 Germán L. Rosano
Germán L. Rosano Eduardo A. Ceccarelli
Eduardo A. Ceccarelli- 1Instituto de Biología Molecular y Celular de Rosario, Consejo Nacional de Investigaciones Científicas y Técnicas, Rosario, Argentina
- 2Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Rosario, Argentina
Escherichia coli is one of the organisms of choice for the production of recombinant proteins. Its use as a cell factory is well-established and it has become the most popular expression platform. For this reason, there are many molecular tools and protocols at hand for the high-level production of heterologous proteins, such as a vast catalog of expression plasmids, a great number of engineered strains and many cultivation strategies. We review the different approaches for the synthesis of recombinant proteins in E. coli and discuss recent progress in this ever-growing field.
Introduction
There is no doubt that the production of recombinant proteins in microbial systems has revolutionized biochemistry. The days where kilograms of animal and plant tissues or large volumes of biological fluids were needed for the purification of small amounts of a given protein are almost gone. Every researcher that embarks on a new project that will need a purified protein immediately thinks of how to obtain it in a recombinant form. The ability to express and purify the desired recombinant protein in a large quantity allows for its biochemical characterization, its use in industrial processes and the development of commercial goods.
At the theoretical level, the steps needed for obtaining a recombinant protein are pretty straightforward. You take your gene of interest, clone it in whatever expression vector you have at your disposal, transform it into the host of choice, induce and then, the protein is ready for purification and characterization. In practice, however, dozens of things can go wrong. Poor growth of the host, inclusion body (IB) formation, protein inactivity, and even not obtaining any protein at all are some of the problems often found down the pipeline.
In the past, many reviews have covered this topic with great detail (Makrides, 1996; Baneyx, 1999; Stevens, 2000; Jana and Deb, 2005; Sorensen and Mortensen, 2005). Collectively, these papers gather more than 2000 citations. Yet, in the field of recombinant protein expression and purification, progress is continuously being made. For this reason, in this review, we comment on the most recent advances in the topic. But also, for those with modest experience in the production of heterologous proteins, we describe the many options and approaches that have been successful for expressing a great number of proteins over the last couple of decades, by answering the questions needed to be addressed at the beginning of the project. Finally, we provide a troubleshooting guide that will come in handy when dealing with difficult-to-express proteins.
First Question: Which Organism to Use?
The choice of the host cell whose protein synthesis machinery will produce the precious protein will initiate the outline of the whole process. It defines the technology needed for the project, be it a variety of molecular tools, equipment, or reagents. Among microorganisms, host systems that are available include bacteria, yeast, filamentous fungi, and unicellular algae. All have strengths and weaknesses and their choice may be subject to the protein of interest (Demain and Vaishnav, 2009; Adrio and Demain, 2010). For example, if eukaryotic post-translational modifications (like protein glycosylation) are needed, a prokaryotic expression system may not be suitable (Sahdev et al., 2008). In this review, we will focus specifically on Escherichia coli. Other systems are described in excellent detail in accompanying articles of this series.
The advantages of using E. coli as the host organism are well known. (i) It has unparalleled fast growth kinetics. In glucose-salts media and given the optimal environmental conditions, its doubling time is about 20 min (Sezonov et al., 2007). This means that a culture inoculated with a 1/100 dilution of a saturated starter culture may reach stationary phase in a few hours. However, it should be noted that the expression of a recombinant protein may impart a metabolic burden on the microorganism, causing a considerable decrease in generation time (Bentley et al., 1990). (ii) High cell density cultures are easily achieved. The theoretical density limit of an E. coli liquid culture is estimated to be about 200 g dry cell weight/l or roughly 1 × 1013 viable bacteria/ml (Lee, 1996; Shiloach and Fass, 2005). However, exponential growth in complex media leads to densities nowhere near that number. In the simplest laboratory setup (i.e., batch cultivation of E. coli at 37°C, using LB media), <1 × 1010 cells/ml may be the upper limit (Sezonov et al., 2007), which is less than 0.1% of the theoretical limit. For this reason, high cell-density culture methods were designed to boost E. coli growth, even when producing a recombinant protein (Choi et al., 2006). Being a workhorse organism, these strategies arose thanks to the wealth of knowledge about its physiology. (iii) Rich complex media can be made from readily available and inexpensive components. (iv) Transformation with exogenous DNA is fast and easy. Plasmid transformation of E. coli can be performed in as little as 5 min (Pope and Kent, 1996).
Second Question: Which Plasmid Should Be Chosen?
The most common expression plasmids in use today are the result of multiple combinations of replicons, promoters, selection markers, multiple cloning sites, and fusion protein/fusion protein removal strategies (Figure 1). For this reason, the catalog of available expression vectors is huge and it is easy to get lost when choosing a suitable one. To make an informed decision, these features have to be carefully evaluated according to the individual needs.
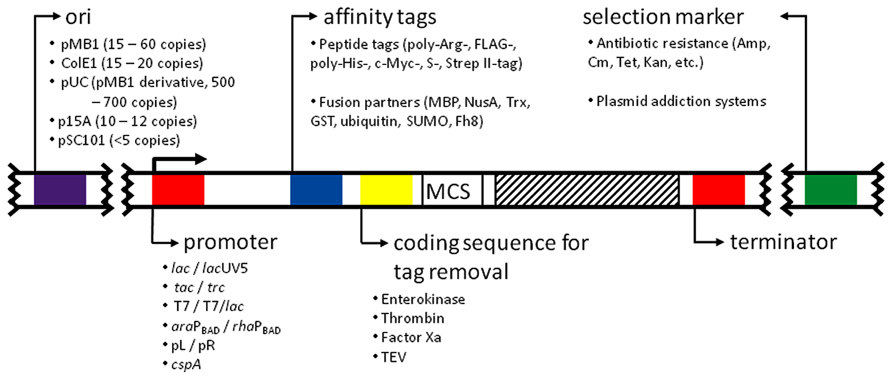
FIGURE 1. Anatomy of an expression vector. The figure depicts the major features present in common expression vectors. All of them are described in the text. The affinity tags and coding sequences for their removal were positioned arbitrarily at the N-terminus for simplicity. MCS, multiple cloning site. Striped patterned box: coding sequence for the desired protein.
Replicon
Genetic elements that undergo replication as autonomous units, such as plasmids, contain a replicon. It consists of one origin of replication together with its associated cis-acting control elements. An important parameter to have in mind when choosing a suitable vector is copy number. The control of copy number resides in the replicon (del Solar and Espinosa, 2000). It is logical to think that high plasmid dosage equals more recombinant protein yield as many expression units reside in the cell. However, a high plasmid number may impose a metabolic burden that decreases the bacterial growth rate and may produce plasmid instability, and so the number of healthy organisms for protein synthesis falls (Bentley et al., 1990; Birnbaum and Bailey, 1991). For this reason, the use of high copy number plasmids for protein expression by no means implies an increase in production yields.
Commonly used vectors, such as the pET series, possess the pMB1 origin (ColE1-derivative, 15–60 copies per cell; Bolivar et al., 1977) while a mutated version of the pMB1 origin is present in the pUC series (500–700 copies per cell; Minton, 1984). The wild-type ColE1 origin (15–20 copies per cell; Lin-Chao and Bremer, 1986; Lee et al., 2006) can be found in the pQE vectors (Qiagen). They all belong to the same incompatibility group meaning that they cannot be propagated together in the same cell as they compete with each other for the replication machinery (del Solar et al., 1998; Camps, 2010). For the dual expression of recombinant proteins using two plasmids, systems with the p15A ori are available (pACYC and pBAD series of plasmids, 10–12 copies per cell; Chang and Cohen, 1978; Guzman et al., 1995). Though rare, triple expression can be achieved by the use of the pSC101 plasmid. This plasmid is under a stringent control of replication, thus it is present in a low copy number (<5 copies per cell; Nordstrom, 2006). The use of plasmids bearing this replicon can be an advantage in cases where the presence of a high dose of a cloned gene or its product produces a deleterious effect to the cell (Stoker et al., 1982; Wang and Kushner, 1991). Alternatively, the use of the Duet vectors (Novagen) simplifies dual expression by allowing cloning of two genes in the same plasmid. The Duet plasmids possess two multiple cloning sites, each preceded by a T7 promoter, a lac operator and a ribosome binding site. By combining different compatible Duet vectors, up to eight recombinant proteins can be produced from four expression plasmids.
Promoter
The staple in prokaryotic promoter research is undoubtedly the lac promoter, key component of the lac operon (Müller-Hill, 1996). The accumulated knowledge in the functioning of the system allowed for its extended use in expression vectors. Lactose causes induction of the system and this sugar can be used for protein production. However, induction is difficult in the presence of readily metabolizable carbon sources (such as glucose present in rich media). If lactose and glucose are present, expression from the lac promoter is not fully induced until all the glucose has been utilized. At this point (low glucose), cyclic adenosine monophosphate (cAMP) is produced, which is necessary for complete activation of the lac operon (Wanner et al., 1978; Postma and Lengeler, 1985). This positive control of expression is known as catabolite repression. In accordance, cAMP levels are low in cells growing in lac operon-repressing sugars, and this correlates with lower rates of expression of the lac operon (Epstein et al., 1975). Also, glucose abolishes lactose uptake because lactose permease is inactive in the presence of glucose (Winkler and Wilson, 1967). To achieve expression in the presence of glucose, a mutant that reduces (but does not eliminate) sensitivity to catabolite regulation was introduced, the lacUV5 promoter (Silverstone et al., 1970; Lanzer and Bujard, 1988). However, when present in multicopy plasmids, both promoters suffer from the disadvantage of sometimes having unacceptably high levels of expression in the absence of inducer (a.k.a. “leakiness”) due to titration of the low levels of the lac promoter repressor protein LacI from the single chromosomal copy of its gene (about 10 molecules per cell; Müller-Hill et al., 1968). Basal expression control can be achieved by the introduction of a mutated promoter of the lacI gene, called lacIQ, that leads to higher levels of expression (almost 10-fold) of LacI (Calos, 1978). The lac promoter and its derivative lacUV5 are rather weak and thus not very useful for recombinant protein production (Deuschle et al., 1986; Makoff and Oxer, 1991). Synthetic hybrids that combine the strength of other promoters and the advantages of the lac promoter are available. For example, the tac promoter consists of the -35 region of the trp (tryptophan) promoter and the -10 region of the lac promoter. This promoter is approximately 10 times stronger than lacUV5 (de Boer et al., 1983). Notable examples of commercial plasmids that use the lac or tac promoters to drive protein expression are the pUC series (lacUV5 promoter, Thermo Scientific) and the pMAL series of vectors (tac promoter, NEB).
The T7 promoter system present in the pET vectors (pMB1 ori, medium copy number, Novagen) is extremely popular for recombinant protein expression. This is not surprising as the target protein can represent 50% of the total cell protein in successful cases (Baneyx, 1999; Graumann and Premstaller, 2006). In this system, the gene of interest is cloned behind a promoter recognized by the phage T7 RNA polymerase (T7 RNAP). This highly active polymerase should be provided in another plasmid or, most commonly, it is placed in the bacterial genome in a prophage (λDE3) encoding for the T7 RNAP under the transcriptional control of a lacUV5 promoter (Studier and Moffatt, 1986). Thus, the system can be induced by lactose or its non-hydrolyzable analog isopropyl β-D-1-thiogalactopyranoside (IPTG). Basal expression can be controlled by lacIQ but also by T7 lysozyme co-expression (Moffatt and Studier, 1987). T7 lysozyme binds to T7 RNAP and inhibits transcription initiation from the T7 promoter (Stano and Patel, 2004). In this way, if small amounts of T7 RNAP are produced because of leaky expression of its gene, T7 lysozyme will effectively control unintended expression of heterologous genes placed under the T7 promoter. T7 lysozyme is provided by a compatible plasmid (pLysS or pLysE). After induction, the amount of T7 RNAP produced surpasses the level of polymerase that T7 lysozyme can inhibit. The “free” T7 RNAP can thus engage in transcription of the recombinant gene. Yet another level of control lies in the insertion of a lacO operator downstream of the T7 promoter, making a hybrid T7/lac promoter (Dubendorff and Studier, 1991). All three mechanisms (tight repression of the lac-inducible T7 RNAP gene by lacIQ, T7 RNAP inhibition by T7 lysozyme and presence of a lacO operator after the T7 promoter) make the system ideal for avoiding basal expression.
The problem of leaky expression is a reflection of the negative control of the lac promoter. Promoters that rely on positive control should have lower background expression levels (Siegele and Hu, 1997). This is the case of the araPBAD promoter present in the pBAD vectors (Guzman et al., 1995). The AraC protein has the dual role of repressor/activator. In the absence of arabinose inducer, AraC represses translation by binding to two sites in the bacterial DNA. The protein–DNA complex forms a loop, effectively preventing RNA polymerase from binding to the promoter. Upon addition of the inducer, AraC switches into “activation mode” and promotes transcription from the ara promoter (Schleif, 2000, 2010). In this way, arabinose is absolutely needed for induction.
Another widely used approach is to place a gene under the control of a regulated phage promoter. The strong leftward promoter (pL) of phage lambda directs expression of early lytic genes (Dodd et al., 2005). The promoter is tightly repressed by the λcI repressor protein, which sits on the operator sequences during lysogenic growth. When the host SOS response is triggered by DNA damage, the expression of the protein RecA is stimulated, which in turn catalyzes the self-cleavage of λcI, allowing transcription of pL-controlled genes (Johnson et al., 1981; Galkin et al., 2009). This mechanism is used in expression vectors containing the pL promoter. The SOS response (and recombinant protein expression) can be elicited by adding nalidixic acid, a DNA gyrase inhibitor (Lewin et al., 1989; Shatzman et al., 2001). Another way of activating the promoter is to control λcI production by placing its gene under the influence of another promoter. This two-stage control system has already been described for T7 promoter/T7 RNAP-based vectors. In the pLEX series of vectors (Life Technologies), the λcI repressor gene was integrated into the bacterial chromosome under the control of the trp promoter. In the absence of tryptophan, this promoter is always “on” and λcI is continuously produced. Upon addition of tryptophan, a tryptophan-TrpR repressor complex is formed that tightly binds to the trp operator, thereby blocking λcI repressor synthesis. Subsequently, the expression of the desired gene under the pL promoter ensues (Mieschendahl et al., 1986).
Transcription from all promoters discussed so far is initiated by chemical cues. Systems that respond to physical signals (e.g., temperature or pH) are also available (Goldstein and Doi, 1995). The pL promoter is one example. A mutant λcI repressor protein ( λcI857) is temperature-sensitive and is unstable at temperatures higher than 37°C. E. coli host strains containing the λcI857 protein (either integrated in the chromosome or into a vector) are first grown at 28–30°C to the desired density, and then protein expression is induced by a temperature shift to 40–42°C (Menart et al., 2003; Valdez-Cruz et al., 2010). The industrial advantage of this system lies in part in the fact that during fermentation, heat is usually produced and increasing the temperature in high density cultures is easy. On the other hand, genes under the control of the cold-inducible promoter cspA are induced by a downshift in temperature to 15°C (Vasina et al., 1998). This temperature is ideal for expressing difficult proteins as will be explained in another section. The pCold series of plasmids have a pUC118 backbone (a pUC18 derivative; Vieira and Messing, 1987) with the cspA promoter (Qing et al., 2004; Hayashi and Kojima, 2008). In the original paper, successful expression was achieved for more than 30 recombinant proteins from different sources, reaching levels as high as 20–40% of the total expressed proteins (Qing et al., 2004). However, it should be noted that in various cases the target proteins were obtained in an insoluble form.
Selection Marker
To deter the growth of plasmid-free cells, a resistance marker is added to the plasmid backbone. In the E. coli system, antibiotic resistance genes are habitually used for this purpose. Resistance to ampicillin is conferred by the bla gene whose product is a periplasmic enzyme that inactivates the β-lactam ring of β-lactam antibiotics. However, as the β-lactamase is continuously secreted, degradation of the antibiotic ensues and in a couple of hours, ampicillin is almost depleted (Korpimaki et al., 2003). Under this situation, cells not carrying the plasmid are allowed to increase in number during cultivation. Although not experimentally verified, selective agents in which resistance is based on degradation, like chloramphenicol (Shaw, 1983) and kanamycin (Umezawa, 1979), could also have this problem. For this reason, tetracycline has been shown to be highly stable during cultivation (Korpimaki et al., 2003), because resistance is based on active efflux of the antibiotic from resistant cells (Roberts, 1996).
The cost of antibiotics and the dissemination of antibiotic resistance are major concerns in projects dealing with large-scale cultures. Much effort has been put in the development of antibiotics-free plasmid systems. These systems are based on the concept of plasmid addiction, a phenomenon that occurs when plasmid-free cells are not able to grow or live (Zielenkiewicz and Ceglowski, 2001; Peubez et al., 2010). For example, an essential gene can be deleted from the bacterial genome and then placed on a plasmid. Thus, after cell division, plasmid-free bacteria die. Different subtypes of plasmid-addiction systems exist according to their principle of function: (i) toxin/antitoxin-based systems, (ii) metabolism-based systems, and (iii) operator repressor titration systems (Kroll et al., 2010). While this promising technology has been proved successful in large-scale fermentors (Voss and Steinbuchel, 2006; Peubez et al., 2010), expression systems based on plasmid addiction are still not widely distributed.
Affinity Tags
When devising a project where a purified soluble active recombinant protein is needed (as is often the case), it is invaluable to have means to (i) detect it along the expression and purification scheme, (ii) attain maximal solubility, and (iii) easily purify it from the E. coli cellular milieu. The expression of a stretch of amino acids (peptide tag) or a large polypeptide (fusion partner) in tandem with the desired protein to form a chimeric protein may allow these three goals to be straightforwardly reached (Nilsson et al., 1997).
Being small, peptide tags are less likely to interfere when fused to the protein. However, in some cases they may provoke negative effects on the tertiary structure or biological activity of the fused chimeric protein (Bucher et al., 2002; Klose et al., 2004; Chant et al., 2005; Khan et al., 2012). Vectors are available that allow positioning of the tag on either the N-terminal or the C-terminal end (the latter option being advantageous when a signal peptide is positioned at the N-terminal end for secretion of the recombinant protein, see below). If the three-dimensional structure of the desired protein is available, it is wise to check which end is buried inside the fold and place the tag in the solvent-accessible end. Common examples of small peptide tags are the poly-Arg-, FLAG-, poly-His-, c-Myc-, S-, and Strep II-tags (Terpe, 2003). Since commercial antibodies are available for all of them, the tagged recombinant protein can be detected by Western blot along expression trials, which is extremely helpful when the levels of the desired proteins are not high enough to be detected by SDS-PAGE. Also, tags allow for one-step affinity purification, as resins that tightly and specifically bind the tags are available. For example, His-tagged proteins can be recovered by immobilized metal ion affinity chromatography using Ni2+ or Co2+-loaded nitrilotriacetic acid-agarose resins (Porath and Olin, 1983; Bornhorst and Falke, 2000), while anti-FLAG affinity gels (Sigma-Aldrich) are used for capturing FLAG fusion proteins (Hopp et al., 1988).
On the other hand, adding a non-peptide fusion partner has the extra advantage of working as solubility enhancers (Hammarstrom et al., 2002). The most popular fusion tags are the maltose-binding protein (MBP; Kapust and Waugh, 1999), N-utilization substance protein A (NusA; Davis et al., 1999), thioredoxin (Trx; LaVallie et al., 1993), glutathione S-transferase (GST; Smith and Johnson, 1988), ubiquitin (Baker, 1996) and SUMO (Butt et al., 2005). The reasons why these fusion partners act as solubility enhancers remain unclear and several hypothesis have been proposed (reviewed in Raran-Kurussi and Waugh, 2012). In the case of MBP, it was shown that it possesses an intrinsic chaperone activity (Kapust and Waugh, 1999; Raran-Kurussi and Waugh, 2012). In comparison studies, GST showed the poorest solubility enhancement capabilities (Hammarstrom et al., 2006; Bird, 2011). NusA, MBP, and Trx display the best solubility enhancing properties but their large size may lead to the erroneous assessment of protein solubility (Costa et al., 2013). Indeed, when these tags are removed, the final solubility of the desired product is unpredictable (Esposito and Chatterjee, 2006). For these reasons, smaller tags with strong solubility enhancing effects are desirable. Recently, the 8-kDa calcium binding protein Fh8 from the parasite Fasciola hepatica was shown to be as good as or better than the large tags in terms of solubility enhancement. Moreover, the recombinant proteins maintained their solubility after tag removal (Costa et al., 2013). MBP and GST can be used to purify the fused protein by affinity chromatography, as MBP binds to amylose–agarose and GST to glutathione–agarose. MBP is present in the pMAL series of vectors from NEB and GST in the pGEX series (GE). A peptide tag must be added to the fusion partner-containing protein if an affinity chromatography step is needed in the purification scheme. MBP and GST bind to their substrates non-covalently. On the contrary, the HaloTag7 (Promega) is based on the covalent capture of the tag to the resin, making the system fast and highly specific (Ohana et al., 2009).
A different group of fusion tags are stimulus-responsive tags, which reversibly precipitate out of solution when subjected to the proper stimulus. The addition of β roll tags to a recombinant protein allows for its selective precipitation in the presence of calcium. The final products presented a high purity and the precipitation protocol only takes a couple of minutes (Shur et al., 2013). Another protein-based stimulus-responsive purification tags are elastin-like polypeptides (ELPs), which consist of tandem repeats of the sequence VPGXG, where X is Val, Ala, or Gly in a 5:2:3 ratio (Meyer and Chilkoti, 1999). These tags undergo an inverse phase transition at a given temperature of transition (Tt). When the Tt is reached, the ELP–protein fusion selectively and reversibly precipitates, allowing for quick enrichment of the recombinant protein by centrifugation (Banki et al., 2005). Precipitation can also be triggered by adjusting the ionic strength of the solution (Ge et al., 2005). These techniques represent an alternative to conventional chromatography-based purification methods and can save production costs, especially in large-scale settings (Fong and Wood, 2010). The main characteristics of the tags mentioned in this section are outlined on Table 1.
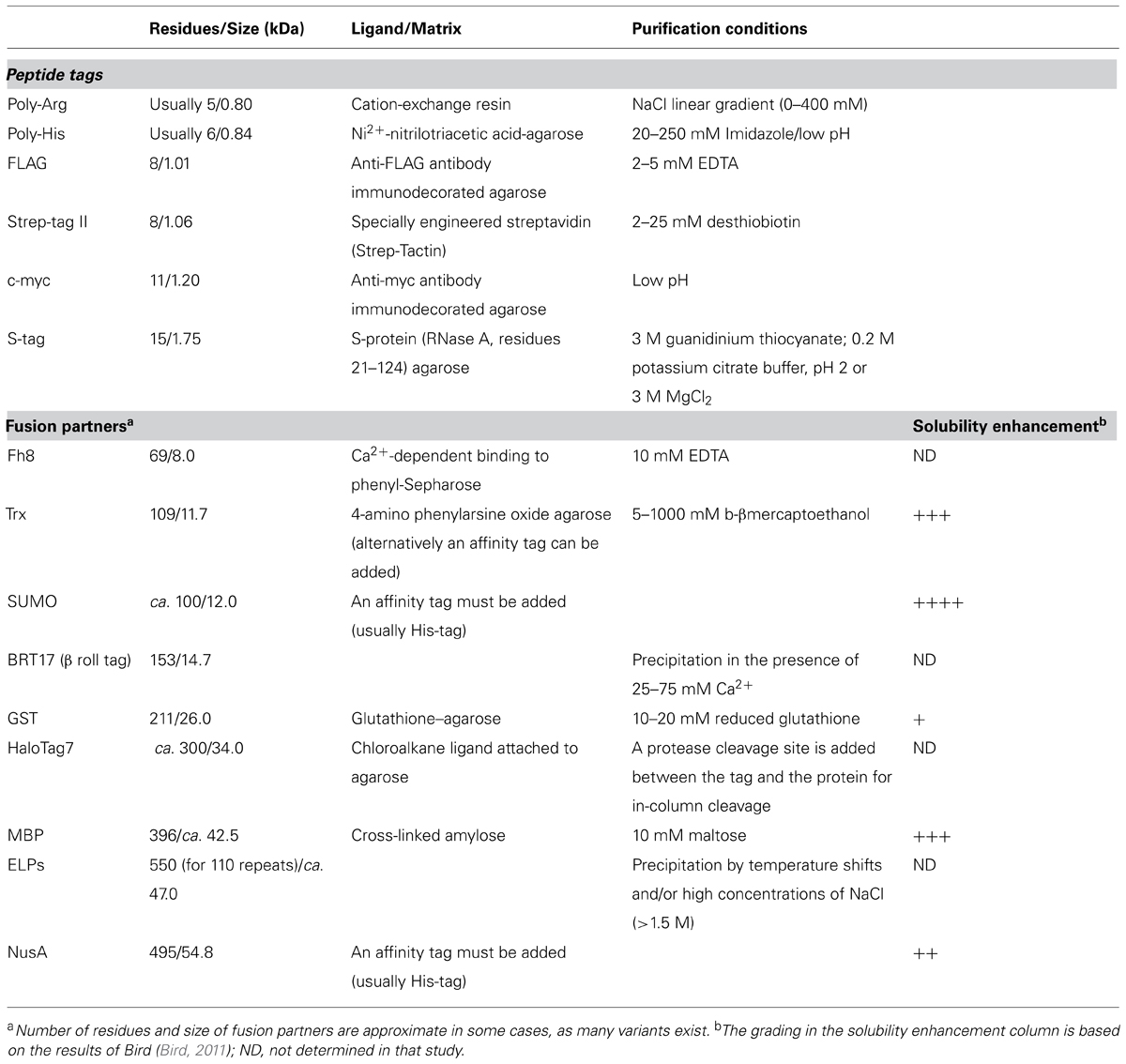
TABLE 1. Main characteristics of protein fusion tags.
Tag Removal
If structural or biochemical studies on the recombinant protein are needed, then the fusion partner must be eliminated from the recombinant protein. Peptide tags should be removed too because they can interfere with protein activity and structure (Wu and Filutowicz, 1999; Perron-Savard et al., 2005), but they can be left in place even for crystallographic studies (Bucher et al., 2002; Carson et al., 2007). Tags can be eliminated by either enzymatic cleavage or chemical cleavage.
In the case of tag removal by enzyme digestion, expression vectors possess sequences that encode for protease cleavage sites downstream of the gene coding for the tag. Enterokinase, thrombin, factor Xa and the tobacco etch virus (TEV) protease have all been successfully used for the removal of peptide tags and fusion partners (Jenny et al., 2003; Blommel and Fox, 2007). Choosing among the different proteases is based on specificity, cost, number of amino acids left in the protein after cleavage and ease of removal after digestion (Waugh, 2011). Enterokinase and thrombin were popular in the past but the use of His-tagged TEV has become an everyday choice due to its high specificity (Parks et al., 1994), it is easy to produce in large quantities (Tropea et al., 2009) and leaves only a serine or glycine residue (or even the natural N-terminus) after digestion (Kapust et al., 2002).
As the name implies, in chemical cleavage the tag is removed by treatment of the fusion protein with a chemical reagent. The advantages of using chemicals for this purpose are that they are easy to eliminate from the reaction mixture and are cheap in comparison with proteolytic enzymes, which makes them an attractive choice in the large-scale production of recombinant proteins (Rais-Beghdadi et al., 1998). However, the reaction conditions are harsh, so their use is largely restricted to purified recombinant proteins obtained from IBs. They also often cause unwanted protein modifications (Hwang et al., 2014). The most common chemical cleavage reagent is cyanogen bromide (CNBr). CNBr cleaves the peptide bond C-terminal to methionine residues, so this amino acid should be present between the tag and the protein of interest (Rais-Beghdadi et al., 1998). Also, the target protein should not contain internal methionines. CNBr cleavage can be performed in common denaturing conditions (6 M guanidinium chloride) or 70% formic acid or trifluoroacetic acid (Andreev et al., 2010). Other chemical methods for protein cleavage can be found in Hwang et al. (2014).
Third Question: Which is the Appropriate Host?
A quick search in the literature for a suitable E. coli strain to use as a host will yield dozens of possible candidates. All of them have advantages and disadvantages. However, something to keep in mind is that many are specialty strains that are used in specific situations. For a first expression screen, only a couple of E. coli strains are necessary: BL21(DE3) and some derivatives of the K-12 lineage.
The history of the BL21 and BL21(DE3) strains was beautifully documented in Daegelen et al. (2009) and we recommend this article to the curious. BL21 was described by Studier in 1986 after various modifications of the B line (Studier and Moffatt, 1986), which in turn Daegelen et al. (2009) traced back to d’Herelle. A couple of genetic characteristics of BL21 are worthy of mention. Like other parental B strains, BL21 cells are deficient in the Lon protease, which degrades many foreign proteins (Gottesman, 1996). Another gene missing from the genome of the ancestors of BL21 is the one coding for the outer membrane protease OmpT, whose function is to degrade extracellular proteins. The liberated amino acids are then taken up by the cell. This is problematic in the expression of a recombinant protein as, after cell lysis, OmpT may digest it (Grodberg and Dunn, 1988). In addition, plasmid loss is prevented thanks to the hsdSB mutation already present in the parental strain (B834) that gave rise to BL21. As a result, DNA methylation and degradation is disrupted. When the gene of interest is placed under a T7 promoter, then T7 RNAP should be provided. In the popular BL21(DE3) strain, the λDE3 prophage was inserted in the chromosome of BL21 and contains the T7 RNAP gene under the lacUV5 promoter, as was explained earlier.
The BL21(DE3) and its derivatives are by far the most used strains for protein expression. Still, there are reports where the K-12 lineage is used for this purpose. The AD494 and OrigamiTM (Novagen) strains are trxB (thioredoxin reductase) mutants, so disulfide bond formation in the cytoplasm is enhanced (the Origami strain also lacks the glutathione reductase gene; Derman et al., 1993). Another widely used strain from the K-12 repertoire is HMS174, a recA mutant (Campbell et al., 1978). This mutation has a positive effect on plasmid stability (Marisch et al., 2013). Plasmid multimer formation, an important cause of instability, relies on the recombination system of E. coli (Summers et al., 1993). All three strains have their λDE3-containing derivative (available at Novagen) so the T7 RNAP system can be used.
Fourth Question: Which is the Combination for Success?
At this point, it should be pretty clear that the number of options when designing an expression system is considerably high. Choosing the perfect combination is not possible a priori, so multiple conditions should be tested to obtain the desired protein. If the project demands expressing two protein constructs, cloned in six different expression vectors, each transformed in three different expression strains, then you are in for 36 expression trials. This number may be even higher when other variables are taken into account. This trial-and-error and time consuming pilot study can be made faster if micro-expression trials are performed before scale-up. Small-scale screens can be performed in 2-ml tubes or 96-well plates (Shih et al., 2002). High throughput protocols adapting automatic liquid handling robots have been described, making it possible for a single person to test more than 1000 culture conditions within a week.
Troubleshooting Recombinant Protein Production
This section of the review covers different strategies for optimizing recombinant protein production in E. coli. Even after careful selection of plasmid and host, it cannot be predicted if the protein will be obtained in high amounts and in a soluble active form. Various situations that impede reaching that goal can be encountered, which unfortunately happen very often. Many things to try in each case are discussed in the following paragraphs and, for convenience of the readers; a summary is included in Table 2.
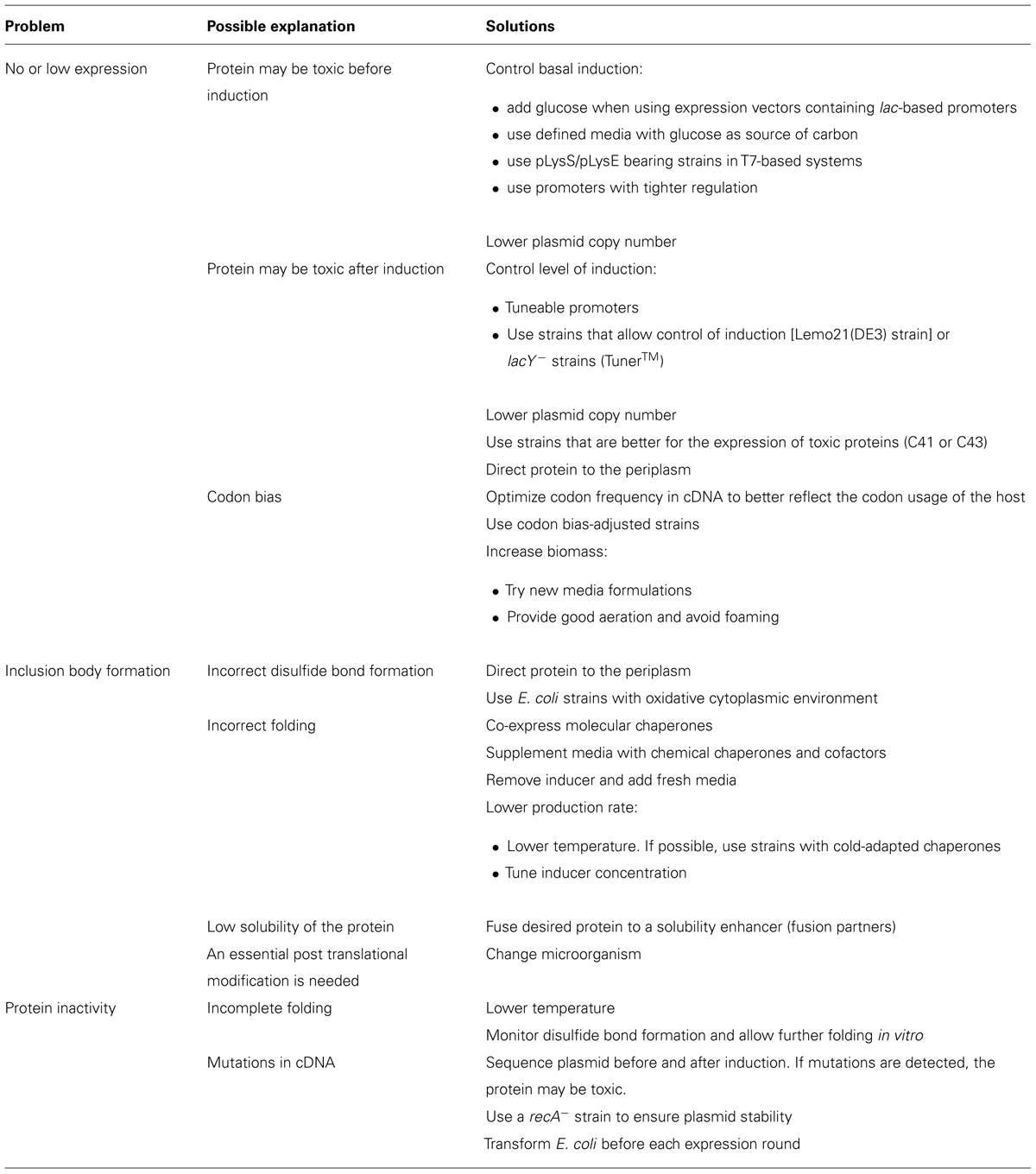
TABLE 2. Strategies for overcoming common problems during recombinant protein expression in E. coli.
No or Low Production
This situation may be regarded as the worst case scenario. When the protein of interest cannot be detected through a sensitive technique (e.g., Western blot) or it is detected but at very low levels (less than micrograms per liter of culture), the problem often lies in a harmful effect that the heterologous protein exerts on the cell (Miroux and Walker, 1996; Dumon-Seignovert et al., 2004).
Protein toxicity
The problem of protein toxicity may arise when the recombinant protein performs an unnecessary and detrimental function in the host cell. This function interferes with the normal proliferation and homeostasis of the microorganism and the visible result is slower growth rate, low final cell density, and death (Doherty et al., 1993; Dong et al., 1995).
As a first measure, cell growth should be monitored before induction. If the growth rate of the recombinant strain is slower compared to an empty-vector bearing strain then two causes may explain the phenotype: gene toxicity and basal expression of the toxic mRNA/protein. Gene toxicity will not be discussed here and the review of Saida et al. (2006) is recommended.
The control of basal synthesis was covered in some detail in Section “Promoter.” As stated, the expression of LacI from lacI or lacIQ represses transcription of lac-based promoters. For high copy number plasmids (>100 copies per cell), lacIQ should be cloned in the expression vector. The pQE vectors from Qiagen utilize two lac operator sequences to increase control of the T5 promoter, which is recognized by the E. coli RNA polymerase (see The QIAexpressionistTM manual from Qiagen). A tighter control can be achieved by the addition of 0.2–1% w/v glucose in the medium as rich media prepared with tryptone or peptone may contain the inducer lactose (Studier, 2005). Another option could be to prepare defined media using glucose as a source of carbon. In T7-based promoters, leaky expression is avoided by co-expression of T7 lysozyme from the pLysS or pLysE plasmids (see above). Use of lower copy number plasmids containing tightly regulated promoters (like the araPBAD promoter) is suggested. An interesting case of copy number control is the one employed in pETcoco vectors (Novagen). These plasmids possess two origins of replication. The oriS origin and its control elements maintain pETcoco at one copy per cell (Wild et al., 2002). However, the TrfA replicator activates the medium-copy origin of replication (oriV) and amplification of copy number is achieved (up to 40 copies per cell). The trfA gene is on the same vector and is under control of the araPBAD promoter, so copy number can be controlled by arabinose (Wild et al., 2002).
After control of basal expression, the culture should grow well until the proper time of induction. At this moment, if the protein is toxic, cell growth will be arrested. In many cases, the level of toxicity of a protein becomes apparent when a certain threshold of host tolerance is reached and exceeded. In such situations, the level of expression should be manipulated at will. Tunable expression can be achieved using the Lemo21(DE3) strain. This strain is similar to the BL21(DE3)pLysS strain, however, T7 lysozyme production from the lysY gene is under the tunable promoter rhaPBAD (Wagner et al., 2008). At higher concentrations of the sugar L-rhamnose, more T7 lysozyme is produced, less active T7 RNAP is present in the cell and less recombinant protein is expressed. Trials using L-rhamnose concentrations from 0 to 2,000 μM should be undertaken to find the best conditions for expression. By contrast, dose-dependent expression when using IPTG as inducer is not possible since IPTG can enter the cell by active transport through the Lac permease or by permease-independent pathways (Fernandez-Castane et al., 2012). Since expression of Lac permease is heterogeneous and the number of active permeases in each cell is highly variable, protein expression does not respond predictably to IPTG concentration. The TunerTM (DE3) strain (Novagen) is a BL21 derivative that possesses a lac permease (lacY) mutation that allows uniform entry of IPTG into all LacY- cells in the population, which produces a concentration-dependent, homogeneous level of induction (Khlebnikov and Keasling, 2002). In the same line of thought, an E. coli strain was constructed by exchanging the wild-type operator by the derivative lacOc, thus converting the lac operon into a constitutive one. This modification avoids the transient non-genetic LacY- phenotype of a fraction of the cells, allowing uniform entry of the inducer lactose. A second modification (gal+) permits the full utilization of lactose as an energy source (Menzella et al., 2003).
A word of caution needs to be said in regard to “tunable promoters” that are inducible by sugars (lactose, arabinose, rhamnose). In the case of the araPBAD promoter, the yields of the target protein can be reproducibly increased over a greater than 100-fold range by supplementing the culture with different sub-maximal concentrations of arabinose (Guzman et al., 1995). This led to the erroneous belief that within each cell, the level of recombinant protein synthesis can be manipulated at will. However, it was shown that the range in protein expression arises from the heterogeneity in the amount of active sugar permeases in each cell, as was also explained for LacY (Siegele and Hu, 1997). So, even though the final protein yield can be controlled, the amount of protein per cell is widely variable, with cells producing massive amounts of protein and others not producing any protein at all. This can be a nuance, since in the case of toxic products; the subpopulation of cells with high-level synthesis may perish (Doherty et al., 1993; Dong et al., 1995).
Some E. coli mutants were specifically selected to withstand the expression of toxic proteins. The strains C41(DE3) and C43(DE3) were found by Miroux and Walker (1996) in a screen designed to isolate derivatives of BL21(DE3) with improved membrane protein overproduction characteristics. It was recently discovered that the previously uncharacterized mutations which prevent cell death during the expression of recombinant proteins in these strains lie on the lacUV5 promoter. In BL21(DE3) cells, the lacUV5 promoter drives the expression of the T7 RNAP, but in the Walker strains two mutations in the -10 region revert the lacUV5 promoter back into the weaker wild-type counterpart. This leads to a lesser (and perhaps more tolerable for the cell) level of synthesis (Wagner et al., 2008).
Another solution could be to remove the protein from the cell. Secretion to the periplasm or to the medium is sometimes the only way to produce a recombinant protein (Mergulhao et al., 2005; de Marco, 2009). The first option for expression in the periplasm is the post-translational Sec-dependent pathway (Georgiou and Segatori, 2005). Routing to the extracytoplasmatic space is achieved by fusing the recombinant protein to a proper leader peptide. The signal peptides of the following proteins are widely used for secretion: Lpp, LamB, LTB, MalE, OmpA, OmpC, OmpF, OmpT, PelB, PhoA, PhoE, or SpA (Choi and Lee, 2004). The co-translational translocation machinery based on the SRP (signal recognition particle) pathway can also be used. SRP recognizes its substrates by the presence of a hydrophobic signal sequence located in the N-terminal end. Following interaction with the membrane receptor FtsY, the complex of nascent chain and ribosome is transferred to the SecYEG translocase (Valent et al., 1998). The signal sequence of disulfide isomerase I (DsbA) has been used to target recombinant proteins to the periplasm via the SRP pathway. Notable examples of recombinant proteins secreted though this system include thioredoxin (Schierle et al., 2003) and the human growth hormone (Soares et al., 2003).
Codon bias
Codon bias arises when the frequency of occurrence of synonymous codons in the foreign coding DNA is significantly different from that of the host. At the moment of full synthesis of the recombinant protein, depletion of low-abundance tRNAs occurs. This deficiency may lead to amino acid misincorporation and/or truncation of the polypeptide, thus affecting the heterologous protein expression levels (which will be low at best) and/or its activity (Gustafsson et al., 2004). To check if codon bias could be an issue when expressing a recombinant protein, a large number of free online apps detect the presence of rare codons in a given gene when E. coli is used as a host (molbiol.ru/eng/scripts/01_11.html, genscript.com/cgibin/tools/rare_codon_analysis, nihserver.mbi.ucla.edu/RACC/, just to name a few). Rare codons were defined as codons used by E. coli at a frequency <1% (Kane, 1995). For example, the AGG codon (Arg) is used in E. coli at a frequency of <0.2%, but it is not rare in plant mRNAs where it can reach frequencies >1.5%.
Two strategies for solving codon usage bias have been used: codon optimization of the foreign coding sequence or increasing the availability of underrepresented tRNAs by host modification (Sorensen and Mortensen, 2005). The rationale behind codon usage optimization is to modify the rare codons in the target gene to mirror the codon usage of the host (Burgess-Brown et al., 2008; Welch et al., 2009; Menzella, 2011). The amino acid sequence of the encoded protein must not be altered in the process. This can be done by site-directed silent mutagenesis or resynthesis of the whole gene or parts of it. Codon optimization by silent mutagenesis is a cumbersome and expensive process, so is not very useful when many recombinant proteins are needed. On the other hand, gene synthesis by design is not a trivial issue since it requires choosing the best sequence from a vast number of possible combinations (Gustafsson et al., 2004). The simplest approach is to replace all instances of a given amino acid in the target gene by the most abundant codon of the host, a strategy called “one amino acid-one codon.” More advanced algorithms, which employ several other optimization parameters such as codon context and codon harmonization, have been described (Gao et al., 2004; Supek and Vlahovicek, 2004; Jayaraj et al., 2005; Angov et al., 2011). Some are freely available as web servers or standalone software. For a comprehensive list, please refer to Puigbo et al. (2007).
Correcting codon usage is a tricky situation. The “one amino acid-one codon” strategy disregards factors other than codon rarity that influence protein expression levels. For example, in bacterial genes enriched in rare codons at the N-terminus, protein expression is actually improved. The cause lies not in codon rarity per se but in the reduction of RNA secondary structure (Goodman et al., 2013). In addition, a recent report has shown that high levels of protein production are mainly (but not only) determined by the decoding speed of the open reading frame (i.e., the time it takes for a ribosome to translate an mRNA), especially if “fast” codons are located at the 5′-end of the mRNA (Chu et al., 2014). This causes a fast ribosome clearance at the initiation site, so that new recruited ribosomes encounter a free start codon and can engage in translation. Finally, some codon combinations can create Shine–Dalgarno-like structures that cause translational pausing by hybridization between the target mRNA and the 16S rRNA of the translating ribosome (Li et al., 2012). Translational pausing along the mRNA has a beneficial effect in protein folding, as it allows for the newly synthesized chain to adopt a well-folded intermediate conformation (Thanaraj and Argos, 1996; Oresic and Shalloway, 1998; Tsai et al., 2008; Yona et al., 2013). All of this new evidence in translational control mechanisms poses a challenge in the rational design of synthetic genes. Newer algorithms should account for 5′ RNA structure, presence of strategically located Shine–Dalgarno-like motifs, ribosome clearance rates at the initiation site and presence of slowly translated regions that are beneficial in co-translational folding.
On the other hand, when the cell is producing massive amounts of proteins (as in the case of recombinant expression of heterologous genes), charged tRNA availability for rare codons does become the major determinant of the levels of produced protein (Pedersen, 1984; Li et al., 2012). Low-abundance tRNA depletion causes ribosome stalling and its subsequent detachment from the RNA strand and thus, failure to generate a full-length product (Buchan and Stansfield, 2007). Several strains carrying plasmids containing extra copies of problematic tRNAs genes can be used to circumvent this issue. The BL21(DE3)CodonPlus strain (Stratagene) contains the pRIL plasmid (p15A replicon, which is compatible with the ColE1 and ColE1-like origins contained in most commonly used expression vectors), which provides extra genes for the tRNAs for AGG/AGA (Arg), AUA (Ile), and CUA (Leu). BL21(DE3)CodonPlus-RP (Stratagene) corrects for the use of AGG/AGA (Arg) and CCC (Pro). The Rosetta(DE3) strains (Novagen) are TunerTM derivatives containing the pRARE plasmid (p15A replicon), supplying tRNAs for all the above-mentioned codons plus GGA (Gly). It should be noted that the use of these strains often improves the levels of protein production but sometimes can cause a decrease in protein solubility. We have found that proteins with higher than 5% content of RIL codons (AGG/AGA, AUA, and CUA) are less soluble when expressed in the CodonPlus strain. In this host, the translational pauses introduced by the RIL codons are probably overridden, increasing translation speed and consequently, protein aggregation (Rosano and Ceccarelli, 2009).
Limiting factors in batch cultivation
When the expression of the recombinant protein is low and cannot be increased by the proposed mechanisms, then the volumetric yield of desired protein can be augmented by growing the culture to higher densities. This can be achieved by changing a few parameters, like medium composition and providing better aeration by vigorous shaking (McDaniel and Bailey, 1969; Cui et al., 2006; Blommel et al., 2007).
LB is the most commonly used medium for culturing E. coli. It is easy to make, it has rich nutrient contents and its osmolarity is optimal for growth at early log phase. All these features make it adequate for protein production and compensate for the fact that it is not the best option for achieving high cell density cultures. Despite being a rich broth, cell growth stops at a relatively low density. This happens because LB contains scarce amounts of carbohydrates (and other utilizable carbon sources) and divalent cations (Sezonov et al., 2007). Not surprisingly, increasing the amount of peptone or yeast extract leads to higher cell densities (Studier, 2005). Also, divalent cation supplementation (MgSO4 in the millimolar range) results in higher cell growth. Adding glucose is of limited help in this regard because acid generation by glucose metabolism overwhelms the limited buffer capacity of LB, at least in shake flasks where pH control can be laborious (Weuster-Botz et al., 2001; Scheidle et al., 2011). If culture acidification poses a problem, the media can be buffered with phosphate salts at 50 mM. 2xYT, TB (Terrific Broth) and SB (Super Broth) media recipes are available elsewhere and have been shown to be superior to LB for reaching higher cell densities (Madurawe et al., 2000; Atlas, 2004; Studier, 2005).
A major breakthrough in media composition came in 2005 by the extensive work of Studier. In that report, the concept of autoinduction was developed (Studier, 2005). In autoinduction media, a mixture of glucose, lactose, and glycerol is used in an optimized blend. Glucose is the preferred carbon source and is metabolized preferentially during growth, which prevents uptake of lactose until glucose is depleted, usually in mid to late log phase. Consumption of glycerol and lactose follows, the latter being also the inducer of lac-controlled protein expression. In this way, biomass monitoring for timely inducer addition is avoided, as well as culture manipulation (Studier, 2014).
As the number of cells per liter increases, oxygen availability becomes an important factor with profound influence on growth (O’Beirne and Hamer, 2000; Losen et al., 2004).Oxygen limitation triggers the expression of more than 200 genes in an attempt to adjust the metabolic capacities of the cell to the availability of oxygen, all of which hinder optimal growth over long culture periods (Unden et al., 1995). The easiest way to increase the amount of available oxygen in shake vessels is to increase shaking speed. For regular flasks, the optimal shaking speed range is 400–450 rpm. More agitation is generated in baffled flasks; under these conditions, 350–400 rpm are enough for good aeration. However, vigorous shaking can induce the formation of foam, which will lower oxygen transfer. For this reason, the addition of an antifoaming agent is recommended, although it was shown that antifoams can affect the growth rate of several microorganisms and the yield of recombinant protein (Routledge et al., 2011; Routledge, 2012). Also, proper aeration depends on the ratio of culture volume to vessel capacity. As a rule of thumb, the culture volume should be less or equal to 10% of the shaking flask capacity, although in our hands, protein production with culture volumes occupying 20% of the flask capacity was possible (Rosano et al., 2011). A strategy that can produce significant increases in cell density is fed-batch fermentation. This approach has a wide availability of tools and methods, but it is beyond the scope of this paper and is addressed elsewhere (Yamanè, 1984; Yee and Blanch, 1992; Moulton, 2013).
Two rarely discussed parameters in the process of recombinant protein production are the preparation of the starting culture and the time of induction. Most protocols call for diluting a saturated overnight preculture (dilution factor 1/100) into the larger culture (Sivashanmugam et al., 2009). However, leaky expression of the chosen system can lead to plasmid instability, which may result in a poor yield of target protein. Also, in the starter culture, cells can be in dissimilar metabolic states. Upon dilution into fresh media, cells will grow at different rates leading to irreproducible induction points (Huber et al., 2009). A proper preculture (cells in an active equalized growing phase) can be prepared by growing the overnight starter culture at 20–25°C or by using a slow-release system for glucose, among other methods (Busso et al., 2008; Huber et al., 2009; Sivashanmugam et al., 2009). After inoculation and further growth, the inducer is often added in mid-log phase because the culture is growing fast and protein translation is maximal. However, induction at early stationary phase is also possible (Ou et al., 2004). In fact, in some cases the target protein was more soluble when inducer was added at this stage (Galloway et al., 2003). Presumably, the reduced rate of protein synthesis may result in less aggregation in IBs, as we describe below.
Inclusion Bodies Formation
When a foreign gene is introduced in E. coli, spatio-temporal control of its expression is lost. The newly synthesized recombinant polypeptide is expressed in the microenvironment of E. coli, which may differ from that of the original source in terms of pH, osmolarity, redox potential, cofactors, and folding mechanisms. Also, in high level expression, hydrophobic stretches in the polypeptide are present at high concentrations and available for interaction with similar regions. All of these factors lead to protein instability and aggregation (Hartley and Kane, 1988; Carrio and Villaverde, 2002). These buildups of protein aggregates are known as IBs. IB formation results from an unbalanced equilibrium between protein aggregation and solubilization. So, it is possible to obtain a soluble recombinant protein by strategies that ameliorate the factors leading to IB formation (Carrio and Villaverde, 2001, 2002). One is to fuse the desired protein to a fusion partner that acts as a solubility enhancer. Some examples were already described in Section “Affinity Tags.” In some cases the generation of IB can be an advantage, especially if the protein can be refolded easily in vitro. If that is the case, conditions can be adjusted to favor the formation IB, providing a simple method for achieving a significant one-step purification of the expressed protein (Burgess, 2009; Basu et al., 2011).
Disulfide bond formation
For many recombinant proteins, the formation of correct disulfide bonds is vital for attaining their biologically active three-dimensional conformation. The formation of erroneous disulfide bonds can lead to protein misfolding and aggregation into IB. In E. coli, cysteine oxidation takes places in the periplasm, where disulfide bonds are formed in disulfide exchange reactions catalyzed by a myriad of enzymes, mainly from the Dsb family (Messens and Collet, 2006). By contrast, disulfide bond formation in the cytoplasm is rare, maybe because cysteine residues are part of catalytic sites in many enzymes. Disulfide bond formation at these sites may lead to protein inactivation, misfolding, and aggregation (Derman et al., 1993). The cytoplasm has a more negative redox potential and is maintained as a reducing environment by the thioredoxin–thioredoxin reductase (trxB) system and the glutaredoxin–glutaredoxin reductase (gor) system (Stewart et al., 1998). This situation has a huge impact in the production of recombinant proteins with disulfide bonds. One option would be to direct the protein to the periplasm, as we have discussed in Section “Protein Toxicity.”
Nevertheless, expression in the cytoplasm is still possible thanks to engineered E. coli strains that possess an oxidative cytoplasmic environment that favors disulfide bond formation (Derman et al., 1993). Worthy of mention are the Origami (Novagen) and SHuffle (NEB) strains. We described earlier the OrigamiTM strain, as having a trxB- gor- genotype in the K-12 background (as this double mutant is not viable, a suppressor mutation in the ahpC gene is necessary to maintain viability; Bessette et al., 1999). OrigamiTM is also available in the BL21(DE3) lacY (TunerTM, Novagen) background. Addition of the pRARE plasmid for the extra advantage of correcting codon bias resulted in the construction of the Rosetta-gamiTM B strain (Novagen). The SHuffle® T7 Express strain [BL21(DE3) background, NEB] goes a little bit further. Besides the trxB- and gor- mutations, it constitutively expresses a chromosomal copy of the disulfide bond isomerase DsbC (Lobstein et al., 2012). DsbC promotes the correction of mis-oxidized proteins into their correct form and is also a chaperone that can assist in the folding of proteins that do not require disulfide bonds. Due to the action of DsbC, less target protein aggregates into IB.
Chaperone co-expression/chemical chaperones and cofactor supplementation
Molecular chaperones lie at the heart of protein quality control, aiding nascent polypeptides to reach their final structure (Hartl and Hayer-Hartl, 2002). Other specialized types of chaperones, like ClpB, can disassemble unfolded polypeptides present in IB. The high level expression of recombinant proteins results in the molecular crowding of the cytosol and quality control mechanisms may be saturated in this situation (Carrio and Villaverde, 2002). One strategy for solving this problem is to stop protein expression by inducer removal after a centrifugation step and addition of fresh media supplemented with chloramphenicol, an inhibitor of protein synthesis. This allows recruitment of molecular chaperones to aid in the folding of newly synthesized recombinant polypeptides (Carrio and Villaverde, 2001; de Marco and De Marco, 2004).
Given their function, it is not surprising that efforts to inhibit IB formation were directed to the co-expression of individual or sets of molecular chaperones (Caspers et al., 1994; Nishihara et al., 2000; de Marco et al., 2007). Commercially, one of the most used systems is the chaperone plasmid set from Takara (Nishihara et al., 1998, 2000). This set consists of five plasmids (pACYC derivatives) which allow overexpression of different chaperones or combinations of them: (i) GroES-GroEL, (ii) DnaK/DnaJ/GrpE, (iii) (i) + (ii), (iv) trigger factor, (v) (i) + (iv). On the other hand, if such a system is not at hand, the natural network of chaperones can be induced by the addition of benzyl alcohol or heat shock, though the latter is not recommended (de Marco et al., 2005).
When proteins are purified from IB, urea-denatured and then refolded in vitro, addition of osmolytes (also called chemical chaperones) in the 0.1–1 M range of concentration increases the yield of soluble protein (Rudolph and Lilie, 1996; Clark, 1998; Tsumoto et al., 2003; Alibolandi and Mirzahoseini, 2011). This situation can be mimicked in vivo by supplementing the culture media with osmolytes such as proline, glycine-betaine, and trehalose (de Marco et al., 2005). Also, the folding pathways that lead to the correct final conformation and stabilization of the proper folded protein may require specific cofactors in the growth media, for example, metal ions (such as iron-sulfur and magnesium) and polypeptide cofactors. Addition of these compounds to the batch culture considerably increases the yield as well as the folding rate of soluble proteins (Sorensen and Mortensen, 2005).
Slowing down production rate
Slower rates of protein production give newly transcribed recombinant proteins time to fold properly. This was previously addressed when we discussed the role of translational pauses at rare codons and their impact in the production of recombinant proteins. Moreover, the reduction of cellular protein concentration favors proper folding. By far, the most commonly used way to lower protein synthesis is reducing incubation temperature (Schein and Noteborn, 1988; Vasina and Baneyx, 1997; Vera et al., 2007). Low temperatures decrease aggregation, which is favored at higher temperatures due to the temperature dependence of hydrophobic interactions (Baldwin, 1986; Makhatadze and Privalov, 1995; Schellman, 1997).
When IB formation is a problem, recombinant protein synthesis should be carried out in the range 15–25°C, though one report described successful expression at 4°C for 72 h (San-Miguel et al., 2013). However, when working at the lower end of the temperature range, slower growth and reduced synthesis rates can result in lower protein yields. Also, protein folding may be affected as the chaperone network may not be as efficient (McCarty and Walker, 1991; Mendoza et al., 2000; Strocchi et al., 2006). The ArticExpressTM (Stratagene) strain (B line) possesses the cold-adapted chaperonin Cpn60 and co-chaperonin Cpn10 from the psychrophilic bacterium Oleispira antarctica (Ferrer et al., 2004). The chaperonins display high refolding activities at temperatures of 4–12°C and confer an enhanced ability for E. coli to grow at lower temperatures (Ferrer et al., 2003).
Protein Inactivity
Obtaining a nice amount of soluble protein is not the end of the road. The protein may still be of bad quality; i.e., it does not have the activity it should. Incomplete folding could be the culprit in this scenario (Gonzalez-Montalban et al., 2007; Martinez-Alonso et al., 2008). In this case, the protein adopts a stable soluble conformation but the exact architecture of the active site is still unsuitable for activity. Some options already addressed can be helpful in these cases. Some proteins require small molecules or prosthetic groups to acquire their final folded conformation. Adding these compounds to the culture media can increase the yield and the quality of the expressed protein significantly (Weickert et al., 1999; Yang et al., 2003). Also, erroneous disulfide bond formation can lead to protein inactivity (Kurokawa et al., 2000). In addition, protein production at lower temperatures has a profound impact on protein quality. Work by the Villaverde lab has shown that conformational quality and functionality of highly soluble recombinant proteins increase when the temperature of the culture is reduced (Vera et al., 2007). This was also the case when the intracellular concentration of the chaperone DnaK was elevated (Martinez-Alonso et al., 2007). This phenomenon calls into question the use of solubility as an indicator of quality. Based on this fact, then it may be wise to express all recombinant proteins at low temperatures or at least, to compare the specific activity of a recombinant protein obtained at different temperatures.
If the activity of the heterologous protein is toxic to the cell, genetic reorganization of the expression vector leading to loss of activity may occur, allowing the host to survive and eventually take over the culture (Corchero and Villaverde, 1998). This structural instability of the plasmid can be detected by DNA sequencing after purification of the plasmid at the end of process. Any point mutation, deletion, insertion, or rearrangement may explain the low activity of a purified recombinant protein (Palomares et al., 2004).
Concluding Remarks
In terms of recombinant expression, E. coli has always been the preferred microbial cell factory. E. coli is a suitable host for expressing stably folded, globular proteins from prokaryotes and eukaryotes. Even though membrane proteins and proteins with molecular weights above 60 kDa are difficult to express, several reports have had success in this regard (our laboratory has produced proteins from plants in the 90–95 kDa range; Rosano et al., 2011). Large-scale protein expression trials have shown that <50% of bacterial proteins and <15% of non-bacterial proteins can be expressed in E. coli in a soluble form, which demonstrates the versatility of the system (Braun and LaBaer, 2003). However, when coming across a difficult-to-express protein, things can get complicated. We hope to have given a thorough list of possible solutions when facing the challenge of expressing a new protein in E. coli. Nevertheless, a word of caution is needed. Many of the approaches described in this review will fail miserably in a lot of cases. This can be explained by the fact that strategies aiming at troubleshooting recombinant protein expression are sometimes protein specific and suffer from positive bias; i.e., things that work get published, all the others, do not. That being said, thanks to the efforts of the scientific community, the general methods available in the literature are no longer anecdotal and can be used systematically. Moreover, the field is always expanding and even after almost 40 years from the first human protein obtained in E. coli (Itakura et al., 1977), there is still much room for improvement.
Author Contributions
Germán L. Rosano and Eduardo A. Ceccarelli wrote the manuscript and approved its final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the reviewers for their insightful comments on the manuscript, as their remarks led to an improvement of the work. Germán L. Rosano and Eduardo A. Ceccarelli are staff members of the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Argentina). Also, Germán L. Rosano is a Teaching Assistant and Eduardo A. Ceccarelli is a Professor of the Facultad de Ciencias Bioquímicas y Farmacéuticas, UNR, Argentina. This study was supported by grants from CONICET and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, Argentina).
References
Adrio, J. L., and Demain, A. L. (2010). Recombinant organisms for production of industrial products. Bioeng. Bugs 1, 116–131. doi: 10.4161/bbug.1.2.10484
Alibolandi, M., and Mirzahoseini, H. (2011). Chemical assistance in refolding of bacterial inclusion bodies. Biochem. Res. Int. 2011:631607. doi: 10.1155/2011/631607
Andreev, Y. A., Kozlov, S. A., Vassilevski, A. A., and Grishin, E. V. (2010). Cyanogen bromide cleavage of proteins in salt and buffer solutions. Anal. Biochem. 407, 144–146. doi: 10.1016/j.ab.2010.07.023
Angov, E., Legler, P. M., and Mease, R. M. (2011). Adjustment of codon usage frequencies by codon harmonization improves protein expression and folding. Methods Mol. Biol. 705, 1–13. doi: 10.1007/978-1-61737-967-3_1
Baker, R. T. (1996). Protein expression using ubiquitin fusion and cleavage. Curr. Opin. Biotechnol. 7, 541–546. doi: 10.1016/S0958-1669(96)80059-0
Baldwin, R. L. (1986). Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. U.S.A. 83, 8069–8072. doi: 10.1073/pnas.83.21.8069
Baneyx, F. (1999). Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 10, 411–421. doi: 10.1016/S0958-1669(99)00003-8
Banki, M. R., Feng, L., and Wood, D. W. (2005). Simple bioseparations using self-cleaving elastin-like polypeptide tags. Nat. Methods 2, 659–662. doi: 10.1038/nmeth787
Basu, A., Li, X., and Leong, S. S. (2011). Refolding of proteins from inclusion bodies: rational design and recipes. Appl. Microbiol. Biotechnol. 92, 241–251. doi: 10.1007/s00253-011-3513-y
Bentley, W. E., Mirjalili, N., Andersen, D. C., Davis, R. H., and Kompala, D. S. (1990). Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol. Bioeng. 35, 668–681. doi: 10.1002/bit.260350704
Bessette, P. H., Aslund, F., Beckwith, J., and Georgiou, G. (1999). Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 96, 13703–13708. doi: 10.1073/pnas.96.24.13703
Bird, L. E. (2011). High throughput construction and small scale expression screening of multi-tag vectors in Escherichia coli. Methods 55, 29–37. doi: 10.1016/j.ymeth.2011.08.002
Birnbaum, S., and Bailey, J. E. (1991). Plasmid presence changes the relative levels of many host cell proteins and ribosome components in recombinant Escherichia coli. Biotechnol. Bioeng. 37, 736–745. doi: 10.1002/bit.260370808
Blommel, P. G., Becker, K. J., Duvnjak, P., and Fox, B. G. (2007). Enhanced bacterial protein expression during auto-induction obtained by alteration of lac repressor dosage and medium composition. Biotechnol. Prog. 23, 585–598. doi: 10.1021/bp070011x
Blommel, P. G., and Fox, B. G. (2007). A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr. Purif. 55, 53–68. doi: 10.1016/j.pep.2007.04.013
Bolivar, F., Rodriguez, R. L., Greene, P. J., Betlach, M. C., Heyneker, H. L., Boyer, H. W., et al. (1977). Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene 2, 95–113. doi: 10.1016/0378-1119(77)90000-2
Bornhorst, J. A., and Falke, J. J. (2000). Purification of proteins using polyhistidine affinity tags. Methods Enzymol. 326, 245–254. doi: 10.1016/S0076-6879(00)26058-8
Braun, P., and LaBaer, J. (2003). High throughput protein production for functional proteomics. Trends Biotechnol. 21, 383–388. doi: 10.1016/S0167-7799(03)00189-6
Buchan, J. R., and Stansfield, I. (2007). Halting a cellular production line: responses to ribosomal pausing during translation. Biol. Cell 99, 475–487. doi: 10.1042/BC20070037
Bucher, M. H., Evdokimov, A. G., and Waugh, D. S. (2002). Differential effects of short affinity tags on the crystallization of Pyrococcus furiosus maltodextrin-binding protein. Acta Crystallogr. D. Biol. Crystallogr. 58, 392–397. doi: 10.1107/S0907444901021187
Burgess, R. R. (2009). Refolding solubilized inclusion body proteins. Methods Enzymol. 463, 259–282. doi: 10.1016/S0076-6879(09)63017-2
Burgess-Brown, N. A., Sharma, S., Sobott, F., Loenarz, C., Oppermann, U., and Gileadi, O. (2008). Codon optimization can improve expression of human genes in Escherichia coli: a multi-gene study. Protein Expr. Purif. 59, 94–102. doi: 10.1016/j.pep.2008.01.008
Busso, D., Stierle, M., Thierry, J. C., and Moras, D. (2008). A comparison of inoculation methods to simplify recombinant protein expression screening in Escherichia coli. Biotechniques 44, 101–106. doi: 10.2144/000112632
Butt, T. R., Edavettal, S. C., Hall, J. P., and Mattern, M. R. (2005). SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 43, 1–9. doi: 10.1016/j.pep.2005.03.016
Calos, M. P. (1978). DNA sequence for a low-level promoter of the lac repressor gene and an ‘up’ promoter mutation. Nature 274, 762–765. doi: 10.1038/274762a0
Campbell, J. L., Richardson, C. C., and Studier, F. W. (1978). Genetic recombination and complementation between bacteriophage T7 and cloned fragments of T7 DNA. Proc. Natl. Acad. Sci. U.S.A. 75, 2276–2280. doi: 10.1073/pnas.75.5.2276
Camps, M. (2010). Modulation of ColE1-like plasmid replication for recombinant gene expression. Recent Pat. DNA Gene Seq. 4, 58–73. doi: 10.2174/187221510790410822
Carrio, M. M., and Villaverde, A. (2001). Protein aggregation as bacterial inclusion bodies is reversible. FEBS Lett. 489, 29–33. doi: 10.1016/S0014-5793(01)02073-7
Carrio, M. M., and Villaverde, A. (2002). Construction and deconstruction of bacterial inclusion bodies. J. Biotechnol. 96, 3–12. doi: 10.1016/S0168-1656(02)00032-9
Carson, M., Johnson, D. H., Mcdonald, H., Brouillette, C., and Delucas, L. J. (2007). His-tag impact on structure. Acta Crystallogr. D. Biol. Crystallogr. 63, 295–301. doi: 10.1107/S0907444906052024
Caspers, P., Stieger, M., and Burn, P. (1994). Overproduction of bacterial chaperones improves the solubility of recombinant protein tyrosine kinases in Escherichia coli. Cell. Mol. Biol. (Noisy-le-grand) 40, 635–644.
Chang, A. C., and Cohen, S. N. (1978). Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134, 1141–1156.
Chant, A., Kraemer-Pecore, C. M., Watkin, R., and Kneale, G. G. (2005). Attachment of a histidine tag to the minimal zinc finger protein of the Aspergillus nidulans gene regulatory protein AreA causes a conformational change at the DNA-binding site. Protein Expr. Purif. 39, 152–159. doi: 10.1016/j.pep.2004.10.017
Choi, J. H., Keum, K. C., and Lee, S. Y. (2006). Production of recombinant proteins by high cell density culture of Escherichia coli. Chem. Eng. Sci. 61, 9. doi: 10.1016/j.ces.2005.03.031
Choi, J. H., and Lee, S. Y. (2004). Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl. Microbiol. Biotechnol. 64, 625–635. doi: 10.1007/s00253-004-1559-9
Chu, D., Kazana, E., Bellanger, N., Singh, T., Tuite, M. F., and Von Der Haar, T. (2014). Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J. 33, 21–34. doi: 10.1002/embj.201385651
Clark, E. D. B. (1998). Refolding of recombinant proteins. Curr. Opin. Biotechnol 9, 157–163. doi: 10.1016/S0958-1669(98)80109-2
Corchero, J. L., and Villaverde, A. (1998). Plasmid maintenance in Escherichia coli recombinant cultures is dramatically, steadily, and specifically influenced by features of the encoded proteins. Biotechnol. Bioeng. 58, 625–632. doi: 10.1002/(SICI)1097-0290(19980620)58:6<625::AID-BIT8>3.0.CO;2-K
Costa, S. J., Almeida, A., Castro, A., Domingues, L., and Besir, H. (2013). The novel Fh8 and H fusion partners for soluble protein expression in Escherichia coli: a comparison with the traditional gene fusion technology. Appl. Microbiol. Biotechnol. 97, 6779–6791. doi: 10.1007/s00253-012-4559-1
Cui, F. J., Li, Y., Xu, Z. H., Xu, H. Y., Sun, K., and Tao, W. Y. (2006). Optimization of the medium composition for production of mycelial biomass and exo-polymer by Grifola frondosa GF9801 using response surface methodology. Bioresour. Technol. 97, 1209–1216. doi: 10.1016/j.biortech.2005.05.005
Daegelen, P., Studier, F. W., Lenski, R. E., Cure, S., and Kim, J. F. (2009). Tracing ancestors and relatives of Escherichia coli B, and the derivation of B strains REL606 and BL21(DE3). J. Mol. Biol. 394, 634–643. doi: 10.1016/j.jmb.2009.09.022
Davis, G. D., Elisee, C., Newham, D. M., and Harrison, R. G. (1999). New fusion protein systems designed to give soluble expression in Escherichia coli. Biotechnol. Bioeng. 65, 382–388. doi: 10.1002/(SICI)1097-0290(19991120)65:4<382::AID-BIT2>3.0.CO;2-I
de Boer, H. A., Comstock, L. J., and Vasser, M. (1983). The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc. Natl. Acad. Sci. U.S.A. 80, 21–25. doi: 10.1073/pnas.80.1.21
del Solar, G., and Espinosa, M. (2000). Plasmid copy number control: an ever-growing story. Mol. Microbiol. 37, 492–500. doi: 10.1046/j.1365-2958.2000.02005.x
del Solar, G., Giraldo, R., Ruiz-Echevarria, M. J., Espinosa, M., and Diaz-Orejas, R. (1998). Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 62, 434–464.
Demain, A. L., and Vaishnav, P. (2009). Production of recombinant proteins by microbes and higher organisms. Biotechnol. Adv. 27, 297–306. doi: 10.1016/j.biotechadv.2009.01.008
de Marco, A. (2009). Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb. Cell Fact. 8, 26. doi: 10.1186/1475-2859-8-26
de Marco, A., and De Marco, V. (2004). Bacteria co-transformed with recombinant proteins and chaperones cloned in independent plasmids are suitable for expression tuning. J. Biotechnol. 109, 45–52. doi: 10.1016/j.jbiotec.2003.10.025
de Marco, A., Deuerling, E., Mogk, A., Tomoyasu, T., and Bukau, B. (2007). Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 7:32. doi: 10.1186/1472-6750-7-32
de Marco, A., Vigh, L., Diamant, S., and Goloubinoff, P. (2005). Native folding of aggregation-prone recombinant proteins in Escherichia coli by osmolytes, plasmid- or benzyl alcohol-overexpressed molecular chaperones. Cell Stress Chaperones 10, 329–339. doi: 10.1379/CSC-139R.1
Derman, A. I., Prinz, W. A., Belin, D., and Beckwith, J. (1993). Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 262, 1744–1747. doi: 10.1126/science.8259521
Deuschle, U., Kammerer, W., Gentz, R., and Bujard, H. (1986). Promoters of Escherichia coli: a hierarchy of in vivo strength indicates alternate structures. EMBO J. 5, 2987–2994.
Dodd, I. B., Shearwin, K. E., and Egan, J. B. (2005). Revisited gene regulation in bacteriophage lambda. Curr. Opin. Genet. Dev. 15, 145–152. doi: 10.1016/j.gde.2005.02.001
Doherty, A. J., Connolly, B. A., and Worrall, A. F. (1993). Overproduction of the toxic protein, bovine pancreatic DNaseI, in Escherichia coli using a tightly controlled T7-promoter-based vector. Gene 136, 337–340. doi: 10.1016/0378-1119(93)90491-K
Dong, H., Nilsson, L., and Kurland, C. G. (1995). Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J. Bacteriol. 177, 1497–1504.
Dubendorff, J. W., and Studier, F. W. (1991). Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J. Mol. Biol. 219, 45–59. doi: 10.1016/0022-2836(91)90856-2
Dumon-Seignovert, L., Cariot, G., and Vuillard, L. (2004). The toxicity of recombinant proteins in Escherichia coli: a comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr. Purif. 37, 203–206. doi: 10.1016/j.pep.2004.04.025
Epstein, W., Rothman-Denes, L. B., and Hesse, J. (1975). Adenosine 3′:5′-cyclic monophosphate as mediator of catabolite repression in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 72, 2300–2304. doi: 10.1073/pnas.72.6.2300
Esposito, D., and Chatterjee, D. K. (2006). Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol. 17, 353–358. doi: 10.1016/j.copbio.2006.06.003
Fernandez-Castane, A., Vine, C. E., Caminal, G., and Lopez-Santin, J. (2012). Evidencing the role of lactose permease in IPTG uptake by Escherichia coli in fed-batch high cell density cultures. J. Biotechnol. 157, 391–398. doi: 10.1016/j.jbiotec.2011.12.007
Ferrer, M., Chernikova, T. N., Yakimov, M. M., Golyshin, P. N., and Timmis, K. N. (2003). Chaperonins govern growth of Escherichia coli at low temperatures. Nat. Biotechnol. 21, 1266–1267. doi: 10.1038/nbt1103-1266
Ferrer, M., Lunsdorf, H., Chernikova, T. N., Yakimov, M., Timmis, K. N., and Golyshin, P. N. (2004). Functional consequences of single: double ring transitions in chaperonins: life in the cold. Mol. Microbiol. 53, 167–182. doi: 10.1111/j.1365-2958.2004.04077.x
Fong, B. A., and Wood, D. W. (2010). Expression and purification of ELP-intein-tagged target proteins in high cell density E. coli fermentation. Microb. Cell Fact. 9, 77. doi: 10.1186/1475-2859-9-77
Galkin, V. E., Yu, X., Bielnicki, J., Ndjonka, D., Bell, C. E., and Egelman, E. H. (2009). Cleavage of bacteriophage lambda cI repressor involves the RecA C-terminal domain. J. Mol. Biol. 385, 779–787. doi: 10.1016/j.jmb.2008.10.081
Galloway, C. A., Sowden, M. P., and Smith, H. C. (2003). Increasing the yield of soluble recombinant protein expressed in E. coli by induction during late log phase. Biotechniques 34, 524–526, 528, 530.
Gao, W., Rzewski, A., Sun, H., Robbins, P. D., and Gambotto, A. (2004). UpGene: application of a web-based DNA codon optimization algorithm. Biotechnol. Prog. 20, 443–448. doi: 10.1021/bp0300467
Ge, X., Yang, D. S. C., Trabbic-Carlson, K., Kim, B., Chilkoti, A., and Filipe, C. D. M. (2005). Self-cleavable stimulus responsive tags for protein purification without chromatography. J. Am. Chem. Soc. 127, 11228–11229. doi: 10.1021/ja0531125
Georgiou, G., and Segatori, L. (2005). Preparative expression of secreted proteins in bacteria: status report and future prospects. Curr. Opin. Biotechnol. 16, 538–545. doi: 10.1016/j.copbio.2005.07.008
Goldstein, M. A., and Doi, R. H. (1995). Prokaryotic promoters in biotechnology. Biotechnol. Annu. Rev. 1, 105–128. doi: 10.1016/S1387-2656(08)70049-8
Gonzalez-Montalban, N., Garcia-Fruitos, E., and Villaverde, A. (2007). Recombinant protein solubility - does more mean better? Nat. Biotechnol. 25, 718–720. doi: 10.1038/nbt0707-718
Goodman, D. B., Church, G. M., and Kosuri, S. (2013). Causes and effects of N-terminal codon bias in bacterial genes. Science 342, 475–479. doi: 10.1126/science.1241934
Gottesman, S. (1996). Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 30, 465–506. doi: 10.1146/annurev.genet.30.1.465
Graumann, K., and Premstaller, A. (2006). Manufacturing of recombinant therapeutic proteins in microbial systems. Biotechnol. J. 1, 164–186. doi: 10.1002/biot.200500051
Grodberg, J., and Dunn, J. J. (1988). ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol. 170, 1245–1253.
Gustafsson, C., Govindarajan, S., and Minshull, J. (2004). Codon bias and heterologous protein expression. Trends Biotechnol. 22, 346–353. doi: 10.1016/j.tibtech.2004.04.006
Guzman, L. M., Belin, D., Carson, M. J., and Beckwith, J. (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130.
Hammarstrom, M., Hellgren, N., Van Den Berg, S., Berglund, H., and Hard, T. (2002). Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 11, 313–321. doi: 10.1110/ps.22102
Hammarstrom, M., Woestenenk, E. A., Hellgren, N., Hard, T., and Berglund, H. (2006). Effect of N-terminal solubility enhancing fusion proteins on yield of purified target protein. J. Struct. Funct. Genomics 7, 1–14. doi: 10.1007/s10969-005-9003-7
Hartl, F. U., and Hayer-Hartl, M. (2002). Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858. doi: 10.1126/science.1068408
Hartley, D. L., and Kane, J. F. (1988). Properties of inclusion bodies from recombinant Escherichia coli. Biochem. Soc. Trans. 16, 101–102.
Hayashi, K., and Kojima, C. (2008). pCold-GST vector: a novel cold-shock vector containing GST tag for soluble protein production. Protein Expr. Purif. 62, 120–127. doi: 10.1016/j.pep.2008.07.007
Hopp, T. P., Prickett, K. S., Price, V. L., Libby, R. T., March, C. J., Pat Cerretti, D., et al. (1988). A short polypeptide marker sequence useful for recombinant protein identification and purification. Nat. Biotechnol. 6, 1204–1210. doi: 10.1038/nbt1088-1204
Huber, R., Scheidle, M., Dittrich, B., Klee, D., and Buchs, J. (2009). Equalizing growth in high-throughput small scale cultivations via precultures operated in fed-batch mode. Biotechnol. Bioeng. 103, 1095–1102. doi: 10.1002/bit.22349
Hwang, P. M., Pan, J. S., and Sykes, B. D. (2014). Targeted expression, purification, and cleavage of fusion proteins from inclusion bodies in Escherichia coli. FEBS Lett. 588, 247–252. doi: 10.1016/j.febslet.2013.09.028
Itakura, K., Hirose, T., Crea, R., Riggs, A. D., Heyneker, H. L., Bolivar, F., et al. (1977). Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science 198, 1056–1063. doi: 10.1126/science.412251
Jana, S., and Deb, J. K. (2005). Strategies for efficient production of heterologous proteins in Escherichia coli. Appl. Microbiol. Biotechnol. 67, 289–298. doi: 10.1007/s00253-004-1814-0
Jayaraj, S., Reid, R., and Santi, D. V. (2005). GeMS: an advanced software package for designing synthetic genes. Nucleic Acids Res. 33, 3011–3016. doi: 10.1093/nar/gki614
Jenny, R. J., Mann, K. G., and Lundblad, R. L. (2003). A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr. Purif. 31, 1–11. doi: 10.1016/S1046-5928(03)00168-2
Johnson, A. D., Poteete, A. R., Lauer, G., Sauer, R. T., Ackers, G. K., and Ptashne, M. (1981). lambda Repressor and cro – components of an efficient molecular switch. Nature 294, 217–223. doi: 10.1038/294217a0
Kane, J. F. (1995). Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr. Opin. Biotechnol. 6, 494–500. doi: 10.1016/0958-1669(95)80082-4
Kapust, R. B., Tozser, J., Copeland, T. D., and Waugh, D. S. (2002). The P1′ specificity of tobacco etch virus protease. Biochem. Biophys. Res. Commun. 294, 949–955. doi: 10.1016/S0006-291X(02)00574-0
Kapust, R. B., and Waugh, D. S. (1999). Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8, 1668–1674. doi: 10.1110/ps.8.8.1668
Khan, F., Legler, P. M., Mease, R. M., Duncan, E. H., Bergmann-Leitner, E. S., and Angov, E. (2012). Histidine affinity tags affect MSP1(42) structural stability and immunodominance in mice. Biotechnol. J. 7, 133–147. doi: 10.1002/biot.201100331
Khlebnikov, A., and Keasling, J. D. (2002). Effect of lacY expression on homogeneity of induction from the P(tac) and P(trc) promoters by natural and synthetic inducers. Biotechnol. Prog. 18, 672–674. doi: 10.1021/bp010141k
Klose, J., Wendt, N., Kubald, S., Krause, E., Fechner, K., Beyermann, M., et al. (2004). Hexa-histidin tag position influences disulfide structure but not binding behavior of in vitro folded N-terminal domain of rat corticotropin-releasing factor receptor type 2a. Protein Sci. 13, 2470–2475. doi: 10.1110/ps.04835904
Korpimaki, T., Kurittu, J., and Karp, M. (2003). Surprisingly fast disappearance of beta-lactam selection pressure in cultivation as detected with novel biosensing approaches. J. Microbiol. Methods 53, 37–42. doi: 10.1016/S0167-7012(02)00213-0
Kroll, J., Klinter, S., Schneider, C., Voss, I., and Steinbuchel, A. (2010). Plasmid addiction systems: perspectives and applications in biotechnology. Microb. Biotechnol. 3, 634–657. doi: 10.1111/j.1751-7915.2010.00170.x
Kurokawa, Y., Yanagi, H., and Yura, T. (2000). Overexpression of protein disulfide isomerase DsbC stabilizes multiple-disulfide-bonded recombinant protein produced and transported to the periplasm in Escherichia coli. Appl. Environ. Microbiol. 66, 3960–3965. doi: 10.1128/AEM.66.9.3960-3965.2000
Lanzer, M., and Bujard, H. (1988). Promoters largely determine the efficiency of repressor action. Proc. Natl. Acad. Sci. U.S.A. 85, 8973–8977. doi: 10.1073/pnas.85.23.8973
LaVallie, E. R., Diblasio, E. A., Kovacic, S., Grant, K. L., Schendel, P. F., and Mccoy, J. M. (1993). A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (N. Y.) 11, 187–193. doi: 10.1038/nbt0293-187
Lee, C., Kim, J., Shin, S. G., and Hwang, S. (2006). Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 123, 273–280. doi: 10.1016/j.jbiotec.2005.11.014
Lee, S. Y. (1996). High cell-density culture of Escherichia coli. Trends Biotechnol. 14, 98–105. doi: 10.1016/0167-7799(96)80930-9
Lewin, C. S., Howard, B. M., Ratcliffe, N. T., and Smith, J. T. (1989). 4-quinolones and the SOS response. J. Med. Microbiol. 29, 139–144. doi: 10.1099/00222615-29-2-139
Li, G. W., Oh, E., and Weissman, J. S. (2012). The anti-Shine–Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484, 538–541. doi: 10.1038/nature10965
Lin-Chao, S., and Bremer, H. (1986). Effect of the bacterial growth rate on replication control of plasmid pBR322 in Escherichia coli. Mol. Gen. Genet. 203, 143–149. doi: 10.1007/BF00330395
Lobstein, J., Emrich, C. A., Jeans, C., Faulkner, M., Riggs, P., and Berkmen, M. (2012). SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 11, 56. doi: 10.1186/1475-2859-11-56.
Losen, M., Frolich, B., Pohl, M., and Buchs, J. (2004). Effect of oxygen limitation and medium composition on Escherichia coli fermentation in shake-flask cultures. Biotechnol. Prog. 20, 1062–1068. doi: 10.1021/bp034282t
Madurawe, R. D., Chase, T. E., Tsao, E. I., and Bentley, W. E. (2000). A recombinant lipoprotein antigen against Lyme disease expressed in E. coli: fermentor operating strategies for improved yield. Biotechnol. Prog. 16, 571–576. doi: 10.1021/bp0000555
Makhatadze, G. I., and Privalov, P. L. (1995). Energetics of protein structure. Adv. Protein Chem. 47, 307–425. doi: 10.1016/S0065-3233(08)60548-3
Makoff, A. J., and Oxer, M. D. (1991). High level heterologous expression in E. coli using mutant forms of the lac promoter. Nucleic Acids Res. 19, 2417–2421. doi: 10.1093/nar/19.9.2417
Makrides, S. C. (1996). Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 60, 512–538.
Marisch, K., Bayer, K., Cserjan-Puschmann, M., Luchner, M., and Striedner, G. (2013). Evaluation of three industrial Escherichia coli strains in fed-batch cultivations during high-level SOD protein production. Microb. Cell Fact. 12, 58. doi: 10.1186/1475-2859-12-58
Martinez-Alonso, M., Gonzalez-Montalban, N., Garcia-Fruitos, E., and Villaverde, A. (2008). The functional quality of soluble recombinant polypeptides produced in Escherichia coli is defined by a wide conformational spectrum. Appl. Environ. Microbiol. 74, 7431–7433. doi: 10.1128/AEM.01446-08
Martinez-Alonso, M., Vera, A., and Villaverde, A. (2007). Role of the chaperone DnaK in protein solubility and conformational quality in inclusion body-forming Escherichia coli cells. FEMS Microbiol. Lett. 273, 187–195. doi: 10.1111/j.1574-6968.2007.00788.x
McCarty, J. S., and Walker, G. C. (1991). DnaK as a thermometer: threonine-199 is site of autophosphorylation and is critical for ATPase activity. Proc. Natl. Acad. Sci. U.S.A. 88, 9513–9517. doi: 10.1073/pnas.88.21.9513
McDaniel, L. E., and Bailey, E. G. (1969). Effect of shaking speed and type of closure on shake flask cultures. Appl. Microbiol. 17, 286–290.
Menart, V., Jevsevar, S., Vilar, M., Trobis, A., and Pavko, A. (2003). Constitutive versus thermoinducible expression of heterologous proteins in Escherichia coli based on strong PR,PL promoters from phage lambda. Biotechnol. Bioeng. 83, 181–190. doi: 10.1002/bit.10660
Mendoza, J. A., Dulin, P., and Warren, T. (2000). The lower hydrolysis of ATP by the stress protein GroEL is a major factor responsible for the diminished chaperonin activity at low temperature. Cryobiology 41, 319–323. doi: 10.1006/cryo.2000.2287
Menzella, H. G. (2011). Comparison of two codon optimization strategies to enhance recombinant protein production in Escherichia coli. Microb. Cell Fact. 10, 15. doi: 10.1186/1475-2859-10-15
Menzella, H. G., Ceccarelli, E. A., and Gramajo, H. C. (2003). Novel Escherichia coli strain allows efficient recombinant protein production using lactose as inducer. Biotechnol. Bioeng. 82, 809–817. doi: 10.1002/bit.10630
Mergulhao, F. J., Summers, D. K., and Monteiro, G. A. (2005). Recombinant protein secretion in Escherichia coli. Biotechnol. Adv. 23, 177–202. doi: 10.1016/j.biotechadv.2004.11.003
Messens, J., and Collet, J. F. (2006). Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 38, 1050–1062. doi: 10.1016/j.biocel.2005.12.011
Meyer, D. E., and Chilkoti, A. (1999). Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat. Biotechnol. 17, 1112–1115. doi: 10.1038/15100
Mieschendahl, M., Petri, T., and Hanggi, U. (1986). A novel prophage independent trp regulated lambda PL expression system. Nat. Biotechnol. 4, 6. doi: 10.1038/nbt0986-802
Minton, N. P. (1984). Improved plasmid vectors for the isolation of translational lac gene fusions. Gene 31, 269–273. doi: 10.1016/0378-1119(84)90220-8
Miroux, B., and Walker, J. E. (1996). Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298. doi: 10.1006/jmbi.1996.0399
Moffatt, B. A., and Studier, F. W. (1987). T7 lysozyme inhibits transcription by T7 RNA polymerase. Cell 49, 221–227. doi: 10.1016/0092-8674(87)90563-0
Moulton, G. G. (2013). Fed-Batch Fermentation: A Practical Guide to Scalable Recombinant Protein Production in Escherichia coli. Cambridge: Woodhead Publishing Limited.
Müller-Hill, B. (1996). The Lac Operon: A Short History of a Genetic Paradigm. Berlin: Walter de Gruyter. doi: 10.1515/9783110879476
Müller-Hill, B., Crapo, L., and Gilbert, W. (1968). Mutants that make more lac repressor. Proc. Natl. Acad. Sci. U.S.A. 59, 1259–1264. doi: 10.1073/pnas.59.4.1259
Nilsson, J., Stahl, S., Lundeberg, J., Uhlen, M., and Nygren, P. A. (1997). Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr. Purif. 11, 1–16. doi: 10.1006/prep.1997.0767
Nishihara, K., Kanemori, M., Kitagawa, M., Yanagi, H., and Yura, T. (1998). Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl. Environ. Microbiol. 64, 1694–1699.
Nishihara, K., Kanemori, M., Yanagi, H., and Yura, T. (2000). Overexpression of trigger factor prevents aggregation of recombinant proteins in Escherichia coli. Appl. Environ. Microbiol. 66, 884–889. doi: 10.1128/AEM.66.3.884-889.2000
Nordstrom, K. (2006). Plasmid R1 – replication and its control. Plasmid 55, 1–26. doi: 10.1016/j.plasmid.2005.07.002
O’Beirne, D., and Hamer, G. (2000). Oxygen availability and the growth of Escherichia coli W3110: a problem exacerbated by scale-up. Bioprocess Eng. 23, 487–494. doi: 10.1007/s004499900185
Ohana, R. F., Encell, L. P., Zhao, K., Simpson, D., Slater, M. R., Urh, M., et al. (2009). HaloTag7: a genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expr. Purif. 68, 110–120. doi: 10.1016/j.pep.2009.05.010
Oresic, M., and Shalloway, D. (1998). Specific correlations between relative synonymous codon usage and protein secondary structure. J. Mol. Biol. 281, 31–48. doi: 10.1006/jmbi.1998.1921
Ou, J., Wang, L., Ding, X., Du, J., Zhang, Y., Chen, H., et al. (2004). Stationary phase protein overproduction is a fundamental capability of Escherichia coli. Biochem. Biophys. Res. Commun. 314, 174–180. doi: 10.1016/j.bbrc.2003.12.077
Palomares, L. A., Estrada-Mondaca, S., and Ramirez, O. T. (2004). Production of recombinant proteins: challenges and solutions. Methods Mol. Biol. 267, 15–52. doi: 10.1385/1-59259-774-2:015
Parks, T. D., Leuther, K. K., Howard, E. D., Johnston, S. A., and Dougherty, W. G. (1994). Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 216, 413–417. doi: 10.1006/abio.1994.1060
Pedersen, S. (1984). Escherichia coli ribosomes translate in vivo with variable rate. EMBO J. 3, 2895–2898.
Perron-Savard, P., De Crescenzo, G., and Le Moual, H. (2005). Dimerization and DNA binding of the Salmonella enterica PhoP response regulator are phosphorylation independent. Microbiology 151, 3979–3987. doi: 10.1099/mic.0.28236-0
Peubez, I., Chaudet, N., Mignon, C., Hild, G., Husson, S., Courtois, V., et al. (2010). Antibiotic-free selection in E. coli: new considerations for optimal design and improved production. Microb. Cell Fact. 9, 65. doi: 10.1186/1475-2859-9-65
Pope, B., and Kent, H. M. (1996). High efficiency 5 min transformation of Escherichia coli. Nucleic Acids Res. 24, 536–537. doi: 10.1093/nar/24.3.536
Porath, J., and Olin, B. (1983). Immobilized metal ion affinity adsorption and immobilized metal ion affinity chromatography of biomaterials. Serum protein affinities for gel-immobilized iron and nickel ions. Biochemistry 22, 1621–1630. doi: 10.1021/bi00276a015
Postma, P. W., and Lengeler, J. W. (1985). Phosphoenolpyruvate: carbohydrate phosphotransferase system of bacteria. Microbiol. Rev. 49, 232–269.
Puigbo, P., Guzman, E., Romeu, A., and Garcia-Vallve, S. (2007). OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 35, W126–W131. doi: 10.1093/nar/gkm219
Qing, G., Ma, L. C., Khorchid, A., Swapna, G. V., Mal, T. K., Takayama, M. M., et al. (2004). Cold-shock induced high-yield protein production in Escherichia coli. Nat. Biotechnol. 22, 877–882. doi: 10.1038/nbt984
Rais-Beghdadi, C., Roggero, M. A., Fasel, N., and Reymond, C. D. (1998). Purification of recombinant proteins by chemical removal of the affinity tag. Appl. Biochem. Biotechnol. 74, 95–103. doi: 10.1007/BF02787176
Raran-Kurussi, S., and Waugh, D. S. (2012). The ability to enhance the solubility of its fusion partners is an intrinsic property of maltose-binding protein but their folding is either spontaneous or chaperone-mediated. PLoS ONE 7:e49589. doi: 10.1371/journal.pone.0049589
Roberts, M. C. (1996). Tetracycline resistance determinants: mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol. Rev. 19, 1–24. doi: 10.1111/j.1574-6976.1996.tb00251.x
Rosano, G. L., Bruch, E. M., and Ceccarelli, E. A. (2011). Insights into the Clp/HSP100 chaperone system from chloroplasts of Arabidopsis thaliana. J. Biol. Chem. 286, 29671–29680. doi: 10.1074/jbc.M110.211946
Rosano, G. L., and Ceccarelli, E. A. (2009). Rare codon content affects the solubility of recombinant proteins in a codon bias-adjusted Escherichia coli strain. Microb. Cell Fact. 8, 41. doi: 10.1186/1475-2859-8-41
Routledge, S. (2012). Beyond de-foaming: the effects of antifoams on bioprocess productivity. Comput. Struct. Biotechnol. J. 3, 1–7. doi: 10.5936/csbj.201210014
Routledge, S. J., Hewitt, C. J., Bora, N., and Bill, R. M. (2011). Antifoam addition to shake flask cultures of recombinant Pichia pastoris increases yield. Microb. Cell Fact. 10, 17. doi: 10.1186/1475-2859-10-17
Sahdev, S., Khattar, S. K., and Saini, K. S. (2008). Production of active eukaryotic proteins through bacterial expression systems: a review of the existing biotechnology strategies. Mol. Cell. Biochem. 307, 249–264. doi: 10.1007/s11010-007-9603-6
Saida, F., Uzan, M., Odaert, B., and Bontems, F. (2006). Expression of highly toxic genes in E. coli: special strategies and genetic tools. Curr. Protein Pept. Sci. 7, 47–56. doi: 10.2174/138920306775474095
San-Miguel, T., Perez-Bermudez, P., and Gavidia, I. (2013). Production of soluble eukaryotic recombinant proteins in is favoured in early log-phase cultures induced at low temperature. Springerplus 2, 89. doi: 10.1186/2193-1801-2-89
Scheidle, M., Dittrich, B., Klinger, J., Ikeda, H., Klee, D., and Buchs, J. (2011). Controlling pH in shake flasks using polymer-based controlled-release discs with pre-determined release kinetics. BMC Biotechnol. 11:25. doi: 10.1186/1472-6750-11-25
Schein, C., and Noteborn, M. (1988). Formation of soluble recombinant proteins in Escherichia coli is favored by lower growth temperature. Nat. Biotechnol. 6, 3. doi: 10.1038/nbt0388-291
Schellman, J. A. (1997). Temperature, stability, and the hydrophobic interaction. Biophys. J. 73, 2960–2964. doi: 10.1016/S0006-3495(97)78324-3
Schierle, C. F., Berkmen, M., Huber, D., Kumamoto, C., Boyd, D., and Beckwith, J. (2003). The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J. Bacteriol. 185, 5706–5713. doi: 10.1128/JB.185.19.5706-5713.2003
Schleif, R. (2000). Regulation of the L-arabinose operon of Escherichia coli. Trends Genet. 16, 559–565. doi: 10.1016/S0168-9525(00)02153-3
Schleif, R. (2010). AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol. Rev. 34, 779–796. doi: 10.1111/j.1574-6976.2010.00226.x
Sezonov, G., Joseleau-Petit, D., and D’Ari, R. (2007). Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 189, 8746–8749. doi: 10.1128/JB.01368-07
Shatzman, A. R., Gross, M. S., and Rosenberg, M. (2001). Expression using vectors with phage lambda regulatory sequences. Curr. Protoc. Mol. Biol. Chapter 16, Unit16.13. doi: 10.1002/0471142727.mb1603s11
Shaw, W. V. (1983). Chloramphenicol acetyltransferase: enzymology and molecular biology. CRC Crit. Rev. Biochem. 14, 1–46. doi: 10.3109/10409238309102789
Shih, Y. P., Kung, W. M., Chen, J. C., Yeh, C. H., Wang, A. H., and Wang, T. F. (2002). High-throughput screening of soluble recombinant proteins. Protein Sci. 11, 1714–1719. doi: 10.1110/ps.0205202
Shiloach, J., and Fass, R. (2005). Growing E. coli to high cell density – a historical perspective on method development. Biotechnol. Adv. 23, 345–357. doi: 10.1016/j.biotechadv.2005.04.004
Shur, O., Dooley, K., Blenner, M., Baltimore, M., and Banta, S. (2013). A designed, phase changing RTX-based peptide for efficient bioseparations. Biotechniques 54, 197–198, 200, 202, 204, 206. doi: 10.2144/000114010
Siegele, D. A., and Hu, J. C. (1997). Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc. Natl. Acad. Sci. U.S.A. 94, 8168–8172. doi: 10.1073/pnas.94.15.8168
Silverstone, A. E., Arditti, R. R., and Magasanik, B. (1970). Catabolite-insensitive revertants of lac promoter mutants. Proc. Natl. Acad. Sci. U.S.A. 66, 773–779. doi: 10.1073/pnas.66.3.773
Sivashanmugam, A., Murray, V., Cui, C., Zhang, Y., Wang, J., and Li, Q. (2009). Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci. 18, 936–948. doi: 10.1002/pro.102
Smith, D. B., and Johnson, K. S. (1988). Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31–40. doi: 10.1016/0378-1119(88)90005-4
Soares, C. R., Gomide, F. I., Ueda, E. K., and Bartolini, P. (2003). Periplasmic expression of human growth hormone via plasmid vectors containing the lambdaPL promoter: use of HPLC for product quantification. Protein Eng. 16, 1131–1138. doi: 10.1093/protein/gzg114
Sorensen, H. P., and Mortensen, K. K. (2005). Advanced genetic strategies for recombinant protein expression in Escherichia coli. J. Biotechnol. 115, 113–128. doi: 10.1016/j.jbiotec.2004.08.004
Stano, N. M., and Patel, S. S. (2004). T7 lysozyme represses T7 RNA polymerase transcription by destabilizing the open complex during initiation. J. Biol. Chem. 279, 16136–16143. doi: 10.1074/jbc.M400139200
Stevens, R. C. (2000). Design of high-throughput methods of protein production for structural biology. Structure 8, R177–R185. doi: 10.1016/S0969-2126(00)00193-3
Stewart, E. J., Aslund, F., and Beckwith, J. (1998). Disulfide bond formation in the Escherichia coli cytoplasm: an in vivo role reversal for the thioredoxins. EMBO J. 17, 5543–5550. doi: 10.1093/emboj/17.19.5543
Stoker, N. G., Fairweather, N. F., and Spratt, B. G. (1982). Versatile low-copy-number plasmid vectors for cloning in Escherichia coli. Gene 18, 335–341. doi: 10.1016/0378-1119(82)90172-X
Strocchi, M., Ferrer, M., Timmis, K. N., and Golyshin, P. N. (2006). Low temperature-induced systems failure in Escherichia coli: insights from rescue by cold-adapted chaperones. Proteomics 6, 193–206. doi: 10.1002/pmic.200500031
Studier, F. W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234. doi: 10.1016/j.pep.2005.01.016
Studier, F. W. (2014). Stable expression clones and auto-induction for protein production in E. coli. Methods Mol. Biol. 1091, 17–32. doi: 10.1007/978-1-62703-691-7_2
Studier, F. W., and Moffatt, B. A. (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130. doi: 10.1016/0022-2836(86)90385-2
Summers, D. K., Beton, C. W., and Withers, H. L. (1993). Multicopy plasmid instability: the dimer catastrophe hypothesis. Mol. Microbiol. 8, 1031–1038. doi: 10.1111/j.1365-2958.1993.tb01648.x
Supek, F., and Vlahovicek, K. (2004). INCA: synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics 20, 2329–2330. doi: 10.1093/bioinformatics/bth238
Terpe, K. (2003). Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 60, 523–533. doi: 10.1007/s00253-002-1158-6
Thanaraj, T. A., and Argos, P. (1996). Ribosome-mediated translational pause and protein domain organization. Protein Sci. 5, 1594–1612. doi: 10.1002/pro.5560050814
Tropea, J. E., Cherry, S., and Waugh, D. S. (2009). Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol. Biol. 498, 297–307. doi: 10.1007/978-1-59745-196-3_19
Tsai, C. J., Sauna, Z. E., Kimchi-Sarfaty, C., Ambudkar, S. V., Gottesman, M. M., and Nussinov, R. (2008). Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J. Mol. Biol. 383, 281–291. doi: 10.1016/j.jmb.2008.08.012
Tsumoto, K., Ejima, D., Kumagai, I., and Arakawa, T. (2003). Practical considerations in refolding proteins from inclusion bodies. Protein Expr. Purif. 28, 1–8. doi: 10.1016/S1046-5928(02)00641-1
Umezawa, H. (1979). Studies on aminoglycoside antibiotics: enzymic mechanism of resistance and genetics. Jpn. J. Antibiot. 32(Suppl.), S1–S14.
Unden, G., Becker, S., Bongaerts, J., Holighaus, G., Schirawski, J., and Six, S. (1995). O2-sensing and O2-dependent gene regulation in facultatively anaerobic bacteria. Arch. Microbiol. 164, 81–90. doi: 10.1007/s002030050238
Valdez-Cruz, N. A., Caspeta, L., Perez, N. O., Ramirez, O. T., and Trujillo-Roldan, M. A. (2010). Production of recombinant proteins in E. coli by the heat inducible expression system based on the phage lambda pL and/or pR promoters. Microb. Cell Fact. 9, 18. doi: 10.1186/1475-2859-9-18
Valent, Q. A., Scotti, P. A., High, S., De Gier, J. W., Von Heijne, G., Lentzen, G., et al. (1998). The Escherichia coli SRP and SecB targeting pathways converge at the translocon. EMBO J. 17, 2504–2512. doi: 10.1093/emboj/17.9.2504
Vasina, J. A., and Baneyx, F. (1997). Expression of aggregation-prone recombinant proteins at low temperatures: a comparative study of the Escherichia coli cspA and tac promoter systems. Protein Expr. Purif. 9, 211–218. doi: 10.1006/prep.1996.0678
Vasina, J. A., Peterson, M. S., and Baneyx, F. (1998). Scale-up and optimization of the low-temperature inducible cspA promoter system. Biotechnol. Prog. 14, 714–721. doi: 10.1021/bp980061p
Vera, A., Gonzalez-Montalban, N., Aris, A., and Villaverde, A. (2007). The conformational quality of insoluble recombinant proteins is enhanced at low growth temperatures. Biotechnol. Bioeng. 96, 1101–1106. doi: 10.1002/bit.21218
Vieira, J., and Messing, J. (1987). Production of single-stranded plasmid DNA. Methods Enzymol. 153, 3–11. doi: 10.1016/0076-6879(87)53044-0
Voss, I., and Steinbuchel, A. (2006). Application of a KDPG-aldolase gene-dependent addiction system for enhanced production of cyanophycin in Ralstonia eutropha strain H16. Metab. Eng. 8, 66–78. doi: 10.1016/j.ymben.2005.09.003
Wagner, S., Klepsch, M. M., Schlegel, S., Appel, A., Draheim, R., Tarry, M., et al. (2008). Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. U.S.A. 105, 14371–14376. doi: 10.1073/pnas.0804090105
Wang, R. F., and Kushner, S. R. (1991). Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199. doi: 10.1016/0378-1119(91)90366-J
Wanner, B. L., Kodaira, R., and Neidhardt, F. C. (1978). Regulation of lac operon expression: reappraisal of the theory of catabolite repression. J. Bacteriol. 136, 947–954.
Waugh, D. S. (2011). An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 80, 283–293. doi: 10.1016/j.pep.2011.08.005
Weickert, M. J., Pagratis, M., Glascock, C. B., and Blackmore, R. (1999). A mutation that improves soluble recombinant hemoglobin accumulation in Escherichia coli in heme excess. Appl. Environ. Microbiol. 65, 640–647.
Welch, M., Govindarajan, S., Ness, J. E., Villalobos, A., Gurney, A., Minshull, J., et al. (2009). Design parameters to control synthetic gene expression in Escherichia coli. PLoS ONE 4:e7002. doi: 10.1371/journal.pone.0007002
Weuster-Botz, D., Altenbach-Rehm, J., and Arnold, M. (2001). Parallel substrate feeding and pH-control in shaking-flasks. Biochem. Eng. J. 7, 163–170. doi: 10.1016/S1369-703X(00)00117-0
Wild, J., Hradecna, Z., and Szybalski, W. (2002). Conditionally amplifiable BACs: switching from single-copy to high-copy vectors and genomic clones. Genome Res. 12, 1434–1444. doi: 10.1101/gr.130502
Winkler, H. H., and Wilson, T. H. (1967). Inhibition of beta-galactoside transport by substrates of the glucose transport system in Escherichia coli. Biochim. Biophys. Acta 135, 1030–1051. doi: 10.1016/0005-2736(67)90073-9
Wu, J., and Filutowicz, M. (1999). Hexahistidine (His6)-tag dependent protein dimerization: a cautionary tale. Acta Biochim. Pol. 46, 591–599.
Yamanè, T., and Shimizu, S. (1984). “Fed-batch techniques in microbial processes,” in Bioprocess Parameter Control, ed. P. Agrawal (Berlin: Springer), 147–194. doi: 10.1007/BFb0006382
Yang, Q., Xu, J., Li, M., Lei, X., and An, L. (2003). High-level expression of a soluble snake venom enzyme, gloshedobin, in E. coli in the presence of metal ions. Biotechnol. Lett. 25, 607–610. doi: 10.1023/A:1023067626846
Yee, L., and Blanch, H. W. (1992). Recombinant protein expression in high cell density fed-batch cultures of Escherichia coli. Biotechnology (N. Y.) 10, 1550–1556. doi: 10.1038/nbt1292-1550
Yona, A. H., Bloom-Ackermann, Z., Frumkin, I., Hanson-Smith, V., Charpak-Amikam, Y., Feng, Q., et al. (2013). tRNA genes rapidly change in evolution to meet novel translational demands. Elife 2:e01339. doi: 10.7554/eLife.01339
Keywords: recombinant protein expression, Escherichia coli, expression plasmid, inclusion bodies, affinity tags, E. coli expression strains
Citation: Rosano GL and Ceccarelli EA (2014) Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 5:172. doi: 10.3389/fmicb.2014.00172
Received: 20 December 2013; Accepted: 29 March 2014;
Published online: 17 April 2014.
Edited by:
Peter Neubauer, Technische Universität Berlin, GermanyReviewed by:
Jose M. Bruno-Barcena, North Carolina State University, USAThomas Schweder, Ernst-Moritz-Arndt-Universität Greifswald, Germany
Copyright © 2014 Rosano and Ceccarelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Germán L. Rosano, Instituto de Biología Molecular y Celular de Rosario, Consejo Nacional de Investigaciones Científicas y Técnicas, Esmeralda y Ocampo, Rosario 2000, Argentina e-mail:cm9zYW5vQGlici1jb25pY2V0Lmdvdi5hcg==