
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 18 January 2021
Sec. Evolutionary and Genomic Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.625961
 Xiaoli Du1†
Xiaoli Du1† Mengyu Wang1,2,3†
Mengyu Wang1,2,3† Haijian Zhou1
Haijian Zhou1 Zhenpeng Li1
Zhenpeng Li1 Jialiang Xu4Zhe Li1
Jialiang Xu4Zhe Li1 Biao Kan1Daoli Chen5Xiaoli Wang6Yujuan Jin7Yan Ren8Yanping Ma9Jiuyin Liu10Yang Luan11
Biao Kan1Daoli Chen5Xiaoli Wang6Yujuan Jin7Yan Ren8Yanping Ma9Jiuyin Liu10Yang Luan11 Zhigang Cui1*†
Zhigang Cui1*† Xin Lu1*†
Xin Lu1*†We compared several identification methods for Aeromonas genus members, including traditional biochemical testing, multiplex-PCR amplification, mass spectrometry identification, whole-genome sequencing, multilocus phylogenetic analysis (MLPA), and rpoD, gyrA, and rpoD-gyrA gene sequencing. Isolates (n = 62) belonging to the Aeromonas genus, which were came from the bacterial bank in the laboratory, were used to assess the identification accuracy of the different methods. Whole-genome sequencing showed that the Aeromonas spp. isolates comprised A. caviae (n = 21), A. veronii (n = 18), A. dhakensis (n = 8), A. hydrophila (n = 7), A. jandaei (n = 5), A. enteropelogenes (n = 2), and A. media (n = 1). Using the whole-genome sequencing results as the standard, the consistency of the other methods was compared with them. The results were 46.77% (29/62) for biochemical identification, 83.87% (52/62) for mass spectrometric identification, 67.74% (42/62) for multiplex-PCR, 100% (62/62) for MLPA typing, 72.58% for gyrA, and 59.68% for rpoD and gyrA-rpoD. MLPA was the most consistent, followed by mass spectrometry. Therefore, in the public health laboratory, both MLPA and whole-genome sequencing methods can be used to identify various Aeromonas species. However, rapid and relatively accurate mass spectrometry is recommended for clinical lab.
Aeromonas, a Gram-negative, opportunistic pathogen bacterium, is prevalent in animals and the environment (Nolla-Salas et al., 2017). Aeromonas is often isolated from marine or aquatic organisms, and as an important fish pathogen, it causes septicemia and death in severe cases (Hossain et al., 2020). In 1963, Aeromonas was isolated from the blood of a girl with leukemia, which suggested its clinical significance (Bulger and Sherris, 1966). With increasing numbers of clinical cases of Aeromonas-related cases, this species is now considered to be a new gastrointestinal disease-causing pathogen in humans and other animals, and infections with it can become serious (Janda and Abbott, 2010). Diarrhea and food poisoning caused by Aeromonas have drawn increasing attention as foodborne illnesses (Pablos et al., 2010; Jamal et al., 2014). Aeromonas was previously classified as Vibrio, but phylogenetic studies have shown that it belongs to the Aeromonas genus. There are at least 18 Aeromonas species (Figueras et al., 2011), including A. hydrophila, A. salmonicida, A. bestiarum, A. sobria, A. trota, A. caviae, A. popoffii, A. media, A. encheleia, A. veronii, A. aquariorum, A. eucrenophila, A. molluscorum, A. schubertii, A. simiae, A. jandaei, A. tecta, and A. bivalvium. Three A. hydrophila subspecies exist among them. In 2010, four new species, A. diversa (Miñana-Galbis et al., 2010), A. rivuli (Figueras et al., 2011), A. taiwanensis (Alperi et al., 2010), and A. sanarellii (Alperi et al., 2010), were proposed. Altogether, 32 Aeromonas species have been identified to date (Martinez-Murcia et al., 2011), among which A. caviae, A. hydrophila, and A. veronii are closely associated with the clinical symptoms of diarrhea (Janda and Duffey, 1988; Parker and Shaw, 2011; Li et al., 2015). However, the specific Aeromonas types are relatively complex, and there is currently a lack of comprehensive and effective identification methods for them.
Traditional biochemical identification is a simple and low-cost method for preliminarily identifying Aeromonas members. However, identifying Aeromonas complex or new species requires supplementary experiments to be performed, so it is currently not possible to confidently distinguish Aeromonas species (Borrell et al., 1998; Martínez-Murcia et al., 2005; Janda and Abbott, 2010). Compared with traditional biochemical identification, mass spectrometry with its fast speed, simplicity, and high accuracy, is increasingly used to identify microorganisms (Bizzini et al., 2010; Benagli et al., 2012). With the development of biotechnology, multiplex-PCR is also commonly used for Aeromonas identification (Del Cerro et al., 2002; LaFrentz et al., 2019). Whole-genome sequencing technology has been widely used in various fields to accurately identify bacterial species by comparing the whole-genome sequences it generates; thus, this technique has become the reference method for bacterial species identification (Jamal et al., 2014; Hughes et al., 2016; Bartkova et al., 2017). MLPA typing is used to identify the characteristics of microbial isolates. This method assesses the degree of bacterial variation in a sample according to the differences existing among house-keeping gene sequences (Maiden et al., 1998; Martínez-Murcia et al., 2016). MLPA provides a strong species description framework for reliable, simple, and rapid identification of Aeromonas species (Navarro and inez-Murcia, 2018).
In this study, we compared the consistency of various techniques (i.e., biochemical detection, mass spectrometry identification, multiplex-PCR, MLPA, and rpoD, gyrA, rpoD-gyrA house-keeping gene amplification) with that of whole-genome sequencing at identifying Aeromonas species. The aim is to provide suggestion to choose the different method for the identification of Aeromonas according to different laboratory conditions.
Isolates belonging to the Aeromonas genus were came from the bacterial bank in the laboratory. Each sample was individually placed into alkaline peptone water broth (Beijing Land Bridge Co., Ltd., China) for 18–24 h at 37°C, and the mixture was inoculated onto an RS selective medium plate (Thermo Fisher Scientific, Massachusetts, USA) for 18–24 h at 37°C. Suspicious colonies on the RS Medium were selected and inoculated onto LB medium. Single colonies were selected and cultured for 18–24 h at 37°C (Soltan Dallal et al., 2016). The SYBR green fluorescence-based PCR method was used to rapidly screen for the presence of Aeromonas (Du et al., 2020).
Pure Aeromonas single colonies that developed within 18–24 h were picked using sterile absorbent cotton sticks dipped in a solution containing sterile 0.45% NaCl and with uniform grinding each was adjusted to 0.5 McNamara turbidity using the VITEK II (BioMerieux, Lyon, France) automatic biochemical identification card for Gram-negative bacteria on the identification apparatus. The quality control strain was E. coli (ATCC700323). The results were read according to the manufacturer's instructions.
Multiplex PCR was conducted in 50 μl volumes, with each reaction containing 25 μl of 2× Taq PCR MasterMix (TaKaRa, Dalian, China), 10 μ mol/L of upstream and downstream primers (Sangon, Shanghai, China), 2 μl of DNA template, with ddH2O used to make up the total volume (Persson et al., 2015).
The matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) system used to type the 62 pure Aeromonas colonies was based on protein “fingerprints” (Lauková et al., 2018), and was performed using a Microflex MALDI-TOF MS mass spectrometer (Antobio, China). To this end, every single colony was mixed with matrix solution and completely dried, and the MALDI-TOF MS instructions were followed for testing. Results were evaluated using the Autof ms1000 (Antobio, China) identification database. The results were exported for local preservation and statistical analysis. An appraisal credibility score of > 95% was considered to be reliable in this study (Jamal et al., 2014).
The Wizard Genomic DNA Purification Kit (Promega, Madison, USA) was used to extract genomic DNA from the cultured strains. The Illumina HiSeq-TM2000 sequencing platform was used to conduct PE-150 double-terminal sequencing, and the size of the inserted fragments was 350 bp.
The published Aeromonas genome assembly sequence (32 Aeromonas species in total) was downloaded from the GenBank database (until July 25, 2019). Altogether, 364 Aeromonas genome sequences were included in the analysis. First, the 364 individually downloaded Aeromonas sequences were used to construct evolutionary trees, and some species were found to be on the same branch (Supplementary Figure 1). The A. hydrophila ATCC 7966 genome sequence (Accession: GCA_000014805-1) was used as the reference. Next, the sequences from our 62 strains and the 364 whole-genome sequences from GenBank were compared with the reference sequence, and the core genome and single-nucleotide polymorphism (SNP) mutation sites were identified using Mummer v3.23 software. Sites in the repeat region that were identified by Blastn v2.2.28 were removed. Based on the identified 103,037 SNPs, Fasttree v2.1.7 software was used to construct a maximum-likelihood tree, which was visualized using Figtree v1.4.3 software and the iTol website.
In this study, the average nucleotide identity (ANI) method was used to assess the sequence similarities among the 62 Aeromonas strains that we sequenced and the 364 whole-genome sequences from GenBank. Gegenees v.2.2.1 was then used to draw a heat map from this data, from which the consistency between the two methods was compared.
Blastn comparison software was used to identify the genomic location information for gyrB, recA, dnaJ, gyrA, dnaX, atpD, and rpoD house-keeping genes in the 426 strains, and the corresponding gene sequences from each strain were extracted using a perl script. We used mafft v7.123b software to perform a multi-sequence comparison on the sequences of the aforementioned seven housekeeping genes and the gyrA gene, rpoD gene, and rpoD-gyrA (Martinez-Murcia et al., 2011). The maximum-likelihood tree constructed by Fasttree v2.1.7 was visualized in Figtree (v1.4.3) software and the iTol website.
We used the Kappa index to analyze the consistency of two qualitative observations from the same subjects (Murphy-Zane and Pyle, 2018). The Kappa index does not only test the consistency of the two results, but also provides a measurement of the degree of consistency. The value range for the Kappa index is 0–1. It is generally believed that a Kappa value ≤ 0.4 shows a poorly consistent result, a value of 0.4 < Kappa ≤ 0.75 shows good consistency, and a Kappa value of ≥0.75 has the best consistency. Statistical significance was defined as P < 0.05. The Kappa index was used to compare the consistency of the Aeromonas identification methods with that of the whole-genome sequencing results.
In this study, the 62 isolates were assessed using the SYBR green fluorescence PCR method, the results of which confirmed that the 62 strains we isolated were Aeromonas spp.
A core genomic SNP tree was constructed on the 364 whole genomes we downloaded from GenBank, along with the genome sequences obtained in this study. According to the cluster generated by MLtree, the species of the 62 isolates from this study were defined as being on the same branch, in accordance with the known species of the 364 strains from GenBank. Finally, seven species in the Aeromonas genus were identified as A. caviae, A. veronii, A. dhakensis, A. hydrophila, A. jandaei, A. enteropelogenes, and A. media (Figure 1). The ANI method showed intraspecies nucleotide similarity rates of >97% (different strains within the same species) (Janda and Abbott, 2010). These results (Figure 2) are consistent with those of the SNP phylogenetic tree.
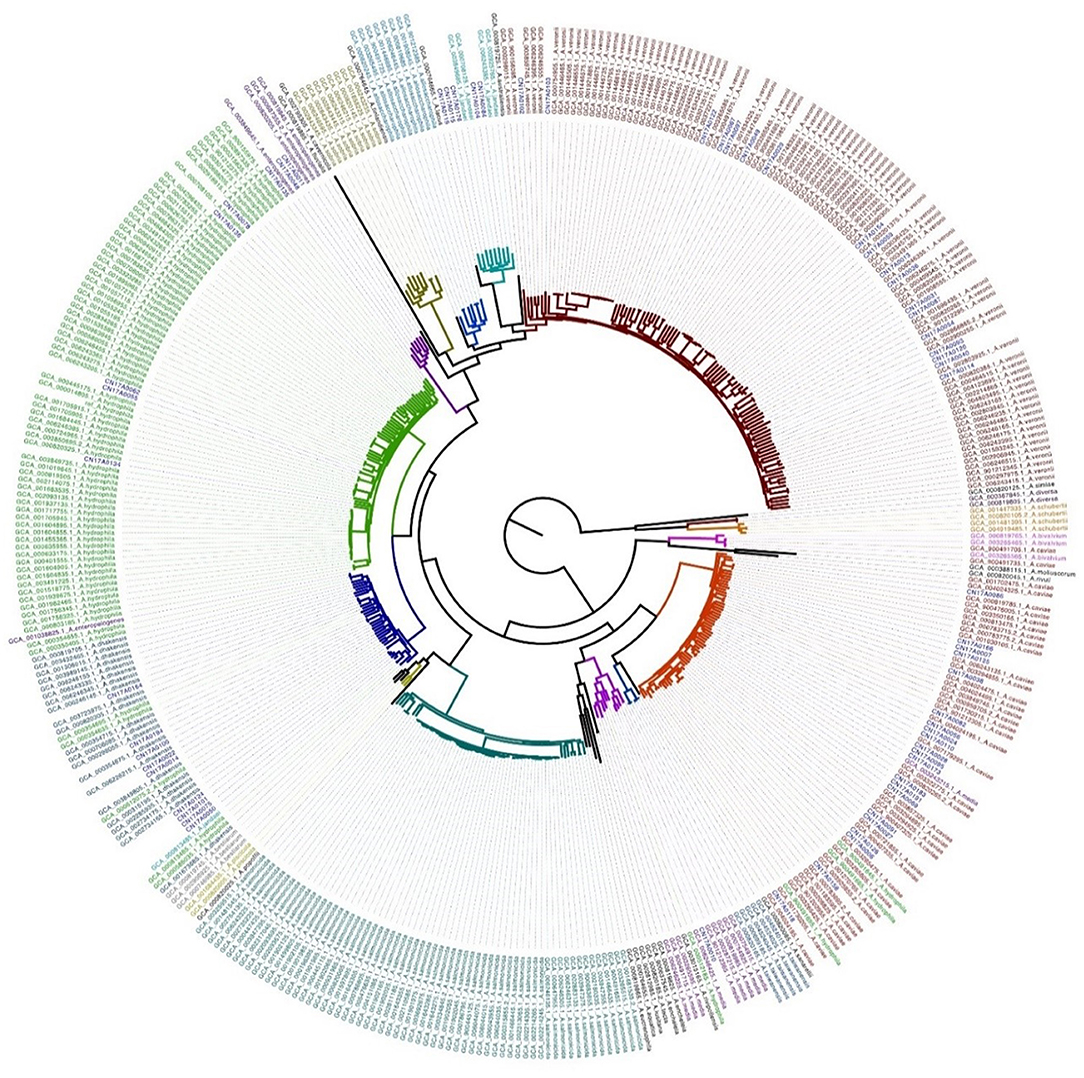
Figure 1. Maximum-likelihood tree based on genome-wide SNPs constructed for the 62 strains sequenced in this study versus 364 strains downloaded from Genbank.
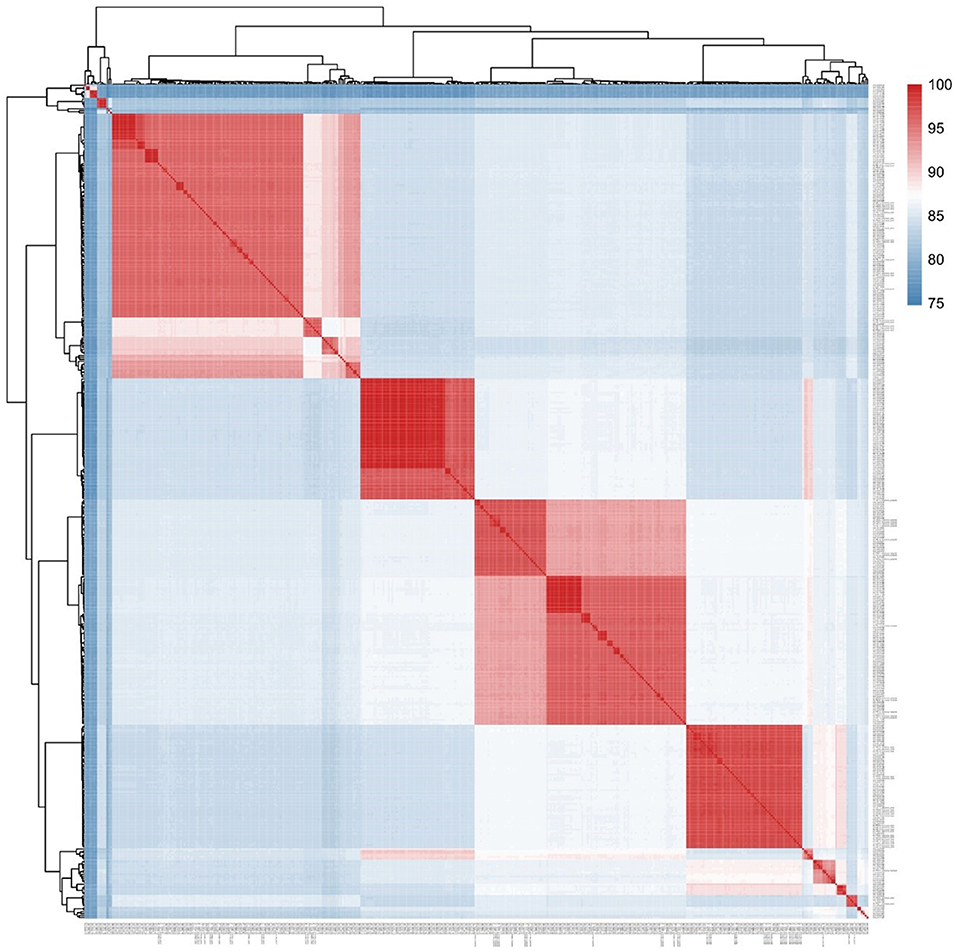
Figure 2. Phylogenomic analysis of the Aeromonas spp. examined in this study. The values generated by the Gegenees software shown in the heat map indicate the percentage similarity between the analyzed genomes. The colors vary from blue (low similarity) to red (high similarity).
Compared with the whole-genome sequencing results, the overall accuracy was 46.77% (29/62) (Table 1). The Kappa index value, which at 0.378 is <0.4, showed poor consistency (Table 2). The best biochemical identification accuracy was for A. hydrophila and A. caviae (100% accuracy), with the worst accuracy being 0% for A. dhakensis. Moreover, A. dhakensis was identified as A. caviae 25% (2/8) or A. hydrophila 75% (6/8). The biochemical identification readily but mistakenly identified A. jandaei, A. veronii, and A. enteropelogenes as A. sobria. Three strains of A. veronii were identified as A. hydrophila, and one was not identifiable (Supplementary Table 1).
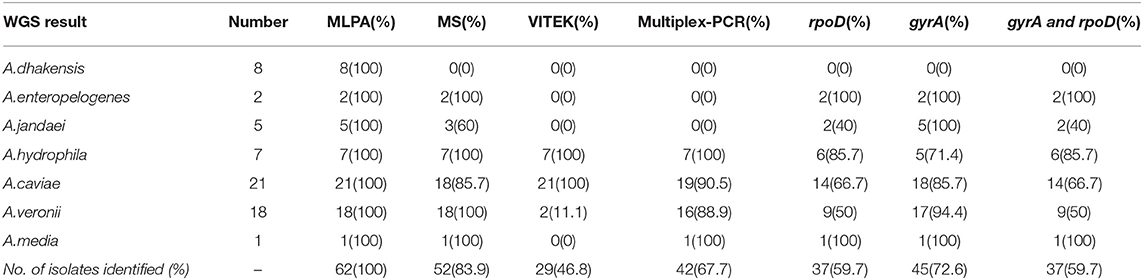
Table 1. Comparison of the identification accuracy of different methods for Aeromonas species.
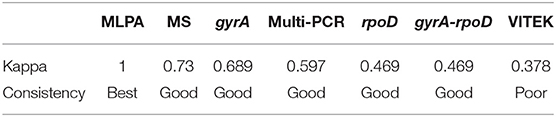
Table 2. Kappa values of the various methods vs. the whole-genome sequencing results.
Compared with the whole-genome sequencing results, the multiplex-PCR accuracy was 67.74% (42/62) (Table 1). At 0.597, the Kappa index is <0.75 but shows good consistency (Table 2). Among them, the identification of A. media and A. hydrophila was 100% accurate. Both A. enteropelogenes and A. jandaei were identified as A. veronii by multiplex PCR, but A. dhakensis was not identified by this method (Supplementary Table 1).
Taking the whole-genome sequencing results as the reference standard, the accuracy of mass spectrometry testing was 83.87% (52/62) (Table 1), and 45 strains scored above 95. At 0.73, the Kappa index is <0.75 but shows good consistency (Table 2). The accuracy of A. veronii identification was 100% (18/18, Supplementary Table 1). Mass spectrometry typed A. caviae as A. hydrophila, and could not identify A. dhakensis at all (Supplementary Table 1).
In this study, the results from rpoD and rpoD-gyrA identification were consistent. Compared with the whole-genome sequencing results, the accuracy of rpoD and gyrA gene identification was 59.7% (37/62) and 72.6% (45/62), respectively (Table 1), and the Kappa index values were 0.469 and 0.689, respectively, which although below 0.75, had good consistency (Table 2). The consistency rate for the gyrA gene method was 100% (5/5) for the identification of A. jandaei (Table 1). Both rpoD-gyrA and rpoD showed 100% consistency at identifying A. enteropelogenes (Table 1), but neither gene distinguished A. jandaei from A. caviae (Supplementary Table 1).
As shown in Table 1, based on the MLPA phylogenetic analysis of the seven house-keeping genes, seven species in the Aeromonas genus were identified. The results of the MLPA typing were 100% consistent with the genome-wide identification results. The Kappa index showed this method to have the best consistency of all the tested methods (Table 2).
Heterogeneity in the phenotypes and genotypes of Aeromonas makes species identification in this genus very complicated (Janda and Abbott, 2010). The emergence of various identification methods has helped with the identification process. Nevertheless, factors relating to the identification method itself and interference from various factors in the identification process have created discrepancies in accuracy among the various methods. In this study, the consistency rates among the multiple methods we used for identifying Aeromonas species were compared with the results from genome-wide identification, and the advantages and disadvantages of these methods in their ability to accurately identify Aeromonas species were evaluated.
The results of the evolutionary tree prepared from the whole-genome sequences showed that our 62 Aeromonas isolates fell into seven species: A. caviae 33.9% (21/62), A. veronii 29.0% (18/62), A. hydrophila 12.9% (8/62), A. dhakensis 11.3% (7/62), A. jandaei 8.1% (5/62), A. enteropelogenes 3.2% (2/62), and A. media 1.6% (1/62). Previous studies have shown MLPA typing to be consistent with the results from whole-genome sequencing (Martinez-Murcia et al., 2011). In the present study, MLPA typing showed the highest degree of consistency with the whole-genome sequencing results when compared with the other methods. rpoD and gyrA genes were used separately to type Aeromonas, and the result for gyrA was more consistent with the whole-genome sequencing results than that of rpoD (Table 1). Li et al. (Xinyue et al., 2016) speculated that this type of result may be related to the fact that rpoD is not good at distinguishing A. allosaccharophila from A. jandaei. Our study found that the results from rpoD-gyrA accorded with those from rpoD alone. Furthermore, the results from gyrA, rpoD, and rpoD-gyrA showed that they could not distinguish A. caviae from A. veronii, a result consistent with that from Persson et al. (2015) and Beaz-Hidalgo et al. (2010), but the identification of A. enteropelogenes was 100% (2/2). Although rpoD could not distinguish A. allosaccharophila from A. jandaei, rpoD had a higher consistency rate than gyrA at identifying A. hydrophila. With A. caviae, the biochemical identification showed 100% agreement with that for the whole genome. Except for A. hydrophila and A. caviae, where the accuracy of the biochemical identification was 100% for both, all other Aeromonas species were 0%, a finding consistent with the conclusion of Zhou (Yanyan et al., 2019).
Mass spectrometry identification is based on the unique map of protein peaks available in a commercial database (Benagli et al., 2012). The consistency between mass spectrometry identification and that of whole-genome sequencing was 83.9%, a result that may be related to the updated commercial database. Because A. dhakensis is a newly identified species, the database was not updated at the time we conducted this study, which led to a failure of identification. Despite multiplex PCR failing to amplify all of the target genes, its concordance rate with the whole-genome sequencing results was between that of biochemical identification and mass spectrometric identification. Multiplex-PCR technology misidentified A. enteropelogenes and A. jandaei as A. veronii. It has been reported that biochemical identification, mass spectrometry, and multiplex-PCR methods can accurately identify A. hydrophila (Bulger and Sherris, 1966; Wang et al., 2008; Elbehiry et al., 2019). In our study, the identification accuracy of A. hydrophila by the biochemical identification and multiplex-PCR methods was also 100%. The mass spectrometry identification method readily misidentified A. caviae as A. hydrophila (error rate, 14.3%), a finding consistent with the conclusion from a published study (Yanyan et al., 2019).
The whole-genome sequencing method used herein redefined A. dhakensis (obtained from human wounds), which was previously wrongly classified as A. hydrophila (Sinclair et al., 2016). When the virulence of A. hydrophila was compared with that of A. dhakensis, it was found that A. dhakensis was more virulent than A. hydrophila (Chen et al., 2013). Use of whole-genome sequencing technology should counteract species identification errors over time, thereby helping to make clinical diagnosis more accurate. Except for MLPA typing, the other identification methods misidentified A. dhakensis as A. hydrophila or A. aquariorum when compared with the whole-genome sequencing results, probably because A. dhakensis was originally considered a closely related subtype of A. hydrophila (Figueras et al., 2011). Furthermore, in one study, the identification rate for A. dhakensis based on its unique protein peak was 96.7% (Chen et al., 2014). In the present study, the mass spectrometer could not distinguish A. dhakensis from A. hydrophila, suggesting that the optimization of the protein peak diagram in the commercial database is conducive to the identification of Aeromonas species by mass spectrometry. A domestic study showed that the accuracy rate for mass spectrometry for A. enteropelogenes was 100 and 96.7% for A. media (Yanyan et al., 2019). In the present study, all the identification methods were able to identify A. media 100% (1/1), except for the biochemical identification method (Table 1). However, this result will need further confirmation because of the small sample size in this study.
When compared with the whole-genome sequencing results, the accuracy of MLPA typing was the highest of all the tested methods, attaining 100% for all 62 of the isolates. Previous studies have shown that the MLPA method achieves results that are consistent with those from whole-genome sequencing, and that the MLPA method can be widely used to screen and species identify isolated bacteria (Navarro and inez-Murcia, 2018). The consistency rate between mass spectrometry identification and whole-genome sequencing identification was 83.87%, somewhat lower than MLPA typing. We also found that the traditional biochemical identification method for A. hydrophila and A. caviae is better than that of mass spectrometry. Because of the difficulty in identifying other Aeromonas species, we suggest that biochemical identification is used for identifying Aeromonas genus members. While multiplex-PCR technology has some ability to identify common Aeromonas species such as A. caviae and A. veronii, the rpoD or gyrA method can be used for uncommon species such as A. enteropelogenes, A. jandaei, and A. media. That one previous study has also reported on a poor consistency of identification from multiple methods (Ørmen et al., 2005) indicates that a variety of identification methods should be combined for Aeromonas species identification. Due to the limitation of sample size in this study, a larger sample size is needed for confirmation.
With differences in accuracy between various methods clearly existing, the whole-genome sequencing method provides a unified standard with which to compare the various methods. Currently, biochemical identification is mainly used in clinical practice to identify isolated bacteria, but it is a time-consuming and laborious method, and the identification results are not accurate enough, which leads to diagnostic misjudgment (Jamal et al., 2014). If the commercial database of mass spectrometry is updated on time, the consistency with whole-genome sequencing results will be improved. What's more, the cost of mass spectrometric identification is reasonable and its operation straightforward, it can be used in the clinical lab to preliminarily identify Aeromonas members. As sequencing technology became more and more convenient in the public health labortory, both MLPA and whole-genome sequencing methods can be used to identify various Aeromonas species. Therefore, choosing an appropriate method for identifying Aeromonas species needs to be situation specific.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI GenBank, accession no: PRJNA685342.
XD and MW: wrote the editorial. HZ, Zhenl, JX, ZheL, and BK: provided technical assistance. DC, XW YJ, YR, YM, JL, and YL: performed data analysis and prepared the resources. ZC and XL: edited the editorial. All the authors read and approved the editorial.
This work was supported by the National Science and Technology Major Project (2018ZX10714002), and the Foundation for Young Scholars from China CDC (2018A103). We thank Sandra Cheesman, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all centers for Disease Control and Prevention for their contributions to this article.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.625961/full#supplementary-material
Alperi, A., Martínez-Murcia, A. J., Ko, W. C., Monera, A., Saavedra, M. J., and Figueras, M. J. (2010). Aeromonas taiwanensis sp. nov. and Aeromonas sanarellii sp. nov., clinical species from Taiwan. Int. J. Syst. Evol. Microbiol. 60, 2048–2055. doi: 10.1099/ijs.0.014621-0
Bartkova, S., Leekitcharoenphon, P., Aarestrup, F. M., and Dalsgaard, I. (2017). Epidemiology of Danish Aeromonas salmonicida subsp. salmonicida in fish farms using whole genome sequencing. Front. Microbiol. 8:2411. doi: 10.3389/fmicb.2017.02411
Beaz-Hidalgo, R., Alperi, A., Buján, N., Romalde, J. L., and Figueras, M. J. (2010). Comparison of phenotypical and genetic identification of Aeromonas strains isolated from diseased fish. Syst. Appl. Microbiol. 33, 149–153. doi: 10.1016/j.syapm.2010.02.002
Benagli, C., Demarta, A., Caminada, A., Ziegler, D., Petrini, O., and Tonolla, M. (2012). A rapid MALDI-TOF MS identification database at genospecies level for clinical and environmental Aeromonas strains. PLoS One 7:e48441. doi: 10.1371/journal.pone.0048441
Bizzini, A., Durussel, C., Bille, J., Greub, G., and Prod'hom, G. (2010). Performance of matrix-assisted laser desorption ionization-time of flight mass spectrometry for identification of bacterial strains routinely isolated in a clinical microbiology laboratory. J. Clin. Microbiol. 48, 1549–1554. doi: 10.1128/JCM.01794-09
Borrell, N., Figueras, M. J., and Guarro, J. (1998). Phenotypic identification of Aeromonas genomospecies from clinical and environmental sources. Can. J. Microbiol. 44, 103–108. doi: 10.1139/cjm-44-2-103
Bulger, R. J., and Sherris, J. C. (1966). The clinical significance of Aeromonas hydrophila. Report of two cases. Arch. Intern. Med. 118, 562–564. doi: 10.1001/archinte.118.6.562
Chen, P.-L., Wu, C.-J., Chen, C.-S., Tsai, P.-J., Tang, H.-J., and Ko, W.-C. (2013). A comparative study of clinical Aeromonas dhakensis and Aeromonas hydrophila isolates in southern Taiwan: A. dhakensis is more predominant and virulent. Clin. Microbiol. Infect. 20, 428–434. doi: 10.1111/1469-0691.12456
Chen, P. L., Lee, T. F., Wu, C. J., Teng, S. H., Teng, L. J., Ko, W. C., et al. (2014). Matrix-assisted laser desorption ionization-time of flight mass spectrometry can accurately differentiate Aeromonas dhakensis from hydrophila A, A. caviae, veronii A. J. Clin. Microbiol. 52, 2625–2628. doi: 10.1128/JCM.01025-14
Del Cerro, A., Marquez, I., and Guijarro, J. A. (2002). Simultaneous detection of Aeromonas salmonicida, Flavobacterium psychrophilum, Yersinia ruckeri. Three major fish pathogens, by multiplex PCR. Appl. Environ. Microbiol. 68, 5177–5180. doi: 10.1128/AEM.68.10.5177-5180.2002
Du, X., Liu, J., Hong, Y., Wang, L., Jiang, L., Chen, J., et al. (2020). Establishment and evaluation of SYBR green fluorescent PCR for detection of Aeromonas. Dis. Surveill. 35, 425–429. doi: 10.3784/j.issn.1003-9961.2020.05.013
Elbehiry, A., Marzouk, E., Abdeen, E., Al-Dubaib, M., Alsayeqh, A., Ibrahem, M., et al. (2019). Proteomic characterization and discrimination of Aeromonas species recovered from meat and water samples with a spotlight on the antimicrobial resistance of Aeromonas hydrophila. Microbiologyopen. 8:e782. doi: 10.1002/mbo3.782
Figueras, M. J., Alperi, A., Beaz-Hidalgo, R., Stackebrandt, E., Brambilla, E., Monera, A., et al. (2011). Aeromonas rivuli sp. nov., isolated from the upstream region of a karst water rivulet. Int. J. Syst. Evol. Microbiol. 61, 242–248. doi: 10.1099/ijs.0.016139-0
Hossain, S., De Silva, B. C. J., Dahanayake, P. S., De Zoysa, M., and Heo, G. J. (2020). Phylogenetic characteristics, virulence properties and antibiogram profile of motile Aeromonas spp. isolated from ornamental guppy (Poecilia reticulata). Arch. Microbiol. 202, 501–509. doi: 10.1007/s00203-019-01762-5
Hughes, H. Y., Conlan, S. P., Lau, A. F., Dekker, J. P., Michelin, A. V., Youn, J. H., et al. (2016). Detection and whole-genome sequencing of carbapenemase-producing Aeromonas hydrophila isolates from routine perirectal surveillance culture. J. Clin. Microbiol. 54, 1167–1170. doi: 10.1128/JCM.03229-15
Jamal, W., Albert, M. J., and Rotimi, V. O. (2014). Real-time comparative evaluation of bioMerieux VITEK MS versus Bruker Microflex MS, two matrix-assisted laser desorption-ionization time-of-flight mass spectrometry systems, for identification of clinically significant bacteria. BMC Microbiol. 14:289. doi: 10.1186/s12866-014-0289-0
Janda, J. M., and Abbott, S. L. (2010). The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23, 35–73. doi: 10.1128/CMR.00039-09
Janda, J. M., and Duffey, P. S. (1988). Mesophilic aeromonads in human disease: current taxonomy, laboratory identification, and infectious disease spectrum. Rev. Infect. Dis. 10, 980–997. doi: 10.1093/clinids/10.5.980
LaFrentz, B. R., Garcia, J. C., and Shelley, J. P. (2019). Multiplex PCR for genotyping Flavobacterium columnare. J. Fish Dis. 42, 1531–1542. doi: 10.1111/jfd.13068
Lauková, A., Kubašová, I., Kandričáková, A., Strompfová, V., Žitnan, R., and Simonová, M. P. (2018). Relation to enterocins of variable Aeromonas species isolated from trouts of Slovakian aquatic sources and detected by MALDI-TOF mass spectrometry. Folia Microbiol. (Praha). 63, 749–755. doi: 10.1007/s12223-018-0616-1
Li, F., Wang, W., Zhu, Z., Chen, A., Du, P., Wang, R., et al. (2015). Distribution, virulence-associated genes and antimicrobial resistance of Aeromonas isolates from diarrheal patients and water, China. J. Infect. 70, 600–608. doi: 10.1016/j.jinf.2014.11.004
Maiden, M. C. J., Bygraves, J. A., and Feil, E. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U. S. A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Martínez-Murcia, A., Beaz-Hidalgo, R., Navarro, A., Carvalho, M. J., Aravena-Román, M., Correia, A., et al. (2016). Aeromonas lusitana sp. nov., isolated from untreated water and vegetables. Curr. Microbiol. 72, 795–803. doi: 10.1007/s00284-016-0997-9
Martinez-Murcia, A. J., Monera, A., Saavedra, M. J., Oncina, R., Lopez-Alvarez, M., Lara, E., et al. (2011). Multilocus phylogenetic analysis of the genus Aeromonas. Syst. Appl. Microbiol. 34, 189–199. doi: 10.1016/j.syapm.2010.11.014
Martínez-Murcia, A. J., Soler, L., Saavedra, M. J., Chacón, M. R., Guarro, J., Stackebrandt, E., et al. (2005). Phenotypic, genotypic, and phylogenetic discrepancies to differentiate Aeromonas salmonicida from Aeromonas bestiarum. Int. Microbiol. 8, 259–269.
Miñana-Galbis, D., Farfán, M., Gaspar Lorén, J., and Carmen Fust,é, M. (2010). Proposal to assign Aeromonas diversa sp. nov. as a novel species designation for Aeromonas group 501. Syst. Appl. Microbiol. 33, 15–19. doi: 10.1016/j.syapm.2009.11.002
Murphy-Zane, M. S., and Pyle, L. (2018). Reliability of the anterior humeral line index compared with the gartland classification for posteriorly hinged supracondylar humerus fractures. Orthopedics. 41, e502–e505. doi: 10.3928/01477447-20180424-06
Navarro, A., and inez-Murcia, A. M. (2018). Phylogenetic analyses of the genus Aeromonas based on housekeeping gene sequencing and its influence on systematics. J. Appl. Microbiol. 125, 622–631. doi: 10.1111/jam.13887
Nolla-Salas, J., Codina-Calero, J., Valles-Angulo, S., Sitges-Serra, A., Zapatero-Ferrandiz, A., Climent, M. C., et al. (2017). Clinical significance and outcome of Aeromonas spp. infections among 204 adult patients. Eur. J. Clin. Microbiol. Infect. Dis. 36, 1393–1403. doi: 10.1007/s10096-017-2945-4
Ørmen, Ø., Granum, PE, Lassen, J., and Figueras, M. J. (2005). Lack of agreement between biochemical and genetic identification of Aeromonas spp. Food Saf. Infect. 113, 203–207. doi: 10.1111/j.1600-0463.2005.apm1130308.x
Pablos, M., Remacha, M. A., Rodriguez-Calleja, J. M., Santos, J. A., Otero, A., and Garcia-Lopez, M. L. (2010). Identity, virulence genes, and clonal relatedness of Aeromonas isolates from patients with diarrhea and drinking water. Eur. J. Clin. Microbiol. Infect. Dis. 29, 1163–1172. doi: 10.1007/s10096-010-0982-3
Parker, J. L., and Shaw, J. G. (2011). Aeromonas spp. clinical microbiology and disease. J. Infect. 62, 109–118. doi: 10.1016/j.jinf.2010.12.003
Persson, S., Al-Shuweli, S., Yapici, S., Jensen, J. N., and Olsen, K. E. (2015). Identification of clinical Aeromonas species by rpoB and gyrB sequencing and development of a multiplex PCR method for detection of hydrophila A, caviae A, A. veronii, media A. J. Clin. Microbiol. 53, 653–656. doi: 10.1128/JCM.01963-14
Sinclair, H. A., Heney, C., Sidjabat, H. E., George, N. M., Bergh, H., Anuj, S. N., et al. (2016). Genotypic and phenotypic identification of Aeromonas species and CphA-mediated carbapenem resistance in Queensland, Australia. Diagn. Microbiol. Infect. Dis. 85, 98–101. doi: 10.1016/j.diagmicrobio.2016.02.005
Soltan Dallal, M. M., Mazaheri Nezhad Fard, R., Kavan Talkhabi, M., Aghaiyan, L., and Salehipour, Z. (2016). Prevalence, virulence and antimicrobial resistance patterns of Aeromonas spp. isolated from children with diarrhea. Germs. 6, 91–96. doi: 10.11599/germs.2016.1094
Wang, Y., Tang, C., Yu, X., Wang, Y., and Yue, H. (2008). [Detecting pathogenic Aeromonas hydrophila in fish by triplex PCR]. Wei Sheng Wu Xue Bao. 48, 947–951.
Xinyue, L., Fengjuan, L., Pengcheng, D., Ruibai, W., and Duochun, W. (2016). Comparative analysis on identification of Aeromonas by rpoD and gyrA genes. Dis. Surveill. 31, 200–204. doi: 10.3784/j.issn.1003-9961.2016.03.006
Keywords: Aeromonas, whole-genome sequencing, mass spectrometry, multilocus phylogenetic analysis (MLPA), traditional biochemical testing, multiplex-PCR
Citation: Du X, Wang M, Zhou H, Li Z, Xu J, Li Z, Kan B, Chen D, Wang X, Jin Y, Ren Y, Ma Y, Liu J, Luan Y, Cui Z and Lu X (2021) Comparison of the Multiple Platforms to Identify Various Aeromonas Species. Front. Microbiol. 11:625961. doi: 10.3389/fmicb.2020.625961
Received: 04 November 2020; Accepted: 18 December 2020;
Published: 18 January 2021.
Edited by:
Vasco Ariston De Carvalho Azevedo, Federal University of Minas Gerais, BrazilReviewed by:
Maria José Saavedra, Universidade de Trás os Montes e Alto Douro, PortugalCopyright © 2021 Du, Wang, Zhou, Li, Xu, Li, Kan, Chen, Wang, Jin, Ren, Ma, Liu, Luan, Cui and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhigang Cui, Y3VpemhpZ2FuZ0BpY2RjLmNu; Xin Lu, bHV4aW5AaWNkYy5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.