 Ying Han
Ying Han Yikai Hu1
Yikai Hu1 Yingchao Zhang
Yingchao Zhang- 1School of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao, China
- 2Hebei Province Key Laboratory of Deep Remediation of Heavy Metals in Water and Resource Utilization, Yanshan University, Qinhuangdao, China
Rapid urbanization and other human activities have exacerbated the global human health risks associated with antibiotic resistance genes (ARGs). Swimming in contaminated waters is one of important pathways by which humans can be exposed to ARGs. However, little is currently known about the overall levels of ARGs in recreational waters and the forces driving their occurrence. In this study, we analyzed the ARGs and bacterial communities in water samples taken from five recreational coastal waters (RCWs) and their adjacent estuaries in the Qinhuangdao area of Bohai Bay, China, using high-throughput quantitative PCR and 16S rRNA gene amplicon sequencing technique. The relative abundances of ARGs in RCWs varied greatly, and occurred at levels close to, or even higher than, those in the neighboring estuaries, ranging from 9.4×10-2 to 2.5 copies/16S rRNA gene. A total of 159 different ARGs able to express resistance to almost all antibiotics were detected in all of the water samples. β-lactamase resistance genes (blaTEM gene) were the most abundance of all ARGs. Patterns of ARG occurrence were significantly correlated with those of bacterial communities (Mantel test, P < 0.05). A partial redundancy analysis showed that differences in bacterial communities accounted for 50.3% of the total ARG variations, which was much higher than the sum of the contribution of mobile genetic elements (18.1%) and the other selected environmental factors (8.3%). Principal coordinate analysis based on Bray-Curtis distance revealed the similarities between ARG profiles in the RCWs and in their adjacent estuaries. To our knowledge, this is the first study to report the ARG profiles in RCWs using high throughput qPCR technology. These results implied that it is necessary to develop proper regulations and monitoring strategies of RCWs for reducing risks of ARGs on human health.
Introduction
The overuse of antibiotics has accelerated the emergence and spread of antibiotic resistance genes (ARGs) in the environment (Wen et al., 2015) . ARGs have been found in a wide variety of environments including agricultural soils (Sun et al., 2020), urban wastewater treatment plants (Freeman et al., 2018), lakes (Y. Q. Wang et al., 2021), rivers (Pruden et al., 2006) and aquacultural facilities (P. Gao et al., 2012). Microorganisms in the environment can contribute to, or acquire, ARGs from human pathogenic bacteria, as demonstrated by the significant similarities in their sequences (Tomova et al., 2015; Jing Yang et al., 2013). Increased levels of ARGs in the environment have led to growing concerns about an impending global public health crisis (Pruden et al., 2006). It has been estimated that the number of deaths arising from antibiotic resistant pathogens may exceed 10 million by the end of 2050 (O' Neil, 2014), and that ARGs will surpass cancer as the most serious risk factor for human health. As emerging contaminants (A. Pruden et al., 2006), ARGs replicate not only through bacterial growth but also can be transferred between different bacterial species mediated by mobile genetic elements (MGEs) (von Wintersdorff et al., 2016). The amount of ARGs can increase and spread if they are selected by antibiotic pollutions or are co-selected by ecologically favorable determinant present in the same MGEs (Blahova et al., 1999). Therefore, identifying environmental reservoirs of ARGs and exploring their routes of transmission are critical for the prevention of ARG related infections.
Estuaries occur at the boundary between terrestrial/freshwater and coastal ecosystems (Stewart et al., 2008; Marti et al., 2014). Approximately half of the world’s population lives within 100 kilometers of the coastline (Culliton, 1998). Generally, estuaries receive mass discharges of waste from industry and other human activities (Wright et al., 2008), such as wastewater treatment plants (WWTP) (Wu et al., 2018; Bi Weihong et al., 2022), medical wastewater (Rodriguez-Mozaz et al., 2015), livestock rearing, aquaculture and agricultural wastewater (Jang et al., 2018). Traditional WWTPs are not generally effective in removing ARGs and the ARG abundance in the effluent can be as high as 3.13×106 copies/mL (H. Chen & Zhang, 2013; H. W. Huang et al., 2019). Terrigenous pollutants, including heavy metals, antibiotics, ARGs and drug-resistant bacteria, are also washed into rivers and then into estuaries. Surface waters also receive wastewater and municipal sewage from nearby factories and cities. It has been shown that rivers in China generally contain various ARGs, including the Yangtze, Yellow, Haihe and Huaihe Rivers (Zhuang et al., 2021). Moreover, the specificity of the water quality environment and the increasing development of tourism can also affect the evolution of microbial resistance (Su et al., 2022). The resistant bacteria will be brought into the coastal tourist area near the shore with the rain, tide, and currents. Swimming in recreational coastal waters (RCWs) is an important route by which humans are exposed to pollutants in the coastal environment. Many studies have reported various pollutants in RCWs, including heavy metals (Bigus et al., 2016), nitrogen, phosphorus, viruses (Wei et al., 2020), microplastics (F. L. Gao et al., 2021), and pollutants resulting in elevated chemical oxygen demands, which may facilitate the spread of ARGs (Stepanauskas et al., 2006; Tandukar et al., 2013; Forsberg et al., 2014). However, the overall profiles, influent factors, and driving mechanisms of ARGs in RCWs are still not fully understood.
The dynamics of ARG occurrence are far more complex than the selective pressure of antibiotics (Tamminen et al., 2011). A growing number of studies have reported that shifts in bacterial communities are the main driver of ARG accumulation in the environment (Zhang et al., 1800; Su et al., 2015). Horizontal gene transfer (HGT) mediated by MGEs is also an important mechanism of ARG propagation (K. Zhang et al., 2021). Environmental microorganism, particularly pathogenic bacteria, can acquire ARGs from different species of bacteria via HGT to express antibiotic resistant and even become the “superbugs” endangering public health (von Wintersdorff et al., 2016). Environmental factors, such as nitrogen and phosphorus concentrations (Ali et al., 2016), pH (H. N. Huang et al., 2017), salinity (Y.-J. Zhang et al., 2019), and temperature (J. Yang et al., 2020), may also be predominant factors governing the distribution patterns of environmental ARGs. Therefore, it is necessary to explore the overall ARG profiles in RCWs as well as all of the possible factors affecting their occurrence to discover the predominant drivers and to control their propagation and thus reduce potential health risks.
The Bohai Sea has a coastline of about 3,800 kilometers and is fed by large number of rivers. Qinhuangdao, located in the north part of the Bohai Sea, is one of the most important tourist cities in northern China, and is greatly impacted by human activities (L. H. Zhu et al., 2014). The “Qinhuangdao Recreational Coastal Water Management Regulations” stipulate the minimum straight-line distance between RCWs and the nearest estuary, fishing port, tourist wharf, fishing boat berth and shallow sea aquacultural area to avoid the spread of pollutants (Qinhuangdao Municipal People’s Government, 2019). However, it is unclear whether the above provisions can prevent the ARG impacts of estuaries on RCWs. In this study, water samples were collected from the estuaries of typical rivers and their adjacent RCWs around Qinhuangdao: (1) to investigate the ARG profiles and bacterial communities in estuaries and their RCWs; (2) to determine the influencing factors and driving mechanisms of ARG occurrence in estuaries and their adjacent RCWs; and (3) to explore the potential antibiotic resistance risks to human health under current regulations.
Methods and materials
Sample collection, water chemistry and DNA extraction
Water samples were collected from the estuaries and their adjacent RCWs around Qinhuangdao City, located on the north coast of Bohai Bay, China. Detailed descriptions of the sampling sites are shown in Figure S1. Water samples (~5L) were taken during December 2020 on three consecutive days without rainfalls during the preceding week. All water samples were stored on ice in sterile containers and brought back to the laboratory within six hours for further treatment.
The bacteria cells in each water sample were collected by filtering through 0.22 mm mixed cellulose esters filters (Millipore, USA) and stored at -20°C for subsequent total extraction of genomic DNA using a QIAGEN Extraction Kit (Qiagen Instruments, USA), following the manufacturer’s instructions. The purity and integrity of the extracted DNA were assessed using a Nano Drop spectrophotometer (Thermo Fisher Scientific, USA) and agar gel electrophoresis, respectively. The extracted DNA was stored at -20°C. Temperature, pH and salinity were measured in situ using a multiparameter water quality analyzer (Hydrolab DS5, Hach Company, Loveland, USA). Ammonia-nitrogen (NH4+-N), nitrate nitrogen (NO3+-N) (Rider and Mellon, 1946), nitrite nitrogen (NO2–N) (J. Yang et al., 2020), and phosphate (PO4–P) were measure according to standard methods (B. Wu et al., 2014).
High-throughput quantitative PCR of ARGs
HT-qPCR was performed using a Warfergen SmartChip Real-time PCR System (Anhui Microanaly Gene Technologies Co., Ltd, China) to detect and quantify ARGs, as described previously (Ouyang et al., 2015). Quality assurance/quality control was conducted following the standard protocols provided by Wafergen Biosystems. A total of 296 primers sets were used targeting at 285 ARGs, eight transposases, three integrons and one 16S rRNA gene. Detailed information on the primers is listed in Table S1. The 285 ARGs could express antibiotic resistance to almost all antibiotics by 4 mechanisms. HT-qPCR assay was performed in a 100 nL reaction system consisting of a 1 × LightCycler 480 SYBR Green I Master mix, 0.1 mg/mL albumin from bovine serum, 500 nM of each primer and 2 ng/μL DNA template. A non-template control was included on each chip for each primer set. The qPCR reaction mixture was first added to a microarray using nanoscale multi-sample point sampling instrument, and then qPCR reaction was performed using a cycler with the thermal cycles: 95°C for 10 min, 40 cycles of 95°C for 30 s, and 60°C for 30 s, ending with a melting curve analysis auto-generated by the Wafergen program.
The HT-qPCR results were analyzed using SmartChip qPCR (v 2.7.0.1) software to remove the results with multi-peak melting curve and with amplification efficiencies beyond the range of 1.8-2.2. A threshold cycle (CT) value of 31 was used as the detection limit. All qPCR reactions were conducted in triplicate, and if more than two were amplified, the result was regarded as positive. Relative copy numbers of ARGs was calculated according to the following equation:
Gene copy number = 10 ^[(31-CT)/(10/3)]
Relative ARG copy number = ARG copy number/16S rRNA gene copy number
Where, CT is the cycle threshold.
The result of the primer sets targeting at the same gene were combines. For example, blaOXA10-01 and blaOXA10-02 both target at the blaOXA10 gene according to their nomenclature (Jacoby, 2006).
Bacterial 16S rRNA gene sequencing and data processing
Bacterial communities were investigated using a 16S rRNA gene amplicon sequencing technology (Anhui Microanaly Gene Technologies Co., Limited, China). The V3-V4 region of the 16S rRNA gene was amplified using barcodes and the primer sets 341F (5’-CCTACGGGNGGCWGCAG-3’) and 765R (5’-GACTACNVGGGTATCTAAT-3’). The genomic DNA was used as templates to conduct PCR using Taq DNA Polymerase (Vazyme, Nanjing, China). PCR products were purified using a QIAquick Gel Extraction Kit (QIAGEN, USA) and pooled in equimolar for sequencing using an Illumina MiSeq PE300 platform. Paired-end sequences were joined after the barcode and primers were removed using FLASH (Magoc and Salzberg, 2011). FLASH software was also used to remove short reads, chimeras and ambiguous N (Magoc and Salzberg, 2011). The sequences were clustered into Operational Taxonomic Units (OTUs) with a threshold of 97% similarity using the VSEARCH method (Rognes et al, 2016). Representative sequences were selected for further OTU annotation using a default method involving assignment to the Greengenes Database taxonomy (Wang et al., 2007). Species diversity and α-diversity of the bacterial communities were estimated with the observed species, Chao1, Shannon, Simpson and PD_whole_tree index, which were calculated using a script in QIIME software. Bacterial communities in different groups (β-diversity) were compared using the unweighted pair group method with arithmetic mean (UPGMA), principal coordinate analysis (PCoA) based on Bray-Curtis distances, and the Adonis test.
Network analysis
The co-occurrence patterns among ARGs, MGEs and bacterial communities were revealed by network analysis using Cytoscape (3.8.2) with the CoNet plug-in (Sarkar and Chang, 1997). Pairwise correlations were calculated by the Mutual information, Spearman’s correlation, Pearson’s correlation, Bray-Curtis dissimilarity and Kullback-Leibler dissimilarity tests. Correlations with a P value above 0.9 and a significance level below 0.05 were considered significant. ARGs or bacterial taxa detected in less than three samples were discarded in correlation calculations. Correlation efficiencies and P values were analyzed using the Simes method implemented with the CoNet plug-in to avoid false-positive correlations (Sarkar and Chang, 1997). The network topology was visualized in Gephi (0.9.2) with the Frucherman Reingold algorithm (Dalcin and Jackson, 2018).
Statistical analyses
Averages and standard deviations of all data were determined using Microsoft Excel 2010 (Microsoft Office 2010, Microsoft, U.S.A.). The normalized ARG abundances were plotted with OriginPro 2017 (OriginLab, U.S.A.). Heat maps analyzing the ARGs data of water samples were generated with using the Pheatmap package in R (Version 4.1.2). Mantel test, PcoAs based on Bray-Curtis distance, Redundancy analyses (RDA) and partial RDAs were performed using R4.1.2 with the vegan package. Aggregated boosted tree (ABT) analysis was performed using R2.7.0 with the gbmplus package. Significant differences between estuaries and RCWs were tested using the Student−Newman−Keuls (S−N−K) test (SPSS V20, IBM, U.S.A.). Pearson’s correlation coefficients among ARG and MGE abundances were calculated using SPSS.
Result
Abundance and diversity of ARGs
A total of 159 ARGs and 5 MGEs were detected in the 10 water samples from RCWs and estuaries in Qinhuangdao, China. An average of 114 ARGs were detected in the estuaries and 94 in the RCWs. However, there were not necessarily fewer ARGs in the RCWs than in their adjacent estuaries, such as the Nandai and Yang Rivers (Figure S2). Aminoglycoside and β-Lactamase were the most dominant types of resistant genes, followed by Macrolide-Lincosamide-Streptogramin B (MLSB) and tetracycline resistant genes, accounted for 30.8%-83.9% of the total ARGs in each sample (Figure S2). ARGs potentially conferring resistance to vancomycin were widely distributed among the estuaries and RCWs, with occurrence ranging from 2.34%-6.94%. It should be noted that vancomycin is critically important for human medicine and is considered the last line of treatment for bacterial infections. In the 159 ARGs, antibiotic deactivation, efflux pump, and cellular protection are the most predominant resistance mechanisms (Figure S2).
The relative abundances of ARGs in water samples from the Tang and Xinkai Rivers are significantly higher than those in other samples (P < 0.01), as shown in Figure 1A. In contrast to the other estuaries, the most abundant type of ARG in the Tang River was aminoglycoside (17.3%). The relative abundance of ARGs in water samples from the Tang and Xinkai Rivers ranged from 1.3-2.5 copies/16S rRNA copies, about 20 times higher than those of the other samples (0.21~0.29copies/16S rRNA gene). A total of 10 MGEs were detected, including eight transposases and two integrases with relative abundances ranging from 2.7%-28.5%. As shown in Figure 1B, the relative abundance of MGEs in all RCWs and estuaries ranged from 5.6×10-3 -8.5×10-1 copies/16S rRNA genes. The normalized copy number of transposases and integrases was significantly different among sampling sites. For example, integrase accounted for 96.0% of MGEs in the RCW adjacent to the Nandai River estuary, but only transposase genes were detected in the RCW close to estuary of the Tang River (0.11copies/16S rRNA genes).
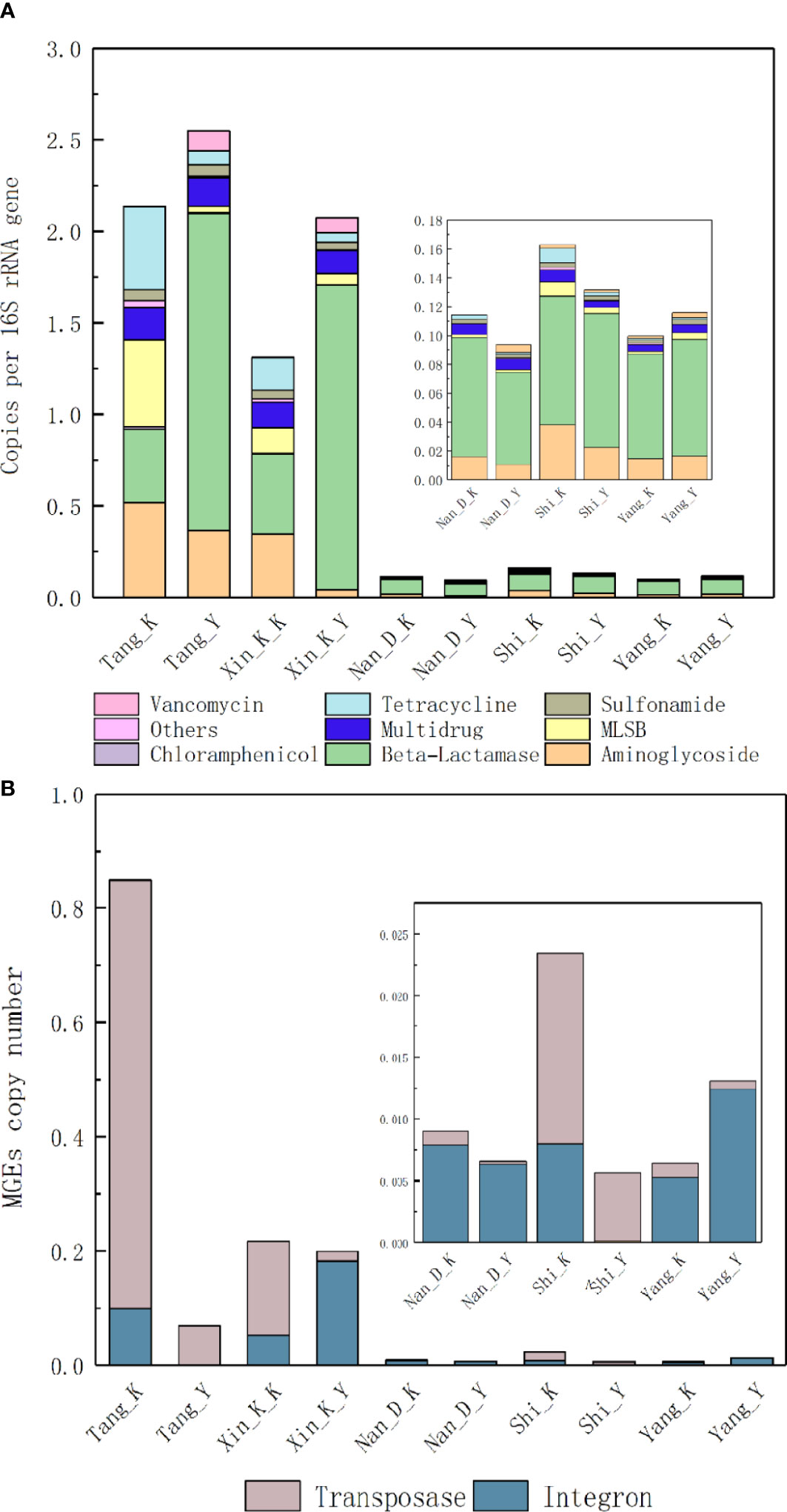
Figure 1 Normalized abundance of (A) ARGs and (B) MGEs in sediment of estuaries and recreational coastal waters. Normalized copy number of ARGs presented as number of ARGs per 16S rRNA genes. Multidrug resistance genes are defined as the resistance to multiple antibiotics with different structures and antimicrobial mechanisms.
There were significant differences in the overall structure of ARGs in the different water samples (Adonis test, P < 0.05). PCoA based on Bray-Curtis distance (Figure 2A) showed that water samples from the Nandai, Yang and Shi Rivers clustered together and separated from the other samples. The first two PCs explained a total of 68.1% of the variance in ARGs, with PC1 explaining more than half (50.5%) of the variation. Samples from different locations were generally separated along the first PC, and samples from the same estuary and its adjacent RCW were separated along PC2, which explained 17.6% of the variation, indicating the minor differences between ARG profiles of an estuary and its adjacent RCWs. Hierarchical clustering analysis further verified such variances in ARG profiles (Figure 3). The estuary samples from the Tang and Xinkai Rivers were separated from the other water samples, which clustered together. As shown in Figure 3, all of the ARGs detected could be divided into three subgroups: (A) ARGs with high relative abundance in all water samples, such as the blaTEM、folA、blaOXA10 and aadA5 genes; (B) ARGs with high relative abundance in the estuaries of Tang and Xinkai Rivers, such as the strB and aadA genes; and (C) ARGs with high relative abundance in other sampling sites, such as the VanC and mphA genes. A detailed table of ARG abundance with names can be found in the Supplementary Materials (Table S2).
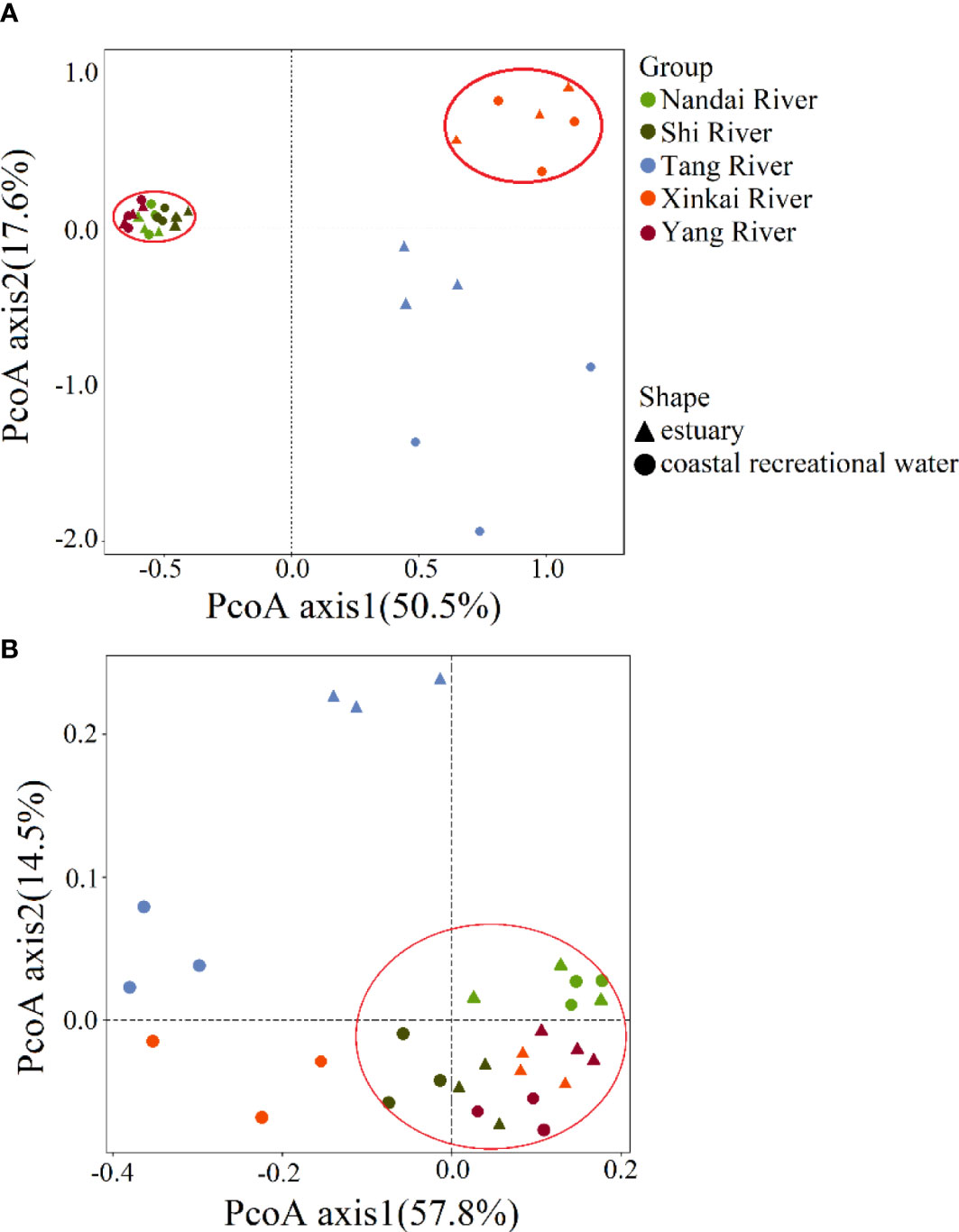
Figure 2 Principal coordinates analysis based on Bray-Curtis distance showing the overall distribution pattern of (A) ARGs and (B) microbial communities in water samples of estuaries and recreational coastal.
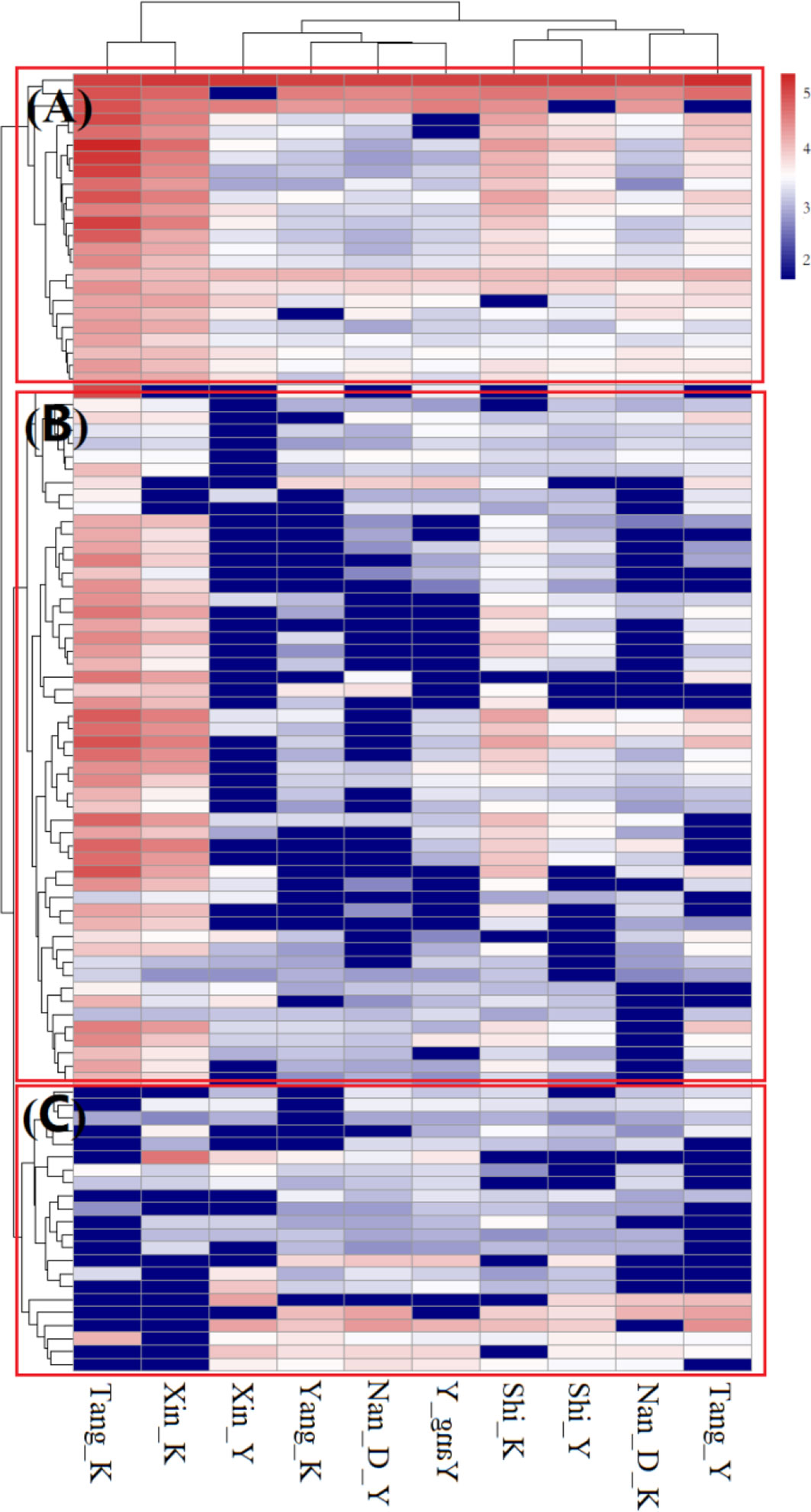
Figure 3 ARG profiles in sediment of estuaries and recreational coastal waters. Each column is labeled with a sample name. K represents water samples from estuaries and Y represents water samples from recreational coastal water. Plotted values are the logarithm-transformed relative abundance of each ARGs. Columns and rows were clustered based on Bray-Curtis distance. (A) ARGs with high relative abundance in all of the water samples; (B) ARGs with high relative abundance in estuaries of Tang and Xinkai Rivers; (C) ARGs with high relative abundance at the other sampling sites.
Characteristics of microbial community structure
After assembling and quality filtering, a total of 640,000 high-quality sequences were obtained from 10 samples, ranging from 45,663-95,376 sequences per sample. These sequences were assigned to 8,675 OTUs at the 97% similarity level, with the number of OTUs in each single sample ranging from 638-1,112. Species accumulation curves and rank abundance curves indicated that the bacteria species in the estuarine samples were generally higher than those in the adjacent RCWs (Figure S3), Proteobacteria and Bacteriodetes were the dominant phyla in all samples, accounting for 57.1-83.5% of the bacterial communities. Gammaproteobacteria and Alphaproteobacteria were the major classes of Proteobacteria, while Flavobacteriia was the major class of Bacteroidetes. The compositions of the bacterial communities at different levels are shown in Figure S4. PCoA analysis based on Bray-Curtis distance showed that estuarine bacterial communities and their corresponding RCWs were relatively similar except in the Tang and Xinkai Rivers. As shown in Figure 2B, the first two PCs explained 57.8% and 14.5% of the differences in bacterial community composition, respectively. The genus-level UPGMA hierarchical analysis confirmed that the bacterial communities in the Shi and Yang River regions were similar at the genus level (Figure S5). Psychrobacer, Litoreibacter, Sulfitobacter, Planktomarina and Loktanella were the most dominant bacteria, accounting for 29.5-49.6% of the total genera. Interestingly, the bacterial communities in samples from the Shi and Yang Rivers were similar even though they are not geographically adjacent, indicating that bacterial communities in coastal environments were affected by more complex factors than mere diffusion between. No significant differences were observed between bacterial communities from estuaries and their adjacent RCW (Anosim, P > 0.05), but certain bacterial species were only observed in estuaries or in RCWs. For example, Campylobacteraceae and Arcobacter were prevalent in estuariesbut rarely detected in RCWs. Whether they can be used as biomarkers to distinguish estuarine samples from RCWs requires further research. It is worth noticing that Arcobacter, a potential human pathogen, was widely distributed in estuaries. Even though its detection limit in RCWs was low in this study, its dissemination and fate in coastal environments requires further surveillance and attention.
Correlations between ARGs, MGEs and microbial community structure
Significant correlations were detected among the all classes of ARGs (Pearson’s correlation), indicating that the spread of various ARGs may share the same dissemination mechanism. The abundance of integrons was not correlated with any particular class of ARG, but the total abundance of transposases genes were significantly correlated with almost all classes of ARG, except for β-Lactamase, sulfonamide, and vancomycin resistance genes, indicating that transposons played an important role in ARG propagation in coastal environment. Several ARGs (blaTEM, strB, blaVEB, sulI genes), generally embedded in transposons, were detected with the highest normalized copy number, indicating that transposons were the most common carrier of ARGs in coastal waters. With the exception of β-Lactamase and vancomycin resistant genes, the normalized abundance of ARGs were correlated with those of MGEs, with correlation coefficients ranging from 0.67-0.99 (Table 1, Person’s correlation, P < 0.05), indicating that ARGs may be readily transferred within bacterial communities. These correlations were based on the normalized abundances of ARGs and MGEs, suggesting that MGEs mediated HGT, contribute to the propagation of ARGs in coastal waters and lead to the enrichment of those ARGs in bacterial communities.
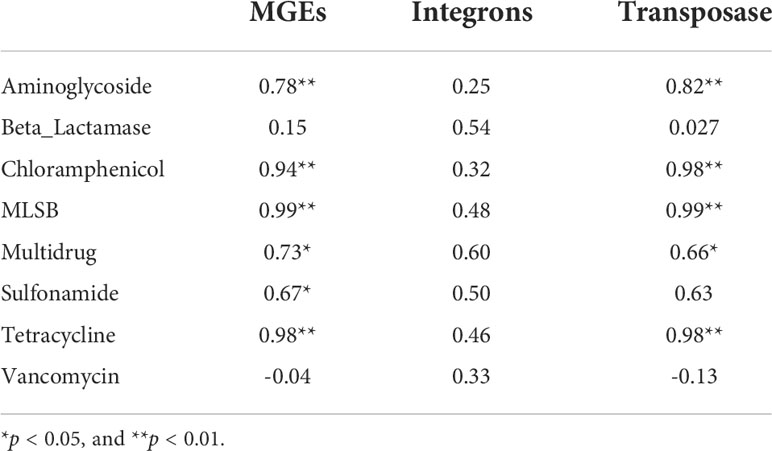
Table 1 Pearson’s correlation analysis among the normalized abundances of ARGs and MGEs.
The patterns of co-occurrence among ARGs and MGEs were analyzed using co-occurrence network analysis (Figure 4) based on the strong and significant correlation between ARGs and MGEs. The network structure consists of 25 nodes and 20 edges, which could be divided into nine modules with a module degree of 0.72. The line in the Figure 4 shows a strong and significant correlation between two nodes (r > 0.9). The most densely connected node was defined as a “hub” in the module. For example, the aadA2 gene is the hub in its module and is significantly correlated with the blaOXA10, catB3, qacEdelta1 and tetM genes. The hubs can be used as indicators for all ARGs in a module to predict dynamic changes of other ARGs in the same module. Most modules contained different types of ARGs or MGEs. For example, module IV includes the aminoglycoside resistant genes (aadA2 gene) and the tetracycline resistance genes (tetR gene), and module VIII includes the vgbB gene and aadD gene, which belong to MLSB and aminoglycoside resistance genes. The correlation between transposon (tnpA gene) and aminoglycoside resistant gene (aac(6’)-Ib(aka aacA4)) further indicated that transposons could contribute to the propagation of ARGs in coastal environments. The correlations between different ARGs suggested that they are carried by the same bacteria or located in the same MGE. We further analyzed the patterns of co-occurrence among ARGs, MGEs and bacterial communities at the genus level. We hypothesized that a strong nonrandom, significant correlation between microorganisms and ARGs (R2 > 0.9, P < 0.05) could indicate the identity of the host of an ARG. For instance, Acinetobacter may be the host cell of an aminoglycoside gene (aac(6’)-Ib(aka aacA4)), and the β-lactamase resistance gene (blaVEB) was probably carried by the genus Phycisphaera.
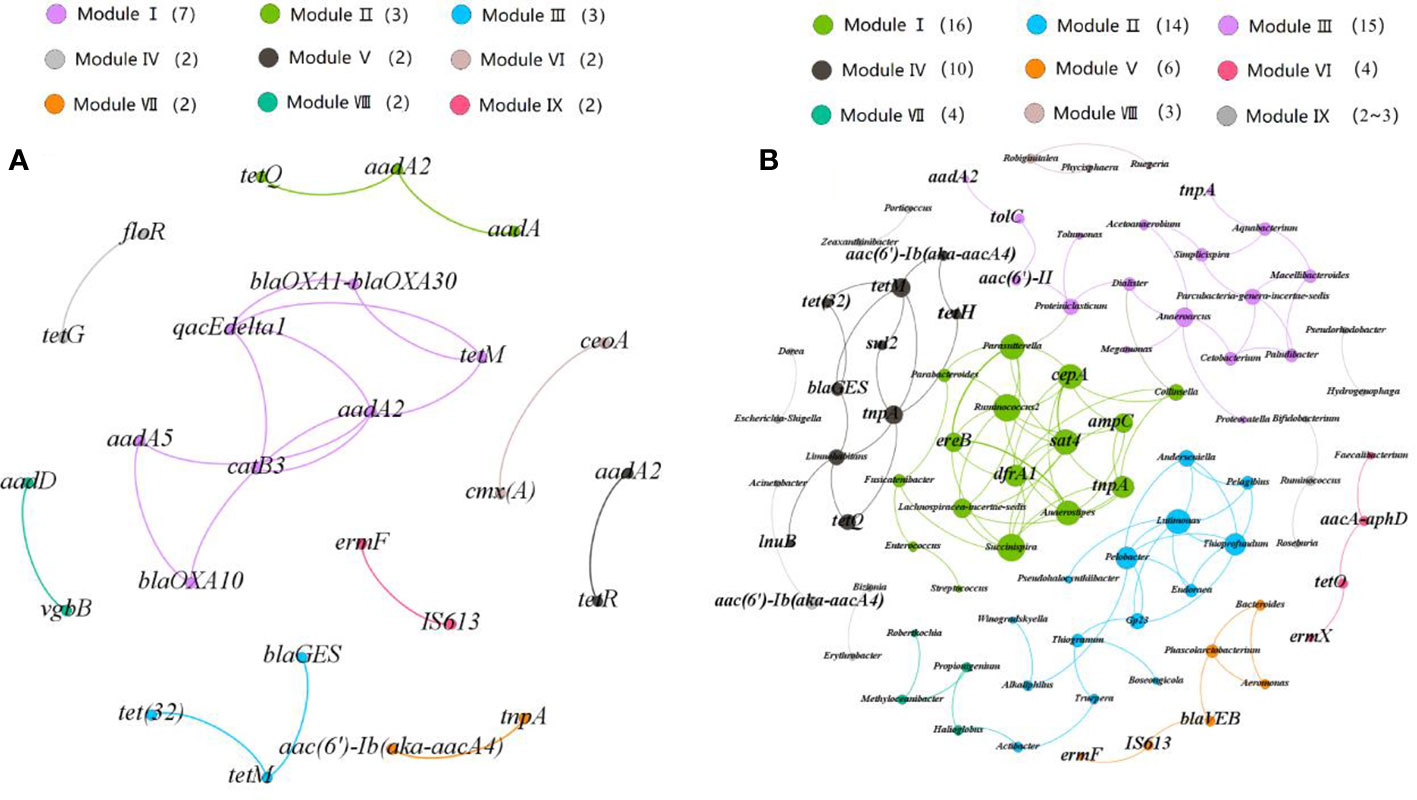
Figure 4 Network analysis revealing (A) co-occurrence patterns among ARGs and (B) co-occurrence patterns between ARGs and microbial taxa at genus level. Nodes are colored according to modularity classes. A connection between nodes indicated a strong (r > 0.8) and (P < 0.01) significant correlation. The size of each node is proportional to the number of connections.
We also studied the differences between the abundances of bacteria that directly correlated with ARGs in RCWs and estuaries. The bacteria which could be recognized as hosts of ARGs were detected with relatively high abundance in estuaries but low abundance in RCWs, and were perhaps present at levels below the limits of detection. The abundance of Succinispira, which is correlated with the trimethoprim resistant gene (dfrA) ranged from 0- 8.3×10-5 in estuaries, but was rarely detected in RCWs. Acinetobacter is a potential human pathogen, and was strongly and significantly associated with aminoglycoside resistant gene (aac(6’)-Ib(aka aacA4)). This potentially resistant pathogen was detected in all of the water samples except for the RCWs adjacent to the Nandai River. Together, these results suggested that environmental bacteria and pathogens had the potential to evolve antibiotic resistance in coastal environments and could spread to humans swimming in these waters so posing serious risks to human health.
Relationships among bacteria communities, mobile genetic elements, environmental factors and antibiotic resistance genes
The relationship between microbial communities, MGEs, environmental factors, and ARGs were explored by RDA analysis (Figure 5A). Taken together, the selected environmental variables (NH4+-N, NO3–N, NO2–N, PO43-, salinity, pH), MGEs, and bacterial communities explained 99.8% of the variation in ARGs. MGEs, salinity, and NH4+-N were significantly positively correlated with the first axis and ARGs from the estuaries of the Tang, Xinkai and Shi Rivers. PO43-, pH, NO3–N, Litoreibacter, and Psychrobacter were significantly positively correlated with ARGs in the RCWs of the Xinkai, Nandai, and Yang Rivers. Partial RDA was performed to further distinguish the effects of environmental factors, MGEs and bacterial communities on antibiotic resistomes in water samples. Based on partial RDA, the bacterial communities accounted for 50.3% of the total ARG variations, much more than the sum of the contribution of MGEs (18.1%) and environmental factors (8.3%). The interaction between environmental factors and microbial community structure, as well as those between MGEs and the bacterial community accounted for 5.9% and 15.2% of total resistome variation, respectively. Mantel correlation based on Bray-Curtis distance showed significant correlations between bacterial communities and antibiotic resistomes in the coastal environments studied (r2 = 0.4591, p = 0.008). The contributions of microbial communities, MGEs and environmental factors to the relative abundance of ARGs in the samples were further explored separately using ABT analysis. Figure S6 shows the relative importance of these indicators. The greater the value, the higher the importance of an indicator. MGEs strongly influenced the occurrence of ARGs in the waters. In addition, the concentration of NO2- (0.011-0.02 mg/L) also had a strong effect on the relative abundance of ARGs. In terms of microbial communities, Acidobacteria had the greatest influence on the resistomes in RCWs and their adjacent estuaries. Coexistence of multiples integrative and conjugative elements has been found in the chromosome of Acidobacteria (Gonçalves and Santana, 2021). Therefore, they could easily become resistant to antibiotics and contribute to the dissemination of ARGs, mediated by the MGEs in estuaries and RCWs.
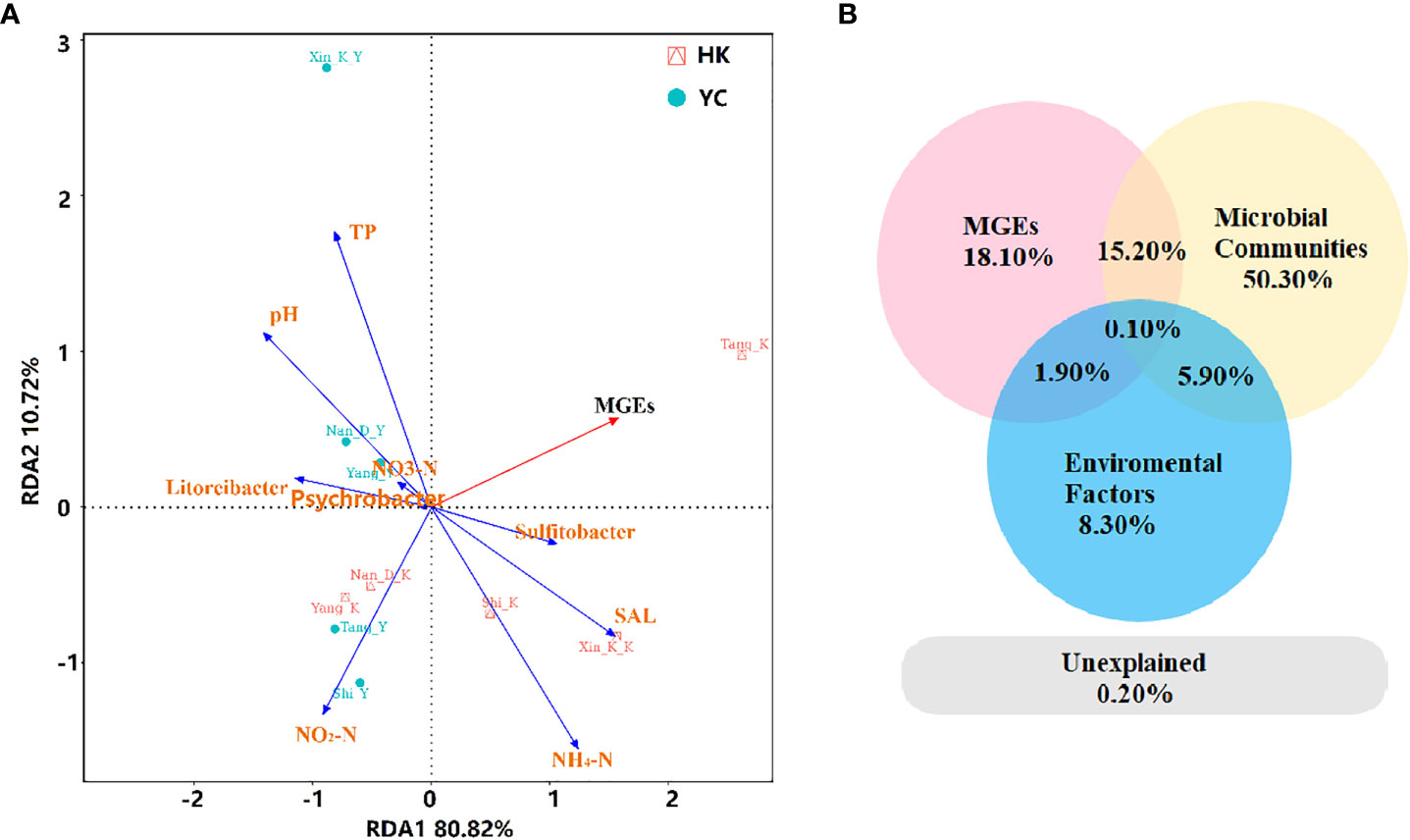
Figure 5 (A) Redundancy analysis (RDA) and (B) partial RDA assess the effects of environmental factors including pH, NH4-N, NO3-N and NO2-N, MGEs and microbial communities (relative abundance of phylum) on the difference of overall ARGs. TP, total phosphorus; SAL, salinity; MGEs, mobile genetic elements.
Discussion
ARGs and MGEs were widely distributed in RCWs
An abundance of diverse ARGs are alarmingly widespread in estuarine environments. Our study detected a broad spectrum of ARGs in RCWs using HT-qPCR techniques, and their normalized copy numbers were close to, or even higher than, those in adjacent estuaries. In particular, 95 and 72 ARGs were detected in the RCWs adjacent to the estuaries of Tang and Xinkai rivers, respectively. The corresponding relative abundances were as high as 2.6 and 1.3/16S rRNA gene, respectively. Clearly, human exposure to these ARGs in RCWs (for example by aerosol inhalation, dermal, eye or ear contact, and oral intake) may represent potential health risks to those swimming in recreational bathing waters, despite there being no currently known infectious dose of antibiotic resistant bacteria or ARGs. In addition, waterborne pathogens also present major threats to public health. Arcobacter spp. were only identified in estuarine samples by the Illumina sequences of their 16S rRNA genes. Whether these species are pathogenic depends on their expression of a few virulence factors. There have been reports of the widespread occurrence of pathogenic marker genes in RCWs, such as the virulence factors of Acanthamoeba spp., Clostridium perfringens, enteropathogenic Escherichia coli, Klebsiella pneumoniae, Vibrio cholera/V. parahaemolyticus and Legionella spp (An et al., 2020). Co-contamination of waters with pathogens and ARGs could probably contribute to the evolution antibiotic resistant pathogenic bacteria. Therefore, the routine monitoring of waterborne ARGs and pathogens is imperative to improve or safeguard human, animal and environmental health, as emphasized in the various “One-Health” initiatives.
The normalized copy numbers of ARGs detected in the RCWs and estuaries of the Tang and Xinkai rivers were even higher than those in some other environmental samples, such as aquaculture waters (P. Gao et al., 2012), estuarine sediments (Y. G. Zhu et al., 2017), and soils (Y. G. Zhu et al., 2013). The high abundance of ARGs in these two estuaries could be explained by the particularly dense populations in these watersheds. Intensive human activities, such as increased sewage discharge and large aquacultural developments might contribute to the relatively high abundance of ARGs in the Tang and Xinkai Rivers and their accumulation in the estuarine environments. Intensive human activity has been reported to facilitate the spread of ARGs, and population density has been positively correlated with the abundance of ARGs (Uyaguari et al., 2013; Berglund, 2015).
ARGs and MGEs associated with clinical setting have been commonly detected in coastal environments, and can often be traced back to particular hospitals and sewage discharges. For example, vancomycin is only used as the “last line” in the treatment of human bacterial infections. Previous studies have detected the vancomycin resistant gene (vanA) in ancient permafrost sediments predating the use of antibiotics (D’Costa et al, 2011), indicating that it occurs naturally. In 2017, the normalized copy number of vancomycin in estuarine sediment around China was as high as 9.3 × 10-3/16S rRNA genes (Zhu et al., 2017). We found vancomycin resistant genes in all of the estuaries and RCWs, with the relative abundances ranging from 3.7 × 10-4 ~ 2.4×10-3 and 1.5 × 10-3 ~ 1.1×10-1/16S rRNA genes, respectively, which could be related to overuse of vancomycin or unregulated wastewater from hospitals or medical facilities. The growing occurrence and abundance of vancomycin resistance presents an urgent problem. The β-Lactamase blaTEM gene had the highest relative abundance in the samples, possibly due to overuse of the antibiotic cephalosporin in clinical settings. It is worth noticing that blaTEM gene has the potential to encode TEM-1 β-lactamase, which requires only a few specific single nucleotide hydrolytic polymorphisms to evolve into a gene encoding an extended-spectrum β-lactamase that degrades all cephalosporins including ceftriaxone (Muhammad et al., 2014). Diversity of clinically relevant antimicrobial resistance genes was found in urban beaches. The clinical intI1 gene commonly detected in RCWs occurs in a variety of clinical pathogenic bacteria and in the human intestinal flora and can be used as a biomarker for municipal sewage pollution and is closely related to human health risks (Zheng et al., 2020). The results were in consistence with previously published paper indicating that diverse clinically relevant ARGs have been found in urban beaches (Furlan et al., 2021). Together, our study indicated that estuaries and RCWs may be polluted by medical and municipal sewage.
ARGs patterns was strongly affected by bacterial phylogenetic compositions in coastal water environments
The RCW samples analyzed in this study were collected along the coastline of Qinhuangdao. However, the two estuaries with the highest normalized copy numbers (the Tang and Xinkai Rivers) were not adjacent to each other, and there was no overall spatial pattern in antibiotic resistomes in the estuarine areas. These results suggested that environmental factors such as tides or latitude did not the drive the occurrence of ARGs in these estuarine environments. Human activities is frequent in this area and people can either acquire ARGs through direct exposure when swimming there and also introduce new ARGs, so promoting their horizontal transfer of ARGs (Chaturvedi et al., 2021). In order to minimize the impact of human activities on the ARGs detected in RCWs, water samples were collected in winter to minimize the anthropogenic variation in the data. ARGs profiles (qPCR data) were significantly correlated with bacterial community (16S rRNA genes, OTU data), as shown by the Mantel test. Environmental factors (NH4+-N, NO3–N, NO2–N, PO43-, salinity, pH), bacterial community compositions (relative abundance of major bacterial phyla), and MGEs accounted for 99.8% of total variation in ARGs profiles in the RCWs. Separation of the variations explained by subsets of these factors indicated that MGEs accounted for 18.1% of the subset variation, while bacterial compositions accounted for a much higher percentage of the variation in ARGs (50.3%, Figure 5B). These data indicated that the particular microbial community was a driving force in the variation of ARGs in these estuarine areas. Thus, any factors that disturb the microbial community, such as salinity, nutrient level, organic pollutants, and storm and runoff levels, may exert previously underestimated increases in abundances of ARGs,.
MGEs are vehicles for horizontal transfer of ARGs, and have been shown to promote transmission and enrichment of ARGs many other environments (Wright et al., 2008; Hoa et al., 2011; K. Zhang et al., 2021). In this study, MGEs accounted for 18.1% of the total variation in ARGs according to the pRDA analysis. Based on the network analysis of ARGs, MGEs, and microbial genera, the correlation between transposons (TnpA) and aminoglycoside resistant gene (aac(6’)-Ib(aka aacA4)) indicated that conjugation may play a role in the dissemination of ARGs in RCWs. Transposons can transfer themselves and related resistant genes at random to a new location within the cell, including plasmids, while integrons can transfer resistant genes between defined sites by site-specific recombination (Cox and Wright, 2013). Although no correlations between ARGs and integrons were detected, the role of MGEs in the enrichment of ARGs in estuarine areas should not be overlooked. It is noteworthy that Acinetobacter, an opportunistic pathogen, was also significantly associated with the aac(6’)-Ib(aka aacA4) gene and may express aminoglycoside resistance. Acinetobacter may cause lung infection through oral inhalation or direct exposure (Wong et al., 2017). In recent years, the occurrence of antibiotic resistant Acinetobacter has been increasing, and ampicillin resistance has remained at a relatively high level (Kutlu et al., 2008). Previous studies have also reported marker genes for Acinetobacter baumanii AB0057 and Acinetobacter oleivorans DR1 in marine recreational beaches around Xiamen City, China (An et al., 2020). Our study showed that coastal environments are ideal places for bacteria or even pathogens to acquire ARGs, and that recreational activities in coastal water may put human beings at risk of antibiotic resistant bacteria infections.
Impacts of estuaries on adjacent recreational coastal waters
PCoA and hierarchical cluster analysis based on Bray-Curtis distance revealed the consistency of ARG occurrences between estuaries and their corresponding RCWs, indicating that estuaries may, to some extent, impact the ARGs in adjacent bathing waters through currents or water exchange. The “Qinhuangdao Recreational Coastal Water Management Regulations”, established to avoid contamination from estuaries, state that the RCWs should be located at least 500 m away from an estuary. However, this distance cannot totally avoid the potential threat of ARGs. ARGs from livestock farms, municipal sewage, medical wastewater, and agricultural sewage outlets may spread to bathing waters through estuaries and threaten human health. Previous studies have reported that if RCWs located downstream from poultry farms or municipal sewage discharge points, people swimming are likely to be exposed to Escherichia coli expressing broad-spectrum β-lactamase (Schijyen et al., 2015). Besides, the diverse and abundant ARGs in RCWs could be due to the fact that frequent human activities contribute to permanent changes in properties of RCWs. For example, changes in environmental factors such as differences in organic matter level (Lin et al., 2019), total nitrogen (J. Chen et al., 2019), and temperature (Liao et al., 2018) have been proven to drive the shift of bacterial communities and promote the propagation of ARGs in aquatic environments. Based on ABT analysis, the concentration of NO2- also had an important effect on the relative abundance of ARGs. We found that NO2—N in RCWs was generally higher than in estuaries. According to Cen et al. (2020), nitrite higher than 1 mg/ml could stimulate free radical-induced RpoS regulators and SOS responses, increase cell membrane permeability, and regulate binding transfer-related genes, thus promoting the horizontal transfer of ARGs. Together, these results indicated that multiple factors could result in the propagation of ARGs and further emphasize the importance of routine surveillance of ARGs in RCWs.
Conclusions
The results of this study showed that the abundance of ARGs in RCWs ranged from 9.9×10-2 to 2.1/16S rRNA gene, higher than in many other environments such as soil, estuarine sediments, and aquacultural developments. The consistency between RCW and their adjacent estuaries indicated that estuaries, to some extent, may have an impact on ARGs in RCWs. Thus, the current rules regulating RCWs cannot avoid the threat of ARGs to human health, and humans may be exposed to ARGs when swimming in RCWs. The Mantel test suggested that microbial community structure is the main force driving the occurrence of ARGs in coastal water environment. Besides the selective pressure of antibiotics, attention should be paid to the spread and enrichment of ARGs that may be overlooked when any perturbation disturbs the microbial composition of swimming waters, such as changes in temperatures, N and P levels, or even algal abundances. Moreover, the consistency between antibiotic resistomes in RCWs and their adjacent estuaries suggested that ARGs from estuaries could contaminate adjacent RCWs. Therefore, proper regulations of bathing areas and reducing the discharge of ARG into terrigenous runoff may be useful in controlling ARG levels in RCWs, and so control the risks to public health. More work is needed to determine the infectious concentrations and risk levels of ARGs to better assess the potential public health risk of ARGs in RCWs. ARG levels and microbial community composition in bathing waters should be routinely detected in real-time.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
YH: Conceptualization, Methodology, Formal analysis, Data curation, Writing – original draft, Writing – review & editing. YKH: Formal analysis, Conceptualization, Writing – review & editing. HXW: Conceptualization, Formal analysis, Writing – review & editing. YWL: Writing – review & editing. JYW: Data curation. YCZ: Methodology. HW: Writing – review & editing. All authors contributed to the article and approved the submitted version.
Funding
This work has been funded by Natural Science Foundation of Hebei Province, China (B2020203005), Cultivation Project for Basic Research and Innovation of Yanshan University (2021LGQN027) and Open Foundation of Key Laboratory of Industrial Ecology and Environmental Engineering, MOE (KLIEEE-20-05).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.976438/full#supplementary-material
References
Ali L., Wang Y. Q., Zhang J., Ajmal M., Xiao Z. R., Wu J. R., et al. (2016). Nutrient-induced antibiotic resistance in enterococcus faecalis in the eutrophic environment. J. Glob. Antimicrob. Resist. 7, 78–83. doi: 10.1016/j.jgar.2016.07.014
An X. L., Wang J. Y., Pu Q., Li H., Pan T., Li H. Q., et al. (2020). High-throughput diagnosis of human pathogens and fecal contamination in marine recreational water. Environ. Res. 190, 109982. doi: 10.1016/j.envres.2020.109982
Berglund B. (2015). Environmental dissemination of antibiotic resistance genes and correlation to anthropogenic contamination with antibiotics. Infect. Ecol. Epidemiol. 5, 28564–28564. doi: 10.3402/iee.v5.28564
Bigus K., Astel A., Niedzielski P. (2016). Seasonal distribution of metals in vertical and horizontal profiles of sheltered and exposed beaches on polish coast. Mar. pollut. Bull. 106 (1-2), 347–359. doi: 10.1016/j.marpolbul.2016.03.009
Bi Weihong Li Yu, Jiancheng S., Yajie J., Pingjie H., Fei Xu (2022). Research review and development trend of marine total organic carbon detection based on spectral technology. J. YanShan Univ. 46 (1), 1–8. doi: 10.3969/j.issn.1007-791X.2022.01.001
Blahova J., Kralikova K., Krcmery V. (1999). Reservoirs, interactions and stability of genetic resistance to antibiotics. the "easy to get–hard to lose" syndrome. [Rezervoary, interakcie a stabilita genov rezistencie na antibiotika. syndrom "easy to get–hard to lose".]. Cas. lek. Cesk. 138 (14), 424–428.
Cen T., Zhang X., Xie S., Li D. (2020). Preservatives accelerate the horizontal transfer of plasmid-mediated antimicrobial resistance genes via differential mechanisms. Environ. Int. 138, 105544. doi: 10.1016/j.envint.2020.105544
Chaturvedi P., Chowdhary P., Singh A., Chaurasia D., Pandey A., Chandra R., et al. (2021). Dissemination of antibiotic resistance genes, mobile genetic elements, and efflux genes in anthropogenically impacted riverine environments. Chemosphere 273, 129693. doi: 10.1016/j.chemosphere.2021.129693
Chen J., Deng W. J., Liu Y. S., Hu L. X., He L. Y., Zhao J. L., et al. (2019). Fate and removal of antibiotics and antibiotic resistance genes in hybrid constructed wetlands. Environ. pollut. 249, 894–903. doi: 10.1016/j.envpol.2019.03.111
Chen H., Zhang M. M. (2013). Effects of advanced treatment systems on the removal of antibiotic resistance genes in wastewater treatment plants from hangzhou, China. Environ. Sci. Technol. 47 (15), 8157–8163. doi: 10.1021/es401091y
Cox G., Wright G. D. (2013). Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. méd. Microbiol. Int. J. Med. Microbiol. 303 (6-7), 287–292. doi: 10.1016/j.ijmm.2013.02.009
Culliton T. J. (1998) Population: Distribution, density, and growth. Available at: https://www.researchgate.net/publication/247207344_Population_Distribution_Density_and_Growth.
D’Costa V. M., King C. E., Kalan L., Morar M., Sung W. W. L., Schwarz C., et al. (2011). Antibiotic resistance is ancient. Nature 377 (7365), 457–461. doi: 10.1038/nature10388
Dalcin E., Jackson P. W. (2018). A network-wide visualization of the implementation of the global strategy for plant conservation in Brazil. Rodriguésia. 69 (4), 1613–1639. doi: 10.1590/2175-7860201869411
Forsberg K. J., Patel S., Gibson M. K., Lauber C. L., Knight R., Fierer N., et al. (2014). Bacterial phylogeny structures soil resistomes across habitats. Nature. 509 (7502), 612–615. doi: 10.1038/nature13377
Freeman C. N., Scriver L., Neudorf K. D., Hansen L. T., Jamieson R. C. (2018). Antimicrobial resistance gene surveillance in the receiving waters of an upgraded wastewater treatment plant. Facets 3, 128–138. doi: 10.1139/facets-2017-0085
Furlan J. P. R., Ramos M. S., dos Santos L. D. R., Gallo I. F. L., Lopes R., Stehling E. G. (2021). Appearance of mcr-9, blaKPC, cfr and other clinically relevant antimicrobial resistance genes in recreation waters and sands from urban beaches, Brazil. Mar. pollut. Bull. 167, 112334. doi: 10.1016/j.marpolbul.2021.112334
Gao F. L., Li J. X., Hu J., Sui B. L., Wang C. A. X., Sun C. J., et al. (2021). The seasonal distribution characteristics of microplastics on bathing beaches along the coast of qingdao, China. Sci. Total Environ. 783, 146969. doi: 10.1016/j.scitotenv.2021.146969
Gao P., Mao D., Luo Y., Wang L., Xu B., Xu L. (2012). Occurrence of sulfonamide and tetracycline-resistant bacteria and resistance genes in aquaculture environment. Water Res. 46 (7), 2355–2364. doi: 10.1016/j.watres.2012.02.004
Gonçalves O. S., Santana M. F. (2021). The coexistence of monopartite integrative and conjugative elements in the genomes of acidobacteria. Gene 777, 145476. doi: 10.1016/j.gene.2021.145476
Hoa P. T. P., Managaki S., Nakada N., Takada H., Shimizu A., Anh D. H., et al. (2011). Antibiotic contamination and occurrence of antibiotic-resistant bacteria in aquatic environments of northern Vietnam. Sci. Total Environ. 409 (15), 2894–2901. doi: 10.1016/j.scitotenv.2011.04.030
Huang H. W., Zeng S. Y., Dong X., Li D., Zhang Y., He M., et al. (2019). Diverse and abundant antibiotics and antibiotic resistance genes in an urban water system. J. Environ. Manage. 231, 494–503. doi: 10.1016/j.jenvman.2018.10.051
Huang H. N., Zheng X., Chen Y. G., Liu H., Wan R., Su Y. L. (2017). Alkaline fermentation of waste sludge causes a significant reduction of antibiotic resistance genes in anaerobic reactors. Sci. Total Environ. 580, 380–387. doi: 10.1016/j.scitotenv.2016.11.186
Jacoby G. A. (2006). β-lactamase nomenclature nomenclature. Antimicrob. Agents Chemother. 50 (4), 1123–1129. doi: 10.1128/AAC.50.4.1123-1129.2006
Jang H. M., Kim Y. B., Choi S., Lee Y., Shin S. G., Unno T., et al. (2018). Prevalence of antibiotic resistance genes from effluent of coastal aquaculture, south Korea. Environ. pollut. 233, 1049–1057. doi: 10.1016/j.envpol.2017.10.006
Kutlu S. S., Sacar S., Suzer T., Cevahir N., Okke D., Caylak S. D., et al. (2008). Successful treatment of a patient with multidrug resistant acinetobacter baumannii meningitis with high dose ampicillin-sulbactam. Mikrobiyol Bul. 42 (2), 353–358.
Liao H., Lu X., Rensing C., Friman V. P., Geisen S., Chen Z., et al. (2018). Hyperthermophilic composting accelerates the removal of antibiotic resistance genes and mobile genetic elements in sewage sludge. Environ. Sci. Technol. 52 (1), 266–276. doi: 10.1021/acs.est.7b04483
Lin W. F., Wan K., Zeng J., Li J. J., Li X., Yu X. (2019). Low nutrient levels as drinking water conditions can reduce the fitness cost of efflux pump-mediated ciprofloxacin resistance in pseudomonas aeruginosa. J. Environ. Sci(China) . 83, 123–132. doi: 10.1016/j.jes.2019.03.022
Magoc T., Salzberg S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 27 (21), 2957–2963. doi: 10.1093/bioinformatics/btr507
Marti E., Variatza E., Balcazar J. L. (2014). The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol. 22 (1), 36–41. doi: 10.1016/j.tim.2013.11.001
Muhammad I., Golparian D., Dillon J. A. R., Johansson A., Ohnishi M., Sethi S., et al. (2014). Characterisation of bla(TEM) genes and types of beta-lactamase plasmids in neisseria gonorrhoeae - the prevalent and conserved bla(TEM-135) has not recently evolved and existed in the Toronto plasmid from the origin. BMC Infect. Dis. BMC Infect. Dis. 14, 454. doi: 10.1186/1471-2334-14-454
O' Neil J. (2014)Review on antimicrobial resistance antimicrobial resistance: tackling a crisis for the health and wealth of nations. In: Review on antimicrobial resistance. Available at: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (Accessed 9 March 2022).
Ouyang W. Y., Huang F. Y., Zhao Y., Li H., Su J. Q. (2015). Increased levels of antibiotic resistance in urban stream of jiulongjiang river, China. Appl. Microbiol. Biotechnol. 99 (13), 5697–5707. doi: 10.1007/s00253-015-6416-5
Pruden A., Pei R. T., Storteboom H., Carlson K. H. (2006). Antibiotic resistance genes as emerging contaminants: Studies in northern Colorado. Environ. Sci. Technol. 40 (23), 7445–7450. doi: 10.1021/es060413l
Qinhuangdao Municipal People’s Government (2019) Regulations of qinhuangdao municipality on the administration of bathing beaches. Available at: http://www.qhd.gov.cn/front_pcthi.do?uuid=8F0D6662565323FFB03AC4C9AE146E42 (Accessed 9 March 2022).
Rider B. F., Mellon M. G. (1946). Colorimetric determination of nitrites. Ind. Eng. Chem. Analytical Ed. 18 (2), 96–99. doi: 10.1021/i560150a003
Rodriguez-Mozaz S., Chamorro S., Marti E., Huerta B., Gros M., Sanchez-Melsio A., et al. (2015). Occurrence of antibiotics and antibiotic resistance genes in hospital and urban wastewaters and their impact on the receiving river. Water Res. 69, 234–242. doi: 10.1016/j.watres.2014.11.021
Rognes T., Flouri T., Nichols B., Quince C., Mahe F. (2016). VSEARCH: a versatile open source tool for metagenomics. Peerj 4, e2584. doi: 10.7717/peerj.2584
Sarkar S. K., Chang C. K. (1997). The simes method for multiple hypothesis testing with positively dependent test statistics. J. Am. Stat. Assoc. 92 (440), 1601–1608. doi: 10.2307/2965431
Schijyen J. F., Blaak H., Schets F. M., Husman A. M., d. R. (2015). Fate of extended-spectrum beta-Lactamase-Producing escherichia coli from faecal sources in surface water and probability of human exposure through swimming. Environ. Sci. Technol. 49 (19), 11825–11833. doi: 10.1021/acs.est.5b01888
Stepanauskas R., Glenn T. C., Jagoe C. H., Tuckfield R. C., Lindell A. H., King C. J., et al. (2006). Coselection for microbial resistance to metals and antibiotics in freshwater microcosms. Environ. Microbiol. 8 (9), 1510–1514. doi: 10.1111/j.1462-2920.2006.01091.x
Stewart J. R., Gast R. J., Fujioka R. S., Solo-Gabriele H. M., Meschke J. S., Amaral-Zettler L. A., et al. (2008). The coastal environment and human health: microbial indicators, pathogens, sentinels and reservoirs. Environ. Health 7, s3. doi: 10.1186/1476-069x-7-s2-s3
Su J., Fan J., Ming H., Guo G., Fu Y., Zhao X., et al. (2022). The municipal sewage discharge may impact the dissemination of antibiotic-resistant escherichia coli in an urban coastal beach. Water 14, 1639. doi: 10.3390/w14101639
Sun J. T., Jin L., He T. T., Wei Z., Liu X. Y., Zhu L. Z., et al. (2020). Antibiotic resistance genes (ARGs) in agricultural soils from the Yangtze river delta, China. Sci. Total Environ. 740, 140001. doi: 10.1016/j.scitotenv.2020.140001
Su J. Q., Wei B., OuYang W. Y., Huang F. Y., Zhao Y., Xu H. J., et al. (2015). Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ. Sci. Technol. 49 (12), 7356–7363. doi: 10.1021/acs.est.5b01012
Tamminen M., Karkman A., Lohmus A., Muziasari W. I., Takasu H., Wada S., et al. (2011). Tetracycline resistance genes persist at aquaculture farms in the absence of selection pressure. Environ. Sci. Technol. 45 (2), 386–391. doi: 10.1021/es102725n
Tandukar M., Oh S., Tezel U., Konstantinidis K. T., Pavlostathis S. G. (2013). Long-term exposure to benzalkonium chloride disinfectants results in change of microbial community structure and increased antimicrobial resistance. Environ. Sci. Technol. 47 (17), 9730–9738. doi: 10.1021/es401507k
Tomova A., Ivanova L., Buschmann A. H., Rioseco M. L., Kalsi R. K., Godfrey H. P., et al. (2015). Antimicrobial resistance genes in marine bacteria and human uropathogenic escherichia coli from a region of intensive aquaculture. Environ. Microbiol. Rep. 7 (5), 803–809. doi: 10.1111/1758-2229.12327
Uyaguari M. I., Scott G. I., Norman R. S. (2013). Abundance of class 1-3 integrons in south Carolina estuarine ecosystems under high and low levels of anthropogenic influence. Mar. pollut. Bull. 76 (1-2), 77–84. doi: 10.1016/j.marpolbul.2013.09.027
von Wintersdorff C. J. H., Penders J., van Niekerk J. M., Mills N. D., Majumder S., van Alphen L. B., et al. (2016). Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00173
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73 (16), 5261–5267. doi: 10.1128/aem.00062-07
Wang Y. Q., Lu S. Y., Liu X. H., Chen J., Han M. Z., Wang Z., et al. (2021). Profiles of antibiotic resistance genes in an inland salt-lake ebinur lake, xinjiang, China: The relationship with antibiotics, environmental factors, and microbial communities. Ecotoxicol. Environ. Saf. 221, 112427. doi: 10.1016/j.ecoenv.2021.112427
Wei Z. L., Miao J., Yang Z. W., Shi D. Y., Wu H. Y., Yang D., et al. (2020). Contamination sources of the enteric virus in recreational marine water shift in a seasonal pattern. Sci. Total Environ. 743, 140641. doi: 10.1016/j.scitotenv.2020.140641
Wen H.-q., Shi J., Xun H., Deng H. P. (2015). Distribution, dissemination and removal of antibiotic resistant genes (ARGs) in the aquatic environment. J. Appl. Ecol. 26 (2), 625–635.
Wong D., Nielsen T. B., Bonomo R. A., Pantapalangkoor P., Luna B., Spellberg B. (2017). Clinical and pathophysiological overview of acinetobacter infections: a century of challenges. Clin. Microbiol. Rev. 30 (1), 409–447. doi: 10.1128/cmr.00058-16
Wright M. S., Baker-Austin C., Lindell A. H., Stepanauskas R., Stokes H. W., McArthur J. V. (2008). Influence of industrial contamination on mobile genetic elements: class 1 integron abundance and gene cassette structure in aquatic bacterial communities. ISME J. 2 (4), 417–428. doi: 10.1038/ismej.2008.8
Wu D., Ma R. Q., Wei H. W., Yang K., Xie B. (2018). Simulated discharge of treated landfill leachates reveals a fueled development of antibiotic resistance in receiving tidal river. Environ. Int. 114, 143–151. doi: 10.1016/j.envint.2018.02.049
Wu B., Song J. M., Li X. G. (2014). Evaluation of potential relationships between benthic community structure and toxic metals in laizhou bay. Mar. pollut. Bull. 87 (1-2), 247–256. doi: 10.1016/j.marpolbul.2014.07.052
Yang J., Wang H., Roberts D. J., Du H. N., Yu X. F., Zhu N. Z., et al. (2020). Persistence of antibiotic resistance genes from river water to tap water in the Yangtze river delta. Sci. Total Environ. 742, 140592. doi: 10.1016/j.scitotenv.2020.140592
Yang J., Wang C., Shu C., Liu L., Geng J., Hu S., et al. (2013). Marine sediment bacteria harbor antibiotic resistance genes highly similar to those found in human pathogens. Microb. Ecol. 65 (4), 975–981. doi: 10.1007/s00248-013-0187-2
Zhang Y. J., Hu H. W., Yan H., Wang J. T., Lam S. K., Chen Q. L., et al. (2019). Salinity as a predominant factor modulating the distribution patterns of antibiotic resistance genes in ocean and river beach soils. Sci. Total Environ. 668, 193–203. doi: 10.1016/j.scitotenv.2019.02.454
Zhang T., Li J., Wang N., Wang H., Yu L. (2021). Metagenomic analysis reveals microbiome and resistome in the seawater and sediments of kongsfjorden (Svalbard, high Arctic). Sci. Total Environ. 809, 151937. doi: 10.1016/j.scitotenv.2021.151937
Zhang K., Xin R., Zhao Z., Li W. P., Wang Y. N., Wang Q., et al. (2021). Mobile genetic elements are the major driver of high antibiotic resistance genes abundance in the upper reaches of huaihe river basin. J. Hazard. Mater 401, 123271. doi: 10.1016/j.jhazmat.2020.123271
Zheng W. L., Huyan J. Q., Tian Z., Zhang Y., Wen X. H. (2020). Clinical class 1 integron-integrase gene - a promising indicator to monitor the abundance and elimination of antibiotic resistance genes in an urban wastewater treatment plant. Environ. Int. 135, 105372. doi: 10.1016/j.envint.2019.105372
Zhuang M., Achmon Y., Cao Y. P., Liang X. M., Chen L., Wang H., et al. (2021). Distribution of antibiotic resistance genes in the environment star. Environ. pollut. 285, 117402. doi: 10.1016/j.envpol.2021.117402
Zhu Y. G., Johnson T. A., Su J. Q., Qiao M., Guo G. X., Stedtfeld R. D., et al. (2013). Diverse and abundant antibiotic resistance genes in Chinese swine farms. P. Natl. Acad. Sci. U.S.A. 110 (9), 3435–3440. doi: 10.1073/pnas.1222743110
Zhu L. H., Wu J. Z., Xu Z. Q., Xu Y. C., Lin J., Hu R. J. (2014). Coastline movement and change along the bohai Sea from 1987 to 2012. J. Appl. Remote. Sens 8, 083585. doi: 10.1117/1.jrs.8.083585
Keywords: antibiotic resistance genes (ARGs), mobile genetic elements (MGEs), bacterial community, recreational coastal water, human public health
Citation: Han Y, Hu Y, Wen H, Wu J, Liu Y, Zhang Y and Wu H (2022) Occurrence and driving mechanism of antibiotic resistance genes in marine recreational water around Qinhuangdao, China. Front. Mar. Sci. 9:976438. doi: 10.3389/fmars.2022.976438
Received: 23 June 2022; Accepted: 24 August 2022;
Published: 15 September 2022.
Edited by:
Songzhe Fu, Dalian Ocean University, ChinaReviewed by:
Ilunga Kamika, University of South Africa, South AfricaJingfeng Fan, National Marine Environmental Monitoring Center, China
Copyright © 2022 Han, Hu, Wen, Wu, Liu, Zhang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Han, aGFueWluZ0B5c3UuZWR1LmNu