 Mahsa Mollapour Sisakht
Mahsa Mollapour Sisakht Mohammad Amir Amirkhani3
Mohammad Amir Amirkhani3
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 09 February 2023
Sec. Dermatology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1096615
This article is part of the Research Topic Cutaneous Oncology and Skin Cancer Genomics View all 15 articles
Therapeutic strategies based on epigenetic regulators are rapidly increasing in light of recent advances in discovering the role of epigenetic factors in response and sensitivity to therapy. Although loss-of-function mutations in genes encoding the SWItch/Sucrose NonFermentable (SWI/SNF) subunits play an important role in the occurrence of ~34% of melanomas, the potential of using inhibitors and synthetic lethality interactions between key subunits of the complex that play an important role in melanoma progression must be considered. Here, we discuss the importance of the clinical application of SWI/SNF subunits as a promising potential therapeutic in melanoma.
The SWI/SNF complex is a large and evolutionarily conserved chromatin remodeler whose epigenomic changes are characterized in most cancers. This complex consists of 15 subunits encoded by 28 genes, including SMARCB1 (also known as SNF5, BAF47, and INI1), SMARCC1/SMARCC2 (also known as BAF155 and BAF170), and one of the two mutually exclusive ATPase subunits, SMARCA4 (also known as BRG1) and SMARCA2 (also known as BRM), which are commonly mutated in 20% of human cancers (1). Although several studies show the function of this complex as a transcriptional regulator, both tumor suppressive and enhancing functions of this complex have been investigated depending on the context. In some patients with hepatocellular carcinoma, ARID1A was strongly expressed in primary tumors but not in metastatic lesions, suggesting that ARID1A may be lost after initiation. Mechanistically, enhancement of ARID1A function promoted initiation by increasing cytochrome P450-mediated oxidative stress, while loss of Arid1a in tumors decreased chromatin accessibility and reduced transcription of genes associated with migration, invasion, and metastasis. At the same time, metastasis was reduced via transcriptional regulation of EMILIN1/MAT1A/LCN2/IL1R1 in vitro. Conversely, loss of ARID1A may increase the risk of steatohepatitis and cancer progression by altering immunity in vivo or tumorigenesis via activation of angiopoietin-2 (ANGPT2) transcription in vitro and angiogenesis in vivo (2). In summary, ARID1A, as a component of the SWI/SNF complex, plays a context-dependent tumor suppressive and oncogenic role in cancer (3). Melanoma results from the malignant transformation of certain cells called melanocytes. These cells are derived from multipotent cells of the neural crest and are responsible for melanin production (4). Metastatic melanoma is a highly aggressive malignancy that responds poorly to chemotherapeutic agents. Although targeted therapy with immune checkpoint inhibitors has resulted in significant improvement in tumor control, many patients do not respond to therapy, making it necessary to identify new therapeutic targets for patients (5). Despite the improvement in therapies developed for melanoma, the 10-year survival rate for patients with advanced melanoma is ~10% (6). The SWI/SNF component has been shown to play a critical role that can be targeted to develop a new therapeutic strategy (7). The synthetic lethal effect of the SWI/SNF subunits, demonstrated in several studies, has opened the possibility for new therapies. In light of the previous study, we attempt in this review to simplify and focus on the major subunits of the SWI/SNF complex in melanomagenesis that influence sensitivity to therapeutic agents. This review summarizes recent publications to highlight the most important SWI/SNF components based on statistical analyses related to melanoma progression and resistance and/or response to current therapies associated with this complex.
SWI/SNF is the first identified ATP-dependent chromatin remodeling multicomponent complex (consisting of 4–17 subunits) (8) that regulates the expression of 5% of genes in yeast (9) and plays an important role in transcription, DNA replication, and repair. This complex has a central catalytic subunit which is SMARCA4 (BRG1) or SMARCA2 (BRM) in the BAF complex, and 10–13 associated subunits (4), SMARCA4 or SMARCA2 function as catalytic subunits of other complexes called canonical (c) BAF, polybromo-associated BAF (PBAF) or non-canonical (nc)BAF (Figure 1). The different biological activity of these complexes is not fully understood, several functions of the biological activity of the complex are described by the genetic deletion of its subunits. The polybromo-associated BAF complex (PBAF) can be distinguished from the cBAF (canonical BAF complex) by the inclusion of BAF200 instead of BAF250A/B and BAF180 (10).
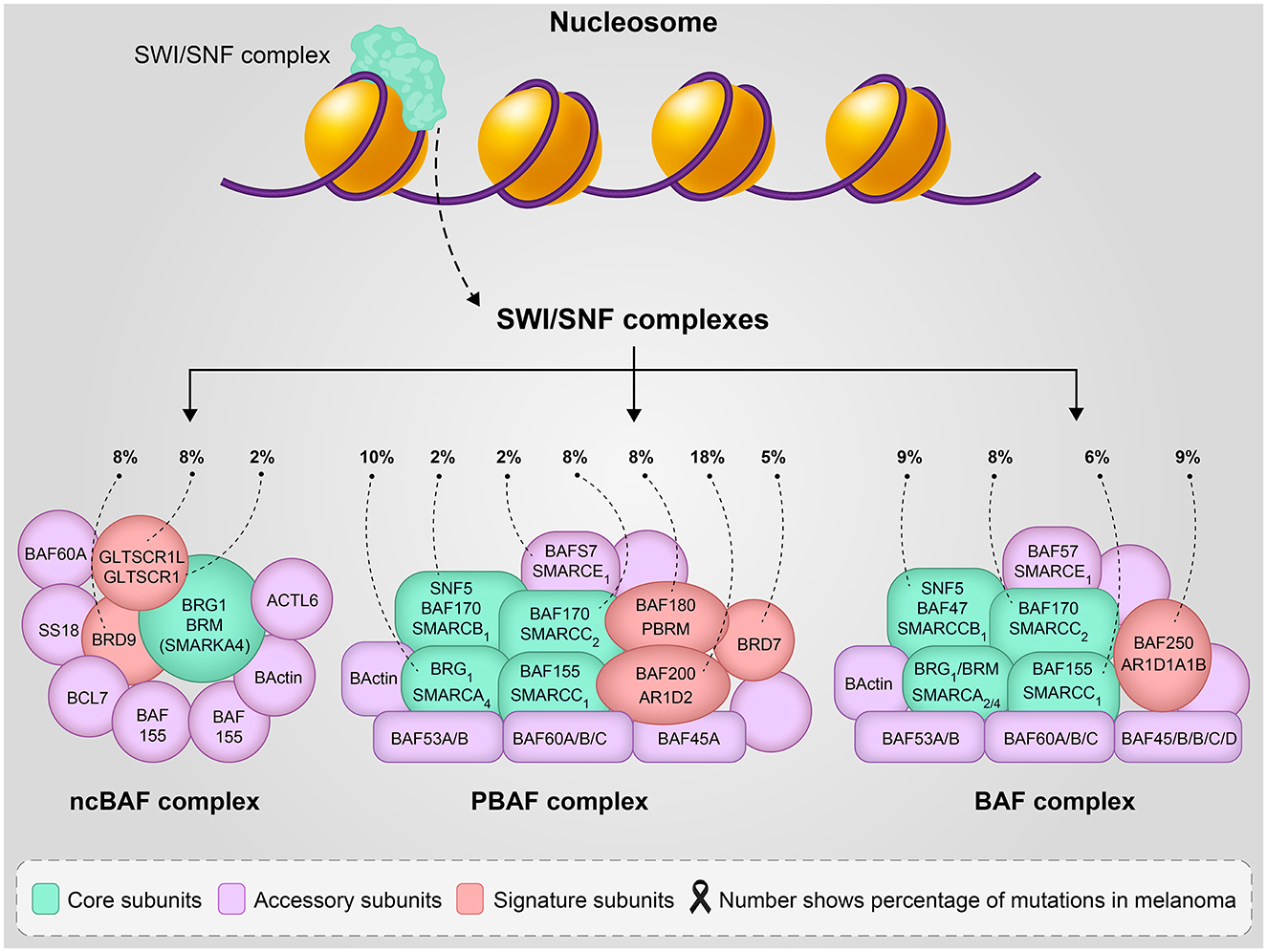
Figure 1. The SWI/SNF complexes structure and function, number shows the percentage of mutation involvement in melanoma reported in reference (5).
The components of the BAF complex can have cell type-specific functions, as evidenced by KD (knock-down) or KO (knock-out) of various subunits that had lethal effects, especially during embryogenesis (11, 12). The function of the BAF complex (esBAF) in pluripotency of embryonic stem cells was demonstrated by HO et al. in 2009 (13), and in 2019, the role of the non-canonical BRD9-containing BAF complex in regulating pluripotency in mouse embryonic stem cells was published (14). In addition, the function of other parts of the BAF complex, such as Baf60c, in the reversion of BAF to heart-specific enhancers during organogenesis (15) and in the differentiation of serotonergic neurons (16) has been published. All SWI/SNF components involve protein-protein or DNA-binding domains that are important for chromatin targeting and remodeling. Based on the biological activity of the complex, this complex contains three functional parts: the actin-related protein (ARP) module as a regulator, Snf2 (ATP-dependent motor that drives the basic DNA translocation reaction), and the substrate recruitment module, SRM. Recently, He and colleagues demonstrated the 12-member SWI/SNF complex in vitro by overexpressing the individual subunits in E. coli using the modified EcoExpress system to show the functional modularity of these complexes (9). Recent genomic studies have shown that the BAF and PBAF complexes are the most frequently mutated in various cancers (17). Non-canonical complexes (ncBAF) localize to the transcriptional repressor CTCF (also known as 11-zinc finger protein or CCCTC-binding factor) and can act as synthetic lethal targets specific for synovial sarcoma (SS) and malignant rhabdoid tumor (MRT) (16) or other cBAF-disrupted cancers.
Loss-of-function mutations in the components of the SWI/SNF complex such as AT-rich interactive domain-containing protein 1A (ARID1A), ARID1B, ARID2, or SMARCA4 are common in melanoma, suggesting that altered chromatin remodeling plays a role in the pathogenesis of this disease (18). A recent comprehensive study published by Dreier et al. using data from the TCGA showed a comparison of the frequency of genetic SWI/SNF alterations in melanoma. Interestingly, the data showed that the ARID2 subunit with missenses and truncating mutations is the most frequently mutated subunit in melanoma and is associated with a UVB mutation signature. Then, ARID1B missense mutations and deep deletions were observed in skin and choroidal melanomas, respectively. SMARCA4 and its paralogue SMARCA2 are almost equally frequently mutated (mostly as missenses) in patients with melanoma (5, 19). Missense mutations in these two tumor suppressors are associated with damage from UV radiation, as the role of SMARCAs is to promote melanin synthesis to protect against damage from solar radiation (20). Based on this study, ARID1A is the third most frequently mutated SWI/SNF, present in 9% of melanoma tumors in the TCGA database and associated with late-stage melanoma and metastasis to the brain (5) (Figure 1). On the other hand, several studies suggest that overexpression of the wild-type of ARID1A in patients with melanoma is more responsive to immune checkpoint inhibitors, while patients with loss of ARID1A may have therapeutic implications by modulating response to immunotherapy (21–23). However, a recent cohort study showed that ARID1A mutations do not have a major impact on survival and particularly on immune checkpoint inhibitors in melanoma. This finding by Griewank et al. (24). showed that ARID1A mutation status in melanoma is not currently relevant to treatment. Recent evidence suggests that ARID2, as a component of the PBAF complex, functions to inhibit invasion in vitro and modulate the response to immunotherapy in vivo (25). The mechanism of ARID2 as a modulator of tumor immunity is still unclear. The 2021 study by Fukumoto et al. showed that ARID2 knockout sensitizes melanoma to immune checkpoint inhibitors (anti-PD-L1 treatment), which is associated with an increase in the infiltration of CD8+ cytotoxic T cells. These results indicate that ARID2 is an immunomodulator and a potential biomarker indicating the efficacy of immune checkpoint inhibitors in patients with melanoma (25). In addition, mutations (C > T-transitions) in ARID2, PPP6C, SNX31, and TACC1 possibly played a role in UVB-induced melanomagenesis (18).
Synthetic lethality is a concept used as one of the most interesting, effective, and safe strategies in cancer treatment. It aims to target alleles of genes with loss-of-function mutations by drug inhibition, deletion, or reduction of expression to induce cell death. One of the best-known agents targeting inhibition of specific DNA repair pathways, based on the synthetic lethal approach, is the use of poly(ADP-ribose) polymerase (PARP) inhibitors to target BRCA1/2 mutated tumors (26). A 2019 systematic review by Kubicek et al. demonstrated strong synthetic-lethal interactions between two factors, SMARCA4-ARID2, in subunits of the SWI/SNF chromatin remodeling complex in melanoma. In view of this theory, cancer cells that have a LOF gene mutation lead to cell death when two genes are lost simultaneously, but not when either gene is lost alone, as we have described above. Thus, the relationship between SMARCA4 and ARID2 would be a potential target for synthetic lethal therapy, as SMARCA4 and ARID2 are frequently mutated in melanoma (27). On the other hand, in cancer cells lacking SMARCA4, SMARCB1, ARID1A, and PBRM1 of the SWI/SNF chromatin remodeling complex, inhibition of EZH2 (subunits of Zeste 2 Polycomb Repressive Complex 2; such as tazemetostat1 or valemetostat) can cause synthetic lethality (19).
ARID1B, as a component of a subset of cBAF complexes, is a homolog of ARID1A that promotes a compensatory pathway in the event of loss of ARID1A in some cancers (28). ARID1A and ARID1B are frequently co-mutated in cancer, but ARID1A-deficient cancers retain at least one functional ARID1B allele. The result suggests that loss of ARID1A and ARID1B alleles (double knockout) together promotes aggressive carcinogenesis, followed by dedifferentiation and hyper-proliferation in the liver and skin (29). Since 10.67% of melanoma patients have altered ARID1A, this may be useful in identifying ARID1B as a potential therapeutic target in patients with ARID1A loss or alteration.
SMARCA4 (BRG1) and SMARCA2 (BRM) are the two critical components of SWI/SNF ATPases that use ATP to generate energy for nucleosome remodeling, which is often mutated or silenced in cancer (30). The first somatic genetic alteration of SMARC2 was found in human non-melanoma cancer. Both act as tumor suppressors by controlling the cell cycle and regulating adhesion between cells. Several studies have also reported a synthetic lethality relationship between these two (19, 31). A 2018 study by Reyes and Martinez reported that high expression of SMARCA4 correlated with aggressive tumors, while high expression of SMARCA2 was associated with benign differentiated tumors. They showed that the expression of these two factors has a high prognostic value and high expression of SMARCA4 was significantly associated with poor prognosis and survival in uveal melanoma (32, 33), with high expression occurring in later stages of metastatic melanoma (34). Furthermore, one of these subunits is required for melanoma tumorigenesis (33, 35).
The master regulator of melanocyte differentiation from progenitor cells and survival, microphthalmia-associated transcription factor (MITF), showed several interactions with the SWI/SNF complex. The MITF gene plays a cooperative role with the subunits of the complex to promote tumorigenesis, and on the other hand, there is evidence that some SWI/SNF subunits are downregulated (4). SMARCA4 regulates MITF, melanin synthesis, protection against UVR damage, and survival in melanocytes and melanoma cells (36). SMARCA2 does compensate for the function of SMARCA4 in the case of a loss-of-function mutation associated with MITF (37), but SMARCA2 only partially compensates for the loss of SMARCA4 when all MITF targets are expressed (38). In addition to SMARCs, a recent study by Trivedi et al. (39) has shown that inhibition of bromodomain and extra-terminal (BET) by JQ1 can lead to decreased expression of MITF targets.
In a recent review article by Guo et al. (40) describing the signaling pathways involved in melanoma, including the mitogen-activated protein kinase (MAPK) pathway, protein kinase B (AKT) pathway, cell cycle regulation pathway, pigmentation-related pathway, and p53 pathway, epigenetic factors were also mentioned as one of the crucial factors in melanoma carcinogenesis. In melanoma, mutations in BRAF (50%−70%), NRAS (15%−30%), NF1, KRAS and HRAS (in 2 and 1% of patients, respectively), and KIT are responsible for MAPK dysregulation. Activation of the MAPK pathway leads to cell proliferation, invasion, metastasis, survival, and angiogenesis (41). BRAF (serine/threonine kinase), which is a member of the Raf family, plays a critical role in MAPK pathway signaling, with a substitution from valine to glutamic acid at codon 600 (V600E) in ~50% of melanomas (40). The FDA approved vemurafenib (PLX4032) and dabrafenib for the treatment of metastatic melanoma in 2011 and 2013, respectively. The drugs specifically target patients whose tumors have the BRAF V600E gene mutation (Table 1). The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway, which has more or less the same functions in cells, is another metabolic pathway that is dysregulated in melanoma, although there is no FDA-approved agent that directly inhibits this pathway in melanoma, however, there are several studies showing that synthetic small molecule compounds have an effect on PI3K/AKt and/or PI3K/Akt/mTOR and the associated RAS/RAF/MEK/ERK or MAPK pathway, such as NVP-BEZ235 (53) and Rapalogs (Everolimus, Deforolimus, and Temsirolimus) (54). c-KIT or CD117 is a transmembrane receptor tyrosine kinase (RTK) identified in hematopoietic cells, germ cells, gastrointestinal tract Cajal cells (GI), melanoma cells, B-cell progenitor cells, and mast cells that sends signals to maintain survival, promote cell proliferation, differentiation and regulate growth and development (55). Approximately 70% of KIT mutations in melanoma are missense mutations (well described in reference (55)). Nilotinib, Dasatinib, Sunitinib, and Masitinib are the small molecules that target cKIT and are currently being tested in clinical trials. It should be noted that pathway inhibitors have shown a potential synergistic effect with other antitumor agents in melanoma such as checkpoint inhibitors (56) and anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4) antibodies (57).
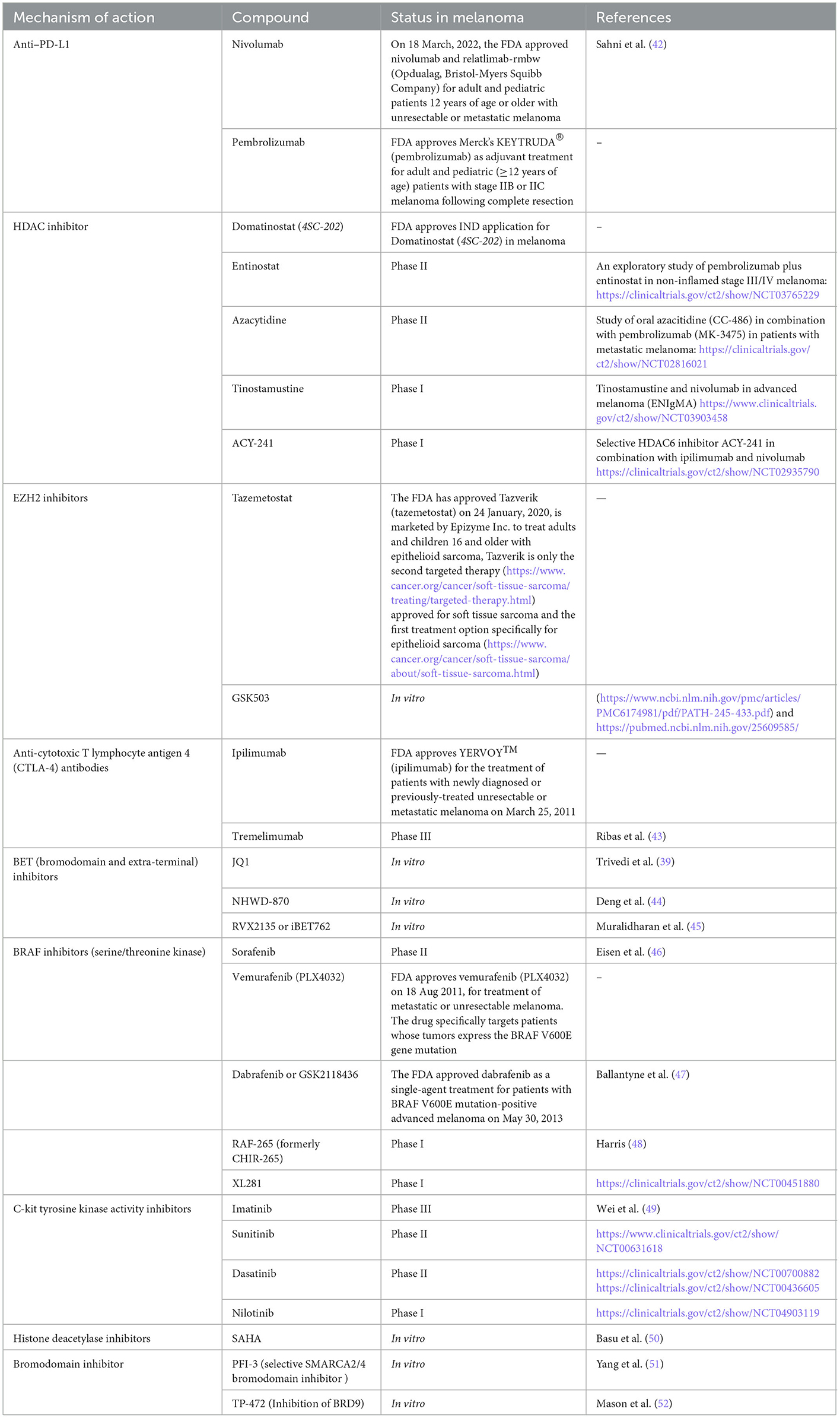
Table 1. Druggable targets in melanoma-the FDA, clinical trial, or assessment status of different compounds in melanoma.
The results of Martí et al. from 2012 show that the potential target therapies in melanoma can be divided into two categories: first, the strategy may target the tumor cell using molecules that can inhibit growth and/or prevent cell death, or molecules responsible for facilitating invasion and/or metastasis. The second category targets structure rather than cells, such as angiogenesis and immune tolerance. They reported BRAF inhibitors (Sorafenib, PLX4032, GSK2118436, RAF-265, XL281), inhibitors of c-kit tyrosine kinase activity (Imatinib, Sunitinib, Dasatinib, Nilotinib), and anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4) antibodies (Ipilimumab and Tremelimumab), which showed the best test results in patients with melanoma (57). Histone deacetylase inhibitors (HDACI) are the enzyme with anticancer activity, the only agents approved in the clinic for melanoma2, which inhibit histone deacetylases (HDAC) to inhibit tumor cell proliferation (58). Domatinostat (59), Entinostat3, Azacytidine4, ACY-241, and Tinostamustine5 can be placed in this category, which has been repeatedly demonstrated in preclinical and clinical studies. Garmpis et al. (58) proposed to investigate the synergistic effect of HDACi (Vorinostat (5AIIA, Zalmza), Romidepsin (Istodox, Depsipeptide), Belinostat and Panobinostat (Farydak) with other inhibitors such as BRAF inhibitors and BET inhibitors, which may lead to melanoma treatment. According to this study, the combination of the HDAC inhibitor LBH589 and the BET inhibitor I-BET151 showed apoptosis of melanoma cells but not of melanocytes via the mitochondrial pathway in the AKT and Hippo/YAP signaling pathways. This study demonstrated the effect of combination therapy on melanomas, including those resistant to BRAF inhibitors (60). In vitro treatment with the histone deacetylase inhibitor Vorinostat (SAHA) resulted in suppression of MITF and cell death in melanocytic nevi (50). A clinical trial of PDR001 and the HDAC inhibitor Panobinostat in patients with metastatic melanoma who have failed prior anti-PD1 or PD-L1 therapy was registered in 2019. The trial was withdrawn by the decision of the sponsor, but another trial evaluating the combination of the HDACi fusion molecule Tinostamustine (EDO-S101) and the anti-PD-1 monoclonal antibody Nivolumab in patients with refractory, locally advanced or metastatic melanoma was initiated in 2019 and is enrolling patients.6 Since 2010, the FDA has approved a number of therapeutic agents and synergistic approaches against melanoma, including Ipilimumab, Nivolumab, Pembrolizumab and the combination of Ipilimumab and Nivolumab, Vemurafenib, Dabrafenib, the combination of Dabrafenib plus Trametinib, Vemurafenib plus Cobimetinib, and Encorafenib plus Binimetinib (well described in reference (40)). Table 1 shows the information on agents tested or approved for melanoma, categorized by the mechanism of action, most of which have been discussed in this article.
The new strategy of combination therapy, especially in combination with BET inhibitors, showed interesting results in overcoming patient relapse and resistance after treatment with approved drugs targeting the MAPK pathway. For example, a recent study (44) showed that secreted phosphoprotein 1 (SPP1) expression can be a melanoma driver, and BET inhibitor NHWD-870 targets the BRD4 subunit and suppresses SPP1 expression and ultimately melanoma progression via the non-canonical NF-κB/SPP1 pathway (44). BET inhibitors (RVX2135 or iBET762) in combination with ATRIs [Ataxia-telangiectasia and Rad3-related (ATR) is a kinase belonging to the PI3 kinase-like family; VE821 or AZ20], showed apoptosis effect on melanoma cells as well as the PDX model of melanoma (45). Another study by Paoluzzi et al. (61) showed that the BET inhibitor JQ1 in combination with the BRAF inhibitorV suppressed tumor growth and significantly improved survival compared to either drug alone. Vemurafenib demonstrated safety and efficacy in both treatment-free and pre-treated BRAF-mutated melanoma patients (62).
In this section, we have attempted to provide an update on agents tested or approved for melanoma, most of which target the SWI/SNF complex. However, it should be noted that some in vitro or preclinical studies have shown the novel potential of new small molecules, e.g., the study by Zingg et al. (63), showed that EZH2 levels are upregulated in cancer cells after anti-CTLA-4 or IL-2 immunotherapies, leading to loss of tumor control. In this study, combination therapy with EZH2i (i.e., GSK503) may restore tumor immunogenicity. In general, and after reviewing various evidence that showed a contrast in the use of EZH2 inhibitors in cancers, especially mesothelioma, it seems that testing the response to EZH2 should be carefully evaluated before using these molecules as therapy (64). Recent research has shown that a novel bromodomain inhibitor called PFI-3, which targets SWI/SNF, and is responsible for repairing double-strand breaks in cancer, synergistically sensitizes many human cancers cell lines against DNA damage caused by chemotherapeutic agents such as doxorubicin (65). PFI-3 is a selective, potent, and cell-permeable SMARCA2/4 bromodomain inhibitor that has been previously characterized in the setting of various cancers (e.g., lung cancer, synovial sarcoma, leukemia, and rhabdoid tumors) (66). SMARCA2/SMARCA4 ATPases simulate synthetic lethality through multiple inhibitors for cancers containing a BRG1 loss-of-function mutation, as described in the 2018 study by Papillon et al. (67). It should be noted that SMARCA4, SMARCA2, BRD7, BRD9, and PBRM1 contain drug-acting bromodomains that showed interaction with synthetic lethality to serve as therapy. TP-472, a compound with selective function and similarity to the bromodomain of BRD7 and BRD9, has antitumor activity on melanoma (68).
Mutations in SWI/SNF's subunits are reported in ~25% of cancers (69). Although this paper is focused on melanoma, this section tries to show the footprint of this complex's mutation in different cancer. A comprehensive review by Centore et al., was published in 2020 based on the large-scale cancer genome-sequencing studies showed targeted therapies in different cancers based on this complex mutation, for example, ARID1A mutation as a hallmark in the bladder, stomach, and endometrial cancers which targeted by ARID1B selective degrader, EZH2 inhibitors and P13K inhibitors. SMARCA4, in nonsmall cell lung carcinoma, was targeted through the synthetic lethal pathway and by targeting SMARCA2 inhibitors (70). In Silico analysis of the SWI/SNF complex shows 70% of mutations with functional impact on lung adenocarcinoma patients (71). SMARCA2 in esophageal, SMARCB1 in malignant rhabdoid tumor and epithelial sarcoma, and PBRM1 in kidney cancer are collectively mutated and reported. These mutations are targeted by SMARCA4-selective inhibitors, BRD9-selective degraders, EZH2 inhibitors, and immune checkpoint inhibitors, respectively (70). On other hand, some of the studies focused on the ATPase part to conduct target therapy, degradation of ATPase subunit of SWI/SNF can disrupt physical chromatin accessibility to disable oncogenic transcription (for instance in prostate cancer) (72), and BRM as a core ATPase subunit is downregulated in hepatocellular carcinoma (HCC), colorectal and gastric cancer, small cell carcinoma of the ovary (SCCOHT), ovarian clear cell carcinoma (OCCC), non-small cell lung cancer (NSCLC), adenocarcinoma of the lung (AD), large cell carcinoma of the lung (LC), pleomorphic carcinoma of the lung (PL), clear cell renal cell carcinoma (ccRCC), and non-melanoma skin cancer (NMSC) (73). It should be noted that BRM as well as some of the therapeutic agents to target these complex acts context-dependent (as we also mentioned above about liver cancer) (74). Considering context dependency, cancer dependent-specific study is required to validate therapeutic agents targeting SWI/SNF complex subunits.
A theme developed from recent studies showed the crucial role of the SWI/SNF complex in defining the therapeutic efficacy of melanoma. In light of accumulated data, ARID2, ARID1B, SMARCA4 (BRG1), and SMARCA2 (BRM) have the most important mutations in melanoma. Considering the important role of epigenetic players in immune therapy resistance in a patient with melanoma. It is crucial to determine how SWI/SNF complex can contribute to melanoma therapy through different subunits. Combinational therapy and synthetic lethality approaches are the well-studied most current findings that show promising clinical responses in melanoma. Of note, further investigations need to be done to elucidate the context-dependent behavior of SWI/SNF subunits, possible off-target inhibition, immunosuppression, and the chance of relapse in target therapy for melanoma. Targeting the druggable SWI/SNF bromodomains (BRD7, BRD9, SMARCA4, SMARCA2), using the BET inhibitors as long as the HDAC inhibitors and identification of synthetic lethal interactions involved in melanoma such as SMARCA4 and ARID2 presents an additional possibility for novel strategies targeting the SWI/SNF subunits toward precise medicine of melanoma. Given the significant role of the SWI/SNF complex in melanoma, future therapeutic approaches must focus on mechanisms of synergic effect and synthetic lethality to enhance the therapeutic benefits of inhibitors, particularly when there is a deficiency in the functional domains mentioned above. We strongly believe an understanding of potential therapeutic vulnerabilities based on SWI/SNF in melanoma is leading to personalized and targeted cures and opening up new areas of clinical investigations.
Conception and design of study: MM and MA. Acquisition of data and revising the manuscript critically for important intellectual content: MA, MN, and MM. Drafting the manuscript: MM. All authors contributed to the article and approved the submitted version.
We would like to thank the skin and stem cell research center, Tehran University of Medical Sciences for technical support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. ^https://clinicaltrials.gov/ct2/show/NCT02601950
2. ^https://clinicaltrials.gov/ct2/show/NCT03022565
3. ^https://clinicaltrials.gov/ct2/show/NCT03765229
4. ^https://clinicaltrials.gov/ct2/show/NCT02816021
1. Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun. (2017) 8:14648. doi: 10.1038/ncomms14648
2. Fang J-Z, Li C, Liu X-Y, Hu T-T, Fan Z-S, Han Z-G. Hepatocyte-specific Arid1a deficiency initiates mouse steatohepatitis and hepatocellular carcinoma. PLoS ONE. (2015) 10:e0143042. doi: 10.1371/journal.pone.0143042
3. Sun X, Wang SC, Wei Y, Luo X, Jia Y, Li L, et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell. (2017) 32:574–589 e6. doi: 10.1016/j.ccell.2017.10.007
4. Mehrotra A, Mehta G, Aras S, Trivedi A, Serna ILD. SWI/SNF chromatin remodeling enzymes in melanocyte differentiation and melanoma. Crit Rev Eukaryot Gene Expr. (2014) 24:151–61. doi: 10.1615/CritRevEukaryotGeneExpr.2014007882
5. Dreier MR, de la Serna IL. SWI/SNF chromatin remodeling enzymes in melanoma. Epigenomes. (2022) 6:10. doi: 10.3390/epigenomes6010010
6. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol. (2019) 30:582–8. doi: 10.1093/annonc/mdz011
7. Rago F, Elliott G, Li A, Sprouffske K, Kerr G, Desplat A, et al. The discovery of SWI/SNF chromatin remodeling activity as a novel and targetable dependency in uveal melanoma. Mol Cancer Ther. (2020) 19:2186–95. doi: 10.1158/1535-7163.MCT-19-1013
8. Tang L, Nogales E, Ciferri C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog Biophys Mol Biol. (2010) 102:122–8. doi: 10.1016/j.pbiomolbio.2010.05.001
9. He Z, Chen K, Ye Y, Chen Z. Structure of the SWI/SNF complex bound to the nucleosome and insights into the functional modularity. Cell Discovery. (2021) 7:28. doi: 10.1038/s41421-021-00262-5
10. Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. (1996) 15:5370–82. doi: 10.1002/j.1460-2075.1996.tb00921.x
11. Menon DU, Shibata Y, Mu W, Magnuson T. Mammalian SWI/SNF collaborates with a polycomb-associated protein to regulate male germline transcription in the mouse. Development. (2019) 146:dev174094. doi: 10.1242/dev.174094
12. Bultman S, Gebuhr T, Yee D, Mantia CL, Nicholson J, Gilliam A, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. (2000) 6:1287–95. doi: 10.1016/S1097-2765(00)00127-1
13. Ho L, Ronan JL, Wu J, Staahl BT, Chen L, Kuo A, et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci U S A. (2009) 106:5181–6. doi: 10.1073/pnas.0812889106
14. Gatchalian J, Malik S, Ho J, Lee D-S, Kelso TWR, Shokhirev MN, et al. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat Commun. (2018) 9:5139. doi: 10.1038/s41467-018-07528-9
15. Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. (2004) 432:107–12. doi: 10.1038/nature03071
16. Weinberg P, Flames N, Sawa H, Garriga G, Hobert O. The SWI/SNF chromatin remodeling complex selectively affects multiple aspects of serotonergic neuron differentiation. Genetics. (2013) 194:189–98. doi: 10.1534/genetics.112.148742
17. Rada-Iglesias ABR, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. (2011) 470:279–83. doi: 10.1038/nature09692
18. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. (2012) 150:251–63. doi: 10.1016/j.cell.2012.06.024
19. Sasaki M, Ogiwara H. Synthetic lethal therapy based on targeting the vulnerability of SWI/SNF chromatin remodeling complex-deficient cancers. Cancer Sci. (2020) 111:774–782 doi: 10.1111/cas.14311
20. Moloney FJ, Lyons JG, Bock VL, Huang XX, Bugeja MJ, Halliday GM. Hotspot mutation of Brahma in non-melanoma skin cancer. J Invest Dermatol. (2009) 129:1012–5. doi: 10.1038/jid.2008.319
21. Li J, Wang W, Zhang Y, Cieślik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. (2020) 130:2712–26. doi: 10.1172/JCI134402
22. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. (2018) 24:556–62. doi: 10.1038/s41591-018-0012-z
23. Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. (2015) 21:231–8. doi: 10.1038/nm.3799
24. Thielmann CM, Matull J, Roth S, Placke J-M, Chorti E, Zaremba A, et al. Genetic and clinical characteristics of ARID1A mutated melanoma reveal high tumor mutational load without implications on patient survival. Cancers. (2022) 14:2090. doi: 10.3390/cancers14092090
25. Fukumoto T, Lin J, Fatkhutdinov N, Liu P, Somasundaram R, Herlyn M, et al. ARID2 deficiency correlates with the response to immune checkpoint blockade in melanoma. J Investig Dermatol. (2021) 141:1564–72.e4. doi: 10.1016/j.jid.2020.11.026
26. Topatana W, Juengpanich S, Li S, Cao J, Hu J, Lee J, et al. Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J Hematol Oncol. (2020) 13:118. doi: 10.1186/s13045-020-00956-5
27. Schick S, Rendeiro AF, Runggatscher K, Ringler A, Boidol B, Hinkel M, et al. Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat Genet. (2019) 51:1399–410. doi: 10.1038/s41588-019-0477-9
28. Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. (2014) 20:251–4. doi: 10.1038/nm.3480
29. Wang Z, Chen K, Jia Y, Chuang J-C, Sun X, Lin Y-H, et al. Dual ARID1A/ARID1B loss leads to rapid carcinogenesis and disruptive redistribution of BAF complexes. Nat Cancer. (2020) 1:909–22. doi: 10.1038/s43018-020-00109-0
30. Caumanns JJ, Wisman GBA, Berns K, van der Zee AGJ, de Jong S. ARID1A mutant ovarian clear cell carcinoma: a clear target for synthetic lethal strategies. Biochim Biophys Acta Rev Cancer. (2018) 1870:176–84. doi: 10.1016/j.bbcan.2018.07.005
31. Morel D, Almouzni G, Soria J-C, Postel-Vinay S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann Oncol. (2017) 28:254–69. doi: 10.1093/annonc/mdw552
32. Guerrero-Martínez JA, Reyes JC. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci Rep. (2018) 8:2043. doi: 10.1038/s41598-018-20217-3
33. Lin H, Wong RPC, Martinka M, Li G. BRG1 expression is increased in human cutaneous melanoma. Br J Dermatol. (2010) 163:502–10. doi: 10.1111/j.1365-2133.2010.09851.x
34. Peng L, Li J, Wu J, Xu B, Wang Z, Giamas G, et al. A pan-cancer analysis of SMARCA4 alterations in human cancers. Front Immunol. (2021) 12:762598. doi: 10.3389/fimmu.2021.762598
35. Keenen B, Qi H, Saladi SV, Yeung M, de la Serna IL. Heterogeneous SWI/SNF chromatin remodeling complexes promote expression of microphthalmia-associated transcription factor target genes in melanoma. Oncogene. (2010) 29:81–92. doi: 10.1038/onc.2009.304
36. Gelmi MC, Houtzagers LE, Strub T, Krossa I, Jager MJ. MITF in normal melanocytes, cutaneous and uveal melanoma: a delicate balance. Int J Mol Sci. (2022) 23:6001. doi: 10.3390/ijms23116001
37. Vachtenheim J, Ondrusová L, Borovanský J. SWI/SNF chromatin remodeling complex is critical for the expression of microphthalmia-associated transcription factor in melanoma cells. Biochem Biophys Res Commun. (2010) 392:454–9. doi: 10.1016/j.bbrc.2010.01.048
38. Saladi SV, Wong PG, Trivedi AR, Marathe HG, Keenen B, Aras S, et al. BRG1 promotes survival of UV-irradiated melanoma cells by cooperating with MITF to activate the melanoma inhibitor of apoptosis gene. Pigment Cell Melanoma Res. (2013) 26:377–91. doi: 10.1111/pcmr.12088
39. Trivedi A, Mehrotra A, Baum CE, Lewis B, Basuroy T, Blomquist T, et al. Bromodomain and extra-terminal domain (BET) proteins regulate melanocyte differentiation. Epigenetics Chromatin. (2020) 13:14. doi: 10.1186/s13072-020-00333-z
40. Guo W, Wang H, Li C. Signal pathways of melanoma and targeted therapy. Signal Transduct Target Ther. (2021) 6:424. doi: 10.1038/s41392-021-00827-6
41. Inamdar GS, Madhunapantula SV, Robertson GP. Targeting the MAPK pathway in melanoma: why some approaches succeed and other fail. Biochem Pharmacol. (2010) 80:624–37. doi: 10.1016/j.bcp.2010.04.029
42. Sahni S, Valecha G, Sahni A. Role of Anti-PD-1 antibodies in advanced melanoma: the era of immunotherapy. Cureus. (2018) 10:e3700. doi: 10.7759/cureus.3700
43. Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. (2013) 31:616–22. doi: 10.1200/JCO.2012.44.6112
44. Deng G, Zeng F, Su J, Zhao S, Hu R, Zhu W, et al. inhibitor suppresses melanoma progression via the noncanonical NF-κB/SPP1 pathway. Theranostics. (2020) 10:11428–43. doi: 10.7150/thno.47432
45. Muralidharan SV, Einarsdottir BO, Bhadury J, Lindberg MF, Wu J, Campeau E, et al. bromodomain inhibitors synergize with ATR inhibitors in melanoma. Cell Death Dis. (2017) 8:e2982. doi: 10.1038/cddis.2017.383
46. Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, et al. Sorafenib in advanced melanoma: a phase II randomised discontinuation trial analysis. Br J Cancer. (2006) 95:581–6. doi: 10.1038/sj.bjc.6603291
47. Ballantyne AD, Garnock-Jones KP. Dabrafenib: first global approval. Drugs. (2013) 73:1367–76. doi: 10.1007/s40265-013-0095-2
49. Wei X, Mao L, Chi Z, Sheng X, Cui C, Kong Y, et al. Efficacy evaluation of imatinib for the treatment of melanoma: evidence from a retrospective study. Oncol Res. (2019) 27:495–501. doi: 10.3727/096504018X15331163433914
50. Basu D, Salgado CM, Bauer B, Hoehl RM, Moscinski CN, Schmitt L, et al. Histone deacetylase inhibitor Vorinostat (SAHA) suppresses micropthalmia transcription factor expression and induces cell death in nevocytes from large/giant congenital melanocytic nevi. Melanoma Res. (2021) 31:319–27. doi: 10.1097/CMR.0000000000000749
51. Yang C, Wang Y, Sims MM, He Y, Miller DD, Pfeffer LM. Targeting the bromodomain of BRG-1/BRM subunit of the SWI/SNF complex increases the anticancer activity of temozolomide in glioblastoma. Pharmaceuticals. (2021) 14:904. doi: 10.3390/ph14090904
52. Mason LD, Chava S, Reddi KK, Gupta R. The BRD9/7 inhibitor TP-472 blocks melanoma tumor growth by suppressing ECM-mediated oncogenic signaling and inducing apoptosis. Cancers. (2021) 13:5516. doi: 10.3390/cancers13215516
53. Calero R, Morchon E, Martinez-Argudo I, Serrano R. Synergistic anti-tumor effect of 17AAG with the PI3K/mTOR inhibitor NVP-BEZ235 on human melanoma. Cancer Lett. (2017) 406:1–11. doi: 10.1016/j.canlet.2017.07.021
54. Strickland LR, Pal HC, Elmets CA, Afaq F. Targeting drivers of melanoma with synthetic small molecules and phytochemicals. Cancer Lett. (2015) 359:20–35. doi: 10.1016/j.canlet.2015.01.016
55. Pham DDM, Guhan S, Tsao H. KIT and melanoma: biological insights and clinical implications. Yonsei Med J. (2020) 61:562–71. doi: 10.3349/ymj.2020.61.7.562
56. Seifert AM, Zeng S, Zhang JQ, Kim TS, Cohen NA, Beckman MJ. PD-1/PD-L1 blockade enhances T-cell activity and antitumor efficacy of imatinib in gastrointestinal stromal tumors. Clin Cancer Res. (2017) 23:454–65. doi: 10.1158/1078-0432.CCR-16-1163
57. Martí RM, Sorolla A, Yeramian A. New therapeutic targets in melanoma. Actas Dermo-Sifiliográficas. (2012) 103:579–90. doi: 10.1016/j.adengl.2012.08.005
58. Garmpis N, Damaskos C, Garmpi A, Dimitroulis D, Spartalis E, Margonis GA, et al. Targeting histone deacetylases in malignant melanoma: a future therapeutic agent or just great expectations? Anticancer Res. (2017) 37:5355–62. doi: 10.21873/anticanres.11961
59. Giunta EFA, Curvietto G, Pappalardo M, Bosso A, Rosanova D, Diana M, et al. Epigenetic regulation in melanoma: facts and hopes. Cells. (2021) 10:2048. doi: 10.3390/cells10082048
60. Heinemann ACC, De Paoli-Iseppi R, Wilmott JS, Gunatilake D, Madore J, Strbenac D, et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget. (2015) 6:21507–21. doi: 10.18632/oncotarget.4242
61. Paoluzzi L, Hanniford D, Sokolova E, Osman I, Darvishian F, Wang J, et al. BET and BRAF inhibitors act synergistically against BRAF-mutant melanoma. Cancer Med. (2016) 5:1183–93. doi: 10.1002/cam4.667
62. Swaika ACJ, Joseph RW. Vemurafenib: an evidence-based review of its clinical utility in the treatment of metastatic melanoma. Drug Des Devel Ther. (2014) 8:775–87. doi: 10.2147/DDDT.S31143
63. Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. (2017) 20:854–67. doi: 10.1016/j.celrep.2017.07.007
64. Schoumacher M, Le Corre S, Houy A, Mulugeta E, Stern MH, Roman-Roman S, et al. Uveal melanoma cells are resistant to EZH2 inhibition regardless of BAP1 status. Nat Med. (2016) 22:577–8. doi: 10.1038/nm.4098
65. Lee D, Lee D-Y, Hwang Y-S, Seo H-R, Lee S-A, Kwon J. The bromodomain inhibitor PFI-3 sensitizes cancer cells to DNA damage by targeting SWI/SNF. Mol Cancer Res. (2021) 19:900–12. doi: 10.1158/1541-7786.MCR-20-0289
66. Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. (2015) 75:3865–78. doi: 10.1158/0008-5472.CAN-14-3798
67. Papillon JPN, Nakajima K, Adair CD, Hempel J, Jouk AO, Karki RG, et al. Discovery of orally active inhibitors of brahma homolog (BRM)/SMARCA2 ATPase activity for the treatment of brahma related gene 1 (BRG1)/SMARCA4-mutant cancers. J Med Chem. (2018) 61:10155–72. doi: 10.1021/acs.jmedchem.8b01318
68. Zhao RL, Wu Y, Li C, Wei M, Niu Y, Yang W H, et al. BRD7 promotes cell proliferation and tumor growth through stabilization of c-Myc in colorectal cancer. Front Cell Dev Biol. (2021) 9:659392. doi: 10.3389/fcell.2021.659392
69. Mittal P, Roberts CWM. The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nat Rev Clin Oncol. (2020) 17:435–48. doi: 10.1038/s41571-020-0357-3
70. Centore RC, Sandoval GJ, Soares LMM, Kadoch C, Chan HM. Mammalian SWI/SNF chromatin remodeling complexes: emerging mechanisms and therapeutic strategies. Trends Genet. (2020) 36:936–50. doi: 10.1016/j.tig.2020.07.011
71. Peinado P, Andrades A, Cuadros M, Rodriguez MI, Coira IF, Garcia DJ, et al. Multi-omic alterations of the SWI/SNF complex define a clinical subgroup in lung adenocarcinoma. Clin Epigenetics. (2022) 14:42. doi: 10.1186/s13148-022-01261-3
72. Xiao L, Parolia A, Qiao Y, Bawa P, Eyunni S, Mannan R, et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature. (2022) 601:434–9. doi: 10.1038/s41586-021-04246-z
73. Jancewicz I, Siedlecki JA, Sarnowski TJ, Sarnowska E, BRM. the core ATPase subunit of SWI/SNF chromatin-remodelling complex—a tumor suppressor or tumor-promoting factor? Epigenetics Chromatin. (2019) 12:68. doi: 10.1186/s13072-019-0315-4
Keywords: melanoma, SWI/SNF enzymes, epigenetics, chromatin remodeling, synthetic lethality, cancer therapy
Citation: Mollapour Sisakht M, Amirkhani MA and Nilforoushzadeh MA (2023) SWI/SNF complex, promising target in melanoma therapy: Snapshot view. Front. Med. 10:1096615. doi: 10.3389/fmed.2023.1096615
Received: 12 November 2022; Accepted: 20 January 2023;
Published: 09 February 2023.
Edited by:
Bahar Dasgeb, The State University of New Jersey, United StatesReviewed by:
Cedric Ng, National Cancer Centre Singapore, SingaporeCopyright © 2023 Mollapour Sisakht, Amirkhani and Nilforoushzadeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahsa Mollapour Sisakht,  bW1vbGxhcG91ckBmYXJhYmkudHVtcy5hYy5pcg==; bS5tb2xhcG91cnNpc2FraHRAZXJhc211c21jLm5s
bW1vbGxhcG91ckBmYXJhYmkudHVtcy5hYy5pcg==; bS5tb2xhcG91cnNpc2FraHRAZXJhc211c21jLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.