 Jovana Vukovic
Jovana Vukovic Dzihan Abazovic2*
Dzihan Abazovic2* Dusan Vucetic
Dusan Vucetic Sanja Medenica
Sanja Medenica- 1Institute for the Application of Nuclear Energy - INEP, University of Belgrade, Belgrade, Serbia
- 2Faculty of Medicine, University of Belgrade, Belgrade, Serbia
- 3Institute for Transfusiology and Hemobiology, Military Medical Academy, Belgrade, Serbia
- 4Faculty of Medicine, University of Montenegro, Podgorica, Montenegro
- 5Department of Endocrinology, Internal Medicine Clinic, Clinical Center of Montenegro, Podgorica, Montenegro
CAR-T therapy has demonstrated great success in treating hematological malignancies, which has led to further research into its potential in treating other diseases. Autoimmune diseases have great potential to be treated with this therapy due to the possibility of specific targeting of pathological immune cells and cells that produce autoantibodies, which could lead to permanent healing and restoration of immunological tolerance. Several approaches are currently under investigation, including targeting and depleting B cells via CD19 in the early stages of the disease, simultaneously targeting B cells and memory plasma cells in later stages and refractory states, as well as targeting specific autoantigens through the chimeric autoantibody receptor (CAAR). Additionally, CAR-engineered T regulatory cells can be modified to specifically target the autoimmune niche and modulate the pathological immune response. The encouraging results from preclinical studies have led to the first successful use of CAR-T therapy in humans to treat autoimmunity. This paved the way for further clinical studies, aiming to evaluate the long-term safety and efficacy of these therapies, potentially revolutionizing clinical use.
1 Introduction
Autoimmune diseases encompass a heterogeneous group of conditions that can be manifested in a particular organ or at the systemic level. Despite their diversity, they share common immunopathogenic mechanisms involving dysregulated T and B cell responses. A combination of factors such as genetic predisposition and environmental triggers mainly influences the development of autoimmune disorders, leading to the breakdown of self-tolerance and pathological immune reactions, causing tissue destruction (1). T and B lymphocytes play crucial roles in the immune system, acting as the main drivers of adaptive immunity. CD4+ T lymphocytes orchestrate immune responses by releasing cytokines and activating other immune cells (2). When CD4+ T lymphocytes malfunction, autoreactive T cells can be generated, leading to persistent inflammation and tissue damage. Similarly, when B lymphocytes are not appropriately regulated, they can start producing autoantibodies that target self-antigens, contributing to tissue damage (1, 3, 4). An in-depth understanding of these processes is essential for developing precisely targeted interventions without damaging healthy tissue.
Ensuring the desired immune response is crucial to re-establishing immune tolerance and effectively mitigating the consequences of the disease. Conventional treatments for these diseases tend to focus on symptom management, often resulting in undesirable side effects and limited effectiveness. However, a more effective strategy would involve treatments that focus on the root cause of the disease, such as chimeric antigen receptor T (CAR-T) cell therapy. Recent advancements in CAR-T therapy hold promise in treating autoimmune diseases.
CAR-T therapy involves genetically engineering a patient’s T cells to express chimeric antigen receptors (CARs) that target specific antigens. CD19 is the most investigated target for CAR-based therapy. It is expressed in normal and neoplastic B cells and maintained at a high level during all stages of B-cell development (5, 6). CD19+ malignancies were the first cancers to be eliminated by CAR-engineered human T cells administered intravenously to mice (7). Various CD19 CARs successfully eliminated B cell tumors, resulting in ongoing clinical trials and FDA approval (8, 9). Similarly, B-cell depletion could also be a promising therapeutic strategy for treating autoimmune diseases. Moreover, other strategies are under investigation, such as restricted B cell depletion by targeting autoantigens, dual targeting, and engineering regulatory T cells (Tregs), which will be further reviewed (Figure 1).
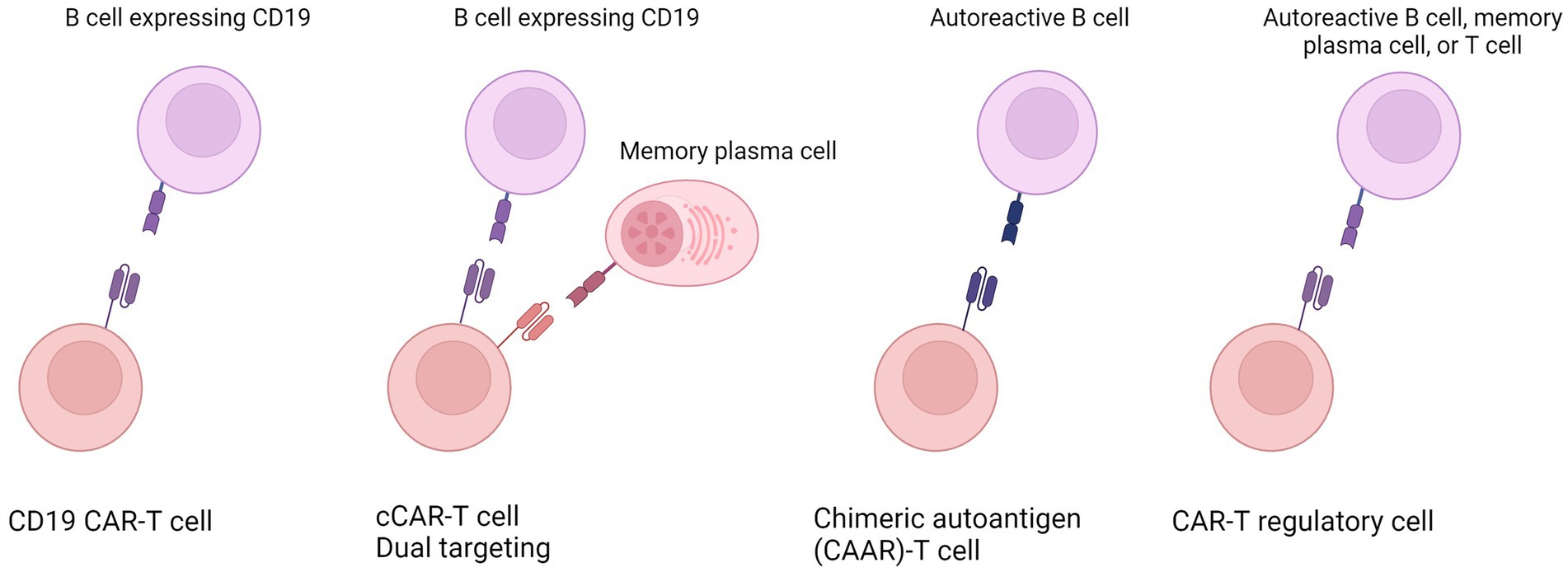
Figure 1. Overview of the various strategies employed in engineering CAR-T cells to tackle autoimmune diseases. These innovative approaches encompass (from left to right) universal targeting of all B cells; the dual targeting or compound CAR engineering targeting B cells and additional cells such as memory plasma cells; restricted B cell depletion or specific targeting of only autoreactive B cells; and the engineering of T regulatory cells. Created in Biorender.
1.1 Chimeric antigen receptor characteristics, design, and manufacturing
Chimeric antigen receptors (CARs) are modified receptors that alter the specificity and activity of T lymphocytes and other immune cells, by bringing together the variable regions of high affinity monoclonal antibodies with intracellular signaling components of the T-cell receptor (TCR) complex. Their modulatory structure consists of four domains: ligand-binding, spacer, transmembrane, and cytoplasmic domains (10). CARs bind to antigens through an extracellular portion composed of a ligand-binding domain and spacer, typically constructed using single-chain variable fragments (scFv) derived from antibodies, Fab fragments from libraries, or natural ligands. Most commonly used, scFvs can function autonomously or as modular units for CAR-T cell therapies, determining the ability of modified T cells to recognize and target desired antigens (11). CAR-mediated recognition is MHC-independent, allowing it to overcome tolerance to self-antigens and target any chosen antigen expressed on the cell surface (12, 13). CARs also control T cell growth and persistence, impacting both efficacy and safety. Associating CARs with costimulatory ligands, chimeric costimulatory receptors, or cytokines can improve T cell efficacy, specificity, and safety (11, 14, 15).
T cells that express first-generation CARs lacking a co-stimulatory domain are insufficient for T cell activation and show limited in vivo effectiveness (16, 17). The second-generation CARs were developed to address this issue by adding a co-stimulatory domain, commonly CD28 and 4-1BB (CD137). Co-stimulatory domains provide additional signals upon antigen recognition, which is crucial for enhanced proliferation, cytokine release, cytotoxic activity, memory formation, and persistence of T cells (18). CAR-T cells with the CD28 intracellular co-stimulatory domain demonstrated significantly increased proliferation and persistence compared to those without a co-stimulatory domain (19). Moreover, CAR-T cells containing the CD28 domain showed better early growth and cytotoxic activity than those containing the 41BB costimulatory domain, which showed better long-term survival (20). Following, the third-generation of CARs incorporates multiple co-stimulatory domains, while fourth-generation CARs are engineered to express additional inducible transgenic elements, typically for inducible cytokine secretion, to improve T cell function and reduce off-target toxicity (14, 21, 22).
The process of CAR-T cell manufacturing begins with harvesting specific T cell subsets from patients via leukapheresis, washing them, and enriching them for specific subsets. Subsequently, T cells undergo activation to ensure adequate transduction of CAR cDNA. CAR gene delivery relies on viral and non-viral gene transfer systems. Following transduction, modified T cells are expanded and, after final concentration, ready to give back to patients (23).
2 CD19-targeted CAR-T cell therapy for autoimmune diseases
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease affecting various organs and tissues. A key role in SLE immunopathogenesis play B cells by producing autoantibodies targeting double-stranded DNA, that cause inflammation and tissue damage. Despite several therapeutic approaches targeting autoreactive B lymphocytes, SLE remains incurable (6). Due to sustained expression of CD19 in B-cell lineage, CD19 CAR-T therapy can potentially treat lupus by offering precision targeting and regulating the overall immune response (5, 6). Modifying T cells with CD19-targeting CAR and targeting CD19+ B cells may restore immune balance, decrease autoantibody production, and reduce the inflammatory response associated with lupus. Preclinical results indicate that anti-CD19 CAR-T cells are effective in depleting CD19+ B cells in a lupus model, eliminating autoantibody production, reversing disease manifestation in target organs, and increasing lifespan, suggesting long-term efficacy of this therapeutic approach (24, 25). Recently, encouraging effects have been shown in humans for anti-CD19 CAR-T treatment of SLE. A patient with severe SLE was treated with autologous CD19 CAR-T cells and exhibited complete and sustained depletion of circulating B cells, followed by the disappearance of dsDNA autoantibodies. The therapy showed no adverse events and suggested wider application in treating severe autoimmune (26). This result was confirmed by another study that treated five refractory SLE patients with anti-CD19 CAR-T. All patients demonstrated sustained remission 1 year following the treatment. Three months following the treatment, naïve B cells reappeared without a relapse of SLE symptoms. This finding suggests that CAR-T in autoimmunity, due to different microenvironments, demonstrates transient persistence and achieves acute B cell aplasia, effectively resetting the immune system (27). In a recent update to its SLE management guidelines, the European League Against Rheumatism (EULAR) included CAR-T as one of the treatment options for patients with refractory SLE (28). However, further clinical trials are needed to evaluate the long-term safety and efficacy of anti-CD19 CAR-T in SLE.
Furthermore, another successful human treatment with anti-CD19 CAR-T therapy has been reported for treating another autoimmune disease, myasthenia gravis, caused by a B-cell-driven dysfunction of neuromuscular transmission, often mediated by anti-acetylcholine receptor (anti-AchR) antibodies. A patient with refractory, anti-AchR positive generalized myasthenia gravis was successfully treated with anti-CD19 CAR-T therapy, followed by reduced anti-Achr antibodies without any CAR-T therapy-related side effects, demonstrating improved muscle strength and fatigue (29).
Additionally, two separate studies reported successfully treating two patients suffering from refractory antisynthetase syndrome with anti-CD19 CAR-T therapy (30, 31). This syndrome is characterized by the presence of autoantibodies directed against aminoacyl transfer RNA synthetase, along with clinical features such as interstitial lung disease, myositis, and arthritis. The therapy completely eliminated circulating B cells as well as pathogenic autoantibodies, and both patients experienced improved disease-related symptoms. One patient had a transient increase in myalgia and creatinine kinase concentration due to one of the most common side effects of CAR-T therapy, cytokine release syndrome (CRS), which resolved within 3 days (30). The second patient also experienced mild CRS, which might have stimulated preexisting autoreactive T cells, as an increased number of CD8+ Temra cells in peripheral blood were found, accompanied by exacerbations of muscle pain a few days following the treatment. Treatment with azathioprine and mycophenolate mofetil normalized Temra subsets and potentially contributed to the patient’s beneficial outcome (31).
3 Dual targeting for optimal treatment of autoimmune diseases
Early in the disease course, CD19-based therapy may prevent autoreactive plasma cell accumulation, but later, memory plasma cells may accumulate and lead to persistent autoantibody production despite B-cell depletion (32, 33). CD19, one of the earliest and most specific markers of B-lineage cells, may not be expressed in all plasma cells. Plasma cells express CD19 heterogeneously, and memory plasma cells are among the CD19-negative plasma cell populations (33–35). Additionally, B cell activating factor (BAFF) cytokine, a member of the TNF superfamily, plays a crucial role in promoting the survival and function of B cells and memory plasma cells (36, 37). BAFF can bind to BAFF-receptor (BAFFR), Transmembrane Activator and CAML-interactor (TACI) and B-Cell Maturation Antigen (BCMA) and these three receptors have distinctive roles in regulating B cells function (37, 38). Overexpression of BAFF leads to the development of autoreactive B cells and displays autoimmune-like symptoms in mice, highlighting the significance of dysregulated BAFF expression in autoimmunity (39). Moreover, autoimmune diseases have been associated with sustained high levels of BAFF, making inhibiting BAFF signaling a promising therapeutic approach (40–43). Further, BCMA has shown to be essential for survival of plasma cells and memory plasma cells and increased expression of BCMA has been observed in SLE patients (44, 45). Therefore, targeting both B and memory plasma cells may be more effective, leading to the complete removal of autoantibodies. By combining various types of CARs with CD19, such as BAFF, BCMA or BAFFR, the effectiveness of CAR-T cells could be improved. This can be achieved by engineering two pools of T cells, each expressing different CARs, or by incorporating multiple antigen recognition domains within a single CAR construct, called a compound CAR (cCAR) (46). Recently, an early phase 1 clinical trial has started to evaluate the effectiveness of CD19-BAFF CAR-T cell therapy for autoimmune diseases (NCT06279923). Nevertheless, several clinical trials have started investigating the safety and efficacy of CD19-BCMA CAR-T cell infusion in various autoimmune conditions (Table 1).
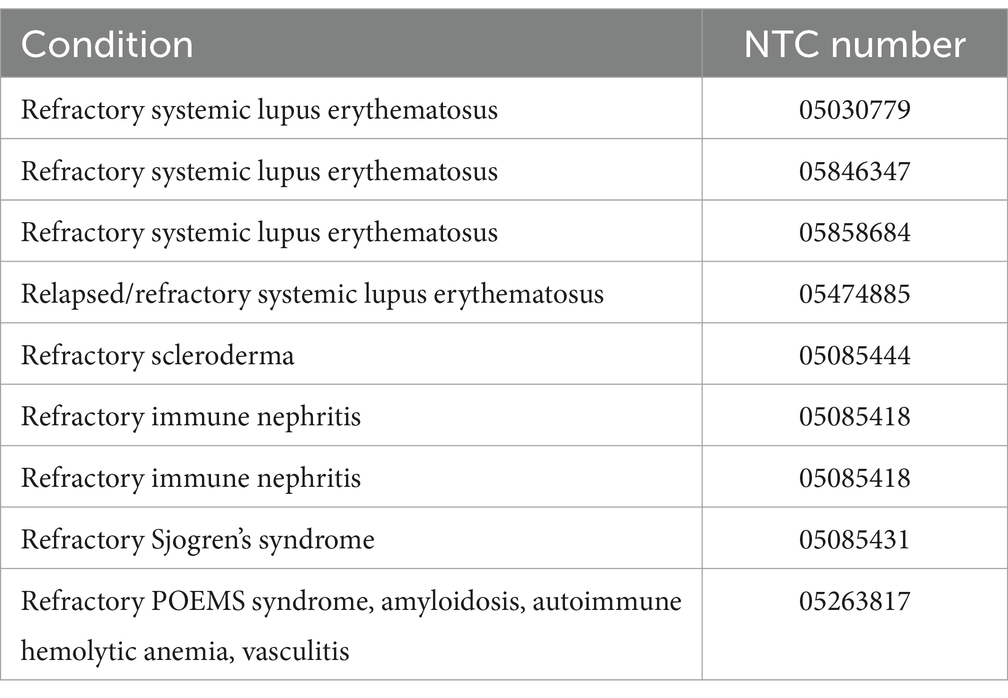
Table 1. Clinical trials using BCMA-CD19-CAR T therapy for autoimmune diseases.
4 Chimeric autoantibody receptor engineering in autoimmune diseases
The use of chimeric autoantibody receptor (CAAR) T cells to precisely target autoantigen-specific B cell subpopulations and overcome complete B cell depletion is an emerging area of research. For autoimmune diseases caused by specific autoantibodies produced by individual B cell clones, these therapeutic T cells can be genetically engineered with a CAR targeting specific antigen of autoantibodies on autoreactive cells to suppress or modulate the immune response without affecting healthy tissues. CAAR-T provides a more targeted and personalized approach compared to traditional immunosuppressive therapies. Similar to CD19-specific CAR-T cells, CAAR-Ts function analogously by specifically targeting autoantigens, leading to the destruction of pathological immune cells. Additionally, natural killer (NK) cells expressing CAAR can also specifically remove pathological B cells in vitro and could be potentially investigated in future clinical trials (47, 48). Preclinical findings have shown the encouraging potential of CAAR-T cells in treating several autoimmune conditions (48–51).
Namely, NMDAR encephalitis is the most common autoimmune encephalitis, leading to psychosis, seizures, and autonomic dysfunction caused by anti-NMDA receptor (NMDAR) autoantibodies. Preclinical research demonstrated the efficacy of NMDAR-specific CAAR-T cells in depletion and sustained reduction of autoantibody levels without off-target toxicity in a mouse model, suggesting phase I/II trials in the treatment of NMDAR encephalitis with NMDAR-CAAR T cells (50). Further, Pemphigus vulgaris (PV) is an autoimmune disease caused by autoantibodies to keratinocyte adhesion protein Dsg3. Eliminating anti-Dsg3 memory B cells could cure PV without general immunosuppression risks. An engineered chimeric autoantibody receptor (CAAR) that targets autoantigen Dsg3 has been shown to guide T cells to target and successfully eliminate autoreactive B cells in vivo in a humanized mouse model. CAAR-Ts demonstrated clear therapeutic potential for antibody-mediated autoimmune diseases without off-target toxicity (51). Two CAAR-T cell therapies for treating human autoimmune diseases have entered clinical trials. A phase I open-label study has begun to determine the maximum tolerated dose, infusion schedule, and safety of Dsg3-CAAR-T cells in patients with mucosal-dominant PV (NCT04422912). Also, another phase 1 study has begun evaluating the safety of MuSK-CAAR-T cell therapy for patients with Muscle-specific tyrosine kinase (MuSK) myasthenia gravis (NCT05451212). Moreover, various combinations of CAARs could be used to enhance effectiveness in conditions where pathogenic autoantibodies target multiple autoantigens. Nevertheless, further studies are needed to demonstrate the long-term safety and efficacy of CAAR-T cells.
5 Engineered Tregs (CAR-Tregs) in autoimmune diseases
Regulatory T lymphocytes (Tregs) are small heterogenous subpopulation of T lymphocytes, that play a vital role in maintaining immunological balance. Tregs suppress the immune response by limiting the ability of antigen-presenting cells to initiate an adaptive immune response, inducing apoptosis of effector T cells, disrupting metabolic pathways, and releasing anti-inflammatory cytokines (52). Dysregulation of these processes can lead to Treg dysfunction, which can also manifest in activation defects. This creates an imbalance in the ratio between resting and activated Tregs, contributing to autoimmune processes (53, 54). Moreover, Treg levels or functional alterations are associated with many autoimmune disorders, and decreased Treg frequencies have been identified in several autoimmune diseases, which may be linked to disease severity (52, 54–57). Altering the low frequency or dysfunction of Tregs is considered a novel approach to treating autoimmune disorders, with the primary goal of decreasing inflammation, facilitating tissue repair and restoring immune tolerance. Phase 1 clinical trials have indicated that infusion of autologous ex vivo-expanded Tregs is safe and well tolerated, with no significant adverse events (58). Antigen-specific Tregs have demonstrated greater efficacy and reduced risk of general immunosuppression compared to polyclonal Tregs in preclinical tests, indicating a potential therapeutic strategy for the future (59, 60). Several autoimmune diseases could benefit from antigen-specific CAR-Tregs, as described below.
Loss of Tregs has been identified as an important factor in the development of SLE (61). Moreover, Tregs in SLE patients have a reduced frequency and suppressive activity as well as an increased apoptosis rate compared to normal controls, suggesting new treatment options focused on Treg function (56). Overactive T cells in SLE can contribute to impaired immunological regulation, increased production of inflammatory cytokines, and an increase in effector T-cell phenotypes. IL-2 is a crucial factor for the expansion of Treg cells, and epigenetic silencing of the IL-2 gene in T cells leads to decreased production of IL-2, contributing to reduced Tregs and a secondary immune deficiency (62–64). Engineered anti-CD19 CAR-Tregs may improve their immune suppressive capabilities, block B cell proliferation, and restore the immune system’s normal composition in inflamed organs in a humanized mouse model of SLE (65).
Further, Tregs are essential in regulating CNS autoimmunity in several experimental autoimmune encephalomyelitis (EAE) models by limiting autoimmune inflammation through controlling cytokine secretion and modulating T effector cell proliferation and migration. Loss of Tregs worsens EAE severity, followed by increased pro-inflammatory cytokine production and proliferation of effector T cells, indicating that modulation of Tregs function could be an effective treatment approach (66). Animal models of EAE showed that CAR-Tregs could prevent, improve, and protect against the disease, effectively reducing or eliminating symptoms (67, 68). Furthermore, CAR-Tregs showed effectiveness in suppressing the manifestations of colitis in mouse models, indicating the viability of a CAR-Treg-based approach (69). Additionally, a study in mice examined antigen-specific CAR-Tregs for treating vitiligo, where GD3-reactive CAR-Tregs were generated. The results indicate that restoring peripheral Tregs can protect against skin depigmentation and that CAR-Tregs lead to increased secretion of IL-10, limit cytotoxicity toward melanocytes and delay depigmentation. This implies that antigen-specific CAR-Treg cells may have a positive effect on the control of long-term progressive depigmentation (70).
Type 1 diabetes mellitus (T1DM) is an autoimmune condition that attacks and destroys β cells in the pancreas. Although the frequency of Tregs in T1DM is normal, they exhibit an activation defect characterized by an increase in resting Tregs and a decrease in activated Tregs. A lower number of activated Tregs has been linked to a more severe clinical course, indicating its clinical significance (54). Therefore, delivering, redirecting, and activating T regulatory cells to target β cells could modulate the immune system and have therapeutic effects. Insulin-specific CAR-Tregs obtained from CD4+ effector T cells have shown promising results in a mouse model of type 1 diabetes. These CAR-Tregs had a normal Treg phenotype, suppressive function, and stability. Although they could not prevent diabetes in mice, they have demonstrated clear therapeutic potential with minimal off-target toxicity (71). Further, a recent study aimed to evaluate the potential of using HPi2 targeting CAR-Tregs to restore immune balance and improve disease in T1D in vitro. HPi2 was hypothesized to target all human endocrine pancreatic islet cell subtypes, and this approach was chosen since autoimmune processes typically destroy most endogenous β cells. However, the study found that anti-HPi2-driven CAR-Tregs were unsuccessful due to their non-specificity for human pancreatic islets and high expression on CD4+ T cells, suggesting future studies should explore alternative islet-specific targets (72).
Rheumatoid arthritis (RA) is a common chronic autoimmune disease causing irreversible joint and bone damage. Tregs have been found to have impaired function or number in RA patients and thus could be used as a promising therapeutic target (73–76). A phase I clinical trial is underway to assess the safety and efficacy of an autologous CAR-Treg cell-based therapy for treating RA (NCT06201416). This therapy specifically targets citrullinated proteins accumulated in the inflamed tissue associated with the disease to reduce inflammation and restore immune system tolerance.
Altogether, additional research is needed to investigate the effectiveness of CAR-Tregs, identify disease-specific targets, improve the manufacturing process by defining suitable sources for Treg isolation, and improve marker selection and proliferation capacity (77).
6 Potential adverse effects and future perspectives
CAR-T cell therapy has shown promise in treating hematological cancers, but it can lead to several potential side effects, including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). CRS is the most common and potentially life-threatening inflammatory response caused by the rapid activation and expansion of CAR-T cells. This can result in the excessive release of cytokines, leading to symptoms such as high fever, hypotension, and in rare cases multi-organ failure. ICANS is linked to CRS but presents distinct symptoms like confusion, seizures, and cerebral edema. CRS often precedes neurotoxic events, suggesting a temporal and mechanistic link (78–80). A recent systematic review and meta-analysis involving 7,604 patients looked into the rate of non-relapse mortality (deaths not caused by cancer progression) in various CAR T-cell trials for lymphoma and multiple myeloma. Non-relapse mortality rates varied from 1 to 10% across trials, with only a minority associated with the most common side effect, CRS. Additionally, 50.9% of non-relapse mortalities were due to infections, underscoring the critical importance of addressing infectious complications after CAR T cell therapy (81).
So far, CAR-T cell therapy have shown encouraging results, with demonstrated feasibility, tolerability, and efficacy in treating autoimmune disorders. However, more long-term assessments are required before they are accepted in wide clinical applications. The FDA recently reported T-cell malignancies in patients treated with BCMA-or CD19-directed autologous CAR T-cell therapies. Secondary malignancy risk is a concern for all approved products in this category. While the advantages of these products still surpass the possible drawbacks of their authorized use, the FDA is examining the potential for severe consequences such as hospitalization and death and is considering regulatory measures (82). Therefore, safety for autoimmune trials must be at a higher rate. The choice of an adequate strategy for cellular engineering to treat autoimmune diseases depends on the pathogenesis underlying the disease, its severity and length, and associated conditions. Additionally, due to the complex and heterogeneous nature of autoimmune diseases, other targets, as well as the efficacy and safety of multitargeting, should be further investigated.
Furthermore, the duration of optimal CAR-T cell activity in the treatment of autoimmune diseases is a subject of debate. In cancer treatment, long-term persistence of CAR T cells is beneficial for maintaining continuous immune monitoring. However, in autoimmune diseases, prolonged CAR T cell activity may result in immunosuppression or organ damage due to excessive elimination of normal immune cells. Therefore, it may be preferable to have limited or controlled expression of CAR T cells in the treatment of autoimmune diseases to minimize potential long-term toxicities. Therefore, future strategies should implement molecular switches or shorter half-life constructs CARs that express transiently or degrade after a specific period to reduce long-term immune suppression. The introduction of molecular switches can help manage immune-mediated toxicities, such as CRS and ICANS. Suicide switches provide a way to rapidly destroy CAR-T cells in cases of severe toxicity, ensuring safety. On/off switches offer external control by activating CAR expression only in the presence of a specific drug, allowing for precise titration of therapy based on patient response. Additionally, logic gate systems like AND and NOT gates improve targeting specificity, activating CAR-T cells only when certain conditions are met, thus minimizing damage to healthy tissues. These innovations enhance safety and efficacy, making CAR-T therapy more adaptable for complex diseases like autoimmunity (83–86).
Shorter half-life constructs CARs such as messenger RNA (mRNA)-based CAR T cells offer an alternative strategy to achieve transient and restricted expression of CARs, providing for more controlled treatment. Moreover, this also allows for in vivo reprogramming of T cells, favoring this approach due to faster production and lower cost (87, 88).
Manufacturing-related issues, including variability in cell quality and production delays, can also affect treatment outcomes. Some drawbacks of CAR-T therapy are the long production time (around 30 days) and high cost (up to half a million dollars) due to the autologous manufacturing process. Thus, alternative strategies focused on resolving these issues are under investigation. Decentralized production or on-site production using a fully automated closed system could significantly shorten the manufacturing process and lower the therapy cost (89, 90).
Recently, the European Society for Blood and Marrow Transplantation (EBMT) and the International Society for Cell and Gene Therapy (ISCT) published expert-based consensus and recommendations on using cellular therapies, including CAR-T, for treating severe and refractory autoimmune diseases. The guidelines emphasize referrals to expert centers with inter-disciplinary interaction, including hematological and autoimmune diseases specialist experience, as well as assessment of the effectiveness and tolerability of the therapies, long-term outcomes, and safety monitoring (91).
In conclusion, CAR-T cell therapy offers significant advantages over conventional treatments for autoimmune diseases by addressing the root cause of the condition: the autoreactive immune cells. Unlike traditional therapies, which broadly suppress the immune system, CAR-engineered T cell therapy holds the potential to specifically target and eliminate the pathogenic immune cells driving the autoimmune response. This precise approach helps preserve overall immune function, reducing the risk of infections and other side effects. Additionally, CAR-T cells can be engineered with molecular switches that allow real-time control over their activity, enhancing both safety and efficacy. This strategy offers the potential for long-term remission by directly targeting the underlying drivers of autoimmunity, rather than merely managing symptoms. Undoubtedly, CAR-T cell therapy stands as a compelling therapeutic approach for various autoimmune diseases, although there are still unresolved challenges that need to be addressed for widespread clinical use.
Author contributions
JV: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. DA: Conceptualization, Funding acquisition, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. DV: Project administration, Resources, Supervision, Writing – review & editing. SM: Formal analysis, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Davidson, A, and Diamond, B. Autoimmune diseases. N Engl J Med. (2001) 345:340–50. doi: 10.1056/NEJM200108023450506
2. Luckheeram, RV, Zhou, R, Verma, AD, and Xia, B. CD4+T cells: differentiation and functions. Clin Dev Immunol. (2012) 2012:925135. doi: 10.1155/2012/925135
3. Viau, M, and Zouali, M. B-lymphocytes, innate immunity, and autoimmunity. Clin Immunol. (2005) 114:17–26. doi: 10.1016/j.clim.2004.08.019
4. Bonilla, FA, and Oettgen, HC. Adaptive immunity. J Allergy Clin Immunol. (2010) 125:S33–40. doi: 10.1016/j.jaci.2009.09.017
5. Wang, K, Wei, G, and Liu, D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. (2012) 1:36. doi: 10.1186/2162-3619-1-36
6. Mei, HE, Schmidt, S, and Dörner, T. Rationale of anti-CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther. (2012) 14:S1. doi: 10.1186/ar3909
7. Brentjens, RJ, Latouche, JB, Santos, E, Marti, F, Gong, MC, Lyddane, C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. (2003) 9:279–86. doi: 10.1038/nm827
8. Halim, L, and Maher, J. CAR T-cell immunotherapy of B-cell malignancy: the story so far. Ther Adv Vaccines Immunother. (2020) 8:2515135520927164. doi: 10.1177/2515135520927164
9. June, CH, and Sadelain, M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
10. Jayaraman, J, Mellody, MP, Hou, AJ, Desai, RP, Fung, AW, Pham, AHT, et al. CAR-T design: elements and their synergistic function. EBioMedicine. (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
11. Sadelain, M, Brentjens, R, and Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
12. Chmielewski, M, Hombach, AA, and Abken, H. Antigen-specific T-cell activation independently of the MHC: chimeric antigen receptor-redirected T cells. Front Immunol. (2013) 4:371. doi: 10.3389/fimmu.2013.00371
13. Sadelain, M, Rivière, I, and Brentjens, R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. (2003) 3:35–45. doi: 10.1038/nrc971
14. Hoyos, V, Savoldo, B, Quintarelli, C, Mahendravada, A, Zhang, M, Vera, J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
15. He, C, Zhou, Y, Li, Z, Farooq, MA, Ajmal, I, Zhang, H, et al. Co-expression of IL-7 improves NKG2D-based CAR T cell therapy on prostate Cancer by enhancing the expansion and inhibiting the apoptosis and exhaustion. Cancers. (2020) 12:1969. doi: 10.3390/cancers12071969
16. Till, BG, Jensen, MC, Wang, J, Chen, EY, Wood, BL, Greisman, HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. (2008) 112:2261–71. doi: 10.1182/blood-2007-12-128843
17. Brocker, T, and Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. (1995) 181:1653–9. doi: 10.1084/jem.181.5.1653
18. Weinkove, R, George, P, Dasyam, N, and McLellan, AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. (2019) 8:e1049. doi: 10.1002/cti2.1049
19. Savoldo, B, Ramos, CA, Liu, E, Mims, MP, Keating, MJ, Carrum, G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. (2011) 121:1822–6. doi: 10.1172/JCI46110
20. Ma, G, Wang, Y, Li, Y, Cui, L, Zhao, Y, Zhao, B, et al. MiR-206, a key modulator of skeletal muscle development and disease. Int J Biol Sci. (2015) 11:345–52. doi: 10.7150/ijbs.10921
21. Chmielewski, M, and Abken, H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
22. Dana, H, Chalbatani, GM, Jalali, SA, Mirzaei, HR, Grupp, SA, Suarez, ER, et al. CAR-T cells: early successes in blood cancer and challenges in solid tumors. Acta Pharm Sin B. (2021) 11:1129–47. doi: 10.1016/j.apsb.2020.10.020
23. Sadeqi Nezhad, M, Seifalian, A, Bagheri, N, Yaghoubi, S, Karimi, MH, and Adbollahpour-Alitappeh, M. Chimeric antigen receptor based therapy as a potential approach in autoimmune diseases: how close are we to the treatment? Front Immunol. (2020) 11:603237. doi: 10.3389/fimmu.2020.603237
24. Kansal, R, and Richardson, N. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med. (2019) 11:eaav1648. doi: 10.1126/scitranslmed.aav1648
25. Jin, X, Xu, Q, Pu, C, Zhu, K, Lu, C, Jiang, Y, et al. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell Mol Immunol. (2021) 18:1896–903. doi: 10.1038/s41423-020-0472-1
26. Mougiakakos, D, Krönke, G, Völkl, S, Kretschmann, S, Aigner, M, Kharboutli, S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. (2021) 385:567–9. doi: 10.1056/NEJMc2107725
27. Mackensen, A, Müller, F, Mougiakakos, D, Böltz, S, Wilhelm, A, Aigner, M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
28. Fanouriakis, A, Kostopoulou, M, Andersen, J, Aringer, M, Arnaud, L, Bae, SC, et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann Rheum Dis. (2024) 83:15–29. doi: 10.1136/ard-2023-224762
29. Haghikia, A, Hegelmaier, T, Wolleschak, D, Böttcher, M, Desel, C, Borie, D, et al. Anti-CD19 CAR T cells for refractory myasthenia gravis. Lancet Neurol. (2023) 22:1104–5. doi: 10.1016/S1474-4422(23)00375-7
30. Müller, F, Boeltz, S, Knitza, J, Aigner, M, Völkl, S, Kharboutli, S, et al. CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet. (2023) 401:815–8. doi: 10.1016/S0140-6736(23)00023-5
31. Pecher, AC, Hensen, L, Klein, R, Schairer, R, Lutz, K, Atar, D, et al. CD19-targeting CAR T cells for myositis and interstitial lung disease associated with Antisynthetase syndrome. JAMA. (2023) 329:2154–62. doi: 10.1001/jama.2023.8753
32. Zhang, W, Feng, J, Cinquina, A, Wang, Q, Xu, H, Zhang, Q, et al. Treatment of systemic lupus erythematosus using BCMA-CD19 compound CAR. Stem Cell Rev and Rep. (2021) 17:2120–3. doi: 10.1007/s12015-021-10251-6
33. Bhoj, VG, Arhontoulis, D, Wertheim, G, Capobianchi, J, Callahan, CA, Ellebrecht, CT, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. (2016) 128:360–70. doi: 10.1182/blood-2016-01-694356
34. Halliley, JL, Tipton, CM, Liesveld, J, Rosenberg, AF, Darce, J, Gregoretti, IV, et al. Long-lived plasma cells are contained within the CD19(−)CD38(hi)CD138(+) subset in human bone marrow. Immunity. (2015) 43:132–45. doi: 10.1016/j.immuni.2015.06.016
35. Landsverk, OJ, Snir, O, and Casado, RB. Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med. (2017) 214:309–17. doi: 10.1084/jem.20161590
36. Smulski, CR, and Eibel, H. BAFF and BAFF-receptor in B cell selection and survival. Front Immunol. (2018) 9:2285. doi: 10.3389/fimmu.2018.02285
37. Benson, MJ, Dillon, SR, Castigli, E, Geha, RS, Xu, S, Lam, KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. (2008) 180:3655–9. doi: 10.4049/jimmunol.180.6.3655
38. Carrillo-Ballesteros, FJ, Oregon-Romero, E, Franco-Topete, RA, Govea-Camacho, LH, Cruz, A, Muñoz-Valle, JF, et al. B-cell activating factor receptor expression is associated with germinal center B-cell maintenance. Exp Ther Med. (2019) 17:2053–60. doi: 10.3892/etm.2019.7172
39. Mackay, F, Woodcock, SA, Lawton, P, Ambrose, C, Baetscher, M, Schneider, P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. (1999) 190:1697–710. doi: 10.1084/jem.190.11.1697
40. Vincent, FB, Kandane-Rathnayake, R, Koelmeyer, R, Hoi, AY, Harris, J, Mackay, F, et al. Analysis of serum B cell-activating factor from the tumor necrosis factor family (BAFF) and its soluble receptors in systemic lupus erythematosus. Clin Transl Immunol. (2019) 8:e01047. doi: 10.1002/cti2.1047
41. Stohl, W, Metyas, S, Tan, SM, Cheema, GS, Oamar, B, Xu, D, et al. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum. (2003) 48:3475–86. doi: 10.1002/art.11354
42. Steri, M, Orrù, V, Idda, ML, Pitzalis, M, Pala, M, Zara, I, et al. Overexpression of the cytokine BAFF and autoimmunity risk. N Engl J Med. (2017) 376:1615–26. doi: 10.1056/NEJMoa1610528
43. Matsushita, T, Hasegawa, M, Yanaba, K, Kodera, M, Takehara, K, and Sato, S. Elevated serum BAFF levels in patients with systemic sclerosis: enhanced BAFF signaling in systemic sclerosis B lymphocytes. Arthritis Rheum. (2006) 54:192–201. doi: 10.1002/art.21526
44. O'Connor, BP, Raman, VS, Erickson, LD, Cook, WJ, Weaver, LK, Ahonen, C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. (2004) 199:91–8. doi: 10.1084/jem.20031330
45. Kim, J, Gross, JA, Dillon, SR, Min, JK, and Elkon, KB. Increased BCMA expression in lupus marks activated B cells, and BCMA receptor engagement enhances the response to TLR9 stimulation. Autoimmunity. (2011) 44:69–81. doi: 10.3109/08916934.2010.509122
46. van der Schans, JJ, van de Donk, N, and Mutis, T. Dual targeting to overcome current challenges in multiple myeloma CAR T-cell treatment. Front Oncol. (2020) 10:1362. doi: 10.3389/fonc.2020.01362
47. Meng, H, Sun, X, Song, Y, Zou, J, An, G, Jin, Z, et al. La/SSB chimeric autoantibody receptor modified NK92MI cells for targeted therapy of autoimmune disease. Clin Immunol. (2018) 192:40–9. doi: 10.1016/j.clim.2018.04.006
48. Seifert, L, Riecken, K, Zahner, G, Hambach, J, Hagenstein, J, Dubberke, G, et al. An antigen-specific chimeric autoantibody receptor (CAAR) NK cell strategy for the elimination of anti-PLA2R1 and anti-THSD7A antibody-secreting cells. Kidney Int. (2024) 105:886–9. doi: 10.1016/j.kint.2024.01.021
49. Oh, S, and Mao, X. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat Biotechnol. (2023) 41:1229–38. doi: 10.1038/s41587-022-01637-z
50. Reincke, SM, von Wardenburg, N, Homeyer, MA, Kornau, HC, Spagni, G, Li, LY, et al. Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell. (2023) 186:5084–5097.e18. doi: 10.1016/j.cell.2023.10.001
51. Ellebrecht, CT, Bhoj, VG, Nace, A, Choi, EJ, Mao, X, Cho, MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
52. Grant, CR, Liberal, R, Mieli-Vergani, G, Vergani, D, and Longhi, MS. Regulatory T-cells in autoimmune diseases: challenges, controversies and--yet--unanswered questions. Autoimmun Rev. (2015) 14:105–16. doi: 10.1016/j.autrev.2014.10.012
53. Miyara, M, Yoshioka, Y, Kitoh, A, Shima, T, Wing, K, Niwa, A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
54. Okubo, Y, Torrey, H, Butterworth, J, Zheng, H, and Faustman, DL. Treg activation defect in type 1 diabetes: correction with TNFR2 agonism. Clin Transl Immunol. (2016) 5:e56. doi: 10.1038/cti.2015.43
55. Xu, WH, Zhang, AM, Ren, MS, Zhang, XD, Wang, F, Xu, XC, et al. Changes of Treg-associated molecules on CD4+CD25 +Treg cells in myasthenia gravis and effects of immunosuppressants. J Clin Immunol. (2012) 32:975–83. doi: 10.1007/s10875-012-9685-0
56. Li, M, Luo, L, Wu, Y, Song, Z, Ni, B, Hao, F, et al. Elevated apoptosis and abnormal apoptosis signaling of regulatory T cells in patients with systemic lupus erythematosus. Lupus. (2022) 31:1441–55. doi: 10.1177/09612033221119455
57. Lyssuk, EY, Torgashina, AV, Soloviev, SK, Nassonov, EL, and Bykovskaia, SN. Reduced number and function of CD4+CD25highFoxP3+ regulatory T cells in patients with systemic lupus erythematosus. Adv Exp Med Biol. (2007) 601:113–9. doi: 10.1007/978-0-387-72005-0_12
58. Bluestone, JA, Buckner, JH, Fitch, M, Gitelman, SE, Gupta, S, Hellerstein, MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
59. Stephens, LA, Malpass, KH, and Anderton, SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol. (2009) 39:1108–17. doi: 10.1002/eji.200839073
60. Tang, Q, Henriksen, KJ, Bi, M, Finger, EB, Szot, G, Ye, J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
61. Li, W, Deng, C, Yang, H, and Wang, G. The regulatory T cell in active systemic lupus erythematosus patients: a systemic review and Meta-analysis. Front Immunol. (2019) 10:159. doi: 10.3389/fimmu.2019.00159
62. Hedrich, CM, Rauen, T, and Tsokos, GC. cAMP-responsive element modulator (CREM)α protein signaling mediates epigenetic remodeling of the human interleukin-2 gene: implications in systemic lupus erythematosus. J Biol Chem. (2011) 286:43429–36. doi: 10.1074/jbc.M111.299339
63. Hofmann, SR, Mäbert, K, Kapplusch, F, Russ, S, Northey, S, Beresford, MW, et al. cAMP response element modulator α induces dual specificity protein phosphatase 4 to promote effector T cells in juvenile-onset. Lupus. (2019) 203:2807–16. doi: 10.4049/jimmunol.1900760
64. Malek, TR. The main function of IL-2 is to promote the development of T regulatory cells. J Leukoc Biol. (2003) 74:961–5. doi: 10.1189/jlb.0603272
65. Doglio, M, Ugolini, A, Bercher, C, Toma, C, Bonini, C, Ciceri, F, et al. CAR-T cells for systemic lupus erythematosus. Blood. (2023) 142:6813. doi: 10.1182/blood-2023-189673
66. Koutrolos, M, Berer, K, Kawakami, N, Wekerle, H, and Krishnamoorthy, G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol Commun. (2014) 2:163. doi: 10.1186/s40478-014-0163-1
67. Fransson, M, Piras, E, Burman, J, Nilsson, B, Essand, M, Lu, B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. (2012) 9:112. doi: 10.1186/1742-2094-9-112
68. Gupta, S, Simic, M, Sagan, SA, Shepherd, C, Duecker, J, Sobel, RA, et al. CAR-T cell-mediated B-cell depletion in central nervous system autoimmunity. Neurol Neuroimmunol Neuroinflamm. (2023) 10:2. doi: 10.1212/NXI.0000000000200080
69. Blat, D, Zigmond, E, Alteber, Z, Waks, T, and Eshhar, Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. (2014) 22:1018–28. doi: 10.1038/mt.2014.41
70. Mukhatayev, Z, Dellacecca, ER, Cosgrove, C, Shivde, R, Jaishankar, D, Pontarolo-Maag, K, et al. Antigen specificity enhances disease control by Tregs in vitiligo. Front Immunol. (2020) 11:581433. doi: 10.3389/fimmu.2020.581433
71. Tenspolde, M, Zimmermann, K, Weber, LC, Hapke, M, Lieber, M, Dywicki, J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun. (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
72. Radichev, IA, Yoon, J, Scott, DW, Griffin, K, and Savinov, AY. Towards antigen-specific Tregs for type 1 diabetes: construction and functional assessment of pancreatic endocrine marker, HPi2-based chimeric antigen receptor. Cell Immunol. (2020) 358:104224. doi: 10.1016/j.cellimm.2020.104224
73. Flores-Borja, F, Jury, EC, Mauri, C, and Ehrenstein, MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA. (2008) 105:19396–401. doi: 10.1073/pnas.0806855105
74. Han, GM, O’Neil-Andersen, NJ, Zurier, RB, and Lawrence, DA. CD4+CD25high T cell numbers are enriched in the peripheral blood of patients with rheumatoid arthritis. Cell Immunol. (2008) 253:92–101. doi: 10.1016/j.cellimm.2008.05.007
75. Kawashiri, SY, Kawakami, A, Okada, A, Koga, T, Tamai, M, Yamasaki, S, et al. CD4+CD25(high)CD127(low/−) Treg cell frequency from peripheral blood correlates with disease activity in patients with rheumatoid arthritis. J Rheumatol. (2011) 38:2517–21. doi: 10.3899/jrheum.110283
76. Jiang, Q, Yang, G, Liu, Q, Wang, S, and Cui, D. Function and role of regulatory T cells in rheumatoid arthritis. Front Immunol. (2021) 12:626193. doi: 10.3389/fimmu.2021.626193
77. Ferreira, LMR, Muller, YD, and Bluestone, JA. Next-generation regulatory T cell therapy. Nat Rev Drug Discov. (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
78. Sheth, VS, and Gauthier, J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. (2021) 56:552–66. doi: 10.1038/s41409-020-01134-4
79. Gust, J, Hay, KA, Hanafi, LA, Li, D, Myerson, D, Gonzalez-Cuyar, LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
80. Dholaria, BR, Bachmeier, CA, and Locke, F. Mechanisms and Management of Chimeric Antigen Receptor T-cell therapy-related toxicities. BioDrugs. (2019) 33:45–60. doi: 10.1007/s40259-018-0324-z
81. Cordas dos Santos, DM, Tix, T, Shouval, R, Gafter-Gvili, A, Alberge, JB, Cliff, ERS, et al. A systematic review and meta-analysis of nonrelapse mortality after CAR T cell therapy. Nat Med. (2024) 30:2667–78. doi: 10.1038/s41591-024-03084-6
82. Nelson, R, and Schumacher, TN. FDA investigating safety risks in CAR T-cell recipients. Lancet. (2023) 402:2181. doi: 10.1016/S0140-6736(23)02747-2
83. Sahillioglu, AC, and Schumacher, TN. Safety switches for adoptive cell therapy. Curr Opin Immunol. (2022) 74:190–8. doi: 10.1016/j.coi.2021.07.002
84. Roybal, KT, Williams, JZ, Morsut, L, Rupp, LJ, Kolinko, I, Choe, JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. (2016) 167:419–432.e16. doi: 10.1016/j.cell.2016.09.011
85. Jan, M, and Scarfò, I. Reversible ON-and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci Transl Med. (2021) 13:575. doi: 10.1126/scitranslmed.abb6295
86. Charrot, S, and Hallam, S. CAR-T cells: future perspectives. Hema. (2019) 3:e188. doi: 10.1097/HS9.0000000000000188
87. Parayath, NN, Stephan, SB, Koehne, AL, Nelson, PS, and Stephan, MT. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. (2020) 11:6080. doi: 10.1038/s41467-020-19486-2
88. Rurik, JG, and Tombácz, I. CAR T cells produced in vivo to treat cardiac injury. Science. (2022) 375:91–6. doi: 10.1126/science.abm0594
89. Ran, T, and Eichmüller, SB. Cost of decentralized CAR T-cell production in an academic nonprofit setting. Int J Cancer. (2020) 147:3438–45. doi: 10.1002/ijc.33156
90. Palani, HK, Arunachalam, AK, Yasar, M, Venkatraman, A, Kulkarni, U, Lionel, SA, et al. Decentralized manufacturing of anti CD19 CAR-T cells using CliniMACS prodigy®: real-world experience and cost analysis in India. Bone Marrow Transplant. (2023) 58:160–7. doi: 10.1038/s41409-022-01866-5
91. Greco, R, Alexander, T, del Papa, N, Müller, F, Saccardi, R, Sanchez-Guijo, F, et al. Innovative cellular therapies for autoimmune diseases: expert-based position statement and clinical practice recommendations from the EBMT practice harmonization and guidelines committee. EClinicalMedicine. (2024) 69:102476. doi: 10.1016/j.eclinm.2024.102476
Keywords: autoimmunity, CAR-T, CAAR-T, CAR-Treg, immunotherapy, cell therapy, regenerative medicine
Citation: Vukovic J, Abazovic D, Vucetic D and Medenica S (2024) CAR-engineered T cell therapy as an emerging strategy for treating autoimmune diseases. Front. Med. 11:1447147. doi: 10.3389/fmed.2024.1447147
Edited by:
Brenda Laky, Austrian Society for Regenerative Medicine (ASRM), AustriaReviewed by:
Oleh Andrukhov, Medical University of Vienna, AustriaXiaolei Li, University of Pennsylvania, United States
Copyright © 2024 Vukovic, Abazovic, Vucetic and Medenica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jovana Vukovic, am92YW5hLnZ1azEyM0BnbWFpbC5jb20=; Dzihan Abazovic, YWR6aWhhbkBnbWFsaS5jb20=