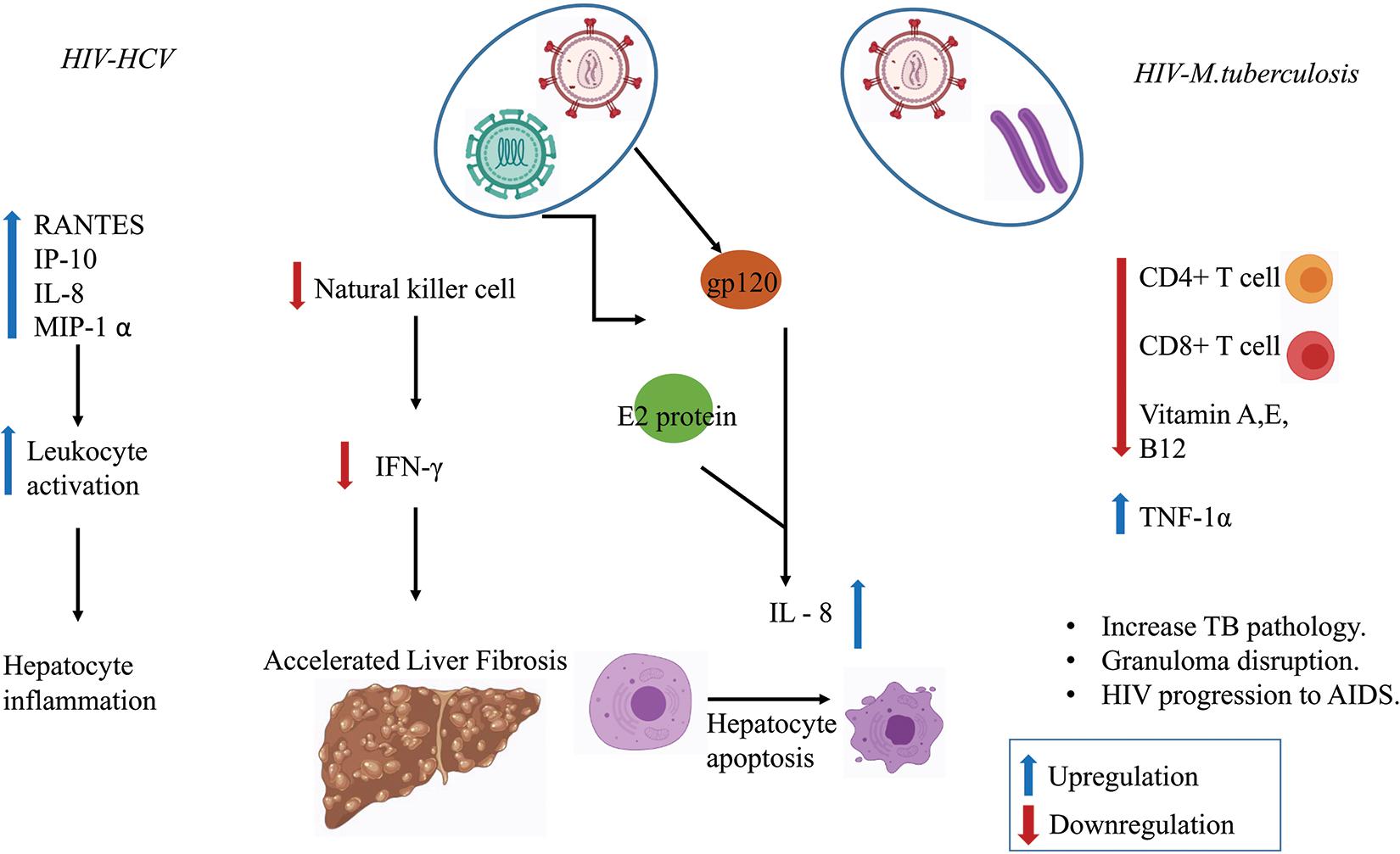
Figure 3. Co-infection in regulating the immune response. Various chemokines and immune cells show dysregulation during co-infection state which enhances disease severity such as liver fibrosis, hepatocyte inflammation, increase in MTb pathology, Granuloma disruption, and the progression of HIV to Acquired Immunodeficiency Syndrome (AIDS).
Several studies have demonstrated the cellular intricacies of the dysregulated immune system in infection and co-infections. While CD4+ T cell depletion is one of the major hallmarks of HIV infection, infection with HIV also results in the depletion of M. tuberculosis-specific CD4+ and CD8+ T cells. This results in a marked decrease in IL-2, IL-4, IL-5, and IL-13 levels in both latent and pulmonary MTb infection, thereby promoting disease progression (Ahmed et al., 2016; Amelio et al., 2019). Moreover, it is known that HIV is a main driver in the re-emergence of primary and reactivation of TB. Reactivation risk of latent TB is increased in patients after acquiring HIV. Interestingly, patients with HIV are more liable to TB, irrespective of CD4+ T cell levels but at the same time, risk increases with decrease in CD4+ T cell levels (Lin et al., 2012). Amelio et al. (2019) reported that serum levels of IL-1 (IL-6, IL-23, IP-10, and CRP in HIV–MTb co-infected patients are reduced compared to MTb infection alone. CD4+ T depletion in immune-compromised HIV infected patients promote the infection of other opportunistic pathogens such as Candida albicans and cytomegalovirus (CMV). Ample of evidences exhibit that HIV infected patients are more susceptible to mucosal candidiasis during early stage but CMV pathogenesis is evident during more severe reduction of CD4+ T cell in late phase of HIV infection (Anglaret et al., 2012).
In HIV infected patients, viral Tat protein stimulates the production of ROS, which may then activate the nuclear factor κB (NF-κB) and increase the viral transcription (Schreck et al., 1991; Sacktor et al., 2004). At the same time, many in vitro and in vivo studies have reported that MTb infection enhances the replication of HIV (Goletti et al., 1998; Hoshino et al., 2007, 2002). However, another study reported inhibition of HIV-1 replication by the MTb infection-induced IL-10 (Goletti et al., 1998; Souriant et al., 2019). In HIV infected patients, restoration or improvement of immune responses is mediated by antiretroviral therapy (ART). ART increases the life expectancy and decreases the mortality rate in HIV infected persons. But initiation of ART exaggerates immune response toward many pathogens that cause the immune reconstitution inflammatory syndrome (IRIS). Innate and adaptive immune system factors such as CD4+ T-cells, CD8+ T-cells, natural killer cells, macrophages, complement system, Toll -like receptors and pro-inflammatory cytokines promote the pathogenesis of IRIS. Risk factors contributing toward development of IRIS are high antigen load, low CD4+ cell counts, and scarcity of time between the start of ART and treatment for opportunistic pathogens. TB is considered among the most frequently reported pathogens linked with IRIS and studies have shown the occurrence of TB-IRIS within 2 months of ART initiation (Nelson et al., 2017; Aggarwal et al., 2018). Most common clinical manifestations of TB-IRIS are fever, lymphadenopathy and severe respiratory cues. Disseminated tuberculosis with associated acute renal failure, tuberculous meningitis, skin or visceral abscesses, osteomyelitis, epididymo-orchitis, abdominal TB-IRIS with non-specific abdominal pain, bowel perforation, and obstructive jaundice are also possible (Cevaal et al., 2019; Stek et al., 2020).
On similar lines, many studies have reported a higher extent of the immune-dysregulation in HIV–HCV co-infection compared with HIV or HCV infection alone. Interferons (IFN) are the key to inducing an antiviral immune response. Flynn et al. (2012) reported a significantly lower IFN-γ response, both in terms of magnitude and breadth, in HIV patients co-infected with HCV, when compared to HCV infection alone. This detrimental effect on IFN-γ in acute HCV infected patients co-infected with HIV correlates with the peripheral CD4+ T cell count. IP-10, an interferon-induced chemokine, is upregulated during HIV–HCV co-infection in comparison to HCV or HIV infection alone and the IP-10 level is correlated with the extent of hepatic inflammation and liver damage (Roe et al., 2007). In HepG2 cells co-infected with HCV and HIV, the HCV envelope protein (HCV-E2) and HIV envelope protein (HIV-gp120) together trigger the p38 mitogen-activated kinase (MAPK) pathway. This stimulates IL-8, a potent chemo-attractant and inducer of diseases such as rheumatoid arthritis, respiratory distress and sepsis. The IL-8 stimulation was found to be higher in the case of HIV–HCV co-infection than in cases of mono-infection (Kunkel et al., 1994; Balasubramanian et al., 2003). Apart from the immune-dysregulation, HIV–HCV co-infection is also linked to poor CD4+ T cell restoration in patients undergoing antiretroviral therapy (ART) (Shmagel and Saidakova, 2018). Taking together, these pieces of evidence of dysregulated immune and inflammatory responses as well as poor response to therapy may partly explain the reason for the rapid development of liver cirrhosis and hepatocarcinoma in HCV patients co-infected with HIV.
Many evidence toward co-infections with SARS-CoV-2 have been discovered during the current COVID-19 pandemic. Among the co-infecting pathogens, rhinovirus, RSV, Chlamydia pneumoniae, IAV, H1N1, Staphylococcus aureus, Pseudomonas aeruginosa, Staphylococcus pneumoniae, and Klebsiella pneumoniae are commonly observed in SARS-CoV-2 positive patients (Chen et al., 2020; Kim D. et al., 2020; Ma et al., 2020; Zhu et al., 2020; Burrel et al., 2021). Several cases of co-infection of MTb and HIV with SARS-CoV-2 have been reported as well, but there is a lack of any clear evidence supporting the role of co-infection in modulating the disease severity (Kanwugu and Adadi, 2020; Stochino et al., 2020). SARS-CoV-2 and M. tuberculosis may interact synergistically and affect the pathology of infection within the host, considering the similar route of cell entry, immune response and immune evasion (Tapela et al., 2020). SARS-CoV-2 and MTb, both infect the same type of cells, which include type II pneumocytes in the lungs and macrophages (Qian et al., 2013; Ganbat et al., 2016; Abassi et al., 2020). A case study by Khayat et al. (2021) reported progression of latent MTb infection (LMTBI) to an active MTb infection following a marked CD4+ T cell depletion associated with COVID-19. Another study involving COVID-19 reported a lower immune response (as measured by IFN-γ response against MTb antigen and SARS-CoV-2 S protein antigen) in MTb-COVID-19 and LMTBI-COVID-19 patients when compared to COVID-19 patients (Petrone et al., 2021). The aforementioned pieces of evidence indicate that depletion of T lymphocytes in COVID-19 patients may be a matter of concern; especially for people with LMTBI. Also, the lower levels of SARS-CoV-2 specific immune response in COVID-19 patients co-infected with MTb may increase the chance of re-infection with SARS-CoV-2.
Cases of HIV co-infection with SARS-CoV-2 have also been reported, wherein SARS-CoV-2 is found to accelerate the T lymphocyte exhaustion in HIV patients. The T lymphocyte depletion was more pronounced in HIV-SARS-CoV-2 co-infected patients without ART. IL-10 and tumor growth factor β (TGFβ) levels were also found to be elevated in patients co-infected with HIV and SARS-CoV-2, but much lower in patients undergoing ART (Sharov, 2021). Hyper-immune activation and resulting cytokine storm is believed to be the key player in COVID-19 disease severity and mortality (Hojyo et al., 2020). However, a case study by Suwanwongse and Shabarek (2020) reported a much higher mortality rate (7 out of 9 patients) in HIV-COVID-19 co-infected patients with a significantly lower CD4+ T cell count, than among COVID-19 patients without HIV. This is in contrast to the idea that the immune suppression by HIV infection may reduce the severe immunological consequences of COVID-19. Also, other studies, including another study by the same group recorded a lower mortality rate among HIV–COVID-19 patients compared to previous studies (Altuntas Aydin et al., 2020; Blanco et al., 2020; Marimuthu et al., 2021; Suwanwongse and Shabarek, 2021). One case study by Wang et al. reported a longer disease course and slow production of specific antibodies, when simultaneously infected by SARS-Cov-2 and HIV, may severely damage the immune system (Wang et al., 2020). The same group suggested that depletion of CD4+ lymphocytes occur and the remaining T-cells may have abnormal response to antigens during such co-infection. Interestingly, it is evident in severe patients contracted with COVID-19 that CD4+ T and CD8+ T levels were low (Zhang and Wu, 2020). Such retrospective study leads to a understanding that irrespective of HIV status, patients with COVID-19 still have low counts of CD4+ T cells. However, some studies reported poorer COVID-19 related outcomes in patients living with HIV (Ceballos et al., 2021; Tesoriero et al., 2021). The divergent findings in the case of HIV–SARS-CoV-2 co-infection warrant a thorough investigation into the immunological aspects of SARS-CoV-2 co-infection with HIV.
As discussed in the previous sections, increased oxidative stress and dysregulated immune function are evident during co-infections. Co-infections by multiple pathogens may play an important role in ameliorating or deteriorating disease outcomes. Opportunistic infection of MTb in HIV patients leads to a higher rate of mortality. The main reason for the compromised immune system in HIV patients is the progressive loss of CD4+ T cells in the lymphoid tissues, blood and mucosa, which significantly contributes to the increased risk of developing active MTb (Bruchfeld et al., 2015). Individuals having latent MTb possess a high propensity for reactivation of MTb if co-infected with HIV (Diedrich and Flynn, 2011). Both HIV and MTb act synergistically and in the process cripple the various arms of the host immune system, leading to a subsequent increase in mortality in the absence of any recommended standard of care (Whalen et al., 1995; Modjarrad and Vermund, 2010). Evidence suggests that HIV–MTb co-infection enhance/synergize each other in individuals and lead to immune dysfunction causing premature death, if left untreated. According to the World Health Organization (WHO) reports, worldwide, an estimated 10 million people were infected with MTb in 2019, among those nearly 8.2% included those living with HIV (Tuberculosis data, 2020). Another WHO report says one-third of the HIV infected patients have MTb and have up to 50 times higher risk of MTb infection compared to HIV negative individuals [Fact Sheet on Tuberculosis (TB), WHO]. An estimated 90% death rate was observed in HIV patients within 1 month of contracting MTb, if not treated properly.
Globally, an estimated 130 million individuals are infected with chronic hepatitis C virus (HCV). Among these, 4–5 million persons are co-infected with HIV (Operskalski and Kovacs, 2011). Infection with HIV enhances the HCV related disease severity such as cirrhosis, fibrosis, and end-stage liver disease (ESLD). Co-infected individuals are three times more prone to develop cirrhosis as compared to mono-infected individuals (Operskalski and Kovacs, 2011). A 20-years prospective study showed that co-infected drug users (DUs) receiving highly active antiretroviral therapy (HAART) had a higher risk of liver-related death compared to HCV mono-infected DUs (Smit et al., 2008). The accelerated liver disease in co-infected individuals is proposed to be due to the dysregulated immune system, increased oxidative stress, direct viral effects and decline of HCV specific T cells responses (Roe and Hall, 2008; Kim and Chung, 2009; Rotman and Liang, 2009). HIV infection is known to dysregulate the levels of cytokines like IL-4, IL-5, and IL-13, which in turn, accelerates liver fibrosis and inflammation (Roe and Hall, 2008). Further exacerbation of liver fibrosis is caused due to the imbalance of extracellular matrix (ECM) characterized by accumulation of ECM and declined deterioration of connective tissue proteins. A large family of zinc-dependent endopeptidases, matrix metalloproteinases (MMPs) control the degradation of ECM. Altered level of MMPs production could cause inflammation, wounds, invasion of cancer cells and liver fibrosis (Friedman, 2008a, b; Zhao et al., 2013). Study revealed that altered level of MMP and tissue inhibitors of metalloproteinases (TIMP) expression are linked to hepatic fibroproliferation in chronic Hepatitis C. HIV/HCV co-infected patients with more advance reduction of CD4+ T cell display enhanced circulating TIMP-1 level and thus promote liver fibrosis (Mastroianni et al., 2014). It is well known fact that low level of hyaluronic acid (HA) is associated with the low risk of liver disease progression. Toward predicting the liver disease progression in HIV/HCV co-infection, detecting the HA plasma detection alone or in combination with other methods, may be a beneficial way of monitoring (Peters et al., 2013). Apoptosis of hepatocytes through a Fas/FasL pathway has been found to be enhanced in HIV–HCV co-infection that results in hastening the liver disease (Kim and Chung, 2009). The accumulation of CD8+ T cells in the liver of co-infected individuals enhances the production of TNF-α and other inflammatory mediators (Kuntzen et al., 2008; Nakamoto et al., 2008; Vali et al., 2008). This further exacerbates the tissue damage and liver fibrosis when compared to HCV infected individuals. Replication of HIV in the hepatocytes and hepatic stellate cells (HSC) stimulates the production of collagen and pro-inflammatory cytokines that further promote the fibrosis (Blackard and Sherman, 2008; Tuyama et al., 2010). HIV–HCV co-infection has been reported to enhance the severity of kidney-related diseases such as acute renal failure and proteinuria when compared to the HIV infection alone (Wyatt et al., 2008). Also, HIV–HCV co-infection related nephropathies increased the mortality rate compared to the HCV mono-infection (Izzedine et al., 2009). Cardiovascular diseases are also found to be more prevalent in the HIV–HCV co-infection when compared to the HIV mono-infection. Co-infection with HCV enhances the chances of acute myocardial infarction in the HIV infected individuals (Bedimo et al., 2010). HIV–HCV co-infected individuals treated with HAART showed higher endothelial dysfunction due to elevated levels of circulating soluble cellular adhesion molecules (CAMS) released by the vascular endothelium, soluble intercellular adhesion molecule-1 (sICAM-1) and vascular adhesion molecule-1 (sVCAM-1) (de Castro et al., 2010).
In contrast to the above-mentioned cases, wherein co-infection deteriorates the disease outcome, co-infection of Influenza A viruses and rhinoviruses, both of which invade the respiratory tract, is found to be associated with reduced disease severity (Greer et al., 2009; Esper et al., 2011; Jain et al., 2015; Nolan et al., 2018). A mouse model-based study by Gonzalez et al. found that prior inoculation of a rhinovirus strain 1B (RV1B) suppressed the severity of influenza infection in a time and dose-dependent manner. The decrease in mortality and disease severity was not associated with a reduction in the viral titer or replication, but prior inoculation with rhinoviruses induced early inflammatory response in the lungs that helped in clearance of influenza virus strain (Gonzalez et al., 2018). Another study by Shinjoh et al. (2000) reported that IAV infected cells when co-infected with RSV had reduced IAV titers, without altering the direct replication of IAV after 12 h post-infection. Ample evidence exhibit role of IAV facilitating the infection of S. pneumoniae in respiratory tract. Type I interferon induced by IAV infection sensitizes the host for other bacterial co-infections like S. pneumoniae and Mtb and often lead to serious consequences (Shahangian et al., 2009; Redford et al., 2014). Although there is no direct involvement of IAV induced IFN in regulating the SARS-CoV-2 co-infection, studies suggested that the higher infectivity of SARS-CoV-2 in IAV infected patients may be due to ACE2 expression, which is IFN-stimulated gene (Ziegler et al., 2020; Bai et al., 2021). Virus promotes the bacterial super-infection by enhancing the availability of receptors. Cryptic receptors for bacterial adherence are facilitated by the influenza virus neuraminidase that cleaves sialic acid and disrupt sialylated mucins that can function as decoy receptors for the bacteria (McCullers and Bartmess, 2003; McCullers, 2004). In addition to dysregulated immune responses and imbalance of ROS, excess of metal such as iron may contribute toward the severity and outcomes of infections. Study revealed that excess amounts of iron promote the viral replication and multiplication of mycobacteria (Lounis et al., 2001; Chao et al., 2019). The proteins involved in virulence and oxidative stress response in MTb are regulated by the iron dependent regulator, IdeR (Kurthkoti et al., 2015). It is reasonable to anticipate that the excess iron may be the cause of more severe outcome during co-infection as excess iron has potential to impact infectivity of different pathogens independently. High serum iron was detected in HIV patients, which, might contribute to further CD4+ T cell depletion (Banjoko et al., 2012; Chang et al., 2015). Elevated hepatic iron level was also reported in HCV infected patients (Maier, 1997; Price and Kowdley, 2009). The elevated iron level was reported to elevate 8-hydroxy-2’-deoxyguanosine (8-OHdG) level, resulting in hepatic oxidative stress and DNA damage, leading to liver fibrosis (Fujita et al., 2008). Iron chelating agents such as silybin and DFO may be a better therapeutic for preventing pathogen multiplication (Agoro and Mura, 2019). The varying outcomes of co-infections necessitate further research into the dynamics of interactions between co-infecting pathogens to enable better management and treatment of infectious diseases.
To understand the drivers of disease severity, clinical implications, diagnosis, and therapeutic management in case of infectious diseases, experimental model systems are required to recapitulate and mimic the patho-physiology of the host-pathogen interactions. Different model systems, as depicted in Figure 4, can be potentially harnessed to decipher the role of co-infections in modulating the disease outcomes.
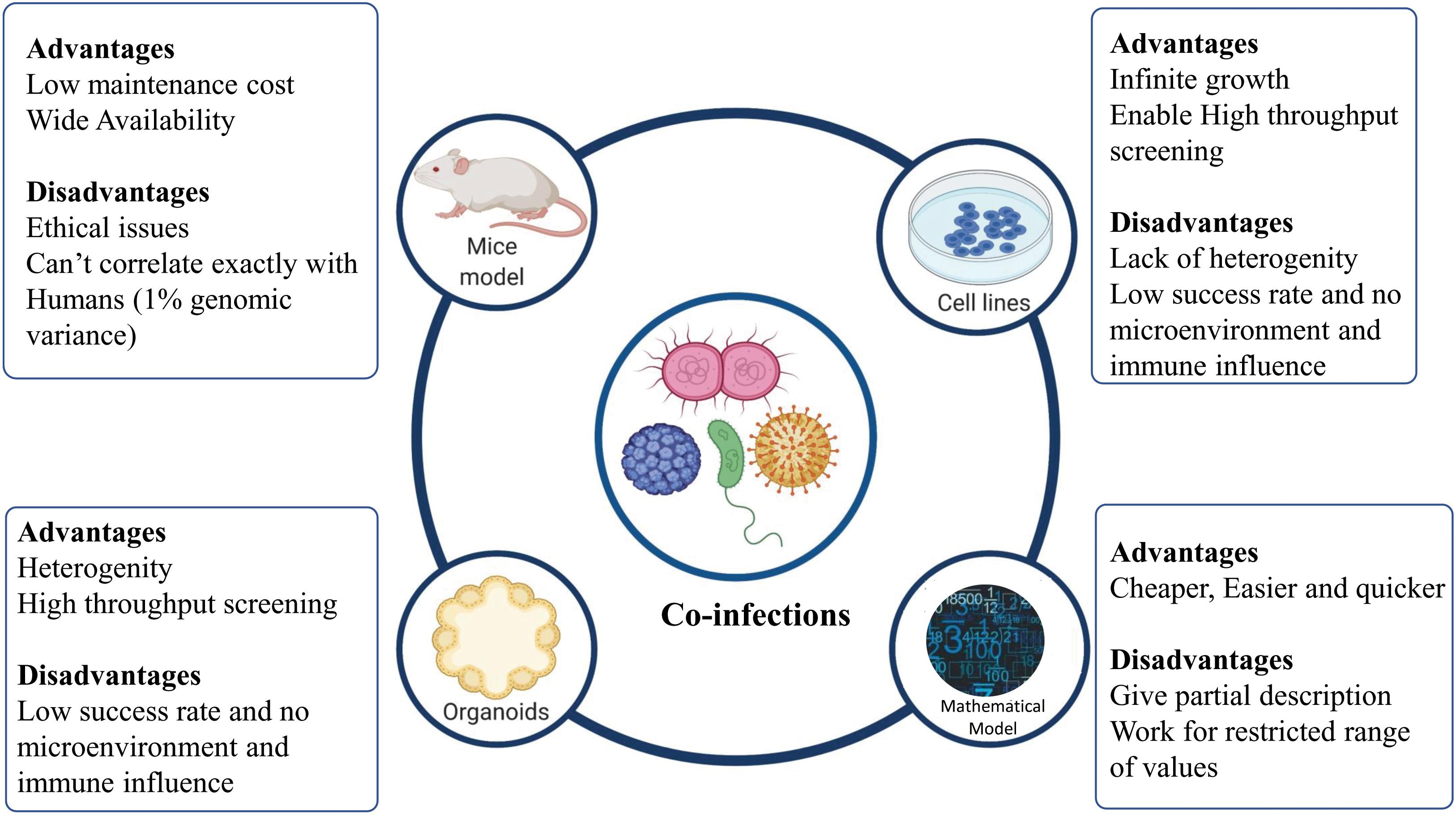
Figure 4. Different models to study co-infection. Various model systems (mice models, cell lines, organoids, and mathematical models) can be potentially harnessed to decipher the role of co-infections. The figure also shows the advantages and disadvantages associated with various model systems mentioned.
Cell lines provide the most rudimentary information about the nature of a host-pathogen interaction, if cells are permissive to their division/replication. For example, the examination of HIV-HBV interactions within hepatic cell lines has been demonstrated by Iser et al. (2010). However, cell lines must be maintained specially to balance selective pressure for growing pathogens. For a more detailed insight into mechanistic details of the infection, a “complex architect of tissue” is the prerequisite. Different animal models are also available, which allow the study of co-infections. Animal models have been used to mimic different diseases caused by co-infecting viruses, bacteria, and virus-bacteria. For example, co-infection of pulmonary tuberculosis with HIV-1 was studied in humanized mice model by Rebecca et al. (Nusbaum et al., 2016). Using animal models allows deciphering the patho-physiology of a particular co-infection for better understanding and therapeutic management of the disease. However, mimicking human diseases in animal hosts presents significant challenges owing to the host-dependent restriction mechanisms of most pathogens (Bakaletz, 2004). An in vitro 3D Organoid model provides a possible means to bridge the gap between the 2D and the in vivo model, to understand the modulators of host-pathogen interaction in deciphering the pathogenesis, defining the species barrier and identifying cellular niches for bacteria or viruses (Rossi et al., 2018; Kim J. et al., 2020).
Organoids are 3D structures, developed from multipotent tissue adult stem cells or most often from induced pluripotent cells. Organoids can be used experimentally to test the new investigational therapeutics against the co-infecting pathogens. However, a lack of the dynamic microenvironment mimicking the human body, blood vessels, nerves and immune influence (inter-organ communication) can be counted as one of the limitations of organoid models of infection (Kim J. et al., 2020). Apart from these experimental model systems, mathematical models have been developed to better understand the disease dynamics and to devise better therapeutic regimens (Esteva and Vargas, 2003; Vickerman et al., 2012; Corson et al., 2013). Different co-infecting pathogens such as RSV, rhinovirus, IAV, Human para-influenza virus, and human metapneumovirus infection dynamics can be studied via a mathematical model devised by Pinky and Dobrovolny (2017). Other than understanding the mechanistic details of co-infection biology, such models can be employed to simulate and understand the disease pathophysiology, decipher host-pathogen interactions and for the screening of drugs and other therapeutic interventions against co-infecting pathogens.
Co-infection biology is known to be modulated by the nature of interaction at multiple hierarchies between interacting microorganisms within the common host, which explains the complex non-linear dynamics of infectious diseases severity and clinical outcome (Figure 5). The degree of facilitation or inhibition between interacting pathogens, dependent upon the co-infecting species, in turn, decides the severity of a disease. A mechanistic understanding of the nature of these interactions, whether synergistic or antagonistic, has major consequences for treatment-specific decision making. The interactions between microbes at the cellular level are driven by a multiplicity of factors. Changes in cell surface receptor presentation may be altered by a pathogen, which may, in turn, lead to super-infection suppression or exclusion. Co-infecting pathogen dynamics within the host are driven by ecological constraints inclusive of resource and space limitation. Co-infecting microbes modulate the infection outcomes by altering the nature and extent of immunological response within the host. A classical case in point is the HIV infection, which compromises the host immune system thereby, making the host more susceptible to co-infection by other opportunistic pathogens. At the host level, inter-pathogen interactions may lead to altered disease epidemiology affecting the mortality and morbidity associated with an infectious disease (Pandey et al., 2021; Figure 5). Co-infection increases the possibility of genetic recombination and reassortment leading to the emergence of novel antigenic variants thus affecting the efficacy of drugs, treatment regimen and vaccines. In the rarest of instances, this may even lead to the evolution of new zoonotic infections. Co-infection biology is also important to evaluate the nature of response to pharmacological interventions. Patients with co-infections, when subjected to antimicrobial chemotherapies, may facilitate the emergence of multi-drug resistant species and propagation of Antimicrobial Resistance (AMR). Population-level consequences of co-infections are, therefore, important to consider for designing, optimizing, and delivering Public Health Interventions in diagnostics, therapeutics, prophylaxis and prognosis of infectious diseases. The non-linear disease outcomes in the case of infectious diseases are important considerations for disease surveillance and public health policymaking at the societal level.
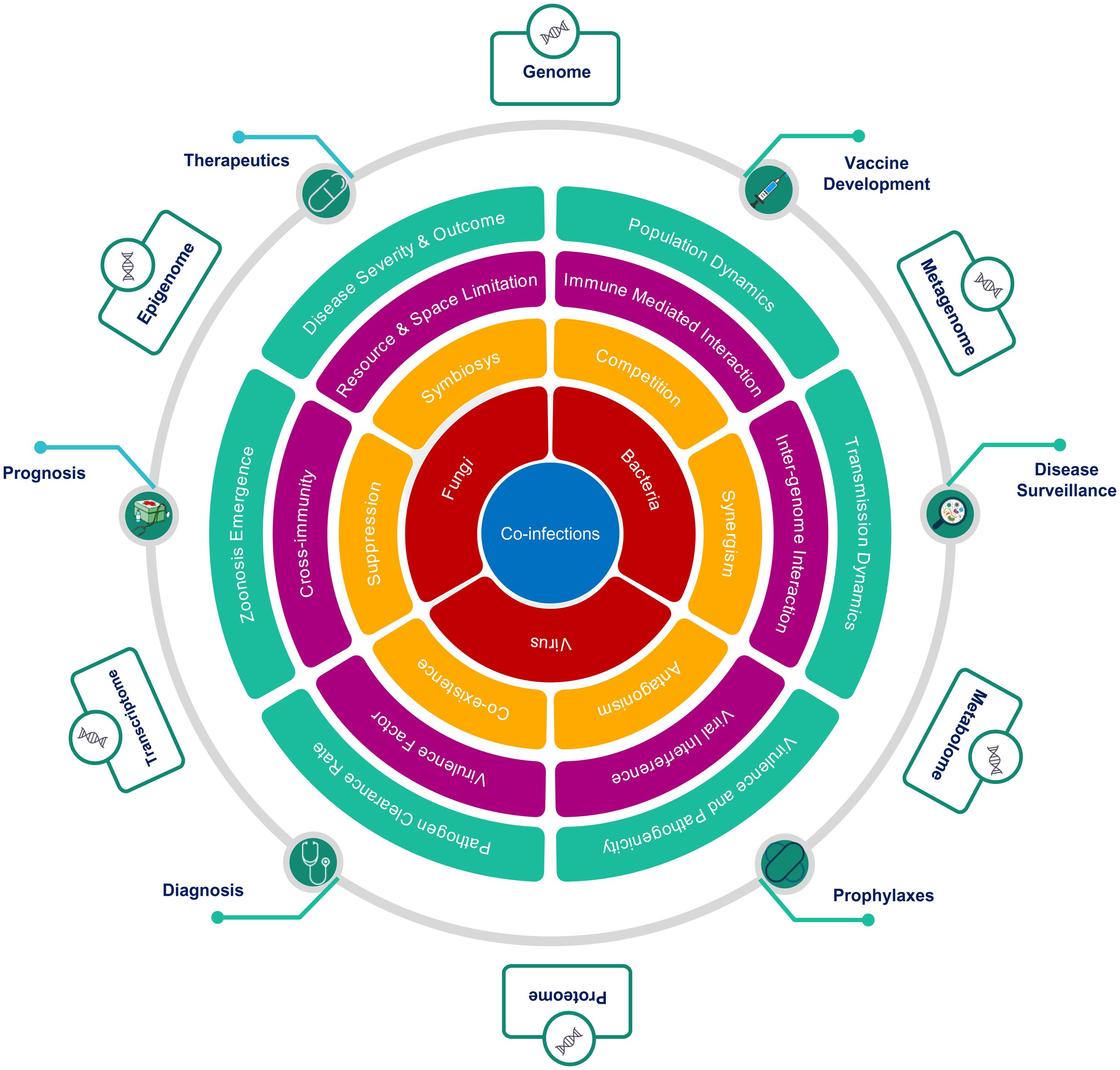
Figure 5. Hierarchical role of co-infections as cause, modulator and effector of clinical outcomes. The figure highlights the importance of an integrative approach across multiple hierarchies to study co-infections, from the perspective of, (A) interacting microorganisms (Red), (B) nature of interactions between them (Yellow), (C) mechanisms mediating interactions (Magenta), (D) macro level effects of co-infections (Green), (E) Public health interventions affected by co-infections (Green circles), and (F) multi-omics approaches to study of co-infections (Outer boxes).
To conclude, infectious disease dynamics has an important component of co-infections or co-presence of microbes (potential pathogens). The degree and nature of this conditioning and the underlying mechanisms remain largely unexplored although its potential has been highlighted by the examples discussed above. Multi-Omics approaches, as depicted in Figure 5, may provide suitable tools and techniques to enable a clearer understanding and elucidation into the role and significance of co-infections in modulating infectious disease outcomes. The co-infection outcome can be infested at multiple hierarchies inclusive of genomic, transcriptomic, epigenomic, metagenomic, proteomic, and metabolomic. This will have functional impact on disease surveillance, diagnosis, detection, treatment regimen, vaccine development and efficacy. Thus, the dynamics of primary infection, secondary infection, pre-existing infection and opportunistic infection in time and space within population/s have important role toward disease trajectory and outcome.
PD, AK, PC, PM, SSa, SSh, and RP wrote the manuscript. RP coordinated the study and funding for the work. All authors contributed to the article and approved the submitted version.
This research was funded by the Council of Scientific and Industrial Research (CSIR), project code (MLP-2005); Fondation Botnar, project code (CLP-0031); Indo-US Science and Technology Forum (IUSSTF), project code (CLP-0033); and Intel funded, project code (CLP-0034).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
PC and PD acknowledge the CSIR for their Research Fellowship. AK, PM, SSa, and SSh acknowledges support from CSIR (MLP-2005).
Abassi, Z., Knaney, Y., Karram, T., and Heyman, S. N. (2020). The lung macrophage in SARS-CoV-2 infection: A friend or a foe? Front. Immunol. 11:1312. doi: 10.3389/fimmu.2020.01312
Aggarwal, N., Barclay, W., and Shinohara, M. L. (2018). Understanding mechanisms underlying the pathology of immune reconstitution inflammatory syndrome (IRIS) by using animal models. Curr. Clin. Microbiol. Rep. 5, 201–209. doi: 10.1007/s40588-018-0099-5
Agoro, R., and Mura, C. (2019). Iron supplementation therapy, A friend and foe of mycobacterial infections? Pharmaceuticals 12:75. doi: 10.3390/ph12020075
Ahmed, A., Rakshit, S., and Vyakarnam, A. (2016). HIV-TB co-infection: mechanisms that drive reactivation of Mycobacterium tuberculosis in HIV infection. Oral Dis. 22(Suppl. 1), 53–60. doi: 10.1111/odi.12390
Altuntas Aydin, O., Kumbasar Karaosmanoglu, H., and Kart Yasar, K. (2020). HIV/SARS-CoV-2 coinfected patients in Istanbul, Turkey. J. Med. Virol. 92, 2288–2290. doi: 10.1002/jmv.25955
Amelio, P., Portevin, D., Hella, J., Reither, K., Kamwela, L., Lweno, O., et al. (2019). HIV infection functionally impairs Mycobacterium tuberculosis-Specific CD4 and CD8 T-Cell Responses. J. Virol. 93:e01728-18. doi: 10.1128/JVI.01728-18
Anglaret, X., Minga, A., Gabillard, D., Ouassa, T., Messou, E., Morris, B., et al. (2012). AIDS and non-AIDS morbidity and mortality across the spectrum of CD4 cell counts in HIV-infected adults before starting antiretroviral therapy in Cote d’Ivoire. Clin. Infect. Dis. 54, 714–723. doi: 10.1093/cid/cir898
Anticoli, S., Amatore, D., Matarrese, P., De Angelis, M., Palamara, A. T., Nencioni, L., et al. (2019). Counteraction of HCV-induced oxidative stress concurs to establish chronic infection in liver cell cultures. Oxid. Med. Cell. Longev. 2019:6452390. doi: 10.1155/2019/6452390
Awodele, O., Olayemi, S. O., Nwite, J. A., and Adeyemo, T. A. (2011). Investigation of the levels of oxidative stress parameters in HIV and HIV-TB co-infected patients. J. Infect. Dev. Ctries. 6, 79–85. doi: 10.3855/jidc.1906
Bai, L., Zhao, Y., Dong, J., Liang, S., Guo, M., Liu, X., et al. (2021). Coinfection with influenza A virus enhances SARS-CoV-2 infectivity. Cell Res. 31, 395–403. doi: 10.1038/s41422-021-00473-1
Bakaletz, L. O. (2004). Developing animal models for polymicrobial diseases. Nat. Rev. Microbiol. 2, 552–568. doi: 10.1038/nrmicro928
Balasubramanian, A., Ganju, R. K., and Groopman, J. E. (2003). Hepatitis C virus and HIV envelope proteins collaboratively mediate interleukin-8 secretion through activation of p38 MAP kinase and SHP2 in hepatocytes. J. Biol. Chem. 278, 35755–35766. doi: 10.1074/jbc.M302889200
Banjoko, S. O., Oseni, F. A., Togun, R. A., Onayemi, O., Emma-Okon, B. O., and Fakunle, J. B. (2012). Iron status in HIV-1 infection: implications in disease pathology. BMC Clin. Pathol. 12:26. doi: 10.1186/1472-6890-12-26
Baum, M. K., Sales, S., Jayaweera, D. T., Lai, S., Bradwin, G., Rafie, C., et al. (2011). Coinfection with hepatitis C virus, oxidative stress and antioxidant status in HIV-positive drug users in Miami. HIV Med. 12, 78–86. doi: 10.1111/j.1468-1293.2010.00849.x
Bedimo, R., Westfall, A. O., Mugavero, M., Drechsler, H., Khanna, N., and Saag, M. (2010). Hepatitis C virus coinfection and the risk of cardiovascular disease among HIV-infected patients. HIV Med. 11, 462–468. doi: 10.1111/j.1468-1293.2009.00815.x
Bekker, L. G., Maartens, G., Steyn, L., and Kaplan, G. (1998). Selective increase in plasma tumor necrosis factor-alpha and concomitant clinical deterioration after initiating therapy in patients with severe tuberculosis. J. Infect. Dis. 178, 580–584. doi: 10.1086/517479
Blackard, J. T., and Sherman, K. E. (2008). HCV/HIV co-infection: time to re-evaluate the role of HIV in the liver? J. Viral Hepat. 15, 323–330. doi: 10.1111/j.1365-2893.2008.00970.x
Blanco, J. L., Ambrosioni, J., Garcia, F., Martínez, E., Soriano, A., Mallolas, J., et al. (2020). COVID-19 in patients with HIV: clinical case series. Lancet HIV 7, e314–e316. doi: 10.1016/S2352-3018(20)30111-9
Bolukbas, C., Bolukbas, F. F., Horoz, M., Aslan, M., Celik, H., and Erel, O. (2005). Increased oxidative stress associated with the severity of the liver disease in various forms of hepatitis B virus infection. BMC Infect. Dis. 5:95. doi: 10.1186/1471-2334-5-95
Bruchfeld, J., Correia-Neves, M., and Källenius, G. (2015). Tuberculosis and HIV coinfection. Cold Spring Harb. Perspect. Med. 5:a017871. doi: 10.1101/cshperspect.a017871
Burrel, S., Hausfater, P., Dres, M., Pourcher, V., Luyt, C.-E., Teyssou, E., et al. (2021). Co-infection of SARS-CoV-2 with other respiratory viruses and performance of lower respiratory tract samples for the diagnosis of COVID-19. Int. J. Infect. Dis. 102, 10–13. doi: 10.1016/j.ijid.2020.10.040
Ceballos, M. E., Ross, P., Lasso, M., Dominguez, I., Puente, M., Valenzuela, P., et al. (2021). Clinical characteristics, and outcomes of people living with HIV hospitalized with COVID-19: a nationwide experience. Int. J. STD AIDS 32, 435–443. doi: 10.1177/0956462420973106
Cevaal, P. M., Bekker, L.-G., and Hermans, S. (2019). TB-IRIS pathogenesis and new strategies for intervention: insights from related inflammatory disorders. Tuberculosis 118:101863. doi: 10.1016/j.tube.2019.101863
Chang, H.-C., Bayeva, M., Taiwo, B., Palella, F. J., Hope, T. J., and Ardehali, H. (2015). Short communication: high cellular iron levels are associated with increased HIV infection and replication. AIDS Res. Hum. Retroviruses 31, 305–312. doi: 10.1089/aid.2014.0169
Chao, A., Sieminski, P. J., Owens, C. P., and Goulding, C. W. (2019). Iron Acquisition in Mycobacterium tuberculosis. Chem. Rev. 119, 1193–1220. doi: 10.1021/acs.chemrev.8b00285
Chen, X., Liao, B., Cheng, L., Peng, X., Xu, X., Li, Y., et al. (2020). The microbial coinfection in COVID-19. Appl. Microbiol. Biotechnol. 104, 7777–7785. doi: 10.1007/s00253-020-10814-6
Corson, S., Greenhalgh, D., Taylor, A., Palmateer, N., Goldberg, D., and Hutchinson, S. (2013). Modelling the prevalence of HCV amongst people who inject drugs: an investigation into the risks associated with injecting paraphernalia sharing. Drug Alcohol Depend. 133, 172–179. doi: 10.1016/j.drugalcdep.2013.05.014
de Castro, I. F., Micheloud, D., Berenguer, J., Guzmán-Fulgencio, M., Catalán, P., Miralles, P., et al. (2010). Hepatitis C virus infection is associated with endothelial dysfunction in HIV/hepatitis C virus coinfected patients. AIDS 24, 2059–2067. doi: 10.1097/QAD.0b013e32833ce54d
De Maria, N., Colantoni, A., Fagiuoli, S., Liu, G. J., Rogers, B. K., Farinati, F., et al. (1996). Association between reactive oxygen species and disease activity in chronic hepatitis C. Free Radic. Biol. Med. 21, 291–295. doi: 10.1016/0891-5849(96)00044-5
de Mendoza, C., Sánchez-Conde, M., Timmermans, E., Buitelaar, M., de Baar, M. P., Gonzalez-Lahoz, J., et al. (2005). Mitochondrial DNA depletion in HIV-infected patients is more pronounced with chronic hepatitis C and enhanced following treatment with pegylated interferon plus ribavirin. Antivir. Ther. 10, 557–561.
Diedrich, C. R., and Flynn, J. L. (2011). HIV-1/Mycobacterium tuberculosis coinfection immunology: how does HIV-1 exacerbate tuberculosis? Infect. Immun. 79, 1407–1417. doi: 10.1128/IAI.01126-10
Esper, F. P., Spahlinger, T., and Zhou, L. (2011). Rate and influence of respiratory virus co-infection on pandemic (H1N1) influenza disease. J. Infect. 63, 260–266. doi: 10.1016/j.jinf.2011.04.004
Esteva, L., and Vargas, C. (2003). Coexistence of different serotypes of dengue virus. J. Math. Biol. 46, 31–47. doi: 10.1007/s00285-002-0168-4
Fact Sheet on Tuberculosis (TB), WHO https://www.who.int/news-room/fact-sheets/detail/tuberculosis.
Farinati, F., Cardin, R., De Maria, N., Della Libera, G., Marafin, C., Lecis, E., et al. (1995). Iron storage, lipid peroxidation and glutathione turnover in chronic anti-HCV positive hepatitis. J. Hepatol. 22, 449–456. doi: 10.1016/0168-8278(95)80108-1
Flynn, J. K., Dore, G. J., Matthews, G., Hellard, M., Yeung, B., Rawlinson, W. D., et al. (2012). Impaired hepatitis C virus (HCV)-specific interferon-γ responses in individuals with HIV who acquire HCV infection: correlation with CD4(+) T-cell counts. J. Infect. Dis. 206, 1568–1576. doi: 10.1093/infdis/jis544
Friedman, S. L. (2008a). Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 88, 125–172. doi: 10.1152/physrev.00013.2007
Friedman, S. L. (2008b). Mechanisms of hepatic fibrogenesis. Gastroenterology 134, 1655–1669. doi: 10.1053/j.gastro.2008.03.003
Fujita, N., Sugimoto, R., Ma, N., Tanaka, H., Iwasa, M., Kobayashi, Y., et al. (2008). Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat. 15, 498–507. doi: 10.1111/j.1365-2893.2008.00972.x
Ganbat, D., Seehase, S., Richter, E., Vollmer, E., Reiling, N., Fellenberg, K., et al. (2016). Mycobacteria infect different cell types in the human lung and cause species dependent cellular changes in infected cells. BMC Pulm. Med. 16:19. doi: 10.1186/s12890-016-0185-5
García-Mediavilla, M. V., Sánchez-Campos, S., González-Pérez, P., Gómez-Gonzalo, M., Majano, P. L., López-Cabrera, M., et al. (2005). Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J. Hepatol. 43, 606–613. doi: 10.1016/j.jhep.2005.04.019
Goletti, D., Weissman, D., Jackson, R. W., Collins, F., Kinter, A., and Fauci, A. S. (1998). The in vitro induction of human immunodeficiency virus (HIV) replication in purified protein derivative-positive HIV-infected persons by recall antigen response to Mycobacterium tuberculosis is the result of a balance of the effects of endogenous interleukin-2 and proinflammatory and antiinflammatory cytokines. J. Infect. Dis. 177, 1332–1338. doi: 10.1086/515276
Gonzalez, A. J., Ijezie, E. C., Balemba, O. B., and Miura, T. A. (2018). Attenuation of influenza A virus disease severity by viral coinfection in a mouse model. J. Virol. 92:e00881-18. doi: 10.1128/JVI.00881-18
Gravier-Hernández, R., and Gil-del Valle, L. (2018). “Oxidative stress and tuberculosis–human immunodeficiency virus coinfection,” in HIV/AIDS, eds V. R. Preedy and R. R. Watson (Amsterdam: Elsevier), 17–27. doi: 10.1016/B978-0-12-809853-0.00002-X
Greer, R. M., McErlean, P., Arden, K. E., Faux, C. E., Nitsche, A., Lambert, S. B., et al. (2009). Do rhinoviruses reduce the probability of viral co-detection during acute respiratory tract infections? J. Clin. Virol. 45, 10–15. doi: 10.1016/j.jcv.2009.03.008
Grønbaek, K., Sonne, J., Ring-Larsen, H., Poulsen, H. E., Friis, H., and Bygum Krarup, H. (2005). Viral load is a negative predictor of antioxidant levels in hepatitis C patients. Scand. J. Infect. Dis. 37, 686–689. doi: 10.1080/00365540410021027
Grootjans, J., Kaser, A., Kaufman, R. J., and Blumberg, R. S. (2016). The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 16, 469–484. doi: 10.1038/nri.2016.62
Hoffman, L. R., Déziel, E., D’Argenio, D. A., Lépine, F., Emerson, J., McNamara, S., et al. (2006). Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 103, 19890–19895. doi: 10.1073/pnas.0606756104
Hojyo, S., Uchida, M., Tanaka, K., Hasebe, R., Tanaka, Y., Murakami, M., et al. (2020). How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 40:37. doi: 10.1186/s41232-020-00146-3
Hoshino, Y., Hoshino, S., Gold, J. A., Raju, B., Prabhakar, S., Pine, R., et al. (2007). Mechanisms of polymorphonuclear neutrophil-mediated induction of HIV-1 replication in macrophages during pulmonary tuberculosis. J. Infect. Dis. 195, 1303–1310. doi: 10.1086/513438
Hoshino, Y., Nakata, K., Hoshino, S., Honda, Y., Tse, D. B., Shioda, T., et al. (2002). Maximal HIV-1 replication in alveolar macrophages during tuberculosis requires both lymphocyte contact and cytokines. J. Exp. Med. 195, 495–505. doi: 10.1084/jem.20011614
Iser, D. M., Warner, N., Revill, P. A., Solomon, A., Wightman, F., Saleh, S., et al. (2010). Coinfection of hepatic cell lines with human immunodeficiency virus and hepatitis B virus leads to an increase in intracellular hepatitis B surface antigen. J. Virol. 84, 5860–5867. doi: 10.1128/JVI.02594-09
Ivanov, A. V., Bartosch, B., and Isaguliants, M. G. (2017). Oxidative stress in infection and consequent disease. Oxid. Med. Cell. Longev. 2017:3496043. doi: 10.1155/2017/3496043
Izzedine, H., Sene, D., Cacoub, P., Jansen, H., Camous, L., Brocheriou, I., et al. (2009). Kidney diseases in HIV/HCV-co-infected patients. AIDS 23, 1219–1226. doi: 10.1097/QAD.0b013e32832ac36a
Jack, C. I., Jackson, M. J., and Hind, C. R. (1994). Circulating markers of free radical activity in patients with pulmonary tuberculosis. Tuber. Lung Dis. 75, 132–137. doi: 10.1016/0962-8479(94)90042-6
Jain, S., Williams, D. J., Arnold, S. R., Ampofo, K., Bramley, A. M., Reed, C., et al. (2015). Community-acquired pneumonia requiring hospitalization among U.S. children. N. Engl. J. Med. 372, 835–845. doi: 10.1056/NEJMoa1405870
Kanwugu, O. N., and Adadi, P. (2020). HIV/SARS-CoV-2 coinfection: a global perspective. J. Med. Virol. 93, 726–732. doi: 10.1002/jmv.26321
Khayat, M., Fan, H., and Vali, Y. (2021). COVID-19 promoting the development of active tuberculosis in a patient with latent tuberculosis infection: a case report. Respir. Med. Case Rep. 32:101344. doi: 10.1016/j.rmcr.2021.101344
Kim, A. Y., and Chung, R. T. (2009). Coinfection with HIV-1 and HCV–a one-two punch. Gastroenterology 137, 795–814. doi: 10.1053/j.gastro.2009.06.040
Kim, D., Quinn, J., Pinsky, B., Shah, N. H., and Brown, I. (2020). Rates of Co-infection Between SARS-CoV-2 and other respiratory pathogens. JAMA 323, 2085–2086. doi: 10.1001/jama.2020.6266
Kim, J., Koo, B.-K., and Knoblich, J. A. (2020). Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584. doi: 10.1038/s41580-020-0259-3
Kitaura, H., Ohara, N., Kobayashi, K., and Yamada, T. (2001). TNF-alpha-mediated multiplication of human immunodeficiency virus in chronically infected monocytoid cells by mycobacterial infection. APMIS 109, 533–540. doi: 10.1111/j.1600-0463.2001.apm090707.x
Kumar, N., Barua, S., Riyesh, T., Chaubey, K. K., Rawat, K. D., Khandelwal, N., et al. (2016). Complexities in isolation and purification of multiple viruses from mixed viral infections: viral interference, persistence and exclusion. PLoS One 11:e0156110. doi: 10.1371/journal.pone.0156110
Kumar, N., Sharma, S., Barua, S., Tripathi, B. N., and Rouse, B. T. (2018). Virological and immunological outcomes of coinfections. Clin. Microbiol. Rev. 31:e00111-17. doi: 10.1128/CMR.00111-17
Kunkel, S. L., Lukacs, N. W., and Strieter, R. M. (1994). The role of interleukin-8 in the infectious process. Ann. N. Y. Acad. Sci. 730, 134–143. doi: 10.1111/j.1749-6632.1994.tb44245.x
Kuntzen, T., Tural, C., Li, B., Feldmann, G., Kupfer, B., Nischalke, H. D., et al. (2008). Intrahepatic mRNA expression in hepatitis C virus and HIV/hepatitis C virus co-infection: infiltrating cells, cytokines, and influence of HAART. AIDS 22, 203–210. doi: 10.1097/QAD.0b013e3282f3553b
Kurthkoti, K., Tare, P., Paitchowdhury, R., Gowthami, V. N., Garcia, M. J., Colangeli, R., et al. (2015). The mycobacterial iron-dependent regulator IdeR induces ferritin (bfrB) by alleviating Lsr2 repression. Mol. Microbiol. 98, 864–877. doi: 10.1111/mmi.13166
Lin, P. L., Rutledge, T., Green, A. M., Bigbee, M., Fuhrman, C., Klein, E., et al. (2012). CD4 T cell depletion exacerbates acute Mycobacterium tuberculosis while reactivation of latent infection is dependent on severity of tissue depletion in cynomolgus macaques. AIDS Res. Hum. Retroviruses 28, 1693–1702. doi: 10.1089/AID.2012.0028
Liou, G.-Y., and Storz, P. (2010). Reactive oxygen species in cancer. Free Radic. Res. 44, 479–496. doi: 10.3109/10715761003667554
Lounis, N., Truffot-Pernot, C., Grosset, J., Gordeuk, V. R., and Boelaert, J. R. (2001). Iron and Mycobacterium tuberculosis infection. J. Clin. Virol. 20, 123–126. doi: 10.1016/s1386-6532(00)00136-0
Ma, L., Wang, W., Le Grange, J. M., Wang, X., Du, S., Li, C., et al. (2020). Coinfection of SARS-CoV-2 and other respiratory pathogens. Infect. Drug Resist. 13, 3045–3053. doi: 10.2147/IDR.S267238
Maier, K. P. (1997). Iron, HCV and the liver. World J. Gastroenterol. 3, 61–63. doi: 10.3748/wjg.v3.i2.61
Marimuthu, J., Kumar, B. S., and Gandhi, P. A. (2021). HIV and SARS CoV-2 coinfection: a retrospective, record-based, case series from South India. J. Med. Virol. 93, 163–165. doi: 10.1002/jmv.26271
Mastroianni, C. M., Lichtner, M., Mascia, C., Zuccalà, P., and Vullo, V. (2014). Molecular mechanisms of liver fibrosis in HIV/HCV coinfection. Int. J. Mol. Sci. 15, 9184–9208. doi: 10.3390/ijms15069184
McArdle, A. J., Turkova, A., and Cunnington, A. J. (2018). When do co-infections matter? Curr. Opin. Infect. Dis. 31, 209–215. doi: 10.1097/QCO.0000000000000447
McCullers, J. A. (2004). Effect of antiviral treatment on the outcome of secondary bacterial pneumonia after influenza. J. Infect. Dis. 190, 519–526. doi: 10.1086/421525
McCullers, J. A., and Bartmess, K. C. (2003). Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J. Infect. Dis. 187, 1000–1009. doi: 10.1086/368163
Medvedev, R., Ploen, D., and Hildt, E. (2016). HCV and Oxidative Stress: implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxid. Med. Cell. Longev. 2016:9012580. doi: 10.1155/2016/9012580
Modjarrad, K., and Vermund, S. H. (2010). Effect of treating co-infections on HIV-1 viral load: a systematic review. Lancet Infect. Dis. 10, 455–463. doi: 10.1016/S1473-3099(10)70093-1
Müller, F., Aukrust, P., Svardal, A. M., Berge, R. K., Ueland, P. M., and Frøland, S. S. (2017). “The thiols glutathione, cysteine, and homocysteine in human immunodeficiency virus (HIV) infection,” in NUTRIENTS and FOODS in AIDS, ed. R. R. Watson (Boca Raton, FL: CRC Press), 35–69. doi: 10.1201/9780203745250-3
Nakamoto, N., Kaplan, D. E., Coleclough, J., Li, Y., Valiga, M. E., Kaminski, M., et al. (2008). Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 134, 1927–1937.e2. doi: 10.1053/j.gastro.2008.02.033
Nelson, A. M., Manabe, Y. C., and Lucas, S. B. (2017). Immune Reconstitution Inflammatory Syndrome (IRIS): What pathologists should know. Semin. Diagn. Pathol. 34, 340–351. doi: 10.1053/j.semdp.2017.04.010
Nolan, V. G., Arnold, S. R., Bramley, A. M., Ampofo, K., Williams, D. J., Grijalva, C. G., et al. (2018). Etiology and impact of coinfections in children hospitalized with community-acquired pneumonia. J. Infect. Dis. 218, 179–188. doi: 10.1093/infdis/jix641
Nusbaum, R. J., Calderon, V. E., Huante, M. B., Sutjita, P., Vijayakumar, S., Lancaster, K. L., et al. (2016). Pulmonary tuberculosis in humanized mice infected with HIV-1. Sci. Rep. 6:21522. doi: 10.1038/srep21522
Operskalski, E. A., and Kovacs, A. (2011). HIV/HCV co-infection: pathogenesis, clinical complications, treatment, and new therapeutic technologies. Curr. HIV/AIDS Rep. 8, 12–22. doi: 10.1007/s11904-010-0071-3
Pace, G. W., and Leaf, C. D. (1995). The role of oxidative stress in HIV disease. Free Radic. Biol. Med. 19, 523–528. doi: 10.1016/0891-5849(95)00047-2
Pandey, R., Mehta, P., Sahni, S., Siddiqui, S., Mishra, N., Sharma, P., et al. (2021). Respiratory Co-infections: modulators of SARS-CoV-2 Patients’ clinical sub-phenotype. Front. Microbiol. 12:653399. doi: 10.3389/fmicb.2021.653399
Paradis, V., Kollinger, M., Fabre, M., Holstege, A., Poynard, T., and Bedossa, P. (1997). In situ detection of lipid peroxidation by-products in chronic liver diseases. Hepatology 26, 135–142. doi: 10.1002/hep.510260118
Parola, M., and Robino, G. (2001). Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 35, 297–306. doi: 10.1016/s0168-8278(01)00142-8
Peters, L., Mocroft, A., Soriano, V., Rockstroh, J., Rauch, A., Karlsson, A., et al. (2013). Hyaluronic acid levels predict risk of hepatic encephalopathy and liver-related death in HIV/viral hepatitis coinfected patients. PLoS One 8:e64283. doi: 10.1371/journal.pone.0064283
Petrone, L., Petruccioli, E., Vanini, V., Cuzzi, G., Gualano, G., Vittozzi, P., et al. (2021). Coinfection of tuberculosis and COVID-19 limits the ability to in vitro respond to SARS-CoV-2. Int. J. Infect. Dis. doi: 10.1016/j.ijid.2021.02.090 [Epub ahead of print].
Pinky, L., and Dobrovolny, H. M. (2017). The impact of cell regeneration on the dynamics of viral coinfection. Chaos 27:063109. doi: 10.1063/1.4985276
Price, L., and Kowdley, K. V. (2009). The role of iron in the pathophysiology and treatment of chronic hepatitis C. Can. J. Gastroenterol. 23, 822–828. doi: 10.1155/2009/290383
Price, T. O., Uras, F., Banks, W. A., and Ercal, N. (2006). A novel antioxidant N-acetylcysteine amide prevents gp120- and Tat-induced oxidative stress in brain endothelial cells. Exp. Neurol. 201, 193–202. doi: 10.1016/j.expneurol.2006.03.030
Qian, Z., Travanty, E. A., Oko, L., Edeen, K., Berglund, A., Wang, J., et al. (2013). Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am. J. Respir. Cell Mol. Biol. 48, 742–748. doi: 10.1165/rcmb.2012-0339OC
Rajopadhye, S. H., Mukherjee, S. R., Chowdhary, A. S., and Dandekar, S. P. (2017). Oxidative stress markers in tuberculosis and HIV/TB Co-infection. J. Clin. Diagn. Res. 11, BC24–BC28. doi: 10.7860/JCDR/2017/28478.10473
Ranjit, S., Sinha, N., Kodidela, S., and Kumar, S. (2018). Benzo(a)pyrene in Cigarette Smoke Enhances HIV-1 Replication through NF-κB Activation via CYP-Mediated Oxidative Stress Pathway. Sci. Rep. 8:10394. doi: 10.1038/s41598-018-28500-z
Rath, P., Huang, C., Wang, T., Wang, T., Li, H., Prados-Rosales, R., et al. (2013). Genetic regulation of vesiculogenesis and immunomodulation in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 110, E4790–E4797. doi: 10.1073/pnas.1320118110
Redford, P. S., Mayer-Barber, K. D., McNab, F. W., Stavropoulos, E., Wack, A., Sher, A., et al. (2014). Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J. Infect. Dis. 209, 270–274. doi: 10.1093/infdis/jit424
Roe, B., Coughlan, S., Hassan, J., Grogan, A., Farrell, G., Norris, S., et al. (2007). Elevated serum levels of interferon- gamma -inducible protein-10 in patients coinfected with hepatitis C virus and HIV. J. Infect. Dis. 196, 1053–1057. doi: 10.1086/520935
Roe, B., and Hall, W. W. (2008). Cellular and molecular interactions in coinfection with hepatitis C virus and human immunodeficiency virus. Expert Rev. Mol. Med. 10:e30. doi: 10.1017/S1462399408000847
Rossi, G., Manfrin, A., and Lutolf, M. P. (2018). Progress and potential in organoid research. Nat. Rev. Genet. 19, 671–687. doi: 10.1038/s41576-018-0051-9
Rotman, Y., and Liang, T. J. (2009). Coinfection with hepatitis C virus and human immunodeficiency virus: virological, immunological, and clinical outcomes. J. Virol. 83, 7366–7374. doi: 10.1128/JVI.00191-09
Sacktor, N., Haughey, N., Cutler, R., Tamara, A., Turchan, J., Pardo, C., et al. (2004). Novel markers of oxidative stress in actively progressive HIV dementia. J. Neuroimmunol. 157, 176–184. doi: 10.1016/j.jneuroim.2004.08.037
Salas-Benito, J. S., and De Nova-Ocampo, M. (2015). Viral interference and persistence in mosquito-borne Flaviviruses. J. Immunol. Res. 2015:873404. doi: 10.1155/2015/873404
Schreck, R., Rieber, P., and Baeuerle, P. A. (1991). Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 10, 2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x
Shahangian, A., Chow, E. K., Tian, X., Kang, J. R., Ghaffari, A., Liu, S. Y., et al. (2009). Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119, 1910–1920. doi: 10.1172/JCI35412
Sharov, K. S. (2021). HIV/SARS-CoV-2 co-infection: T cell profile, cytokine dynamics and role of exhausted lymphocytes. Int. J. Infect. Dis. 102, 163–169. doi: 10.1016/j.ijid.2020.10.049
Shinjoh, M., Omoe, K., Saito, N., Matsuo, N., and Nerome, K. (2000). In vitro growth profiles of respiratory syncytial virus in the presence of influenza virus. Acta Virol. 44, 91–97.
Shmagel, K., and Saidakova, E. (2018). “Immune Disorders in HIV-Infected Patients Coinfected with Hepatitis C Virus,” in Advances in HIV and AIDS Control, ed. S. Okware (Rijeka: IntechOpen). doi: 10.5772/intechopen.76810
Smit, C., van den Berg, C., Geskus, R., Berkhout, B., Coutinho, R., and Prins, M. (2008). Risk of hepatitis-related mortality increased among hepatitis C virus/HIV-coinfected drug users compared with drug users infected only with hepatitis C virus: a 20-year prospective study. J. Acquir. Immune Defic. Syndr. 47, 221–225. doi: 10.1097/QAI.0b013e31815d2f59
Souriant, S., Balboa, L., Dupont, M., Pingris, K., Kviatcovsky, D., Cougoule, C., et al. (2019). Tuberculosis exacerbates HIV-1 Infection through IL-10/STAT3-dependent tunneling nanotube formation in macrophages. Cell Rep. 26, 3586–3599.e7. doi: 10.1016/j.celrep.2019.02.091
Stehbens, W. E. (2004). Oxidative stress in viral hepatitis and AIDS. Exp. Mol. Pathol. 77, 121–132. doi: 10.1016/j.yexmp.2004.04.007
Stek, C., Allwood, B., Du Bruyn, E., Buyze, J., Schutz, C., Thienemann, F., et al. (2020). The effect of HIV-associated tuberculosis, tuberculosis-IRIS and prednisone on lung function. Eur. Respir. J. 55:1901692. doi: 10.1183/13993003.01692-2019
Stochino, C., Villa, S., Zucchi, P., Parravicini, P., Gori, A., and Raviglione, M. C. (2020). Clinical characteristics of COVID-19 and active tuberculosis co-infection in an Italian reference hospital. Eur. Respir. J. 56:2001708. doi: 10.1183/13993003.01708-2020
Suwanwongse, K., and Shabarek, N. (2020). Clinical features and outcome of HIV/SARS-CoV-2 coinfected patients in The Bronx, New York city. J. Med. Virol. 92, 2387–2389. doi: 10.1002/jmv.26077
Suwanwongse, K., and Shabarek, N. (2021). Variation in mortality of HIV/SARS-CoV-2 coinfected patients in the Bronx, New York City. J. Med. Virol. 93, 603–605. doi: 10.1002/jmv.26370
Tapela, K., Ochieng’ Olwal, C., and Quaye, O. (2020). Parallels in the pathogenesis of SARS-CoV-2 and M. tuberculosis: a synergistic or antagonistic alliance? Future Microbiol. 15, 1691–1695. doi: 10.2217/fmb-2020-0179
Tesoriero, J. M., Swain, C.-A. E., Pierce, J. L., Zamboni, L., Wu, M., Holtgrave, D. R., et al. (2021). COVID-19 Outcomes Among Persons Living With or Without Diagnosed HIV Infection in New York State. JAMA Netw. Open 4:e2037069. doi: 10.1001/jamanetworkopen.2020.37069
Tuberculosis data (2020). Available online at: https://www.who.int/teams/global-tuberculosis-programme/data (accessed February 3, 2021).
Tuyama, A. C., Hong, F., Saiman, Y., Wang, C., Ozkok, D., Mosoian, A., et al. (2010). Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatology 52, 612–622. doi: 10.1002/hep.23679
Tyagi, P., Pal, V. K., Agrawal, R., Singh, S., Srinivasan, S., and Singh, A. (2020). Mycobacterium tuberculosis Reactivates HIV-1 via Exosome-Mediated Resetting of Cellular Redox Potential and Bioenergetics. mBio 11:e03293-19. doi: 10.1128/mBio.03293-19
Unsal, E., Aksaray, S., Köksal, D., and Sipit, T. (2005). Potential role of interleukin 6 in reactive thrombocytosis and acute phase response in pulmonary tuberculosis. Postgrad. Med. J. 81, 604–607. doi: 10.1136/pgmj.2004.030544
Vali, B., Yue, F. Y., Jones, R. B., Sheth, P. M., Kaul, R., Betts, M. R., et al. (2008). HIV-specific T-cells accumulate in the liver in HCV/HIV co-infection. PLoS One 3:e0003454. doi: 10.1371/journal.pone.0003454
Vickerman, P., Martin, N. K., and Hickman, M. (2012). Understanding the trends in HIV and hepatitis C prevalence amongst injecting drug users in different settings–implications for intervention impact. Drug Alcohol Depend. 123, 122–131. doi: 10.1016/j.drugalcdep.2011.10.032
Wang, M., Luo, L., Bu, H., and Xia, H. (2020). One case of coronavirus disease 2019 (COVID-19) in a patient co-infected by HIV with a low CD4+ T-cell count. Int. J. Infect. Dis. 96, 148–150. doi: 10.1016/j.ijid.2020.04.060
Whalen, C., Horsburgh, C. R., Hom, D., Lahart, C., Simberkoff, M., and Ellner, J. (1995). Accelerated course of human immunodeficiency virus infection after tuberculosis. Am. J. Respir. Crit. Care Med. 151, 129–135. doi: 10.1164/ajrccm.151.1.7812542
Wyatt, C. M., Malvestutto, C., Coca, S. G., Klotman, P. E., and Parikh, C. R. (2008). The impact of hepatitis C virus coinfection on HIV-related kidney disease: a systematic review and meta-analysis. AIDS 22, 1799–1807. doi: 10.1097/QAD.0b013e32830e0152
Zhang, H., and Wu, T. (2020). CD4+T, CD8+T counts and severe COVID-19: a meta-analysis. J. Infect. 81, e82–e84. doi: 10.1016/j.jinf.2020.06.036
Zhao, Q., Qin, C.-Y., Zhao, Z.-H., Fan, Y.-C., and Wang, K. (2013). Epigenetic modifications in hepatic stellate cells contribute to liver fibrosis. Tohoku J. Exp. Med. 229, 35–43. doi: 10.1620/tjem.229.35
Zhu, X., Ge, Y., Wu, T., Zhao, K., Chen, Y., Wu, B., et al. (2020). Co-infection with respiratory pathogens among COVID-2019 cases. Virus Res. 285:198005. doi: 10.1016/j.virusres.2020.198005
Ziegler, C. G. K., Allon, S. J., Nyquist, S. K., Mbano, I. M., Miao, V. N., Tzouanas, C. N., et al. (2020). SARS-CoV-2 Receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035.e19. doi: 10.1016/j.cell.2020.04.035
Keywords: co-infection, HIV, HCV, MTB, oxidative stress, immune response, disease severity
Citation: Devi P, Khan A, Chattopadhyay P, Mehta P, Sahni S, Sharma S and Pandey R (2021) Co-infections as Modulators of Disease Outcome: Minor Players or Major Players? Front. Microbiol. 12:664386. doi: 10.3389/fmicb.2021.664386
Received: 05 February 2021; Accepted: 16 June 2021;
Published: 06 July 2021.
Edited by:
Karl Kuchler, Medical University of Vienna, AustriaReviewed by:
Radha Gopal, University of Pittsburgh, United StatesCopyright © 2021 Devi, Khan, Chattopadhyay, Mehta, Sahni, Sharma and Pandey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajesh Pandey, cmFqZXNocEBpZ2liLmlu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.