 Jaime Simbaqueba1*
Jaime Simbaqueba1* Edwin A. Rodríguez1
Edwin A. Rodríguez1 Diana Burbano-David1
Diana Burbano-David1 Carolina González1
Carolina González1 Alejandro Caro-Quintero2*
Alejandro Caro-Quintero2*- 1Corporación Colombiana de Investigación Agropecuaria – AGROSAVIA, Centro de Investigación Tibaitatá, Mosquera, Colombia
- 2Department of Biology, Universidad Nacional de Colombia, Bogotá, Colombia
The vascular wilt disease caused by the fungus Fusarium oxysporum f. sp. physali (Foph) is one of the most limiting factors for the production and export of cape gooseberry (Physalis peruviana) in Colombia. A transcriptomic analysis of a highly virulent strain of F. oxysporum in cape gooseberry plants, revealed the presence of secreted in the xylem (SIX) effector genes, known to be involved in the pathogenicity of other formae speciales (ff. spp.) of F. oxysporum. This pathogenic strain was classified as a new f. sp. named Foph, due to its specificity for cape gooseberry hosts. Here, we sequenced and assembled the genome of five strains of F. oxysporum from a fungal collection associated to the cape gooseberry crop (including Foph), focusing on the validation of the presence of SIX homologous and on the identification of putative effectors unique to Foph. By comparative and phylogenomic analyses based on single-copy orthologous, we found that Foph is closely related to F. oxysporum ff. spp., associated with solanaceous hosts. We confirmed the presence of highly identical homologous genomic regions between Foph and Fol that contain effector genes and identified six new putative effector genes, specific to Foph pathogenic strains. We also conducted a molecular characterization using this set of putative novel effectors in a panel of 36 additional stains of F. oxysporum including two of the four sequenced strains, from the fungal collection mentioned above. These results suggest the polyphyletic origin of Foph and the putative independent acquisition of new candidate effectors in different clades of related strains. The novel effector candidates identified in this genomic analysis, represent new sources involved in the interaction between Foph and cape gooseberry, that could be implemented to develop appropriate management strategies of the wilt disease caused by Foph in the cape gooseberry crop.
Introduction
Fusarium oxysporum is a cosmopolitan ascomycete fungus that commonly inhabits agricultural soils. Rather than a single species, it is a species complex of non-pathogenic, plant pathogenic, and human pathogenic strains, termed the Fusarium oxysporum species complex (FOSC) (Di Pietro et al., 2003; Michielse and Rep, 2009; O’Donnell et al., 2009; Ma et al., 2013; Ma, 2014). Several hundred different members of the FOSC are able to penetrate plant roots, colonize xylem vessels and produce vascular wilt diseases in a broad range of host plants, including economically important crops such as banana, cotton, date palm, onion, brassicas, cucurbits, legumes and solanaceous species, such as tomato, eggplant, chili and cape gooseberry, but not grasses (Michielse and Rep, 2009). However, individual pathogenic isolates of F. oxysporum are highly host specific and have therefore been classified into different formae speciales (ff. spp.) according to the host they infect, e.g., strains that infect banana cannot infect tomato plants and vice versa (Lievens et al., 2008; Michielse and Rep, 2009; Ma, 2014). F. oxysporum has no known sexual stage and the mechanism for species diversification has been associated with the parasexual cycle through heterokaryon formation, which enables a mitotic genetic exchange between different nuclei (Glass et al., 2000; Di Pietro et al., 2003).
Comparative genomics of phytopathogens in the genus Fusarium [i.e., F. graminearum, F. verticillioides, and F. oxysporum f. sp. lycopersici (Fol)], revealed the presence of lineage specific (LS) chromosomes and chromosomal regions in Fol that were rich in repetitive elements and contained genes encoding known or putative effector proteins (Ma et al., 2010). Among them, 14 genes were identified that encode small proteins secreted into the xylem sap of tomato plants infected with Fol (called SIX proteins) (Houterman et al., 2007; Schmidt et al., 2013). Three of these SIX genes are avirulence genes (Avr), with resistance (R) gene counterparts identified in tomato (Simons et al., 1998; Rep et al., 2004; Houterman et al., 2008, 2009; Catanzariti et al., 2015, 2017).
Small proteins secreted by a broad range of plant pathogens, including bacteria, fungi, oomycetes and nematodes, that interfere with the cellular structure and function of their hosts are known as effector proteins (Kamoun, 2006; Kamoun et al., 2007; Hogenhout et al., 2009). The low level of homology among fungal effectors makes it difficult to identify common features that allow their classification as a group or protein family (Stergiopoulos and de Wit, 2009; de Guillen et al., 2015; Lo Presti et al., 2015). Nevertheless, many fungal effectors have been identified based on the presence of a signal peptide sequence for secretion, small size of around 300 amino acids or less, and the fact that they are often cysteine rich (Sperschneider et al., 2015). A large-scale search for putative effector genes in 59 strains of various ff. spp., resulted in a set of 104 candidate effectors including the 14 secreted in the xylem (SIX) genes, identified in Fol (Ma et al., 2010; Schmidt et al., 2013; van Dam et al., 2016). From this candidate effector repertoire, strains were classified according to the putative effector sequences they shared. Interestingly, all the cucurbit-infecting ff. spp. (melonis, niveum, cucumerinum, and radicis-cucumerinum), were grouped together in a separate supercluster, sharing an overlapping set of putative effectors and possibly conferring the ability to those ff. spp. to infect cucurbit host species (van Dam et al., 2016). This supercluster includes a substantial overlap with SIX1, SIX6, SIX8, SIX9, SIX11 and SIX13 and largely excluded SIX2, SIX3, SIX4, SIX5, SIX7, SIX10, SIX12, and SIX14. Homologous of Fol SIX genes have been identified in alliaceous, legumes, musaceous, solanaceous, and narcissus infecting ff. spp. of F. oxysporum (Taylor et al., 2016, 2019; Williams et al., 2016; Czislowski et al., 2018; Simbaqueba et al., 2018).
Cape gooseberry (Physalis peruviana) from the Solanaceae family, is a tropical native fruit of South America found typically growing in the Andes. In Colombia, over the last three decades, the cape gooseberry has been transformed from a wild and under-utilized species to an important exotic fruit for national and international markets and represents one of the most exported fruit for Colombia (Simbaqueba et al., 2011; Moreno-Velandia et al., 2019). The cape gooseberry is also appreciated by its nutritional and medicinal properties (Yen et al., 2010; Ramadan, 2011; El-Gengaihi et al., 2013; Ramadan et al., 2015). However, despite its significant value, cape gooseberry production has been limited due to the lack of known cultivars and the absence of adequate phytosanitary measures. One of the most important disease problems in cape gooseberry is the vascular wilt disease caused by F. oxysporum. This disease was first described in 2005 and has become one of the limiting factors for cape gooseberry production and export (Moreno-Velandia et al., 2019). Field observations indicated typical symptoms of a vascular wilt disease with an incidence ranging from 10 to 50% with losses in production of 90% approximately (unofficially reported), in the Cundinamarca central region of Colombia. Consequently, producers moved to other places in the same region, spreading contaminated plant material and seeds (Barrero et al., 2012).
From 2012 to 2015, a total of 136 fungal isolates were obtained from cape gooseberry plants showing wilting disease symptoms, collected from different locations of the central Andean Region of Colombia. The fungal isolates were described as F. oxysporum, using Koch postulates and molecular markers for intergenic spacers (IGS) and the translation elongation factor 1 alpha (EF1α) gene of F. oxysporum (AGROSAVIA, Unpublished results). A strain of F. oxysporum named MAP5, was found to be highly virulent in a number of cape gooseberry plant material, including a commercial variety and different accessions from National Germplasm Bank and different collections (Enciso-Rodríguez et al., 2013; Osorio-Guarín et al., 2016). Further RNAseq analysis was performed to study differential gene expression comparing susceptible and resistant cape gooseberry plants inoculated with MAP5 (AGROSAVIA, Unpublished results). This RNAseq data was used in comparative transcriptomics, identifying eight homologous of effector genes between Fol and MAP5. Thus, describing a newly forma specialis of Fusarium oxysporum that affect cape gooseberry plants, designated as F. oxysporum f. sp. physali (Foph) (Simbaqueba et al., 2018).
In this study, we sequenced the genome of five fungal strains, including Foph_MAP5, three additional pathogenic and one non-pathogenic strains of F. oxysporum in cape gooseberry. We performed comparative genomics using the resulted genome assemblies to infer the phylogenetic relationship of Foph within the F. oxysporum clade. This result showed the polyphyletic origin of Foph and the closer relationship with ff. spp. related to Solanaceous hosts. We also identified putative LS genomic regions specific to virulent strains of Foph, that could be related with pathogenicity and host specificity, as they contain the homologous effectors previously reported for Foph MAP5and eight new putative effector genes identified in this study. We mapped the Foph RNAseq dataset previously reported against the candidate effectors and identified that these novel effectors are expressed during host infection. These results suggest that the new effector candidates, could have a putative role in virulence. Additionally, we tested the presence of the novel effectors by PCR amplification in a panel of 36 F. oxysporum isolates (including MAP5), associated to the cape gooseberry crop and identified that the presence of novel candidates was unique to Foph related strains, suggesting host specificity toward cape gooseberry plants. Furthermore, we conducted a phylogenetic analysis using the EF1alpha sequences available for this panel of F. oxysporum isolates. This result reflects the polyphyletic origin of Foph and suggests the independent acquisition of the novel putative effectors in at least two divergent clades of Foph related strains.
Materials and Methods
Fungal Material
The strains Foph_MAP5, Foph_13, Foph_36, Foph_72, and Foph_117, classified as F. oxysporum, were selected from a collection of 136 fungal isolates obtained from cape gooseberry crops, based on their ability to cause wilting symptoms (Foph_MAP5, Foph_13, and Foph_117), non-pathogenic (Foph_36) on susceptible cape gooseberry plants.
DNA Extraction
The five fungal strains were reactivated in PDA media and incubated at 28°C for 8 days or until enough biomass was obtained for DNA extraction. The DNA of MAP5 strain (Foph), used for genome sequencing, was obtained using the ZR Fungal/Bacterial DNA kit from Zymo research®, according to the protocol proposed by the manufacturer. The DNA of the remaining F. oxysporum isolates used in this study, was extracted from 100 mg of the mycelia, using the cetyltrimethylammonium bromide (CTAB) protocol modified for fungal DNA (Zhang et al., 2010). The quality and DNA concentration using both methodologies were verified in 1% agarose gel using the 1 Kb Plus DNA Ladder (Invitrogen®) and also by Nanodrop DNA/RNA Quantification system.
Foph Genome Sequencing and Assembly
Libraries were generated from purified DNA with the Illumina Nextera XT DNA Sample Preparation Kit (San Diego, California, United States). The resulting libraries were verified in the Bioanalyzer Agilent 2100, using a DNA-HS chip and adjusted to a final concentration of 10 nM. Libraries were then amplified. The sequencing of the libraries was performed using the TruSeq PE Cluster V2 (Illumina, San Diego, CA) kit generating 250 bp pair-end reads in the Illumina MiSeq platform (San Diego, CA, United States) at the Genetics and Antimicrobe Resistance Unit of El Bosque University.
The quality of the reads produced was verified with the software FastQC (Andrews, 2015), and reads were trimmed using the software Trimmomatic (Bolger et al., 2014), with the following parameters “LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:45.” Additionally, adaptor sequences and reads less than 25 bp in length were filtered and removed using the scripts fastq_quality_trimmer and fastq_quality_filter of the FASTX-toolkit platform1. A primary de novo assembly was performed with the pair-end reads overlapped into contigs, using the software, MEGAHIT v1.2.9 (Li et al., 2015), Newbler v 2.0.01.14. (454 Life Sciences), Velvet (Zerbino and Birney, 2008), and SPAdes, v 3.5.0. (Illumina, San Diego CA). The Quality Assessment Tool for Genome Assemblies (QUAST) software (Gurevich et al., 2013), was used to determine the best genome assembly based on the highest N50 parameter.
Gene Prediction and Annotation
Ab initio gene models for the genome sequence of Foph, were predicted using the software Augustus (Stanke and Morgenstern, 2005), using the gene prediction model for Fol4287, as species gene model with the following parameters “–strand = both” and “–uniqueGeneId = true,” other parameters were used with the default settings. The resulted transcripts were annotated by combining predictions using the software HMMER 3.0 (Finn et al., 2011), with the PFAM protein database. The functional annotation of the transcripts was performed with the software eggNOG-mapper v4.5.1 (Huerta-Cepas et al., 2017). Gene models were corroborated with the Foph in planta RNAseq database reported in our previous study.
Comparative Genomics Analysis
The comparative genomic analysis was carried out to establish the gene composition similarity and conserved patterns within phylogenetic clusters of 22 genomes of different F. oxysporum ff. spp. (including Foph), and the genome sequence of F. fujikuroi (Supplementary Table S1). To identify these gene clusters, we used the anvi’o software (Eren et al., 2015), following the pangenomic workflow described before (Delmont and Eren, 2018). In brief, this pipeline generates a genome database that stores DNA and amino acid sequence information of all genomes. Gene clusters were identified by calculating the similarities of each amino acid sequence in every genome against every other amino acid sequence using Blastp (Altschul et al., 1990) and finally hierarchical clustering was performed using the Euclidean distance and Ward clustering algorithm. The distribution of these gene clusters across the genomes was plotted using the anvi’o visualization tool. To reconstruct the phylogenetic relationship of these genomes, the single copy orthologous genes (SCG) were extracted from the pangenome database for all genomes, and a phylogenomic tree was generated using the FastTree 2.1 software (Price et al., 2010) as a component of the anvi’o pipeline. To root the tree, we used the genome sequence of F. fujikoroi as an outgroup (Supplementary Table S1).
Identification of Effector Genes in Foph
To validate the presence of homologous effectors (i.e., SIX, Ave1, and FOXM_16303), identified in our previous Foph-MAP5 transcriptomic analysis, we carried out two search strategies of the homolog effectors in a database that included the 22 genome sequences of the ff. spp. of F. oxysporum used for comparative genomics of Foph. The first strategy consisted in a tBlastn search. The hits with an e-value <0.0001 and identity higher than 50%, in the 50% of the length of the sequence query, were selected for further analysis. In the second strategy, a Blastx search was performed to identify all possible putative peptides of the homologous effectors in the F. oxysporum genome database. The best hits with an e-value <0.0001 were selected for further analysis.
To identify de novo candidate effector genes in Foph, the secretome and effectorome were predicted from the proteome of Foph_MAP5, using the software SignalP v5.0 (Almagro Armenteros et al., 2019) and EffectorP v2.0 (Sperschneider et al., 2018), respectively. In order to discard homologous sequences in other ff. spp., The two BLAST search strategies mentioned above were performed using the protein sequences positive for signal peptide and effector structure (i.e., <300 aa in length and cysteine rich) as a query. An additional search of Miniature Impala Transposable Elements (mimp) was performed in the UTR of the transcripts predicted of Foph, with the regular expression “NNCAGT[GA][GA]G[GAT][TGC]GCAA[TAG]AA,” using a customized Perl script as described by Schmidt et al. (2013) and van Dam and Rep (2017), to determine whether or not the novel candidate could correspond to SIX type genes.
Molecular Characterization of Foph Isolates and PCR Analysis of Candidate Effectors
A panel of 39 F. oxysporum isolates (including the highly virulent Foph_MAP5, 13, 72, 117 and the non-virulent Foph_36), derived from the collection fungal collection mentioned above were selected based on their ability to cause wilting symptoms on susceptible cape gooseberry plants (Supplementary Table S2). The EF1a gene of Fol (GenBank XM_018381269), was used as a molecular marker to characterize the Foph isolates to species level. EF1a sequences for seven out of 39 isolates (including MAP5) were obtained from the GenBank, while the EF1a sequences of Foph_36, 72, 117 were predicted from their genome assemblies (Supplementary Table S2). For the remaining 28 isolates, a fragment of the EF1a gene was amplified and sequenced using the primers reported by Imazaki and Kadota (2015). PCR reactions were conducted with Taq DNA Polymerase (InvitrogenTM, Carlsbad, CA, United States), in a 25 μL reaction volume. The PCR reaction consisted of 0.25 μL Taq Polymerase, 2.5 μL of 10X buffer (InvitrogenTM, Carlsbad, CA, United States), 0.16 μM of each primer, 0.16 mM of dNTP mix, 2 mM MgCl2 and 25 ng of template DNA. PCRs were carried out with an initial denaturing step at 95°C for 2 min followed by 30 cycles of denaturing at 95°C for 45 s, annealing of primers at 59°C (62°C for Forl_155.3) for 45 s and primer extension at 72°C for 45 s. The PCR was completed by a final extension at 72°C for 10 min. PCR products were purified using a QIAquick PCR Purification Kit (Qiagen) and then sequenced by Sanger platform.
EF1a sequences obtained from 27 out of the total of 39 Foph related isolates included in this study, were submitted to the GenBank with accession numbers (MT738937-MT738958 and MW233573-MW233575) and a total of 30 EF1a sequences of Foph related strains were aligned (MUSCLE method) using MEGA version 7 (Kumar et al., 2016). The corresponding EF1a sequence from the selected F. oxysporum ff. spp. mentioned above, were also included for comparison. Phylogenetic analysis was performed using the software BEAST (Bayesian Evolutionary Analysis Sampling Trees) v 2.6.1 (Bouckaert et al., 2019), with default settings. The resulting phylogenetic trees were visualized using the Interactive Tree of Life (iTOL) v4 (Letunic and Bork, 2019). The EF1a from F. fujikuroi was used as an outgroup. To corroborate the presence of the new effectors in the Foph related strains, specific primers for the new candidates were designed and used for PCR amplification (Supplementary Table S3), using the same conditions as mentioned above. DNA from Colombian strains of Fol, Foc R1 and TR4, were provided by Dr. Mauricio Soto (AGROSAVIA), and used as a control for amplification.
Results
Foph Genome Sequencing and Assembly
The genome sequence of four pathogenic strains of Foph (i.e., MAP5, 13, 72, and 117 and one non-virulent named Foph_36 in cape gooseberry plants, were assembled from 250 bp paired end reads Illumina MiSeq. The genome of Foph_MAP5 was assembled into 1,856 contigs with a total size of 44.9 Mb. This genome assembly is smaller, compared to the remaining assemblies of Foph strains obtained in this study ∼46–48 Mb, other Illumina genome assemblies available for Fophy strains (infecting a close related host Physalis philadelphica, known as “husk tomato or tomatillo”), two solanaceous specific strains (Fomel_002 and Fonic_003), and the nearly complete genome assemblies of Fol4287 and FoC_Fus2, included here as reference genomes (Table 1; Ma et al., 2010; van Dam et al., 2016, 2017a; Armitage et al., 2018). The difference in size and contig number in the assembly of Foph_MAP5, might be a consequence of genome fragmentation due to the sequencing based on short reads (250bp) used in this study. Nevertheless, the predicted gene content of Foph_MAP5 (14,897 transcripts), is similar to other Illumina genome assemblies of different ff. spp. of F. oxysporum, available in the GenBank (Table 1 and Supplementary Table S1).
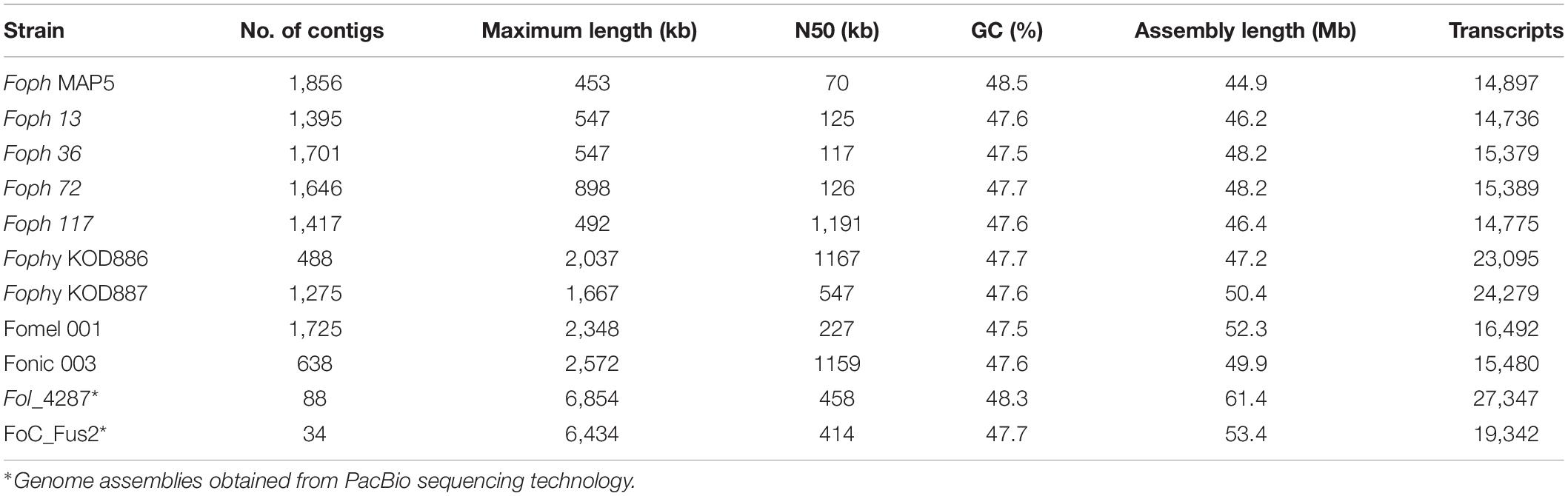
Table 1. Foph genome assembly statistics, compared to other Illumina genome sequences of F. oxysporum strains infecting Solanaceous hosts and two nearly complete genome assemblies of Fol and FoC.
Comparative Genomics of Foph With Other Isolates of Fusarium oxysporum
A total of 14,897 transcripts were predicted from the genome assembly of Foph_MAP5 (used here as a reference of Foph pathogenic strains), from which 14,140 have an orthologous counterpart in the genomes of F. oxysporum compared in this study (Table 1). Using the anvi’o pipeline for pangenome analysis, a set of the single copy orthologous genes (SCG) present in the 22 F. oxysporum genomes were extracted to reconstruct their phylogenetic relationship. We used this phylogenetic reconstruction to test whether Foph could be related to Fophy (i.e., other Physalis infecting strains), or might be grouped in a lineage of strains that infect Solanaceous hosts. The phylogenomic tree showed that Foph shared the same clade with Fonic and Fol_R3 and is closely related to Forl, Fo47 (both strains associated to the tomato crop) and more distantly related to Fophy_KOD886, while the remaining solanaceous infecting strains are grouped on a different clade (Figure 1A), reflecting the polyphyletic origin of infecting strains of F. oxysporum and their host specificity. We also performed a comparative analysis using the SCG shared between Foph and the remaining 21 genomes of F. oxysporum ff. spp., This analysis showed that the majority of Foph SCG (∼14 K), are syntenic with the core chromosomes of Fol (used here as the reference genome sequence of F. oxysporum species). The syntenic SCG might correspond to the core genome of Foph, while the remaining ∼0.5 K of Foph SCG, could correspond to transcripts that are not present in any cluster and their contigs could be part of the LS genomic regions specific of Foph (Figure 1B).
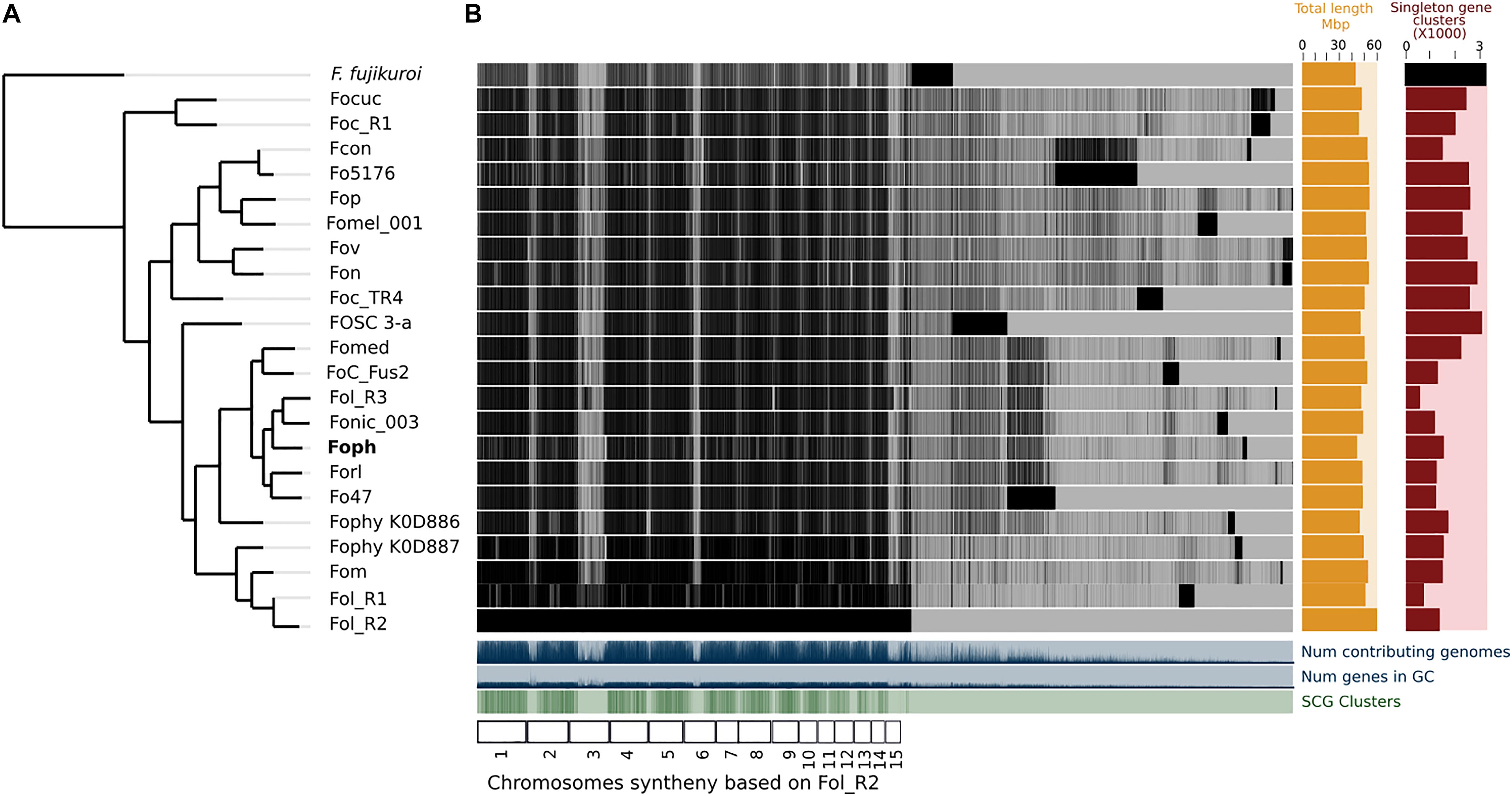
Figure 1. Comparative genomics between Foph and 21 F. oxysporum ff. spp. (A) The phylogenomic tree was inferred with the single copy orthologous genes of the 22 genomes of F. oxysporum used in this analysis. The amino acid sequence of the translated genes was concatenated, and the final alignment consists of a total of 41,84,361 amino acid positions. The phylogenetic tree was constructed using FastTree 2.1. F. fujikoroi (IMI58289), was used as outgroup. (B) Pangenomic analysis of F. oxysporum, showing the core genome the species complex and single copy orthologous genes, possibly forming the LS genome for each forma specialis. The F. oxysporum pan-genome was generated using the anvi’o pangenomic workflow.
Effector Homologous Are Confirmed in Foph Pathogenic Strains
In our previous study, eight homologous effectors were identified in Foph by in planta RNAseq mapping analysis with the LS regions of Fol. Here, we performed a combination of Blastp and Blastx searches of the known SIX effectors and Ave1 effectors in the genome assemblies of the five strains of Foph and the genome sequences of 21 F. oxysporum ff. spp., used for comparative genomics (Supplementary Table S1). This result showed the widespread presence of SIX homologous in different ff. spp. of F. oxysporum and confirmed the presence of highly identical Fol and Fomed homologous effectors with identities from 87 to 100% in their counterparts of Foph virulent strains MAP5, 13, 72, and 117 and absent in the non-pathogenic strain Foph_36 (Table 2). Interestingly, we compared the genome assemblies of Foph strains using the anvi′o software and showed that the putative effectors are located in genomic regions unique to Foph virulent strains (Figure 2), which could correspond to the LS regions of Foph. Furthermore, we also identified a highly identical putative homologous transcript of the Fol SIX13 effector in the genomes Foph virulent strains. This prediction was manually confirmed as the corresponding transcript of SIX13, was fragmented into two contigs (ctg_1292 and ctg_1535) in the genome of Foph_MAP5 (Table 3). We used the Foph_MAP5 prediction of the SIX13 transcript to search the corresponding homologous sequence in the remaining genome sequences of Foph generated in this study and confirmed the presence of SIX13 homolog only in the Foph virulent strains (Figure 2 and Table 2).
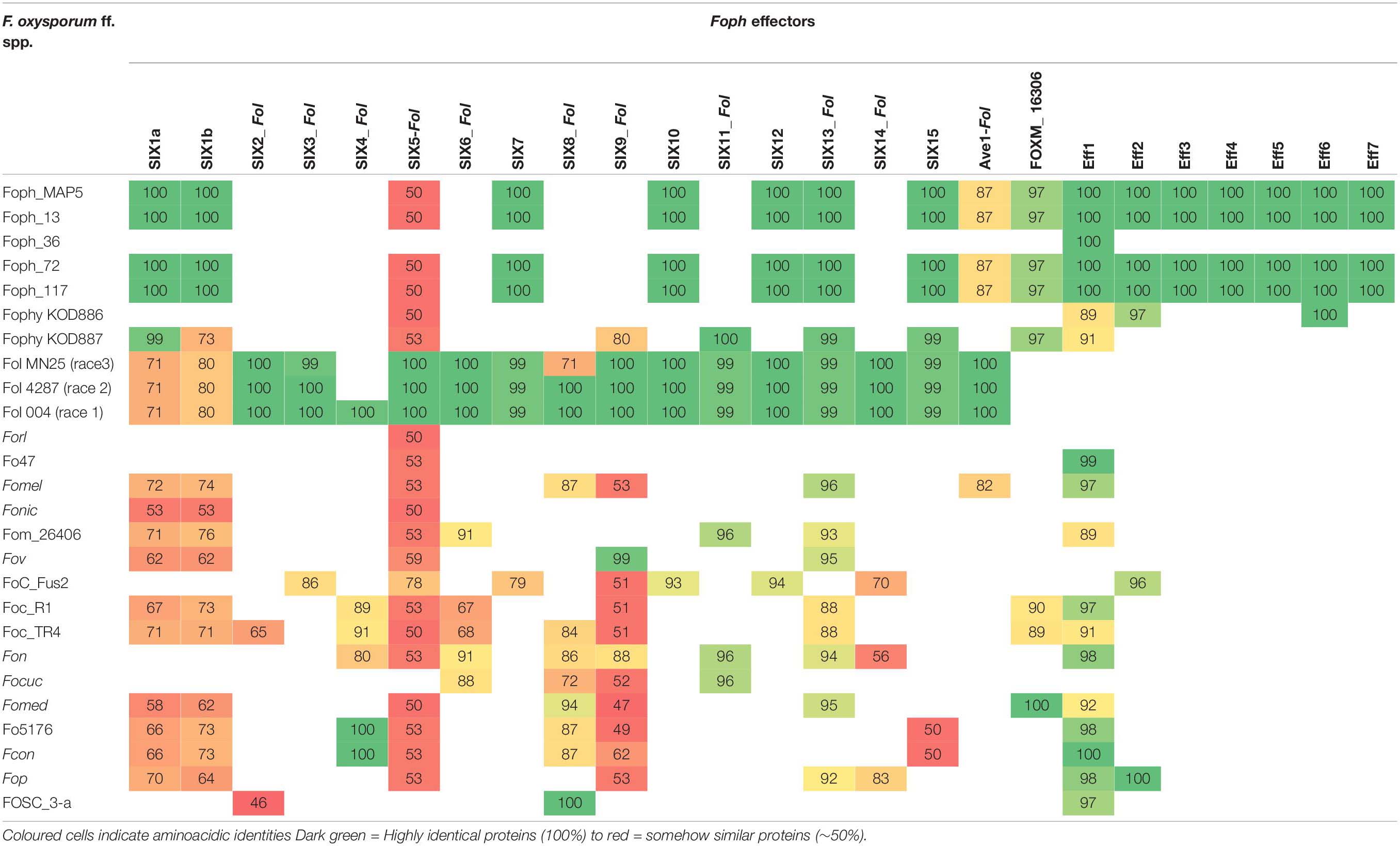
Table 2. tBlastn identities of Foph effectors compared against the F. oxysporum species complex WGS databases.
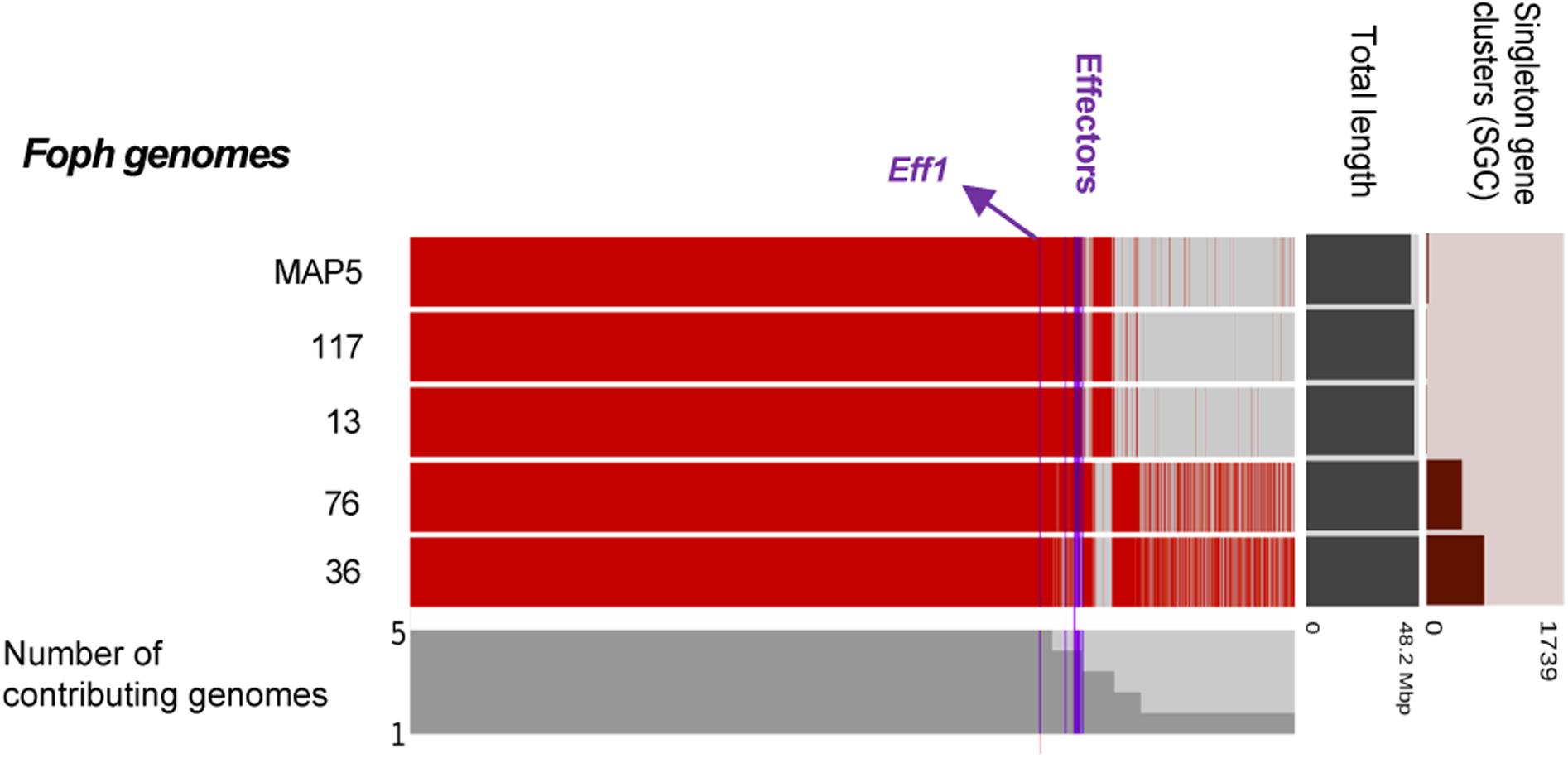
Figure 2. Comparative genomic analysis of Foph isolates. The comparative genomic analysis of four pathogenic strains, Foph_MAP5, Foph_13, Foph_72, and Foph_117, and one non-pathogenic strain, Foph 36, was conducted to establish the possible lineage-specific regions based on the patterns of gene sharing. The cluster of shared and unique genes are shown by the red lines. Gene clusters are organized by their presence in all five genomes to those present in only one genome, indicated by the gray bars. The horizontal bar graphs to the right represent the total length of the genome and the number of singleton gene clusters for each genome. Blue vertical lines show the location of the SIX effectors as well as the putative novel effectors. The majority of putative and known effectors are only present in the gnomes of pathogenic strains, while effector Eff1 is present in all Foph genome assemblies, including the non-pathogenic Foph_36.
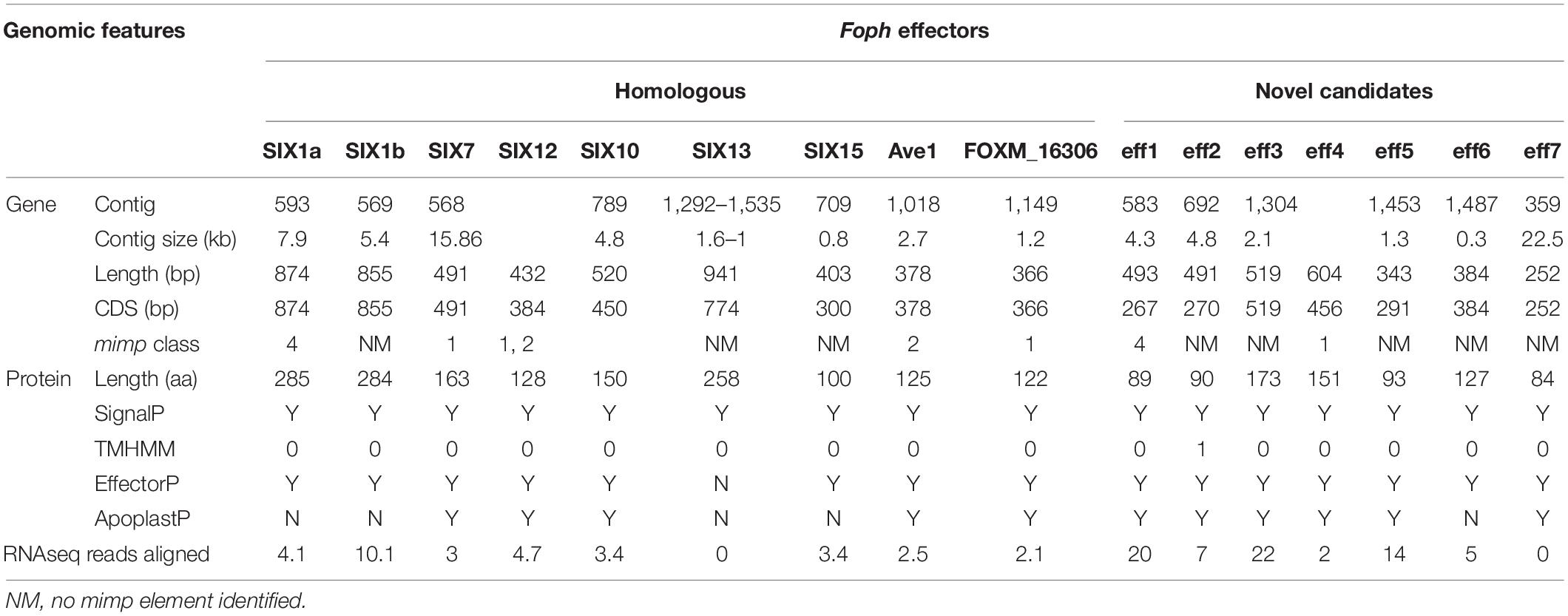
Table 3. Genomic analysis of the effectors identified in Foph_MAP5.
These results also confirmed that the Fol effector gene cluster formed by the SIX7, 12 and 10 (Ma et al., 2010; Schmidt et al., 2013) and partially identified in Foph by in planta transcriptomics (Simbaqueba et al., 2018), is entirely conserved in the genome of Foph. SIX7 and SIX12 homologous are both present in the same contig (ctg_568) while SIX10 is located in another contig (ctg_789) of the Foph_MAP5 genome assembly (Table 3). Thus, we manually inspected the sequences of these contigs and found that both contigs are overlapped by a sequence segment of 22 bp at the proximal 5′ end of the ctg_586 with the distal 3′ end of the ctg_789. This overlapped segment of both contigs correspond to a mimp class 2 sequence in intergenic region between SIX10 and SIX12. The Foph effector gene cluster is 4.7 kb in length and is similar to that formed by the same homologous effectors in Fol (5.2 kb), including the intergenic regions with the approximate same length as Fol (1.8 kb between SIX7 and SIX12 and 1.4 kb between SIX12 and SIX10, respectively), and three mimp elements that flank the effector gene cluster reported by Schmidt et al. (2013) in Fol (Figure 3).
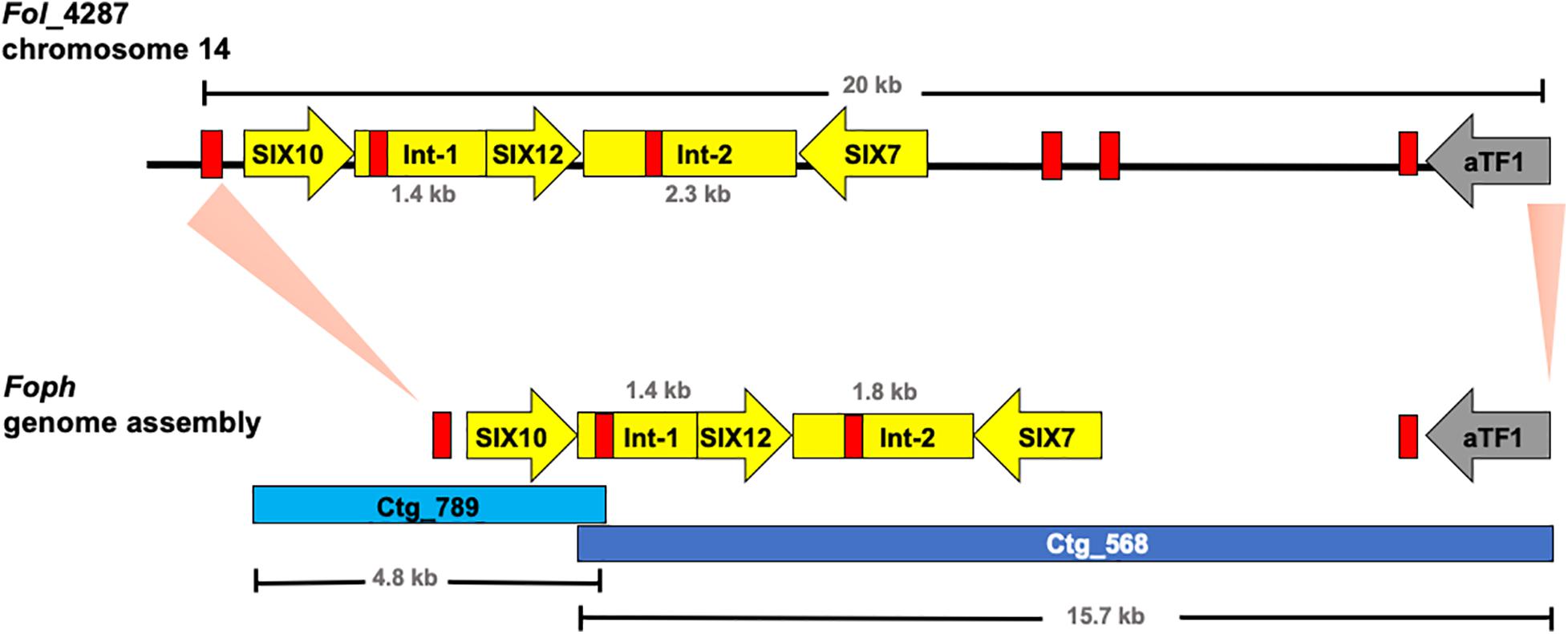
Figure 3. Graphical representation of a 20 kb segment of the chromosome 14 in Fol, containing the cluster of effectors SIX10, 12 and 7, and the aTF1 gene (FOXM_17458) (upper part). The chromosomal segment is conserved in the Foph genomic region (shown by orange pale triangles), corresponding to the overlapped Contigs 568 and 789 (Bottom part), suggesting a highly possible horizontal transfer of a chromosomal segment of 20 kb between both ff. spp. Int-1 = conserved intergenic region between SIX10 and SIX12. Int-2 = conserved intergenic region between SIX12 and SIX7. Red blocks represent Mimp transposable elements flanking the cluster of effector genes shared between Fol and Foph.
Further inspection of the ctg_568 in Foph_MAP5, also confirmed the presence of another highly conserved homologous gene (FOXG_17458) between Foph and Fol, including the corresponding mimp class 1 element in the 5′ UTR (Figure 3). The transcript FOXG_17458 in Fol, encode a transcription factor of the family aTF1—FTF1 (van der Does et al., 2016), and is located 9 kb away from the ORF of the SIX7 (Schmidt et al., 2013). Intriguingly its homologous counterpart presented in the Foph genome is located 7 kb away from the SIX7 ORF (Figure 3). Although, there is a difference of 2 kb that includes two mimp elements presented in Fol, comparing the above-mentioned intergenic regions in both genomes. This finding points out to a probable horizontal transfer event of a chromosomal segment of at least 20 kb in length between Fol and Foph. In Fol, SIX15 is a non-annotated transcript and is located 55 kb away from the aTF1. This chromosome region includes other four annotated transcripts: FOXG_17459, FOXG_17460, FOXG_17461, and FOXG_17462. Thus, we performed a Blastn search using this sequence of 55 kb from Fol as a query and compared with the Foph-MAP5 genome assembly, in order to test whether an extended sequence of the chromosome 14 of Fol might be conserved in Foph. However, no additional chromosomal segment shared between Fol and Foph was identified by comparing both genomic sequences.
Novel Candidates for Effector Genes in Foph
We identified novel effector genes in the Foph_MAP5 genome, by combining the sets of proteins from the secretome and effectorome. We predicted a total of 1,495 secreted proteins, forming the secretome of Foph_MAP5, from which 276 were determined to be effectors, named herein as “Foph effectorome.” Six transcripts of the Foph effectorome (named Eff2 to 7), were identified as putative novel effectors, due to their specificity to Foph virulent strains and their lack (i.e., Eff2, Eff3, Eff4, and Eff7) or low similarity (Eff5 and Eff6) to any protein reported in the public databases (Table 2). Additionally, mimp elements were identified 624and 430 bp upstream from the transcripts Foph_eff2 and Foph_eff5, respectively (Table 3).
The candidate effector Eff1, was the only predicted putative effector present in all the sequenced strains, including the non-pathogenic Foph_36 (Figure 2). In addition, Eff1 showed significant tBLASTn hits with different F. oxysporum ff. spp., including another non-pathogenic strain Fo47. Therefore, this transcript could be excluded as a novel effector gene. The putative effectors Eff3 and Eff4 are clustered in the contig_692 at 700 bp of distance approximately between them in the genome of Foph_MAP5. Furthermore, we predicted a transmembrane domain for protein encoded by Eff3 (Table 3), suggesting a cellular localization and with a possible different function from a secreted protein. Additionally, we performed an RNAseq mapping against the ORF of the novel candidate effectors and found that all six putative effectors are expressed in Foph_MAP5 during cape gooseberry infection at 4 dpi. In this analysis, we also included the homologous of SIX effectors and the homologous transcripts of EF1alpha, β-tubulin and Fusarium extracellular matrix 1 (FEM1) in Foph_MAP5, as housekeeping genes for expression controls. We found that Eff2, Eff4, Eff6, and Eff7, showed higher expression compared to the rest of the transcripts analyzed (Figure 4). Interestingly, Eff2, Eff4, and Eff6, showed higher expression, compared to all three-housekeeping gene controls. This in planta expression evidence of the putative novel effectors, together with their specific presence in the genomes of virulent strains of Foph, suggest that these genes could be involved in Foph pathogenicity toward cape gooseberry hosts.
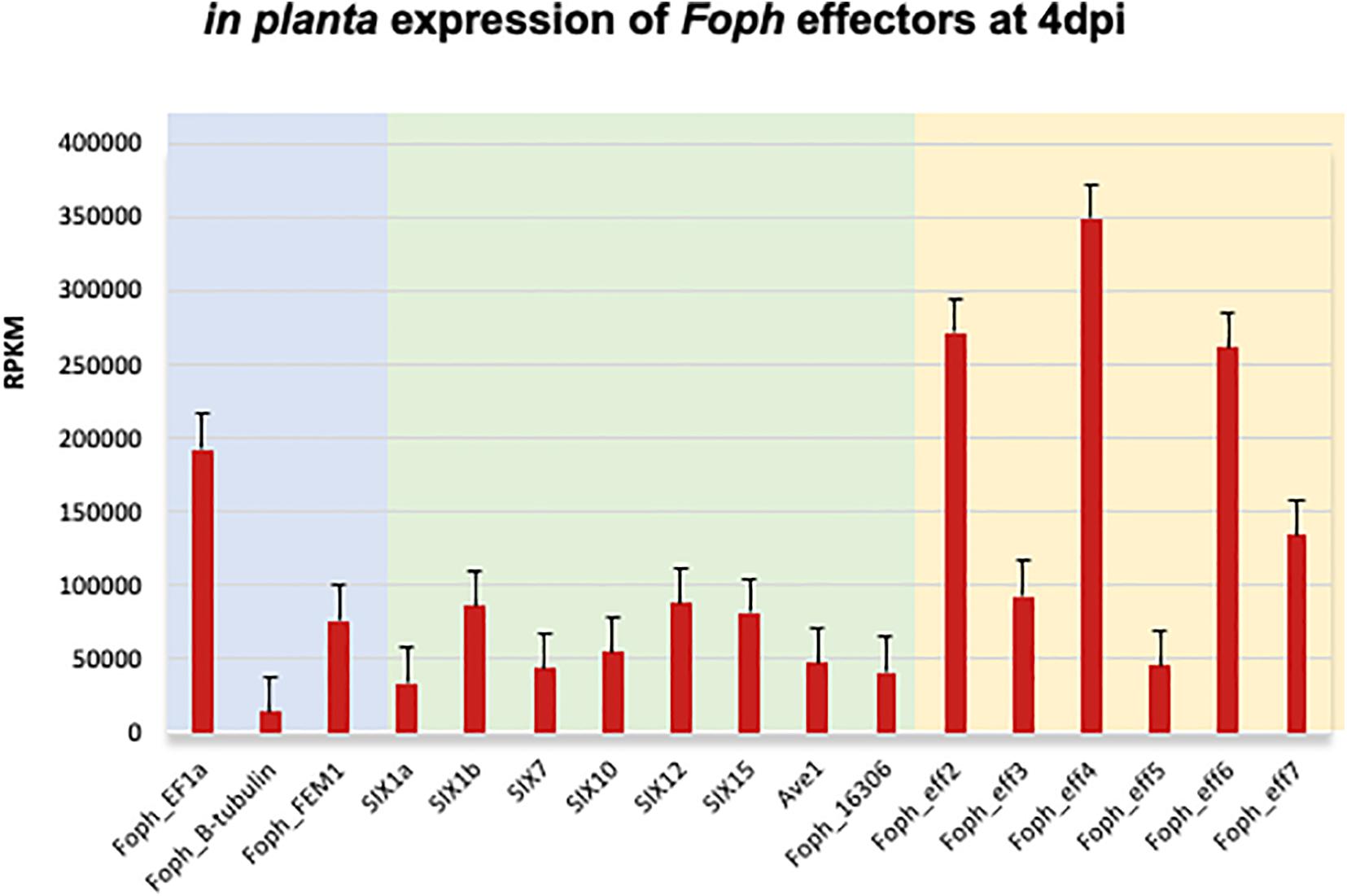
Figure 4. Expression analysis of the effectors identified in the genome sequence of Foph_MAP5, using the RNAseq data form cape gooseberry susceptible plants inoculated with Foph at 4 dpi, reported in our previous in planta transcriptomic analysis. Pale blue panel indicate the genes translation elongation factor alpha (EF1a), tubulin B-chain (B-tubulin), and Fusarium Extracellular Matrix 1 (FEM1), to use as constitutive expressed control genes of Foph during host infection. Pale green indicates the expression of the homologous effectors identified in Foph. Pale yellow indicates the expression of the newly identified effectors in Foph. Six out of seven new effector candidates are expressed during cape gooseberry infection with a higher expression of eff2, eff4, and eff6, compared to the rest of the effectors analyzed. RPKM, reads per kilobase per million of mapped reads. Scale bars indicate standard error.
Novel Effectors Are Present in F. oxysporum Isolates Associated to the Cape Gooseberry Crop
In order to test whether the candidate effectors genes could be used as potential molecular makers for Foph identification in diagnostic strategies, we performed a preliminary screening of the novel candidate effectors by PCR amplification in a panel of 37 F. oxysporum isolates (including Foph-MAP5), obtained from cape gooseberry crops. Thirty-three of them (including the sequenced strains MAP5 and 13), have been classified as pathogenic due to their ability to cause wilting symptoms on a susceptible cape gooseberry genotype, while the remaining four isolates (including the sequenced strain Foph_36), do not cause wilting symptoms on cape gooseberry plants. Thereby, classified as non-pathogenic strains of Foph (Supplementary Table S2). The screening also included DNA isolated from Fol, FocR1, and FocTR4 strains, as control for amplification. We found amplification for all candidates in the majority of F. oxysporum isolates associated with cape gooseberry, including pathogenic and non-pathogenic isolates (Supplementary Table S2 and Supplementary Figure S1). The putative novel effector Eff2 was identified by PCR amplification in Foph_36 (Supplementary Figure S1), but was absent in our genome analysis (Figure 2 and Table 2), contradicting the molecular identification. Although the genomic evidence of the five sequenced strains Foph suggest that the putative novel effectors could be specific to virulent strains of Foph, the PCR screening in the panel of Foph pathogenic and non-pathogenic isolates, showed a more widespread presence of these putative effectors (Supplementary Table S2). Interestingly, we did not identify the presence of the novel effectors Eff3 to 7 in the control strains Fol, FocR1 and FocTR4. Nevertheless, together, these results, would suggest that five of the six putative novel effectors could be specific for F. oxysporum strains associated to the cape gooseberry crop. We also conducted a molecular characterization using the EF1alpha sequence in 30 out of 37 Foph and the additional sequenced pathogenic strains Foph 72 and 117, in order to test whether these isolates associated to cape gooseberry plants, might originated from a single linage. The phylogenetic tree showed that Foph isolates are grouped together in two different lineages, suggesting their polyphyletic origin. Additionally, we found that the pathogenic strains MAP5, 117 and 13 are grouped together in the clade 1 and intriguingly, the non-pathogenic strain Foph_36 was grouped on a different clade out of Foph (Figure 5).
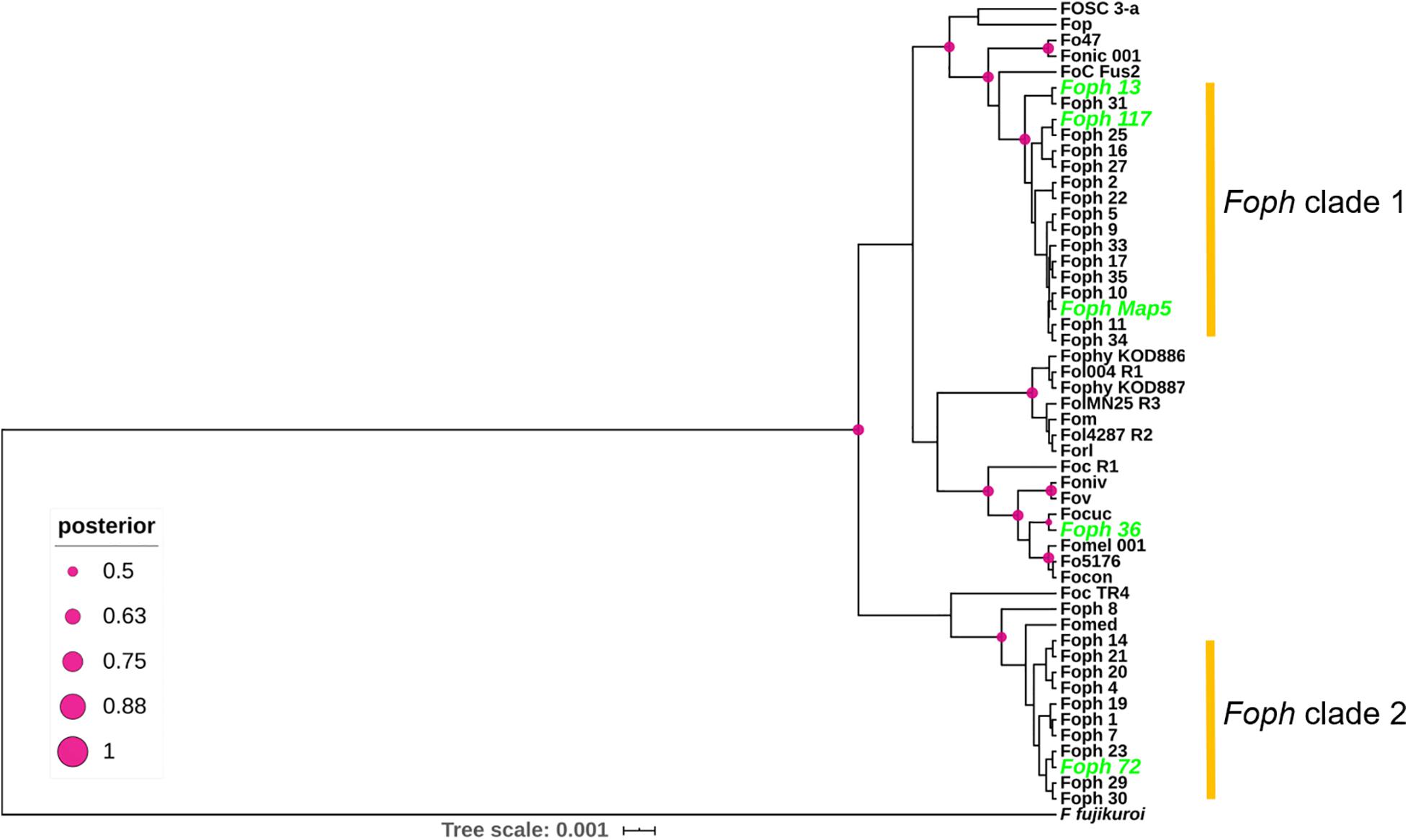
Figure 5. Phylogenetic tree of a partial sequence of the EF1a gene from the genome sequences of 24 ff. spp. of F. oxysporum (Supplementary Table S1) and 29 F. oxysporum isolates obtained from cape gooseberry crops (Supplementary Table S2). The phylogenetic analysis was conducted using BEAST. Node shapes indicate the bootstrapping support, indicated as Bayesian posterior probabilities. The scale bar indicates time in millions of years.
Discussion
Foph Genome and Phylogenetic Relationship With Other ff. spp.
In Colombia, the cape gooseberry crop is severely affected by pathogenic strains of Foph, with losses of nearly 90%. In this pathosystem, SNPs associated to resistant cape gooseberry genotypes, Foph pathogenic strains and homologous effectors have been identified (Osorio-Guarín et al., 2016; Simbaqueba et al., 2018). However, there is a need to implement genomic approaches to corroborate these findings and to identify new sources associated to the interaction between Foph and cape gooseberry. These approaches could be used in the development of disease management strategies and plant breeding programs in the cape gooseberry crop. Here we sequenced and assembled the genomes of four virulent strains of Foph (MAP5, 13, 72, and 117) and one non-virulent strain (Foph_36), aiming to identify novel candidates for effector genes unique to virulent strains, that could be characterized in further studies and implemented in diagnostic strategies. Comparative and functional genomics of F. oxysporum that infect cucurbit species, suggested that their host range could be determined by the close phylogenetic relationship associated to their homolog effector gene content (van Dam et al., 2016, 2017b). This hypothesis is supported by additional evidence on the formae speciales radicis-cucumerinum (Forc) and melonis (Fom), showing that a syntenic LS chromosome region is highly related to the expansion formae speciales range (van Dam et al., 2017b; Li et al., 2020a). Recent genome analysis with a chromosome-scale assembly of the brassicas infecting f. sp. Fo5176, showed a similar pattern of phylogenetic relationship possibly associated to the expansion of their host range (Fokkens et al., 2020). Here, we performed comparative genomics using the Foph_MAP5 genome assembly in order to test whether a set of available genomes of Solanaceous-infecting formae speciales including Foph, could show a similar phylogenetic related pattern. Nevertheless, our analysis showed that the tested strains have a different ancestry (Figures 1A, 5), despite the close phylogenetic relationship of Foph with tomato infecting ff. spp. Fol-R3, Fo47, Forl, and tobacco Fonic_003. Resequencing of the genomes including Foph, Fophy, Fonic, and Fomel, using long reads, will help to gain a deeper understanding of the phylogenetic relationship among Solanaceous-infecting ff. spp.
Confirmation of Homologous and Identification of New Putative Effectors
Homologous of Fol SIX genes have been identified in other ff. spp. of F. oxysporum and other Fusarium species (Meldrum et al., 2012; Thatcher et al., 2012; Li et al., 2016; Rocha et al., 2016; Schmidt et al., 2016; Taylor et al., 2016, 2019; van Dam et al., 2016, 2017a; Williams et al., 2016; Armitage et al., 2018; Simbaqueba et al., 2018). The presence of the SIX homologous might be a consequence of horizontal transfer of genes or segments of pathogenicity chromosomes between different strains of F. oxysporum and/or fungal phytopathogenic species. In our previous study, we identified homologous of the SIX, Ave1 and FOXM_16306 effectors, analyzing an in planta RNAseq of Foph. Despite the fragmentation of this genome assembly (i.e., no scaffolds/chromosomes scale), we corroborated the presence of complete sequences of the homologous effectors SIX, Ave1 and FOXM_16306, in the genome sequences of Foph virulent strains, contained in different contigs that could correspond to the LS genomic regions of Foph (Figure 2 and Tables 2, 3).
We also found a homolog transcript of the Fol SIX13 in the genome of Foph_MAP5, fragmented into two contigs and corroborated in the other Foph sequenced genomes of virulent strains. In Foph_MAP5, this homolog was not expressed at 4 dpi and therefore, it was not identified in our previous transcriptomics study. SIX13 homologous are present in legume, cucurbits, musaceous and solanaceous infecting ff. spp. of F. oxysporum (van Dam et al., 2016; Williams et al., 2016; Czislowski et al., 2018). The later mentioned ff. spp., are highly identical at the protein level (96% in Fomel and 99% in Foph and Fophy, respectively) (Table 2). In cucurbits infecting ff. spp. of F. oxysporum, a suit of effectors was found to be associated with host specificity (van Dam et al., 2016). Thus, the highly identical SIX13 homologous in the Solanaceous-infecting ff. spp., could be related to their specificity for these group of host species. Moreover, the majority of the SIX genes in Fol are located on the chromosome 14 (i.e., pathogenicity chromosome), except for SIX13, which is found in the LS chromosome 6 (Schmidt et al., 2013). Similarly, SIX13 corresponding homologous of Fomed and Foph are located on putative LS regions (Williams et al., 2016; Figure 2). In Foc, SIX13 homologous, have been associated to the differentiation of TR4 and R4 and are currently used in molecular based diagnostic of TR4 in banana crops (Carvalhais et al., 2019). Together, this evidence suggests that SIX13 could play a role in pathogenicity or host specificity. Future functional analysis of on Foph-SIX13 is necessary to confirm this hypothesis.
Furthermore, we performed a manual inspection of the contigs 568 and 789 of the Foph_MAP5 genome and confirmed the presence of a highly conserved chromosomal segment of 20kb of Fol that includes a cluster of physically linked effector genes (SIX7, SIX10, SIX12, and extended transcription factor αTF1). This shared region also included their corresponding flanking mimp elements (Figure 3; Schmidt et al., 2013; Simbaqueba et al., 2018). This finding suggests a highly probable horizontal acquisition of an entire genomic segment of 20kb from an ancestor of Fol or Foph. Miniature impala (mimp) transposable elements (TEs), have been identified in the genome sequences of different phytopathogenic fungi of the Fusarium genus (Schmidt et al., 2013; van Dam and Rep, 2017). In F. oxysporum, mimp elements have been associated to the gain or loss of effector genes, presumably acting as an evolutionary mechanism of emergence of new phytopathogenic strains (van Dam et al., 2017b). The presence highly identical mimp elements, flanking the homologous effector gene cluster in both Fol and Foph (Figure 3), suggests that these TEs could play a role in the lateral transference of this homolog genomic region between Foph and Fol.
Functional analysis of SIX effectors in Fol, showed that mutant strains with a large deletion (0.9 Mb) of chromosome 14, including the candidate effector genes SIX6, SIX9, and SIX11 did not show any loss of virulence compared to wild type Fol on tomato plants (Vlaardingerbroek et al., 2016). Recent evidence revealed by another set of Fol mutant strains with chromosomal deletions that include the SIX10, SIX12, and SIX7 gene cluster, showed no loss of virulence on tomato plants (Li et al., 2020a). These findings indicate that the genes located in these chromosomal segments (including the SIX genes with homologous in Foph), could be dispensable for pathogenicity, while the remaining segments could be sufficient for tomato infection (Vlaardingerbroek et al., 2016; Li et al., 2020b). Although neither of the SIX7, SIX10, and SIX12 effector genes have a role in Fol virulence, the presence of the highly identical homologous between Fol and Foph, suggests that this segment could be undergoing adaptation to another environment (i.e., different host plant). Therefore, it might be possible that SIX7, SIX10, and SIX12 have a role in Foph pathogenicity. Future investigation about the function of this conserved genomic region between these two Solanaceous-infecting ff. spp., is required. Crossed pathogenicity assays inoculating tomato and cape gooseberry with Fol and Foph and knock out of the gene cluster in Foph could be performed to support these hypotheses.
In this study, we confirmed that homologous of Ave1 have been only identified in the solanaceous infecting ff. spp. Fol, Foph, Fomel001 (Table 2) and in the f. sp. gladioli of F. oxysporum (Simbaqueba et al., 2018). Ave1 could also be present in putative conditional dispensable segments on the Foph genome (Table 2 and Figures 1B, 2). The presence of less conserved homologous of Fol including SIX1 and Ave1, which are also located on Fol chromosome 14 (Schmidt et al., 2013), suggests that these effectors may have a different ancestry, via acquisition of different segments of the pathogenicity chromosome at different times in the evolution of Fol or Foph.
In the tomato pathogen Verticillium dahliae, Ave1 is involved in pathogenicity, while there is no evidence that its homolog present in Fol has a role in virulence (de Jonge et al., 2012; Schmidt et al., 2013). Furthermore, Fol-Ave1 is not expressed during tomato infection (Catanzariti, personal communication). Conversely, we found that Foph-Ave1 was expressed during cape gooseberry infection (Figure 4). This finding suggests that Ave1 might have a role in Foph pathogenicity. Therefore, functional analyses are required by generating gene knockout strains in Foph. In both V. dahliae and Fol, Ave1 could act as avirulence factors since they are recognized by the tomato receptor Ve1 (de Jonge et al., 2012). The Ave1 homolog of Foph is highly similar at the protein level to its counterparts in F. oxysporum (Fol, Fomel, and Fogla), and less similar to V. dahliae Ave1 (Table 2; Simbaqueba et al., 2018). The presence of Ave1 in Foph, suggests that the avirulence function of Fol Ave1 might be conserved. This hypothesis needs further investigation e.g., by testing for recognition of Foph Ave1 by tomato Ve1 or a homolog in cape gooseberry.
Novel candidate effectors in F. oxysporum have been reported for other ff. spp., including Fom, Foc_Fus2, Fonar and legume infecting strains (Schmidt et al., 2016; Taylor et al., 2016, 2019; Williams et al., 2016; van Dam et al., 2017a; Armitage et al., 2018), based on the analysis of their genome sequences to identify transcripts that encode for small proteins with a secretion signal peptide and the proximity of mimp to the start codon. Here, we used the predicted transcripts from the genome assembly of Foph_MAP5 to identify novel effectors, based on the effectorome andsecretome repertoires, and the absence or low similarity to any predicted or non-predicted protein sequences compared to F. oxysporum genomes available in the public databases and additional genome assemblies generated in this study of four different Foph strains (Figure 2 and Tables 2, 3). Three highly expressed novel effectors during infection (Foph_eff2, eff4 and eff7), are unique Foph candidate effectors, while the other highly expressed candidate Foph_eff6, have identical homologous proteins in the genomes of Fomel and FoC_Fus2 (Table 2). Furthermore, the homologous counterpart identified in FoC_Fus2 is located in a lineage specific region (Armitage et al., 2018). These findings suggest that Foph_eff6 and its homologous, may have a putative role in pathogenicity and represent a subject for future functional analysis.
Presence of Effectors in Foph Compared to Other ff. spp. of F. oxysporum
Foph pathogenic strains are responsible for the wilting disease that affect cape gooseberry crops in Colombia. Thus, appropriate disease management strategies are needed to be implemented (Barrero et al., 2012). However, the development of those strategies has been largely limited due to the lack of knowledge of the wilting disease caused by Foph, and accurate identification of pathogenic strains. Detection methods based on the use of effector genes as molecular markers are highly desirable for precise identification of pathogenic strains in disease management programs of soilborne pathogens due to their limited sequence diversity between members of the same f. sp. (Rocha et al., 2016; Gordon, 2017), thus providing a solid and sensitive identification of pathogenic strains of soilborne pathogens including F. oxysporum (van Dam et al., 2017a; Carvalhais et al., 2019; Taylor et al., 2019).
Comparative genomics have been performed to design molecular markers based on candidate effector genes and successfully tested for the identification of cucurbit and Narcissus Infecting ff. spp. of F. oxysporum (van Dam et al., 2016; Taylor et al., 2019). In this study, we used the highly conserved novel candidate effectors found by comparative genomics in Foph, to explore their usefulness as potential molecular markers specific for pathogenic strains. The presence of homologous effectors suggests a functional redundancy between different ff. spp. (Taylor et al., 2019). Here, we identified that the candidate novel effector Foph_eff1 has homologous in other ff. spp. (Table 2). We also identified the presence of Eff1 in all tested strains, including the non-pathogenic strain sequenced Foph_36 (Figure 2), Fol and Foc. Thus, the role of Eff1 in pathogenicity may be dispensable due to its presence in different F. oxysporum strains and could be discarded for diagnostic purposes. The remaining novel effectors showed a clear pattern of amplification in F. oxysporum strains associated to the cape gooseberry crop, compared to the highly pathogenic Fol and Foc in tomato and banana respectively (Supplementary Figure S1 and Supplementary Table S2). Although the novel effectors we predicted were only identified in the genome sequences of Foph virulent strains (Figure 2), we did not find an amplification pattern associated in three out of the e four non-pathogenic strains compared in this analysis. A similar inconsistent pattern of presence/absence between pathogenic and non-pathogenic cucurbit infecting strains of F. oxysporum was observed for some of the effectors-based markers developed by van Dam et al. (2017a). These results might be supported by the fact that effectors show limited sequence diversity between strains of the same f. sp. (van Dam et al., 2017a; Taylor et al., 2019). An alternative explanation could be related to the limited number of effectors-based markers identified in this fragmented genome assembly of Foph-MAP5. New markers associated to Foph pathogenicity will be predicted in future studies, enlarging effectorome repertoire by resequencing of the Foph genomes with long reads sequencing technologies as performed for Forc and FoC_Fus2 (van Dam et al., 2017b; Armitage et al., 2018).
Data Availability Statement
The data presented in the study are deposited in the NCBI repository under the BioProject accession number PRJNA640423. GenBank accession numbers: MT738929 –Foph_MAP5 SIX13, MT38930-MT38936-Foph_MAP5 eff1 to eff7, MT738937, MW160862-MW160910 Foph_01, 04, 72 and 117 –eff1 to eff7 and homologues of SIX effectors – MT738958, MW233573- MW233575, EF1a sequences of F. oxysporum strains associated to cape gooseberry crops. “Access to these sequences must be requested to the Ministry of Environment and Development of Colombia. Foph strains used in this work were collected under the framework collection permit No.1466 from 2014 of AGROSAVIA and registered in the National Collections Registry (RNC129) of Colombia.”
Author Contributions
JS planned and carried out the Foph genome analysis, planned the experiments, analyzed the data, created figures, and drafted, wrote and edited the manuscript. ER and DB-D carried out the experiments with Foph isolates. CG obtained funding, planned experiments, contributed and edited the manuscript. AC-Q obtained funding, planned and carried out the Foph genome sequencing, analysis and all bioinformatics, created figures, drafted and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the resources from the internal research agenda (TV15, project ID: 601) and from AGROSAVIA-Los Andes University Agreement (TV18-01, project ID:1000930).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JS was supported by a Postdoctoral Fellowship from the Ministry of Science, Technology and Innovation (MINCIENCIAS), Colombia. We thank to Johan David Barbosa for his contribution to the results obtained in the molecular and pathogenic characterization of Foph strains, reflected in the Supplementary Table S2. We thank to Dr. Mauricio Soto (AGROSAVIA) for provided DNA from Colombian strains of Fol, Foc R1, and TR4, used as PCR amplification controls. We thank grateful to Ministry of Agriculture and Rural Development of Colombia for the financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.593915/full#supplementary-material
Footnotes
References
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews, S. (2015). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed November, 2019).
Armitage, A. D., Taylor, A., Sobczyk, M. K., Baxter, L., Greenfield, B. P. J., Bates, H. J., et al. (2018). Characterisation of pathogen-specific regions and novel effector candidates in Fusarium oxysporum f. sp. cepae. Sci. Rep. 8:13530. doi: 10.1038/s41598-018-30335-7
Barrero, L. S., Bernal, A., Navas, A., Rodríguez, A., López, C., González, C., et al. (2012). “Generación de valor para el desarrollo competitivo del cultivo de la uchuva como modelo de bioprospección de frutas en Colombia,” in Bioprospección Para el Desarrollo del Sector Agropecuario de Colombia, eds A. M. Cotes, L. S. Barrero, F. Rodriguez, M. V. Zuluaga, and H. Arevalo, (Bogota: Corporación Colombiana de Investigación Agropecuaria – CORPOICA), 120–162.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., et al. (2019). BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15:e1006650. doi: 10.1371/journal.pcbi.1006650
Carvalhais, L. C., Henderson, J., Rincon-Florez, V. A., O’Dwyer, C., Czislowski, E., Aitken, E. A. B., et al. (2019). Molecular diagnostics of banana Fusarium wilt targeting Secreted-in-xylem genes. Front. Plant Sci 10:547. doi: 10.3389/fpls.2019.00547
Catanzariti, A.-M., Do, H. T. T., Bru, P., de Sain, M., Thatcher, L. F., Rep, M., et al. (2017). The tomato I gene for Fusarium wilt resistance encodes an atypical leucine-rich repeat receptor-like protein whose function is nevertheless dependent on SOBIR1 and SERK3/BAK1. The Plant Journal 89, 1195–1209. doi: 10.1111/tpj.13458
Catanzariti, A.-M., Lim, G. T. T., and Jones, D. A. (2015). The tomato I-3 gene: a novel gene for resistance to Fusarium wilt disease. New Phytol. 207, 106–118. doi: 10.1111/nph.13348
Czislowski, E., Fraser-Smith, S., Zander, M., O’Neill, W. T., Meldrum, R. A., Tran-Nguyen, L. T. T., et al. (2018). Investigation of the diversity of effector genes in the banana pathogen, Fusarium oxysporum f. sp. cubense, reveals evidence of horizontal gene transfer. Mol. Plant Pathol. 19, 1155–1171. doi: 10.1111/mpp.12594
de Guillen, K., Ortiz-Vallejo, D., Gracy, J., Fournier, E., Kroj, T., and Padilla, A. (2015). Structure analysis uncovers a highly Diverse but structurally conserved effector family in phytopathogenic fungi. PLoS Pathog. 11:e1005228. doi: 10.1371/journal.ppat.1005228
de Jonge, R., van Esse, H. P., Maruthachalam, K., Bolton, M. D., Santhanam, P., Saber, M. K., et al. (2012). Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 109, 5110–5115. doi: 10.1073/pnas.1119623109
Delmont, T. O., and Eren, E. M. (2018). Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 2018:e4320. doi: 10.7717/peerj.4320
Di Pietro, A., Madrid, M. P., Caracuel, Z., Delgado-Jarana, J., and Roncero, M. I. G. (2003). Fusarium oxysporum: exploring the molecular arsenal of a vascular wilt fungus. Mol. Plant Pathol. 4, 315–325. doi: 10.1046/j.1364-3703.2003.00180.x
El-Gengaihi, S. E., Hassan, E. E., Hamed, M. A., Zahran, H. G., and Mohammed, M. A. (2013). Chemical composition and biological evaluation of Physalis peruviana root as hepato-renal protective agent. J. Diet. Suppl. 10, 39–53. doi: 10.3109/19390211.2012.760509
Enciso-Rodríguez, F. E., González, C., Rodríguez, E. A., López, C. E., Landsman, D., Barrero, L. S., et al. (2013). Identification of immunity related genes to study the Physalis peruviana – Fusarium oxysporum Pathosystem. PLoS One 8:e68500. doi: 10.1371/journal.pone.0068500
Eren, A. M., Esen, O. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi‘o: an advanced analysis and visualization platform for ’omics data. PeerJ 2015:e1319. doi: 10.7717/peerj.1319
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Fokkens, L., Dora, S., Sánchez-Rodríguez, C., and Croll, D. (2020). A chromosome-scale genome assembly for the Fusarium oxysporum strain Fo5176 to establish a model Arabidopsis-fungal pathosystem. BioRxiv [Preprint]. doi: 10.1101/2020.05.07.082867
Glass, N. L., Jacobson, D. J., and Shiu, P. K. T. (2000). The genetics of hyphal fusion and vegetative incompatibility in filamentous ascomycete fungi. Annu. Rev. Genet. 34, 165–186. doi: 10.1146/annurev.genet.34.1.165
Gordon, T. R. (2017). Fusarium oxysporum and the Fusarium wilt syndrome. Ann. Rev. Phytopathol. 55, 23–39. doi: 10.1146/annurev-phyto-080615-095919
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hogenhout, S. A., Van Der Hoorn, R. A. L., Terauchi, R., and Kamoun, S. (2009). Emerging concepts in effector biology of plant-associated organisms. Mol. Plant Microbe Interact. 22, 115–122. doi: 10.1094/MPMI-22-2-0115
Houterman, P. M., Speijer, D., Dekker, H. L., De Koster, C. G., Cornelissen, B. J. C., and Rep, M. (2007). The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants: short communication. Mol. Plant Pathol. 8, 215–221. doi: 10.1111/j.1364-3703.2007.00384.x
Houterman, P. M., Cornelissen, B. J. C., and Rep, M. (2008). Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathogens 4:e1000061. doi: 10.1371/journal.ppat.1000061
Houterman, P. M., Ma, L., Van Ooijen, G., De Vroomen, M. J., Cornelissen, B. J. C., Takken, F. L. W., et al. (2009). The effector protein Avr2 of the xylem-colonizing fungus Fusarium oxysporum activates the tomato resistance protein I-2 intracellularly. Plant J. 58, 970–978. doi: 10.1111/j.1365-313X.2009.03838.x
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Imazaki, I., and Kadota, I. (2015). Molecular phylogeny and diversity of Fusarium endophytes isolated from tomato stems. FEMS Microbiol. Ecol. 91:98. doi: 10.1093/femsec/fiv098
Kamoun, S. (2006). A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 44, 41–60. doi: 10.1146/annurev.phyto.44.070505.143436
Kamoun, S., Win, J., Morgan, W., Oh, S. K., Young, C., Jiang, R., et al. (2007). The Effector Secretome of Phytophthora Infestans: Structure and Function. 85. Available online at: https://research.wur.nl/en/publications/the-effector-secretome-of-phytophthora-infestans-structure-and-fu-2 (accessed March 20-25, 2007).
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Letunic, I., and Bork, P. (2019). Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, E., Wang, G., Xiao, J., Ling, J., Yang, Y., and Xie, B. (2016). A SIX1 homolog in Fusarium oxysporum f. sp. conglutinans is required for full virulence on cabbage. PLoS One 11:e0152273. doi: 10.1371/journal.pone.0152273
Li, J., Fokkens, L., van Dam, P., and Rep, M. (2020a). Related mobile pathogenicity chromosomes in Fusarium oxysporum determine host range on cucurbits. Mol. Plant Pathol. 21, 761–776. doi: 10.1111/mpp.12927
Li, J., Fokkens, L., Conneely, L. J., and Rep, M. (2020b). Partial pathogenicity chromosomes in Fusarium oxysporum are sufficient to cause disease and can be horizontally transferred. Environ. Microbiol. 22, 4985–5004. doi: 10.1111/1462-2920.15095
Lievens, B., Rep, M., and Thomma, B. P. H. J. (2008). Recent developments in the molecular discrimination of formae speciales of Fusarium oxysporum. Pest Manag. Sci. 64, 781–788. doi: 10.1002/ps.1564
Lo Presti, L., Lanver, D., Schweizer, G., Tanaka, S., Liang, L., Tollot, M., et al. (2015). Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 66, 513–545. doi: 10.1146/annurev-arplant-043014-114623
Ma, L. J. (2014). Horizontal chromosome transfer and rational strategies to manage Fusarium vascular wilt diseases. Mol. Plant Pathol. 15, 763–766. doi: 10.1111/mpp.12171
Ma, L.-J., Geiser, D. M., Proctor, R. H., Rooney, A. P., O’Donnell, K., Trail, F., et al. (2013). Fusarium Pathogenomics. Annu. Rev. Microbiol. 67, 399–416. doi: 10.1146/annurev-micro-092412-155650
Ma, L. J., Van Der Does, H. C., Borkovich, K. A., Coleman, J. J., Daboussi, M. J., Di Pietro, A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. doi: 10.1038/nature08850
Meldrum, R. A., Fraser-Smith, S., Tran-Nguyen, L. T. T., Daly, A. M., and Aitken, E. A. B. (2012). Presence of putative pathogenicity genes in isolates of Fusarium oxysporum f. sp. cubense from Australia. Australas. Plant Pathol. 41, 551–557. doi: 10.1007/s13313-012-0122-x
Michielse, C. B., and Rep, M. (2009). Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol. 10, 311–324. doi: 10.1111/j.1364-3703.2009.00538.x
Moreno-Velandia, C. A., Izquierdo-García, L. F., Ongena, M., Kloepper, J. W., and Cotes, A. M. (2019). Soil sterilization, pathogen and antagonist concentration affect biological control of Fusarium wilt of cape gooseberry by Bacillus velezensis Bs006. Plant Soil 435, 39–55. doi: 10.1007/s11104-018-3866-4
O’Donnell, K., Sutton, D. A., Rinaldi, M. G., Gueidan, C., Crous, P. W., and Geiser, D. M. (2009). Novel multilocus sequence typing scheme reveals high genetic diversity of human pathogenic members of the Fusarium incarnatum-F. equiseti and F. chlamydosporum species complexes within the United States. J. Clin. Microbiol. 47, 3851–3861. doi: 10.1128/JCM.01616-09
Osorio-Guarín, J. A., Enciso-Rodríguez, F. E., González, C., Fernández-Pozo, N., Mueller, L. A., and Barrero, L. S. (2016). Association analysis for disease resistance to Fusarium oxysporum in cape gooseberry (Physalis peruviana L). BMC Genomics 17:248. doi: 10.1186/s12864-016-2568-7
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Ramadan, M. F. (2011). Bioactive phytochemicals, nutritional value, and functional properties of cape gooseberry (Physalis peruviana): an overview. Food Res. Int. 44, 1830–1836. doi: 10.1016/j.foodres.2010.12.042
Ramadan, M. M., El-Ghorab, A. H., and Ghanem, K. Z. (2015). Volatile compounds, antioxidants, and anticancer activities of Cape gooseberry fruit (Physalis peruviana L.): an in-vitro study. J. Arab. Soc. Med. Res. 10, 56–64.
Rep, M., Van Der Does, H. C., Meijer, M., Van Wijk, R., Houterman, P. M., Dekker, H. L., et al. (2004). A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 53, 1373–1383. doi: 10.1111/j.1365-2958.2004.04177.x
Rocha, L. O., Laurence, M. H., Ludowici, V. A., Puno, V. I., Lim, C. C., Tesoriero, L. A., et al. (2016). Putative effector genes detected in Fusarium oxysporum from natural ecosystems of Australia. Plant Pathol. 65, 914–929. doi: 10.1111/ppa.12472
Schmidt, S. M., Houterman, P. M., Schreiver, I., Ma, L., Amyotte, S., Chellappan, B., et al. (2013). MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genom. 14:119. doi: 10.1186/1471-2164-14-119
Schmidt, S. M., Lukasiewicz, J., Farrer, R., van Dam, P., Bertoldo, C., and Rep, M. (2016). Comparative genomics of Fusarium oxysporum f. sp. melonis reveals the secreted protein recognized by the Fom-2 resistance gene in melon. New Phytol. 209, 307–318. doi: 10.1111/nph.13584
Simbaqueba, J., Catanzariti, A. M., González, C., and Jones, D. A. (2018). Evidence for horizontal gene transfer and separation of effector recognition from effector function revealed by analysis of effector genes shared between cape gooseberry- and tomato-infecting formae speciales of Fusarium oxysporum. Mol. Plant Pathol. 19, 2302–2318. doi: 10.1111/mpp.12700
Simbaqueba, J., Sánchez, P., Sanchez, E., Núñez Zarantes, V. M., Chacon, M. I., Barrero, L. S., et al. (2011). Development and characterization of microsatellite markers for the cape gooseberry Physalis peruviana. PLoS One 6:e26719. doi: 10.1371/journal.pone.0026719
Simons, G., Groenendijk, J., Wijbrandi, J., Reijans, M., Groenen, J., Diergaarde, P., et al. (1998). Dissection of the Fusarium I2 Gene cluster in tomato reveals six homologs and one active gene copy. Plant Cell 10, 1055–1068. doi: 10.1105/tpc.10.6.1055
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Manners, J. M., Singh, K. B., and Taylor, J. M. (2015). Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 11:e1004806. doi: 10.1371/journal.ppat.1004806
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Singh, K. B., and Taylor, J. M. (2018). Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 19, 2094–2110. doi: 10.1111/mpp.12682
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467. doi: 10.1093/nar/gki458
Stergiopoulos, I., and de Wit, P. J. G. M. (2009). Fungal effector proteins. Annu. Rev. Phytopathol. 47, 233–263. doi: 10.1146/annurev.phyto.112408.132637
Taylor, A., Armitage, A. D., Handy, C., Jackson, A. C., Hulin, M. T., Harrison, R. J., et al. (2019). Basal rot of Narcissus: understanding pathogenicity in Fusarium oxysporum f. sp. narcissi. Front. Microbiol. 10:2905. doi: 10.3389/fmicb.2019.02905
Taylor, A., Vágány, V., Jackson, A. C., Harrison, R. J., Rainoni, A., and Clarkson, J. P. (2016). Identification of pathogenicity-related genes in Fusarium oxysporum f. sp. cepae. Mol. Plant Pathol. 17, 1032–1047. doi: 10.1111/mpp.12346
Thatcher, L. F., Gardiner, D. M., Kazan, K., and Manners, J. M. (2012). A highly conserved effector in Fusarium oxysporum is required for full virulence on Arabidopsis. Mol. Plant Microbe Interact. 25, 180–190. doi: 10.1094/MPMI-08-11-0212
van Dam, P., Fokkens, L., Schmidt, S. M., Linmans, J. H. J., Corby Kistler, H., Ma, L. J., et al. (2016). Effector profiles distinguish formae speciales of Fusarium oxysporum. Environ. Microbiol. 18, 4087–4102. doi: 10.1111/1462-2920.13445
van Dam, P., de Sain, M., Ter Horst, A., van der Gragt, M., and Rep, M. (2017a). Use of comparative genomics-based markers for discrimination of host specificity in Fusarium oxysporum. Appl. Environ. Microbiol. 84:e01868–17. doi: 10.1128/AEM.01868-17
van Dam, P., Fokkens, L., Ayukawa, Y., van der Gragt, M., Ter Horst, A., Brankovics, B., et al. (2017b). A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Sci. Rep. 7:9042. doi: 10.1038/s41598-017-07995-y
van Dam, P., and Rep, M. (2017). The distribution of miniature impala elements and SIX genes in the Fusarium genus is suggestive of horizontal gene transfer. J. Mol. Evol. 85, 14–25. doi: 10.1007/s00239-017-9801-0
van der Does, H. C., Fokkens, L., Yang, A., Schmidt, S. M., Langereis, L., Lukasiewicz, J. M., et al. (2016). Transcription factors encoded on core and accessory chromosomes of Fusarium oxysporum induce expression of effector genes. PLoS Genet. 12:1006401. doi: 10.1371/journal.pgen.1006401
Vlaardingerbroek, I., Beerens, B., Rose, L., Fokkens, L., Cornelissen, B. J. C., and Rep, M. (2016). Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum. Environ. Microbiol. 18, 3702–3713. doi: 10.1111/1462-2920.13281
Williams, A. H., Sharma, M., Thatcher, L. F., Azam, S., Hane, J. K., Sperschneider, J., et al. (2016). Comparative genomics and prediction of conditionally dispensable sequences in legume-infecting Fusarium oxysporum formae speciales facilitates identification of candidate effectors. BMC Genomics 17:191. doi: 10.1186/s12864-016-2486-8
Yen, C.-Y., Chiu, C.-C., Chang, F.-R., Chen, J. Y.-F., Hwang, C.-C., Hseu, Y.-C., et al. (2010). 4β-Hydroxywithanolide E from Physalis peruviana (golden berry) inhibits growth of human lung cancer cells through DNA damage, apoptosis and G2/M arrest. BMC Cancer 10:46. doi: 10.1186/1471-2407-10-46
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: Fusarium oxysporum f. sp. physali, cape gooseberry, effector genes, pathogenicity, vascular wilt disease
Citation: Simbaqueba J, Rodríguez EA, Burbano-David D, González C and Caro-Quintero A (2021) Putative Novel Effector Genes Revealed by the Genomic Analysis of the Phytopathogenic Fungus Fusarium oxysporum f. sp. physali (Foph) That Infects Cape Gooseberry Plants. Front. Microbiol. 11:593915. doi: 10.3389/fmicb.2020.593915
Received: 11 August 2020; Accepted: 28 December 2020;
Published: 18 January 2021.
Edited by:
Gustavo Henrique Goldman, University of São Paulo, BrazilReviewed by:
Orlando Borras-Hidalgo, Qilu University of Technology, ChinaGeorgios Tzelepis, Swedish University of Agricultural Sciences, Sweden
Hyeon-Su Ro, Gyeongsang National University, South Korea
Copyright © 2021 Simbaqueba, Rodríguez, Burbano-David, González and Caro-Quintero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaime Simbaqueba, anNpbWJhcXVlYmFAZ21haWwuY29t; Alejandro Caro-Quintero, YWNhcm9xQHVuYWwuZWR1LmNv