 Qiang Li
Qiang Li Lijiao Li1
Lijiao Li1 Chuan Xiong
Chuan Xiong Xu Wang
Xu Wang Wenli Huang
Wenli Huang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 May 2021
Sec. Evolutionary and Genomic Microbiology
Volume 12 - 2021 | https://doi.org/10.3389/fmicb.2021.646567
This article is part of the Research Topic Mitochondrial Genomes and Mitochondrion Related Gene Insights to Fungal Evolution View all 11 articles
In this study, the mitogenome of Hannaella oryzae was sequenced by next-generation sequencing (NGS) and successfully assembled. The H. oryzae mitogenome comprised circular DNA molecules with a total size of 26,444 bp. We found that the mitogenome of H. oryzae partially deleted the tRNA gene transferring cysteine. Comparative mitogenomic analyses showed that intronic regions were the main factors contributing to the size variations of mitogenomes in Tremellales. Introns of the cox1 gene in Tremellales species were found to have undergone intron loss/gain events, and introns of the H. oryzae cox1 gene may have different origins. Gene arrangement analysis revealed that H. oryzae contained a unique gene order different from other Tremellales species. Phylogenetic analysis based on a combined mitochondrial gene set resulted in identical and well-supported topologies, wherein H. oryzae was closely related to Tremella fuciformis. This study represents the first report of mitogenome for the Hannaella genus, which will allow further study of the population genetics, taxonomy, and evolutionary biology of this important phylloplane yeast and other related species.
Hannaella is a basidiomycetous yeast genus belonging to the order Tremellales, phylum Basidiomycota. It was proposed to accommodate species closely related to the genera Derxomyces and Dioszegia, which belong to the Bullera sinensis clade of the Luteolus lineage of the Tremellales based on a series of molecular markers (Wang and Bai, 2008; Kaewwichian et al., 2015; Han et al., 2017). So far, about 12 species have been described in this genus, including Hannaella coprosmaensis, Hannaella dianchiensis, Hannaella kunmingensis, Hannaella luteola, Hannaella phyllophila, Hannaella phetchabunensis, Hannaella pagnoccae, Hannaella surugaensis, Hannaella sinensis, Hannaella siamensis, Hannaella zeae, and Hannaella oryzae (Landell et al., 2014; Kaewwichian et al., 2015; Han et al., 2017). These species are found widely distributed on the leaf surfaces of various plants, including rice, wheat, and fruit trees (Han et al., 2017). As an important phyllosphere-inhabiting yeast, H. oryzae was considered to play an important role in promoting plant growth and biocontrol. This genus is considered to be monophyletic in nature, and H. kunmingensis showed genotypic and phenotypic variability (Dayo-Owoyemi et al., 2013). Both the basidiomycetous and the ascomycetous yeasts have been found colonizing on phylloplane from various regions of the world (Glushakova et al., 2007; Landell et al., 2010; Molnarova et al., 2014). These basidiomycetous genera were found to be the most common phylloplane yeasts, including Sporobolomyces, Rhodotorula, Cryptococcus, Trichosporon, and Hannaella (de Azeredo et al., 1998; Kaewwichian et al., 2015). The mitochondrial genomic characteristics of representative species of the two phylloplane yeast genera (Cryptococcus and Trichosporon) have been published, which facilitated our understanding of phylloplane yeasts (Yang et al., 2012; Gomes et al., 2018; Yan et al., 2018). However, no mitochondrial genome has been published on the genus Hannaella or the family Bulleribasidiaceae. H. oryzae has been isolated from the phylloplane of various plants. The report of its mitochondrial genome will help us understand the genetic characteristics of this important phylloplane yeast.
Mitochondrial genomes are effective tools for analyzing the phylogenetic and genetic evolution of eukaryotes because they contain many available molecular markers (Burger et al., 2003; Bullerwell and Lang, 2005; Cheng et al., 2021). Besides, the arrangement of mitochondrial genes, their transfer RNA (tRNA) structure, their codon usage, and the dynamic changes of introns can also be used to infer the evolutionary status of eukaryotes (Li et al., 2018a; Wang et al., 2018, 2020b; Wu et al., 2021). With the rapid development of next-generation sequencing technologies in recent years, more and more mitochondrial genomes have been obtained (du Toit et al., 2017; Yang et al., 2018; Zhang et al., 2018; Wang et al., 2020c). However, the mitochondrial genomes of fungi are less studied than those of animals. So far, less than 130 basidiomycete mitochondrial genomes have been reported1, which limits our understanding of the “second genome” (mitochondrial genome) of fungi. Studies on the fungal mitogenomes have shown that fungal mitogenomes exhibited significant variations in gene order, introns, intergenic regions, genome size, and repetitive sequences (Zhang et al., 2016; Zhang S. et al., 2017; Zhang Y.J. et al., 2017; Fourie et al., 2018; Wang et al., 2020a; Li et al., 2021). Despite enormous variations in the fungal genome, the 15 protein-coding genes, including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3, have been detected in most basidiomycete mitochondrial genomes, which were considered to be core protein-coding genes (PCGs) in the basidiomycete mitochondrial genomes.
In the present study, the complete mitochondrial genome of H. oryzae was sequenced and assembled by next-generation sequencing technology. The content, organization, and structure of the mitochondrial genes were revealed. We compared the mitochondrial genome of H. oryzae with its closely related species to identify variations and similarities in the gene content, genome organization, and gene order. The dynamic changes of introns were also revealed in H. oryzae and other basidiomycete species. In addition, the phylogenetic relationships among various basidiomycete species based on combined mitochondrial gene sets were analyzed. The mitochondrial genome of H. oryzae will allow further study of the population genetics, taxonomy, and evolutionary biology of this important phylloplane yeast and other related species.
The H. oryzae strain s11 was isolated from the surface of corn leaves collected in Sichuan, China, using the improved ballistoconidia-fall method as described by Kaewwichian et al. (2015). The morphological, biochemical, and physiological characteristics of the collected yeast strains were examined according to standard methods described by Kurtzman et al. (2011). The total genomic DNA was extracted using the method described by Wang and Bai (2008). H. oryzae was further identified based on the internal transcribed spacer (ITS) sequence and the mitochondrial cob gene. Whole-genome sequencing libraries were constructed using NEBNext Ultra II DNA Library Prep Kits (NEB, Beijing, China) with the extracted genomic DNA following the manufacturer’s instructions. Whole-genome sequencing was carried out on an Illumina HiSeq 2500 Platform (Illumina, San Diego, CA, United States). To verify the accuracy and integrity of our assembled genome, we further sequenced the genomic DNA using the Pacbio RSII platform (Pacific Biosciences, CA, United States). A 40-kb SMRTbell DNA library was prepared to perform the Pacbio sequencing.
Illumina PCR adapter reads and low-quality reads from the paired-end reads were filtered using custom scripts. Clean reads were obtained after the quality control step. The mitogenome of H. oryzae was assembled by CANU v1.6 (Koren et al., 2017) using the Pacbio long reads. The assembled contigs were further polished using the paired-end Illumina reads with Pilon v1.22 (Walker et al., 2014). The obtained H. oryzae complete mitogenome was annotated according to the methods previously described (Li et al.,2020b,c). Briefly, the complete mitogenome of H. oryzae was firstly annotated based on the results of MITOS (Bernt et al., 2013) and MFannot (Valach et al., 2014). At this step, PCGs, ribosomal RNA (rRNA) genes, and tRNA genes were initially annotated. Open reading frames (ORFs) were modified or predicted with the NCBI Open Reading Frame Finder [ORFs less than 100 amino acids (aa) were excluded] (Coordinators, 2017) and annotated with BLASTp searches against the NCBI non-redundant protein sequence database (Bleasby and Wootton, 1990). tRNA genes were also predicted with tRNAscan-SE v1.3.1 (Lowe and Chan, 2016). The graphical map of the complete mitogenome was drawn with OGDraw v1.2 (Lohse et al., 2013).
The base composition of the H. oryzae mitogenome was analyzed using DNASTAR Lasergene v7.12 software. Strand asymmetry of the H. oryzae mitogenome was assessed using the following formulas: AT skew = [A − T]/[A + T] and GC skew = [G − C]/[G + C] (Wang et al., 2017). The non-synonymous substitution rate (Ka) and the synonymous substitution rate (Ks) for the core PCGs in the four Tremellales mitogenomes were calculated using DnaSP v6.10.01 (Rozas et al., 2017). We used MEGA v6.06 (Caspermeyer, 2016) to calculate the overall mean genetic distances between each pair of the 15 core PCGs (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3) using the Kimura-2-parameter (K2P) model. The genome synteny of the closely related mitogenomes was analyzed using Mauve v2.4.0 (Darling et al., 2004). The introns of the cox1 gene in 33 basidiomycete species were classified into different position classes (Pcls) and named according to previously described methods (Zhang and Zhang, 2019).
In order to investigate the phylogenetic status of H. oryzae among the Basidiomycota phylum, we constructed a phylogenetic tree of 33 species based on the combined mitochondrial gene set (15 core PCGs + two rRNA genes) (Li et al., 2018d). The individual mitochondrial gene was first aligned using MAFFT v7.037 (Katoh et al., 2019). Then, these alignments were concatenated in SequenceMatrix v1.7.8 (Vaidya et al., 2011). In order to detect potential phylogenetic conflicts between different genes, we carried out a preliminary partition homogeneity test. Phylogenetic trees were constructed using both Bayesian inference (BI) and maximum likelihood (ML) methods. Best-fit models of evolution and partitioning schemes for the gene set were determined according to PartitionFinder 2.1.1 (Lanfear et al., 2017). BI analysis was performed with MrBayes v3.2.6 (Ronquist et al., 2012). The RAxML v 8.0.0 software (Stamatakis, 2014) was used for ML analysis.
The newly sequenced H. oryzae mitogenome was deposited in the GenBank database under the following Accession No.: MH732752.
The complete mitochondrial genome of H. oryzae was assembled into a circular DNA molecule with a total size of 26,444 bp (Figure 1). The GC content of the H. oryzae mitogenome was 38.98% (Supplementary Table 1). Both the GC skew and the AT skew of the H. oryzae mitochondrial genome were positive. Fifteen conserved PCGs were detected in the H. oryzae mitogenome, including 14 core PCGs for energy metabolism and one rps3 gene (Supplementary Table 2). Eight introns were detected in the mitogenome of H. oryzae distributed in seven host genes, including cox1, atp6, rnl, atp9, nad3, nad1, and nad5. The cox1 gene contained two introns, while the other six genes each contained one intron. All these introns belonged to group I, which could catalyze their own splicing. One intronic ORF (orf195) was found in the H. oryzae mitochondrial genome, which encoded LAGLIDADG homing endonuclease. Of the 42 genes detected in the H. oryzae mitogenome, 19 were on the direct strand and the other 23 were on the reverse strand.
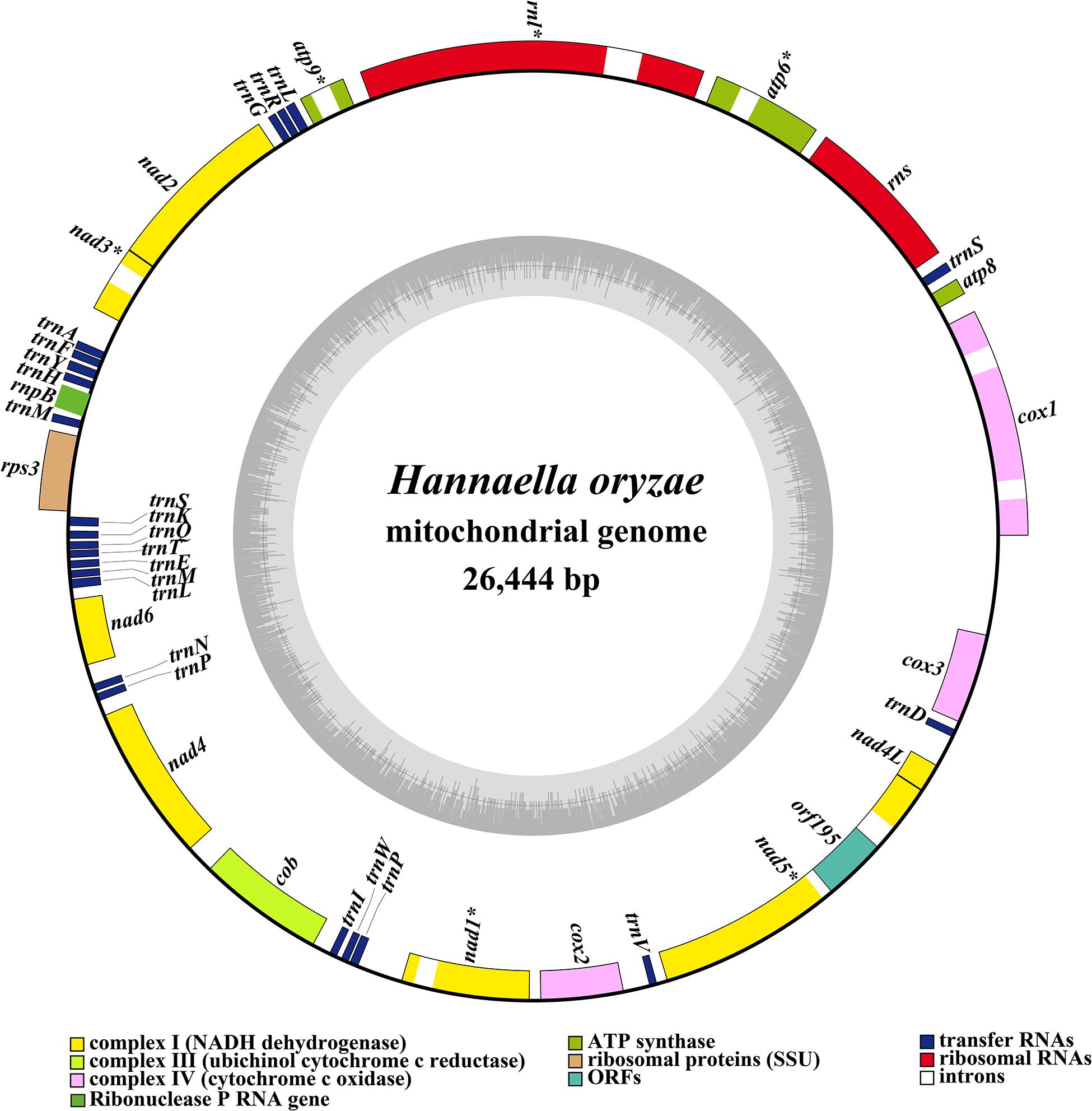
Figure 1. Circular map of the mitochondrial genome of Hannaella oryzae. Genes are represented by different colored blocks. Colored blocks outside each ring indicate that the genes are on the direct strand, while colored blocks within the ring indicate that the genes are located on the reverse strand. The inner grayscale bar graph shows the GC content of the mitochondrial sequences. The circle inside the GC content graph marks the 50% threshold. The graphical map of the complete mitogenome was drawn with OGDraw v1.2 (Lohse et al., 2013).
The mitogenome of H. oryzae contained two rRNA genes, namely the small subunit ribosomal RNA (rns) and the large subunit ribosomal RNA (rnl) (Supplementary Table 2). Twenty-three tRNA genes were detected in the H. oryzae mitogenome, encoding for 19 standard amino acids. The H. oryzae mitogenome lacked a tRNA gene that encoded for amino acid cysteine. Interestingly, we found that all four species from the order Tremellales lacked the tRNA gene to transfer cysteine. We detected about 40 bases of homology to Cys-tRNAs in the four mitogenomes. In addition, the H. oryzae mitogenome contained two tRNAs with the same anticodons coding for methionine and proline (Supplementary Figure 1). However, these two tRNAs varied in length and base composition, indicating frequent gene mutations in tRNA genes. The H. oryzae mitogenome also contained two tRNAs with different anticodons that coded for leucine and serine. The mitogenome of H. oryzae lost a tRNA gene with different anticodons that coded for arginine. The length of individual tRNAs ranged from 71 to 85 bp, which was mainly due to the variations in length of the extra arm. A ribonuclease P RNA gene (rnpB) was found in the H. oryzae mitogenome with a length of 211 bp.
The sizes of the five mitogenomes varied greatly, ranging from 24,874 to 35,058 bp. Correlation analysis showed that the intronic regions had the greatest impact on the size variations of Tremellales and Trichosporonales mitogenomes, with Pearson’s correlation coefficient of 0.907 (P < 0.05; Supplementary Figure 2). The results suggested that the intronic region was the main factor promoting the size variations of mitogenomes in Tremellales and Trichosporonales.
Introns were considered as the main factors leading to variations of the mitogenome size in Tremellales, Trichosporonales, and other basidiomycetes. So the dynamic changes of the introns in H. oryzae and other basidiomycetes were analyzed in depth in the present study. The cox1 gene was found to be the largest host gene of introns in basidiomycetes (Ferandon et al., 2010). Introns in the cox1 gene could be divided into different Pcls according to the insertion sites in the coding region. The introns from the same Pcls were considered homologous introns with high sequence similarities (Ferandon et al., 2010). Introns belonging to different Pcls were considered to be non-homologous and contained low sequence similarities. In the present study, a total of 246 introns of the cox1 gene were detected in the 33 basidiomycete species we tested, seven of which belonged to group II and the others belonged to group I (Figure 2). The 239 group I introns could be classified into 31 Pcls, 26 of which were reported in previous studies (Ferandon et al., 2010), while the other five were novel Pcls found in the 33 basidiomycetes. Among these Pcls, Pcl P386 was the most widely distributed, distributed in 21 of 33 basidiomycete species. Pcls P615 and P709 were also common Pcls in basidiomycetes, both of which existed in 16 of the 33 basidiomycete species. Pcls P196, P221, and P941 were found only in one of the 33 basidiomycete species, which were considered as rare introns in basidiomycetes. Eight introns were detected in the cox1 gene of five species from Tremellales and Trichosporonales. Cryptococcus neoformans and Tremella fuciformis lost introns in the cox1 gene, while the cox1 gene of Cryptococcus gattii contained the most introns among Tremellales and Trichosporonales, indicating that species in Tremellales and Trichosporonales had experienced intron loss/gain events during evolution. In addition, H. oryzae was found to contain two novel intron Pcls at sites 103 and 425 aa (Supplementary Table 3). No homologous introns were found in the four closely related species and other basidiomycete species, indicating that H. oryzae had undergone a unique intron origin.
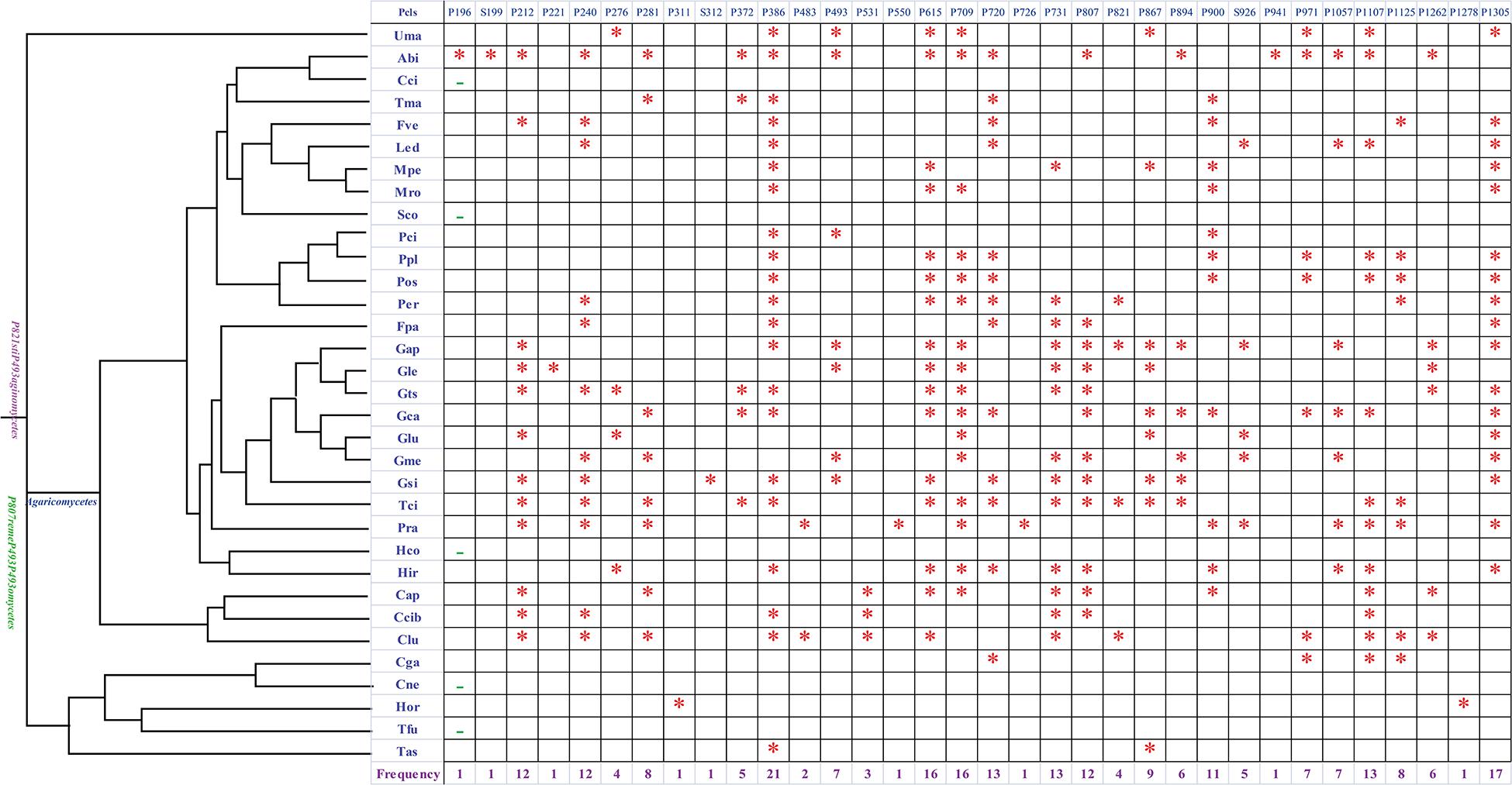
Figure 2. Position class (Pcl) information of cox1 introns in 33 basidiomycete species. The phylogenetic tree on the left was constructed using the Bayesian inference (BI) and maximum likelihood (ML) methods based on a combined mitochondrial gene set (Bayesian posterior probabilities and bootstrap support values are shown in Figure 5). Pcls are named according to previously described methods (Zhang and Zhang, 2019). Species IDs for the mitogenomes used in the intron analysis are provided in Supplementary Table 4. Asterisk indicates the presence of the Pcl in the species and an en dash indicates the absence of any Pcl in the species. The number at the bottom indicates the frequency of occurrence of the Pcl in the 33 basidiomycete species.
Of the 15 core PCGs detected in the four Tremellales mitogenomes, 12 PCGs were found to vary in length among the four species, except for atp8, atp9, and nad4L (Supplementary Figure 3). Nine genes of the H. oryzae mitogenome had unique lengths among the four Tremellales mitogenomes, including the cob, cox1, cox3, nad2, nad3, nad4, nad5, nad6, and rps3 genes. The GC contents of the core PCGs in the four Tremellales mitogenomes also varied, indicating frequent base variations in the core PCGs of the Tremellales mitogenomes. Among the 15 core PCGs detected, atp9 contained the highest GC content in all four Tremellales mitogenomes. The AT and GC skews of most of the core PCGs were negative in the four mitogenomes. The GC skews of the 15 core PCGs in the H. oryzae mitogenome were negative. The rps3 gene contained a positive AT skew in the H. oryzae mitogenome.
Of the 15 core PCGs detected, the rps3 gene had the highest mean K2P genetic distance among the four Tremellales mitogenomes, followed by nad3 (Supplementary Figure 4). The mean genetic distance of atp9 in the four mitogenomes was the smallest among the 15 core PCGs, indicating that this gene was highly conserved across the mitogenomes. nad4L contained the highest mean Ks among the 15 core PCGs detected, while atp9 had the lowest rate. The highest Ka was observed in the rps3 gene, while nad4L exhibited the lowest Ka value among the 15 PCGs detected. The Ka/Ks values for all 15 PCGs were less than 1, indicating that these genes were subject to purifying selection.
The mitogenome size of H. oryzae was the second smallest among all published Basidiomycota mitochondrial genomes (see text footnote 1), only higher than that of the basidiomycete yeast C. neoformans (Yan et al., 2018; Supplementary Table 1). The mitogenome size of H. oryzae was smaller than that of mushroom-forming species, such as Pleurotus spp. (Li et al., 2018a), Ganoderma spp. (Li et al., 2018d, 2019c), Coprinopsis cinereal (Stajich et al., 2010), and Agaricus bisporus (Ferandon et al., 2013), and its closely related species, T. fuciformis (MF422647); smaller than that of the ectomycorrhizal fungi Tricholoma matsutake (Yoon et al., 2016), Lactarius spp. (Li et al., 2019b), Russula spp.(Li et al., 2018c), and Cantharellus spp. (Li et al., 2018b); and also smaller than that of the basidiomycete yeast C. gattii (Yadav et al., 2018). The GC content of the H. oryzae mitogenome was relatively high (38.98%), which was only lower than that of Rhodotorula mucilaginosa (40.43%) (Gan et al., 2017), among all the published Basidiomycota mitochondrial genomes detected. There were seven, seven, two, and four introns detected in the mitogenomes of T. fuciformis, C. gattii, C. neoformans, and Trichosporon asahii, which contained five, six, one, and four introns, respectively. However, eight introns were detected in the mitogenome of H. oryzae, but only one of them contained intronic ORF, suggesting that the mitogenome introns of H. oryzae were undergoing constriction.
The gene arrangement in the mitogenomes of five closely related species in Tremellales and Trichosporonales was highly variable (Figure 3). Of the 18 genes detected in the five closely related species, including the 15 core PCGs, two rRNA genes, and one rnpB gene, the relative positions of the 16 genes varied among the five mitogenomes. The gene arrangements of C. gattii and C. neoformans were highly conserved between the two species. However, at the class level, the arrangement of the mitochondrial genes was highly variable. H. oryzae contained a unique gene arrangement in the order Tremellales, indicating that gene rearrangements have occurred during the evolution of H. oryzae, involving protein-encoded genes, rRNA genes, and the rnpB gene.
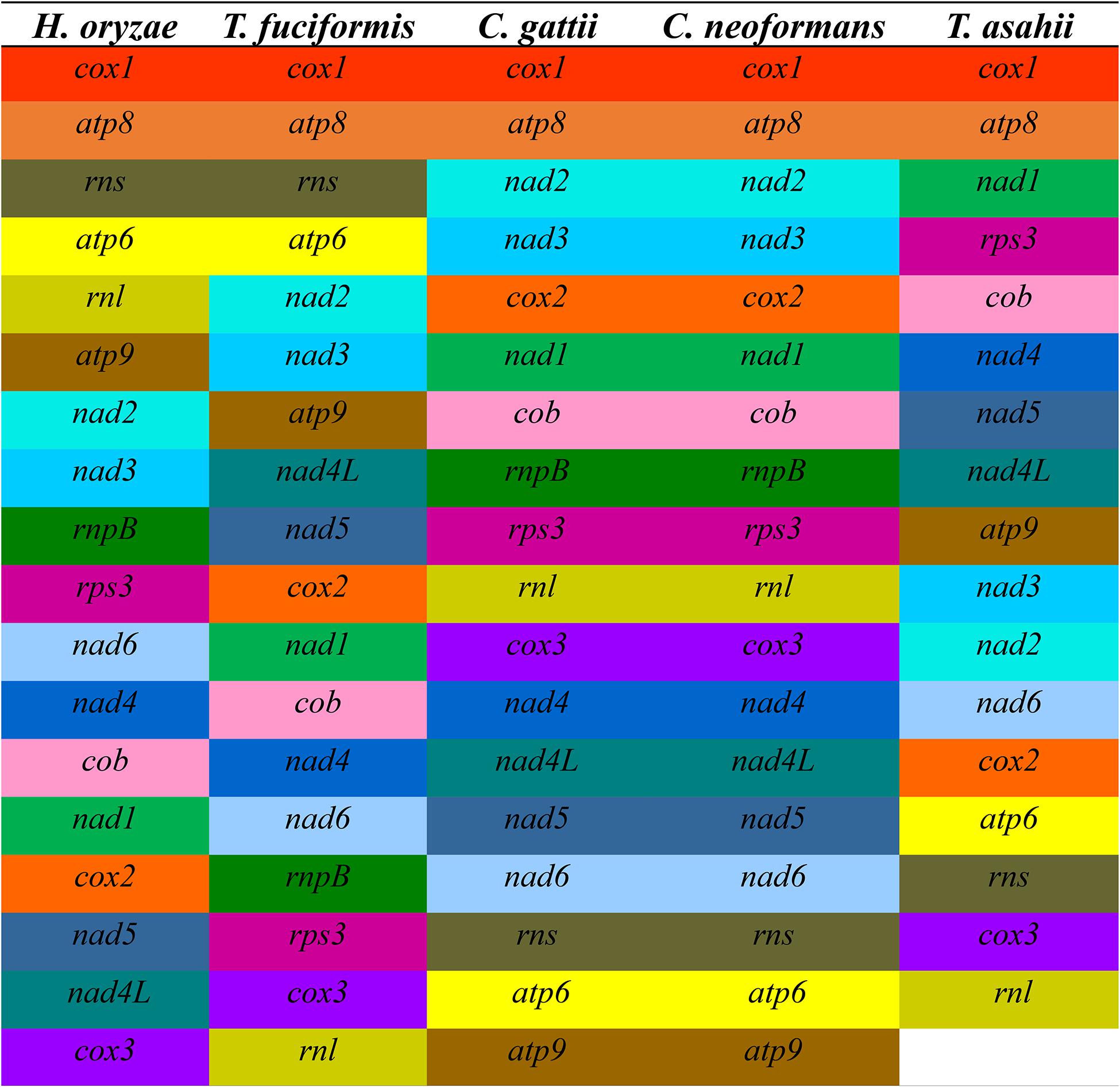
Figure 3. Comparison of the conserved gene order among the mitochondrial genomes of the five closely related species in Tremellales and Trichosporonales, including Cryptococcus gattii, Cryptococcus neoformans, Hannaella oryzae, Tremella fuciformis, and Trichosporon asahii. Fifteen core protein-coding genes (PCGs) and two rRNA genes were included in this analysis. Genes are represented by different colored blocks.
The genome collinearity analysis showed that the five mitogenomes of Tremellales and Trichosporonales could be divided into 18 homologous regions (Figure 4). The relative positions of these homologous regions were highly variable among the five Tremellales mitogenomes. Out of the 18 homologous regions, the relative positions of 17 homologous regions varied among the five mitogenomes. The relative positions of the homologous regions were identical between C. gattii and C. neoformans.
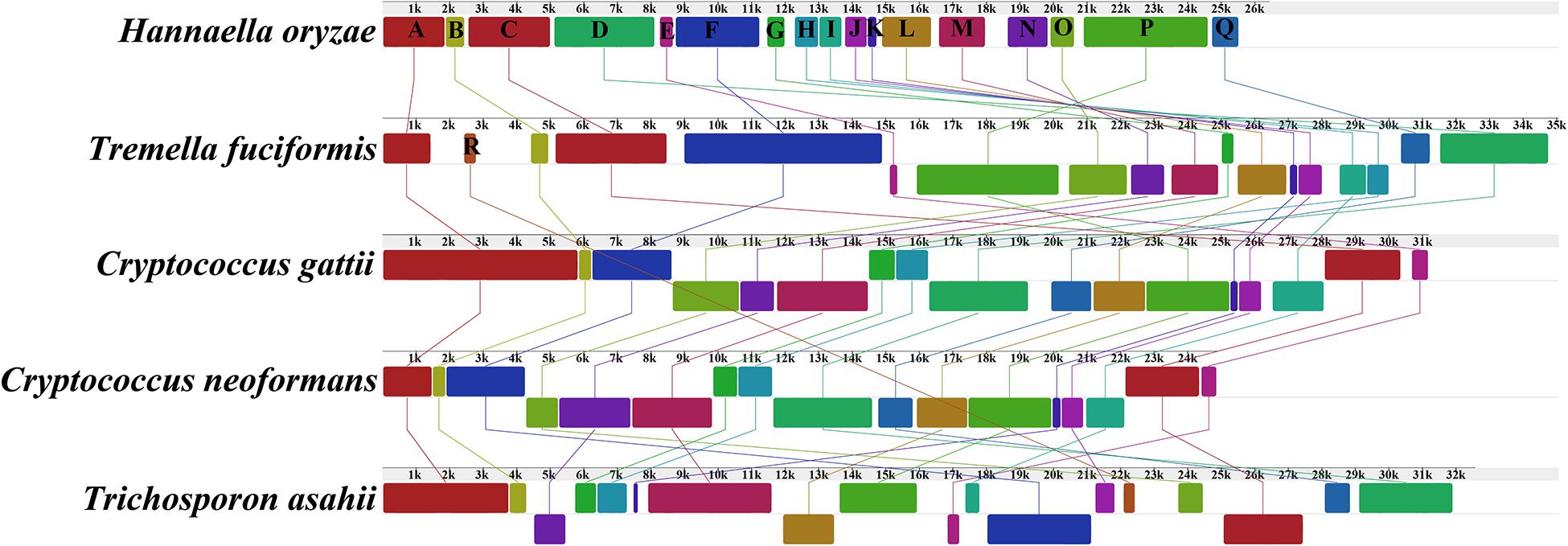
Figure 4. Collinearity analysis of the mitochondrial genomes of the five closely related species in Tremellales and Trichosporonales, including Cryptococcus gattii, Cryptococcus neoformans, Hannaella oryzae, Tremella fuciformis, and Trichosporon asahii. Eighteen homologous regions were detected among the five mitogenomes. The sizes and the relative positions of the homologous regions varied among the mitogenomes. The genome synteny of the closely related mitogenomes was analyzed using Mauve v2.4.0 (Darling et al., 2004).
We obtained identical and well-supported tree topologies using both BI and ML methods based on the combined mitochondrial gene set (15 core PCGs + two rRNA genes) (Figure 5). All major clades of the trees were well supported [Bayesian posterior probability (BPP) = 1.00, bootstrap (BS) = 100]. Based on the phylogenetic analyses, the 33 Basidiomycota species could be divided into seven major clades corresponding to the orders Ustilaginales, Agaricales, Polyporales, Russulales, Cantharellales, Tremellales, and Trichosporonales (Supplementary Table 4). The four species from Tremellales could be divided into two groups: one group was composed of two species in the Cryptococcus genus and the second group was composed of H. oryzae and T. fuciformis. H. oryzae was identified as a sister species to T. fuciformis. The results showed that the combined mitochondrial gene set was suitable as a reliable molecular marker for the analysis of the phylogenetic relationships among Basidiomycota species.
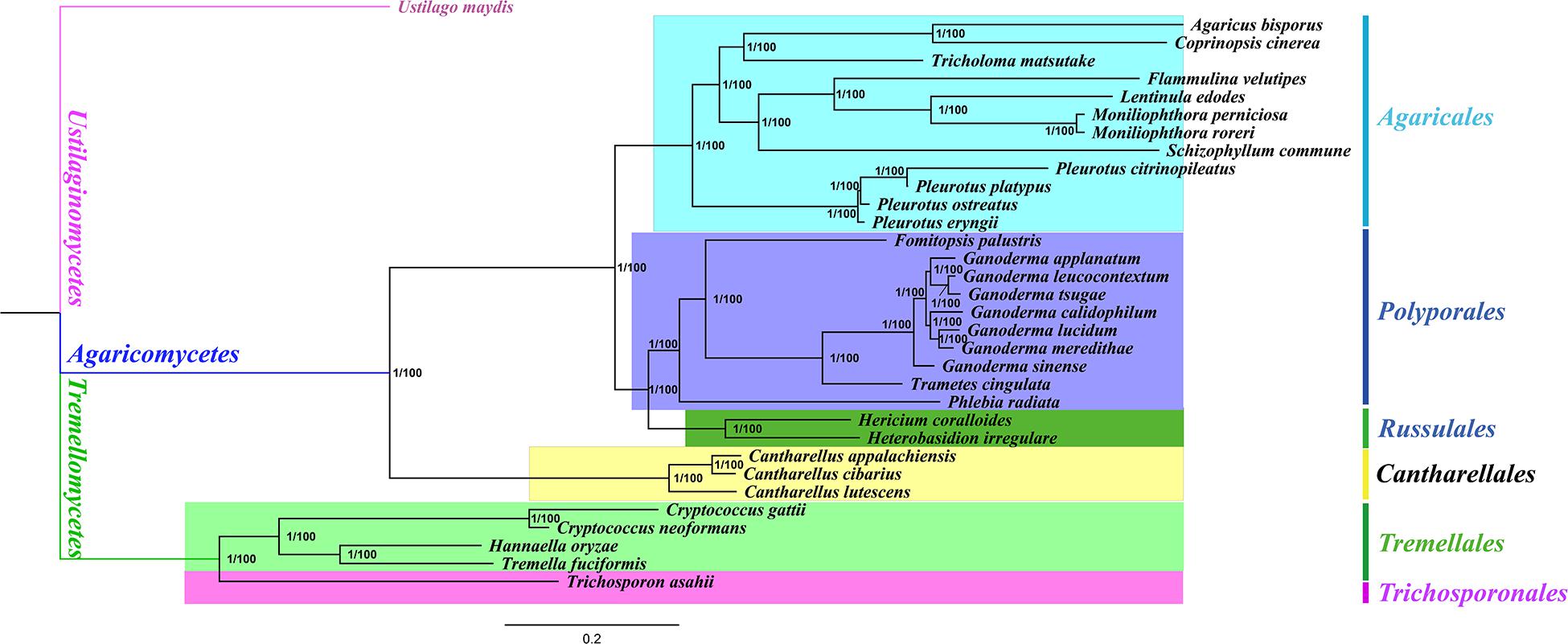
Figure 5. Molecular phylogeny of 33 Basidiomycota species based on both Bayesian inference (BI) and maximum likelihood (ML) analyses of 15 core protein-coding genes and two rRNA genes. Support values are Bayesian posterior probabilities (BPP; before the slash) and bootstrap (BS; after the slash). The species and the NCBI accession numbers for the genomes used in the phylogenetic analysis are provided in Supplementary Table 4.
As the “second genome” of eukaryotes, the mitochondrial genome plays an important role in regulating the growth and development, energy metabolism, aging, and stress resistance of eukaryotes (Bhargava and Schnellmann, 2017; Scheede-Bergdahl and Bergdahl, 2017). With the development of the next-generation sequencing technology, more and more mitochondrial genomes have been obtained, which has promoted our understanding of the origin, evolution, and taxonomy of eukaryotes (du Toit et al., 2017; Yang et al., 2018; Zhang et al., 2018). However, compared with animals, the mitochondrial genomes of fungi were less studied, especially that of Basidiomycota. It is estimated that there are more than 30,000 species of Basidiomycota in nature. However, up to now, less than 100 complete mitochondrial genomes of Basidiomycota are available in the public database (see text footnote 1). Only four mitochondrial genomes of basidiomycete yeasts were reported. This report of the mitochondrial genome of the important phylloplane yeast H. oryzae will broaden our understanding of basidiomycete mitochondrial genomes. The mitochondrial genome size of H. oryzae is the second smallest among all published Basidiomycota mitochondrial genomes, smaller than those of many mushroom-forming fungi (Stajich et al., 2010; Yadav et al., 2018), ectomycorrhizal fungi (Li et al., 2018b, 2020c), and that of its related species, T. fuciformis. However, the GC content of the H. oryzae mitogenome is the second highest among all published Basidiomycota mitochondrial genomes, indicating its unique mitogenome characteristics. Comparative mitogenomic analysis showed that the intron region had the greatest effect on the variations of the mitochondrial genome size of Tremellales, followed by the intergenic region and the protein-coding region, which was consistent with previous studies (Li et al., 2018b). Introns of the Tremellales cox1 gene were found to have undergone intron loss/gain events, which resulted in the size and organization variations of the Tremellales mitogenomes. In addition, the cox1 gene of H. oryzae contained two novel introns, which showed different origins from the known introns in basidiomycetes. Interestingly, we did not find any intron homologous to the introns of the H. oryzae cox1 gene in the NCBI database by BlastN search. More mitochondrial genomes need to be obtained to reveal the origin of the introns in H. oryzae or other basidiomycete species. Mitochondrial genes of H. oryzae were found on both strands as compared with the ascomycete mitochondrial genes, which are usually on the same strand (Aguileta et al., 2014).
It was reported that the mitochondrial genome of eukaryotes was obtained from a common alpha-proteobacterium ancestor (Lang et al., 1999). As evolution progresses, most mitochondrial genes were transferred to the nuclear genome, which was considered to have many advantages (Adams et al., 2002; Adams and Palmer, 2003). However, most fungi retain 14 conserved protein-coding genes for energy metabolism and one rps3 gene, which were called the core PCGs of the fungal mitogenome (Ye et al., 2020). In this study, these core PCGs were found to have high variation rates in the length and base composition even among the closely related species. Nine genes of H. oryzae were found to have unique lengths in the five closely related species, indicating the unique evolutionary characteristics of H. oryzae. Of all core PCGs, atp9 was found the most conserved among the five closely related species we studied. The genetic distance of the rps3 gene was the largest among the five closely related species we examined, which was consistent with the report in other literature (Li et al., 2018b, 2020c). Interestingly, we found that the atp8 gene in Tremellomycetes and Ustilaginomycetes had 12 nucleotides missing compared with that in Agaricomycetes species; the effect of the base variation of PCGs on the energy metabolism of species needs to be examined. In addition, we found that the core PCGs of the five closely related species in Tremellales and Trichosporonales were subject to purifying selection.
The arrangement of mitochondrial genes can serve as an important reference for revealing the evolutionary position of species (Li et al., 2018a; Zheng et al., 2018). Mitochondrial gene arrangement has been extensively studied in animals, and several models have been proposed to explain the rearrangement of mitochondrial genes in animals (Boore, 1999; Perseke et al., 2008). Compared with animals, the arrangement of fungal mitochondrial genes is highly variable (Hamari et al., 2001; Aguileta et al., 2014; Li et al., 2018a), and the gene rearrangement of the fungal mitochondrial genome has not been fully understood. Previous studies have shown that the accumulation of repetitive sequences in the fungal mitogenome has caused the fungal mitogenome rearrangement (Aguileta et al., 2014). In the present study, we found that the arrangement of mitochondrial genes was highly variable in the five closely related species, and the mitogenome of H. oryzae contained a unique gene order. In addition, the gene order varied at different taxonomic levels and is highly conserved between two species of the Cryptococcus genus. However, large-scale gene rearrangements have been observed at the class level, suggesting that Tremellales species undergo large-scale rearrangement events during evolution. Mitochondrial genes are also widely used as important molecular markers in the study of evolution, phylogeny, and population genetics (Bronstein and Kroh, 2018; Dai et al., 2018; Doyle et al., 2018; Li et al., 2020a). In this study, we divided 33 Basidiomycota species into seven clades based on a phylogenetic tree with high support for 15 core PCGs and two rRNA genes. Because there are few morphological features for fungi to be identified, this leads to confusion in fungal taxonomy and affects the phylogenetic research and utilization of fungi. The introduction of the mitochondrial genome will promote the understanding of fungal taxonomy and genetic evolution and can be used as a reliable tool for the analysis of fungal phylogeny (Li et al., 2018c, 2019a). More fungal mitochondrial genomes need to be uncovered to reconstruct the phylogenetic tree of fungi and to clarify the phylogenetic status of the Basidiomycota species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
QL and WH conceived and designed experiments. QL, LL, HF, WT, ZB, CX, and XW analyzed the data. QL, YQ, and WH wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by the Outstanding Youth Fund of Sichuan Province (2019JDJQ0034), the Liangshan Science and Technology and Intellectual Property Office Project (17NYCX0023), the Talent Fund of Sichuan Academy of Agricultural Sciences (2019QYXK003), and the High Tech Field Expansion Project of Sichuan Academy of Agricultural Sciences (2018GXTZ-001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.646567/full#supplementary-material
Supplementary Figure 1 | Putative secondary structures of the 23 tRNA genes identified in the mitogenome of H. oryzae. All genes are shown in order of occurrence in the mitogenome of H. oryzae, starting from trnS.
Supplementary Figure 2 | Pearson correlation analyses between mitochondrial genome sizes and different mitochondrial components of five closely related species in Tremellales and Trichosporonales.
Supplementary Figure 3 | Variation in the length and base composition of each of 15 core protein-coding genes (PCGs) among the mitochondrial genomes of the four closely related species in Tremellales. (A) PCG length variation; (B) GC content of the PCGs; (C) AT skew; (D) GC skew.
Supplementary Figure 4 | Genetic analysis of 15 core protein-coding genes conserved in the four closely related species in Tremellales. K2P, the Kimura-2-parameter distance; Ka, the mean number of non-synonymous substitutions per non-synonymous site; Ks, the mean number of synonymous substitutions per synonymous site.
Adams, K. L., and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/s1055-7903(03)00194-5
Adams, K. L., Qiu, Y. L., Stoutemyer, M., and Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. U.S.A. 99, 9905–9912. doi: 10.1073/pnas.042694899
Aguileta, G., de Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Bernt, M., Donath, A., Juhling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Bhargava, P., and Schnellmann, R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646. doi: 10.1038/nrneph.2017.107
Bleasby, A. J., and Wootton, J. C. (1990). Construction of validated, non-redundant composite protein sequence databases. Protein Eng. 3, 153–159. doi: 10.1093/protein/3.3.153
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Bronstein, O., and Kroh, A. (2018). The first mitochondrial genome of the model echinoid Lytechinus variegatus and insights into Odontophoran phylogenetics. Genomics 111, 710–718. doi: 10.1016/j.ygeno.2018.04.008
Bullerwell, C. E., and Lang, B. F. (2005). Fungal evolution: the case of the vanishing mitochondrion. Curr. Opin. Microbiol. 8, 362–369. doi: 10.1016/j.mib.2005.06.009
Burger, G., Gray, M. W., and Lang, B. F. (2003). Mitochondrial genomes: anything goes. Trends Genet. 19, 709–716. doi: 10.1016/j.tig.2003.10.012
Caspermeyer, J. (2016). MEGA evolutionary software re-engineered to handle today’s big data demands. Mol. Biol. Evol. 33:1887.
Cheng, J., Luo, Q., Ren, Y. H., Luo, Z., Liao, W. L., Wang, X., et al. (2021). Panorama of intron dynamics and gene rearrangements in the phylum Basidiomycota as revealed by the complete mitochondrial genome of Turbinellus floccosus. Appl. Microbiol. Biotechnol. 105, 2017–2032. doi: 10.1007/s00253-021-11153-w
Coordinators, N. R. (2017). Database resources of the National Center for biotechnology information. Nucleic Acids Res. 44, D7–D19.
Dai, L. S., Kausar, S., Abbas, M. N., and Wang, T. T. (2018). Complete sequence and characterization of the Ectropis oblique mitochondrial genome and its phylogenetic implications. Int. J. Biol. Macromol. 107, 1142–1150. doi: 10.1016/j.ijbiomac.2017.09.093
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dayo-Owoyemi, I., Rodrigues, A., Landell, M. F., Valente, P., Mueller, U. G., Ramos, J. P., et al. (2013). Intraspecific variation and emendation of Hannaella kunmingensis. Mycol. Prog. 12, 157–165. doi: 10.1007/s11557-012-0846-6
de Azeredo, L. A., Gomes, E. A., Mendonca-Hagler, L. C., and Hagler, A. N. (1998). Yeast communities associated with sugarcane in Campos, Rio de Janeiro, Brazil. Int. Microbiol. 1, 205–208.
Doyle, J. M., Bell, D. A., Bloom, P. H., Emmons, G., Fesnock, A., Katzner, T. E., et al. (2018). New insights into the phylogenetics and population structure of the prairie falcon (Falco mexicanus). BMC Genomics 19:233. doi: 10.1186/s12864-018-4615-z
du Toit, Z., du Plessis, M., Dalton, D. L., Jansen, R., Paul Grobler, J., and Kotze, A. (2017). Mitochondrial genomes of African pangolins and insights into evolutionary patterns and phylogeny of the family Manidae. BMC Genomics 18:746. doi: 10.1186/s12864-017-4140-5
Ferandon, C., Moukha, S., Callac, P., Benedetto, J. P., Castroviejo, M., and Barroso, G. (2010). The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS One 5:e14048. doi: 10.1371/journal.pone.0014048
Ferandon, C., Xu, J., and Barroso, G. (2013). The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 55, 85–91. doi: 10.1016/j.fgb.2013.01.009
Fourie, G., Van der Merwe, N. A., Wingfield, B. D., Bogale, M., Wingfield, M. J., and Steenkamp, E. T. (2018). Mitochondrial introgression and interspecies recombination in the Fusarium fujikuroi species complex. IMA Fungus 9, 37–48. doi: 10.5598/imafungus.2018.09.01.04
Gan, H. M., Thomas, B. N., Cavanaugh, N. T., Morales, G. H., Mayers, A. N., Savka, M. A., et al. (2017). Whole genome sequencing of Rhodotorula mucilaginosa isolated from the chewing stick (Distemonanthus benthamianus): insights into Rhodotorula phylogeny, mitogenome dynamics and carotenoid biosynthesis. PeerJ 5:e4030. doi: 10.7717/peerj.4030
Glushakova, A. M., Iurkov, A. M., and Chernov, I. (2007). [Massive isolation of anamorphous ascomycete yeasts Candida oleophila from plant phyllosphere]. Mikrobiologiia 76, 896–901.
Gomes, F., Arantes, T. D., Fernandes, J. A. L., Ferreira, L. C., Romero, H., Bosco, S. M. G., et al. (2018). Polymorphism in mitochondrial group I introns among Cryptococcus neoformans and Cryptococcus gattii genotypes and its association with drug susceptibility. Front. Microbiol. 9:86. doi: 10.3389/fmicb.2018.00086
Hamari, Z., Juhasz, A., Gacser, A., Kucsera, J., Pfeiffer, I., and Kevei, F. (2001). Intron mobility results in rearrangement in mitochondrial DNAs of heterokaryon incompatible Aspergillus japonicus strains after protoplast fusion. Fungal Genet. Biol. 33, 83–95. doi: 10.1006/fgbi.2001.1272
Han, L., Li, Z. Y., Guo, X. F., Tan, J. L., He, S. Z., Cui, X. L., et al. (2017). Hannaella dianchiensis sp. nov., a basidiomycetous yeast species isolated from lake water. Int. J. Syst. Evol. Microbiol. 67, 2014–2018. doi: 10.1099/ijsem.0.001908
Kaewwichian, R., Jindamorakot, S., Am-In, S., Sipiczki, M., and Limtong, S. (2015). Hannaella siamensis sp. nov. and Hannaella phetchabunensis sp. nov., two new anamorphic basidiomycetous yeast species isolated from plants. Int. J. Syst. Evol. Microbiol. 65, 1297–1303. doi: 10.1099/ijs.0.000101
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kurtzman, C. P., Fell, F. W., Boekhout, T., and Robert, V. (2011). “Methods for isolation, phenotypic characterization and maintenance of yeasts,” in The Yeasts, a Taxonomic Study, eds C. P. Kurtzman, J. W. Fell, and T. Boekhout (Amsterdam: Elsevier), 87–110. doi: 10.1016/b978-0-444-52149-1.00007-0
Landell, M. F., Billodre, R., Ramos, J. P., Leoncini, O., Vainstein, M. H., and Valente, P. (2010). Candida aechmeae sp. nov. and Candida vrieseae sp. nov., novel yeast species isolated from the phylloplane of bromeliads in Southern Brazil. Int. J. Syst. Evol. Microbiol. 60, 244–248. doi: 10.1099/ijs.0.011577-0
Landell, M. F., Brandao, L. R., Barbosa, A. C., Ramos, J. P., Safar, S. V., Gomes, F. C., et al. (2014). Hannaella pagnoccae sp. nov., a tremellaceous yeast species isolated from plants and soil. Int J Syst Evol Microbiol 64, 1970–1977. doi: 10.1099/ijs.0.059345-0
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773.
Lang, B. F., Gray, M. W., and Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi: 10.1146/annurev.genet.33.1.351
Li, Q., Chen, C., Xiong, C., Jin, X., Chen, Z., and Huang, W. (2018a). Comparative mitogenomics reveals large-scale gene rearrangements in the mitochondrial genome of two Pleurotus species. Appl. Microbiol. Biotechnol. 102, 6143–6153. doi: 10.1007/s00253-018-9082-6
Li, Q., He, X., Ren, Y., Xiong, C., Jin, X., Peng, L., et al. (2020a). Comparative mitogenome analysis reveals mitochondrial genome differentiation in Ectomycorrhizal and Asymbiotic Amanita species. Front. Microbiol. 11:1382. doi: 10.3389/fmicb.2020.01382
Li, Q., Liao, M., Yang, M., Xiong, C., Jin, X., Chen, Z., et al. (2018b). Characterization of the mitochondrial genomes of three species in the ectomycorrhizal genus Cantharellus and phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 118, 756–769. doi: 10.1016/j.ijbiomac.2018.06.129
Li, Q., Ren, Y., Shi, X., Peng, L., Zhao, J., Song, Y., et al. (2019a). Comparative mitochondrial genome analysis of two ectomycorrhizal fungi (Rhizopogon) reveals dynamic changes of intron and phylogenetic relationships of the Subphylum Agaricomycotina. Int. J. Mol. Sci. 20:5167. doi: 10.3390/ijms20205167
Li, Q., Ren, Y., Xiang, D., Shi, X., Zhao, J., Peng, L., et al. (2020b). Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 11:12.
Li, Q., Wang, Q., Chen, C., Jin, X., Chen, Z., Xiong, C., et al. (2018c). Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 119, 792–802. doi: 10.1016/j.ijbiomac.2018.07.197
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019b). Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 121, 249–260. doi: 10.1016/j.ijbiomac.2018.10.029
Li, Q., Wu, P., Li, L., Feng, H., Tu, W., Bao, Z., et al. (2021). The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 172, 560–572. doi: 10.1016/j.ijbiomac.2021.01.087
Li, Q., Xiang, D., Wan, Y., Wu, Q., Wu, X., Ma, C., et al. (2019c). The complete mitochondrial genomes of five important medicinal Ganoderma species: features, evolution, and phylogeny. Int. J. Biol. Macromol. 139, 397–408. doi: 10.1016/j.ijbiomac.2019.08.003
Li, Q., Yang, L., Xiang, D., Wan, Y., Wu, Q., Huang, W., et al. (2020c). The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 145, 974–984. doi: 10.1016/j.ijbiomac.2019.09.188
Li, Q., Yang, M., Chen, C., Xiong, C., Jin, X., Pu, Z., et al. (2018d). Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus. Sci. Rep. 8:9104.
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581.
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57.
Molnarova, J., Vadkertiova, R., and Stratilova, E. (2014). Extracellular enzymatic activities and physiological profiles of yeasts colonizing fruit trees. J. Basic Microbiol. 54(Suppl. 1), S74–S84.
Perseke, M., Fritzsch, G., Ramsch, K., Bernt, M., Merkle, D., Middendorf, M., et al. (2008). Evolution of mitochondrial gene orders in echinoderms. Mol. Phylogenet. Evol. 47, 855–864. doi: 10.1016/j.ympev.2007.11.034
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol Biol Evol 34, 3299–3302. doi: 10.1093/molbev/msx248
Scheede-Bergdahl, C., and Bergdahl, A. (2017). Adaptation of mitochondrial expression and ATP production in dedifferentiating vascular smooth muscle cells. Can. J. Physiol. Pharmacol. 95, 1473–1479. doi: 10.1139/cjpp-2017-0227
Stajich, J. E., Wilke, S. K., Ahren, D., Au, C. H., Birren, B. W., Borodovsky, M., et al. (2010). Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. U.S.A. 107, 11889–11894.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Vaidya, G., Lohman, D. L., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Valach, M., Burger, G., Gray, M. W., and Lang, B. F. (2014). Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 42, 13764–13777. doi: 10.1093/nar/gku1266
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, J., Zhang, L., Zhang, Q. L., Zhou, M. Q., Wang, X. T., Yang, X. Z., et al. (2017). Comparative mitogenomic analysis of mirid bugs (Hemiptera: Miridae) and evaluation of potential DNA barcoding markers. PeerJ 5, e3661. doi: 10.7717/peerj.3661
Wang, Q. M., and Bai, F. Y. (2008). Molecular phylogeny of basidiomycetous yeasts in the Cryptococcus luteolus lineage (Tremellales) based on nuclear rRNA and mitochondrial cytochrome b gene sequence analyses: proposal of Derxomyces gen. nov. and Hannaella gen. nov., and description of eight novel Derxomyces species. FEMS Yeast Res. 8, 799–814. doi: 10.1111/j.1567-1364.2008.00403.x
Wang, X., Jia, L., Wang, M., Yang, H., Chen, M., Li, X., et al. (2020a). The complete mitochondrial genome of medicinal fungus Taiwanofungus camphoratus reveals gene rearrangements and intron dynamics of Polyporales. Sci Rep 10:16500.
Wang, X., Song, A., Wang, F., Chen, M., Li, X., Li, Q., et al. (2020b). The 206 kbp mitochondrial genome of Phanerochaete carnosa reveals dynamics of introns, accumulation of repeat sequences and plasmid-derived genes. Int. J. Biol. Macromol. 162, 209–219. doi: 10.1016/j.ijbiomac.2020.06.142
Wang, X., Wang, Y. J., Yao, W., Shen, J. W., Chen, M. Y., Gao, M., et al. (2020c). The 256 kb mitochondrial genome of Clavaria fumosa is the largest among phylum Basidiomycota and is rich in introns and intronic ORFs. IMA Fungus 11:26.
Wang, Z., Wang, Z., Shi, X., Wu, Q., Tao, Y., Guo, H., et al. (2018). Complete mitochondrial genome of Parasesarma affine (Brachyura: Sesarmidae): Gene rearrangements in Sesarmidae and phylogenetic analysis of the Brachyura. Int. J. Biol. Macromol. 118, 31–40. doi: 10.1016/j.ijbiomac.2018.06.056
Wu, P., Bao, Z., Tu, W., Li, L., Xiong, C., Jin, X., et al. (2021). The mitogenomes of two saprophytic Boletales species (Coniophora) reveals intron dynamics and accumulation of plasmid-derived and non-conserved genes. Comput. Struct. Biotechnol. J. 19, 401–414. doi: 10.1016/j.csbj.2020.12.041
Yadav, V., Sun, S., Billmyre, R. B., Thimmappa, B. C., Shea, T., Lintner, R., et al. (2018). RNAi is a critical determinant of centromere evolution in closely related fungi. Proc. Natl. Acad. Sci. U.S.A. 115, 3108–3113. doi: 10.1073/pnas.1713725115
Yan, Z., Li, Z., Yan, L., Yu, Y., Cheng, Y., Chen, J., et al. (2018). Deletion of the sex-determining gene SXI1alpha enhances the spread of mitochondrial introns in Cryptococcus neoformans. Mob DNA 9, 24.
Yang, H., Xia, J., Zhang, J. E., Yang, J., Zhao, H., Wang, Q., et al. (2018). Characterization of the complete mitochondrial genome sequences of three croakers (Perciformes, Sciaenidae) and novel insights into the phylogenetics. Int. J. Mol. Sci. 19:1741. doi: 10.3390/ijms19061741
Yang, R. Y., Li, H. T., Zhu, H., Zhou, G. P., Wang, M., and Wang, L. (2012). Draft genome sequence of CBS 2479, the standard type strain of Trichosporon asahii. Eukaryot. Cell 11, 1415–1416. doi: 10.1128/ec.00237-12
Ye, J., Cheng, J., Ren, Y., Liao, W., and Li, Q. (2020). The first mitochondrial genome for Geastrales (Sphaerobolus stellatus) reveals intron dynamics and large-scale gene rearrangements of basidiomycota. Front. Microbiol. 11:1970. doi: 10.3389/fmicb.2020.01970
Yoon, H., Kong, W. S., Kim, Y. J., and Kim, J. G. (2016). Complete mitochondrial genome of the ectomycorrhizal fungus Tricholoma matsutake. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 3855–3857. doi: 10.3109/19401736.2014.958699
Zhang, S., Wang, X. N., Zhang, X. L., Liu, X. Z., and Zhang, Y. J. (2017). Complete mitochondrial genome of the endophytic fungus Pestalotiopsis fici: features and evolution. Appl. Microbiol. Biotechnol. 101, 1593–1604. doi: 10.1007/s00253-017-8112-0
Zhang, S., and Zhang, Y. J. (2019). Proposal of a new nomenclature for introns in protein-coding genes in fungal mitogenomes. IMA Fungus 10:15.
Zhang, Y., Sun, J., Rouse, G. W., Wiklund, H., Pleijel, F., Watanabe, H. K., et al. (2018). Phylogeny, evolution and mitochondrial gene order rearrangement in scale worms (Aphroditiformia, Annelida). Mol. Phylogenet. Evol. 125, 220–231. doi: 10.1016/j.ympev.2018.04.002
Zhang, Y. J., Zhang, H. Y., Liu, X. Z., and Zhang, S. (2017). Mitochondrial genome of the nematode endoparasitic fungus Hirsutella vermicola reveals a high level of synteny in the family Ophiocordycipitaceae. Appl. Microbiol. Biotechnol. 101, 3295–3304. doi: 10.1007/s00253-017-8257-x
Zhang, Y. J., Zhang, S., and Liu, X. Z. (2016). The complete mitochondrial genome of the nematode endoparasitic fungus Hirsutella minnesotensis. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 2693–2694. doi: 10.3109/19401736.2015.1046126
Zheng, B. Y., Cao, L. J., Tang, P., van Achterberg, K., Hoffmann, A. A., Chen, H. Y., et al. (2018). Gene arrangement and sequence of mitochondrial genomes yield insights into the phylogeny and evolution of bees and sphecid wasps (Hymenoptera: Apoidea). Mol. Phylogenet. Evol. 124, 1–9. doi: 10.1016/j.ympev.2018.02.028
Keywords: Hannaella, mitochondrial genome, protein coding gene, repeat sequence, gene rearrangement, phylogenetic analysis 3
Citation: Li Q, Li L, Feng H, Tu W, Bao Z, Xiong C, Wang X, Qing Y and Huang W (2021) Characterization of the Complete Mitochondrial Genome of Basidiomycete Yeast Hannaella oryzae: Intron Evolution, Gene Rearrangement, and Its Phylogeny. Front. Microbiol. 12:646567. doi: 10.3389/fmicb.2021.646567
Received: 27 December 2020; Accepted: 19 April 2021;
Published: 28 May 2021.
Edited by:
Georg Hausner, University of Manitoba, CanadaReviewed by:
Sajeet Haridas, Joint Genome Institute, United StatesCopyright © 2021 Li, Li, Feng, Tu, Bao, Xiong, Wang, Qing and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Wang, eHV3YW5nQGhlbmF1LmVkdS5jbg==; Yuan Qing, NjQ4OTk4NjdAcXEuY29t; Wenli Huang, d2VubGloMTFAMTI2LmNvbQ==
†Present address: Wenli Huang, Sichuan Academy of Agricultural Sciences, Chengdu, China
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.