 Atinuke M. Olajide
Atinuke M. Olajide
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Microbiol., 11 January 2021
Sec. Food Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.596828
This article is part of the Research TopicFood Storage, Spoilage and Shelf Life: Recent Developments and InsightsView all 18 articles
As close relatives, Bacillus paralicheniformis is often wrongly identified as Bacillus licheniformis. In this study, two genetic markers are presented based on fenC and fenD from the fengycin operon of B. paralicheniformis to rapidly distinguish it from B. licheniformis. The fengycin operon is one of the few present in B. paralicheniformis but absent in B. lichenformis up to date. Using these markers, two presumptive B. paralicheniformis isolates each were recovered from a set of isolates previously identified as B. licheniformis by Matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) or identified only to genus level as Bacillus by 16S ribosomal RNA (rRNA) gene sequencing, respectively. Whole genome sequencing of the four isolates confirmed their identity as B. paralicheniformis having the closest similarity with B. paralicheniformis ATCC 9945a (GenBank: CP005965.1) with a 7,682 k-mer score and 97.22% Average Nucleotide Identity (ANI). ANI of 100% suggests that the four isolates are highly similar. Further analysis will be necessary to determine if finer differences exist among these isolates at the level of single nucleotide polymorphisms.
In the Bacillus genus, many species within the assigned groups are very closely related so that 16S ribosomal RNA (rRNA) gene sequences cannot be used to distinguish them (Branquinho et al., 2014a). For instance, in the Bacillus cereus group, more than 97% sequence similarity exists among the 16S rRNA genes of Bacillus anthracis, B. cereus, Bacillus weihenstephanensis, Bacillus thuringiensis, Bacillus mycoides, Bacillus pseudomycoides, Bacillus cytotoxicus, Bacillus gaemokensis, and Bacillus manliponensis (Guinebretière et al., 2013). Furthermore, in the Bacillus pumilus group, B. pumilus, Bacillus safensis, Bacillus altitudinis, Bacillus stratosphericus, Bacillus aerophilus, Bacillus xiamenensis, and Bacillus invictae have over 99.5% similarity in their 16S rRNA gene sequences (Satomi et al., 2006; Liu et al., 2013; Lai et al., 2014; Branquinho et al., 2014b). Likewise, based on the comparative analysis of the 16S rRNA gene, Bacillus paralicheniformis strain KACC 18426 (KJ-16T) is 99.5% similar to Bacillus sonorensis KCTC-13918T and 99.4% similar to Bacillus licheniformis DSM 13T (Dunlap et al., 2015).
Due to the close relatedness of B. paralicheniformis to B. licheniformis, the former has been wrongly identified as B. licheniformis up until 2015. The phylogenetic analysis of nine B. paralicheniformis and 46 B. licheniformis showed that most of the former belonged to lineage P while the latter clustered together in lineage L (Du et al., 2019). However, the researchers observed that a few B. lichenformis clustered with B. paralicheniformis in lineage P rather than in lineage L. The genome comparative study by Du et al. (2019) revealed differences existing in the operons implicated in secondary metabolite synthesis among many Bacillus species. Some examples are the fengycin, paralichenicidin, and bacitracin operons, encoded by the genomes of B. paralicheniformis but absent in those of B. licheniformis analyzed to date (Harwood et al., 2018; Du et al., 2019).
Fengycins belong to a group of non-ribosomally synthesized antifungal lipopeptides, produced by certain strains of Bacillus species, such as B. subtilis, B. velezensis, B. paralicheniformis, and B. amyloliquefaciens (Steller et al., 1999; Stein, 2005; Harwood et al., 2018; Du et al., 2019). Also, fengycin lowered the numbers of Staphylococcus aureus in the human intestine (Piewngam et al., 2018). Fengycin synthetase consists of five non-ribosomal peptide synthetases (NRPSs) FenA – FenE encoded by fenA – fenE (Figure 1; Vanittanakom et al., 1986; Steller et al., 1999; Chen et al., 2007). Fengycin is a cyclic lipodecapeptide (CLP) containing a β-hydroxy fatty acid side chain of up to 19 carbon atoms (Steller et al., 1999) while the peptide portion contains at least four amino acids (Steller et al., 1999). The structure and function of the fengycin synthetase of B. subtilis origin have been reviewed previously by Steller et al. (1999).
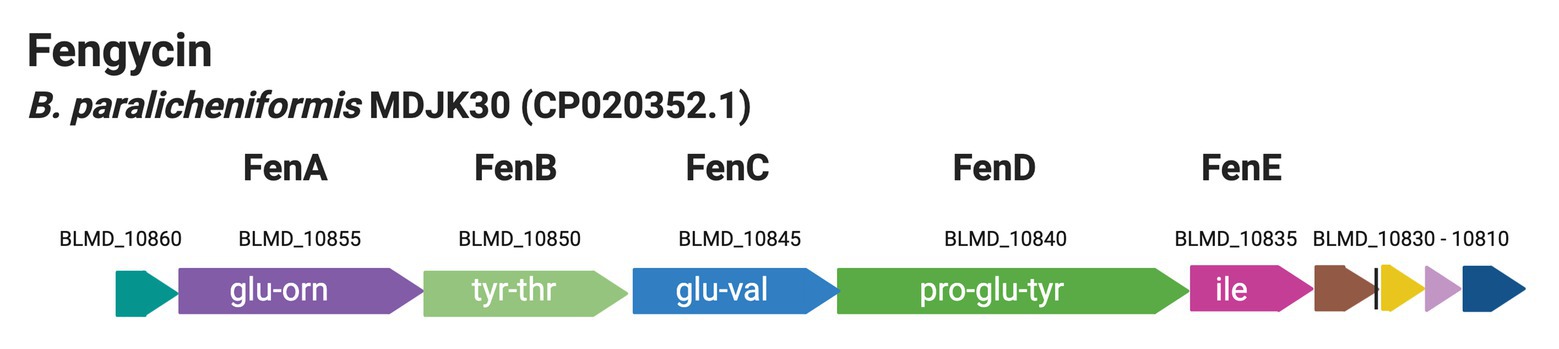
Figure 1. Genetic organization of the cluster coding for fengycin biosynthesis from the genome of B. paralicheniformis MDJK30 (Du et al., 2019).
Although Dunlap et al. (2015) attempted to differentiate B. paralicheniformis from B. licheniformis based on 16S rRNA gene phylogenetic and phenotypic analyses; there are no genetic markers that have been used to rapidly achieve this. To adequately identify Bacillus species, known to cause spoilage in foods including dairy, it is important to develop markers that work rapidly and efficiently in distinguishing these close relatives. Hence, B. paralicheniformis specific markers were designed based on the fenC gene (7,640 bp) and the fenD (10,780 bp). The markers were used to screen a collection of isolates previously identified as B. licheniformis or Bacillus spp. by 16S rRNA gene sequencing, matrix-assisted laser desorption ionization–time of flight (MALDI-TOF), or B. licheniformis specific primers. To the best of our knowledge, dairy B. paralicheniformis isolates of Canadian origin have not yet been reported.
Three hundred and nineteen (319) isolates were obtained after spore pasteurization (80°C for 12 min) or laboratory pasteurization (63°C for 30 min) of samples (raw milk, HT-milk, whey, curds, and environmental swabs) from a Cheddar cheese making plant and incubation on BHI agar (Oxoid) at 30°C for 48 h (aerobic). The identification was carried out by Sanger sequencing of the 16S rRNA gene, MALDI-TOF mass spectroscopy or B. licheniformis specific primers by de Almeida (2014). Sanger sequencing of at least 750 bp of the 16S rRNA gene was carried out using the FD1 forward primer (5' AGAGTTTGATCCTGGCTCAG-3') and the RD1 reverse primer (5' AAGGAGGTGATCCAGCCGCA-3'; Weisburg et al., 1991). Colony PCR was carried out with the condition as follows: 94°C for 2 min, 35 cycles of 94°C for 15 s, 55°C for 30 s, 72°C for 1.5 min and finally, and 72°C for 10 min. After amplification, the amplicons were separated by agarose gel electrophoresis (1% w/v, 70 v for 30 min) and visualized using a ChemiDoc Imaging System (Bio-Rad, Canada). Once the appropriate bands were purified, the products were sequenced at Eurofins (Canada). The sequences obtained per isolate were assembled (using DNA Baser v4) and compared against the National Center for Biotechnology Information (NCBI) database using the Ribosomal Database Project (Michigan State University) to obtain identification (> 99% similarity to species level).
Matrix-assisted laser desorption ionization–time of flight was used to identify isolates at the Animal Health Laboratory, University of Guelph (Guelph, ON, Canada) on the MALDI Biotyper (Bruker, Canada) with the software Compass v 4.1.80 (PYTH) 1022017-08-226_04-55-52. Each isolate to be identified was struck on a plate (Brain Heart Infusion agar at 30°C for 1 day) to obtain single and distinct colonies. After incubation, the isolates were maintained at room temperature until they were transported to the MALDI-TOF analysis facility. A bacterial colony was directly transferred to a 96-spot stainless steel target plate. One microliter of HCCA matrix (α-Cyano-4-hydroxycinnamic acid) was applied to each spot and left to dry (at room temperature). One microliter 70% formic acid was applied to the spot and allowed to dry followed by the addition of another 1 μl HCCA matrix after which it was left to dry. Identification was completed using a Bruker MALDI Biotyper and the software “Compass version 4.1.80 (PYTH) 1022017-08-226_04-55-52.” Calibration of each run was completed by applying a Bacterial Test Standard (BTS) to each target. The BTS was an Escherichia coli extract spiked with two high molecular weight proteins that was developed for the quality control process of the MALDI Biotyper System. The specific composition of the BTS covered the entire mass range of proteins used for precise identification of microorganisms. This control was run at every MALDI identification procedure, and the calibration must pass in flex control with a score greater than 2.0. A MALDI-TOF spectrum was automatically generated by the software and instantly matched against the reference library to give identification. For taxonomy allocation, if an organism obtained a score (x): x < 1.7, the ID was reported as “not reliable identification”; a score between 1.7 ≤ x ≤ 1.99 was reported to genus level only and with a score x ≥ 2.0, the ID was reliably reported to species level.
Lastly, four B. licheniformis specific primer sets (in pairs per PCR) were applied to crude DNA from Bacillus isolates with undetermined species as described by de Almeida (2014) (Table 1) using DNA from B. licheniformis ATCC 14580 and B. paralicheniformis KACC 18426 as positive and negative control, respectively. The PCR reaction mixture (23.5 μl) contained 1x PCR Supermix (Invitrogen), 0.25 μl (12.5 μM) of each primer, and 10 μl of template DNA. The amplification was as follows: initial denaturation for 5 min at 95°C, 35 cycles of 30 s at 95°C, 30 s at 58°C, and 90 s at 72°C followed by a final extension step for 10 min at 72°C. The amplification products were separated on a 2% agarose gel at 100 V for 35 min. Isolates for which amplicons were obtained by all four primer sets were assigned to B. licheniformis.
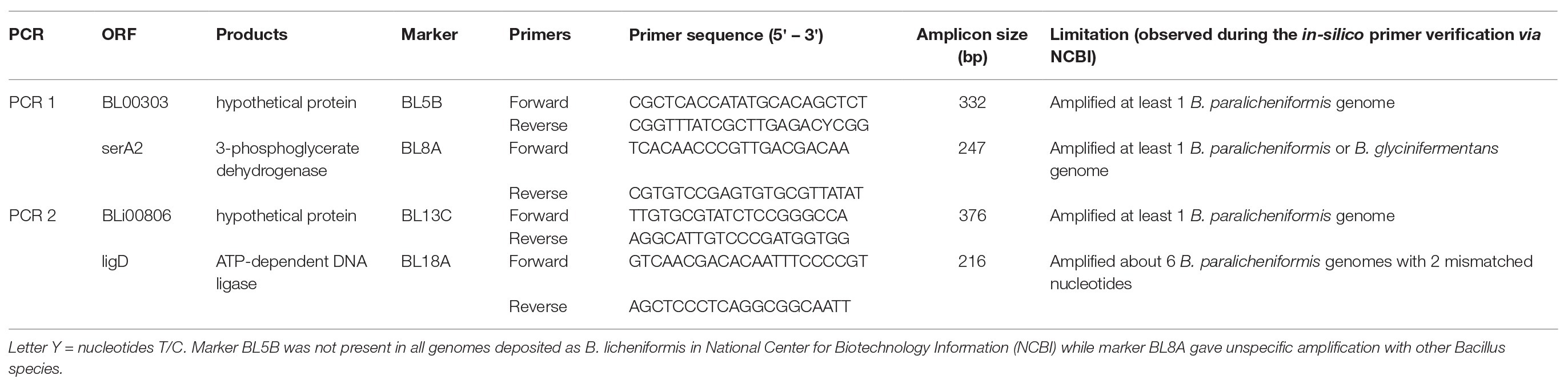
Table 1. Bacillus licheniformis specific primer sequences and amplification conditions.
The genes making up the fengycin operon were searched using BLAST within the six B. paralicheniformis whole genomes present in the NCBI GenBank (NZ_CP023666.1, NZ_CP023168.1, NC_021362.1, NZ_CP020352.1, NZ_CP023665.1, and NZ_CP033389.1). Genes were located using their arrangement in the genome of strain MDJK30 as a reference. The two longest genes (fenC and fenD) of the operon were chosen for designing primers. The sequences of these two genes from the six genomes listed above were aligned (MEGA 7; default settings) and observed to be 100% identical.
A primer set for each of fenC and fenD genes were designed (using PrimerQuest tool on IDT.com) based on the B. paralicheniformis MDJK30 genome sequence (GenBank: NZ_CP020352.1). Once specificity for B. paralicheniformis was ascertained in silico, primers were synthesized by Eurofins (Canada) and the PCR conditions were optimized using B. paralicheniformis KACC 18426.
Crude DNA was extracted from all the isolates identified as B. licheniformis isolates and Bacillus spp. isolates using Instagene matrix according to the manufacturers’ recommendations (Bio-Rad, Canada) and screened (Table 2). The PCR reaction mix of 23.5 μl contained 1× PCR Supermix (Invitrogen), 10 μM (0.5 μl) each of forward and reverse primers and 10 μl of template DNA (approx. 10–100 ng). The PCR cycle was carried out on Biometra professional thermocycler as follows: 2 min at 95°C, 40 cycles of 30 s at 95°C, 30 s at 53°C, and 1.5 min at 72°C, followed by a final extension step at 72°C for 5 min.
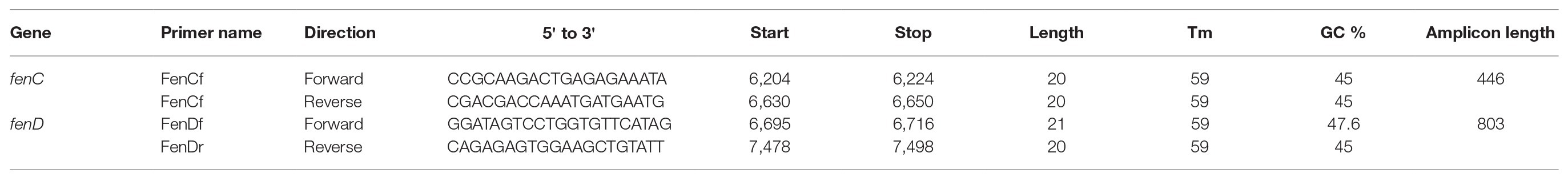
Table 2. PCR primer sequences and amplification conditions for fenC and fenD.
The isolates which gave amplicons (of correct sizes) with the two sets of B. paralicheniformis specific primers were sequenced using the Sanger method which was carried out at Eurofins (Canada). Also, whole genome sequencing of the presumptive B. paralicheniformis isolates was carried out for confirmation of their identity and diversity. For each isolate, genomic DNA was extracted from a single colony (picked from freshly prepared plates) using the Ultraclean Microbial DNA Isolation Kit (Qiagen, Canada), following the manufacturer’s protocol. The sequencing was carried out at the Laboratory Services, University of Guelph (Guelph, Ontario). The sequence library preparation was done using the Nextera DNA Flex library preparation kit (Illumina, Canada) following the manufacturer’s protocol. A MiSeq sequencer was used for sequencing with a MiSeq V2 reagent kit (Illumina) and 2 × 250 paired-end cycles, according to the manufacturer’s recommendation.
The MiSeq sequencer system software v3.1 (Illumina) was used to process the raw sequence reads. Furthermore, short sequences were filtered using FastQC 1.0.0 in BaseSpace.1 The sequences that passed a quality score of 30 were assembled via de novo assembly following an overlap-layout-consensus method using SPAdes v3.9.0 Genome Assembler (Bankevich et al., 2012) to generate contigs and scaffolds. Bacterial Analysis Pipeline v1.0.4 was used to predict the bacterial species following a k-mer based approach (Larsen et al., 2014). An Average Nucleotide Identity (ANI) calculator on EZBioCloud (https://www.ezbiocloud.net/; Yoon et al., 2017) was used to determine the percentage of nucleotide identity. NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v4.10 was used to predict, name, and annotate genes using the best-placed reference protein set method (Tatusova et al., 2016). Default parameters were used for all software.
Using the genomes of B. subtilis (168 and ATCC 13952), B. velezensis (UCMB5033 and BZB42), B. amyloliquefaciens (IT-45 and DSM 7), and B. paralicheniformis MDJK30 and a presumptive B. paralicheniformis isolate, a maximum likelihood dendrogram was constructed. Consensus sequences of the fengycin operon were extracted from each genome and aligned in the CLC workbench genomics (v10.0), after which the tree was created using MEGA (K2 + I model).
Out of 319 isolates, 124 were identified as B. licheniformis while 116 were identified to the genus level as Bacillus by 16S rRNA sequencing and MALDI-TOF. Using the species-specific primers, 17 additional B. licheniformis isolates were obtained (Figure 2) out of the 116 isolates previously identified to the genus level as Bacillus. However, the negative control, B. paralicheniformis KACC 18426, was amplified by three out of the four primer sets (BL13C, BL18A, and BL8A). Overall, 141 B. licheniformis and 85 Bacillus spp. were identified, resulting in a total of 226 isolates for screening.

Figure 2. Agarose gel showing the amplicons from 17 isolates identified as B. licheniformis using the four B. licheniformis specific markers (de Almeida, 2014). (A) Multiplex 1: primer sets 18A and 13C; (B) Multiplex 2: primer sets 5B and 8A. Lane 1 contains the GeneRuler Low range DNA ladder (ThermoScientific). Lanes 2–18 represent the additional B. licheniformis isolates while lane 19 contains B. licheniformis ATCC 14580 (positive control), lane 20 contains B. paralicheniformis KACC 18426 (negative control), and lane 21 contains the no template control.
Out of the 226 isolates tested, four (9MF010A, 9MF10B, 7CS50, and 7WI3) gave amplicons with both the fenC and fenD gene primer sets (Table 3). On all occasions, DNA from B. licheniformis ATCC 14580 (negative control) and the no-template control were not amplified by both sets of primers (Figure 3). Sanger sequencing of the amplicons from fenD confirmed that they were 99% identical to B. paralicheniformis ATCC 9945a as top match.
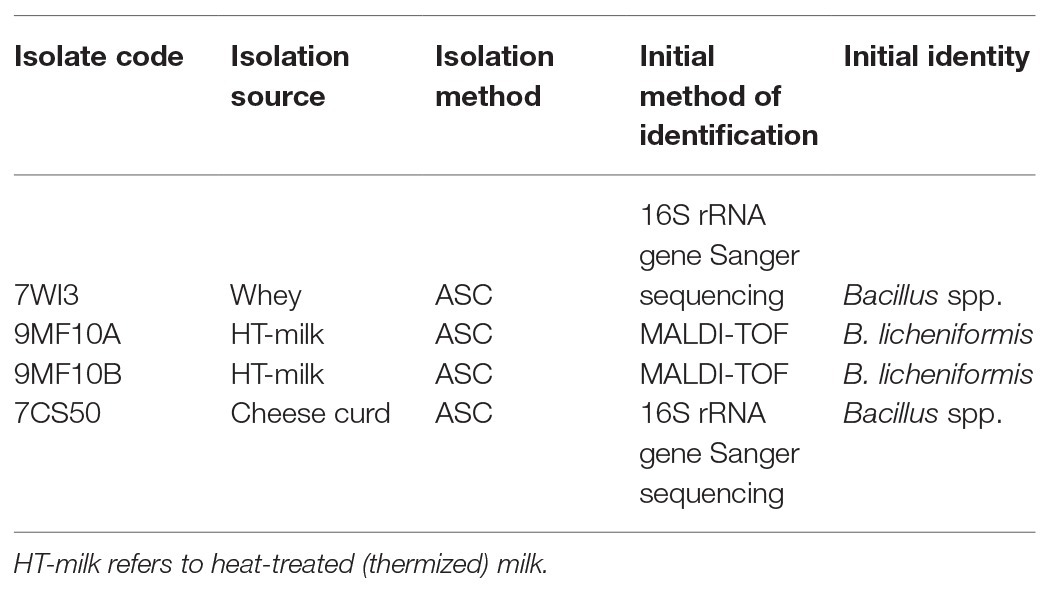
Table 3. Isolates newly identified as Bacillus paralicheniformis.
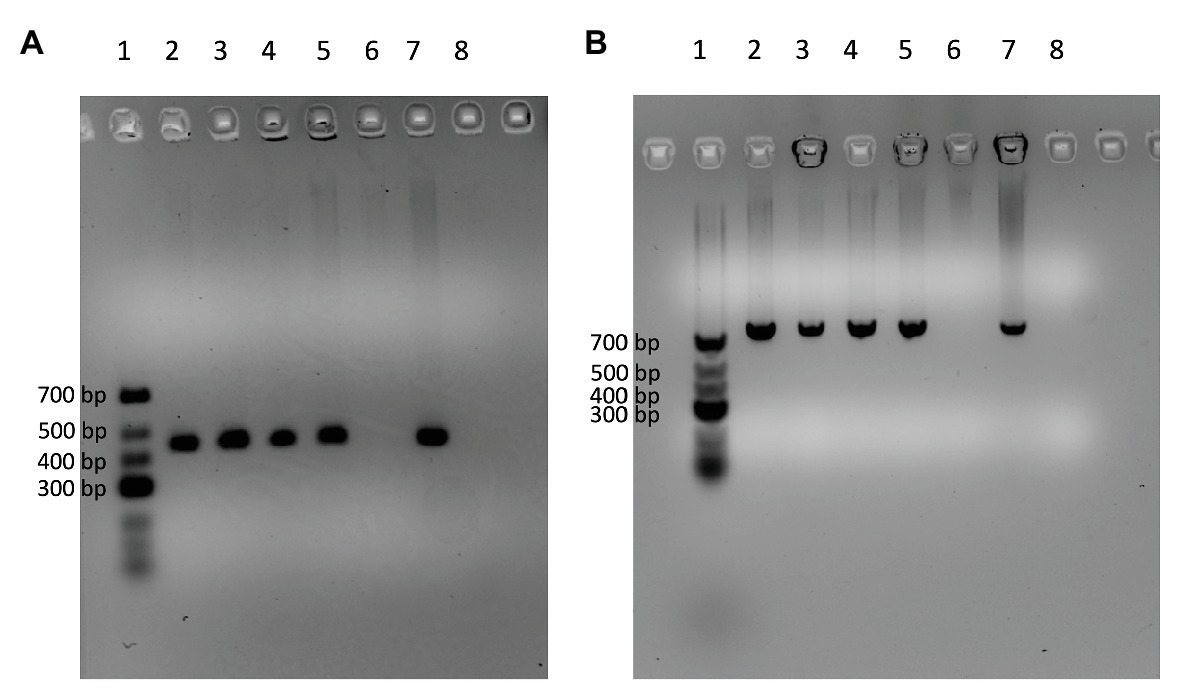
Figure 3. Agarose gels showing four isolates (Lanes 2–5) which were amplified by the DNA markers [fenC (A) and fenD (B)] designed specifically for B. paralicheniformis using B. licheniformis ATCC 14580 as negative control (Lane 6) and B. paralicheniformis KACC 18426 (NRRL – B65293) as positive control (Lane 7). Lane 8 contains the no-template control of the PCR reaction. Lane 1 contains the GeneRuler Low range DNA ladder (ThermoScientific).
Whole genome sequencing of the four B. paralicheniformis isolates showed an ANI of 100%, therefore, only one genome (7CS50) is presented here. The genome was closest to B. paralicheniformis ATCC 9945a (GenBank: CP005965.1) with a k-mer score of 7,682 and ANI of 97.22% was identified as B. paralicheniformis. With 137x coverage, 2,302,435 reads were generated for strain 7CS50 which were trimmed to 250 bp and assembled into 26 contigs. Strain 7CS50 has an estimated genome size of 4,199,730 bp and a 46.07% GC content (Figure 4). In total, 3,981 protein-coding sequences, 111 RNAs and 4,217 genes were found. The fengycin operon in B. paralicheniformis 7CS50 is closest in overall nucleotide identity to strain MDJK30 (97%) and farthest from the other species (78–87%; Figure 5).
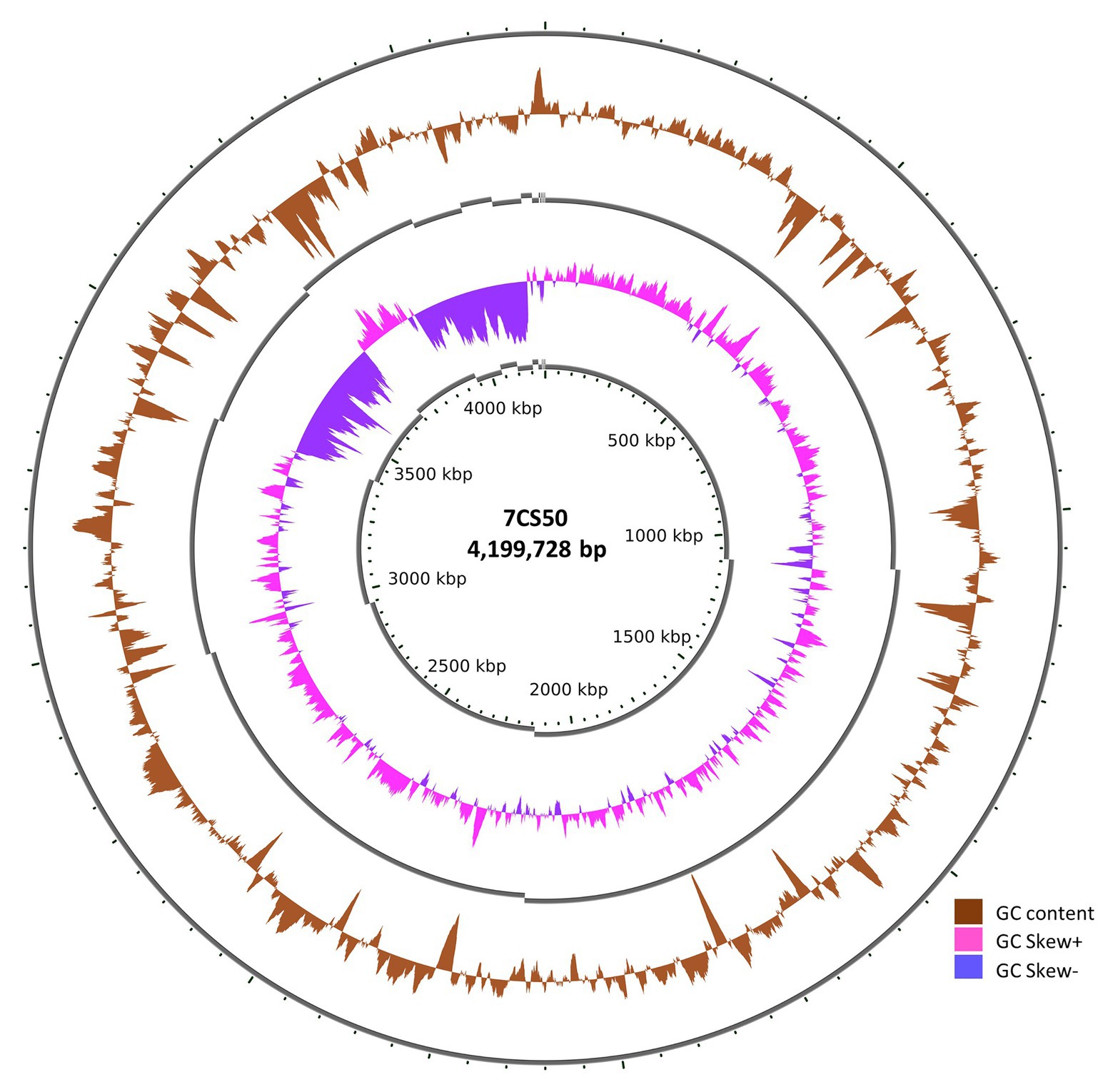
Figure 4. The circular genome of B. paralicheniformis 7CS50 using the CGViewer and a command line (https://github.com/paulstothard/cgview) for multiple contigs (Grant and Stothard, 2008).
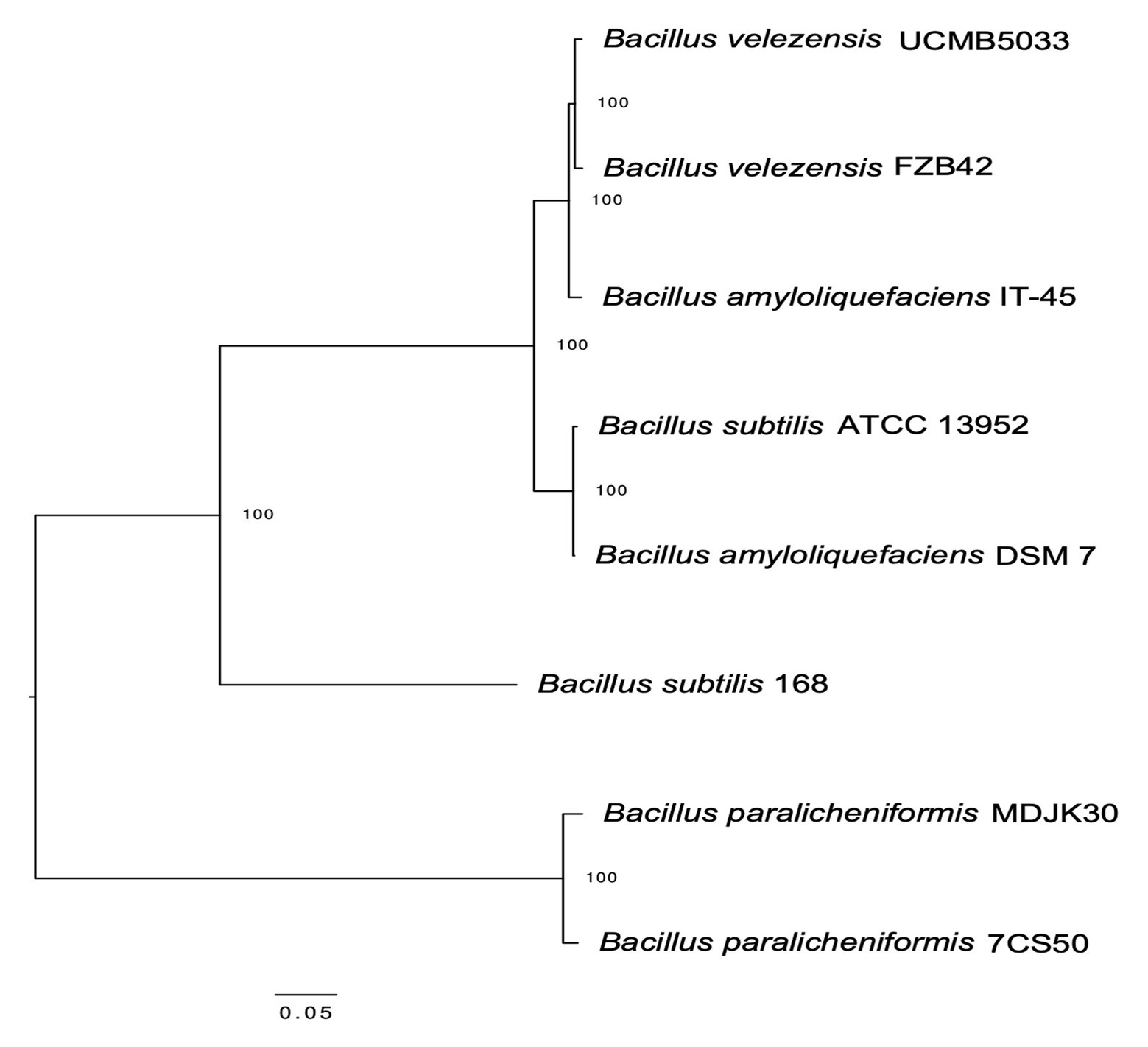
Figure 5. Maximum likelihood dendrogram based on the fengycin operon alignment from B. paralicheniformis 7CS50 and seven other Bacillus reference sequences via MEGA (K2 + I model) using the CLC workbench genomics v 10.
Fengycin was selected for the design of the B. paralicheniformis specific primers due to the ease of locating the operon in at least five publicly available genomes coupled with the ease of primer design. As most relevant literature target B. licheniformis for marker design, developing markers based on B. paralicheniformis should be explored. Of the five large proteins in the fengycin operon, only fenC and fenD were present in the six genomes with complete genome sequence data in the NCBI GenBank database.
The four isolates which were amplified by the designed primers were confirmed to be B. paralicheniformis using B. paralicheniformis ATCC 9945a as the reference genome. The four genomes encode the fengycin A–D operon, specific to B. paralicheniformis (Du et al., 2019). Two of the isolates were obtained from winter cheese and whey samples from separate vats while the other two were from a heat-treated (HT) milk samples collected in spring, all from a cheese making plant. These isolates were obtained after spore pasteurization (80°C for 12 min) of dairy samples obtained from a cheese processing system during the aerobic spore former count (ASC). These four isolates were highly similar as their sequences showed an ANI score of 100%. Bacillus licheniformis ATCC 14580 DNA was not amplified by the primer sets, implying that the sets were indeed specific for B. paralicheniformis.
Bacillus paralicheniformis specific markers have not been reported to date, to the best of our knowledge. These markers provide a fast detection of B. paralicheniformis among presumptive B. licheniformis isolates or even isolates identified to genus level as Bacillus. These markers are very promising as they provide a better approach to distinguishing this closely related Bacillus in comparison to previous phylogenetic and the phenotypic approaches. The four new B. paralicheniformis isolates had been previously identified as B. licheniformis by MALDI-TOF or as Bacillus spp. by Sanger sequencing of the 16S rRNA gene. It can be inferred that these approaches cannot adequately identify this bacterial species, as seen with many other close relatives among the genera Bacillus, Anoxybacillus, and Geobacillus (Randazzo et al., 2009; Weng et al., 2009; Burgess et al., 2010; Branquinho et al., 2014a). The misidentification of B. paralicheniformis by MALDI-TOF may have been due to either the quality and size of the protein database itself or due to the instability of the proteins produced by the isolates (Stîngu et al., 2008; Seng et al., 2009; Starostin et al., 2015). Adding the protein profile of these isolates to the MALDI-TOF database will improve future discrimination of the species using this method. Although 16S rRNA gene sequencing has been the gold standard for bacterial identification and for inferring phylogenetic relationships, it shows insufficient variation between closely related species (Zeigler, 2005). Similarly, species-specific primers of B. licheniformis that are not designed based on operons lacking in B. paralicheniformis will likely amplify the latter. This probably means that many of the primers previously designed specifically for B. lichenformis might also detect B. paralicheniformis. Even though fenC and fenD are not housekeeping genes which are alternative options for providing accurate taxonomic resolution of close relatives, they may be suitable to provide presumptive species identification. These markers could be used singly or in duplex to distinguish these two closely related species.
Across Bacillus species that possess a fengycin operon, the level of homology in terms of protein identity was low, ranging from 62 to 74%, indicating divergence from a common ancestor rather than horizontal transfer of these genes (Du et al., 2019). The fengycin operon from strain 7CS50 shows less than 90% nucleotide identity with the fengycin operons from seven other Bacillus species, which also suggests divergence, not horizontal transfer. Horizontal gene transfer (HGT) can occur in three main ways: transformation, transduction, and conjugation (Thomas and Nielsen, 2005). Genes (even if they are not accompanied by mobile elements) can be integrated into genomes if there is an active recombination system in the strain. Similarly, cells must be competent to take up DNA and integrate it into their genome (Johnston et al., 2014). Therefore, if fenC or fenD were transferred to B. licheniformis, then the strain would become indistinguishable from B. paralicheniformis with these markers. To mitigate this problem, it would be prudent to combine these markers with additional types of taxonomic identification or other markers based on secondary metabolites such as paralichenicidin (specific to B. paralicheniformis) or lichenicidin (specific to B. licheniformis). There appears to be a lower risk of HGT with the diverse polyketide synthase (pks) and NRPS-type operons (Grubbs et al., 2017), even though they are often found on genomic islands. Other approaches can be considered, such as Single Nucleotide Polymorphisms (SNPs) or a k-mer based approach as well as the use of smaller individual genes.
Structures, such as transposons, genomic islands, and integrases could suggest the possibility of HGT (Bose and Grossman, 2010). At this time, an in silico search using the IslandViewer online tool (v4 – www.pathogenomics.sfu.ca/islandviewer/query.php) predicted no genomic island close to the fengycin operon in the three B. paralicheniformis genomes analyzed (NZ_CP020352.1, NZ_CP023168.1, and NZ_CP023666.1; Bertelli et al., 2017). However, in the B. amyloliquefaciens FZB42 genome (now B. velezensis FZB42), there is a Tn1546 transposase encoded close to the fengycin operon located on genomic island 10 (Rückert et al., 2011). Similarly, the bmy (bacillomycin D) operon (located on a genomic island) is closely inserted by the fengycin operon in the FZB42 genome (Koumoutsi et al., 2004). A low (74%) nucleotide similarity between the fengycin-related gene (fenD) in B.velezensis (amyloliquefaciens) FZB42 and B. paralicheniformis MDJK30 suggests that they are only distantly related. For these markers to remain relevant, the possible occurrence of horizontal gene transfer of specific fengycin operons from B. paralicheniformis to B. licheniformis accompanied by recombination should continue to be monitored.
Two markers for the reliable and quick differentiation of B. licheniformis from B. paralicheniformis were established. They can be useful for presumptive identification of B. paralicheniformis. A new dairy B. paralicheniformis strain of Canadian origin was discovered using the fenC and fenD markers. Unlike lanthipeptides, such as bacitracin, the operons encoding fengycin-type NRPS metabolites do not show any evidence of HGT as yet but display a significant level of diversity among Bacillus species. The draft genome assembly of strain 7CS50 provides more data for future research on the species such as exploring the genomic diversity and functions of dairy B. paralicheniformis strains in comparison with those obtained from other environments.
The draft genome data were deposited at DDBJ/EMBL/GenBank (BioProject ID PRJNA580480) under the accession number WLVZ00000000 with SRA accession number SRX7224652 (Olajide et al., 2019).
AO: methodology, investigation, writing, original draft and revising, formal analysis, and visualization. SC: supervision, resources, formal analysis, writing, reviewing, and editing. GL: conceptualization, supervision, project administration, funding acquisition, resources, writing – review and editing, and data curation. All authors contributed to the article and approved the submitted version.
Funding for this research was provided by Genome Canada [Genomic Applications Partnership Program (GAPP) Round 7] and the NSERC/Dairy Farmers of Ontario Industrial Research Chair in Dairy Microbiology held by GL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The ARS Culture Collection is appreciated for providing us with B. paralicheniformis NRRL B65293 (KACC 18426 = KJ-16T), the type strain of B. paralicheniformis free of charge in this study. We are grateful for the help of Myra Siddiqi in constructing the sequence alignment dendrogram.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bose, B., and Grossman, A. D. (2010). Regulation of horizontal gene transfer in Bacillus subtilis by activation of a conserved site-specific protease. J. Bacteriol. 193, 22–29. doi: 10.1128/JB.01143-10
Branquinho, R., Meirinhos-Soares, L., Carriço, J. A., Pintado, M., and Peixe, L. V. (2014a). Phylogenetic and clonality analysis of Bacillus pumilus isolates uncovered a highly heterogeneous population of different closely related species and clones. FEMS Microbiol. Ecol. 90, 689–698. doi: 10.1111/1574-6941.12426
Branquinho, R., Sousa, C., Lopes, J., Pintado, M. E., Peixe, L. V., and Osorio, H. (2014b). Differentiation of Bacillus pumilus and Bacillus safensis using MALDI-TOF-MS. PLoS One 9:e110127. doi: 10.1371/journal.pone.0110127
Burgess, S. A., Lindsay, D., and Flint, S. H. (2010). Thermophilic Bacilli and their importance in dairy processing. Int. J. Food Microbiol. 144, 215–225. doi: 10.1016/j.ijfoodmicro.2010.09.027
Chen, X. H., Koumoutsi, A., Scholz, R., Eisenreich, A., Schneider, K., and Heinemeyer, I. (2007). Comparative analysis of the complete genome sequence of the plant growth—promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 25, 1007–1014. doi: 10.1038/nbt1325
de Almeida, E. M. M. (2014). Bacillus licheniformis specific DNA markers for identification and culture independent monitoring. Master thesis. Portugal: Departamento de Biologia, Universidade do Porto.
Du, Y., Ma, J., Yin, Z., Liu, K., Yao, G., Xu, W., et al. (2019). Comparative genomic analysis of Bacillus paralicheniformis MDJK30 with its closely related species reveals an evolutionary relationship between B. paralicheniformis and B. licheniformis. BMC Genomics 20:283. doi: 10.1186/s12864-019-5646-9
Dunlap, C. A., Kwon, S. -W., Rooney, A. P., and Kim, S. -J. (2015). Bacillus paralicheniformis sp. nov., isolated from fermented soybean paste. Int. J. Syst. Evol. Microbiol. 65, 3487–3492. doi: 10.1099/ijsem.0.000441
Grant, J. R., and Stothard, P. (2008). The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. doi: 10.1093/nar/gkn179
Grubbs, K. J., Bleich, R. M., Santa Maria, K. C., Allen, S. E., and Farag, S. AgBiome Team, et al. (2017). Large-scale bioinformatics analysis of Bacillus genomes uncovers conserved roles of natural products in bacterial physiology. mSystems 2, e00040–e00057. doi: 10.1128/mSystems.00040-17
Guinebretière, M. H., Auger, S., Galleron, N., Contzen, M., De Sarrau, B., De Buyser, M., et al. (2013). Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus group occasionally associated with food poisoning. Int. J. Syst. Evol. Microbiol. 63, 31–40. doi: 10.1099/ijs.0.030627-0
Harwood, C. R., Mouillon, J., Pohl, S., and Arnau, J. (2018). Secondary metabolite production and the safety of industrially important members of the Bacillus subtilis group. FEMS Microbiol. Rev. 42, 721–738. doi: 10.1093/femsre/fuy028
Johnston, C., Martin, B., Fichant, G., Polard, P., and Claverys, J. -P. (2014). Bacterial transformation: distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 12, 181–196. doi: 10.1038/nrmicro3199
Koumoutsi, A., Chen, X., Henne, A., Liesegang, H., Hitzeroth, G., Franke, P., et al. (2004). Structural and functional characterization of gene clusters directing nonribosomal synthesis of bioactive cyclic lipopeptides in Bacillus amyloliquefaciens strain FZB42. J. Bacteriol. 186, 1084–1096. doi: 10.1128/JB.186.4.1084–1096.2004
Lai, Q., Liu, Y., and Shao, Z. (2014). Bacillus xiamenensis sp. nov., isolated from intestinal tract contents of a flathead mullet (Mugil cephalus). Antonie Van Leeuwenhoek 105, 99–107. doi: 10.1007/s10482-013-0057-4
Larsen, M. V., Cosentino, S., Lukjancenko, O., Saputra, D., Rasmussen, S., Hasman, H., et al. (2014). Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 52, 1529–1539. doi: 10.1128/jcm.02981-13
Liu, Y., Lai, Q., Dong, C., Sun, F., Wang, L., Li, G., et al. (2013). Phylogenetic diversity of the Bacillus pumilus group and the marine ecotype revealed by multilocus sequence analysis. PLoS One 8:e800097. doi: 10.1371/journal.pone.0085660
Olajide, A. M., Chen, S., Mitterboeck, T. F., and LaPointe, G. (2019). Data from Bacillus paralicheniformis 7CS50 whole genome shotgun sequencing project draft genome data. NCBI Database (GenBank). 1.0. Available at: https://www.ncbi.nlm.nih.gov/nuccore/WLVZ00000000 (Accessed December 16, 2020).
Piewngam, P., Zheng, Y., Nguyen, T. H., Dickey, S. W., Joo, H. -S., Villaruz, A. E., et al. (2018). Pathogen elimination by probiotic Bacillus via signalling interference. Nature 562, 532–537. doi: 10.1038/s41586-018-0616-y
Randazzo, C., Caggia, C., and Neviani, E. (2009). Application of molecular approaches to study lactic acid bacteria in artisanal cheeses. J. Microbiol. Methods 78, 1–9. doi: 10.1016/j.mimet.2009.04.001
Rückert, C., Blom, J., Chenb, X., Reva, O., and Borriss, R. (2011). Genome sequence of B. amyloliquefaciens type strain DSM7T reveals differences to plant-associated B. amyloliquefaciens FZB42. J. Biotechnol. 155, 78–85. doi: 10.1016/j.jbiotec.2011.01.006
Satomi, M., La Duc, M. T., and Venkateswaran, K. (2006). Bacillus safensis sp. nov., isolated from spacecraft and assembly-facility surfaces. Int. J. Syst. Evol. Microbiol. 56, 1735–1740. doi: 10.1099/ijs.0.64189-0
Seng, P., Drancourt, M., Gouriet, F., La Scola, B., Fournier, P., Rolain, J. M., et al. (2009). Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectometry. Clin. Infect. Dis. 49, 543–551. doi: 10.1086/600885
Starostin, K. V., Demidov, E. A., Bryanskaya, A. V., Efimov, V. M., Rozanov, A. S., and Peltek, S. E. (2015). Identification of Bacillus strains by MALDI TOF MS using geometric approach. Sci. Rep. 5:16989. doi: 10.1038/srep16989
Stein, T. (2005). Bacillus subtilis antibiotics: structures, syntheses and specific functions. Mol. Microbiol. 56, 845–857. doi: 10.1111/j.1365-2958.2005.04587.x
Steller, S., Vollenbroich, D., Leenders, F., Stein, T., Conrad, B., and Hofemeister, J. (1999). Structural and functional organization of the fengycin synthetase multienzyme system from Bacillus subtilis b213 and A1/3. Chem. Biol. 6, 31–41. doi: 10.1016/S1074-5521(99)80018-0
Stîngu, C. S., Rodloff, A. C., Jentsch, H., Schaumann, R., and Eschrich, K. (2008). Rapid identification of oral anaerobic bacteria cultivated from subgingival biofilm by MALDI-TOFMS. Oral Microbiol. Immunol. 23, 372–276. doi: 10.1111/j.1399-302X.2008.00438.x
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Thomas, C., and Nielsen, K. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721. doi: 10.1038/nrmicro1234
Vanittanakom, N., Loeffler, W., Koch, U., and Jung, G. (1986). Fengycin—a novel antifungal lipopeptide antibiotic produced by Bacillus subtilis F-29-3. J. Antibiot. 39, 888–901. doi: 10.7164/antibiotics.39.888
Weisburg, W., Barns, S., Pelletier, D., and Lane, D. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703. doi: 10.1128/JB.173.2.697-703.1991
Weng, F. Y., Chiou, C. S., Lin, P. H. P., and Yang, S. S. (2009). Application of recA and rpoB sequence analysis on phylogeny and molecular identification of Geobacillus species. J. Appl. Microbiol. 107, 452–464. doi: 10.1111/j.1365-2672.2009.04235.x
Yoon, S. H., Ha, S. M., Lim, J. M., Kwon, S. J., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Keywords: Bacillus paralicheniformis, markers, fengycin, dairy, draft genome
Citation: Olajide AM, Chen S and LaPointe G (2021) Markers to Rapidly Distinguish Bacillus paralicheniformis From the Very Close Relative, Bacillus licheniformis. Front. Microbiol. 11:596828. doi: 10.3389/fmicb.2020.596828
Edited by:
Anindya Chanda, Mycologics LLC, United StatesReviewed by:
Colin Harwood, Newcastle University, United KingdomCopyright © 2021 Olajide, Chen and LaPointe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gisèle LaPointe, glapoint@uoguelph.ca; Atinuke M. Olajide, atinuke@uoguelph.ca
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.