 Kristine Griffett
Kristine Griffett Thomas P. Burris
Thomas P. Burris- 1Department of Anatomy, Physiology and Pharmacology, College of Veterinary Medicine, Auburn University, Auburn, AL, United States
- 2The University of Florida Genetics Institute, Gainesville, FL, United States
Activation of LXR activity by synthetic agonists has been the focus of many drug discovery efforts with a focus on treatment of dyslipidemia and atherosclerosis. Many agonists have been developed, but all have been hindered due to their ability to efficaciously stimulate de novo lipogenesis. Here, we review the development of LXR inverse agonists that were originally optimized for their ability to enable recruitment of corepressors leading to silencing of genes that drive de novo lipogenesis. Such compounds have efficacy in animal models of MAFLD, dyslipidemia, and cancer. Several classes of LXR inverse agonists have been identified and one is now in clinical trials for treatment of severe dyslipidemia.
1. Introduction
Metabolic-associated fatty liver disease (MAFLD) is a relatively new classification of disease to incorporate the metabolic dysfunction that often occurs within patients presenting with non-alcoholic fatty liver disease (NAFLD) (1). NAFLD has been deemed the world’s leading chronic liver disease leading to liver transplantation or death (2). Currently, there are no universally approved therapies for NAFLD, and its heterogenous pathology often makes it difficult to identify and treat. The incorporation of metabolic dysfunction (e.g., high plasma triglycerides, prediabetes/diabetes, and increased blood pressure) into the definition of NAFLD has a widespread impact on patients and physicians alike and will improve treatment options for those with the disease (1, 3, 4). The current requirements for the diagnosis of MAFLD includes: 1) hepatic steatosis and 2) overweight/obesity, type 2 diabetes mellitus (T2DM), or metabolic dysfunction. Thus, MAFLD diagnosis partly overlaps with NAFLD but is independent of alcohol intake and co-existing causes of liver diseases. MAFLD is considered independent of other liver disease etiologies and allows for the identification of fatty liver in patients displaying other metabolic disorders (2, 3, 5). This review describes some of the processes that contribute to the development of MAFLD, processes that are involved in both MAFLD and NAFLD, and the novelty of targeting the liver X receptor (LXR) pathway using tissue selective inverse agonists to alleviate this disease.
2. Pathophysiological processes in the development of MAFLD
The pathophysiology of chronic liver diseases is quite complex, and with the multitude of factors than may contribute to the development of fatty liver, the term MAFLD allows physicians to distinguish between the term “non-alcohol” and be inclusive of key metabolic factors contributing to the disease and potential therapeutic options (6, 7).
2.1. Genetic factors involved in MAFLD development
Scientific evidence suggests that genetic factors strongly influence the development of MAFLD, and these factors overlap with those identified as factors for NAFLD and NASH (non-alcoholic steatohepatitis) (1, 5, 8). The patatin-like phospholipase domain-containing protein 3 gene (PNPLA3), Membrane bound O-acyltransferase domain containing 7 gene (MBOAT7), transmembrane 6 superfamily member 2 gene (TM6SF2), and glucokinase regulator gene (GCKR) have been the most recognized genes involved in the pathogenesis of fatty liver diseases (9, 10). PNPLA3 was the first NAFLD-related genetic variant (rs738409; I148M) identified that displays a robust association with the development and severity of NAFLD. This gene is highly expressed in the liver and white adipose tissues and is regulated by insulin signaling via LXR and sterol regulatory binding protein 1c (SREBP1c) pathways (5, 9, 11, 12). Normally, this protein hydrolyzes triglycerides and retinyl esters, however the genetic variant results in impairment of the hydrolase activity, leading to hepatic lipid accumulation.
MBOAT7 functions to remodel phosphatidyl inositol with arachidonic acid and is primarily expressed in the liver (13–15). The rs641738 mutation (C > T), increases the risk of developing not only MAFLD, but an entire spectrum of liver diseases (NASH, cirrhosis, and hepatocellular carcinoma) (16, 17). The TM6SF2 protein facilities hepatic secretion of triglyceride-rich lipoproteins through the VLDL secretion pathway. The variant associated with the development of MAFLD/NAFLD involves a C-to-T substitution at nucleotide 499, which causes a glutamate to lysine change and results in decreased expression of TM6SF2 (18–20). Reduced expression of TM6SF2 results in an increase in hepatic lipid content and is associated with increased hepatic fibrosis in patients.
GCKR regulates glucose influx into hepatocytes to control de novo lipogenesis (9). There have been several variants of GCKR associated with the development of liver diseases, and the most severe variants lead to overexpression of GCKR, enhancement of hepatic glucose uptake, and increased hepatic lipogenesis (21, 22). Interestingly, this often leads to reduced serum glucose levels (attributed to the enhanced hepatic uptake), and while it may be beneficial by lowering T2DM risk, can alter insulin signaling and further contribute to the progression of MAFLD (16, 23). While these are not the sole genetic factors that contribute to MAFLD and liver disease development, they are currently the most common and well-characterized of the genetic factors. Interestingly, a commonality among these factors is that they either enhance or inhibit pathways involved with insulin and glucose signaling, de novo lipogenesis, or lipoprotein secretion and/or packaging.
2.2. De novo lipogenesis in MAFLD
Because of metabolic dysfunction during MAFLD progression, adiponectin levels are often decreased which leads to the decrease in free fatty acid (FFA) oxidation, which can stimulate de novo lipogenesis (DNL) in the liver. DNL is the metabolic pathway that synthesizes saturated fatty acids and monounsaturated fatty acids (MUFAs) from acetyl-coA (5, 19). In patients with MAFLD, the rate of hepatic DNL in greatly increased due to enhanced expression of DNL pathway enzymes that are regulated by the transcription factors sterol regulatory element binding protein-1 (SREBP1) and carbohydrate response element-binding protein (ChREBP) (19). These transcription factors can be activated via glucose flux and insulin signaling, demonstrating how metabolic dysfunction caused by hyperglycemia and/or hyperinsulinemic conditions promotes DNL and steatosis in MAFLD.
Within hepatocytes, FFAs can be esterified to produce TGs, which are either stored as lipid droplets in the liver or packaged as VLDLs into circulation. Because of this, MAFLD patients often present with pro-atherogenic lipid profiles (e.g., low HDL-C and elevated LDL-C, TG, and apolipoprotein B) (11, 19, 22, 24). Humans have a compensatory mechanism to reduce hepatic fat content through the activity of cholesterol ester transfer protein (CETP), which exchanges TG and cholesterol esters between VLDL, HDL, and LDL cholesterol (25–27). However, this mechanism often results in abnormally high HDL cholesterol metabolism and leads to undesirable alterations in lipid profiles in patients. Like NAFLD, dyslipidemia is not constant across the stages of MAFLD (19). Typically, circulating levels of VLDL and LDL are increased in earlier stages, then as MAFLD progresses, patients will develop hepatic fibrosis and circulating levels of apoB-containing lipoproteins decrease (4, 11, 25, 28). Therefore, research indicates that dyslipidemia in MAFLD appears the most pronounced at the earlier stages of the disease.
While DNL contributes to hepatic steatosis, it also is linked to very low-density lipoprotein (VLDL) production via ChREBP activation of microsomal triglyceride transfer protein (MTTP) and TM6SF2. Several studies have described an increase in VLDL particle size and number due to increased ChREBP and SREBP1 activity, which was observed due to pharmacological activation of the nuclear receptor LXR, a known regulator of DNL (29, 30). Stimulation of the DNL pathway and an increase in the quantity and particle size of VLDL via LXR activation was further confirmed in a study that investigated the stearoyl-CoA desaturase (SCD) enzyme which is involved in the synthesis of MUFAs (29, 30). In MAFLD patients, SCD activity is increased, leading to increased VLDL secretion as well as increased plasma and hepatic triglyceride (TG) levels. Inhibition of SCD, which is a direct target gene of LXR, can suppress hypertriglyceridemia (5, 22).
Other enzymes within the DNL pathway have been identified as targets for alleviating dyslipidemia and MAFLD. The inhibition of fatty acid synthase (FASN), which is the enzyme that synthesizes palmitate from acetyl-CoA and malonyl-CoA, can suppress hepatic steatosis in a variety of mouse models of fatty liver disease (5, 31, 32). While it has yet to be determined whether specifically inhibiting FASN is a valid approach for the treatment of MAFLD, it is also a direct target gene of LXR, suggesting that targeting this nuclear receptor for MAFLD will have beneficial effects in several physiological pathways involved in the pathogenesis of this disease.
2.3. Altered lipoprotein processing in the liver attributes to MAFLD
Lipoprotein processing and signaling play an important role in the development of metabolic dysfunction and MAFLD. As mentioned earlier, genetic anomalies and altered DNL processes can contribute to lipoprotein processing defects in MAFLD (5, 16, 19). For example, alterations in VLDL secretion leads to increased lipid content in hepatocytes. However, one area that should be discussed is the role of lipoprotein receptors in the development of MAFLD. The major receptor for cholesterol enriched APOB containing lipoproteins is the LDL receptor (LDLR) (33, 34). Decades of studies have demonstrated that the functional loss of this receptor induces severe hypercholesterolemia and plays a key role in the development of several cardiovascular diseases including atherosclerosis (35–37). Ldlr knockout rodents are prone to develop hepatic steatosis particularly when fed a western or high fat diet (38, 39). The role that this receptor plays in MAFLD in humans is unclear, but mutations in the LDLR gene are relatively common (40).
ApoE-deficient rodents are another model of hypercholesterolemia and cardiovascular disease commonly used to evaluate therapeutics for atherosclerosis (41–43). Like the Ldlr-deficient mice, ApoE-deficient mice and rats also exhibit steatohepatitis regardless of diet and are likely an important model of MAFLD for drug discovery. APOE in human and mice, affects hepatic lipid balance via VLDL secretion. This altered balance signals for the activation of resident Kupffer cells and infiltration of peripheral macrophages, leading to progression of MAFLD and hepatic fibrosis. As VLDL balance is implicated in the development of MAFLD, there is a role for the VLDL receptor (VLDLR) in this disease as well. VLDLR is typically expressed at low levels in healthy liver and mediates the clearance of triglyceride-rich particles. During MAFLD development, in both mouse models and humans, the expression levels of VLDLR in liver increases enhancing the development of the disease. Recent data demonstrated that in mice lacking the proprotein convertase subtilisin/kexin type 9 (PCSK9) protein have increased expression of VLDLR, LDLR, and fatty acid transporters including CD36, which enhance the development of MAFLD. Loss of PCSK9 also results in increased hepatic lipid accumulation, impaired beta cell function, and decreased plasma insulin levels.
MAFLD is a highly complex and systemic disease associated with a variety of metabolic changes and has similarities in the development of progression between mouse models and humans. Like NAFLD, MAFLD often begins with the accumulation of lipids in the hepatocytes, driven by a variety of biological factors (e.g., genetic alterations, nutrition, etc.) which is often mediated by VLDL secretion, fatty acid and lipoprotein uptake and processing, and DNL. These altered metabolic signaling processes and downstream effects on insulin signaling/regulation share features with NAFLD, cardiovascular diseases, and T2DM. While there is likely no single therapeutic target that can fully alleviate the complex metabolic dysfunction occurring in MAFLD, the nuclear receptors have proven to be a rich target class for targeting metabolic and cardiovascular diseases.
3. Nuclear receptors
Nuclear receptors (NRs) are ligand-regulated transcription factors that orchestrate numerous physiological processes including metabolism, immunity, and development (44). In humans, there are 48 members of the NR superfamily, which include receptors for steroid hormones, retinoic acid, thyroid hormones, fatty acids, and cholesterol metabolites or oxysterols (44–46). Many of the NRs are categorized as orphans since their natural ligands are not yet known. These signaling molecules regulate target gene transcriptional activity through a common mechanism enhanced by their modular structures. NRs have a highly conserved N-terminal DNA-binding domain (DBD) and a C-terminal ligand-binding domain (LBD) connected by a variable (in size and sequence) hinge region. While the LBD is involved in determining ligand specificity, this region also contains a ligand-dependent transactivation function 2 (AF-2) domain, which allows the NRs to recruit co-factors for transcriptional regulation of target genes. These transcriptional co-factors include coactivators that mediate activation of transcription as well as corepressors that mediate silencing of target gene transcription (47–49). While many receptors are considered either exclusively activators (recruit coactivators) or repressors (recruit corepressors) of transcription, several receptors can recruit either coactivators or corepressors depending on the context of a physiological situation.
Several NRs respond to changes in cellular levels of lipids and other metabolic signals including LXRs, farnesoid X receptor (FXR), and peroxisome proliferated-activated receptors (PPARs), and have been identified as therapeutic targets for a variety of metabolic diseases (44, 50). FXR ligands (obeticholic acid, bile acid analogs, etc.) have therapeutic potential for the treatment of NASH with fibrosis (51). PPAR agonists (PPARα and PPARγ) have been clinically used for many years as treatments for diabetes and dyslipidemia while mixed (PPARα/δ/γ) agonists have been evaluated for efficacy against NASH (52–55). Here, we will focus on the LXRs, as they are master regulators of hepatic lipogenesis and are intricately involved in a variety of processes that lead to the development of MAFLD.
3.1. Liver X receptors
LXRα and LXRβ were originally identified as orphan members of the NR superfamily (56, 57). Both isoforms form heterodimers with obligate partner Retinoid X Receptor (RXR) and share the conserved domain structure with other NR members including a central DNA-binding domain (DBD) and carboxy-terminal ligand-binding domain (LBD). LXRα is primarily expressed in the liver, kidneys, intestines, and adipose tissues while LXRβ is widely expressed (56, 57). LXRs function as ligand-dependent transcription factors and bind directly to specific DNA sequences known as LXR 4esponse elements (LXREs). Following the discovery of the LXRs in the 1990s, oxysterols were identified as the direct ligands for both receptor proteins (56, 57). Since oxysterols are metabolites of cholesterol and have been shown to be key signaling molecules that indicate sterol levels, it has been elucidated that LXRs function as cholesterol sensors. LXRs can detect relative cholesterol levels through oxysterol metabolites and alter cell physiology as appropriate. LXRs have been shown to regulate cholesterol efflux and transport, as well as regulate lipogenesis and glucose metabolism. Synthetic LXR agonists (T0901317, GW3965) have been shown to display anti-atherogenic properties due to their effects on reverse cholesterol transport mediated by increased cholesterol efflux from peripheral tissues (58–60). However, the activation of LXR by synthetic ligands results in deleterious effects due to increased hepatic lipogenesis and the development of hepatic steatosis (61, 62). This has led to significant difficulties in the development of tissue selective LXR agonists for the treatment of atherosclerosis. The stimulation of hepatic lipogenesis by LXR agonists is due to the increased expression of lipogenic enzymes including FASN, SCD1, and SREBP1c that are direct target genes of LXR (61). LXR expression has been correlated with the degree of hepatic lipid accumulation, as well as hepatic fibrosis and inflammation in patients with liver diseases.
LXRs have a significant role in the regulation of physiological processes involved in the development of MAFLD. As described earlier, numerous physiological pathways can contribute to MAFLD in both mice and humans. This disease is systemic in its development and pathogenesis, beginning with altered lipid storage and metabolism, and progressing in part, due to abnormal metabolic functioning in a variety of tissues and cell types (i.e., T2DM, obesity, inflammation, lipoprotein processing, etc.). Here, we will focus on the physiological processes that LXR regulates, that are distinctively known for enhancing the development and progression of MAFLD.
3.2. LXRs are involved in cholesterol and fatty acid metabolism
The role of LXRs functioning as “cholesterol sensors” was confirmed utilizing Lxrα-null mice, which accumulated significant amounts of cholesterol in the liver when challenged with a high cholesterol diet due to their inability to activate an LXR-dependent mechanism for excess cholesterol to be converted to bile acids (63). Subsequent studies have identified that LXRs enhance hepatobiliary cholesterol excretion through the direct activation of target genes, Abcg5 and Abcg8 (64). Reverse cholesterol transport (RCT) is the process by which excess cholesterol in the periphery is transferred to HDL and transported to the liver for bile acid synthesis and excretion. This process is mediated by the ATP-binding cassette transporter ABCA1 and ABCG1 in macrophages, both of which are direct target genes of LXR (65). Activation of LXRs also induces the expression of several apolipoproteins and genes involved in lipoprotein remodeling including phospholipid transfer protein (PLTP), lipoprotein lipase (LPL), and CETP (66–70). LXR’s role in regulating cholesterol homeostasis via cholesterol transport into and out of the liver has a significant physiological impact on the development of MAFLD. Interestingly, this is not the only component of cholesterol metabolism that is regulated by LXR and has a direct effect on the pathogenesis of MAFLD and other dyslipidemic diseases.
It is well known that high levels of LDL cholesterol (LDL-C) contribute to the development of cardiovascular diseases including atherosclerosis. LDLR is responsible for the uptake of LDL-C and maintenance of systemic cholesterol levels and can be regulated at both the transcriptional and post-transcriptional levels. The sterol regulatory element-binding protein 2 (SREBP2) is the main transcription factor that regulates the expression of LDLR and is activated in response to low cholesterol levels in the cells. LXR however, can control the post-transcriptional regulation of LDLR through its direct target gene, inducible degrader of LDLR (IDOL) (71–73). The IDOL protein functions as an E3 ubiquitin protein ligase that directly leads to the degradation of LDLR, as well as VLDLR and other related proteins. Studies have shown that treatment with LXR agonists (T0901317 or GW3965) reduces LDLR expression and raises LDL-C plasma levels through an IDOL-dependent mechanism in humans and non-human primates. Genome-wide association studies have identified polymorphisms in the LDLR locus that leads to severe forms of statin-resistant hyperlipidemia (Familial hypercholesterolemia; FH). Patients with FH often are also diagnosed with some form of fatty liver disease.
LXRs are not only important in maintaining cholesterol homeostasis, but they are intricately involved in the regulation of DNL in the liver. Numerous studies have demonstrated the enhancement of fatty acid biosynthesis and VLDL secretion due to LXR agonist treatment. LXR directly controls the transcription of SREBP1c, FASN, and SCD1, and modulates the expression of ChREBP, all of which are directly involved in the pathogenesis of MAFLD and have been discussed earlier.
3.3. Synthetic LXR modulators
LXRs are master regulators of lipid and cholesterol metabolism and have remarkable anti-inflammatory activities. Because of their multiple roles, they are very interesting drug targets. The major classes of LXR modulators are agonists and antagonists. Agonists bind the LBD of the receptor and recruit coactivator proteins leading to receptor activation and increased expression of downstream target genes. Three LXR agonists are currently in clinical trials for the treatment of atopic dermatitis and advanced solid tumors and lymphoma. LXR antagonists block the binding of agonists and have yet to demonstrate therapeutic utility. A third type of modulator is LXR inverse agonists that was first developed by our group. LXRs have been demonstrated to recruit either coactivators or corepressors depending on the physiological context. We envisioned that development of a LXR ligand that bound to the LBD and selectively enhanced the ability of the receptor to recruit corepressor and suppress the expression of LXR target genes, such as those encoding the DNL enzyme genes, would have the potential to be used in the treatment of metabolic disorders such as MAFLD. The LXR antagonist scaffold (74) was used as an initial point to develop and optimize two novel LXR inverse agonists, SR9238 and SR9243 (Figure 1), that display potent activity for both LXRα and LXRβ and function to very efficaciously recruit corepressor proteins (75–77).
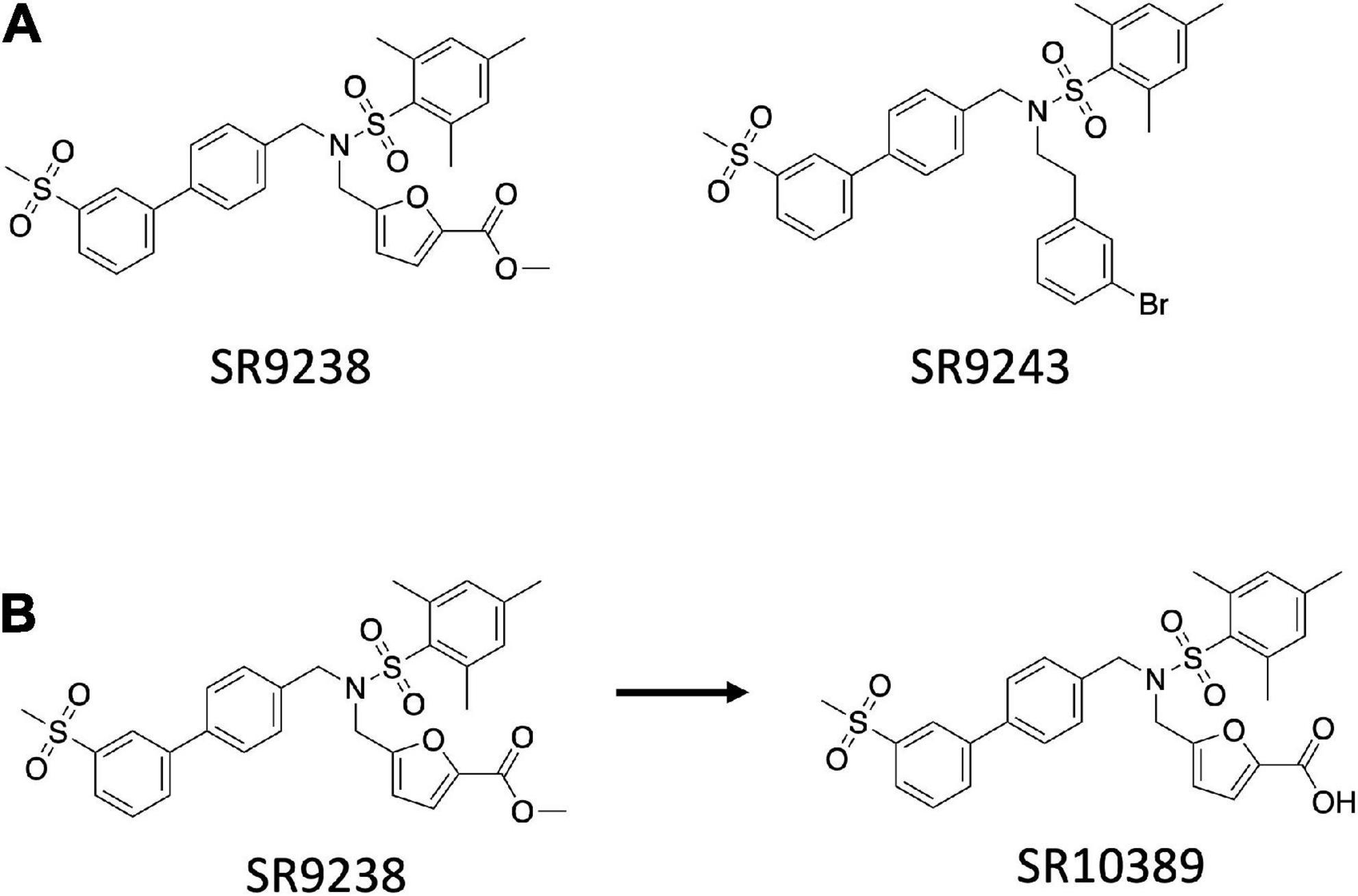
Figure 1. Structure of the SR series of LXR inverse agonists. (A) Chemical structure of SR9243 and SR9238. (B) Rapid metabolism of the ester functionality of SR9238 to the acid yields a liver specific LXR agonist.
SR9238 exhibits the ability to suppress basal transcriptional activity of LXRα (IC50 = 210 nM) and LXRβ (IC50 = 40 nM) in a co-transfection assay with a multimerized LXRE luciferase reporter in HEK293T cells (76). In biochemical assays, SR9238 binding to LXRα or LXRβ resulted in recruitment of corepressor NCoR CoRNR box peptides consistent with the ligand functioning as an inverse agonist. Although both NCoR ID1 and ID2 peptides were recruited in a SR9238-dependent manner, there was clear preference for the ID1 peptide for both receptors (76). Treatment of HepG2 cells with SR9238 resulted in significant decreases in the expression of FASN and SREBF1c, which are key drivers of DNL (76).
Our goal was to develop a LXR inverse agonist that could be used in vivo to test our hypothesis that such compounds may hold utility in treated NAFLD. However, a significant concern we had about this type of compound was that if it may decrease RCT even though it may have a beneficial effect on hepatic DNL. Thus, we designed SR9238 with this in mind and SR9238 is a compound with a labile ester group that is rapidly metabolized to a carboxylic acid (Figure 1B). We had noticed previously that certain LXR antagonists would display significant activity with an ester substitution that was lost with hydrolysis of the ester to an acid (78). We observed a similar paradigm with SR9238 as its acid analog (SR10389) was inactive (76). When administered i.p. SR9238 displayed intestinal and hepatic exposure, but no SR9238 was detected in the plasma, skeletal muscle, or brain. Thus, SR9238 provided a tool to assess the ability to target the liver without adversely affecting LXR target genes in peripheral tissues that drive RCT.
When administered to diet induced obese (DIO) mice, SR9238 (i.p.) drove a decrease in expression of Fasn, Srebf1c, and Scd1, which was associated with a significant reduction in hepatic steatosis (76). The reduction in hepatic fat accumulation was accompanied by a decrease in expression of inflammatory genes including Tnfa and Il1b and a decrease in hepatic F4/80 + cells was also noted. Markers of hepatocellular injury in the plasma (ALP, ALT, and AST) were also significantly reduced consistent with SR9238 improving hepatic function.
The classic DIO mouse provides a model of NAFLD, but hepatic fibrosis (associated with NASH) is not typically noted in this model. However, mice provided a diet high in cholesterol, fructose and trans-fat do develop NASH (79) and we examined the effect of SR9238 in this model as well. This NASH model has been utilized in both C57Bl6 mice and the ob/ob leptin deficient mice with similar results, but the disease is accelerated in ob/ob mice possibly due to their increased intake of the diet. In the ob/ob mice provided this NASH diet, we observed similar effects of SR9238 as we did in the DIO mice with decreased expression of genes encoding DNL enzymes and decreased hepatic steatosis (80). Hepatic weight was decreased, and plasma liver enzymes were also substantially decreased. Importantly, hepatic inflammation was significantly suppressed and hepatic fibrosis decreased by 75% as assessed by collagen staining (80). Although the mechanism of suppression of hepatic fibrosis is not clear, we hypothesized that this was due to the reduction of hepatic steatosis due to suppression of de novo lipogenesis leading to reduced inflammation and thus, reduced fibrosis. One interesting point that we noticed in both the NAFLD, and NASH models was that plasma LDL-cholesterol levels (LDL-C) were lowered significantly. In the DIO mice there was a ∼20% decrease whereas in the NASH model there was a ∼50% decrease. We also observed that SR9238 analog, SR9243, that has systemic exposure also displayed similar effects on plasma LDL-C in mice on a normal chow diet (∼50% decrease) (81). At this time, we did not have a proposed mechanism underlying the reduction in LDL-C, but interestingly, a later study of a LXR agonist showed an increase in LDL-C in both non-human primates and in clinical studies (82). This suggested that the LXR inverse agonist mediated decrease in LDL-C we observed in mouse models may be clinically relevant. An independent group assessed the activity of SR9243 in distinct models of NASH including the bile-duct ligation and carbon tetrachloride treatment (83). Huang et al. observed that SR9243 treatment reduced hepatic fibrosis and liver enzymes in both models (83). LDL-C levels were also substantially reduced.
Alcohol consumption is another major driver of liver disease and ethanol also induces hepatic DNL leading to inflammation and fibrosis. Chronic ethanol consumption by mice (Lieber-DiCarli (LD) diet) leads to substantial hepatic steatosis but does not lead to significant hepatic fibrosis. However, addition of “binge” ethanol doses near the end of the chronic ethanol consumption does lead to fibrosis and is a model of alcoholic hepatosteatosis (ASH). We assessed the effects of SR9238 in both models and observed that the drug reduced both fat content in the liver and inflammation (and fibrosis in the ASH model) (84). Like in the NASH model, SR9238 treatment resulted in substantial decrease in the expression of Srebpf1c and Fasn. Interestingly, treatment also led to an increase in expression of ethanol metabolizing enzymes Cyp2e1, Adh2, and Adh3, suggesting that not only did the LXR inverse agonist suppress DNL but also increased ethanol clearance (84). Given that many patients with steatohepatitis that is driven by both a high fat diet and ethanol consumption, we developed a diet that is composed of both the high cholesterol/trans-fat/fructose and ethanol (WASH diet – western diet and alcohol steatohepatitis). We found that the high cholesterol/trans-fat/fructose diet synergistically acted with the ethanol to enhance hepatic steatosis, inflammation, and fibrosis (85). Importantly, SR9238 treatment was able to suppress the severity of the effects on the liver (85).
As indicated above, one interesting observation we had made with any of the LXR inverse agonists we had tested in vivo was that there was a significant decrease in LDL-C. When examining the expression of intestinal genes that changed with SR9238 or SR9243 treatment (i.p.) we found that sterol O-acyltransferase 2 (Soat2) gene expression was suppressed by ∼95% (86). This intrigued us given that SOAT2 has been a target for development of drugs to treat hypercholesterolemia and atherosclerosis (87). SOAT2 is an enzyme that converts cholesterol to cholesterol esters and drives intestinal cholesterol absorption (88). Mice with an intestine specific KO of Soat2 are resistant to development of elevated plasma LDL-C on a high cholesterol diet (89) suggesting that targeting intestinal SOAT2 function or expression may be sufficient to provide this benefit. With our knowledge that SR9243 had no significant oral bioavailability we treated Ldlr null mice on a high cholesterol diet and observed that even though there was no liver or plasma exposure when SR9243 was administered orally, LDL-C was substantially decreased and was associated with repression of intestinal Soat2 expression and increased fecal cholesterol elimination (86). These data provided us with a clear mechanism that was driving the reduction in plasma LDL-C that we consistently observed as well as suggested that such compounds may hold utility in treatment of hypercholesterolemia, particularly in individuals that have mutations in the LDL receptor driving familial hypercholesterolemia.
After the description of the SR9238/SR9243 series of LXR inverse agonists additional chemical scaffolds with similar pharmacological profiles have been described. Burton et al. discovered that several cholestenoic acid analogs displayed LXR inverse agonist activity (Figure 2) (90, 91). These compounds showed the ability to suppress basal transcription in LXR cotransfection assays as well as suppress the expression of LXR target genes (Fasn, Srebf1c, and Abcg1) in HepG2 cells. These compounds drove the recruitment of corepressor proteins to the LXRs, but they did not appear to be very potent as doses greater than 1 μM were required for activity) (90, 91). This group also identified certain fluorinated oxysterol agonists as LXR inverse agonists based on their activity in LXR cotransfection assays in HEK293 cells, but these were also relatively low potency (Figure 2) (92). Chen et al. identified several non-steroidal LXR inverse agonists based on a screen of a compound library that was designed based on co-crystal structures of LXRβ in complex with spiro[pyrrolidine-3,3′-oxindole] agonists (93). These compounds displayed a significant degree of LXRβ selectivity (as much as 100-fold) and the most potent compound was approximately 3.5-fold less potent than SR9238 in a LXRβ cotransfection assay (93). Their most potent compound, 10rr (Figure 2), effectively suppressed SREBF1c, ACC, FASN, and SCD1 expression in both 3T3-LI adipocytes and HepG2 cells. Compound 10rr suppressed DNL in HepG2 cells consistent with the effects on gene expression and suppressed hyperlipidemia in the Triton WR-1339 induced mouse model (93). Working from the SR9238/SR9243 scaffold, Phenex Pharmaceuticals, developed additional LXR inverse agonists based on a published patent application (94). This intellectual property was licensed by Orsobio, Inc., and a compound (TLC-2716) is currently in phase I clinical trials for treatment of severe dyslipidemia (ClinicalTrials.gov NCT05483998). The structure of TLC-2,716 is not directly disclosed.
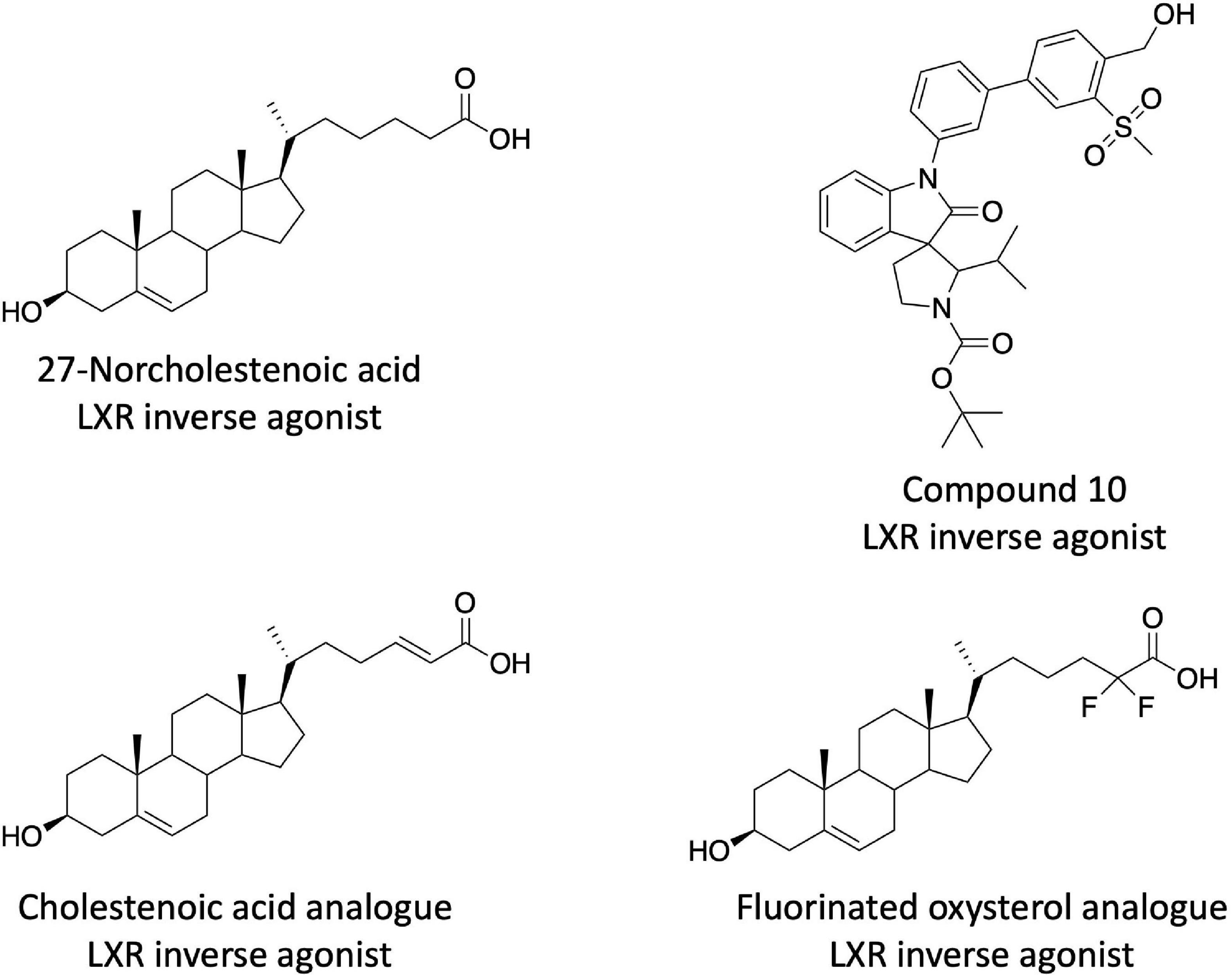
Figure 2. Examples of additional steroidal and non-steroidal LXR inverse agonists.
4. Conclusions and perspectives
LXRs have long been the focus of synthetic ligand development for the purpose of treating dyslipidemia and atherosclerosis. However, this focus has almost entirely been on the development of LXR agonists attempting to drive RCT. It was rapidly determined that such compounds had limiting on-target toxicity associated stimulating DNL resulting in hepatic steatosis and hypertriglyceridemia. Work from our lab focused on utilizing this observed side effect to develop and characterize LXR inverse agonists that actively silence LXR target genes, particularly those that drive de novo lipogenesis. With these compounds in hand we were able to demonstrate that were effective in treatment of NASH, ASH, hypercholesterolemia, and cancer in animal models. Several LXR inverse agonist chemical scaffolds have now been identified that display similar pharmacology and even one has entered phase I clinical trials for treatment of severe dyslipidemia.
Author contributions
KG and TB wrote and edited the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was funded by Auburn University and the University of Florida.
Acknowledgments
We acknowledge the our colleagues who aided in the development and characterization of the SR9238/SR2943 inverse agonist compounds.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Garcia-Compean D, Jimenez-Rodriguez A. NAFLD VS MAFLD. The evidence-based debate has come. Time to change? Ann Hepatol. (2022) 27:100765. doi: 10.1016/j.aohep.2022.100765
2. Zhu X, Xia M, Gao X. Update on genetics and epigenetics in metabolic associated fatty liver disease. Ther Adv Endocrinol Metab. (2022) 13:20420188221132138. doi: 10.1177/20420188221132138
3. Alem SA, Fouad Y, AbdAllah M, Attia D, Kamal I, Said E, et al. Hepatologists’ awareness and knowledge toward NAFLD and the familiarity with renaming from NAFLD to MAFLD. Endocr Metab Immune Disord Drug Targets. (2022). doi: 10.2174/1871530323666221028154159 [Epub ahead of print].
4. Perdomo C, Nunez-Cordoba J, Ezponda A, Mendoza F, Ampuero J, Bastarrika G, et al. Cardiometabolic characterization in metabolic dysfunction-associated fatty liver disease. Front Med. (2022) 9:1023583. doi: 10.3389/fmed.2022.1023583
5. Badmus O, Hillhouse S, Anderson C, Hinds T, Stec D. Molecular mechanisms of metabolic associated fatty liver disease (MAFLD): functional analysis of lipid metabolism pathways. Clin Sci. (2022) 136:1347–66. doi: 10.1042/CS20220572
6. Seko Y, Yamaguchi K, Yano K, Takahashi Y, Takeuchi K, Kataoka S, et al. The additive effect of genetic and metabolic factors in the pathogenesis of nonalcoholic fatty liver disease. Sci Rep. (2022) 12:17608. doi: 10.1038/s41598-022-22729-5
7. Theofilis P, Vordoni A, Kalaitzidis R. Interplay between metabolic dysfunction-associated fatty liver disease and chronic kidney disease: epidemiology, pathophysiologic mechanisms, and treatment considerations. World J Gastroenterol. (2022) 28:5691–706. doi: 10.3748/wjg.v28.i39.5691
8. Li Y, Zhou Y, Wang L, Lin X, Mao M, Yin S, et al. Emerging trends and hotspots in the links between the gut microbiota and MAFLD from 2002 to 2021: a bibliometric analysis. Front Endocrinol. (2022) 13:990953. doi: 10.3389/fendo.2022.990953
9. Kim D, Park J. Genetic risk factors associated with NAFLD. Hepatoma Res. (2020) 6:85. doi: 10.20517/2394-5079.2020.96
10. Wang S, Yan J, Chan Y. Advancements in MAFLD modeling with human cell and organoid models. Int J Mol Sci. (2022) 23:11850. doi: 10.3390/ijms231911850
11. Luo Z, Liu Y, Li H, Zhou Y, Peng Y, Lin X, et al. Associations of PNPLA3 rs738409 polymorphism with plasma lipid levels: a systematic review and meta-analysis. Horm Metab Res. (2022) 54:686–95. doi: 10.1055/a-1929-1677
12. Bashir A, Duseja A, Verma A, De A, Tiwari P. Lysosomal acid lipase activity in non-alcoholic fatty liver disease as a novel diagnostic and therapeutic target: a systematic literature review of current evidence and future directions. J Clin Exp Hepatol. (2022) 12:1535–46. doi: 10.1016/j.jceh.2022.04.011
13. Lee J, Shamim A, Park J, Jang J, Kim J, Kwon J, et al. Functional and structural changes in the membrane-bound O-acyltransferase family member 7 (MBOAT7) protein: the pathomechanism of a novel MBOAT7 variant in patients with intellectual disability. Front Neurol. (2022) 13:836954. doi: 10.3389/fneur.2022.836954
14. Li X, Wang Z, Meng H, Meng F, Yang P. Expression of membrane bound O-acyltransferase domain containing 7 after myocardial infarction and its role in lipid metabolism in vitro. Int J Med Sci. (2022) 19:609–17. doi: 10.7150/ijms.70614
15. Quek S, Tan E, Ren Y, Muthiah M, Loo E, Tham E, et al. Factors early in life associated with hepatic steatosis. World J Hepatol. (2022) 14:1235–47. doi: 10.4254/wjh.v14.i6.1235
16. Sharma D, Mandal P. NAFLD: genetics and its clinical implications. Clin Res Hepatol Gastroenterol. (2022) 46:102003. doi: 10.1016/j.clinre.2022.102003
17. Varadharajan V, Massey W, Brown J. Membrane-bound O-acyltransferase 7 (MBOAT7)-driven phosphatidylinositol remodeling in advanced liver disease. J Lipid Res. (2022) 63:100234. doi: 10.1016/j.jlr.2022.100234
18. Holmer M, Ekstedt M, Nasr P, Zenlander R, Wester A, Tavaglione F, et al. Effect of common genetic variants on the risk of cirrhosis in non-alcoholic fatty liver disease during 20 years of follow-up. Liver Int. (2022) 42:2769–80. doi: 10.1111/liv.15438
19. Heeren J, Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab. (2021) 50:101238. doi: 10.1016/j.molmet.2021.101238
20. Liao S, An K, Liu Z, He H, An Z, Su Q, et al. Genetic variants associated with metabolic dysfunction-associated fatty liver disease in western China. J Clin Lab Anal. (2022) 36:e24626. doi: 10.1002/jcla.24626
21. Fanlo-Maresma M, Esteve-Luque V, Pinto X, Padro-Miquel A, Corbella E, Candas-Estebanez B. Study of common hypertriglyceridaemia genetic variants and subclinical atherosclerosis in a group of women with SLE and a control group. Lupus Sci Med. (2022) 9:e000774. doi: 10.1136/lupus-2022-000774
22. Esteve-Luque V, Fanlo-Maresma M, Padro-Miquel A, Corbella E, Rivas-Regaira M, Pinto X, et al. Polygenic risk of hypertriglyceridemia is modified by BMI. Int J Mol Sci. (2022) 23:9837. doi: 10.3390/ijms23179837
23. Wang K, Shi M, Yang A, Fan B, Tam C, Lau E, et al. GCKR and GCK polymorphisms are associated with increased risk of end-stage kidney disease in Chinese patients with type 2 diabetes: the Hong Kong diabetes register (1995-2019). Diabetes Res Clin Pract. (2022) 193:110118. doi: 10.1016/j.diabres.2022.110118
24. Muthiah M, Ng C, Chan K, Fu C, Lim W, Tan D, et al. Type 2 diabetes mellitus in metabolic-associated fatty liver disease vs. type 2 diabetes mellitus non-alcoholic fatty liver disease: a longitudinal cohort analysis. Ann Hepatol. (2022) 28:100762. doi: 10.1016/j.aohep.2022.100762
25. Liu C, Schonke M, Zhou E, Li Z, Kooijman S, Boon M, et al. Pharmacological treatment with FGF21 strongly improves plasma cholesterol metabolism to reduce atherosclerosis. Cardiovasc Res. (2022) 118:489–502. doi: 10.1093/cvr/cvab076
26. Meroni M, Longo M, Paolini E, Tria G, Ripolone M, Napoli L, et al. Expanding the phenotypic spectrum of non-alcoholic fatty liver disease and hypertriglyceridemia. Front Nutr. (2022) 9:967899. doi: 10.3389/fnut.2022.967899
27. Perez-Robles M, Campos-Perez W, Rivera-Valdés JJ, Franco-Topete RA, Navarrete-Medina EM, Maldonado-González M, et al. Elevated serum low-density lipoproteins-cholesterol levels and B1B2/B2B2 CETP genotype are positively associated with nonalcoholic fatty liver disease in women with gallstone disease. Metab Syndr Relat Disord. (2022). doi: 10.1089/met.2022.0010 [Epub ahead of print].
28. Wen W, Li H, Wang C, Chen C, Tang J, Zhou M, et al. Metabolic dysfunction-associated fatty liver disease and cardiovascular disease: a meta-analysis. Front Endocrinol. (2022) 13:934225. doi: 10.3389/fendo.2022.934225
29. Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol. (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
30. Wang B, Tontonoz P. Phospholipid remodeling in physiology and disease. Annu Rev Physiol. (2019) 81:165–88. doi: 10.1146/annurev-physiol-020518-114444
31. Sandoval-Rodriguez A, Torre A, Armendariz-Borunda J. Editorial: liver fibrosis and MAFLD: from molecular aspects to novel pharmacological strategies. Front Med. (2022) 9:892495. doi: 10.3389/fmed.2022.892495
32. St Aubin C, Fisher A, Hernandez J, Broderick T, Al-Nakkash L. Mitigation of MAFLD in high fat-high sucrose-fructose fed mice by a combination of genistein consumption and exercise training. Diabetes Metab Syndr Obes. (2022) 15:2157–72. doi: 10.2147/DMSO.S358256
33. Ma S, Sun W, Gao L, Liu S. Therapeutic targets of hypercholesterolemia: HMGCR and LDLR. Diabetes Metab Syndr Obes. (2019) 12:1543–53. doi: 10.2147/DMSO.S219013
34. Jeon H, Blacklow S. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. (2005) 74:535–62. doi: 10.1146/annurev.biochem.74.082803.133354
35. Soutar A, Naoumova R. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. (2007) 4:214–25. doi: 10.1038/ncpcardio0836
36. Austin M, Hutter C, Zimmern R, Humphries S. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. (2004) 160:407–20. doi: 10.1093/aje/kwh236
37. Berberich A, Hegele R. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol. (2019) 16:9–20. doi: 10.1038/s41569-018-0052-6
38. Kampschulte M, Stockl C, Langheinrich A, Althohn U, Bohle R, Krombach G, et al. Western diet in ApoE-LDLR double-deficient mouse model of atherosclerosis leads to hepatic steatosis, fibrosis, and tumorigenesis. Lab Invest. (2014) 94:1273–82. doi: 10.1038/labinvest.2014.112
39. Subramanian S, Goodspeed L, Wang S, Kim J, Zeng L, Ioannou G, et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J Lipid Res. (2011) 52:1626–35. doi: 10.1194/jlr.M016246
40. Abifadel M, Boileau C. Genetic and molecular architecture of familial hypercholesterolemia. J Intern Med. (2023) 293:144–65.
41. Zhao Y, Yang Y, Xing R, Cui X, Xiao Y, Xie L, et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis. (2018) 271:26–35. doi: 10.1016/j.atherosclerosis.2018.02.015
42. Martinez H, Quinones M, Jimenez F, Estrada C, Clark K, Muscogiuri G, et al. Critical role of chemokine (C-C motif) receptor 2 (CCR2) in the KKAy + Apoe -/- mouse model of the metabolic syndrome. Diabetologia. (2011) 54:2660–8. doi: 10.1007/s00125-011-2248-8
43. Camargo F, Matos S, Araujo L, Carvalho C, Amaral A, Camporez J. Western diet-fed ApoE knockout male mice as an experimental model of non-alcoholic steatohepatitis. Curr Issues Mol Biol. (2022) 44:4692–703. doi: 10.3390/cimb44100320
44. Burris T, Solt L, Wang Y, Crumbley C, Banerjee S, Griffett K, et al. Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev. (2013) 65:710–78. doi: 10.1124/pr.112.006833
45. Burris T, Busby S, Griffin P. Targeting orphan nuclear receptors for treatment of metabolic diseases and autoimmunity. Chem Biol. (2012) 19:51–9. doi: 10.1016/j.chembiol.2011.12.011
46. Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. (2010) 72:247–72. doi: 10.1146/annurev-physiol-021909-135917
47. Savkur R, Burris T. The coactivator LXXLL nuclear receptor recognition motif. J Peptide Res. (2004) 63:207–12. doi: 10.1111/j.1399-3011.2004.00126.x
48. Lonard D, O’Malley B. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. (2012) 8:598–604. doi: 10.1038/nrendo.2012.100
49. Savkur R, Bramlett K, Clawson D, Burris T. Pharmacology of nuclear receptor-coregulator recognition. Vitamins Horm. (2004) 68:145–83. doi: 10.1016/S0083-6729(04)68005-8
50. Gronemeyer H, Gustafsson J, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. (2004) 3:950–64. doi: 10.1038/nrd1551
51. George J, Liddle C. Nonalcoholic fatty liver disease: pathogenesis and potential for nuclear receptors as therapeutic targets. Mol Pharm. (2008) 5:49–59. doi: 10.1021/mp700110z
52. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. (2015) 62:720–33. doi: 10.1016/j.jhep.2014.10.039
53. Li Y, Cao C, Zhou Y, Nie Y, Cao J, Zhou Y. The roles and interaction of FXR and PPARs in the pathogenesis of nonalcoholic fatty liver disease. Arab J Gastroenterol. (2020) 21:162–8. doi: 10.1016/j.ajg.2020.04.018
54. Welch R, Billon C, Losby M, Bedia-Diaz G, Fang Y, Avdagic A, et al. Emerging role of nuclear receptors for the treatment of NAFLD and NASH. Metabolites. (2022) 12:238. doi: 10.3390/metabo12030238
55. Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. (2012) 1821:809–18. doi: 10.1016/j.bbalip.2011.10.016
56. Tontonoz P, Mangelsdorf D. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. (2003) 17:985–93. doi: 10.1210/me.2003-0061
57. Schulman I. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. (2017) 591:2978–91. doi: 10.1002/1873-3468.12702
58. Joseph S, McKilligin E, Pei L, Watson M, Collins A, Laffitte B, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U.S.A. (2002) 99:7604–9. doi: 10.1073/pnas.112059299
59. Levin N, Bischoff ED, Daige C, Thomas D, Vu C, Heyman R, et al. Macrophage liver x receptor is required for antiatherogenic activity of LXR agonists. Arterioscler Thromb Vasc Biol. (2005) 25:135–42. doi: 10.1161/01.ATV.0000150044.84012.68
60. Joseph S, Castrillo A, Laffitte B, Mangelsdorf D, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. (2003) 9:213–9. doi: 10.1038/nm820
61. Schultz J, Tu H, Luk A, Repa J, Medina J, Li L, et al. Role of LXRs in control of lipogenesis. Genes Dev. (2000) 14:2831–8. doi: 10.1101/gad.850400
62. Grefhorst A, Elzinga B, Voshol P, Plosch T, Kok T, Bloks V, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem. (2002) 277:34182–90. doi: 10.1074/jbc.M204887200
63. Peet D, Turley S, Ma W, Janowski B, Lobaccaro J, Hammer R, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. (1998) 93:693–704. doi: 10.1016/S0092-8674(00)81432-4
64. Repa J, Berge K, Pomajzl C, Richardson J, Hobbs H, Mangelsdorf D. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J Biol Chem. (2002) 277:18793–800. doi: 10.1074/jbc.M109927200
65. Costet P, Luo Y, Wang N, Tall A. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem. (2000) 275:28240–5. doi: 10.1074/jbc.M003337200
66. Mak P, Kast-Woelbern H, Anisfeld A, Edwards P. Identification of PLTP as an LXR target gene and apoE as an FXR target gene reveals overlapping targets for the two nuclear receptors. J Lipid Res. (2002) 43:2037–41. doi: 10.1194/jlr.C200014-JLR200
67. Laffitte B, Joseph S, Chen M, Castrillo A, Repa J, Wilpitz D, et al. The phospholipid transfer protein gene is a liver X receptor target expressed by macrophages in atherosclerotic lesions. Mol Cell Biol. (2003) 23:2182–91. doi: 10.1128/MCB.23.6.2182-2191.2003
68. Luo Y, Tall A. Sterol upregulation of human CETP expression in vitro and in transgenic mice by an LXR element. J Clin Invest. (2000) 105:513–20. doi: 10.1172/JCI8573
69. Cao G, Beyer T, Yang X, Schmidt R, Zhang Y, Bensch W, et al. Phospholipid transfer protein is regulated by liver X receptors in vivo. J Biol Chem. (2002) 277:39561–5. doi: 10.1074/jbc.M207187200
70. Zhang Y, Repa J, Gauthier K, Mangelsdorf D. Regulation of lipoprotein lipase by the oxysterol receptors, LXRalpha and LXRbeta. J Biol Chem. (2001) 276:43018–24. doi: 10.1074/jbc.M107823200
71. Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. (2009) 325:100–4. doi: 10.1126/science.1168974
72. Hong C, Duit S, Jalonen P, Out R, Scheer L, Sorrentino V, et al. The E3 ubiquitin ligase IDOL induces the degradation of the low density lipoprotein receptor family members VLDLR and ApoER2. J Biol Chem. (2010) 285:19720–6. doi: 10.1074/jbc.M110.123729
73. Scotti E, Hong C, Yoshinaga Y, Tu Y, Hu Y, Zelcer N, et al. Targeted disruption of the idol gene alters cellular regulation of the low-density lipoprotein receptor by sterols and liver x receptor agonists. Mol Cell Biol. (2011) 31:1885–93. doi: 10.1128/MCB.01469-10
74. Zuercher W, Buckholz R, Campobasso N, Collins J, Galardi C, Gampe R, et al. Discovery of tertiary sulfonamides as potent liver X receptor antagonists. J Med Chem. (2010) 53:3412–6. doi: 10.1021/jm901797p
75. Flaveny C, Griffett K, El-Gendy B, Kazantzis M, Sengupta M, Amelio A, et al. Broad anti-tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer Cell. (2015) 28:42–56.
76. Griffett K, Solt L, El-Gendy Bel D, Kamenecka T, Burris TP. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem Biol. (2013) 8:559–67. doi: 10.1021/cb300541g
77. Elgendy B, Griffett K, Hegazy L, Di Fruscia P, Sample K, Schoepke E, et al. Synthesis and structure activity relationship of the first class of LXR inverse agonists. Bioorg Chem. (2022) 119:105540. doi: 10.1016/j.bioorg.2021.105540
78. Thomas J, Bramlett K, Montrose C, Foxworthy P, Eacho P, McCann D, et al. A chemical switch regulates fibrate specificity for peroxisome proliferator-activated receptor alpha (PPARalpha) versus liver X receptor. J Biol Chem. (2003) 278:2403–10. doi: 10.1074/jbc.M209629200
79. Trevaskis J, Griffin P, Wittmer C, Neuschwander-Tetri B, Brunt E, Dolman C, et al. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G762–72. doi: 10.1152/ajpgi.00476.2011
80. Griffett K, Welch R, Flaveny C, Kolar G, Neuschwander-Tetri B, Burris T. The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol Metab. (2015) 4:353–7. doi: 10.1016/j.molmet.2015.01.009
81. Flaveny C, Griffett K, El-Gendy Bel D, Kazantzis M, Sengupta M, Amelio A, et al. Broad anti-tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer Cell. (2015) 28:42–56.
82. Kirchgessner T, Sleph P, Ostrowski J, Lupisella J, Ryan C, Liu X, et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab. (2016) 24:223–33. doi: 10.1016/j.cmet.2016.07.016
83. Huang P, Kaluba B, Jiang X, Chang S, Tang X, Mao L, et al. Liver X receptor inverse agonist SR9243 suppresses nonalcoholic steatohepatitis intrahepatic inflammation and fibrosis. Biomed Res Int. (2018) 2018:8071093. doi: 10.1155/2018/8071093
84. Sengupta M, Griffett K, Flaveny C, Burris T. Inhibition of hepatotoxicity by a LXR inverse agonist in a model of alcoholic liver disease. ACS Pharmacol Transl Sci. (2018) 1:50–60. doi: 10.1021/acsptsci.8b00003
85. Sengupta M, Abuirqeba S, Kameric A, Cecile-Valfort A, Chatterjee A, Griffett K, et al. A two-hit model of alcoholic liver disease that exhibits rapid, severe fibrosis. PLoS One. (2021) 16:e0249316. doi: 10.1371/journal.pone.0249316
86. Griffett K, Hayes M, Bedia-Diaz G, Appourchaux K, Sanders R, Boeckman M, et al. Antihyperlipidemic activity of gut-restricted LXR inverse agonists. ACS Chem Biol. (2022) 17:1143–54. doi: 10.1021/acschembio.2c00057
87. Kobayashi K, Ohshiro T, Tomoda H, Yin F, Cui H, Chouthaiwale P, et al. Discovery of SOAT2 inhibitors from synthetic small molecules. Bioorg Med Chem Lett. (2016) 26:5899–901. doi: 10.1016/j.bmcl.2016.11.008
88. Anderson R, Joyce C, Davis M, Reagan J, Clark M, Shelness G, et al. Identification of a form of acyl-CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J Biol Chem. (1998) 273:26747–54. doi: 10.1074/jbc.273.41.26747
89. Zhang J, Kelley K, Marshall S, Davis M, Wilson M, Sawyer J, et al. Tissue-specific knockouts of ACAT2 reveal that intestinal depletion is sufficient to prevent diet-induced cholesterol accumulation in the liver and blood. J Lipid Res. (2012) 53:1144–52. doi: 10.1194/jlr.M024356
90. Alvarez L, Dansey M, Grinman D, Navalesi D, Samaja G, Del Fueyo M, et al. Destabilization of the torsioned conformation of a ligand side chain inverts the LXRbeta activity. Biochim Biophys Acta. (2015) 1851:1577–86. doi: 10.1016/j.bbalip.2015.09.007
91. Alvarez L, Dansey M, Ogara M, Pena C, Houtman R, Veleiro A, et al. Cholestenoic acid analogues as inverse agonists of the liver X receptors. J Steroid Biochem Mol Biol. (2020) 199:105585. doi: 10.1016/j.jsbmb.2020.105585
92. Rodriguez C, Alvarez L, Dansey M, Paolo L, Veleiro A, Pecci A, et al. Fluorinated oxysterol analogues: synthesis, molecular modelling and LXRbeta activity. J Steroid Biochem Mol Biol. (2017) 165(Pt B):268–76. doi: 10.1016/j.jsbmb.2016.07.001
93. Chen Z, Chen H, Zhang Z, Ding P, Yan X, Li Y, et al. Discovery of novel liver X receptor inverse agonists as lipogenesis inhibitors. Eur J Med Chem. (2020) 206:112793. doi: 10.1016/j.ejmech.2020.112793
Keywords: MAFLD, NAFLD, liver X receptors, dyslipidemia, hypercholesterolemia, cirrhosis, hepatocellular carcinoma, pharmacology
Citation: Griffett K and Burris TP (2023) Development of LXR inverse agonists to treat MAFLD, NASH, and other metabolic diseases. Front. Med. 10:1102469. doi: 10.3389/fmed.2023.1102469
Received: 18 November 2022; Accepted: 16 January 2023;
Published: 02 February 2023.
Edited by:
Ana Sandoval-Rodriguez, University of Guadalajara, MexicoReviewed by:
Moshe Levi, Georgetown University, United StatesRohini Mehta, Quest Diagnostics Nichols Institute, United States
Copyright © 2023 Griffett and Burris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas P. Burris,  YnVycmlzLnRob21hc0B1ZmwuZWR1
YnVycmlzLnRob21hc0B1ZmwuZWR1