
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med., 26 August 2021
Sec. Ophthalmology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.708717
This article is part of the Research TopicWomen in Science - Ophthalmology 2021View all 12 articles
 Justyna Paprocka1*
Justyna Paprocka1* Magdalena Nowak2
Magdalena Nowak2 Maria Nieć2
Maria Nieć2 Izabela Janik2
Izabela Janik2 Małgorzata Rydzanicz3
Małgorzata Rydzanicz3 Śmigiel Robert4
Śmigiel Robert4 Magdalena Klaniewska4
Magdalena Klaniewska4 Karolina Rutkowska3
Karolina Rutkowska3 Rafał Płoski3
Rafał Płoski3 Aleksandra Jezela-Stanek5*
Aleksandra Jezela-Stanek5*Germline variants in tumor necrosis factor receptor-associated factor 7 (TRAF7) gene have recently been described in about 50 patients with developmental delay and cardiac, facial, and digital anomalies (CAFDADD). We aimed to depict further the clinical and genetic spectrum associated with TRAF7 germline variants in two additional patients, broaden the mutational spectrum, and support the characteristic clinical variety to facilitate the diagnostics of the syndrome among physician involved in the evaluation of patients with developmental delay/congenital malformations.
TRAF7 (Tumor necrosis factor receptor-associated factor 7) is a multifunctional intracellular protein belonging to the TRAF family (tumor necrosis factor receptor - TNF-R), which consists of seven proteins (TRAF1-7) (1, 2). It is encoded by a homonymous gene - TRAF7 – encompassing 21 exons, located within the chromosomal region 16p13.3 (2). A modular structure characterizes all members of the TRAF family. TRAF7 670-amino acid protein contains an N-terminal RING finger domain (aa 125–160), a zinc finger domain (aa 221–287) and a coiled-coil domain, but instead of the classical TRAF domain, it has seven WD40 repeats at the C-terminus (1, 3).
TRAF proteins are active in many biological processes, including embryonic development, tissue homeostasis as well as innate and adaptive humoral immune responses (4, 5). TRAF7 is involved in the process of protein ubiquitination. It is one of the critical mediators of NF-κB (nuclear factor-κB) and MAPK (mitogen-activated protein kinase) signaling pathways. It, therefore, plays an important role in many cellular processes (1–3, 6). The interaction of TRAF7 and MEKK3 (mitogen-activated protein kinase kinase kinase 3) through the WD40 domain leads to the activation of JNK and p38 MAP signaling pathways, which are responsible for cell survival, proliferation and differentiation (1–3). TRAF7 also plays a specific role in muscle function. Reduced TRAF7 expression under the influence of MyoD1 in growing myoblasts causes a decrease in NF-κB ubiquitination, exit from the cell cycle and acceleration of myogenesis (3, 6).
As a result of research conducted toward a closer understanding of TRAF7, a relationship between the protein and the development of neoplasms was demonstrated. Somatic mutations in TRAF7 have been described in meningiomas, mesotheliomas, perineural tumors of soft tissues, as well as in glandular neoplasms of the genital tract (1, 7–9). In turn, germline mutations in this gene have been described in the neurodevelopmental syndrome, encompassing cardiac, facial, and digital anomalies with developmental delay, thus termed CAFDADD (OMIM #6181640), characterized by facial dysmorphism with specific anomalies within the eyes (ptosis, epicanthic folds), congenital defects of the heart (aortic coarctation, hypoplastic left heart, double outlet right ventricle) and skeletal system (distal contractures, overlapping digits), and psychomotor delay. The described to date germline variants are heterozygous missense mutations, including frequently repeated c.1964G>A (p. Arg655Gln) and occur de novo (2, 5, 10).
We present two patients with characteristic facial dysmorphic features in whom, as a result of performed Whole Exome Sequencing (WES), heterozygous de novo variants were detected in TRAF7 gene: the c.1708C>G (p.His570Asp) in case 1 and c.1783C>G (p.Leu595Val) in case 2. Our aim is to delineate the phenotype and to underline the specific ocular findings that may facilitate the clinical diagnosis.
A 2-month-old boy was admitted to the Department of Pediatric Neurology for the diagnosis of dysmorphic and ophthalmoplegic features. The boy was born in the 41st week of an uncomplicated pregnancy with the Apgar score of 10, bodyweight of 3,100 g and head circumference of 32 cm. The examination revealed a short two-vessel umbilical cord. He developed neonatal jaundice, which was treated with phototherapy. Transfontanelle ultrasound showed an asymmetric ventricular system. The patient's family history was unremarkable.
The child was diagnosed with facial dysmorphism, including swollen eyelids, small sunken eyes, which initially opened only 2–3 mm, as well as a wide nose with an upturned tip, a receding mandible and asymmetrical auricles; head circumference was 36 cm (<3rd percentile) and the frontal fontanel, sized 2 × 3 cm, was below the level of the skull bone. Moreover, the physical examination revealed a narrow chest with widely spaced nipples and significant binary stenosis (Figure 1).
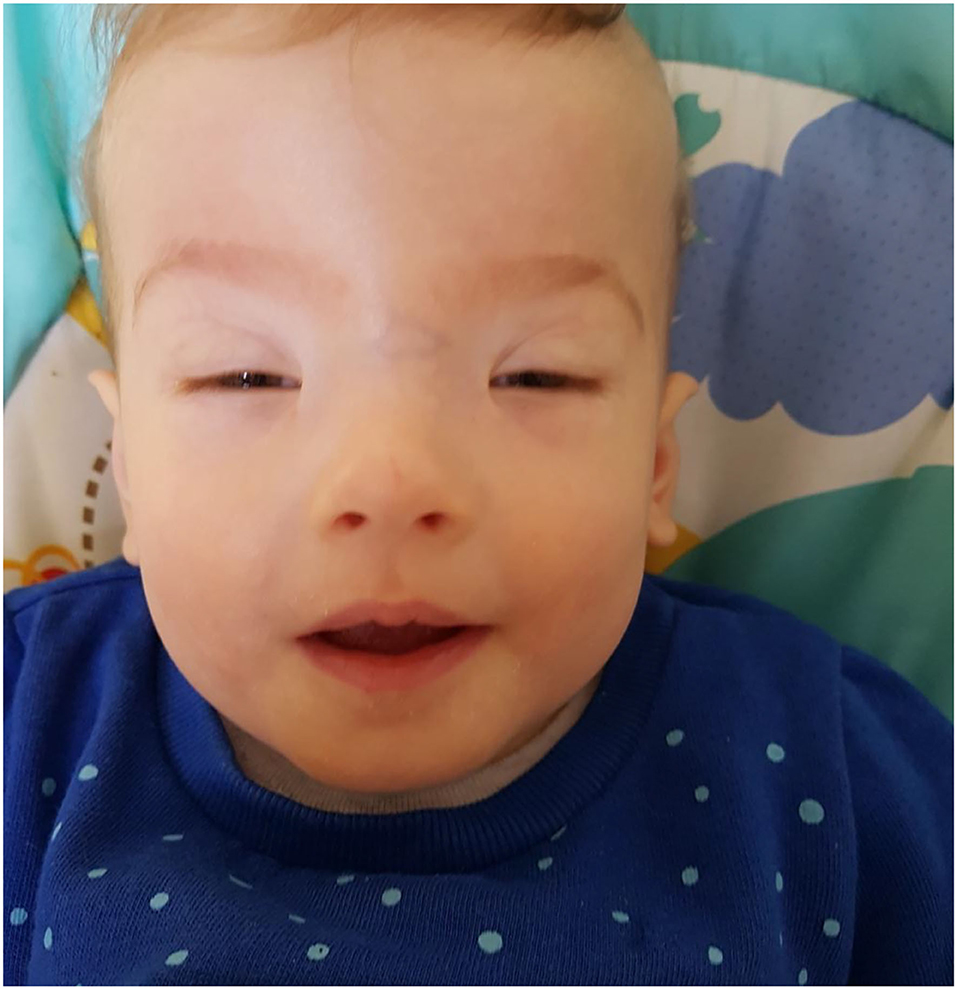
Figure 1. The facial phenotype of Case 1 at 11 months. Note the significant short palpebral fissures, ptosis, epicanthic folds and high forehead with flat, broad nasal bridge.
Correct tendon-periosteal reflexes and symmetrical posture reflexes were found. Positional asymmetry with shortening of the left side and reduced muscle tension in the head-torso axis were revealed, and, for this reason, physical rehabilitation was recommended. Ophthalmoplegia was diagnosed by an ophthalmologist. The results of basic laboratory tests (morphology, urinalysis, capillary gasometry, liver function assessment, ionogram) were within the normal range.
MRI of the head and eye sockets was performed, which showed no abnormalities apart from the asymmetry of the bones of the skull cover. Suspecting the myogenic background of the described ptosis, the diagnostics was extended to include an electrostimulation test for measuring muscle fatigue, during which the right axillary nerve was stimulated with the registration of a response from the right deltoid muscle. No decrease in amplitude was found; therefore, the test result was determined as negative. In addition, the test for antibodies against acetylcholine receptors was performed, and the result was normal.
Echocardiography showed patent foramen ovale/atrial septal defect (PFO/ASD II) and patent ductus arteriosus (PDA) with a left-right shunt. The ultrasound of the abdomen and retroperitoneal space was also performed, which visualized the enlarged caliceal-pelvic system of the left kidney. E. Coli 104 was obtained from the urine culture, and therefore a nephrological consultation was recommended. The patient was discharged with recommendations for further care in specialist units: cardiology, nephrology, ophthalmology, genetic counseling and physical rehabilitation clinics.
The patient was consulted by a geneticist, who recommended an array comparative genomic hybridization (aCGH) test. The result was normal. Thus, whole-exome sequencing (WES) was recommended for the suspected blepharophimosis, ptosis, epicanthus in vs. syndrome (BPES).
An almost 2-year-old boy is currently under the care of the Genetic Clinic because of a delay in psychomotor development and facial dysmorphic features. The boy was born by cesarean section at 40 weeks of gestation, with Apgar score of 8 points and birth weight of 4,120 g. The pregnancy was complicated by maternal infection and threatened preterm delivery. Family history was unremarkable.
After birth, no malformations of internal organs were found. Conductive hearing loss on the left side was identified. Due to clubfoot, the boy was referred to orthopedic consultation. The feet were successively plastered and splinted with good results. Cardiac ultrasound showed a bicuspid tricuspid valve. Ultrasound of the abdominal cavity showed a left-sided duplication of the caliceal-pelvic system. Cerebral NMR and angio-NMR showed hypoplasia of the middle and anterior segment of the sagittal sinus with peripheral venous circulation and slight dilatation of the supra-ventricular system as well as dilatation of brain fissures in the frontal and temporal regions and basal cisterns. TSH and CPK levels were normal. There were no abnormalities in the ophthalmological examination.
Physical examination at the age of 16 months revealed specific features of craniofacial dysmorphia such as increased head circumference (OFC 50 cm – 97 percentile), prominent forehead and prominent veins on the forehead, two hair whirls as well as wide nasal bridge, short palpebral fissures, blepharophimosis and mild ptosis of both eyelids (Figure 2).
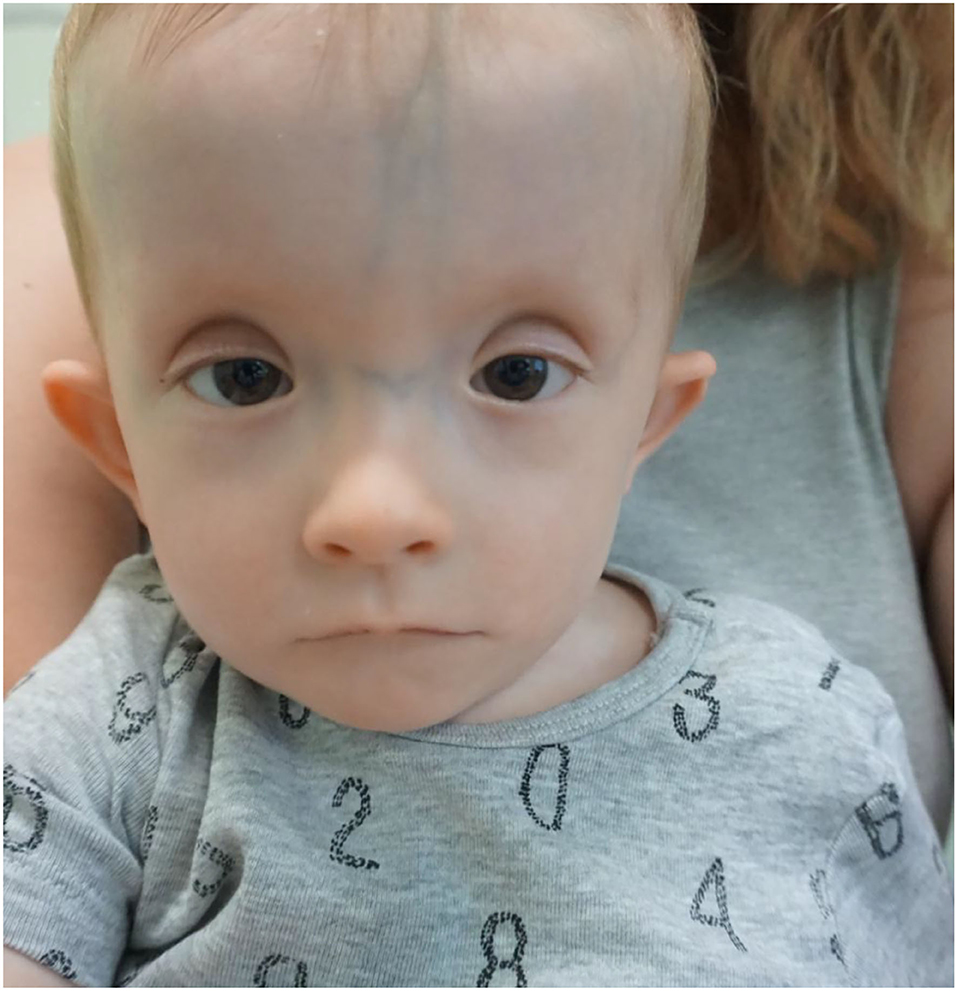
Figure 2. The facial phenotype of Case 2 and 16 months. Not the high and broad forehead, flat nasal bridge, short palpebral fissures and ptosis, thin lips.
Palms and fingers were normal; however, the toes overlap. At 16 months of life, the weight was 10kg (25 percentile) and height 78 cm (25–50 percentile). Hypotonia was also found. The boy is intensively rehabilitated. He sat up by himself at the age of 12 months and started walking at the age of 2 years. He does not speak; however, according to his parents, he understands speech.
Cytogenetic examination showed normal male karyotype. Array CGH did not reveal any chromosomal aberrations. Clinically dysmorphic syndrome belonging to the RASopathies group was suspected in the child. Therefore, a whole-exome sequencing (WES) was performed.
For both probands, WES analysis was performed using SureSelectXT Human kit All Exon v7 - Case 1 and v5 – Case 2 (Agilent, Agilent Technologies, Santa Clara, CA). Enriched libraries were paired-end sequenced (2 × 100 bp) on HiSeq 1,500 (Illumina, San Diego, CA, USA) and analyzed as previously described (11). In brief, raw sequence readouts were initially analyzed with bcl2fastq software (Illumina) to generate reads in fastq format. After the quality control steps, including adapter trimming and low-quality read removal, reads were aligned to the GRCh38 (hg38) reference genome with Burrows-Wheeler Alignment Tool (http://bio-bwa.sourceforge.net/), and processed further by Picard (http://broadinstitute.github.io/picard/) and Genome Analysis Toolkit (https://software.broadinstitute.org/gatk/). Identified variants were annotated with functional information, frequency in population (including gnomAD (http://gnomad.broadinstitute.org/), and an in-house database of >3,500 Polish exomes), and known association with clinical phenotypes, based on both ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD (http://www.hgmd.cf.ac.uk) databases. In silico pathogenicity prediction was performed based on Varsome pathogenicity and conservation scores (12) and by MetaSVM (13).
Variants passing a default quality were further filtered to include only those with <1% minor allele frequency in all tested databases (gnomAD, in house database of >3,500 Polish exomes), and to exclude deep intronic variants. The final list of variants were screened against known pathogenic mutations listed in ClinVar an dHGMD databases, and then searched for biallelic mutations consistent with autosomal recessive inheritance and monoallelic variants potentially causative of an autosomal dominant. All prioritized variants were manually inspected with Integrative Genomics Viewer (14).
Enriched libraries were paired-end sequenced (2 × 100 bp) on HiSeq 1,500 (Illumina, San Diego, CA, USA). Raw sequencing data and variants prioritization were performed as previously described (11). For variants considered as disease-causing segregation analysis in probands' families was performed by amplicon deep sequencing (ADS) performed by Nextera XT Kit (Illumina) and sequenced as described for WES.
In Case 1, ultra-rare variants in LEMD3, RAI1 and TRAF7 genes were prioritized for further validation. Variants in LEMD3, RAI1 were found to be inherited from proband's healthy mother and were classified as benign (data not shown). Whereas, missense heterozygous variant in TRAF7 gene [(hg38, chr16:g.002175915-C>G, NM_032271.3: c.1708C>G, p.(His570Asp)] was absent in both parents (Figure 3A) and considered as de novo event.
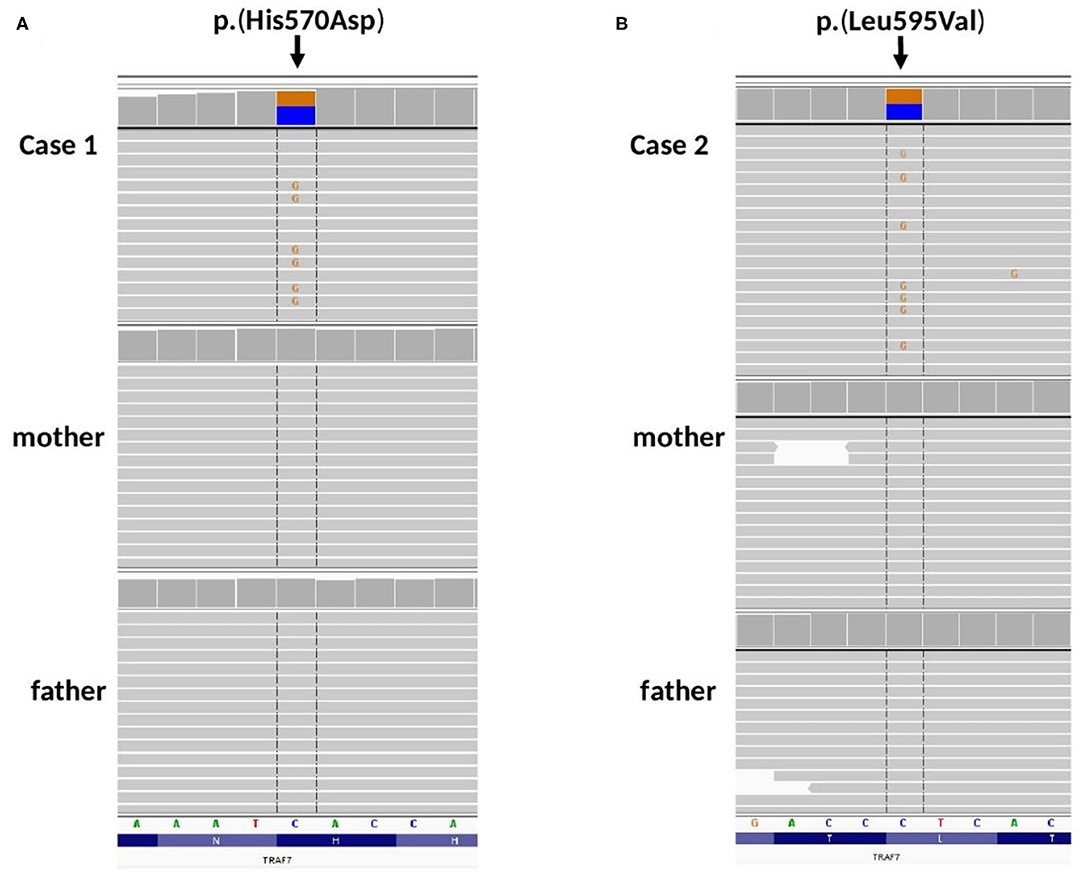
Figure 3. Amplicon deep sequencing validation of TRAF7 gene variants identified by WES in (A) Case 1, de novo p.(His570Asp), (B) Case 2, de novo p.(Leu595Val). Integrative Genomic Viewer screenshots are presented.
In Case 2, ultra-rare variants in CHD1, NAV2, KIF3B and TRAF7 genes were prioritized for segregation study. Variants in CHD1, NAV2, KIF3B were inherited from healthy parents (CHD1 from a father, NAV2, KIF3B from a mother, date not shown) and were considered as benign. Missense heterozygous variant in TRAF7 gene [(hg38, chr16:g.002176085-C>G, NM_032271.3: c.1783C>G, p.(His595Asp)] was absent in both parents (Figure 3B) and considered as de novo event.
Both p.(His570Asp) and p.(Leu595Val) have 0 frequency in all tested databases (including the in-house database of >4,500 Polish individuals examined by WES). According to ACMG classification (15) p.(His570Asp) variant is classified as “Likely Pathogenic,” while p.(Leu595Val) as “Variant of Unknown Significance.” Additionally, c.1783C>G (p.Leu595Val) was predicted as damaging/pathogenic by 16 out of 21 pathogenicity predictors implemented by Varsome (including MutationTaster, SIFT, FATHMM), and as damaging by MetaSVM.
Germline mutations in TRAF7 and the associated clinical symptoms were first described in 2018 by Tokita et al. De novo missense mutations in TRAF7 were found in 7 patients with diagnosed developmental delay, congenital defects and dysmorphic features (2). Castilla-Vallmanya et al., on the basis of the identification of 45 patients, defined this disease entity as TRAF7 syndrome characterized by specific features of facial dysmorphia, especially eyelid fissure defects, congenital heart and skeletal defects, as well as motor retardation and intellectual disability (10). To date, 54 cases of patients with a germline mutation in TRAF7 have been described in the literature (2, 5, 10).
De novo variant p.(His570Asp) identified in Case 1 was previously reported in two independent CAFDADD patients (10). While p.(Leu595Val) identified in Case 2 has not been described in the literature so far.
We present a case of a patient with a de novo heterozygous TRAF7 mutation. The pathogenic nature of this variant and its participation in the clinical picture of the patient is indicated by the characteristic clinical symptoms consistent with the phenotypes of patients with diagnosed TRAF7 germinal mutations (2, 10), the applied bioinformatics programs and the exclusion of its occurrence in both parents.
The phenotype revealed in the patient, and primarily the occurrence of blepharophimosis and epicanthic folds, highlight the intensity of dysmorphic features within the eyes, which is characteristic of the CAFDADD syndrome. In addition, the described features such as broad nasal base, short neck, dysmorphic, asymmetrically placed auricles, and a retracted mandible (retrognathia) complete the picture of typical phenotype features characteristic of TRAF7 germline mutations (2, 10). The skeletal defects, manifested by significant bilateral narrowing of the skull, raise the suspicion of craniosynostosis, which is one of the most frequently described skull defects in patients with TRAF7 mutations (5, 10). Therefore, the CAFDADD syndrome should be included in the spectrum of genetic defect syndromes, in which the neurosurgical assessment of the patient and constant monitoring of their condition and development is of key importance in order to avoid possible complications of craniosynostosis through properly implemented treatment (16–18). The patent ductus arteriosus and patent foramen ovale diagnosed in the patient are typical cardiac manifestations of the CAFDADD syndrome, which indicates the need for constant cardiological supervision (2, 10). Due to the previously described developmental disorders such as psychomotor retardation, intellectual disability and speech disorders, the support of early child development as well as physical rehabilitation, aimed at compensating for frequently occurring muscle tension disorders, is of key importance (2, 5, 10).
Primarily, the CAFDADD syndrome phenotype includes distinctive dysmorphic features in the eye area, which can often be the first abnormalities noticed. Thus, it is of importance for child ophthalmologists to keep the syndrome in mind during the evaluation of the patients with such features having other developmental anomalies (physical as well as delayed psychomotor milestones). The patient's disorders of eye-opening indicated a clinical picture characteristic of ophthalmic forms of myasthenia gravis (19). The exclusion of myasthenia gravis by means of a negative result of the electrostimulation-induced muscle fatigue test led to further diagnostics toward a genetic syndrome with severe ptosis, narrow eyelid cracks and epicanthic folds.
Initially, the blepharophimosis-ptosis-epicanthus invertus syndrome (BPES) was suspected in the presented Case 1. BPES is a complex eyelid defect syndrome characterized by four main features: blepharophimosis, eyelid ptosis, epicanthus invertus and telecanthus (BPES II), as well as premature ovarian failure in BPES I (20). In both BPES and CAFDADD, a palpebral fissure defect is one of the characteristic clinical features. If BPES is suspected, it is necessary to exclude CAFDADD, in which there are both systemic complications, especially the heart and skeletal defects, as well as possible predisposition to cancer development and premature aging, absent in BPES.
In patients with a suspected TRAF7 mutation in the differential diagnosis, Ohdo's syndrome, which is characterized by blepharophimosis, ptosis, congenital disorders of the heart and limbs, and developmental delay, should also be considered (21, 22). In previously described patients, RASopathy group diseases were also suspected, e.g., Noonan syndrome and Costello syndrome (2, 5, 10). Of note, the RASopathy was initially suspected in presented Case 2. The common pathomechanism of RASopathy associated with dysregulation of the Ras/MAPK signaling pathways causes a characteristic clinical picture: features of craniofacial dysmorphia, heart defects, ocular and musculoskeletal disorders (23). The overlapping phenotypes of RASopathy and the CAFDADD syndrome and the suspected involvement of TRAF7 in the regulation of MAPK signaling pathways suggest a possible correlation in the pathomechanisms (1, 2, 5).
Somatic mutations in TRAF7 have been detected in such neoplasms as meningiomas, mesotheliomas, perineural tumors of soft tissues or glandular neoplasms of the genital tract (1, 23). Most of them are missense mutations located within the WD40 domain. The germline mutations in TRAF7 are also mostly clustered within the WD40 domain (1, 10). For this reason, constant supervision in aging patients is required to determine whether they have a higher risk of cancer development and should be included in the oncological surveillance (10).
The detected de novo variant c.1708C>G [p.(His570Asp)] in TRAF7 and the described phenotype correlating with it allow to extend of the genetic spectrum of the very rare CAFDADD syndrome (Cardiac, facial and digital anomalies with developmental delay). Moreover, the presented known variant - c.1783C>G, p.(His595Asp) – support the previous findings concerning the phenotypic spectrum of TRF7 germline variants. A TRAF7 mutation should be suspected in patients with characteristic dysmorphic features, especially within the palpebral fissure (blepharophimosis and/or ptosis), congenital defects of the heart and skeleton, and psychomotor delay. As we proved herein, the clinical manifestation may differ and overlaps with other, more frequent genetic condition as Noonan syndrome (RASopathies). Constant ophthalmic, neurological and cardiological assessment, as well as early development support and motor rehabilitation, are essential in the management of patients with the syndrome resulting from variants in TRAF7.
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Written informed consent was obtained from the parents for the publication of any potentially identifiable images or data included in this article.
Conceptualization was performed by JP and AJ-S. Methodology, formal analysis, investigation, resources, data curation, and project administration were performed by JP, MNo, MNi, IJ, MR, ŚR, RP, and AJ-S. Software by validation was performed by JP, MNo, MNi, IJ, and AJ-S. Writing-original draft preparation, writing-review and editing, and visualization were performed by JP, MNo, MNi, IJ, MR, ŚR, MK, KR, RP, and AJ-S. Supervision was performed by AJ-S. Funding acquisition was performed by JP. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the patients and their parents for their cooperation in this study.
1. Zotti T, Scudiero I, Vito P, Stilo R. The emerging role of TRAF7 in tumor development. J Cell Physiol. (2017) 232:1233–8. doi: 10.1002/jcp.25676
2. Tokita MJ, Chen CA, Chitayat D, Macnamara E, Rosenfeld JA, Hanchard N, et al. De novo missense variants in TRAF7 cause developmental delay, congenital anomalies, and dysmorphic features. Am J Hum Genet. (2018) 103:154–62. doi: 10.1016/j.ajhg.2018.06.005
3. Zotti T, Vito P, Stilo R. The seventh ring: exploring TRAF7 functions. J Cell Physiol. (2012) 227:1280–4. doi: 10.1002/jcp.24011
4. Park HH. Structure of TRAF family: current understanding of receptor recognition. Front Immunol. (2018) 9:1999. doi: 10.3389/fimmu.2018.01999
5. Accogli A, Scala M, Pavanello M, Severino M, Gandolfo C, De Marco P, et al. Sinus pericranii, skull defects, and structural brain anomalies in TRAF7-related disorder. Birth Defects Res. (2020) 112:1085–92. doi: 10.1002/bdr2.1711
6. Tsikitis M, Acosta-Alvear D, Blais A, Campos EI, Lane WS, Sánchez I, et al. Traf7, a MyoD1 transcriptional target, regulates nuclear factor-κ B activity during myogenesis. EMBO Rep. (2010) 11:969–76. doi: 10.1038/embor.2010.154
7. Klein CJ, Wu Y, Jentoft ME, Mer G, Spinner RJ, Dyck PJ, et al. Genomic analysis reveals frequent TRAF7 mutations in intraneural perineuriomas. Ann Neurol. (2017) 81:316–21. doi: 10.1002/ana.24854
8. Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. (2013) 339:1077–80. doi: 10.1126/science.1233009
9. Scudiero I, Zotti T, Ferravante A, Vessichelli M, Reale C, Masone MC, et al. Tumor necrosis factor (TNF) receptor-associated factor 7 is required for TNFα-induced Jun NH2-terminal kinase activation and promotes cell death by regulating polyubiquitination and lysosomal degradation of c-FLIP protein. J Biol Chem. (2012) 287:6053–61. doi: 10.1074/jbc.M111.300137
10. Castilla-Vallmanya L, Selmer KK, Dimartino C, Rabionet R, Blanco-Sánchez B, Yang S, et al. Phenotypic spectrum and transcriptomic profile associated with germline variants in TRAF7. Genet Med. (2020) 22:1215–26. doi: 10.1038/s41436-020-0792-7
11. Smigiel R, Biela M, Szmyd K, Błoch M, Szmida E, Skiba P, et al. Rapid whole-exome sequencing as a diagnostic tool in a neonatal/pediatric intensive care unit. J Clin Med. (2020) 9:2220. doi: 10.3390/jcm9072220
12. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
13. Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. (2015) 24:2125–37. doi: 10.1093/hmg/ddu733
14. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. (2013) 14:178–92. doi: 10.1093/bib/bbs017
15. Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, et al. Modeling the ACMG/AMP variant classification guidelines as a bayesian classification framework. Genet Med. (2018) 20:1054–60. doi: 10.1038/gim.2017.210
16. Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of craniosynostosis. Semin Pediatr Neurol. (2007) 14:150–61. doi: 10.1016/j.spen.2007.08.008
17. Kajdic N, Spazzapan P, Velnar T. Craniosynostosis-recognition, clinical characteristics, and treatment. Bosn J Basic Med Sci. (2018) 18:110–6. doi: 10.17305/bjbms.2017.2083
18. Governale LS. Craniosynostosis. Pediatr Neurol. (2015) 53:394–401. doi: 10.1016/j.pediatrneurol.2015.07.006
19. Nair AG, Patil-Chhablani P, Venkatramani DV, Gandhi RA. Ocular myasthenia gravis: a review. Indian J Ophthalmol. (2014) 62:985–91. doi: 10.4103/0301-4738.145987
20. Méjécase C, Nigam C, Moosajee M, Bladen JC. The genetic and clinical features of FOXL2-related blepharophimosis, ptosis and epicanthus inversus syndrome. Genes. (2021) 12:1–14. doi: 10.3390/genes12030364
21. Verloes A, Bremond-Gignac D, Isidor B, David A, Baumann C, Leroy MA, et al. Blepharophimosis-mental retardation (BMR) syndromes: a proposed clinical classification of the so-called ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet Part A. (2006) 140:1285–96. doi: 10.1002/ajmg.a.31270
22. Campeau PM, Lu JT, Dawson BC, Fokkema IFAC, Robertson SP, Gibbs RA, et al. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum Mutat. (2012) 33:1520–5. doi: 10.1002/humu.22141
Keywords: blepharophimosis, ptosis, developmental delay, TRAF7 variants, facial features, dysmorphic features
Citation: Paprocka J, Nowak M, Nieć M, Janik I, Rydzanicz M, Robert Ś, Klaniewska M, Rutkowska K, Płoski R and Jezela-Stanek A (2021) Case Report: Blepharophimosis and Ptosis as Leading Dysmorphic Features of Rare Congenital Malformation Syndrome With Developmental Delay – New Cases With TRAF7 Variants. Front. Med. 8:708717. doi: 10.3389/fmed.2021.708717
Received: 12 May 2021; Accepted: 27 July 2021;
Published: 26 August 2021.
Edited by:
Filippo M. Santorelli, Fondazione Stella Maris (IRCCS), ItalyReviewed by:
Diego Lopergolo, University of Siena, ItalyCopyright © 2021 Paprocka, Nowak, Nieć, Janik, Rydzanicz, Robert, Klaniewska, Rutkowska, Płoski and Jezela-Stanek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justyna Paprocka, anBhcHJvY2thQHN1bS5lZHUucGw=; Aleksandra Jezela-Stanek, amV6ZWxhQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.