
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 05 April 2019
Sec. Extreme Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00528
This article is part of the Research TopicMicrobial Life Under Stress: Biochemical, Genomic, Transcriptomic, Proteomic, Bioinformatics, Evolutionary Aspects and Biotechnological Applications of Poly-Extremophilic Bacteria View all 27 articles
 Roberto E. Durán1
Roberto E. Durán1 Valentina Méndez1
Valentina Méndez1 Laura Rodríguez-Castro1
Laura Rodríguez-Castro1 Bárbara Barra-Sanhueza1
Bárbara Barra-Sanhueza1 Francisco Salvà-Serra2,3,4,5
Francisco Salvà-Serra2,3,4,5 Edward R. B. Moore2,3,4
Edward R. B. Moore2,3,4 Eduardo Castro-Nallar6
Eduardo Castro-Nallar6 Michael Seeger1*
Michael Seeger1*Alcaligenes aquatilis QD168 is a marine, aromatic hydrocarbon-degrading bacterium, isolated from an oil-polluted sediment of Quintero Bay, an industrial-coastal zone that has been chronically impacted by diverse pollutants. The aims of this study were to characterize the phylogenomic positions of Alcaligenes spp. and to characterize the genetic determinants and the physiological response of A. aquatilis QD168 to model environmental stressors (benzene, oxidizing agents, and salt). Phylogenomic analyses, using 35 housekeeping genes, clustered A. aquatilis QD168 with four other strains of Alcaligenes spp. (A. aquatilis BU33N, A. faecalis JQ135, A. faecalis UBA3227, and A. faecalis UBA7629). Genomic sequence analyses of A. aquatilis QD168 with 25 Alcaligenes spp., using ANIb, indicated that A. aquatilis BU33N is the closest related strain, with 96.8% ANIb similarity. Strain QD168 harbors 95 genes encoding proteins of seven central catabolic pathways, as well as sixteen peripheral catabolic pathways/reactions for aromatic compounds. A. aquatilis QD168 was able to grow on 3-hydroxybenzoate, 4-hydroxybenzoate, benzoate, benzene, 3-hydroxycinnamate, cinnamate, anthranilate, benzamide, 4-aminobenzoate, nicotinate, toluene, biphenyl and tryptophan, as sole carbon or nitrogen source. Benzene degradation was further analyzed by growth, metabolite identification and gene expression analyses. Benzene strongly induced the expression of the genes encoding phenol hydroxylase (dmpP) and catechol 1,2-dioxygenase (catA). Additionally, 30 genes encoding transcriptional regulators, scavenging enzymes, oxidative damage repair systems and isozymes involved in oxidative stress response were identified. Oxidative stress response of strain QD168 to hydrogen peroxide and paraquat was characterized, demonstrating that A. aquatilis QD168 is notably more resistant to paraquat than to H2O2. Genetic determinants (47 genes) for osmoprotective responses were identified, correlating with observed high halotolerance by strain QD168. The physiological adaptation of A. aquatilis QD168 to environmental stressors such as pollutants, oxidative stress and salinity may be exploited for bioremediation of oil-polluted saline sites.
The Alcaligenes genus, belonging to the Alcaligenaceae family of the order Burkholderiales (Garrity et al., 2005), was first described in 1911 during the study of a bacterium isolated from a fecal sample and identified as A. faecalis (Kühnemann, 1911). Strains of Alcaligenes spp. have been isolated from diverse environments, such as water (Regar et al., 2016), agricultural soil (Liu et al., 2016), crude oil-polluted sites (Singha et al., 2017), industrial processes (Qiu et al., 2017; Basharat et al., 2018), insects (Quiroz-Castañeda et al., 2015) and human clinical samples (Winslow et al., 1920; Al Laham et al., 2017). Strains of Alcaligenes spp. have been used for biotechnological applications, such as bioremediation and biocontrol. Strains of Alcaligenes spp. are capable of degrading a wide range of xenobiotics, including crude-oil (Alcaligenes spp.), polyaromatic hydrocarbons (PHAs) (A. faecalis BDB4), pesticides (Alcaligenes sp. EGD-AK7), dyes (A. faecalis subsp. phenolicus MB207), 4-hydroxyacetophenone (Alcaligenes sp. 4HAP) and phenols (A. faecalis subsp. phenolicus DSM 16503T and A. faecalis JF339228) (Kiyohara et al., 1982; Hopper and Kaderbhai, 1999; Rehfuss and Urban, 2005; Adebusoye et al., 2007; Kumar et al., 2013; Sagarkar et al., 2014; Regar et al., 2016; Singha et al., 2017; Basharat et al., 2018).
Whole-genome sequencing and genome mining are useful for characterizing bacteria and to discover their biotechnological capabilities. A comparative genomic analysis of A. faecalis subsp. phenolicus MB207 and 10 other strains of Alcaligenes spp. identified biosynthetic clusters for butyrolactone, ectoine, resorcinol, terpene, non-ribosomal peptides and polyketide synthases (Basharat et al., 2018). Strain MB207 is able to degrade azo dyes and possesses tolerance to heavy metals, although, its genomic and physiological stress determinants have not been analyzed. Currently, four complete genome sequences are publicly available for this genus, including those of A. faecalis ZD02 (Ju et al., 2016), A. faecalis DSM 30030T (Genome accession: GCA_002443155.1), A. faecalis JQ135 (Zhang et al., 2018) and A. faecalis P156 (Genome accession: GCA_001641975.2), but no whole-genome bioprospecting analyses have been performed previously for any of these strains.
Marine ecosystems are characterized by a wide range of multiple stressors, such as salinity, hypoxia, low and high temperatures, acidic pH, ultraviolet radiation, pollution and scarcity of nutrients. Microorganisms from these locations are well adapted to such environmental pressures. Marine ecosystems are valuable sources for pollutant-degrading and stress-resistant bacterial strains. Although isolates of marine Alcaligenes spp. have been described less frequently than their soil counterparts, degradation capabilities and biotechnological applications of marine and coastal strains have been reported. Alcaligenes sp. strains ASW3 and ASS-1 were isolated from crude oil-polluted seawater and were applied in an immobilized bacterial consortium for biodegradation of crude-oil (Chen et al., 2017). Mercury-resistant strains of A. faecalis were isolated from various locations along the Indian Coast, exhibiting capabilities for cadmium and lead detoxification (De and Ramaiah, 2007).
Quintero Bay is located in an industrial-coastal zone in Valparaíso Region, Central Chile, which has been subjected to extensive and long-term pollution. Quintero is a highly industrialized area for its small population. A crude-oil harbor, copper refinery, thermoelectric power plant, liquefied gas plant and chemical industries are located in this sector. Oil-spills have been documented in this bay several times during the last two decades (Palma-Fleming et al., 2008; Piraino et al., 2017).
Monoaromatic compounds, such as benzene, toluene, ethylbenzene and the three-xylene isomers, commonly known as BTEX, are often present in industrial and crude oil-polluted sites (Fuentes et al., 2014). Benzene is the most toxic and hazardous BTEX compound, being the only carcinogenic pollutant of this group. Marine bacterial strains of species of Bacillus, Comamonas, Exiguobacterium, Marinobacterium, Pseudomonas and Rhodococcus genera that are capable of degrading BTEX hydrocarbons have been described (Wang et al., 2008; Jiang et al., 2015; Baek et al., 2018). However, information on the genes and catabolism of BTEX compounds by strains of Alcaligenes spp. is limited.
Bacterial communities are able to thrive in polluted environments despite the harsh conditions encountered (Michas et al., 2017; Ahmed et al., 2018). Isolation and characterization of multistress-tolerant bacteria from severe environments, lead to potential discovery of novel strains for the clean-up of polluted sites (Head et al., 2006; Fuentes et al., 2014; Orellana et al., 2018). Diverse bacteria, including extremophiles, have been used as biocatalysts for bioremediation of soils and water contaminated with persistent organic pollutants and heavy metals (Harayama et al., 2004; Saavedra et al., 2010; Rojas et al., 2011; Fuentes et al., 2016; Méndez et al., 2017; Orellana et al., 2018).
A. aquatilis QD168 was isolated from crude oil-polluted coastal sediments of the industrial zone of Quintero Bay, Central Chile (Durán et al., 2019). The aims of this study were to elucidate the phylogenomic positions of strains of Alcaligenes spp., and to characterize the genetic determinants and the physiological responses to model environmental stressors (benzene, oxidizing agents, and salt) of the novel hydrocarbonoclastic marine bacterium A. aquatilis QD168. To our knowledge, this is the first phylogenomic study reported for Alcaligenes spp. which included 26 strains from diverse environments. Additionally, this is the first genomic characterization of an A. aquatilis strain that reveals the genetic potential for aromatic catabolism and physiological responses to endure diverse environmental stresses.
Benzoate, 3-hydroxybenzoate, 4-hydroxybenzoate, cinnamate, 3-hydroxycinnamate, salicylate, nicotinate, anthranilate, 4-aminobenzoate, 2-aminophenol, benzamide, simazine, tryptophan, and methyl viologen dichloride hydrate (paraquat, PQ) were obtained from Sigma-Aldrich (St. Louis, MO, United States). Benzene (>99.7% purity), toluene, biphenyl, naphthalene, phenanthrene, anthracene, fluorene, and 30% (w v-1) hydrogen peroxide (H2O2) were purchased from Merck (Darmstadt, Germany).
A. aquatilis QD168 was isolated by selective enrichment. 10 g (wet weight) of polluted marine sediment from Quintero Bay, Chile (32°45′49.5″S 71°29′13.5″W) and 90 mL of 0.85% (w v-1) NaCl were mixed in a Heidolph blender at high speed for 2 h at room temperature. Bushnell-Haas (BH) mineral medium (1 g L-1 KH2PO4, 1 g L-1 K2HPO4, 1 g L-1 NH4NO3, 0.2 g L-1 MgSO4, g L-1 CaCl2 0.02, 0.05 g L-1 FeCl3 [pH 7.0]) (Bushnell and Haas, 1941) supplemented with 1% v v-1 commercial diesel as sole carbon source, was used for enrichment. Specifically, 45 mL of Bushnell-Haas broth with artificial seawater (23.47 g L-1 NaCl, 3.92 g L-1 Na2SO4, 10.64 g L-1 MgCl2 × 6H2O, 1.457 g L-1 CaCl2 × 2H2O, 0.192 g L-1 NaHCO3, 0.664 g L-1 KCl, 0.096 g L-1 KBr, 0.026 g L-1 H3BO3, 0.0404 g L-1 SrCl2 × 6H2O, 0.003 g L-1 NaF) (BH-saline), supplemented with 200 μg mL-1 cycloheximide, in order to inhibit eukaryotic cell growth, in a 250 mL Erlenmeyer flask, was inoculated with 5 mL of supernatant. The enrichment was obtained by 10 consecutive subcultures (1-week intervals) at 20°C with rotary shaking at 180 rpm. The isolation of hydrocarbon degrading bacteria was carried out by serial dilutions. A. aquatilis QD168 was obtained by spread-plating on BH agar, supplemented with artificial sea water (ASW) and 200 μg mL-1 cycloheximide and subsequently purified by streak-plating. The BH agar plate contained a paper filter with diesel in the lid as the sole carbon source and was incubated at 20°C. Bacterial isolates were stored in 15% (v v-1) glycerol at -80°C.
All the genome sequences of the genus Alcaligenes that were available in GenBank in September 2018 (Accession numbers in Supplementary Table S1, including a status of complete, scaffolds, or contigs). The resulting 26 genomes were organized into 24 described species plus 2 unclassified at the species level. All the genome sequences (including the A. aquatilis strain QD168 from this study), were re-annotated using a combination of ab initio and similarity methods as implemented in Prokka version 1.10 (Seemann, 2014) and NCBI Prokaryotic Genome Annotation Pipeline version 4.6 (Tatusova et al., 2016).
Genome-wide nucleotide identity trends were explored in the genome dataset by estimating all-against-all pairwise Average Nucleotide Identity (ANI). The ANIb approach which uses BLAST, was used to align the input sequences as implemented in JSpeciesWS v.3.0.2 (Richter et al., 2016). Heat maps were generated, using pheatmap V 1.0.8 R package (Kolde, 2015). Genomic clusters were defined, using hierarchical clustering (method: average), based on a 95% ANI threshold, which represents a conservative boundary for species level classification (Richter and Rosselló-Móra, 2009).
Twenty-eight phylogenetic gene markers implemented in AMPHORA2 were used (dnaG, frr, infC, nusA, pgk, pyrG, rplB, rplC, rplD, rplE, rplF, rplM, rplN, rplP, rplS, rplT, rpoD, rpmA, rpsB, rpsC, rpsE, rpsI, rpsJ, rpsK, rpsM, rpsS, smpB, and tsf) (Wu and Scott, 2012), as well as seven other genetic markers (atpD, gltA, gyrB, nirK, pnp, recA, and thrC), from the data set. All nucleotide sequences were aligned, using MAFFT version 7.407 (Katoh and Standley, 2013). The alignments were manually trimmed, using the alignment editor AliView version 1.24 (Larsson, 2014) and concatenated, using the Bio.Nexus module, as implemented in Biopython version 1.72 (Cock et al., 2009). The best partitioning scheme was identified, using the program PartitionFinder version 2.1.1 (Lanfear et al., 2017). A distribution of probable trees was obtained by Bayesian Inference as implemented in MrBayes 3.2.6 (Ronquist et al., 2012). Two separate runs of 20 million generations were executed (four chains each run; sampling every 1,000 generations). Visualization and editing of phylogenetic trees were performed, using the FigTree v. 1.4.2 software1.
CGview version 2 (Stothard and Wishart, 2005) was used to generate a circular map of the A. aquatilis QD168 genome. Genes were classified according to the Clusters of Orthologous Groups (COG) nomenclature (Galperin et al., 2015), using the eggNOG database version 4.5.1 (Huerta-Cepas et al., 2016).
Previously identified coding sequences were submitted to a local alignment, using the BLASTP and TBLASTN tools of BLAST 2.7.1+ software (Camacho et al., 2009), against a database of enzymes, transcriptional regulators and transporters involved in aromatic compound catabolism, oxidative stress and osmotic stress response reported in UniProtKB-Swissprot (Bateman et al., 2015). An e-value cutoff <10-10, identity >30% and coverage >50% were used to filter the outcome. Critical residues and domains for protein function were identified by DELTA-BLAST (Camacho et al., 2009).
OxyR and SoxR amino acid sequences with experimental evidence were retrieved from UniProtKB Swiss-Prot/TrEMBL database (Bateman et al., 2015). All nucleotide sequences were aligned, using MAFFT version 7.407 (Katoh and Standley, 2013). The alignments were manually trimmed, using the alignment editor AliView version 1.24 (Larsson, 2014). The best partitioning scheme was identified, using the program PartitionFinder version 2.1.1 (Lanfear et al., 2017). A distribution of probable trees was obtained, by Bayesian Inference as implemented in MrBayes 3.2.6 (Ronquist et al., 2012). Two separate runs of one million generations were executed (two chains each run; sampling every 1,000 generations). Visualization and editing of phylogenetic trees were performed using the FigTree v. 1.4.2 software (see footnote 1).
To assess catabolic potential of A. aquatilis QD168, growth assays were performed in M9 minimal medium (6.78 g L-1 Na2HPO4, 3 g L-1 KH2PO4, 0.5 g L-1 NaCl, 1 g L-1 NH4Cl) with trace solutions [154 mg L-1 MgSO4⋅7H2O, 13.4 mg L-1 MgCl2⋅6H2O, 11.9 mg L-1 FeSO4⋅7H2O, 2.5 mg L-1 CaCO3, 1.8 mg L-1 ZnSO4⋅7H2O, 1.4 mg L-1 MnSO4⋅H2O, 0.35 mg L-1 CoCl2⋅6H2O, 0.3 mg L-1 CuSO4⋅5H2O, 0.075 mg L-1 H3BO3, and 0.0024% (v v-1) HCl] (Overwin et al., 2012; Romero-Silva et al., 2013; Agulló et al., 2017) or the nitrogen free M8 minimal medium (6.78 g L-1 Na2HPO4, 3 g L-1 KH2PO4, 0.5 g L-1 NaCl) with trace solutions supplemented with 1.75% (w v-1) NaCl, and an aromatic compound as sole carbon or nitrogen source.
Protocatechuate (1.33 mM), catechol (1.33 mM), gentisate (1.33 mM), salicylate (1.14 mM), benzoate (1.14 mM), phenol (1.33 mM), 3-hydroxybenzoate (1.14 mM), 4-hydroxybenzoate (1.14 mM), 3-toluate (1.00 mM), cinnamate (0.89 mM), 3-hydroxycinnamate (0.89 mM), benzene (1.33 mM), phenanthrene (0.57 mM), fluorene (0.62 mM) and anthracene (0.57 mM) were tested as sole carbon source for bacterial growth. Anthranilate (1.14 mM), tryptophan (0.72 mM), nicotinate (1.33 mM), 2-aminophenol (1.33 mM), 4-aminobenzoate (1.14 mM), benzamide (1.14 mM), and simazine (1.14 mM) were used in M8 minimal medium as sole carbon and nitrogen source for growth. Bacterial growth was determined using a 2-mL squared 96-well plate by measuring turbidity (600 nm), and using closed 15-mL glass tubes by colony-forming units (CFU) (Méndez et al., 2011). Values were calculated as the mean ± SD of the results of, at least, three independent experiments.
A. aquatilis QD168 was cultured in M9 minimal medium with benzene (5 mM) as sole carbon source. For this, cells were previously adapted with successive passages in M9 medium with 0.5, 2.0, and 5.0 mM benzene as carbon source. After adaptation, cells were cultured at 200 rpm and 30°C in M9 medium with 5 mM benzene as sole carbon source. To avoid benzene volatilization, 250-mL glass flasks closed using PTFE/silicone-screw caps were used. Growth was monitored by measuring turbidity at 600 nm until 120 h. Values were calculated as the mean ± SD of results of, at least, three independent experiments.
Resting cells (turbidity600 nm ∼ 8.0) were incubated in sodium phosphate buffer (50 mM, pH 7.2) with benzene (5 mM). Aliquots of cell suspensions were taken at different incubation times and centrifuged (10,000 × g for 10 min). Assays with boiled cells and without cells were used as controls. Resting cell assays were performed in triplicate.
For benzene and phenol quantification, an organic extraction was performed of samples grown in benzene with one volume of ethyl acetate. The mixture was centrifuged at 10,000 × g for 5 min. The organic fraction was used for quantification by reverse phase chromatography with a Jasco liquid chromatograph equipped with a diode array detector and a Whatman C-18 column (25 cm by 4.6 mm ID) (Chirino et al., 2013). Methanol:acetonitrile (30:70) was used as the mobile phase with a flux of 1 mL min-1.
A. aquatilis QD168 was grown until exponential phase in M9 minimal medium with succinate as sole carbon and energy source. 100-μL of the culture were spread on Petri dishes. 6-mm diffusion disks containing 15 μL of hydrogen peroxide (H2O2; 1, 5, 10, and 20 mM) or paraquat (PQ; 1, 20, 100, and 500 mM) were deposited on the plates. Growth inhibition diameters were measured after 24 h incubation at 30°C (Sandoval et al., 2011). Values were calculated as the mean ± SD of results of, at least, three independent experiments.
A. aquatilis QD168 was grown until exponential phase in M9 minimal medium with succinate as carbon source and washed three times with 2% (w v-1) NaCl. Cells were exposed to H2O2 (0.1, 0.5, 1, and 4 mM) or PQ (25, 50, 100, 200 and 500 mM) during 1 h. Aliquots were taken to determine CFU (Ponce et al., 2011). The percentage of survival was determined as the ratio of CFU of cells exposed to oxidizing agents and CFU of untreated cells. Values were calculated as the mean ± SD of results of, at least, three independent experiments.
A. aquatilis QD168 was grown until exponential phase (turbidity600 nm ∼ 0.6) in M9 minimal medium with succinate as sole carbon and energy source. 20-μL of the culture was plated on Reasoner’s 2A (R2A) medium with 0, 2, 4, 6, 8, and 10% (w v-1) NaCl. The growth was monitored after 24 and 48 h of incubation at 30°C (Gallardo-Cerda et al., 2018). Growth assays to assess halotolerance were performed in three independent experiments.
To analyze the expression of genes involved in benzene catabolism, A. aquatilis QD168 was grown until exponential phase in M9 minimal medium with benzene or succinate as sole carbon and energy sources. To analyze the expression of genes involved in oxidative stress response, A. aquatilis QD168 was grown until exponential phase in M9 minimal medium with succinate (2 mM) as sole carbon and energy sources, washed three times with 0.85% (w v-1) NaCl, and incubated in 1.75% (w v-1) NaCl in absence (untreated cells) or presence of H2O2 (0.5 mM), PQ (50 mM) during 1 h (Ponce et al., 2011).
Bacterial cells were harvested, and RNA was extracted, using QIAGEN RNeasy purification kit (Qiagen; Hilden, Germany), following the manufacturer’s instructions. Contaminant DNA was removed, employing the TURBO DNA-free Kit (Thermo Fisher Scientific; Waltham, MA, United States). RNA quality was verified with the NanoDrop One spectrometer (Thermo Fisher Scientific; Waltham, MA, United States). RNA integrity was checked in a 1% (w v-1) agarose gel. Amplification of the 16S rRNA gene by qPCR was performed as control for DNA contamination. cDNA was synthetized with the First Strand cDNA Synthesis Kit (Thermo Fisher Scientific; Waltham, MA, United States). The qRT-PCRs were carried out with the KAPA SYBR FAST qPCR Master Mix Kit (Kapa Biosystems; Boston, MA, United States), following the manufacturer’s instructions. The genes 16S rRNA and ftsZ were employed as reference genes. The results were analyzed, using Hellemans method (Hellemans et al., 2007). Succinate-grown and untreated cells were used as control. Negative and positive controls were included in each RT-PCR assay. At least three independent RNA samples were analyzed at each condition and two independent RT-PCR reactions for each sample were done to assess reproducibility. Genes and primers employed are listed in Table 1.

Table 1. Oligonucleotides designed and used in this study.
Statistical analyses were performed using one-way ANOVA and Tukey HSD test to assess differences in mean values from each experiment. Differences are significant at α = 0.05.
To investigate the phylogenomic relationships of strains of Alcaligenes spp., a more comprehensive set of housekeeping gene markers was obtained from strain QD168 and 25 publicly available genomes of Alcaligenes spp. (Supplementary Table S1), to perform a multi-locus sequence analysis (MLSA). AMPHORA2 was used to evaluate the presence of single copy genes encoding ribosomal proteins, as well as some other classic bacterial marker genes, in all the genomes evaluated. This analysis revealed 28 genetic markers and seven additional housekeeping genes were manually incorporated to obtain a total of 35 housekeeping genes, including the gyrB and nirK genes, which have been assessed in the taxonomic identification of A. pakistanensis (Abbas et al., 2015). The following genome sequences available at GenBank were not included in this, and the following analyses: A. faecalis GZAF2 (accession no.: GCA_002119985.1) and A. faecalis GZAF4 (GCA_002120025.1), due to the absence of relevant genetic markers (gyrB, dnaG or nirK) selected for the MLSA. Figure 1A, shows the phylogenetic relationships between strains of Alcaligenes spp. Interestingly, strains of Alcaligenes spp. clustered in five distinct clades, which did not correlate with their previous species identification. A. aquatilis QD168 clustered in close proximity with four other Alcaligenes strains: A. aquatilis BU33N, the only other A. aquatilis genome available (Genome accession: GCA_0030765151.1); A. faecalis JQ135, a 5-hydroxypicolinic acid and nicotinic acid degrader (Qiu et al., 2017; Zhang et al., 2018); A. faecalis UBA3227 and A. faecalis UBA7629, whose genomes were obtained from subway metagenomes (Figure 1A).
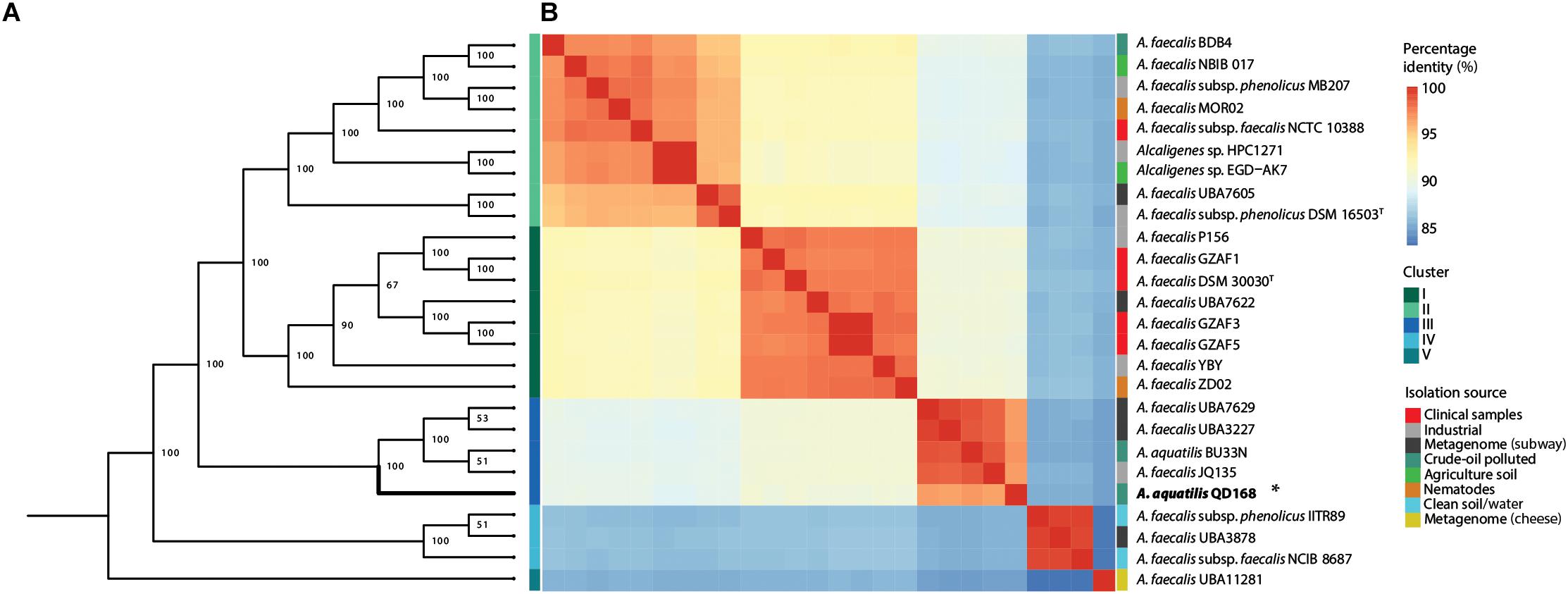
Figure 1. Phylogenetic analysis of representative Alcaligenes spp. strains including A. aquatilis QD168. (A) MLSA-based clustering of 26 strains of Alcaligenes spp. based on 35 housekeeping genes. Mid-rooted phylogeny showed five distinctive clades. (B) ANIb analysis of 26 strains of Alcaligenes spp. The first column indicates 95% ANI clustering (75% alignment). Isolation source is indicated in the last column. A. aquatilis QD168 belongs to cluster III.
To further study the genome-genome relationships between strains of Alcaligenes spp., an average nucleotide identity based on BLAST (ANIb) analysis was performed with A. aquatilis QD168 and 25 strains of Alcaligenes spp. from the dataset (Figure 1B). The most similar strain to A. aquatilis QD168 is A. aquatilis BU33N (96.8%), suggesting that both strains belong to the same species. A. aquatilis BU33N was isolated from a petroleum-polluted sediment from Tunisia (BioProject: PRJNA386470). Using the proposed boundary for species delineation (ANIb value >95%, Richter and Rosselló-Móra (2009)) the 26 strains of Alcaligenes spp. were grouped in five genomic clusters correlating with the information obtained by the MLSA analysis (Figure 1B). The cluster I comprised eight strains (A. faecalis P156, A. faecalis DSM 30030T, A. faecalis GZAF1, A. faecalis UBA7622, A. faecalis GZAF3, A. faecalis GZAF5, A. faecalis ZD02, and A. faecalis YBY) containing the type strain for A. faecalis species (DSM 30030T) and including most of the clinical isolates from the dataset. The cluster II, which belonged to a wide range of isolation source environments, included nine strains (A. faecalis BDB4, A. faecalis NBIB-017, A. faecalis subsp. phenolicus MB207, A. faecalis MOR02, A. faecalis subsp. phenolicus 10388, Alcaligenes sp. HPC1271, Alcaligenes sp. EGD-AK7, A. faecalis UBA7605 and A. faecalis subsp. phenolicus DSM 16503T) containing the type strain for the subsp. phenolicus of A. faecalis species (DSM 16503T). The cluster III included A. aquatilis QD168, and four other strains (A. faecalis UBA3227, A. faecalis UBA7629, A. aquatilis BU33N and A. faecalis JQ135). Three of these strains were isolated from polluted environments, and the genomes of two strains were retrieved from subway metagenomes. The cluster IV, contained three strains (A. faecalis subsp. phenolicus IITR89, A. faecalis UBA3878 and A. faecalis subsp. faecalis NCIB 8687); two members were isolated from clean soil or water sources. Finally, cluster V, consisted of a singleton containing A. faecalis UBA11281 (Figure 1B). The genome of strain UBA11281 was retrieved from metagenome data of a cheese sample.
The genomes of strains of Alcaligenes spp. included in this study have a size range between 3.68 and 4.44 Mbp (average 4.14 Mbp), and possess a GC content between 55.4 and 57.6% (average 56.6%) (Supplementary Table S1). A. aquatilis QD168 has a genome size of 4,323,879 bp, with a GC content of 56.4%, assembled in one circular chromosome. The genome of strain QD168 contains 3,892 coding sequences (CDS) (Durán et al., 2019). The functions of 3,557 genes of A. aquatilis QD168 were annotated, based on Clusters of Orthologous Groups (COG) categories (Galperin et al., 2015), corresponding to 91.4% of total CDS (Figure 2). The three most abundant categories were: unknown function (S; 23.67%), transcription (K; 7.95%), and amino acid transport and metabolism (E; 7.40%). A COG comparison analysis was performed between the 26 strains of Alcaligenes spp. The functions of the 26 predicted proteomes, obtained by Prokka annotation of the genomic dataset, were classified based on COG categories. Figure 3 shows the percentage of each COG category for the proteomes of eight specific Alcaligenes members. From outer to inner circle: A. aquatilis QD168; A. aquatilis BU33N; the reference genome for the species A. faecalis, A. faecalis ZD02; the type strain of A. faecalis species A. faecalis DSM 30030T, the type strain A. faecalis subsp. phenolicus DSM 16503T; the nicotinic acid-degrader A. faecalis JQ135; the azo dye-degrader A. faecalis subsp. phenolicus MB207; the clinical human isolate A. faecalis GZAF5, and the atrazine-degrader Alcaligenes sp. EGD-AK7 (Supplementary Table S2). In general, the distribution of COG categories among the genus is highly similar, obtaining the highest average percentages for (S) function unknown (21.6%), (K) transcription (8.1%), and (E) amino acid transport and metabolism (8.0%) categories (Figure 3, dashed lines). A. aquatilis QD168 possessed a higher percentage of orthologous genes compared to the average of all the strains of Alcaligenes spp. included in this study on the categories functions: (S) function unknown (S: 23.27%) in comparison to 21.6 ± 1.20%; (L) replication, recombination and repair (L: 5.11%) compared to 3.62 ± 0.47% and the category (U) intracellular trafficking, secretion, and vesicular transport (U: 2.68%) in comparison to 2.02 ± 0.36% (Figure 3).
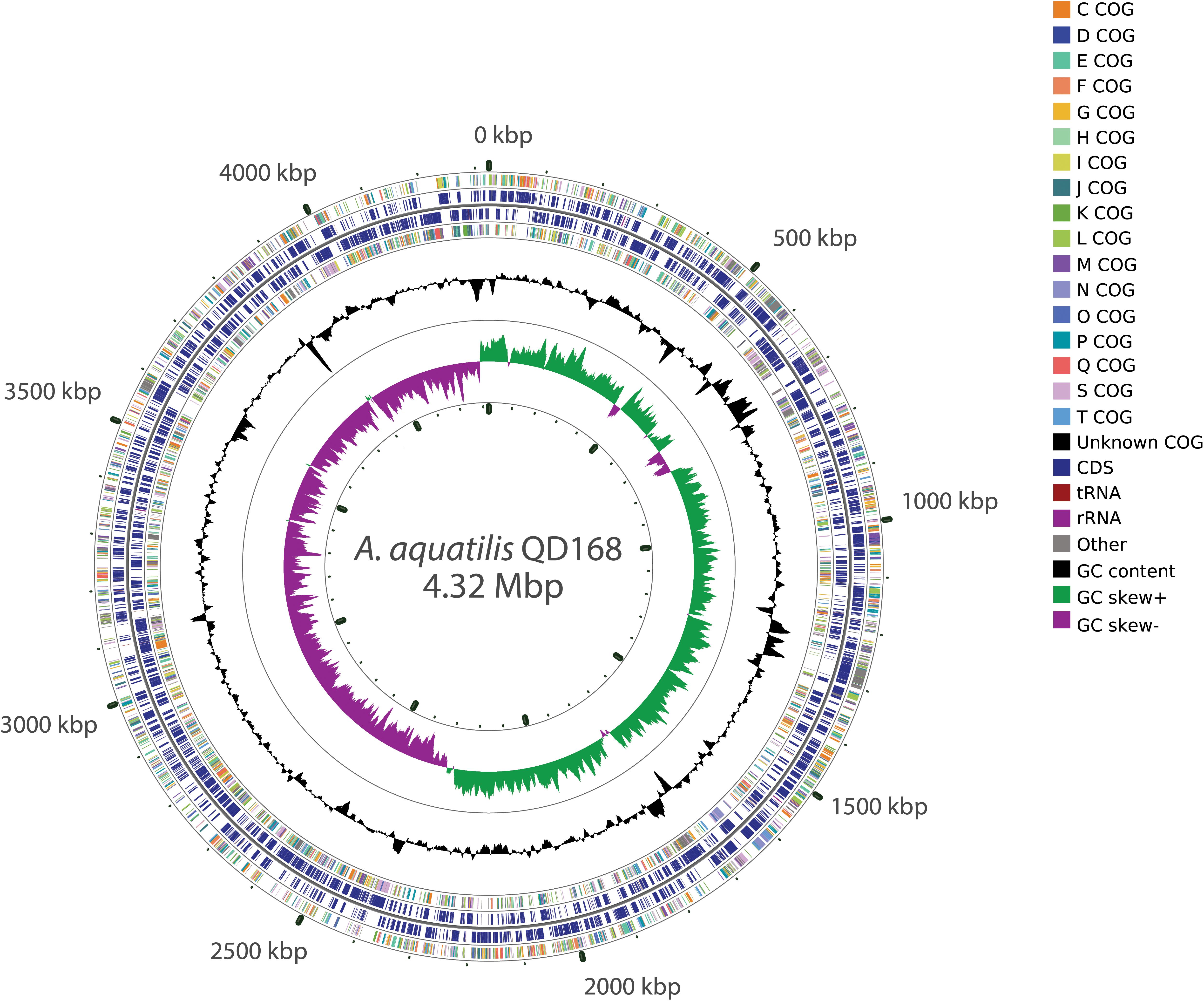
Figure 2. Schematic representation of the genome of A. aquatilis QD168. Alcaligenes aquatilis QD168 genome has a size of 4,323,879 bp and contains 3,892 coding sequences, 9 rRNA and 58 tRNA. Rings from inside to outside: (1) G + C skew; (2) G + C content; (3) QD168 genes colored by functional COG class on the reverse strand; (4) predicted QD168 genes on the reverse strand; (5) predicted QD168 genes on the forward strand; (6) QD168 genes colored by functional COG class on the forward strand. Gene functions were annotated based on COG categories. The whole genome map was generated using CGView.
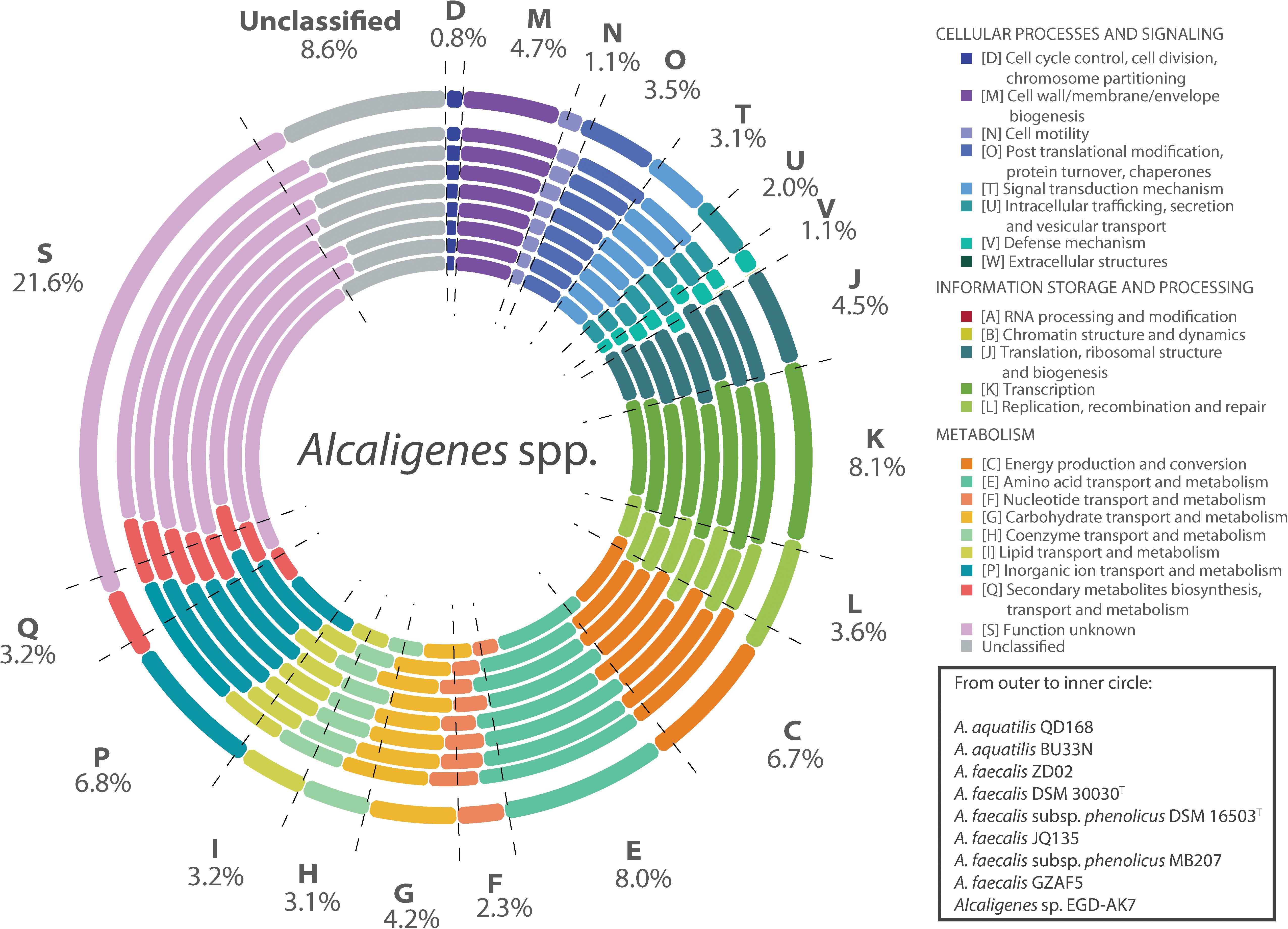
Figure 3. Cluster of orthologous groups comparison of A. aquatilis QD168 and representative Alcaligenes strains. Cluster of Orthologs (COG) classification of eight Alcaligenes spp. strains. For each COG entry the average percentage of hits among 26 Alcaligenes spp. strains has been indicated by values and dashed lines. From outer to inner circle: A. aquatilis QD168, A. aquatilis BU33N A. faecalis ZD02, A. faecalis DSM 30030T A. faecalis subsp. phenolicus DSM 16503T A. faecalis JQ135, A. faecalis subsp. phenolicus MB207, A. faecalis GZAF5, Alcaligenes sp. EGD-AK7.
In order to predict the catabolic potential for aromatic compounds of A. aquatilis QD168, a genomic search for genes of aromatic catabolic pathways was performed. The genes of seven aromatic central pathways, and sixteen peripheral pathways/reactions were identified in the QD168 genome. Central pathways included the catechol (cat), protocatechuate (lig), homogentisate (hmg), gentisate (nag), phenylacetyl-CoA (paa), 3-(2,3-dihydroxyphenyl)-propionate (mhp), and 2,5-dihydroxynicotinate (nic) (Figure 4).
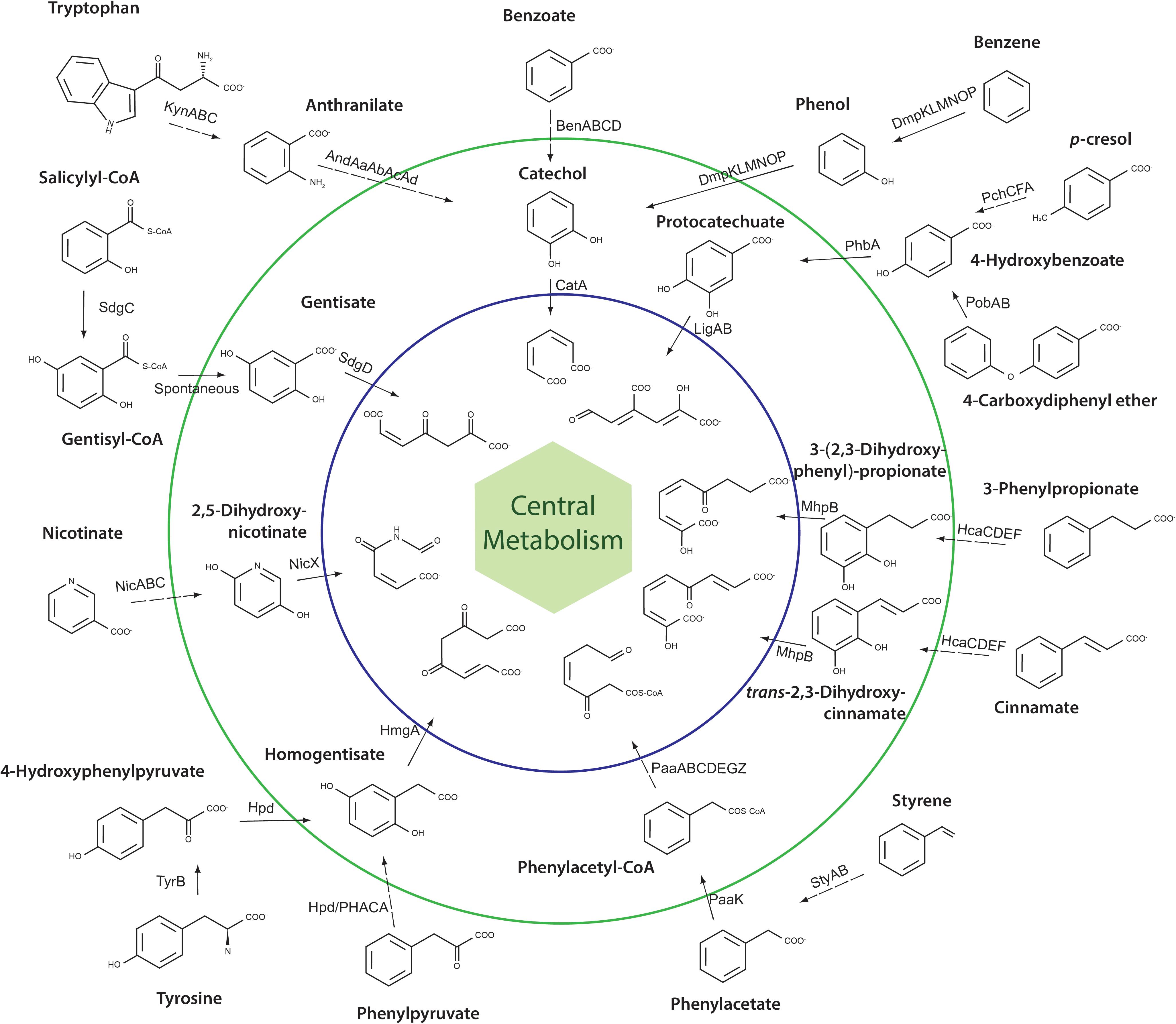
Figure 4. Schematic representation of all central aromatic catabolic pathways and peripheral aromatic catabolic pathways/reactions present in A. aquatilis QD168. The inner circle (blue) includes ring-cleavage product structures of central aromatic catabolic pathways. The outer circle (green) includes the structure of aryl-CoA and dihydroxylated ring-cleavage intermediates. Dashed lines indicate multiple steps.
The genes encoding the catechol branch of the β-ketoadipate pathway were found clustered (catRBAIJDC), whereas the genes encoding the protocatechuate branch (pca; ortho-cleavage pathway) were not present in strain QD168. The cat gene products of A. aquatilis QD168 were highly similar to proteins of the catechol branch that has been characterized mainly in bacteria of Acinetobacter and Pseudomonas genera (Supplementary Table S3). Interestingly, strain QD168 possessed the genes encoding the peripheral reactions for benzoate (benMABCD), benzene and phenol (dmpRKLMNOP), anthranilate (andAaAbAcAd), and tryptophan (kynABU) that converge into catechol as a central intermediate (Figure 4 and Supplementary Table S4). The ben, dmp, and kyn gene products of strain QD168 were highly similar to the corresponding characterized proteins of bacteria of Acinetobacter, Burkholderia, and Pseudomonas genera (Supplementary Table S4).
The genes encoding the protocatechuate meta-cleavage (ligCBAIKJ) were found clustered in the QD168 genome. The ligCBA gene products possessed high identity (>58%) to the proteins of Sphingomonas paucimobilis SYK-6 (Supplementary Table S3). The ligIKJ gene products of strain QD168 possessed high identity (70%) to the proteins of Comamonas sp. E6, Pseudomonas straminea and Acidovorax sp. T1 (Supplementary Table S3). Interestingly, the gene D3M96_07080, encoding a 4-hydroxybenzoate transporter (PcaK), was found within this genomic context. The genes encoding peripheral reactions for 4-hydroxybenzoate (phbA), p-cresol (pchCFA), and 4-carboxy-diphenyleter (pobAB) that channel into the protocatechuate meta-cleavage pathway were identified in the QD168 genome (Supplementary Table S4). The phbA and pobAB gene products of strain QD168, were highly similar to the PhbA, PobA, and PobB proteins of strains of Pseudomonas spp. The pchCFA gene products of strain QD168, which encode enzymes for p-cresol conversion into 4-hydroxybenzoate and further protocatechuate formation, possessed high identities with the proteins of P. putida NCIMB 9869, and were located within the neighborhood of the lig genes (protocatechuate central pathway).
The paa genes encoding the phenylacetyl-CoA central pathway, were grouped in the paaABCDEZGI gene cluster next to the paaK gene that encodes the enzyme of the peripheral reaction for phenylacetate degradation into phenylacetyl-CoA. The paa gene products of strain QD168 were highly similar to the corresponding proteins of Escherichia coli K-12 (Supplementary Table S3). Additionally, the genes encoding enzymes of peripheral reactions for styrene degradation (styAB) via phenylacetate that is funneled into phenylacetyl-CoA central pathway, were identified in the QD168 genome (Supplementary Table S4).
Catabolic genes encoding for tyrosine (tyrB), phenylpyruvate (hpd), and 4-hydroxyphenylpyruvate (hpd) degradation via the homogentisate central pathway were identified in the QD168 genome. The gene products possessed >55% identity with the Hpd and TyrB proteins of Pseudomonas and E. coli strains (Supplementary Table S4). Although the hmgA, hmgB, and hmgC genes, encoding the proteins of the homogentisate central pathway are located in an operon in Gammaproteobacteria, such as E. coli and P. putida, the hmg genes were found distributed in A. aquatilis QD168 genome. Interestingly, the hmg genes products of strain QD168 were highly similar to the HmgA and HmgB proteins of Bordetella genus (>65% identity) and HmgC of P. aeruginosa (47% identity) (Supplementary Table S3).
The sdgD, nagL, and nagK genes encoding the enzymes of gentisate central pathway, were distributed within the QD168 genome. The enzyme of the peripheral reaction for salicylyl-CoA (SdgC) that is channeled into gentisyl-CoA, and then to the gentisate central pathway, was identified in strain QD168, and possessed 40% identity to the characterized SdgC protein of Streptomyces sp. WA46 (Supplementary Table S4).
The mhpBCDFE genes encoding the 3-(2,3-dihydroxyphenyl)-propionate/2,3-dihydroxy-cinnamate central catabolic pathway, were found clustered and located next to the hcaREFCBC gene cluster, which encodes the 3-phenylpropionate and cinnamate peripheral pathways (Figure 4 and Supplementary Table S4). The genes encoding the 2,5-dihydroxynicotinate central catabolic pathway (nicDXF) in strain QD168, were similar (>43% identity) to the corresponding enzymes of P. putida KT2440. Two copies of the nicDXF gene cluster, were found in the QD168 genome. The nicAB gene products of strain QD168 involved in the degradation of nicotinate into the 2,5-dihydroxynicotinate central catabolic pathway, were also similar (>41% identity) to the NicA and NicB enzymes of P. putida KT2440 (Supplementary Table S3).
To assess the degradation capabilities of strain QD168, growth assays on aromatic compounds as sole carbon or nitrogen source were performed. Twenty-five aromatic compounds were tested. A. aquatilis QD168 was able to grow in minimal medium on thirteen aromatic compounds. Strain QD168 was able to grow on 3-hydroxybenzoate, 4-hydroxybenzoate, benzoate, benzene, 3-hydroxycinnamate, cinnamate, anthranilate, benzamide, 4-aminobenzoate, nicotinate, tryptophan, toluene, and biphenyl as sole carbon or nitrogen source (Table 2). No color formation, that may indicate meta-cleavage of the aromatic ring, was observed during growth on aromatic compounds.
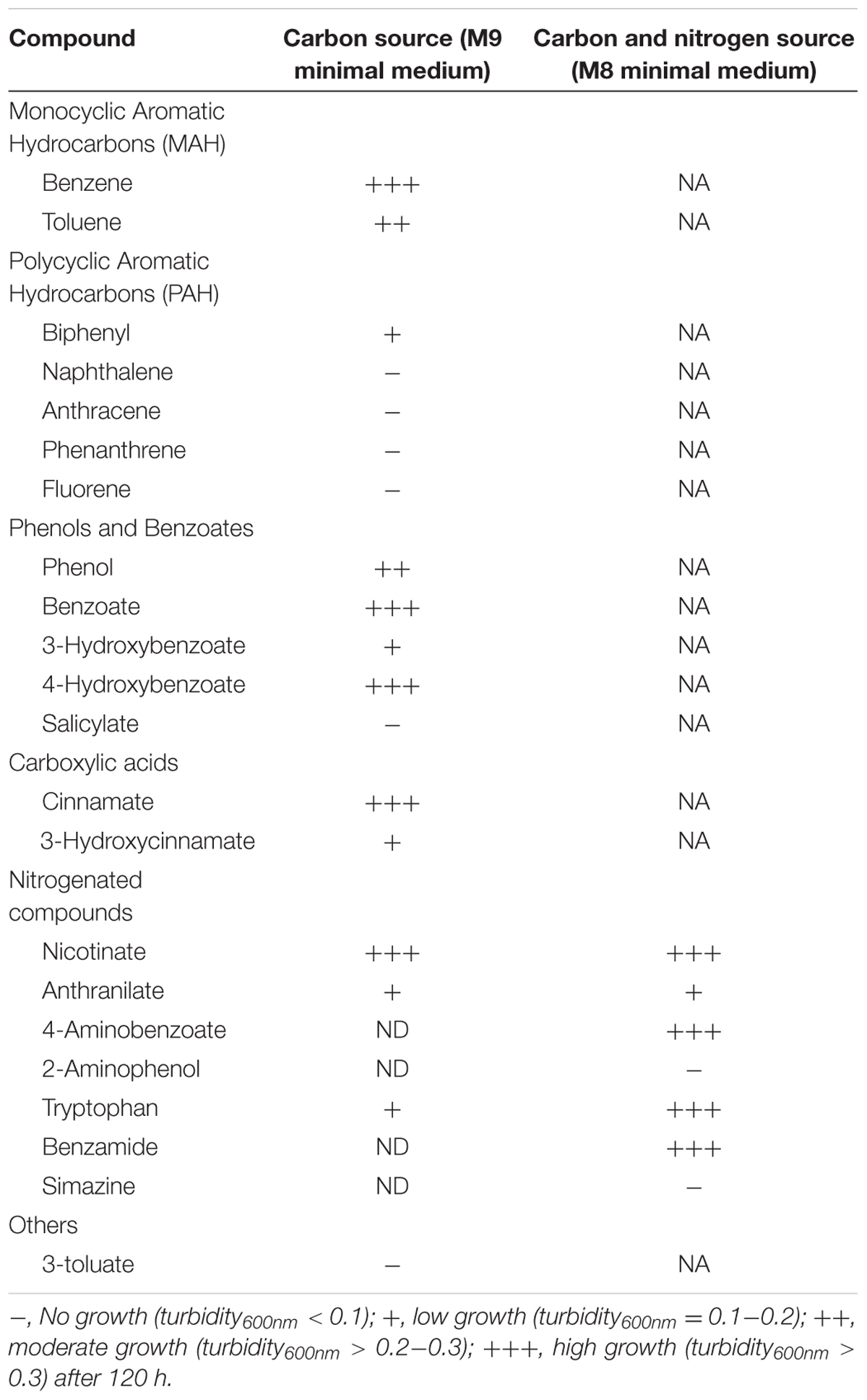
Table 2. Growth of A. aquatilis QD168 on aromatic compounds.
The degradation of the model aromatic compound benzene was further analyzed due to its wide distribution as part of the highly toxic BTEX compounds and its major environmental concern. A. aquatilis QD168 possesses the genes for benzene degradation via phenol into catechol (Figure 4). The dmpRKLMNOP gene cluster that encodes the multicomponent phenol hydroxylase was identified in the QD168 genome (Figure 5A). The benMABCDE gene cluster was located downstream to the dmp cluster. The benABC genes encode a multicomponent benzoate 1,2-dioxygenase, and the benD gene encodes a cis-diol dehydrogenase that are involved in benzoate degradation via catechol (Figure 4). Two coding sequences were identified between the dmp and ben clusters, the bedC1 gene that encodes a benzene 1,2-dioxygenase alpha subunit, and the D3M96_09770 gene with unknown function (Figure 5A). However, the bedA and bedB genes encoding the benzene 1,2-dioxygenase reductase and ferredoxin subunits, respectively, were not found in the QD168 genome. The cat genes encoding the catechol ortho-cleavage pathway were found in the QD168 genome (Figure 5A), while only the catechol 2,3-dioxygenase encoded by the catE gene (D3M96_10490) of the meta-cleavage pathway was identified. The catE gene product of strain QD168 showed low identity (26% identity) with the catechol 2,3-dioxygenase of Cupriavidus pinatubonensis JMP222. A. aquatilis QD168 was able to grow in minimal medium in presence of 1.75% (w v-1) NaCl and benzene (5 mM) as sole carbon source, attaining a turbidity of ∼ 0.5 at 600 nm after 74 h (Figure 5B). After 60 min incubation of resting cells, benzene was not detected by HPLC, strongly suggesting that strain QD168 is able to degrade benzene. Phenol was identified by HPLC as a metabolic intermediate during benzene degradation. To correlate bioinformatic predictions and functional assays, transcriptional analysis of key catabolic genes for benzene degradation was performed. The transcription of catechol dioxygenase-encoding genes for meta- and ortho-cleavage (catA and catE), and the phenol hydroxylase-encoding gene for benzene monooxygenation reaction (dmpP gene) was determined. An up-regulation of dmpP (>930-fold), catA (>610-fold), and catE (>3-fold) genes was observed in exponentially growing cells of strain QD168 on benzene, compared to succinate-grown cells (Figure 5C). These results suggest that benzene degradation in strain QD168 occurs mainly via phenol formation as metabolic intermediate and catechol ortho-cleavage, by the phenol hydroxylase and the catechol 1,2-dioxygenase enzymes, respectively. Figure 5D illustrates the proposed benzene catabolic pathway in A. aquatilis QD168.
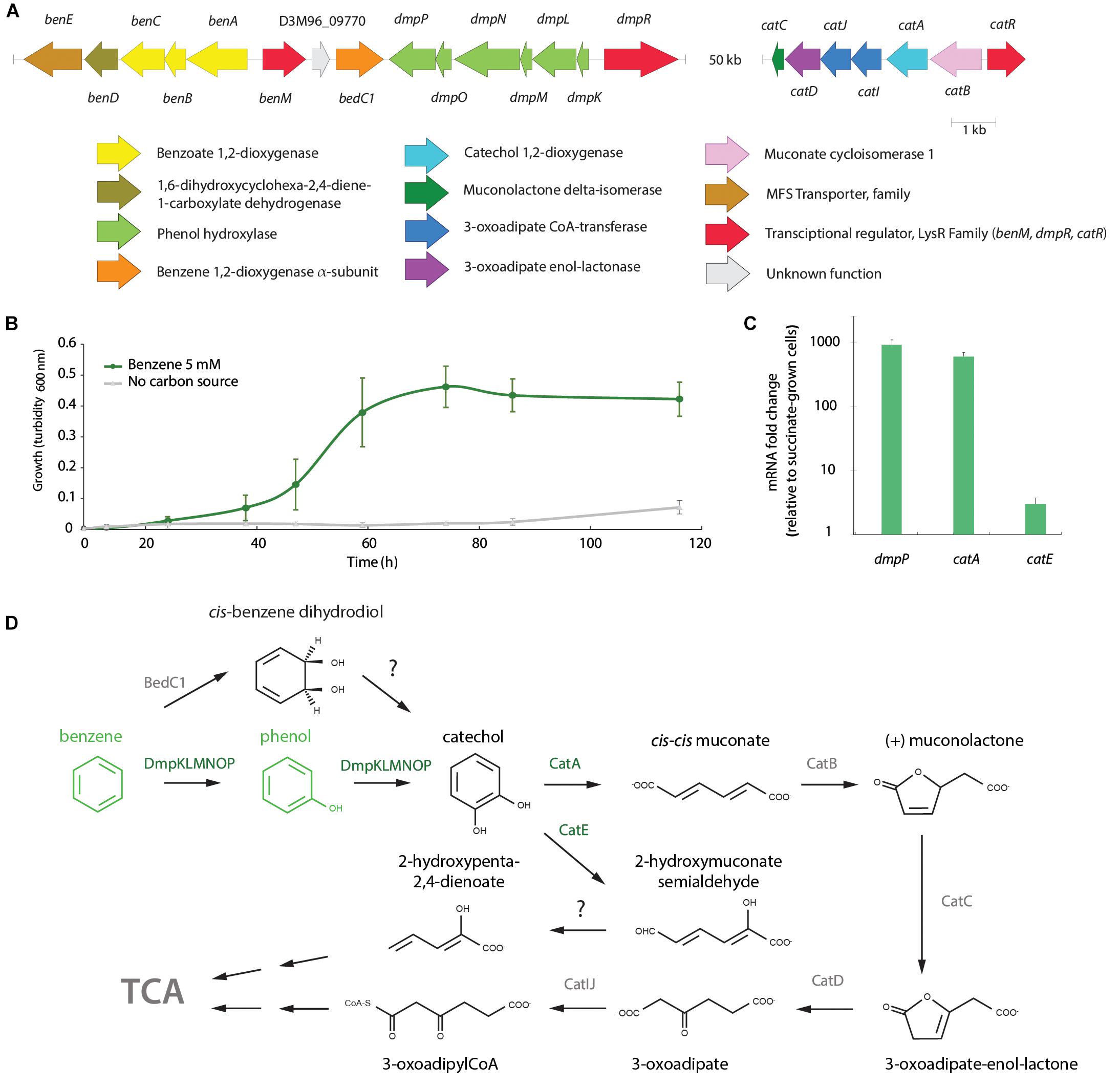
Figure 5. Functional analyses of benzene degradation by A. aquatilis QD168. (A) Predicted ben, dmp and cat genes encoding the catechol branch of the β-ketoadipate catabolic pathway and peripheral reactions on A. aquatilis QD168 genome. Genes and intergenic regions sizes are on scale (B) A. aquatilis QD168 growth on benzene (5 mM) as sole carbon and energy sources. Turbidity values were calculated as the mean ± SD of results of, at least, three independent experiments. (C) Expression of dmpP, catA and catE genes during QD168 growth on benzene (5 mM). 16S rRNA and ftsZ genes were used as reference genes. At least three independent RNA samples were collected at each condition and two independent RT-PCR reactions for each sample were done to assess reproducibility. (D) Proposed benzene degradation pathway of A. aquatilis QD168. Enzymes and compounds with experimental data are shown in green.
Genome analyses indicate that thirty genes of A. aquatilis QD168 may participate in scavenging of reactive oxygen species (ROS), resistance to endogenous redox imbalance and regulation of oxidative stress response (Supplementary Table S5). Genes encoding a thioredoxin/thioredoxin reductase system (seven genes), a glutaredoxin/glutathione peroxidase system (four genes), alkyl hydroperoxide reductases (four genes), catalases (two genes), and superoxide dismutases (three genes) were identified in the QD168 genome (Figure 6A). Additionally, five genes encoding transcriptional regulators for oxidative stress response were found. Genes encoding for the SoxR redox-sensitive transcriptional regulator (soxR1, soxR2), the H2O2-sensing transcriptional regulator OxyR (oxyR), the transcriptional regulator (perR), and the organic hydroperoxide resistance transcriptional regulator OhrR (ohrR) were identified.
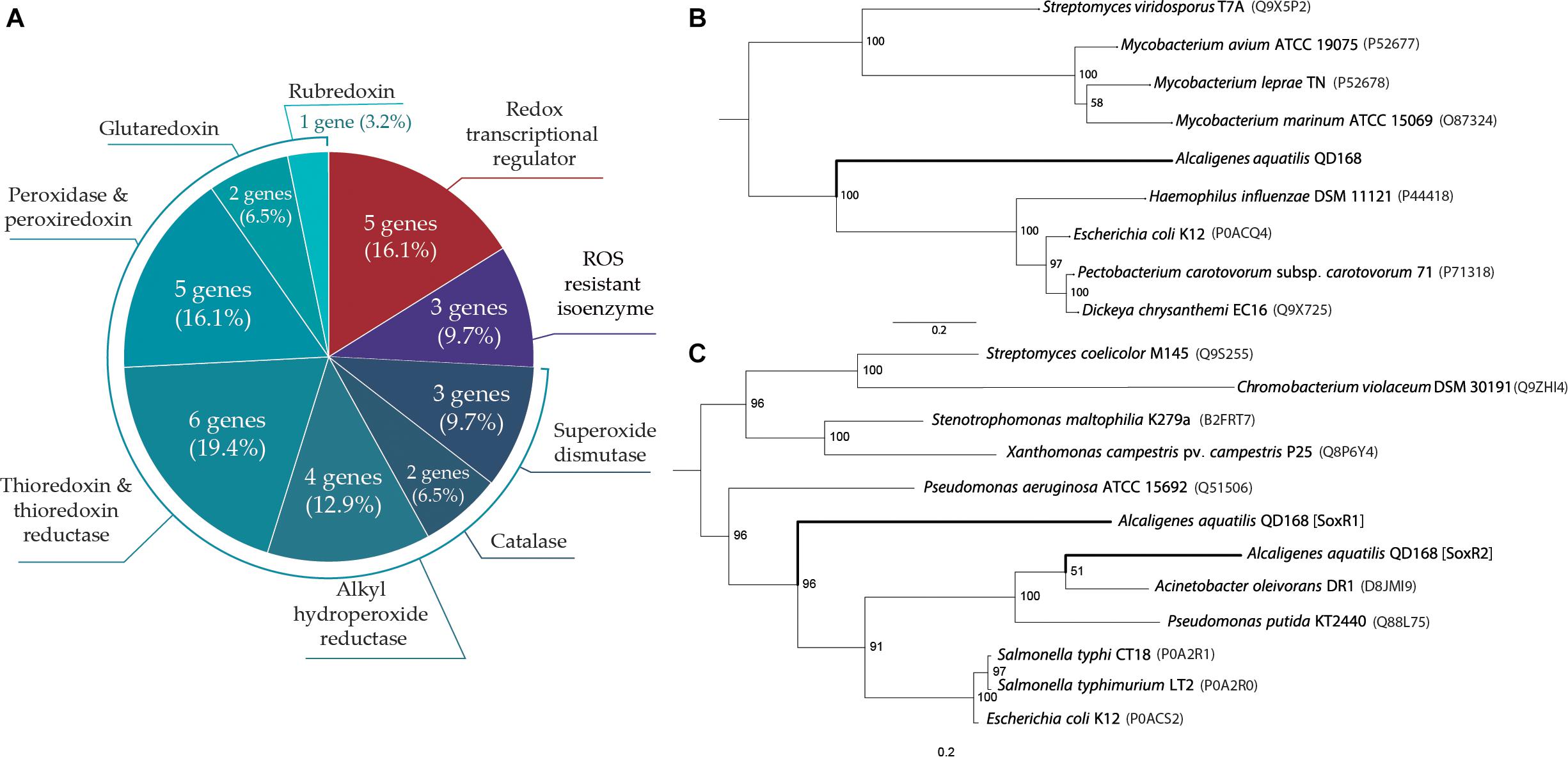
Figure 6. Genetic determinants of A. aquatilis QD168 for oxidative stress response and phylogenetic relationships of OxyR and SoxR transcriptional regulators. (A) Genetic determinants involved in oxidative stress response identified by genome mining. (B) Phylogenetic tree of OxyR protein orthologous: mid-rooted phylogeny showed the clade of QD168 OxyR (C) Phylogenetic tree of SoxR orthologous proteins: mid-rooted phylogeny showed five distinctive clades.
To further study oxidative stress response in strain QD168, the phylogeny of OxyR, SoxR1 and SoxR2 transcriptional regulators of A. aquatilis QD168 was studied (Figure 6). For OxyR (Figure 6B), the amino acid sequence of nine OxyR regulators with experimental evidence in bacteria of Actinobacteria and Gammaproteobacteria classes were used for bioinformatic analyses. This analysis revealed three distinctive clades, wherein OxyR of A. aquatilis QD168 formed a singleton. OxyR orthologs of Actinobacteria clustered together, while OxyR orthologs from Gammaproteobacteria were grouped in the third clade (Figure 6B). For SoxR1 and SoxR2 analyses (Figure 5C), the amino acid sequence of 10 SoxR transcriptional regulators with experimental evidence of Gammaproteobacteria, Actinobacteria (Streptomyces coelicolor M145) and Betaproteobacteria (Chromobacterium violaceum DSM 30191) classes were used. SoxR1 of A. aquatilis QD168 formed a singleton ungrouped with other SoxR orthologs. SoxR2 of strain QD168 grouped with Gammaproteobacteria orthologs. This phylogenetic clade, consisted of a subgroup of SoxR orthologs of enteric bacteria (E. coli K12, Salmonella typhi CT18 and S. typhimurium LT2), and another subgroup of non-enteric bacteria (P. putida KT2440 and A. oleovorans DR1) that included SoxR2 ortholog of A. aquatilis QD168.
The response of A. aquatilis QD168 to H2O2, and the redox-cycling compound paraquat (PQ) was investigated. To study strain QD168 susceptibility to oxidative stress inducers, cells grown until exponential phase were incubated in presence of H2O2 or PQ. Strain QD168 showed an increase of the growth inhibition zone in response to increasing concentrations of H2O2 (1, 5, 10, and 20 mM) (Figure 7A). During strain QD168 cells exposure to 1 and 20 mM PQ, no significant differences were detected. A small increase in the inhibition zone was observed in strain QD168 growth after exposure to higher concentrations of PQ (100 and 500 mM) (Figure 7C).
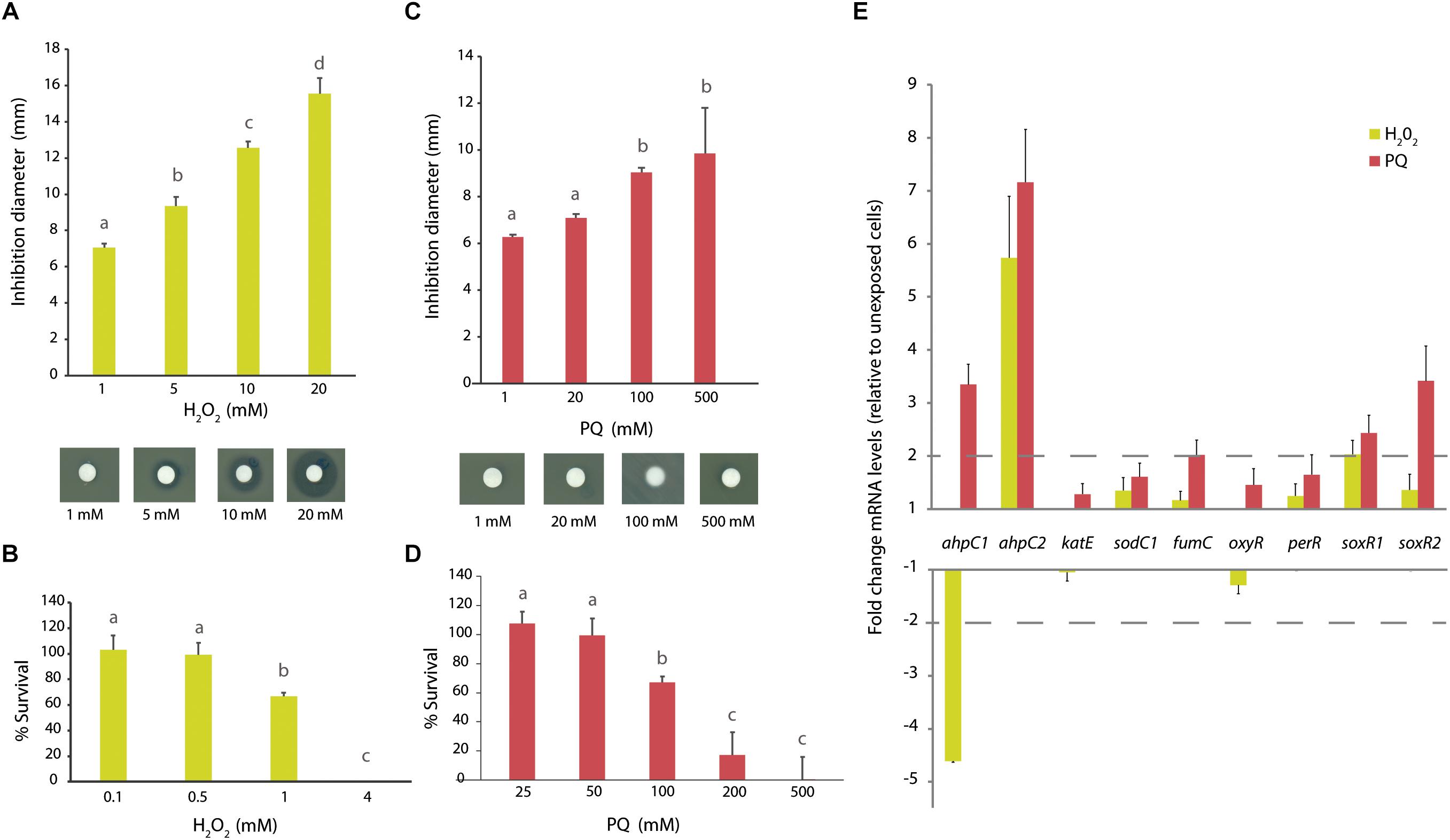
Figure 7. Oxidative stress response of A. aquatilis QD168 to H2O2 and paraquat. (A) Growth inhibition diameter of A. aquatilis QD168 after 60 min exposure to H2O2 (1–20 mM). (B) Survival of A. aquatilis QD168 after 60 min exposure to H2O2 (0.1–4 mM). (C) Growth inhibition diameter of A. aquatilis QD168 after 60 min exposure to PQ (1–100 mM). (D) Survival of A. aquatilis QD168 after 60 min exposure to PQ (25–500 mM). Values for growth inhibition diameter and survival of strain QD168 were calculated as the mean ± SD of results of, at least, three independent experiments. Different letters indicate significant differences between conditions (HSD Tukey test, α = 0.05) (E) Expression of oxidative stress genes of A. aquatilis QD168 cells after 60 min exposure to H2O2 (500 μM) and PQ (50 mM). 16S rRNA and ftsZ were used as reference genes. At least three independent RNA samples were collected at each condition and two independent RT-PCR reactions for each sample were done to assess reproducibility.
To study A. aquatilis QD168 survival during exposure to oxidizing agents, exponentially growing cells were incubated with H2O2 or PQ for 1 h. After exposure to 0.1 and 0.5 mM H2O2, strain QD168 survival was almost 100%, but it significantly decreased at 1 mM H2O2. After exposure to 4 mM H2O2, no viable cells were observed (Figure 7B). Interestingly, after exposure to 25 and 50 mM PQ, cell viability was not affected. In contrast, 100 mM PQ caused a significant decrease in cell survival (Figure 7D). After exposure to 200 and 500 mM PQ, cell viability was highly affected, but no significant differences were observed between both concentrations. Overall, these results indicate that A. aquatilis QD168 is notably more resistant to PQ than to H2O2.
To further characterize the bacterial response to oxidizing agents, a gene expression analysis was carried out. For this assay, nine genes were selected: genes encoding the antioxidant enzymes alkyl hydroperoxide reductase (ahpC1 and ahpC2), catalase E (katE), superoxide dismutase (sodC1), a ROS-resistant isoform of fumarate hydratase (fumC), and four transcriptional regulators involved in oxidative stress response (oxyR, perR, soxR1, and soxR2) (Figure 7E). In response to H2O2 (0.5 mM), the ahpC1 gene was down-regulated (>4-fold), while the ahpC2 gene was up-regulated (>5-fold) compared to control cells. SoxR1 was the only transcriptional regulator whose expression was up-regulated (2-fold) by H2O2. In response to PQ (50 mM), both genes encoding alkyl hydroperoxide reductase proteins (ahpC1 and ahpC2) displayed an increase of >3- and 7-fold, respectively. An up-regulation (>2-fold) by PQ of the fumC gene encoding the enzyme fumarate hydratase C, which is involved in Krebs cycle, was observed. In response to PQ, the genes encoding the transcriptional regulators SoxR1 and SoxR2 were up-regulated >2- and 3-fold, respectively (Figure 7E).
Forty-seven genes that may participate in osmoprotection (synthesis and transport of compatible solutes) were identified in A. aquatilis QD168 genome (Supplementary Table S6). Genes involved in the synthesis (ectABCD) and transport (ehuBCDA) of ectoine/hydroxyectoine, synthesis (18 genes) and transport (4 genes) of glutamate, synthesis (glnE) and transport (glnHPQ and bztDCBA) of glutamine, synthesis (proA, proBC, and argF) and transport (putP) of proline, transport of glycine betaine/choline (ousVW), and synthesis (iscS, sufS, and sufE) of alanine were identified in strain QD168 (Figure 8A). To study the halotolerance of A. aquatilis QD168, cells grown in LB medium until exponential phase were plated on R2A medium supplemented with NaCl ranging from 0 to 10% (w v-1). Bacterial cell growth was observed at all salt concentration assessed after 48 h of incubation at 30°C (Figure 8B). These results indicate that A. aquatilis QD168 is a halotolerant bacterium.
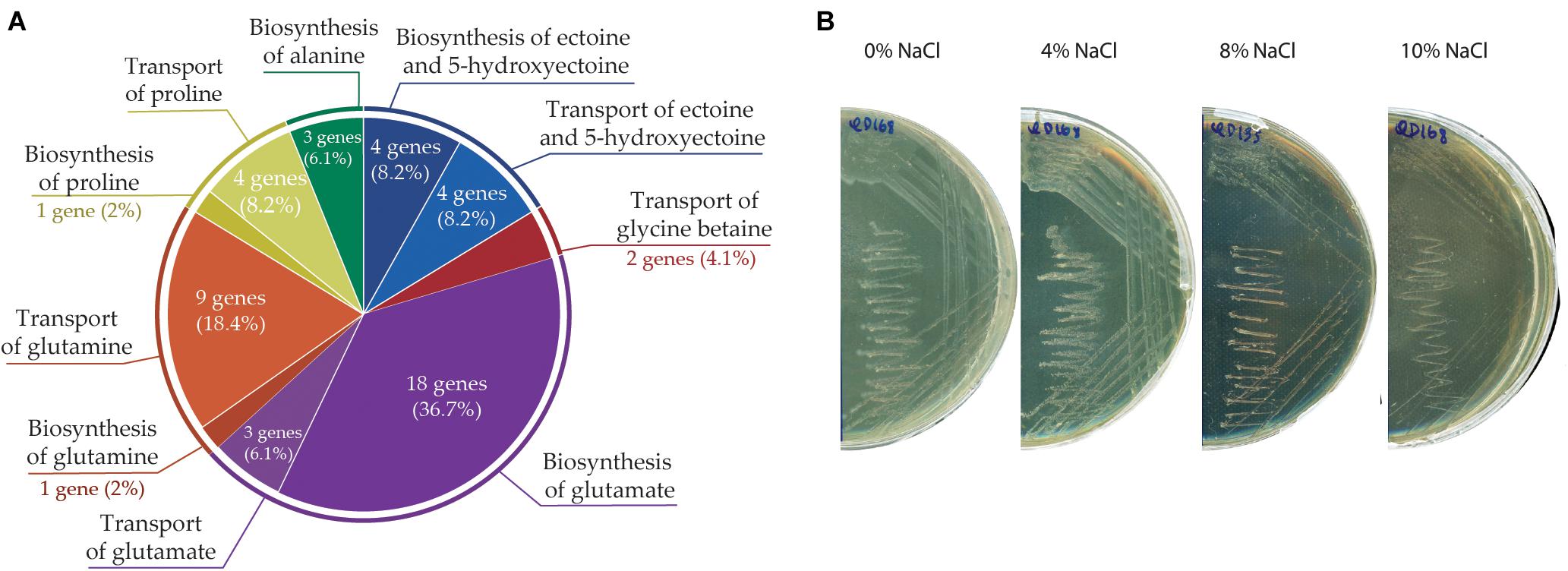
Figure 8. Osmotolerance response of A. aquatilis QD168 to salt stress. (A) Genetic determinants involved in biosynthesis and transport of osmo-protectant metabolites. (B) Salt tolerance of A. aquatilis QD168 in R2A medium supplemented with 0 to 10% (w v-1) NaCl.
Strains of the Alcaligenes spp. have been associated with diverse ecological niches, suggesting genetic diversity and physiological potential to encounter severe conditions, such as the presence of toxic organic compounds, heavy metals, oxidative stress and high salinity.
The phylogenomic analysis, in this study, of strains of Alcaligenes spp. revealed five clustering profiles (clusters I–V), which were distributed, in general, according to their isolation sources (Figure 1). The phylogenomic analysis, placing A. aquatilis QD168 within the Alcaligenes genus, also revealed the same clustering pattern of strains by the MLSA of 35 housekeeping genes and by using ANIb similarities (Konstantinidis and Tiedje, 2005; Richter and Rosselló-Móra, 2009). Genomes corresponding to the Alcaligenes genus were highly represented by the species, A. faecalis, comprising approximately 84.6% of all genomes of Alcaligenes spp. analyzed. However, the presence of, at least, one strain classified as A. faecalis in every phylogenomic cluster identified is unusual, indicating probable misclassifications of these strains.
A. faecalis represents the best-studied species of this genus; three subspecies have been described and validly published: A. faecalis subsp. faecalis; A. faecalis subsp. parafaecalis (Schroll et al., 2001); and A. faecalis subsp. phenolicus (Rehfuss and Urban, 2005). The low ANIb values obtained (<92.3%) and MLSA analysis between A. faecalis subsp. faecalis (DSM 30030T) and A. faecalis subsp. phenolicus (DSM 16503T) indicate that they belong to two different phylogenetic lineages. These results suggest that A. faecalis subsp. phenolicus should be reclassified as a representative of a different species. Cluster III is represented by A. aquatilis QD168 and the strains A. aquatilis BU33N (Genome accession: GCA_003076515.1), A. faecalis JQ135 (Qiu et al., 2017, 2018; Zhang et al., 2018), A. faecalis UBA7629 and A. faecalis UBA3227 (Parks et al., 2017). Accordingly, identification of strains QD168 and BU33N by 16S rRNA gene sequence analysis showed >99.9% similarity with A. aquatilis LMG 22996T (Durán et al., 2019). Although the type strain of A. aquatilis has not yet been sequenced, these data indicate the phylogenomic placement of the A. aquatilis species to be in the Alcaligenes genomic cluster III. A. aquatilis QD168 was isolated from hydrocarbon-polluted sediments from Quintero Bay (Valparaiso Region, Chile). In agreement with the hostile conditions for life found in this crude oil-polluted environment (i.e., high salinity, high levels of persistent organic pollutants, and heavy metals), the genome sequence of A. aquatilis QD168 revealed a high number of genetic determinants for the degradation of aromatic compounds, response to oxidative stress, and osmoprotection.
Proteobacteria isolated from polluted sites are known to degrade a broad spectrum of aromatic compounds (Chain et al., 2006; McLeod et al., 2006; Pérez-Pantoja et al., 2008, 2012). Genomic sequence analyses of, so-called, “environmental” bacteria, such as P. putida KT2440, Paraburkholderia xenovorans LB400 and C. pinatubonensis JMP134, have revealed versatile metabolisms and a wide enzymatic repertoire for degradation of aromatic compounds (Jiménez et al., 2002; Chain et al., 2006; Pérez-Pantoja et al., 2008). To our knowledge, this is the first genome mining report of A. aquatilis, describing degradative potential for a number of aromatic compounds. The remarkable number of genes encoding catabolic aromatic pathways found in strain QD168 revealed its metabolic potential to overcome oil-polluted environments. Seven central catabolic pathways and sixteen peripheral catabolic pathways/reactions for the degradation of aromatic compounds were identified in the QD168 genome. Central catabolic pathways included the catechol, protocatechuate, 3-(2,3-dihydroxyphenyl)-propionate, phenylacetyl-CoA, homogentisate, gentisate, and 2,5-dihydroxynicotinate. Interestingly, strain QD168 was able to grow on benzoate, 3-hydroxybenzoate, 4-hydroxybenzoate, benzene, cinnamate, 3-hydroxycinnamate, nicotinate, tryptophan, anthranilate, 4-aminobenzoate, benzamide, toluene, and biphenyl as sole carbon sources. In agreement with these results, prediction of benzoate (benABCD), cinnamate (hcaBCFE), nicotinate (nicABC), tryptophan (kynABU), anthranilate (andAaAbAcAd), and benzene (dmpKLMNOP) peripheral catabolic pathways were identified in the QD168 genome. However, bioinformatic analyses did not identify genes for 3-hydroxybenzoate, 3-hydroxycinnamate, benzamide, 4-aminobenzoate, biphenyl, and toluene pathways, which are substrates for QD168 growth. In addition, 18 other catabolic genes (i.e., monooxygenases, hydroxylases, and aromatic ring-hydroxylating dioxygenases) could not be affiliated to a specific catalytic function, although they may play key roles in the degradations of these compounds. Further functional analyses are needed to reveal the catabolic roles of these genes of strain QD168 in the degradation of aromatic compounds. Even though, genes encoding toluene catabolic pathway were not found in the QD168 genome, the presence of dmp genes (phenol hydroxylase) likely explains QD168 growth on toluene. In Arthrobacter sp. W1 and Comamonas testosteroni R5, a phenol hydroxylase, encoded by dmp genes, catalyzes the oxidation of benzene and toluene (Teramoto et al., 1999; Ma et al., 2013).
Among the Alcaligenes spp. analyzed in this study, five strains of A. faecalis (JQ135, BDB4, P156, DSM 30030T, and ZD02) possessed the dmpKLMNOP gene cluster. Strains QD168, BU33N and JQ135, members of the A. aquatilis phylogenomic cluster (cluster III), possess the dmp gene cluster. The presence of the dmpRKLMNOP gene cluster, encoding a multicomponent phenol hydroxylase, and a DmpR transcriptional regulator in strain QD168, may explain its ability to degrade benzene. Accordingly, the dmpP gene that encodes the phenol hydroxylase protein P5, was up-regulated during growth of A. aquatilis QD168 on benzene (Figure 5C). Detection of phenol during benzene-degradation, suggests a monooxygenation catalytic step and subsequent oxidation to catechol, as a central intermediate (Figure 5D). In agreement with the proposed catechol ortho-cleavage catabolic route, a significant up-regulation of the catA gene was observed (Figure 5C). The phenol catabolic pathway via meta-cleavage, encoded by the dmpKMNLOPQBCDEFGHI gene cluster, has been reported in Pseudomonas sp. CF600 (Shingler et al., 1992). The DmpR transcriptional activator of strain CF600 is regulated by benzene, phenol and catechol (Shingler and Moore, 1994). An ortho-cleavage pathway for phenol- and benzene-degradation, encoded by the phc gene cluster, has been described in Pseudomonas sp. M1 (Santos and Sá-Correia, 2007). The phc gene cluster is regulated by the transcription factor, PhcR, possessing high similarity to the DmpR transcriptional regulator of strain CF600 (Shingler and Moore, 1994). Similarly, in strain QD168, degradation of benzene probably occurs via an ortho-cleavage pathway (Figure 4D).
Although several strains of Alcaligenes spp. have been described for their ability to degrade some aromatic compounds, information about the physiological and stress response of Alcaligenes spp. in the presence of environmental pollutants is scarce. Oxidative stress during the degradation of aromatic compounds has been reported (Agulló et al., 2007; Ponce et al., 2011; Kim and Park, 2014). In this study, a broad repertoire of genes encoding antioxidant and detoxifying enzymes were identified in A. aquatilis strain QD168, suggesting an efficient response to redox imbalance. Genes-encoding scavenging enzymes were highly represented by catalases (two cat genes), alkyl hydroperoxide reductases (four ahp genes), thioredoxin (six trx genes), and peroxiredoxin/peroxidase (six genes). Moreover, five transcriptional regulators involved in oxidative stress response were identified in the QD168 genome. In E. coli, the regulation of the cat, ahp and trx genes during exposure to H2O2 is mainly mediated by the transcriptional regulator, OxyR (Imlay, 2013). An ortholog of OxyR regulator was identified in the QD168 genome, as well as two orthologs of the SoxR transcriptional regulator. In bacteria, the redox-sensitive transcriptional activator SoxR responds to redox-active compounds, such as PQ (Pomposiello and Demple, 2001). The OhrR organic hydroperoxide resistance repressor was identified in the QD168 genome, which has been described in Bacillus subtilis as a repressor involved in the organic peroxide response (Fuangthong et al., 2001). Moreover, the HTH-type transcriptional regulator, PerR, identified in strain QD168, has been further studied in B. subtilis as an inducible transcriptional regulator under H2O2 exposure (Fuangthong et al., 2002). These results suggest a complex regulatory strategy of strain QD168 responding to oxidative stress under environmental pressures. The phylogeny lineage of OxyR revealed three distinctive OxyR clades, wherein OxyR of A. aquatilis QD168 formed a singleton. The SoxR1 and SoxR2 phylogenetic analyses revealed that SoxR1 of A. aquatilis QD168 formed a singleton ungrouped with other SoxR orthologs. Interestingly, SoxR2 from strain QD168 grouped with Gammaproteobacteria orthologous proteins. Within this main phylogenetic clade, two subgroups of SoxR, including those in enteric (E. coli K-12, S. typhi CT18 and S. typhimurium LT2) and non-enteric bacteria (P. putida KT2440 and A. oleovorans DR1), were clearly distinguished. In enteric bacteria, the oxidized form of the transcription factor, SoxR, induces the expression of the transcription factor, SoxS, activating more than 100 genes. In non-enteric bacteria, a different response mechanism occurs, in which SoxS is not present. SoxR directly activates, not only the expression of genes involved in antioxidant defense, but also up-regulates mono- and dioxygenases-encoding genes (Dietrich and Kiley, 2011; Kim et al., 2017). Accordingly, SoxR2 of A. aquatilis QD168 clustered with proteins from non-enteric bacteria and the soxS gene is not present in its genome.
In agreement with the broad repertoire of identified stress response genes, an unusual resistance to PQ (0.2 M) and tolerance to H2O2 (1 mM) was observed in strain QD168. Notably, inhibitory concentrations of H2O2 and PQ in strain QD168 were significantly higher compared to those previously reported in E. coli. Inhibition of E. coli growth has been reported to occur at low H2O2 concentrations (0.03 mM), inactivating key enzymes containing active-site sulfhydryl residues (Seaver and Imlay, 2001). Resistance to H2O2 and PQ in strain QD168 may correlate with the number of genes-encoding ROS scavenging enzymes, and transcriptional regulators identified for redox balance. Stress-related transcriptional regulators found in strain QD168 probably regulate the oxidative stress response to H2O2 and PQ.
One of the most important types of marine environmental stress is a change in the external osmotic conditions. Consequently, bacterial cells have developed a broad and sophisticated repertoire to counteract drastic changes in external osmotic conditions (Krämer, 2010). A mechanism to maintain osmotic balance is the production of compatible solutes as betaines (glycine betaine and derivatives), amino acids (proline, glutamate, glutamine, and alanine) and ectoines (ectoine and hydroxyectoine). A. aquatilis QD168 possesses four genes encoding ectoine/5-hydroxyectoine synthesis (ectABCD), and four genes encoding transports for these solutes (ehuBCDA), which may explain its resistance to 10% (w v-1) NaCl (Figure 8). Alcaligenes spp. tolerance to NaCl has been reported previously (Lu et al., 2017). A. aquatilis CCTCC 2015279T showed a salt tolerance to 7% (w v-1) NaCl, while A. faecalis subsp. faecalis CGMCC 11786T, A. faecalis subsp. parafaecalis DSM 13975T and A. faecalis subsp. phenolicus DSM 16503T tolerate 8% (w v-1) NaCl. A. endophyticus AER10T showed tolerance to 10% (w v-1) NaCl (Lu et al., 2017). The ectoine biosynthetic cluster has been reported in A. faecalis subsp. phenolicus MB207 (Basharat et al., 2018). The presence of ectoine hydroxylase (ectD) gene within the ect gene cluster of strain QD168 is probably also involved in its high tolerance to salt stress. The production of 5-hydroxyectoine by ectoine hydroxylase confers additional protective properties (Pastor et al., 2010).
Overall, this study describes a phylogenetic analysis of the marine strain A. aquatilis QD168 isolated from a highly polluted environment in Quintero Bay, Central Chile. Genomic and physiological analyses elucidated the adaptive mechanisms of strain QD168 to overcome this hostile environment. A. aquatilis QD168 possesses an unusually wide range of enzymatic machinery to survive environmental stressors, including toxic compounds, high salinity, and oxidative stress, which can be suitable for bioremediation processes in unfavorable conditions.
RD, VM, BB-S, EC-N, and MS conceived and designed the experiments. RD performed the experiments. RD, VM, BB-S, LR-C, FS-S, EM, EC-N, and MS analyzed the data. MS, EC-N, and BB-S contributed reagents, materials, and analysis tools. RD, VM, BB-S, and LR-C prepared figures and/or tables. RD, VM, BB-S, LR-C, FS-S, EM, and MS authored or reviewed drafts of the manuscript.
This study was supported by FONDECYT 1151174 (MS), 11160905 (EC-N) (http://www.fondecyt.cl), CONICYT Programa de Investigación Asociativa (PIA) Anillo GAMBIO ACT172128 (MS) and USM (MS) (http://www.usm.cl) grants. FS-S was supported by stipends for Basic and Advanced Research from the CCUG, through the Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00528/full#supplementary-material
Abbas, S., Ahmed, I., Iida, T., Lee, Y.-J., Busse, H.-J., Fujiwara, T., et al. (2015). A heavy-metal tolerant novel bacterium, Alcaligenes pakistanensis sp. nov., isolated from industrial effluent in Pakistan. Antonie Van Leeuwenhoek 108, 859–870. doi: 10.1007/s10482-015-0540-1
Adebusoye, S. A., Ilori, M. O., Amund, O. O., Teniola, O. D., and Olatope, S. O. (2007). Microbial degradation of petroleum hydrocarbons in a polluted tropical stream. World J. Microbiol. Biotechnol. 23, 1149–1159. doi: 10.1007/s11274-007-9345-3
Agulló, L., Cámara, B., Martínez, P., Latorre, V., and Seeger, M. (2007). Response to (chloro)biphenyls of the polychlorobiphenyl-degrader Burkholderia xenovorans LB400 involves stress proteins also induced by heat shock and oxidative stress. FEMS Microbiol. Lett. 267, 167–175. doi: 10.1111/j.1574-6968.2006.00554.x
Agulló, L., Romero-Silva, M. J., Domenech, M., and Seeger, M. (2017). p-Cymene promotes its catabolism through the p-cymene and the p-cumate pathways, activates a stress response and reduces the biofilm formation in Burkholderia xenovorans LB400. PLoS One 12:e0169544. doi: 10.1371/journal.pone.0169544
Ahmed, A. M., Lyautey, E., Bonnineau, C., Dabrin, A., and Pesce, S. (2018). Environmental concentrations of copper, alone or in mixture with arsenic, can impact river sediment microbial community structure and functions. Front. Microbiol. 9:1852. doi: 10.3389/fmicb.2018.01852
Al Laham, N., Chavda, K. D., Cienfuegos-Gallet, A. V., Kreiswirth, B. N., and Chen, L. (2017). Genomic characterization of VIM metallo-β-lactamase-producing Alcaligenes faecalis from Gaza, Palestine. Antimicrob. Agents Chemother. 61:e01499-17. doi: 10.1128/AAC.01499-17
Baek, K., Bae, S. S., Jung, J., and Chung, D. (2018). Complete genome sequence of Marinobacterium aestuarii ST58-10 T, a benzene-degrading bacterium isolated from Estuarine Sediment. Microbiol. Resour. Announc. 7:e00971-18. doi: 10.1128/MRA.00971-18
Basharat, Z., Yasmin, A., He, T., and Tong, Y. (2018). Genome sequencing and analysis of Alcaligenes faecalis subsp. phenolicus MB207. Sci. Rep. 8:3616. doi: 10.1038/s41598-018-21919-4
Bateman, A., Martin, M. J., O’Donovan, C., Magrane, M., Apweiler, R., Alpi, E., et al. (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Bushnell, L. D., and Haas, H. F. (1941). The utilization of certain hydrocarbons by microorganisms. J. Bacteriol. 41, 653–673.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chain, P. S. G., Denef, V. J., Konstantinidis, K. T., Vergez, L. M., Agullo, L., Reyes, V. L., et al. (2006). Burkholderia xenovorans LB400 harbors a multi-replicon, 9.73-Mbp genome shaped for versatility. Proc. Natl. Acad. Sci. U.S.A. 103, 15280–15287. doi: 10.1073/pnas.0606924103
Chen, Q., Li, J., Liu, M., Sun, H., and Bao, M. (2017). Study on the biodegradation of crude oil by free and immobilized bacterial consortium in marine environment. PLoS One 12:e0174445. doi: 10.1371/journal.pone.0174445
Chirino, B., Strahsburger, E., Agulló, L., González, M., and Seeger, M. (2013). Genomic and functional analyses of the 2-aminophenol catabolic pathway and partial conversion of its substrate into picolinic acid in Burkholderia xenovorans LB400. PLoS One 8:e75746. doi: 10.1371/journal.pone.0075746
Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
De, J., and Ramaiah, N. (2007). Characterization of marine bacteria highly resistant to mercury exhibiting multiple resistances to toxic chemicals. Ecol. Indic. 7, 511–520. doi: 10.1016/j.ecolind.2006.05.002
Dietrich, L. E. P., and Kiley, P. J. (2011). A shared mechanism of SoxR activation by redox-cycling compounds. Mol. Microbiol. 79, 1119–1122. doi: 10.1111/j.1365-2958.2011.07552.x
Durán, R. E., Barra-Sanhueza, B., Salvà-Serra, F., Méndez, V., Jaén-Luchoro, D., Moore, E. R. B., et al. (2019). Complete genome sequence of the marine hydrocarbon degrader Alcaligenes aquatilis QD168, isolated from crude oil-polluted sediment of Quintero Bay, Central Chile. Microbiol. Resour. Announc. 8:e01664-18. doi: 10.1128/MRA.01664-18
Fuangthong, M., Atichartpongkul, S., Mongkolsuk, S., and Helmann, J. D. (2001). OhrR is a repressor of ohrA, a key organic hydroperoxide resistance determinant in Bacillus subtilis. J. Bacteriol. 183, 4134–4141. doi: 10.1128/JB.183.14.4134-4141.2001
Fuangthong, M., Herbig, A. F., Bsat, N., and Helmann, J. D. (2002). Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J. Bacteriol. 184, 3276–3286. doi: 10.1128/JB.184.12.3276-3286.2002
Fuentes, S., Barra, B., Caporaso, J. G., and Seeger, M. (2016). From rare to dominant: a fine-tuned soil bacterial bloom during petroleum hydrocarbon bioremediation. Appl. Environ. Microbiol. 82, 888–896. doi: 10.1128/AEM.02625-15
Fuentes, S., Méndez, V., Aguila, P., and Seeger, M. (2014). Bioremediation of petroleum hydrocarbons: catabolic genes, microbial communities, and applications. Appl. Microbiol. Biotechnol. 98, 4781–4794. doi: 10.1007/s00253-014-5684-9
Gallardo-Cerda, J., Levihuan, J., Lavín, P., Oses, R., Atala, C., Torres-Díaz, C., et al. (2018). Antarctic rhizobacteria improve salt tolerance and physiological performance of the Antarctic vascular plants. Polar Biol. 41, 1973–1982. doi: 10.1007/s00300-018-2336-z
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Garrity, G. N., Bell, J. A., and Lilburn, T. (2005). “Order Burkholderiales ord. nov,” in Bergeys Manual of Systematic Bacteriology, eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York, NY: Springer), 575. doi: 10.1007/0-387-29298-5_2
Harayama, S., Kasai, Y., and Hara, A. (2004). Microbial communities in oil-contaminated seawater. Curr. Opin. Biotechnol. 15, 205–214. doi: 10.1016/j.copbio.2004.04.002
Head, I. M., Jones, D. M., and Röling, W. F. M. (2006). Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 4, 173–182. doi: 10.1038/nrmicro1348
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F., and Vandesompele, J. (2007). qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8:R19. doi: 10.1186/gb-2007-8-2-r19
Hopper, D. J., and Kaderbhai, M. A. (1999). 2,4’-dihydroxyacetophenone dioxygenase (EC 1.13.11.41) from Alcaligenes sp. 4HAP: a novel enzyme with an atypical dioxygenase sequence. Biochem. J. 344, 397–402. doi: 10.1042/0264-6021:3440397
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). EGGNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Imlay, J. A. (2013). The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol. 11, 443–454. doi: 10.1038/nrmicro3032
Jiang, B., Zhou, Z., Dong, Y., Tao, W., Wang, B., Jiang, J., et al. (2015). Biodegradation of benzene, toluene, ethylbenzene, and o-, m-, and p-xylenes by the newly isolated bacterium Comamonas sp. JB. Appl. Biochem. Biotechnol. 176, 1700–1708. doi: 10.1007/s12010-015-1671-6
Jiménez, J. I., Miñambres, B., García, J. L., and Díaz, E. (2002). Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ. Microbiol. 4, 824–841. doi: 10.1046/j.1462-2920.2002.00370.x
Ju, S., Zheng, J., Lin, J., Geng, C., Zhu, L., Guan, Z., et al. (2016). The complete genome sequence of Alcaligenes faecalis ZD02, a novel potential bionematocide. J. Biotechnol. 218, 73–74. doi: 10.1016/j.jbiotec.2015.12.001
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, J., Park, C., Imlay, J. A., and Park, W. (2017). Lineage-specific SoxR-mediated regulation of an endoribonuclease protects non-enteric bacteria from redox-active compounds. J. Biol. Chem. 292, 121–133. doi: 10.1074/jbc.M116.757500
Kim, J., and Park, W. (2014). Oxidative stress response in Pseudomonas putida. Appl. Microbiol. Biotechnol. 98, 6933–6946. doi: 10.1007/s00253-014-5883-4
Kiyohara, H., Nagao, K., Kouno, K., and Yano, K. (1982). Phenanthrene-degrading phenotype of Alcaligenes faecalis AFK2. Appl. Environ. Microbiol. 43, 458–461.
Kolde, R. (2015). pheatmap: Pretty Heatmaps in R package. R Package. version 1.0.8. Available at: https://CRAN.R-project.org/package=pheatmap
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Krämer, R. (2010). Bacterial stimulus perception and signal transduction: response to osmotic stress. Chem. Rec. 10, 217–229. doi: 10.1002/tcr.201000005
Kühnemann, G. (1911). Zur Identifizierung des Bacillus faecalis alcaligenes. Zentr. Bakt. Parasitenk. 57, 469–471.
Kumar, A., Bhunia, B., Dasgupta, D., Mandal, T., Dey, A., Datta, S., et al. (2013). Optimization of culture condition for growth and phenol degradation by Alcaligenes faecalis JF339228 using Taguchi methodology. Desalin. Water Treat. 51, 3153–3163. doi: 10.1080/19443994.2012.749021
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). Partitionfinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278. doi: 10.1093/bioinformatics/btu531
Liu, X., Huang, D., Wu, J., Yu, C., Zhou, R., Liu, C., et al. (2016). The genome sequence of Alcaligenes faecalis NBIB-017 contains genes with potentially high activities against Erwinia carotovora. Genome Announc. 4:e00222-16. doi: 10.1128/genomeA.00222-16
Lu, C. Y., Li, Y. Q., Tian, Y., Han, M. X., Rao, M. P. N., Li, Y. R., et al. (2017). Alcaligenes endophyticus sp. nov., isolated from roots of Ammodendron bifolium. Int. J. Syst. Evol. Microbiol. 67, 939–943. doi: 10.1099/ijsem.0.001719
Ma, F., Shi, S. N., Sun, T. H., Li, A., Zhou, J. T., and Qu, Y. Y. (2013). Biotransformation of benzene and toluene to catechols by phenol hydroxylase from Arthrobacter sp. W1. Appl. Microbiol. Biotechnol. 97, 5097–5103. doi: 10.1007/s00253-012-4301-z
McLeod, M. P., Warren, R. L., Hsiao, W. W. L., Araki, N., Myhre, M., Fernandes, C., et al. (2006). The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. U.S.A. 103, 15582–15587. doi: 10.1073/pnas.0607048103
Méndez, V., Agulló, L., González, M., and Seeger, M. (2011). The homogentisate and homoprotocatechuate central pathways are involved in 3- and 4-hydroxyphenylacetate degradation by Burkholderia xenovorans LB400. PLoS One 6:e17583. doi: 10.1371/journal.pone.0017583
Méndez, V., Fuentes, S., Morgante, V., Hernández, M., González, M., Moore, E., et al. (2017). Novel hydrocarbon-degrading and heavy metal-resistant Acinetobacter, Kocuria and Pseudomonas strains isolated from a crude oil polluted soil in Central Chile. J. Soil Sci. Plant Nutr. 17, 1074–1087. doi: 10.4067/S0718-95162017000400017
Michas, A., Vestergaard, G., Trautwein, K., Avramidis, P., Hatzinikolaou, D. G., Vorgias, C. E., et al. (2017). More than 2500 years of oil exposure shape sediment microbiomes with the potential for syntrophic degradation of hydrocarbons linked to methanogenesis. Microbiome 5:118. doi: 10.1186/s40168-017-0337-8
Orellana, R., Macaya, C., Bravo, G., Dorochesi, F., Cumsille, A., Valencia, R., et al. (2018). Living at the frontiers of life: extremophiles in Chile and their potential for bioremediation. Front. Microbiol. 9:2309. doi: 10.3389/fmicb.2018.02309
Overwin, H., González, M., Méndez, V., Seeger, M., Wray, V., and Hofer, B. (2012). Dioxygenation of the biphenyl dioxygenation product. Appl. Environ. Microbiol. 78, 4529–4532. doi: 10.1128/AEM.00492-12
Palma-Fleming, H., Cornejo, C., González, M., Pérez, V., González, M., Gutierrez, E., et al. (2008). Polycyclic aromatic hydrocarbons and polychlorinated biphenyls in coastal environments of Valdivia and Valparaíso, Chile. J. Chil. Chem. Soc. 53, 1393–1398. doi: 10.4067/S0717-97072008000200020
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-7
Pastor, J. M., Salvador, M., Argandoǹa, M., Bernal, V., Reina-Bueno, M., Csonka, L. N., et al. (2010). Ectoines in cell stress protection: uses and biotechnological production. Biotechnol. Adv. 28, 782–801. doi: 10.1016/j.biotechadv.2010.06.005
Pérez-Pantoja, D., De La Iglesia, R., Pieper, D. H., and González, B. (2008). Metabolic reconstruction of aromatic compounds degradation from the genome of the amazing pollutant-degrading bacterium Cupriavidus necator JMP134. FEMS Microbiol. Rev. 32, 736–794. doi: 10.1111/j.1574-6976.2008.00122.x
Pérez-Pantoja, D., Donoso, R., Agulló, L., Córdova, M., Seeger, M., Pieper, D. H., et al. (2012). Genomic analysis of the potential for aromatic compounds biodegradation in Burkholderiales. Environ. Microbiol. 14, 1091–1117. doi: 10.1111/j.1462-2920.2011.02613.x
Piraino, E., Owens, E., Rios, J., and Graham, A. (2017). Oil behaviour and the response to a sunken oil spill of slurry in Quintero Bay, Chile. Int. Oil Spill Conf. Proc. 2017, 124–133. doi: 10.7901/2169-3358-2017.1.124
Pomposiello, P. J., and Demple, B. (2001). Redox-operated genetic switches: the SoxR and OxyR transcription factors. Trends Biotechnol. 19, 109–114. doi: 10.1016/S0167-7799(00)01542-0
Ponce, B. L., Latorre, V. K., González, M., and Seeger, M. (2011). Antioxidant compounds improved PCB-degradation by Burkholderia xenovorans strain LB400. Enzyme Microb. Technol. 49, 509–516. doi: 10.1016/j.enzmictec.2011.04.021
Qiu, J., Liu, B., Zhao, L., Zhang, Y., Cheng, D., Yan, X., et al. (2018). A novel degradation mechanism for pyridine derivatives in Alcaligenes faecalis JQ135. Appl. Environ. Microbiol. 84, e910–e918. doi: 10.1128/AEM.00910-18
Qiu, J., Zhang, J., Zhang, Y., Wang, Y., Tong, L., Hong, Q., et al. (2017). Biodegradation of picolinic acid by a newly isolated bacterium Alcaligenes faecalis Strain JQ135. Curr. Microbiol. 74, 508–514. doi: 10.1007/s00284-017-1205-2
Quiroz-Castañeda, R. E., Mendoza-Mejía, A., Obregón-Barboza, V., Martínez-Ocampo, F., Hernández-Mendoza, A., Martínez-Garduño, F., et al. (2015). Identification of a new Alcaligenes faecalis Strain MOR02 and assessment of its toxicity and pathogenicity to insects. Biomed Res. Int. 2015:570243. doi: 10.1155/2015/570243
Regar, R. K., Gaur, V. K., Mishra, G., Jadhao, S., Kamthan, M., and Manickam, N. (2016). Draft genome sequence of Alcaligenes faecalis strain IITR89, an indole-oxidizing bacterium. Genome Announc. 4:e00067-16. doi: 10.1128/genomeA.00067-16
Rehfuss, M., and Urban, J. (2005). Alcaligenes faecalis subsp. phenolicus subsp. nov. a phenol-degrading, denitrifying bacterium isolated from a graywater bioprocessor. Syst. Appl. Microbiol. 28, 421–429. doi: 10.1016/j.syapm.2005.03.003
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rojas, L. A., Yañez, C., González, M., Lobos, S., Smalla, K., and Seeger, M. (2011). Characterization of the metabolically modified heavy metal-resistant Cupriavidus metallidurans strain MSR33 generated for mercury bioremediation. PLoS One 6:e17555. doi: 10.1371/journal.pone.0017555
Romero-Silva, M. J., Méndez, V., Agulló, L., and Seeger, M. (2013). Genomic and functional analyses of the gentisate and protocatechuate ring-cleavage pathways and related 3-hydroxybenzoate and 4-hydroxybenzoate peripheral pathways in Burkholderia xenovorans LB400. PLoS One 8:e56038. doi: 10.1371/journal.pone.0056038
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Saavedra, M., Acevedo, F., González, M., and Seeger, M. (2010). Mineralization of PCBs by the genetically modified strain Cupriavidus necator JMS34 and its application for bioremediation of PCB in soil. Appl. Microbiol. Biotechnol. 87, 1543–1554. doi: 10.1007/s00253-010-2575-6
Sagarkar, S., Bhardwaj, P., Yadav, T. C., Qureshi, A., Khardenavis, A., Purohit, H. J., et al. (2014). Draft genome sequence of atrazine-utilizing bacteria isolated from indian agricultural soil. Genome Announc. 2:e01149-13. doi: 10.1128/genomeA.01149-13
Sandoval, J. M., Arenas, F. A., and Vásquez, C. C. (2011). Glucose-6-phosphate dehydrogenase protects Escherichia coli from tellurite-mediated oxidative stress. PLoS One 6:e25573. doi: 10.1371/journal.pone.0025573
Santos, P. M., and Sá-Correia, I. (2007). Characterization of the unique organization and co-regulation of a gene cluster required for phenol and benzene catabolism in Pseudomonas sp. M1. J. Biotechnol. 131, 371–378. doi: 10.1016/j.jbiotec.2007.07.941
Schroll, G., Busse, H. J., Parrer, G., Rölleke, S., Lubitz, W., and Denner, E. B. (2001). Alcaligenes faecalis subsp. parafaecalis subsp. nov., a bacterium accumulating poly-beta-hydroxybutyrate from acetone-butanol bioprocess residues. Syst. Appl. Microbiol. 24, 37–43. doi: 10.1078/0723-2020-00001
Seaver, L. C., and Imlay, J. A. (2001). Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 183, 7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shingler, V., and Moore, T. (1994). Sensing of aromatic compounds by the DmpR transcriptional activator of phenol-catabolizing Pseudomonas sp. strain CF600. J. Bacteriol. 176, 1555–1560. doi: 10.1128/jb.176.6.1555-1560.1994
Shingler, V., Powlowski, J., and Marklund, U. (1992). Nucleotide sequence and functional analysis of the complete phenol/3,4-dimethylphenol catabolic pathway of Pseudomonas sp. strain CF600. J. Bacteriol. 174, 711–724. doi: 10.1128/jb.174.3.711-724.1992
Singha, L. P., Kotoky, R., and Pandey, P. (2017). Draft genome sequence of Alcaligenes faecalis BDB4, a polyaromatic hydrocarbon-degrading bacterium isolated from crude oil-contaminated soil. Genome Announc. 5:e01346-17. doi: 10.1128/genomeA.01346-17
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Teramoto, M., Futamata, H., Harayama, S., and Watanabe, K. (1999). Characterization of a high-affinity phenol hydroxylase from Comamonas testosteroni R5 by gene cloning, and expression in Pseudomonas aeruginosa PAO1c. Mol. Gen. Genet. 262, 552–558. doi: 10.1007/s004380051117
Wang, L., Qiao, N., and Shao, Z. (2008). Isolation, gene detection and solvent tolerance of benzene, toluene and xylene degrading bacteria from nearshore surface water and Pacific Ocean sediment. Extremophiles 12, 335–342. doi: 10.1007/s00792-007-0136-4
Winslow, C. E., Broadhurst, J., Buchanan, R. E., Krumwiede, C., Rogers, L. A., and Smith, G. H. (1920). The families and genera of the bacteria: final report of the committee of the Society of American Bacteriologists on characterization and classification of bacterial types. J. Bacteriol. 5, 191–229.
Wu, M., and Scott, A. J. (2012). Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034. doi: 10.1093/bioinformatics/bts079
Keywords: Alcaligenes aquatilis, Alcaligenes, aromatic catabolism, benzene, oxidative stress, osmotolerance, phylogenomics, Quintero Bay
Citation: Durán RE, Méndez V, Rodríguez-Castro L, Barra-Sanhueza B, Salvà-Serra F, Moore ERB, Castro-Nallar E and Seeger M (2019) Genomic and Physiological Traits of the Marine Bacterium Alcaligenes aquatilis QD168 Isolated From Quintero Bay, Central Chile, Reveal a Robust Adaptive Response to Environmental Stressors. Front. Microbiol. 10:528. doi: 10.3389/fmicb.2019.00528
Received: 03 November 2018; Accepted: 01 March 2019;
Published: 05 April 2019.
Edited by:
Gloria Paz Levicán, Universidad de Santiago de Chile, ChileReviewed by:
Kim Yrjälä, Zhejiang Agriculture and Forestry University, ChinaCopyright © 2019 Durán, Méndez, Rodríguez-Castro, Barra-Sanhueza, Salvà-Serra, Moore, Castro-Nallar and Seeger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Seeger, bWljaGFlbC5zZWVnZXJAZ21haWwuY29t; bWljaGFlbC5zZWVnZXJAdXNtLmNs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.