 Hai-Feng Xia
Hai-Feng Xia Xiao-Yu Jia3
Xiao-Yu Jia3 Zong-Jun Du
Zong-Jun Du Da-Shuai Mu
Da-Shuai Mu Guan-Jun Chen
Guan-Jun Chen- 1Marine College, Shandong University, Weihai, China
- 2State key Laboratory of Microbial Technology, Shandong University, Qingdao, China
- 3Department of Pulmonary and Critical Care Medicine, Weihai Municipal Hospital, Cheeloo College of Medicine. Shandong University, Weihai, China
Introduction: Macroalgae contain various polysaccharides that serve as nutrient sources Introduction: Macroalgae contain various polysaccharides that serve as nutrient sources for marine bacteria. Carbohydrate-active enzymes (CAZymes) are the primary feature of marine bacteria that utilize these polysaccharides. In this study, we describe Tamlana sp. S12, a novel strain of marine flavobacteria that can degrade alginate and Laminaria japonica biomass, isolated from the intestines of the sea cucumber Apostichopus japonicas collected at Weihai coast.
Methods: We sequenced the entire genome of strain S12 and constructed a phylogenetic tree using the core genome sequences of related strains. We determined the enzymatic activity of strain S12 using the DNS method and measured its growth curve under different carbon sources using spectrophotometry.
Results: Strain S12 degraded dehydrated L. japonica fragments as the sole nutrient source within 48h. Strain S12 harbors a diverse array of CAZymes at multiple polysaccharide utilization loci (PUL). One PUL encoding lyases from PL6, 7, and 17 families may be used for the degradation of alginate. Additionally, strain S12 harbors PULs encoding carrageenan- and agar-targeting CAZymes. Comparative analysis with related flavobacteria from Algibacter, Maribacter, and Zobellia showed shared CAZymes among these strains, potentially derived from a common ancestor and stably maintained within strains. Genomic signatures, algal degradation ability, and CAZyme patterns suggest that strain S12 has the potential to degrade complex algal polysaccharides.
Conclusion: These results expand our knowledge of CAZymes and enrich our understanding of how marine Flavobacteriaceae adapt to marine algal polysaccharide environments. The availability of the genome of Tamlana sp. S12 will be beneficial for further analyses of marine Flavobacteriaceae.
1 Introduction
In marine ecosystems, significant amounts of organic matter are produced through photosynthesis by pelagic phytoplankton and benthic macroalgae (Hehemann et al., 2014; Arnosti et al., 2021). These organisms are the primary sources of polysaccharides in the marine environment (Arnosti et al., 2021), which are important nutrient sources for marine bacteria. For example, brown algae contain carboxylated polysaccharides (alginate), anionic sulfated polysaccharides (fucoidan), and laminarin, none of which are present in land plants (Zhu et al., 2016).
Polysaccharide-degrading bacteria (PDB) specialize in the degradation of algal polysaccharides and colonization of algal surfaces, which are critical events in the global carbon cycle and algal biomass utilization (Ferrer-González et al., 2021). PDB mainly comprises two classes: gammaproteobacteria in the phylum Proteobacteria and flavobacteria in the phylum Bacteroidetes. Marine bacteria have developed specific degradation mechanisms to utilize these polysaccharides, which differ significantly from the degradation of polysaccharides from terrestrial plants (Chernysheva et al., 2021). Marine flavobacteria are vital in the degradation of these complex polysaccharides (Thomas et al., 2017). Polysaccharide degradation characteristics mainly rely on dedicated genomic regions called polysaccharide utilization loci (PUL), which encode proteins that are necessary for the utilization of a specific polysaccharide (Dudek et al., 2020). Flavobacteria carbohydrate-active enzymes (CAZymes) are typically clustered with susCD genes in PUL for orchestrated uptake and degradation (Grondin et al., 2017). Maribacter, Algibacter, and Zobellia, members of the marine flavobacteria, comprise the core community of the cultivable surface microbiota of brown algae, with the ability to degrade algal polysaccharides (Martin et al., 2015). Cha et al. (2021) compared alginate utilization pathways in culturable bacteria isolated from Arctic and Antarctic marine environments, clarifying the function of alginate utilization pathways in Bacteroidetes. Wolter et al. (2021) compared Maribacter dokdonensis 62–1 with related strains at the genomic level, revealing Maribacter to be a versatile polysaccharide-degrading flavobacterium (Wolter et al., 2021). The study by Chernysheva et al. (2019) demonstrated the specificity of Zobellia in the degradation of algal polysaccharides. Zobellia members have been extensively studied for their utilization of algal polysaccharides such as agar, porphyrin, mannitol, and alginate, improving our understanding of the molecular mechanisms behind this process (Jam et al., 2005; Thomas et al., 2017; Groisillier et al., 2015; Dudek et al., 2020). Brunet et al. (2022) compared the ability of Z. galactanivorans with other Zobellia spp. to decompose large fresh algae, evaluating its unique role in the decomposition of large fresh algae. However, research on the ability of marine Flavobacteriaceae to attack algal tissues is still limited.
The genus Tamlana is classified under the family Flavobacteriaceae, and was first described by Lee (2007). Currently, there are nine species that have been validly published under the genus Tamlana. Tamlana species have been isolated from various sources, including seawater (Yoon et al., 2008; Liu et al., 2015; Jung et al., 2019), algae (Li et al., 2020; Li et al., 2023), marine sediment (Lee, 2007; Romanenko et al., 2014), and the gut of abalone (Cao et al., 2021). Research conducted by Li et al. (2023) has shown that members of Tamlana possess a wide range of CAZymes that are associated with the utilization of algal polysaccharides. Tamlana laminarinivorans and Tamlana haliotis can degrade alginate, Tamlana sargassicola can degrade laminarin, and Tamlana crocina can degrade utilizing ulvan (Li et al., 2023). Furthhermore, production of κ-carrageenase and alginate lyase have been reported (Sun et al., 2010; Mostafa et al., 2020; Yin et al., 2021). These findings suggest a clear preference among Tamlana strains for the utilization of algal polysaccharides.
The sea cucumber, Apostichopus japonicus, is an economically important fishery resource in the coastal areas of East Asia (Zhang et al., 2019). These organisms have a significant impact on the cycling of benthic organic matter by consuming various types of organic debris, bottom sediments, microorganisms, algae, and aquatic animal debris (Wang et al., 2018). Flavobacteriaceae, as a component of A. japonicus intestine microbiota, enhances the utilization of algal polysaccharides (Zhang et al., 2019), thus benefiting the polysaccharide metabolism of A. japonicus.
In this study, the novel marine Flavobacteriaceae strain S12 was isolated from the intestine of A. japonicus. Based on whole genome analysis and physiological characteristics, we classified strain S12 as a member of the genus Tamlana. Analysis also revealed that strain S12 has the potential to utilize various algal polysaccharides, and effectively degrades L. japonica biomass. The degradation of polysaccharides may play an important ecological function in the genus Tamlana (Mcbride, 2014; Jung et al., 2019).
Furthermore, we describe the CAZyme content and hydrolytic capacities of strain S12. In the pan-genomic context, we compared the CAZyme characteristics of Tamlana (including strain S12) and other related members of marine flavobacteria (Algibacter, Maribacter, and Zobellia) to illustrate the role of CAZyme and PUL-related genes in niche specialization among the four genera. Algibacter is most closely related to Tamlana, while Maribacter and Zobellia are widely referenced in research on algal polysaccharides (Wolter et al., 2021; Brunet et al., 2022). The genomic machinery for the degradation of alginate as well as red algal polysaccharides were compared to further understand the utilization ability and degradation mechanism of strain S12 for seaweed polysaccharides. Our findings establish an eco-evolutionary perspective on CAZyme-related niche specialization among marine flavobacteria and its connection to biogeochemical processes.
2 Materials and methods
2.1 Isolation and cultivation
Strain S12 (Yin et al., 2021) was isolated from the intestinal tract of A. japonicus collected from a pond in Weihai, Shandong, China (37°30′7″N, 122°07′24″E) in March 2013. The strain was cultured on marine agar 2216 (MA; Difco, Detroit, MI, USA) at 28° for 72 h. After several rounds of sub-culture, the 16S rRNA gene of the isolate was amplified using PCR with universal primers 27F and 1492R (Jung et al., 2019), following the protocol described in previous reports (Leblond-Bourget et al., 1996). The 16S rRNA sequence of strain S12 was extracted and aligned using EzBioCloud to preliminarily determine its taxonomic status (Yoon et al., 2017).
2.2 Phenotypic tests
Commercial systems API ZYM, 20NE, and 50CHB (BioMérieux) were used to evaluate the following enzymatic activities: Amylase (Starch), Esterase (C4) (2-naphthyl butyrate), α- Galactosidase (6-Br-2-naphthyl-αD-galactopyranoside), N-acetyl-β-glucosamindase (1-naphthyl-N-acetyl-βD-glucosaminide), Lipase (C14) (2-naphthyl myristate), α- Fucosidase (2-naphthyl-αL-fucopyranoside), α-Chymotrypsin (N-glutaryl-phenylalanine-2-naphthylamide), Cystine arylamidase (L-cystyl-2-naphthylamide), α- Mannosidase (6-Br-2-naphthyl-αD-mannopyranoside). Bacterial suspensions in a 3% NaCl solution (w/v) were inoculated onto the test plates and cultivated at 28° according to the instructions. To prepare the enzymatic reaction, 200 μL of a 1% solution of beechwood xylan (Megazyme International Ireland, Bray, Ireland) or a 0.2% solution of agar (sigma-aldrich, USA) was mixed with 300 μL of cell-free supernatant obtained from strain S12 fermentation broth after 24 hours of growth in MA medium. The supernatant was obtained by centrifugation at 12000 rpm for 6 minutes. The substrate and enzyme mixture was thoroughly mixed before initiating the enzymatic reaction. The reaction was conducted at 28°C for 30 minutes, and the change in reducing sugar before and after the reaction was determined using the DNS method. As a reference strain, Tamlana agarivorans KCTC 22176 was tested under the same conditions as strain S12. Alginate utilization was analyzed in optimized basic salt medium (OBSM; 1 L distilled water, 25 g NaCl, 2 g K2HPO4, 2 g MgSO4·7H2O, 0.1 g FeSO4·7H2O, nitrogen source: 5 g NaNO3, pH 7.2 – 7.4). Additionally, 15g of sodium alginate (Cat. no. A2158; Sigma-Aldrich, St. Louis, MO, USA) was supplemented to achieve a concentration of 1.5% in the medium. To assess glucose utilization in strain S12, OBSM was utilized with a final concentration of 1.5% glucose. The seed solution was produced from a single colony grown in marine broth 2216 (MB) for 16 hours at 28° with an inoculation rate of 1% (v/v). L. japonica utilization was analyzed in a culture medium prepared from a mixture of 1% (w/v) dehydrated L. japonica and sterile seawater. The strains used in this experiment (except strain S12) were purchased from the Korean Collection for Type Cultures, the Japan Collection of Microorganisms, and the German Collection of Microorganisms and Cell Cultures (DSMZ). The information on experimental strains is listed in Table S1. One milliliter of the seed solution was collected, centrifuged (10000 rpm, 2 min), and washed with sterile seawater 3 times. It was then adjusted to an optical density measured at 600 nm (OD600) of 0.1. The medium was inoculated with 1% (v/v) washed bacteria in triplicate, followed by cultivation at 28°C and 100 rpm for 120 h with regular photometric measurements (diluted if OD600 >0.4). L. japonica residues were well precipitated by low-speed centrifugation at 4000 rpm for 5 seconds was used to precipitate L. japonica residues and remove impurities while the bacterial cells remained suspended.
2.3 Biofilm measurement
To determine biofilm formation, 100 μl of 2216E liquid medium (Qingdao Hope Bio-technology Co., Qingdao, China) and OBSM+1.5% alginate solution were added into the wells of a polystyrene microplate. Next, 10 μl of an overnight culture of strain S12 in 2216E liquid medium was inoculated into each well. The plate was then incubated at 28°C for 36 h. After discarding the contents of the wells, 200μl of sterile water was used to wash the wells and remove non-adherent bacteria. The biofilm was fixed with 250 μL of 96% ethanol and air-dried. Then, 100 μl of 1% (v/v) crystal violet (Merck, Germany) was added to each well for staining for 5 minutes. After washing off the excess dye, the plate was air-dried, and then 100 μl of 33% (v/v) glacial acetic acid was added to dissolve the crystal violet. The washed samples were measured for OD570, and the average value was taken as the test value (Gomes et al., 2013). The un-inoculated bacterial medium was used as a negative control, and the negative value was taken twice as the limit value.
2.4 Genome sequencing and taxonomy
Genomic DNA was extracted using a DNA extraction kit (TaKaRa Bio, Shiga, Japan) following the manufacturer’s instructions. DNA sequencing was performed using both the Illumina HiSeq and SMRT platforms. The Illumina HiSeq platform was used to construct the Illumina PE library, while the SMRT platform was used for Pacbio library construction. Subsequently, the sequencing data obtained from the Illumina HiSeq platform was utilized to refine the assembly of the SMRT platform-generated data. This optimization procedure resulted in an improved quality of the assembly results. (Mu et al., 2020). The genomic DNA was fragmented using a 26G needle, and fragments > 20 kB were selected by the BluePippin system (Sage Science, Beverly, MA, USA) to prepare the DNA library. Quality control was performed using fastp (quality value threshold: 30, filter length threshold: 50bp) for Illumina and mecat2 (default parameter) for PacBio Sequel (Xiao et al., 2017). After quality control of the samples, de novo genome assembly was performed using sequencing data from two platforms with Unicycler (https://github.com/rrwick/Unicycler), followed by error correction using Arrow (Archibald, 2017), and gene prediction using Glimmer3 and Prodigal (Delcher et al., 2007; Hyatt et al., 2010). The complete genome sequence is available at the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/) under NZ_CP068547.
A phylogenetic analysis of 48 selected species (Supplementary Table S1) based on 120 core genes using GTDB-Tk (Chaumeil et al., 2019) was performed, including Flavobacterium aquatile ATCC 11947 as an outgroup, with the remaining strains derived from Tamlana, Algibacter (the genus most closely related to Tamlana), and Maribacter and Zobellia (which are widely referenced in research on algal polysaccharides). Strain genome assemblies were obtained from the NCBI GenBank facility, and the genome accession numbers of strains were listed in Table S1. RAxML (version 8.2.11) was used to construct a maximum likelihood (ML) phylogenetic tree with 1,000 bootstrap replicates (Tian et al., 2020), using the general time reversible-GTR was used as the model for DNA sequence evolution. An interactive tree of life (iTOL) (https://itol.embl.de) was used for phylogenetic tree display and annotation (Letunic and Bork, 2021).
Phylogenetic analysis of the PL6, PL7, and PL17 was performed using MEGA-X (version 10.1.8). Multiple amino acid sequence alignments were conducted using Clustal W (Kumar S. et al., 2018). Three methods were used to verify the phylogenetic tree, including the Jones–Taylor–Thornton (JTT), p-distance, and subtree-pruning-regrafting (SPR) models, which were used to construct the ML (Chatukuta and Rey, 2020), neighbour-joining (NJ) (Malik et al., 2021), and maximum parsimony (MP) trees (Kumar A. et al., 2018), respectively. If there were no obvious differences, the NJ tree was shown.
2.5 Genome comparison
The bioinformatic software used to compare the genomes of strain S12 and related strains (Supplementary Table S1) included Pyani (https://github.com/widdowquinn/pyani) (Mu et al., 2020) for calculating average nucleotide identities (ANIs), and Genome-to-genome distance calculator 3.0 (GGDC) (https://ggdc.dsmz.de/home.php) (Meier-Kolthoff et al., 2022) for digital DNA-DNA (dDDH) hybridization. The dDDH comparison was performed between the genome of strain S12 and all others from Tamlana, with a species boundary threshold of 70% using GGDC (Meier-Kolthoff et al., 2022). Pan-genome analyses were conducted using the Bacterial Pan Genome Analysis (BPGA) tool v1.3 (Chaudhari et al., 2016), with clustering performed using USEARCH with a 50% sequence identity cut-off. Genomic analysis results were obtained by identifying Cluster of Orthologous Groups of proteins (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. The BPGA tool was also used to identify genes whose GC % deviated from the average GC % of strain S12 by more than twice the value, and to calculate the GC % of protein-coding genes of strain S12.
CAZymes were identified using blastp and dbCAN2 (Zhang H. et al., 2018), and genomes were annotated by HMMER: dbCAN (E-Value < 1e-15, coverage > 0.35), DIAMOND: CAZy (E-Value < 1e-102), and HMMER: dbCAN-sub (E-Value < 1e-15, coverage > 0.35). The density of alginate lyase genes was determined by calculating the number of alginate lyase genes present in every 1000 open reading frames (ORFs). Gene annotations were done on the CAZy database and UniProtKB-SwissProt database, and PUL boundaries were determined based on the gene annotation results and the analysis of similar PULs in the PULDB (Lombard et al., 2014; Terrapon et al., 2018; UniProt Consortium, 2021). Genes were assigned to the KEGG classes and pathways using the KofamKOALA and KEGG Mapper (Aramaki et al., 2020; Kanehisa and Sato, 2020).
PUL comparison was analyzed using PULDB, and protein function was analyzed using PDB (Burley et al., 2018). Pairwise comparisons of PULs were performed using the BLASTn (BLAST version 2.11.0+) run in EasyFig (version 2.2.5) (Sullivan et al., 2011). SignalP v5.0 was used to predict the signal peptides (Almagro Armenteros et al., 2019).
2.6 Statistical analysis
Barbato et al. (2022) identified polysaccharide lyases (PL) and glycoside hydrolases (GH) as the primary enzymes responsible for degrading algal polysaccharides. Table S2 presents data on PLs + GHs, while Table S1 shows the strain codes. Principal coordinate analysis (PCoA) was conducted at the CAZymes level using the Bray-Curtis distance. The Bray-Curtis matrix distance was built by log-transforming the relative abundance of PLs + GHs. The PCoA plot was generated using ImageGP (Chen et al., 2022). Finally, a grouped percentage stacked bar chart was used to illustrate the relative proportion of different CAZyme components in various strains and genera. This chart displays the percentage contribution of each CAZyme component across different bacterial strains and genera.
3 Results and discussion
3.1 Genomic features and taxonomic analysis of Strain S12
Strain S12 was isolated from the intestine of A. japonicus. Colonies on the solid medium were round, smooth, and yellowish. The strain S12 has a whole genome sequence of 3.57 Mb with a GC content of 39%, which includes 224 predicted CAZymes (Supplementary Table S1), corresponding to 7.1% of all protein-encoding genes. This value was higher than the average value for Tamlana (6.7%). According to genome annotation, three 16S rRNA gene copies were found in the genome of strain S12, all of which were very similar to the 16S rRNA gene of Tamlana agarivorans JW-26T (accession EU221275). The sequence identities, lengths, and coverage were 99.2% (1519 bp, 97% cover), 98.8% (1521 bp, 97% cover), and 98.7% (1521 bp, 97% cover), respectively. The ANI and dDDH between strain S12 and T. agarivorans JD-26 were 95.0% and 67.7% (Supplementary Figure S1), respectively. Core genome-based phylogeny revealed clear assignment to Tamlana within Flavobacteriaceae (Figure 1). According to the genomic taxonomy parameters, strain S12 might represent a novel species of Tamlana.
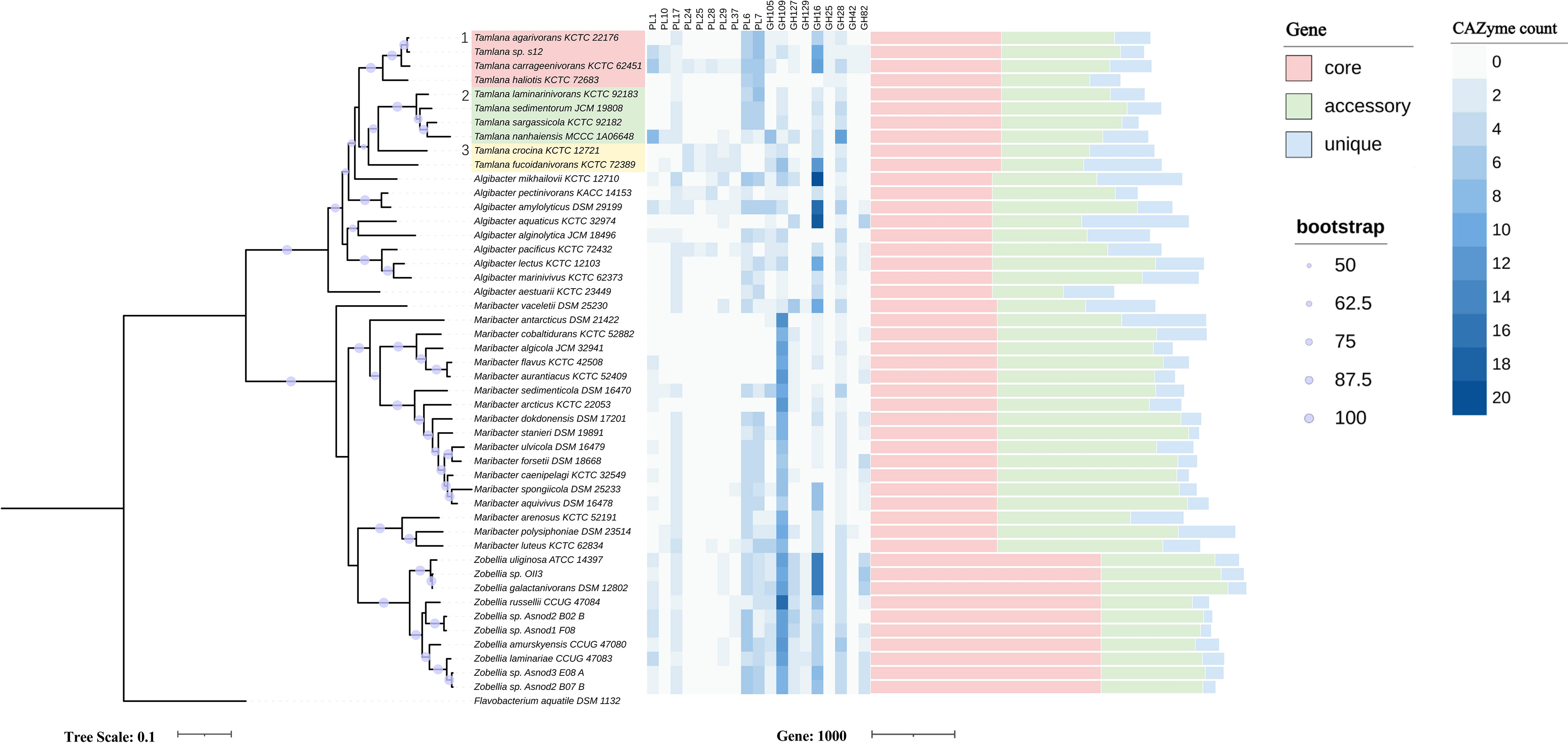
Figure 1 Maximum-likelihood phylogenetic tree based on 120 core genes (left panel), numbers of genes from selected carbohydrate-active enzymes (CAZyme) families (middle panel) and pan-genome (right panel) in Tamlana sp. S12 and related strains. Flavobacterium aquatile DSM 1132 served as outgroup. The three resolved Tamlana lineages are numbered and colored. PL, polysaccharide lyase; GH, glycoside hydrolase.
3.2 Interspecific difference comparison and Phylogenetic reconstruction in the context of CAZymes
To compare the differences in CAZyme composition between strain S12 and other related flavobacteria, 47 publicly available genomes from the four genera were compared using phylogenetic analysis in the context of CAZymes. The four genera include Tamlana, to which strain S12 belongs, Algibacter, which is most closely related to Tamlana, as well as Maribacter and Zobellia, which are widely referenced in research on algal polysaccharides. In terms of protein-encoding genes, Maribacter exhibited the lowest average proportion (5.4%) of CAZyme-encoding genes compared to the other three genera, while Algibacter (7.1%) and Zobellia (7.5%) had the highest average proportions (Table 1; Supplementary Table S1). We further calculated the standard deviation of the proportion of CAZyme among the four genera members and found that the deviation of Zobellia spp. was the smallest (0.4%), and the deviation of the other three genera exceeded 1%. Zobellia spp. showed a stable proportion of CAZyme-encoding genes, whereas the other three genera members displayed higher variability in this regard.
Table 1 CAZyme families encoded by Tamlana sp. S12.
The PCoA analysis demonstrated differences in the CAZyme composition between strains (Figure S2). According to PCoA grouping, Tamlana and Algibacter exhibited similar CAZyme profiles. Conversely, Zobellia and Maribacter strains formed clusters and displayed distinct CAZyme profiles between the two genera. These findings are consistent with their evolutionary relationships, as supported by the phylogenetic tree (Figure 1).
We analyzed CAZyme patterns in the context of phylogenetic relationships (Figure 1, Supplementary Table S2). Core genome-based phylogeny resolved three lineages of Tamlana (Figure 1). Lineages 1 and 2 exhibited different PLs compared to lineage 3, including PL6, 7, and 17, which are involved in alginate degradation. Moreover, PL1 and 10 encoding pectinases were identified in lineages 1 and 2 concurrently, while PL10 was not detected in most genomes outside lineages 1 and 2. GH25 encoding lysozyme was exclusively present in lineage 1. In contrast, PL24, 25, 28, 29, and 37 encoding ulvan lyases were common features of lineage 3 (Figure 1).
PL7 and PL6 accounted for the highest proportion of CAZymes in Tamlana, while GH16 and GH2 proportions dominated in Algibacter. The proportion of GH109 in the CAZymes of Zobellia and Maribacter was higher than that in other genera (Figure 2A). GH109 is involved an NAD+-dependent hydrolysis mechanism that cleaves N-acetylgalactosamine residues from various substrates, such as glycolipids, glycopeptides, and glycoproteins (Malinski and Singh, 2019; Gavriilidou et al., 2020). These substrates are abundant in the macroalgal matrix (Kalisch et al., 2016). GH109 in Zobellia and Maribacter may provide adaptive mechanisms for these substrates.
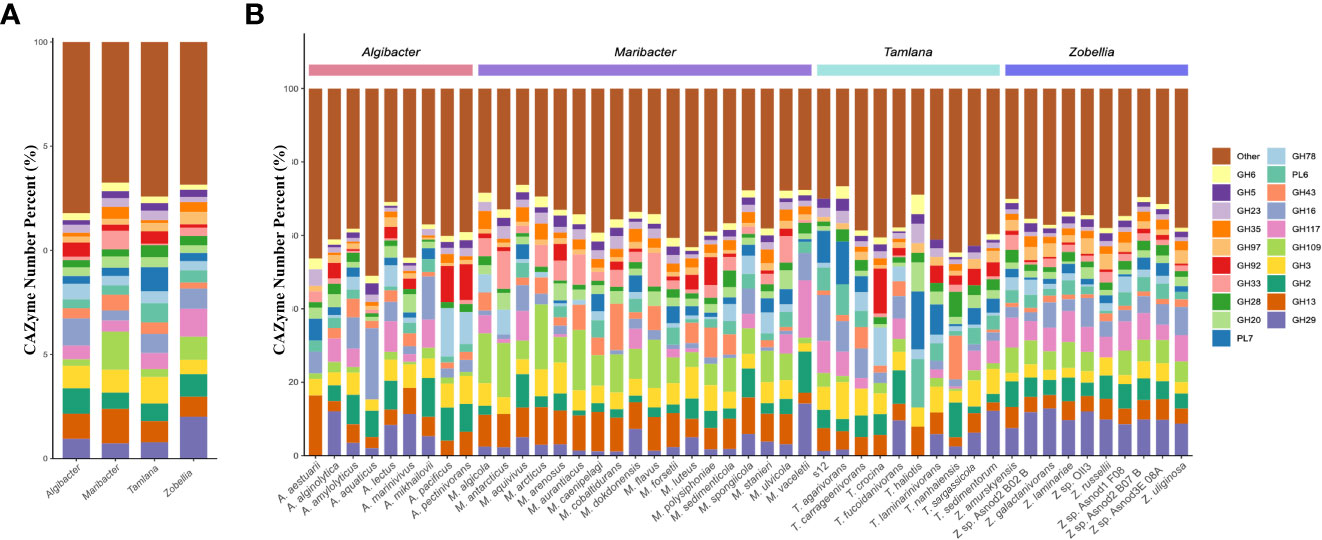
Figure 2 Comparison of glycoside hydrolase (GH) and polysaccharide lyase (PL) families composition diversity in the four genera studied. (A) The histogram and (B) showed the proportion of 20 PL and GH families with the highest abundance at the level of species and genera. Different color represents different CAZyme families.
3.3 Pan-genomic analysis of the four genera
The pan-genome refers to the complete genomic repertoire of a given phylogenetic clade, encoding all possible lifestyles of its organisms (Tian et al., 2016). To compare the genomic information of the four genera, we analyzed of the pan-genomes of each genus using BPGA.
In an attempt to understand the relationships between the pan-genome sizes and number, the pan- genomic numbers combined with the phylogenetic tree are shown in Figure 1, and the data are shown in Supplementary Table S3. The number of new orthologue clusters is directly proportional to the number of genomes, indicating that the four genera have open pan-genomes (Figure S3, Supplementary Table S4). The four genera share only a limited number of genes (833 core genes), with an average percentage of 23.7% for the core genes. Pan-genome analysis revealed that Zobellia had the highest number and average proportion of core genes (2614, 65.2%) and the lowest average proportion of unique genes (5.0%) in terms of the pan-genome. A large average proportion of accessory genes was found in the members of Maribacter (50.0%), while the average proportion of accessory genes in the Tamlana and Algibacter was relatively similar (38.8%, 39.8%). Compared to the other two genera, Tamlana and Algibacter were relatively similar, with a higher average proportion of unique genes (14.8%, 19.4%) (Supplementary Table S3). A significant number of hypothetical genes were identified among the unique genes found in Tamlana. This may be due to the fact that not all HGT events may have biological significance (Arnold et al., 2022).
We also utilized BPGA to identify and extract genes whose GC content deviated from the average GC content of strain S12 by more than twice the standard deviation (Chaudhari et al., 2016). These genes are typically acquired through HGT (Liu et al., 2009; Ravenhall et al., 2015; Chaudhari et al., 2016). We then calculated the proportion of genes with atypical GC content in the unique genome, core genome, and accessory genome of the strains. Our findings revealed that the unique genome had the highest proportion of genes with atypical GC content (Supplementary Table S3), which was significantly higher than their proportion in the core genome and accessory genome. However, the deviation between species was substantia (Supplementary Table S3).
Among the genes with atypical GC content, one (WP_068604863.1) showed 95.2% amino acid identity towards a carrageenase GH16 gene (WP_102994428) from T. carrageenivorans. To explore the homology of the two sequences, we selected 18 sequences with the highest amino acid sequence identity from NCBI to construct a phylogenetic tree. These two sequences clustered together in the same branch (Figure 3A). Although Formosa haliotis and T. agarivorans are distantly related in Flavobacteriaceae (Tanaka et al., 2015; García-López et al., 2019), GH16 sequences from these species (WP_066225175.1 and WP_216067801.1, respectively) are homologous (70.96% amino acid identity). One PUL-encoded protein from the putative GH30 family with atypical GC content was identified in the genome of strain S12. According to NCBI annotation, two GH30s in PUL were identified as glucosylceramidase, which are speculated to play a role in transglycosylation and synthesizing oligosaccharide and glycoconjugate (Ben Bdira et al., 2018). The homologous gene clusters also existed in T. carrageenivorans (NCBI assembly GCA_002893765.1) (>95%) and Hyunsoonleella jejuensis (NCBI assembly GCA_000060345.1) (>75%) (Figure 3B).
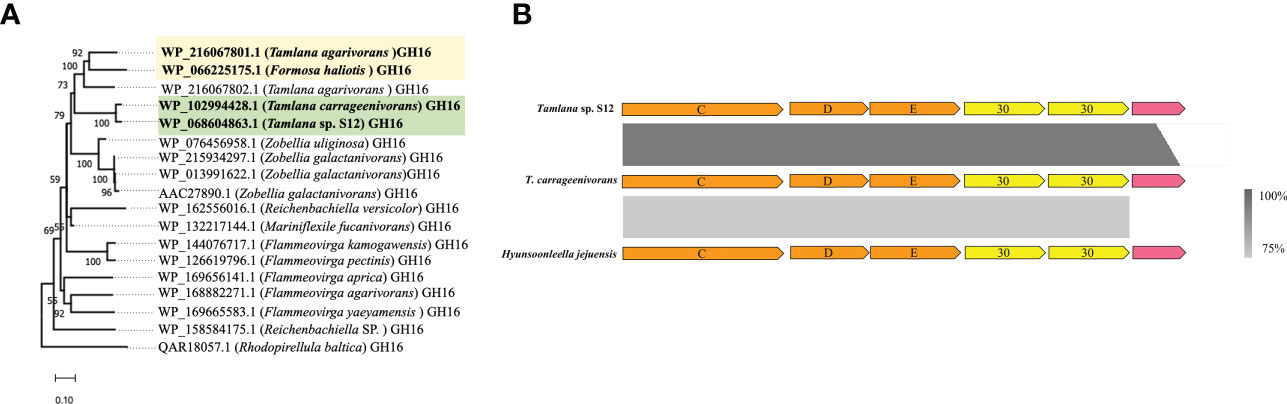
Figure 3 Detection of possible horizontal gene transfer (HGT) candidate genes using atypical GC% content and sequence homology analyzis. (A) Phylogenetic tree constructed with two GH16 (WP_102994428 and WP_216067801.1) and their closest amino acid sequences. For proteins, locus tags are listed. The organism names are listed in brackets. Bootstrap values lower than 50 are not indicated. Colored boxes mark the HGT candidate and their closest homologous protein sequences. (B) Comparison of three homologous polysaccharide utilisation loci (PUL). From top to bottom: a PUL with atypical GC% genes including two GH30 detected in Tamlana sp. S12 (top) and homologous clusters in T. carragenivorans (center) and Hyunsoonleella jejuensis (bottom).
A cluster for the biosynthesis of exopolysaccharides (EPS) was found in strain S12 (Figure 4A, Supplementary Table S5). The EPS biosynthesis gene polyprenyl glycosylphosphotransferase (JJL45_RS08315) and O-antigen (JJL45_RS08305) were identified as core genes, indicating the existence of EPS in Tamlana. Five unique genes in the gene cluster are labelled in Figure 4A. Genes with atypical GC content were identified as members of GT2 (JJL45_RS08185) and GT4 (JJL45_RS08285). Based on the analysis, EPS production may be an adaptive feature among Tamlana strains, potentially mediating distinct surface adhesion and host interaction (Decho and Gutierrez, 2017).
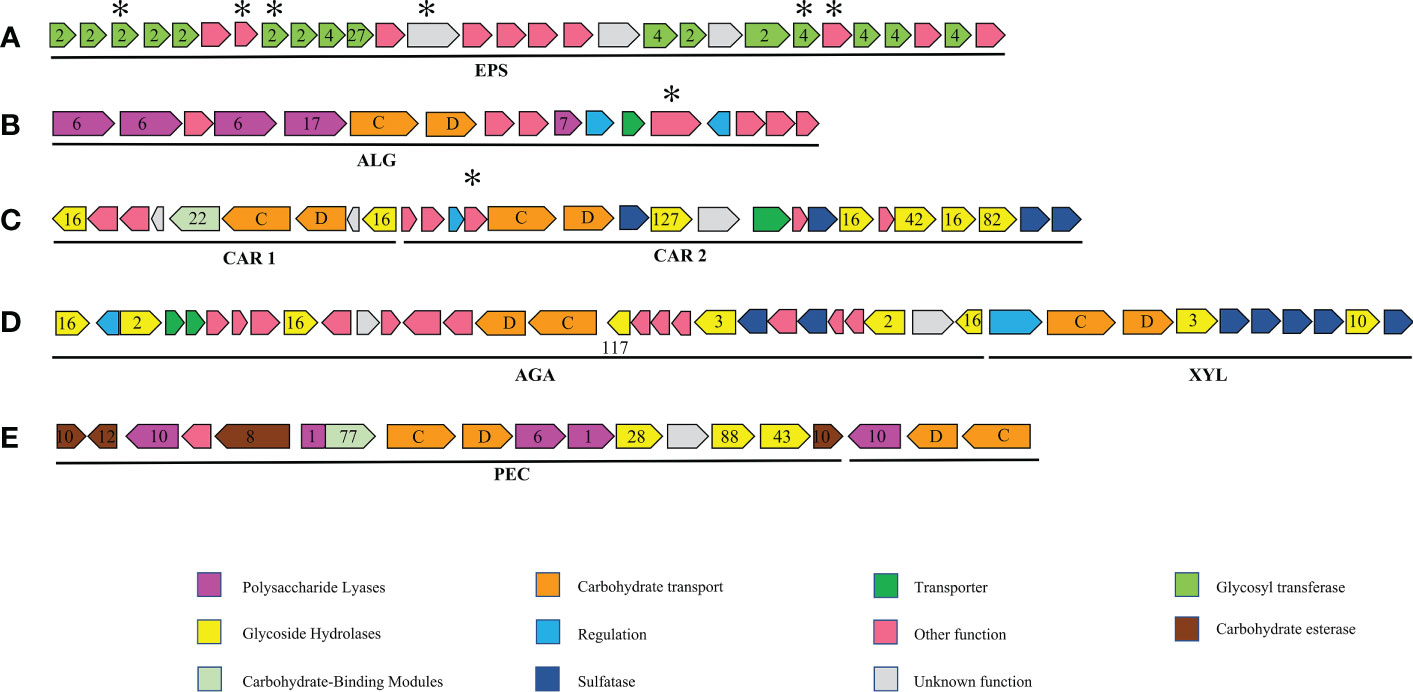
Figure 4 Major polysaccharide utilisation loci (PUL) and gene clusters mentioned in this study. numbers designate CAZyme families, letters susCD genes. and asterisks, unique genes of strain S12. (A) Exopolysaccharide (EPS) gene cluster: putative exopolysaccharide biosynthesis gene cluster only in Tamlana sp. S12, T. carrageenivorans, T. agarivorans; (B) ALG PUL: putative PUL with alginate hydrolysis potential, similar gene clusters also exist in T. haliotis, T. agarivorans; (C) CAR PUL1 and CAR PUL2: putative PUL with carrageenan and porphyrin hydrolysis potential, no other similar gene clusters have been found in other genomes of the four genera; (D) XLY PUL and AGA PUL: putative PULs with hydrolysis potential of xylan and agar, a similar gene cluster also exists in T. agarivorans; (E) PEC PUL: putative PUL with pectin hydrolysis potential, no other similar gene clusters have been found in other genomes of the four genera.
3.4 Alginolytic PUL in strain S12
We compared the genome sizes, PUL numbers, and the number and density of alginate lyase genes (Figure 5). Compared to the other 46 strains, strain S12 had a smaller genome size (Figure 5A), and PUL density (Figure 5D), but it displayed a high density of alginate lyases (Figure 5B), indicating specialization toward alginate. The growth curve of strain S12 in various culture media showed growth with 1.5% alginate (w/v) as sole nutrient source (Figure 6). This concentration is much higher than typically used concentrations (0.05 to 0.4%), confirming its ability to utilize alginate (Figure 6). Strain S12 exhibited delayed growth with glucose after being inoculated from the MB seed culture (Figure 6B). This was potentially linked to removal of accumulated mRNA before switching to glucose metabolism (Lin et al., 2011; Ammar et al., 2018; Iosub et al., 2021). This is also supported by the fact that pre-culturing in OBSM+1.5% glucose prior to transfer weakened the growth delay phenomenon of strain S12 caused by glucose. Further research is needed to explore this phenomenon.
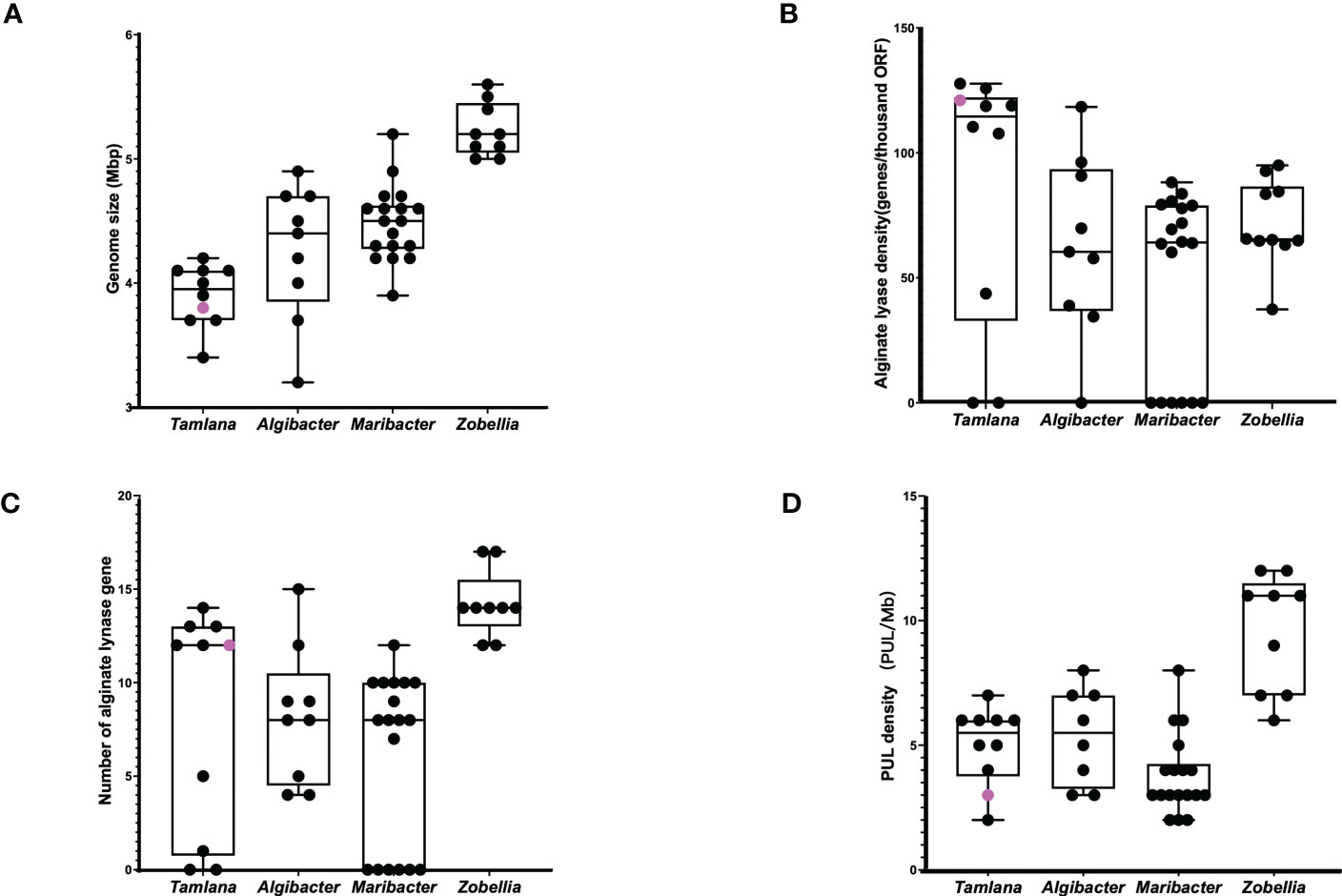
Figure 5 (A) Genome size, (B) alginate lyase density, (C) alginate lynase numbers and (D) PUL density of Tamlana sp. S12 (pink point) and related strains (black point).
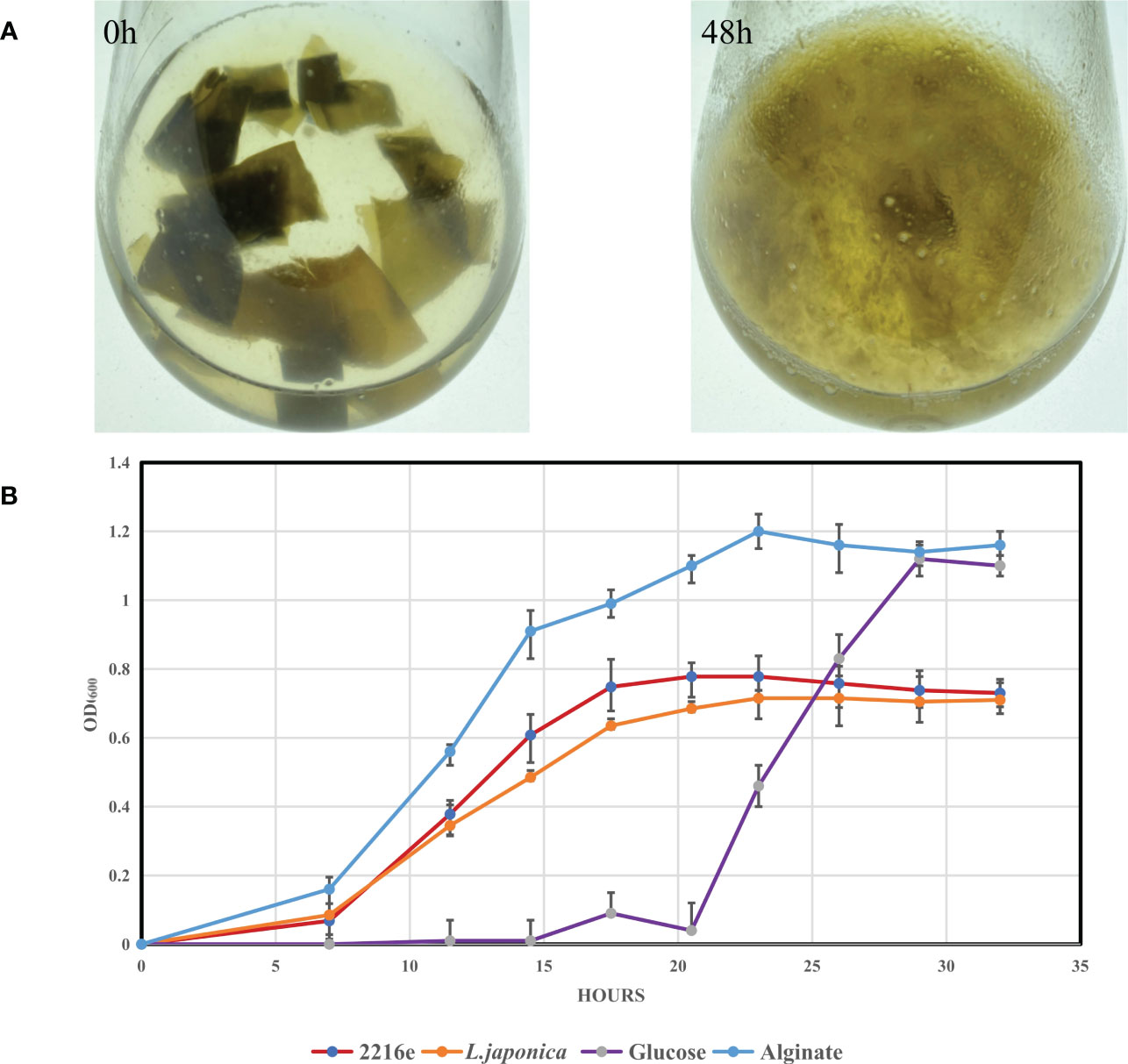
Figure 6 Degradation of L. japonica by Tamlana sp. S12 and growth analysis of Tamlana sp. S12 on different mediums. (A) Degradation of L. japonica by strain S12 in minimal medium with L. japonica pieces at 0 h and 48 h post-inoculation, (B) Shows the growth curve of strain S12 in four medium.
Strain S12 is characterized by high proportions of PL7 and PL6 (Figure 2B), which are polysaccharide lyases specialized towards alginate. The genes encoding these alginate lyases are present in one PUL, named ALG PUL (Figure 4B), while seven additional single alginate lyase genes are dispersed throughout the genome. ALG PUL contains five adjacent lyase genes, PL6-6-6-17-7, which co-localize with a susCD pair. T. laminarinivorans, T. sargassicola, and T. sedimentorum all contain two similar PULs related to alginate utilization. These PULs contain a similar set of CAZymes as ALG PUL, consisting of PL6, 7, and 17 (Li et al., 2023). However, their gene cluster structure and arrangement differ significantly from ALG PUL, which is consistent with the lineage and phylogenetic tree results (Figure 1).
The ALG PUL encodes a metabolic cascade that involves the complete breakdown of external polymers (PL6 and 7) and hydrolysis of oligosaccharides (PL17) to monomers. The interactions between PLs with catalytic diversity may facilitate the cleavage of alginate. The signal peptide in the 5 PLs (Supplementary Table S6) indicates that one PL6 (JJL45_RS04310) is active intracellularly, while the others are secreted extracellularly. This complementary localization may contribute to the efficient degradation of alginate.
According to genomic analysis, it is hypothesized that strain S12 depolymerizes alginate in a stepwise manner using extracellular lyases PL6 (JJL45_RS04315), PL6 (JJL45_RS04325), and PL7 (JJL45_RS04355). Surface PKD (protein kinase domain) and susD proteins facilitate the recruitment of oligosaccharides from the environment and transfer them to the periplasm via susC. There, PL17 alginate lyases (JJL45_RS04330) further degrade the oligosaccharides to unsaturated monoaldehydes. The major facility superfamily permease (JJL45_RS04365) (Grondin et al., 2017) transfers alginate oligomers and unsaturated monouronates to the cytoplasm. S12 does not encode a kdgF gene, indicating that unsaturated monouronates are converted into 4-deoxy-l-erythro-5-hexoseulose uronate (DEH) via an alternative (possibly non-enzymatic) mechanism. (Hobbs et al., 2016; Nakata et al., 2019). Subsequently, short-chain dehydrogenase (dehR) and 2-keto-3-deoxy-d-gluconate kinase (KdgK) catalyze their conversion in the cytoplasm. Finally, 2-dehydro-3-deoxyphosphogluconate (KDPG) is assimilated into the central metabolic pathway through the Entner–Doudoroff pathway. All relevant enzymes and pathways have been annotated in strain S12 (Supplementary Table S5).
According to the sequence identity of catalytic domains, the PL7 family is subdivided into seven subfamilies (SF1–SF7) (Lombard et al., 2010; Thomas et al., 2013; Pei et al., 2019; Chernysheva et al., 2021). Similarly, PL17 and PL6 are divided into two (SF1–SF2) and three (SF1–SF3) subfamilies, respectively (Mathieu et al., 2018; Violot et al., 2021). To classify the predicted alginate lyases from the four genera within the ALG PULs, three phylogenetic trees were constructed using characterized PL6, 7, and 17 alginate lyases available in the CAZy database. The representative alginate lyases for each subfamily, along with their target sequences (Figure S4), were included in the analysis.
The phylogenetic tree revealed that seven out of eight Tamlana representatives encoded PL7-SF6 lyases, which clustered together as presumptive orthologues. T. nanhaiensis PK35_RS15195, T. laminarinivorans LG649_RS00180, T. sargassicola LG651_RS06140, and T. sedimentorum PW52_RS01745 were classified as PL7-SF5 lyases (Figure S4A). Only PL7 from ALG PUL was classified as SF6. To date, only two enzymes in PL7-SF6 have been studied and characterized as endolytic enzymes with a preference for polymannuronate (Inoue et al., 2014; Pei et al., 2019; Chernysheva et al., 2021).
Three PL6 alginate lyases from strain S12 belonged to SF1 and SF2 (Figure S4B), which clustered together with those of T. agarivorans, T. carrageenivorans, and T. haliotis as presumptive orthologues. Twenty-two PL6 alginate lyases from Zobellia and Maribacter fell strictly into SF1. Mathieu et al. (2016) reported that PL6-SF2 has strict endo-poly-MG lyase activity, whereas PL6-SF1 has either endo-poly-MG/exo-poly-G lyase, endo-poly-MG/endo-poly-G lyase, or strict exo-poly-G lyase activity. The combination of different PL6 subfamilies in the ALG PUL may improve degradation efficiency and substrate adaptability.
PL17 (JJL45_RS04330) alginate lyases from strain S12 showed high amino acid identity (80.8–96.7%) with those of T. agarivorans, T. carrageenivorans, and T. haliotis (A6I66_RS06905, C1A40_RS05220, and F6U93_RS07740) (Figure S4C) and were classified as SF2. However, due to the lack of nearly all the required catalytic and functionally critical residues, PL17-SF2 showed activity towards oligosaccharides containing an unsaturated sugar at the nonreducing end (Jouanneau et al., 2021). Hence, PL17 on ALG PUL was considered to have exolytic oligoalginate lyase activity. PL17 alginate lyases from T. haliotis F6U93_RS07745 and A. marinivivus GQR97_RS04345 formed a separate branch in addition to PL17-SF1 which may represent a new subfamily. This branch was consistent through the three phylogenetic algorithms (ML, NJ, MP).
Alginate lyases from the PL7 and PL17 families have mainly been isolated from marine organisms (Hehemann et al., 2014; Xu et al., 2020). PL7 from SF6 appears to be conserved only in marine representatives of the Flavobacteriaceae (Chernysheva et al., 2021), and the results also supports this finding.
3.5 Other PULs related to polysaccharide utilization
The genes encoding putative carrageenases and porphyranase were found in two adjacent large gene clusters (one ∼19,000 and ∼28,000 bp), which are dedicated to carrageenan and porphyrin degradation (Figure 4C). We have named these two gene clusters CAR PUL1 and CAR PUL2, respectively, which encode carrageenolytic and porphyranolytic enzymes (Figure 4C; Supplementary Table S5). The identity level between the putative GHs encoded by CAR PULs and sequences in the Protein Data Bank or Swiss-Prot databases ranged from 24% – 97% (amino acid identity).
Their systems for degrading polysaccharides were represented by GH16.Two putative GH16 (JJL45_RS04765, 04640) showed 62.5% and 45.7% amino acid identity to GH16 κ-carrageenases (ZOBGAL_RS23455) from Z. galactanivorans. Additionally, one putative GH82 (JJL45_RS04770) displayed 44.6% identity towards ι-carrageenases from Z. laminariae (D9V96_RS20855). GH16 sequences (JJL45_RS04680, 04745) were queried against the sequences with known functions in the Swiss-Prot Database, revealing 25.2% and 24.7% identity, respectively, to β-porphyranases from Z. galactanivorans (ZOBGAL_RS04805 and 12330). Blastp analysis also detected these two putative β-porphyranases in Seonamhaeicola marinus (NCBI Sequence Number: WP_148539743.1, WP_148539726.1) (77.9% and 74.1% identity, respectively). However, they were not detected in the genomes of the other members of the four genera, suggesting a potential genetic material transfer event between Seonamhaeicola and Tamlana.
BLASTp analysis revealed that JJL45_RS04760 was conserved in T. carrageenivorans, T. agarivorans, and T. fucoidanivorans (80.4%–97.0% amino acid identity). Based on the PDB database, the protein sequence of JJL45_RS04760 was found to be similar to those of β-galactosidases characterized as GH42, with a high amino acid identity (77.3%) compared to the S. marinus sequence (NCBI Sequence Number: WP_187388126.1). A BLASTp search also identified sequences similar to GH42 in several marine bacteria, including Paraglaciecola hydrolytica S66T (NCBI Sequence Number: WP_068375692.1) (60.9% amino acid identity), which encodes β-galactosidasecan (Schultz-Johansen et al., 2018), suggesting the presence of oligosaccharide enzyme activity of GH42.
Z. galactanivorans was found to harbor three genes encoding GH127 with α-1,3-(3,6)-anhydro-D-galactosidase activity, which play a role in the degradation of carrageenan (Ficko-Blean et al., 2017). JJL45_RS04720 (Supplementary Table S5) showed homology with the three GH127 from Z. galactanivorans (ZOBGAL_RS14860, ZOBGAL_RS14875, and ZOBGAL_RS14865), with BLASTp identities ranging from 44.9% to 74.9%. The presence of carrageenan significantly induced the expression of GH127 in Z. galactanivorans (Thomas et al., 2017), suggesting that GH127 in CAR PUL2 has 1,3-(3,6)-anhydro-D-galactosidase activity.
CAR PUL1 was found to contain a carbohydrate-binding module family 22 (CBM22) with xylan binding function (Supplementary Table S5) (Liu and Ding, 2016), which is only found in 10 genomes, four of which belong to Zobellia. However, CBM22 was not found in Tamlana except in strain S12. In red seaweeds, cellulose fibers interact with unusual hemicelluloses embedded in a matrix of sulfated galactans (agars or carrageenans) (Popper et al., 2011). Thomas et al. (2017) showed that one gene coding CBM22 in Z. galactanivorans was induced in the presence of agar or porphyrin. Given the presence of two possible porphyranases (JJL45_RS04680, 04745) in CAR PULs, the CBM22 in CAR PUL1 may be induced to act together with porphyranases in the presence of porphyran. However, the role of CBM22 in the utilization of red algal polysaccharides requirs to further study.
Due to the absence of GH127 and/or GH129 for the hydrolysis of anhydrogalactose (AHG) (Supplementary Table S7), it has been suggested that Maribacter absorbs hydrolysis products from primary degraders to utilize AHG in red macroalgae (Wolter et al., 2021). CAR PUL 1 and 2 contain one GH127 and a variety of carrageenases and porphyranase, indicating the potential of strain S12 to degrade complex polysaccharides found in red algae. With the assistance of sulfatase, the GHs in CAR PUL1 and 2 are each specific for a particular part of carrageenans and porphyrin, releasing oligosaccharides that can be further desulphated through the action of sulfatases. Further degradation of these oligosaccharides can be performed by GH42 (JJL45_RS04760), resulting in neocarrabiose. Finally, the GH127 α-1,3-(3,6-anhydro)-d-galactosidase homologue (JJL45_RS04720) can release d-AHG from the nonreducing end of neocarrabiose saccharides, leading to depolymerization of carrageenans (Ficko-Blean et al., 2017).
One cluster containing two PULs was found in strain S12 and named AGA PUL and XYL PUL according to the functional annotation. The significant number of GH glycoside hydrolases associated with the utilization of agar polysaccharides in AGA PUL indicates that strain S12 has the potential to degrade these polysaccharides (Figure 4D, Supplementary Table S5). The function of XYL PUL is determined by GH10 encoding for xylanase and GH3 encoding glucosidase. We tested the substrate activity and compared it with T. agarivorans KCTC 22176 (Table 2). The results were consistent with the functional prediction. Both strain S12 and T. agarivorans can use agar and xylan (Table 2). Additionally, a similar gene cluster was also found in T. agarivorans.
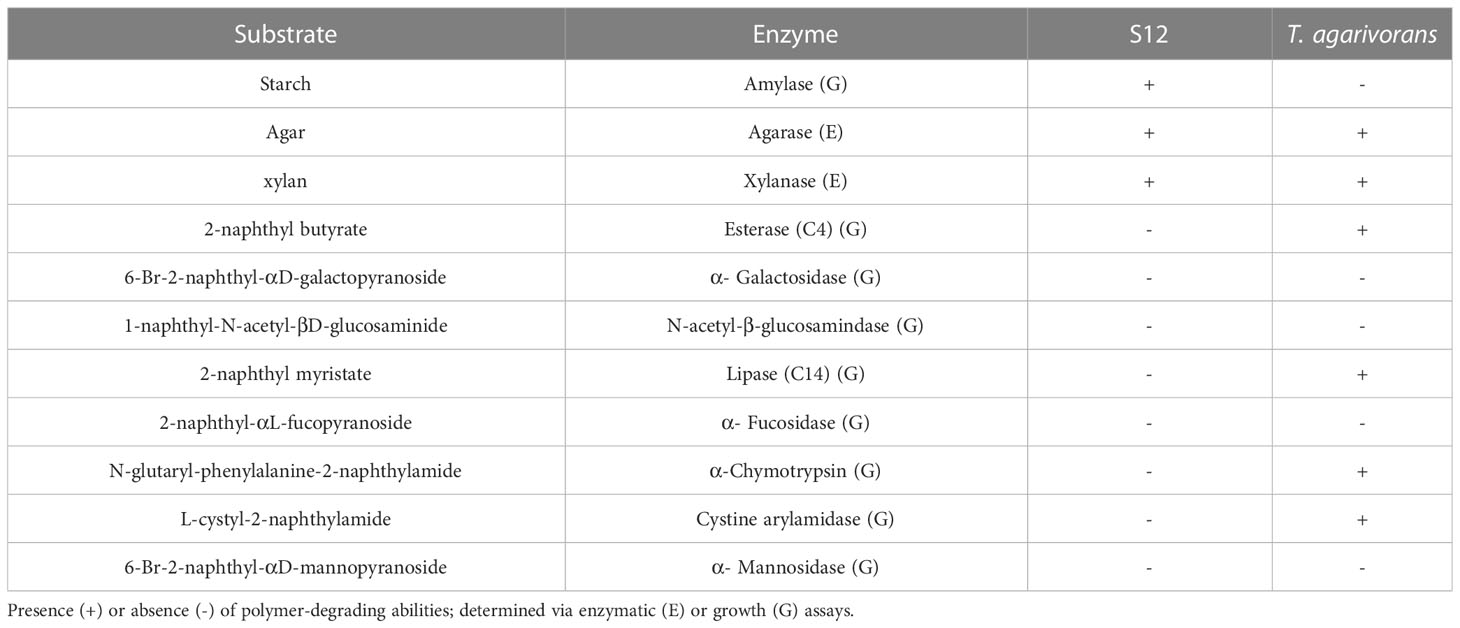
Table 2 Comparison between the novel strain, Tamlana sp. S12 and closely related taxon Tamlana agarivorans KCTC 22176.
AGA PUL contains three GH16 β-agarases (JJL45_RS12010, JJL45_RS12050, JJL45_RS12150) and a GH117 (JJL45_RS12090) that encodes 3,6 anhydro-α-L-galactosidases. GH117 is capable of breaking down neoagarooligosaccharides and generating L-AHG (Jin et al., 2021), playing a role in the degradation of agarose. Additionally, two potential GH2 genes encoding β-galactosidases were found in AGA PUL. These potential GH2s may encode β-galactosidases that remove the non-reducing end of galactose from agarooligosaccharides (Lee et al., 2014). A gene cluster containing GH16, GH117, and GH2 was identified as a putative agarolytic cluster in the human gut bacterium (Pluvinage et al., 2018). The results indicate that the GH16-degraded products of agarose are cyclically degraded into monosaccharides through the collaborative action of GH117 and GH2.
The PL1-PL10 pair exists as accessory genes in a few strains of the four genera (Figure 1). This pair was found in strain S12 and located in two clusters named PEC PUL (Figure 4E), which presumably contributes to pectin utilization, supported by the existence of genes of the families GH28 and 105 that participate in pectinolytic activities (Hobbs et al., 2019). GH28 can encode α-D-galacturonosidase, while GH105 encodes unsaturated uronyl hydrolases, and the catalytic mechanism of GH105 is believed to parallel that of GH88 (Jongkees and Withers, 2011). Pectin is mainly composed of α-(1, 4)-galacturonate and is abundant in terrestrial plants. However, pectin-like polysaccharides also exist in marine microalgae, red and green algae, and seagrasses (Popper et al., 2011; Hehemann et al., 2017). Galacturonate levels can reach 0.3 µM during phytoplankton blooms (Sperling et al., 2017).
In the presence of carbohydrate esterases (CEs) (Supplementary Table S5), specifically CE8 (JJL45_RS15625), 10 (JJL45_RS15605, JJL45_RS15675), and 12 (JJL45_RS15610), the de-esterification of pectin occurs (Hobbs et al., 2019). Additionally, pectinases encoded by PL1 and PL10 may cleave the pectin backbone. This results in the formation of oligouronates with 4,5-unsaturated nonreducing ends. Thees oligouronates can then be further degraded into disaccharides and trisaccharides by exo-poly-α-D-galacturonosidase GH28 (JJL45_RS15655), while GH88 (JJL45_RS15665) results in the direct release of monosaccharides. KDPG is a key intermediate in both pectin and alginate degradation (Koch et al., 2019). The enzyme essential for both pathways is encoded in ALG PUL and may potentially be shared among them (short-chain dehydrogenase, JJL45_RS04370; KdgK, JJL45_RS04385; KDPG aldolase, JJL45_RS04395) (Supplementary Table S5). The PL1-PL10 pair in PEC PUL may also give strain S12 wider substrate adaptability. However, further discussion is needed to fully understand their ecological significance.
3.6 Potential role of S12 in the gut of marine invertebrates
According to Mfilinge and Tsuchiya (2016), sea cucumbers feed on algal fragments (including L. japonica) and bacteria. The metabolic activity of Bacteroidetes may enhance the production of carbohydrates, and facilitate the absorption of polysaccharides in the intestine of A. japonicus. We sought to investigate the characteristics of strain S12, which enable it to survive in such a marine environment and might contribute to the host’s metabolism.
The formation of gut microbiota in marine invertebrates is influenced by bacterial substrate preference and their ability to adhere to the host (Harris, 1993). Utilization of alginate and L. japonica biomass as sole nutrient source (Figure 6, Supplementary Figure S5) suggests that strain S12 may contribute to the release of polysaccharides derived from L. japonica, facilitating their digestion and absorption within the intestine of A. japonicus (Zhang et al., 2019).
Additionally, biofilm assays demonstrated the ability of strain S12 to form biofilms (Figure S6), with poly(β-D-mannuronate) O-acetylase (JJL45_RS08230; Supplementary Table S5) potentially enhancing biofilm formation (Nivens et al., 2001). Biofilm-forming abilities and potential EPS production may facilitates establishment of S12 in the A. japonicus gut (Stapper et al., 2004). The formation of a microenvironment around the strain by EPS could potentially reduce the diffusion of secreted CAZymes, which may be advantageous for enhancing degradation efficiency, preserving hydrolysis products (Wolter et al., 2021), and facilitating their absorption within the gut of marine invertebrates. This may provide an added advantage for colonization, reducing competition while preventing excretion along with fecal matter (Harris, 1993; Boyd and Chakrabarty, 1995).
Furthermore, we searched for CAZyme genes that might benefit the invertebrate host, including a GH25 (JJL45_RS06375) encoding a lysozyme. Previous studies have suggested that over 70% of the energy required by herbivorous or omnivorous marine invertebrates is related to bacteria (Zhou et al., 2009). Lysozyme activity has been detected in the gut bacteria of invertebrates. This enzyme has the ability to lyse microbes, and the resulting breakdown products may contribute to extra digestive capacity by providing active enzymes to the host’s gut (Harris, 1993). Furthermore, cellulase has been widely detected in the gut microbiota of marine invertebrates (Harris, 1993; Kim et al., 2011), indicating that it may play a role in food preprocessing and improving the host’s ability to utilize cellulose (Harris, 1993). Given the promoting effect of cellulase on L. japonica degradation (Sun et al., 2020), XYL PUL may facilitate the preprocessing of algal polysaccharides and provide additional enzyme activity to the host’s intestine upon L. japonica ingestion.
4 Conclusions
The diversity of CAZymes and PUL targeting alginate, carrageenan, and other algal carbohydrates in strain S12 highlights its strong predisposition for polysaccharide degradation. This is evident from its distinct growth in L. japonica and alginate as the sole nutrient source, indicating considerable hydrolytic activity in marine systems. In comparison to Zobellia, Algibacter, and Maribacter, Tamlana is primarily isolated from marine invertebrates or sediments (Supplementary Table S3). It remains to be determined whether the predisposition of strain S12 towards algal polysaccharides corresponds to the co-occurrence with macroalgae. Growth with L. japonica biomass indicated that some members of Tamlana, Zobellia, Maribacter, and Algibacter are capable of initiating the release of polysaccharides from algal tissue. Comparative genomic analysis and evidence related to HGT have demonstrated the widespread occurrence of HGT events involving CAZymes among bacteria. The CAZyme diversity in strain S12 adds to the understanding of CAZymes in flavobacteria and the hydrolytic activity of Tamlana, with implications for the cycling of algal polysaccharides in the ocean and associated ecological dynamics.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA224116.
Author contributions
H-FX conducted genomic analyses and contributed to writing. X-YJ contributed to pan-genome analyses. Y-XZ performed isolation of strain S12 and growth experiments. Z-JD carried out the work of genome data collection and collation. D-SM performed genome sequencing. G-JC designed research and wrote the manuscript. All authors contributed to the final version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (41876166).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.985514/full#supplementary-material
References
Almagro Armenteros J. J., Tsirigos K. D., Sønderby C. K., Petersen T. N., Winther O., Brunak S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37 (4), 420–423. doi: 10.1038/s41587-019-0036-z
Ammar E. M., Wang X., Rao C. V. (2018). Regulation of metabolism in Escherichia coli during growth on mixtures of the non-glucose sugars: arabinose, lactose, and xylose. Sci. Rep. 8, 609. doi: 10.1038/s41598-017-18704-0
Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., et al. (2020). KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36 (7), 2251–2252. doi: 10.1093/bioinformatics/btz859
Archibald A. L. (2017). Exploiting long read sequencing technologies to establish high quality highly contiguous pig reference genome assemblies (W137: International Plant and Animal Genome.
Arnold B. J., Huang I. T., Hanage W. P. (2022). Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Microbiol. 20 (4), 206–218. doi: 10.1038/s41579-021-00650-4
Arnosti C., Wietz M., Brinkhoff T., Hehemann J. H., Amann R. (2021). The biogeochemistry of marine polysaccharides: sources, inventories, and bacterial drivers of the carbohydrate cycle. Ann. Rev. Mar. Sci. 13, 81–108. doi: 10.1146/annurev-marine-032020-012810
Barbato M., Vacchini V., Engelen A. H., Patania G., Mapelli F., Borin S., et al. (2022). What lies on macroalgal surface: diversity of polysaccharide degraders in culturable epiphytic bacteria. AMB Express. 12 (1), 98. doi: 10.1186/s13568-022-01440-8
Ben Bdira, Artola M., Overkleeft H. S., Ubbink M., Aerts J. (2018). Distinguishing the differences in β-glycosylceramidase folds, dynamics, and actions informs therapeutic uses. J. Lipid Res. 59 (12), 2262–2276. doi: 10.1194/jlr.R086629
Boyd A., Chakrabarty A. M. (1995). Pseudomonas aeruginosa biofilms: role of the alginate exopolysaccharide. J. Ind. Microbiol. 15 (3), 162–168. doi: 10.1007/BF01569821
Brunet M., Le Duff N., Barbeyron T., Thomas F. (2022). Consuming fresh macroalgae induces specific catabolic pathways, stress reactions and type IX secretion in marine flavobacterial pioneer degraders. ISME J. 16 (8), 2027–2039. doi: 10.1038/s41396-022-01251-6
Burley S. K., Berman H. M., Christie C., Duarte J. M., Feng Z., Westbrook J., et al. (2018). RCSB protein data bank: sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 27 (1), 316–330. doi: 10.1002/pro.3331
Cao W. R., Liu B. T., Sun X. K., Sun Y. Y., Jiang M. Y., Du Z. J. (2021). Tamlana haliotis sp. nov., isolated from the gut of the abalone Haliotis rubra. Arch. Microbiol. 203 (5), 2357–2364. doi: 10.1007/s00203-021-02216-7
Cha Q. Q., Wang X. J., Ren X. B., Li D., Wang P., Li P. Y., et al. (2021). Comparison of alginate utilization pathways in culturable bacteria isolated from Arctic and Antarctic marine environments. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.609393
Chatukuta P., Rey M. E. C. (2020). A cassava protoplast system for screening genes associated with the response to South African cassava mosaic virus. Virol. J. 17 (1), 184. doi: 10.1186/s12985-020-01453-4
Chaudhari N. M., Gupta V. K., Dutta C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6, 24373. doi: 10.1038/srep24373
Chaumeil P. A., Mussig A. J., Hugenholtz P., Parks D. H. (2019). GTDB-tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36 (6), 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen T., Liu Y.-X., Huang L. (2022). ImageGP: An easy-to-use data visualization web server for scientific researchers. iMeta 1, e5. doi: 10.1002/imt2.5
Chernysheva N., Bystritskaya E., Likhatskaya G., Nedashkovskaya O., Isaeva M. (2021). Genome-wide analysis of PL7 alginate lyases in the genus Zobellia. Molecules 26 (8), 2387. doi: 10.3390/molecules26082387
Chernysheva N., Bystritskaya E., Stenkova A., Golovkin I., Nedashkovskaya O., Isaeva M. (2019). Comparative genomics and CAZyme genome repertoires of marine Zobellia amurskyensis KMM 3526T and Zobellia laminariae KMM 3676T. Mar. Drugs 17 (12), 661. doi: 10.3390/md17120661
Decho A. W., Gutierrez T. (2017). Microbial extracellular polymeric substances (EPSs) in ocean systems. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00922
Delcher A. L., Bratke K. A., Powers E. C., Salzberg S. L. (2007). Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics 23 (6), 673–679. doi: 10.1093/bioinformatics/btm009
Dudek M., Dieudonné A., Jouanneau D., Rochat T., Michel G., Sarels B., et al. (2020). Regulation of alginate catabolism involves a GntR family repressor in the marine flavobacterium Zobellia galactanivorans DsijT. Nucleic Acids Res. 48 (14), 7786–7800. doi: 10.1093/nar/gkaa533
Ferrer-González F., Widner B., Holderman N. R., Glushka J., Moran M. A. (2021). Resource partitioning of phytoplankton metabolites that support bacterial heterotrophy. ISME J. 15 (3), 762–773. doi: 10.1038/s41396-020-00811-y
Ficko-Blean E., Préchoux A., Thomas F., Rochat T., Larocque R., Zhu Y., et al. (2017). Carrageenan catabolism is encoded by a complex regulon in marine heterotrophic bacteria. Nat. Commun. 8 (1), 1685. doi: 10.1038/s41467-017-01832-6
García-López M., Meier-Kolthoff J. P., Tindall B. J., Gronow S., Woyke T., Kyrpides N. C., et al. (2019). Analysis of 1,000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02083
Gavriilidou A., Gutleben J., Versluis D., Forgiarini F., Sipkema D. (2020). Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genomics 21 (1), 569. doi: 10.1186/s12864-020-06971-7
Gomes L. C., Moreira J. M., Miranda J. M., Simões M., Melo L. F., Mergulhão F. J. (2013). Macroscale versus microscale methods for physiological analysis of biofilms formed in 96-well microtiter plates. J. Microbiol. Methods 95 (3), 342–349. doi: 10.1016/j.mimet.2013.10.002
Groisillier A., Labourel A., Michel G., Tonon T. (2015). The mannitol utilization system of the marine bacterium zobellia galactanivorans. Appl. Environ. Microbiol. 81 (5), 1799–1812. doi: 10.1128/AEM.02808-14
Grondin J. M., Tamura K., Déjean G., Abbott D. W., Brumer H. (2017). Polysaccharide utilization loci: fueling microbial communities. J. Bacteriol. 199 (15), e00860–e00816. doi: 10.1128/JB.00860-16
Harris J. M. (1993). The presence, nature, and role of gut microflora in aquatic invertebrates: a synthesis. Microb. Ecol. 25 (3), 195–231. doi: 10.1007/BF00171889
Hehemann J. H., Boraston A. B., Czjzek M. (2014). A sweet new wave: structures and mechanisms of enzymes that digest polysaccharides from marine algae. Curr. Opin. Struct. Biol. 28, 77–86. doi: 10.1016/j.sbi.2014.07.009
Hehemann J. H., Truong L. V., Unfried F., Welsch N., Kabisch J., Heiden S. E., et al. (2017). Aquatic adaptation of a laterally acquired pectin degradation pathway in marine gammaproteobacteria. Environ. Microbiol. 19 (6), 2320–2333. doi: 10.1111/1462-2920.13726
Hobbs J. K., Hettle A. G., Vickers C., Boraston A. B. (2019). Biochemical reconstruction of a metabolic pathway from a marine bacterium reveals its mechanism of pectin depolymerization. Appl. Environ. Microbiol. 85 (1), e02114–e02118. doi: 10.1128/AEM.02114-18
Hobbs J. K., Lee S. M., Robb M., Hof F., Barr C., Abe K. T., et al. (2016). KdgF, the missing link in the microbial metabolism of uronate sugars from pectin and alginate. PNAS 113 (22), 6188–6193. doi: 10.1073/pnas.1524214113
Hyatt D., Chen G. L., Locascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Inoue A., Takadono K., Nishiyama R., Tajima K., Kobayashi T., Ojima T. (2014). Characterization of an alginate lyase, FlAlyA, from flavobacterium sp. strain UMI-01 and its expression in Escherichia coli. Mar. Drugs 12 (8), 4693–4712. doi: 10.3390/md12084693
Iosub I. A., Marchioretto M., van Nues R. W., McKellar S., Viero G., Granneman S. (2021). The mRNA derived MalH sRNA contributes to alternative carbon source utilization by tuning maltoporin expression in E. coli. RNA Biol. 18 (6), 914–931. doi: 10.1080/15476286.2020.1827784
Jam M., Flament D., Allouch J., Potin P., Thion L., Kloareg B., et al. (2005). The endo-beta-agarases AgaA and AgaB from the marine bacterium Zobellia galactanivorans: two paralogue enzymes with different molecular organizations and catalytic behaviours. Biochem. J. 385 (Pt 3), 703–713. doi: 10.1042/BJ20041044
Jin Y., Yu S., Kim D. H., Yun E. J., Kim K. H. (2021). Characterization of neoagarooligosaccharide hydrolase Bp GH117 from a human gut bacterium Bacteroides plebeius. Mar. Drugs 19 (5), 271. doi: 10.3390/md19050271
Jongkees S. A. K., Withers S. G. (2011). Glycoside cleavage by a new mechanism in unsaturated glucuronyl hydrolases. J. Am. Chem. Soc. 133 (48), 19334–19337. doi: 10.1021/ja209067v
Jouanneau D., Klau L. J., Larocque R., Jaffrennou A., Duval G., Duff Le, et al. (2021). Structure-function analysis of a new PL17 oligoalginate lyase from the marine bacterium Zobellia galactanivorans DsijT. Glycobiology 31 (10), 1364–1377. doi: 10.1093/glycob/cwab058
Jung J., Bae S. S., Chung D., Baek K. (2019). Tamlana carrageenivorans sp. nov., a carrageenan-degrading bacterium isolated from seawater. Int. J. Syst. Evol. Microbiol. 69 (5), 1355–1360. doi: 10.1099/ijsem.0.003318
Kalisch B., Dörmann P., Hölzl G. (2016). DGDG and glycolipids in plants and algae. Subcell Biochem. 86, 51–83. doi: 10.1007/978-3-319-25979-6_3
Kanehisa M., Sato Y. (2020). KEGG mapper for inferring cellular functions from protein sequences. Protein Sci. 29 (1), 28–35. doi: 10.1002/pro.3711
Kim D., Kim S. N., Baik K. S., Park S. C., Lim C. H., Kim J. O., et al. (2011). Screening and characterization of a cellulase gene from the gut microflora of abalone using metagenomic library. J. Microbiol. 49 (1), 141–145. doi: 10.1007/s12275-011-0205-3
Koch H., Dürwald A., Schweder T., Noriega-Ortega B., Vidal-Melgosa S., Hehemann J. H., et al. (2019). Biphasic cellular adaptations and ecological implications of alteromonas macleodii degrading a mixture of algal polysaccharides. Isme J. 13 (1), 92–103. doi: 10.1038/s41396-018-0252-4
Kumar A., Jaiswal V., Kumar V., Dey A., Kumar A. (2018). Functional redundancy in echinocandin b in-cluster transcription factor ecdB of Emericella rugulosa NRRL 11440. Biotechnol. Rep. (Amst). 19, e00264. doi: 10.1016/j.btre.2018.e00264
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi: 10.1093/molbev/msy096
Leblond-Bourget N., Philippe H., Mangin I., Decaris B. (1996). 16S rRNA and 16S to 23S internal transcribed spacer sequence analyses reveal inter- and intraspecific bifidobacterium phylogeny. Int. J. Syst. Bacteriol. 46 (1), 102–111. doi: 10.1099/00207713-46-1-102
Lee S. D. (2007). Tamlana crocina gen. nov., sp. nov., a marine bacterium of the family Flavobacteriaceae, isolated from beach sediment in Korea. Int. J. Syst. Evol. Microbiol. 57 (Pt 4), 764–769. doi: 10.1099/ijs.0.64720-0
Lee C. H., Kim H. T., Yun E. J., Lee A. R., Kim S. R., Kim J. H., et al. (2014). A novel agarolytic β-galactosidase acts on agarooligosaccharides for complete hydrolysis of agarose into monomers. Appl. Environ. Microbiol. 80 (19), 5965–5973. doi: 10.1128/AEM.01577-14
Letunic I., Bork P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W296. doi: 10.1093/nar/gkab301
Li J., Liang Y., He Z., An L., Liu Y., Zhong M., et al. (2023). Tamlana laminarinivorans sp. nov. and tamlana sargassicola sp. nov., two novel species isolated from Sargassum, show genomic and physiological adaptations for a Sargassum-associated lifestyle. Int. J. Syst. Evol. Microbiol. 73 (3). doi: 10.1099/ijsem.0.005706
Li J., Xu Y., Feng J., Zhong M., Xie Q., Peng T., et al. (2020). Tamlana fucoidanivorans sp. nov., isolated from algae collected in China. Int. J. Syst. Evol. Microbiol. 70 (3), 1496–1502. doi: 10.1099/ijsem.0.003850
Lin H. H., Hsu C. C., Yang C. D., Ju Y. W., Chen Y. P., Tseng C. P. (2011). Negative effect of glucose on ompA mRNA stability: a potential role of cyclic AMP in the repression of hfq in Escherichia coli. J. Bacteriol. 193 (20), 5833–5840. doi: 10.1128/JB.05359-11
Liu S., Ding S. (2016). Replacement of carbohydrate binding modules improves acetyl xylan esterase activity and its synergistic hydrolysis of different substrates with xylanase. BMC Biotechnol. 16 (1), 73. doi: 10.1186/s12896-016-0305-6
Liu X., Lai Q., Du Y., Li G., Sun F., Shao Z. (2015). Tamlana nanhaiensis sp. nov., isolated from surface seawater collected from the south China Sea. Antonie Van Leeuwenhoek 107 (5), 1189–1196. doi: 10.1007/s10482-015-0410-x
Liu M., Siezen R. J., Nauta A. (2009). In silico prediction of horizontal gene transfer events in Lactobacillus bulgaricus and Streptococcus thermophilus reveals protocooperation in yogurt manufacturing. Appl. Environ. Microbiol. 75 (12), 4120–4129. doi: 10.1128/AEM.02898-08
Lombard V., Bernard T., Rancurel C., Brumer H., Coutinho P., Henrissat B. (2010). A hierarchical classification of polysaccharide lyases for glycogenomics. Biochem. J. 432 (3), 437–444. doi: 10.1042/BJ20101185
Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42 (Database issue), D490–D495. doi: 10.1093/nar/gkt1178
Malik S., Rashid M., Javid A., Hussain A., Bukhari S. M., Suleman S., et al. (2021). Genetic variations and phylogenetic relationship of genus Uromastyx from punjab Pakistan. Braz. J. Biol. 84, e254253. doi: 10.1590/1519-6984.254253
Malinski T. J., Singh H. (2019). Enzymatic conversion of RBCs by α -N-Acetylgalactosaminidase from spirosoma linguale. Enzyme Res. 2019, 6972835. doi: 10.1155/2019/6972835
Martin M., Barbeyron T., Martin R., Portetelle D., Michel G., Vandenbol M. (2015). The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal-Polysaccharide-Degrading bacteria. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.01487
Mathieu S., Henrissat B., Labre F., Skjåk-Bræk G., Helbert W. (2016). Functional exploration of the polysaccharide lyase family PL6. PloS One 11 (7), e0159415. doi: 10.1371/journal.pone.0159415
Mathieu S., Touvrey-Loiodice M., Poulet L., Drouillard S., Vincentelli R., Henrissat B., et al. (2018). Ancient acquisition of "alginate utilization loci" by human gut microbiota. Sci. Rep. 8 (1), 8075. doi: 10.1038/s41598-018-26104-1
Mcbride M. J. (2014). The family flavobacteriaceae (Berlin Heidelberg: Springer), 643–676. doi: 10.1007/978-3-642-38954-2_130
Meier-Kolthoff J. P., Sardà Carbasse J., Peinado-Olarte R. L., Göker M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acid Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Mfilinge P. L., Tsuchiya M. (2016). Changes in sediment fatty acid composition during passage through the gut of deposit feeding holothurians: Holothuria atra (Jaeger 1883) and Holothuria leucospilota (Brandt 1835). J. Lipids. 2016, 4579794. doi: 10.1155/2016/4579794
Mostafa Y. S., Alrumman S. A., Otaif K. A., Alamri S. A., Mostafa M. S., Sahlabji T. (2020). Production and characterization of bioplastic by polyhydroxybutyrate accumulating Erythrobacter aquimaris isolated from mangrove rhizosphere. Molecules 25 (1), 179. doi: 10.3390/molecules25010179
Mu D. S., Wang S., Liang Q. Y., Du Z. Z., Tian R., Ouyang Y., et al. (2020). Bradymonabacteria, a novel bacterial predator group with versatile survival strategies in saline environments. Microbiome 8 (1), 126. doi: 10.1186/s40168-020-00902-0
Nakata S., Murata K., Hashimoto W., Kawai S. (2019). Uncovering the reactive nature of 4-deoxy-L-erythro-5-hexoseulose uronate for the utilization of alginate, a promising marine biopolymer. Sci. Rep. 9 (1), 17147. doi: 10.1038/s41598-019-53597-1
Nivens D. E., Ohman D. E., Williams J., Franklin M. J. (2001). Role of alginate and its O -acetylation in formation of pseudomonas aeruginosa microcolonies and biofilms. J. Bacteriol 183, 1047–1057. doi: 10.1128/JB.183.3.1047-1057.2001
Pei X., Chang Y., Shen J. (2019). Cloning, expression and characterization of an endo-acting bifunctional alginate lyase of marine bacterium wenyingzhuangia fucanilytica. Protein Expr Purif. 154, 44–51. doi: 10.1016/j.pep.2018.09.010
Pluvinage B., Grondin J. M., Amundsen C., Klassen L., Moote P. E., Xiao Y., et al. (2018). Molecular basis of an agarose metabolic pathway acquired by a human intestinal symbiont. Nat. Commun. 9 (1), 1043. doi: 10.1038/s41467-018-03366-x
Popper Z. A., Michel G., Hervé C., Domozych D. S., Willats W. G., Tuohy M. G., et al. (2011). Evolution and diversity of plant cell walls: from algae to flowering plants. Annu. Rev. Plant Biol. 62, 567–590. doi: 10.1146/annurev-arplant-042110-103809
Ravenhall M., Škunca N., Lassalle F., Dessimoz C. (2015). Inferring horizontal gene transfer. PloS Comput. Biol. 11 (5), e1004095. doi: 10.1371/journal.pcbi.1004095
Romanenko L. A., Tanaka N., Kurilenko V. V., Svetashev V. I. (2014). Tamlana sedimentorum sp. nov., isolated from shallow sand sediments of the Sea of Japan. Int. J. Syst. Evol. Microbiol. 64 (Pt 8), 2891–2896. doi: 10.1099/ijs.0.061812-0
Schultz-Johansen M., Bech P. K., Hennessy R. C., Glaring M. A., Barbeyron T., Czjzek M., et al. (2018). A novel enzyme portfolio for red algal polysaccharide degradation in the marine bacterium Paraglaciecola hydrolytica S66(T) encoded in a sizeable polysaccharide utilization locus. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00839
Sperling M., Piontek J., Engel A., Wiltshire K. H., Niggemann J., Gerdts G., et al. (2017). Combined carbohydrates support rich communities of particle-associated marine bacterioplankton. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00065
Stapper A. P., Narasimhan G., Ohman D. E., Barakat J., Hentzer M., Molin S., et al. (2004). Alginate production affects Pseudomonas aeruginosa biofilm development and architecture, but is not essential for biofilm formation. J. Med. Microbiol. 53 (Pt 7), 679–690. doi: 10.1099/jmm.0.45539-0
Sullivan M. J., Petty N. K., Beatson S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27 (7), 1009–1010. doi: 10.1093/bioinformatics/btr039
Sun F. X., Ma Y. X., Wang Y., Liu Q. (2010). Purification and characterization of novel κ-carrageenase from marine tamlana sp. HC4. Chin. J. Oceanology&Limnology 28 (6), 7. doi: 10.1007/s00343-010-9012-7
Sun C., Zhou J., Duan G., Yu X. (2020). Hydrolyzing Laminaria japonica with a combination of microbial alginate lyase and cellulase. Bioresour Technol. 311, 123548. doi: 10.1016/j.biortech.2020.123548
Tanaka, Cleenwerck I., Mizutani Y., Iehata S., Shibata T., Miyake H., et al. (2015). Formosa Haliotis sp. nov., a brown-alga-degrading bacterium isolated from the gut of the abalone haliotis gigantea. Int. J. Syst. Evol. Microbiol. 65 (12), 4388–4393. doi: 10.1099/ijsem.0.000586
Terrapon N., Lombard V., Drula É., Lapébie P., Al-Masaudi S., Gilbert H. J., et al. (2018). PULDB: the expanded database of polysaccharide utilization loci. Nucleic Acids Res. 46 (D1), D677–D683. doi: 10.1093/nar/gkx1022
Thomas F., Bordron P., Eveillard D., Michel G. (2017). Gene expression analysis of Zobellia galactanivorans during the degradation of algal polysaccharides reveals both substrate-specific and shared transcriptome-wide responses. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01808
Thomas F., Lundqvist L., Jam M., Jeudy A., Barbeyron T. (2013). Comparative characterization of two marine alginate lyases from zobellia galactanivorans reveals distinct modes of action and exquisite adaptation to their natural substrate. J. Biol. Chem. 288 (32), 23021–23037. doi: 10.1074/jbc.M113.467217
Tian R., Ning D., He Z., Zhang P., Spencer S. J., Gao S., et al. (2020). Small and mighty: adaptation of superphylum patescibacteria to groundwater environment drives their genome simplicity. Microbiome 8 (1), 51. doi: 10.1186/s40168-020-00825-w
Tian X., Zhang Z., Yang T., Chen M., Li J., Chen F., et al. (2016). Comparative genomics analysis of streptomyces species reveals their adaptation to the marine environment and their diversity at the genomic level. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00998
UniProt Consortium (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49 (D1), D480–D489. doi: 10.1093/nar/gkaa1100
Violot S., Galisson F., Carrique L., Jugnarain V., Conchou L., Robert X., et al. (2021). Exploring molecular determinants of polysaccharide lyase family 6-1 enzyme activity. Glycobiology 31 (11), 1557–1570. doi: 10.1093/glycob/cwab073
Wang L., Zhao X., Xu H., Bao X., Liu X., Chang Y., et al. (2018). Characterization of the bacterial community in different parts of the gut of sea cucumber (Apostichopus japonicus) and its variation during gut regeneration. Aquac Res. 491987–1996. doi: 10.1111/are.13654
Wolter L. A., Mitulla M., Kalem J., Daniel R., Wietz M. (2021). CAZymes in Maribacter dokdonensis 62-1 from the Patagonian shelf: genomics and physiology compared to related flavobacteria and a co-occurring Alteromonas strain., (2020). Front. Microbiol. 12. doi: 10.3389/fmicb.2021.628055
Xiao C. L., Chen Y., Xie S.-Q., Chen K.-N., Wang Y., Han Y., et al. (2017). MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 14, 1072–1074. doi: 10.1038/nmeth.4432
Xu F., Chen X. L., Sun X. H., Dong F., Li C. Y., Li P. Y., et al. (2020). Structural and molecular basis for the substrate positioning mechanism of a new PL7 subfamily alginate lyase from the arctic. J. Biol. Chem. 295 (48), 16380–16392. doi: 10.1074/jbc.RA120.015106
Yin R., Yi Y. J., Chen Z., Wang B. X., Li X. H., Zhou Y. X. (2021). Characterization of a new biofunctional, exolytic alginate lyase from tamlana sp. s12 with high catalytic activity and cold-adapted features. Mar. Drugs 19 (4), 191. doi: 10.3390/md19040191
Yoon S. H., Ha S. M., Kwon S., Lim J., Kim Y., Seo H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67 (5), 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon J. H., Kang S. J., Lee M. H., Oh T. K. (2008). Tamlana agarivorans sp. nov., isolated from seawater off jeju island in Korea. Int. J. Syst. Evol. Microbiol. 58 (Pt 8), 1892–1895. doi: 10.1099/ijs.0.65704-0
Zhang H., Wang Q., Liu S., Huo D., Zhao J., Zhang L., et al. (2019). Genomic and metagenomic insights into the microbial community in the regenerating intestine of the Sea cucumber Apostichopus japonicus. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01165
Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46 (W1), W95–W101. doi: 10.1093/nar/gky418
Keywords: marine flavobacteria, Tamlana, alginate, Laminaria japonica, PUL, EPS, Apostichopus japonicus, pan-genome
Citation: Xia H-F, Jia X-Y, Zhou Y-X, Du Z-J, Mu D-S and Chen G-J (2023) Comparative genomics reveal distinct potential of Tamlana sp. S12 for algal polysaccharide degradation. Front. Mar. Sci. 10:985514. doi: 10.3389/fmars.2023.985514
Received: 04 July 2022; Accepted: 02 May 2023;
Published: 15 May 2023.
Edited by:
Hongbin Liu, Hong Kong University of Science and Technology, Hong Kong SAR, ChinaReviewed by:
Heng-Lin Cui, Jiangsu University, ChinaMatthias Wietz, Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research (AWI), Germany
Copyright © 2023 Xia, Jia, Zhou, Du, Mu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guan-Jun Chen, Z3Vhbmp1bkBzZHUuZWR1LmNu; Da-Shuai Mu, ZGFzaHVhaS5tdUBzZHUuZWR1LmNu