 Peleg Itay1Eli Shemesh1Maya Ofek-Lalzar2Nadav Davidovich1,3Yael Kroin1Shlomi Zrihan1Nir Stern4Arik Diamant1Natascha Wosnick5Dalit Meron1Dan Tchernov1Danny Morick1,6*
Peleg Itay1Eli Shemesh1Maya Ofek-Lalzar2Nadav Davidovich1,3Yael Kroin1Shlomi Zrihan1Nir Stern4Arik Diamant1Natascha Wosnick5Dalit Meron1Dan Tchernov1Danny Morick1,6*- 1Morris Kahn Marine Research Station, Department of Marine Biology, Leon H. Charney School of Marine Sciences, University of Haifa, Haifa, Israel
- 2Bioinformatics Services Unit, University of Haifa, Haifa, Israel
- 3Aquatic Animals Health, Israeli Veterinary Services, Bet Dagan, Israel
- 4National Institute of Oceanography, Israel Oceanographic and Limnological Research, Haifa, Israel
- 5Programa de Pós-graduação em Zoologia, Universidade Federal do Paraná, Curitiba, Paraná, Brazil
- 6Hong Kong Branch of Southern Marine Science and Engineering, Guangdong Laboratory (Guangzhou), Hong Kong, Hong Kong SAR, China
Bacterial diseases of marine fish inflict significant economic damage to fisheries and aquaculture and pose an increasing risk to public health. When addressing fish disease, an accumulating body of research suggests adding another factor to the classic epidemiological triangle of host-environment-pathogen: the microbiome. The gills, being a gateway into the fish body and bearing an important role in fish homeostasis, have been found to be a proxy of the gut microbiota as well as reflecting the microbial communities of surrounding water. In this study, 16S rRNA amplicons of bacterial DNA extracted from the gills of 89 asymptomatic specimens of three wild fish species (Pagrus caeruleostictus, Scomber colias and Saurida lessepsianus) were sequenced using Next Generation Sequencing methodology (NGS). Data analyses revealed the presence of 41 potentially pathogenic species, including several zoonotic agents. Five genera known to include widespread and potentially pathogenic species were chosen for further investigation: Photobacterium, Shewanella, Staphylococcus, Streptococcus and Vibrio. Of these, Photobacterium and Shewanella proved the most prevalent and abundant, making up 30.2% and 11.3% of the Bluespotted seabream (P. caeruleostictus) gill microbiome alone. Photobacterium damselae and Shewanella baltica were most common at the species level. The remaining genera - Vibrio, Staphylococcus and Streptococcus – were less prevalent, and at a species level were comprised of only 1−4% potentially pathogenic representatives. Gill microbiomes exhibited host species specificity, with strong correlations between certain bacterial taxonomic groups. No definite obligatory pathogenic bacteria were found in this study, and it was suggested that pathogenic species are present as either covert pathobionts or as opportunists of the fish found to host them.
Introduction
In many areas of the world, one of the most common horizontal transmission routes of pathogens into wild fish is the rapidly growing mariculture cage-farm industry (Arechavala-Lopez et al., 2013; Barrett et al., 2019; Shea et al., 2020). A reverse pattern of pathogen transmission can also be observed from wild fish to farm stocks (Arechavala-Lopez et al., 2013). Fish cage farms have become ecological hotspots, releasing a steady source of residual uneaten feed and providing a refuge for small-bodied fish species in an otherwise unsheltered open sea habitat. The farms also present an opportunity for replenishment for migratory fish (Shea et al., 2020) and become an attraction for predators (Papastamatiou et al., 2010; Piroddi et al., 2011; Barash et al., 2018). Today, over a quarter of globally farmed fish species are non-native to their rearing environment (Atalah and Sanchez-jerez, 2020), which means in addition to competing with native wild populations over local resources, mariculture escapees pose a risk for introduction of alien pathogens to naïve hosts. The effects of mariculture are coupled with many other anthropogenic factors that increase risk of disease outbreaks in wild fish, including increases in sea surface temperature, pollution and structural alterations to ecosystems through development and industry (Harvell et al., 1999; Halpern et al., 2008; Lejeusne et al., 2010; Nguyen and Liou, 2019). Human-driven changes in the ocean environment directly impact the health of fish. When faced with physiochemical conditions outside of their optimal range, fish may become stressed and immunosuppressed, lowering their defenses against agents of infectious disease (Johnson et al., 1992; Conte, 2004). The epidemiological triangle, describing such interactions between a host, a pathogen, and their environment (King et al., 2019), forms the basis of research aimed at understanding the effects of disease on marine animals (Andrade et al., 2017; Elarabany et al., 2017; Wang et al., 2018; Genin et al., 2020; Zarantoniello et al., 2021).
In recent years, there has been great interest in adding the contribution of the microbiome to this complex interplay, applying the concept of the holobiont, a host with all of its associated microorganisms, to disease research. The intimate partnership between hosts and their symbiotic microbiota plays a significant role in host maintenance and well-being, contributing to metabolism, immune system maturation, and additional defenses against pathogenic invaders (Aschenbrenner et al., 2016; Ramsey et al., 2016; Apprill, 2017; Vorburger and Perlman, 2018). Shifts in this microbial composition due to external pressures from natural or anthropogenic changes in the environment (Halpern et al., 2008; Pérez-Ruzafa et al., 2018; Nguyen and Liou, 2019), or internal physiological pressures (Yildirimer and Brown, 2018), may lead to substantial consequences for the host, and maintaining the balance of the microbiome has been shown to be of importance for maintenance of fish health (Llewellyn et al., 2014). Microbiota composition shifts may serve as early-warning bioindicators, enabling assessment of the host’s health even before clinical signs become visible. Combined with data on shifts in pathogen prevalence, these aspects become key factors in understanding the intricacies of host-pathogen relations.
Previous fish microbiome studies focused mostly on skin and intestinal microbiota (Ni et al., 2013; Liu et al., 2016; Egerton et al., 2018; Tarnecki et al., 2019; Krotman et al., 2020), and occasionally on other organs, such as kidneys and liver (Sevellec et al., 2014; Meron et al., 2020). These studies found that gut, kidney microbiota are deeply influenced by fish trophic levels and diversity of their prey, while skin microbiota is both highly adaptive and affected by qualities of the ambient water. Gills, however, are receiving increasing attention, as they may be sampled non-destructively from live fish (Merrifield and Rodiles, 2015; Mohammed and Arias, 2015). In addition to their role in gas and waste exchange (Evans et al., 2005), gills are a gateway into the fish body and an important site of mucosal immunity (Salinas, 2015), constantly in direct interaction with the aquatic environment and its associated microbes. To a certain extent, the gill microbiome reflects the microbial composition of the water, including pathogens present (Kuang et al., 2020). Understanding the mechanisms of pathogen adherence to and entry through the gill mucosal barrier, and the potential impact of the microbiome and surrounding environment on this process, is an ongoing challenge in aquaculture. To date, only a handful of studies aimed at farmed (Brown et al., 2019; Rosado et al., 2019; Minich et al., 2020) or wild fish (Hess et al., 2015; Minich et al., 2020) have been published on the gill microbiome. Pratte et al., (2018) studied reef fish and found that gill and intestinal microbiomes from the same individual showed greater similarity than respective gill or intestinal microbiomes from different individuals, and these authors concluded the presence of a core microbiome amidst the intra and inter-species variances. The gill microbial community, then, could be a representative metric for the total fish microbiome.
Due to its cost-effectiveness and relative accuracy (Caporaso et al., 2011; Vayssier-Taussat et al., 2013; Walters et al., 2015), the use of 16S rRNA amplicon sequencing is considered common practice in such studies aiming to elucidate the composition of bacterial communities (Sevellec et al., 2014; Mohammed and Arias, 2015; Pratte et al., 2018; Krotman et al., 2020; Meron et al., 2020; Minich et al., 2020). A certain tradeoff exists between accuracy at the species and subspecies level and the ability to comprehensively screen bacterial communities (Ghyselinck et al., 2013; Martínez-porchas et al., 2016). Improvements in primer design and bioinformatics tools have helped bridge that gap, and enabled both higher reliability of NGS screening and the additional benefit of discovering novel species (Al-Hebshi et al., 2015; Johnston et al., 2017; Abu Fanas et al., 2021; Greay et al., 2021). In the present study, we use 16S rRNA NGS screening to provide an analysis of the gill microbiome of three fish species, in order to assess community composition and the presence of potential pathogens. Furthermore, this study aims to show that this method is useful in detecting multiple fish pathogens in parallel and finding correlations between pathogenic species residing together.
Materials and methods
Fish collection
All fish samples used were collected during a trawler survey conducted during May−June 2020, at depths of 20−80m, as part of a biannual survey, conducted by the Israeli Oceanographic and Limnological Research center (IOLR), in Haifa, Israel. The surveys are carried out at constant locations southwest of Ashdod, and eight kilometers away from cultured fish cages. Fish were immediately placed on ice and transferred to the lab. Eighty-nine fish individuals were collected: Atlantic chub mackerel (Scomber colias; n = 40), Bluespotted seabream (Pagrus caeruleostictus; n = 25) and Lessepsian lizardfish (Saurida lessepsianus; n = 24). Some of the samples were dissected fresh while others were frozen at −20°C to be later thawed and necropsied.
Tissue sampling
Frozen specimens were gradually thawed in small batches, weighed and measured, and were then dissected aseptically according to an established fish necropsy protocol (Yanong, 2003). Gills tissue samples were gently removed and placed in predesignated test tubes, then frozen at a temperature of −80°C until undergoing DNA extraction. P. caeruleostictus samples ranged in length between 11.4-20.4cm and in weight between 23.4-153.4g. S. lessepsianus samples ranged in length between 15.2-30.2cm and in weight between 21.7-230.7g. S. colias samples ranged in length between 13.2-19.5cm and in weight between 18.6-71.8g.
DNA extraction
Extractions of DNA were done using the GeneMATRIX Soil DNA Purification Kit (EURx, Gdańsk, Poland), following the manufacturer instructions for tissue lysates, with an additional two hour incubation at 55°C following suspension of the sample tissue in the kit-provided lysis buffer. DNA quality was examined by NanoDrop spectrophotometry analysis and agarose gel-electrophoresis.
PCR amplification and amplicon sequencing
Total DNA extracts were used as template for amplification of partial 16S rRNA gene sequences, at the V4 hypervariable region. Amplicons were generated using a two-stage PCR amplification protocol as described previously (Naqib et al., 2018). Each of the first stage PCR reactions consisted of a total of 50μl in volume and included: 25μl of GoTaq Green Master mix (Promega, Fitchburg, WI, USA), 2μl of mixed forward and reverse primers (in a concentration of 1nM each), 2μl of bovine serum albumin (BSA), 18μl of ultra-purified water (UPW) and 3μl of 80ng/μl template DNA. The primers contained 5’ common sequence tags (known as common sequence 1 and 2, CS1 and CS2) compatible with Access Array™ primers for Illumina sequencers (Fluidigm, South San Francisco, CA, USA) (Caporaso et al., 2012). The primers used for amplification were (linker sequences in bold): CS1_518F: 5’ –ACACTGACGACATGGTTCTACACCAGCAGCCGCGGTAATACG – 3’ (Nakasaki et al., 2009) and CS2_806Rc: 5’ – TACGGTAGCAGAGACTTGGTCTGGACTACNVGGGTWTCT – 3’ (Walters et al., 2015).
The PCR conditions were as follows: 10 cycles of denaturation at 95°C for 15s, annealing at 60°C for 15s and elongation at 72°C for 30s; followed by 10 cycles of denaturation at 95°C (15s), annealing at 55°C (15s) and elongation at 72°C (30s); continued with 10 more cycles at 95°C (15s)/50°C (15s)/72°C (30s); and then 5 additional cycles with yet another change of annealing temperature, performed at 62°C. The PCR concluded with 2 minutes of incubation at 72°C, before being lowered to 4°C for one hour (or until samples were removed). Amplicons were sent to UIC Sequencing Core (Chicago, IL, USA), in which a second PCR amplification was performed in 10 microliter reactions in 96-well plates using MyTaq HS 2X mastermix (Bioline, Taunton, MA, USA). Each well received a separate primer pair with a unique 10-base barcode, obtained from the Access Array Barcode Library for Illumina (Fluidigm, South San Francisco, CA; Item# 100-4876). One microliter of PCR product from the first stage amplification was used as template for the 2nd stage, without cleanup. Cycling conditions were 95°C for 5 minutes, followed by 8 cycles of 95°C for 30”, 60°C for 30” and 72°C for 30”. Libraries were then pooled and sequenced with a 20% phiX spike-in on an Illumina Miniseq sequencer employing a mid-output flow cell (2x150 paired-end reads). Final library preparation, pooling, and sequencing were performed at the Genome Research Core (GRC) at the University of Illinois at Chicago (UIC).
Sequence data processing
Detailed information regarding the sequence data processing is provided in the Supplementary Information File. In brief, sequence data was analyzed using the Dada2 pipeline (Callahan et al., 2016) using R package ‘dada2’ (version 1.14.1). Error rate estimation was carried out in order to sample nucleotides and reads for model building randomly across all samples. The dada2 algorithm was implemented for error correction and a count table containing the amplicon sequence variants and counts per sample was produced. For each amplicon sequence variant (ASV), taxonomy (up to the species level) was inferred by alignment to the Silva non-redundant small subunit ribosomal RNA database (version 138) using dada2 commands ‘assignTaxonomy’ and “addSpecies” with minimum bootstrap value set to 80%.
Data analysis
All data filtering parameter settings are detailed in the Supplementary Information File. In short, for data analysis and generation of figures, the online tool MicrobiomeAnalyst (https://www.microbiomeanalyst.ca/MicrobiomeAnalyst/home.xhtml) was used (Dhariwal et al., 2017; Chong et al., 2020). Taxonomy labels were assigned using the SILVA taxonomic framework (https://www.arb-silva.de/documentation/silva-taxonomy/). Initial analyses provided 189 bacterial ASVs identified to the taxonomic level of species, with 177 unique values (i.e., species). All 177 species were searched in the literature using their species name separately and together with conjugations of ‘Pathogen’, ‘Infection’ or ‘Disease’, with and without reference to fish/humans. In addition, sequences belonging to “pathogenic” genera were run through BLAST. This enabled identifying four more species and raised the total number of species to 181. Forty-one species were categorized as pathogenic to marine animals and/or humans. A literature-based scale was built applying several categories for their range of pathogenicity: from ‘Unknown’ to ‘Rarely’, ‘Pathobiont’, ‘Opportunistic’, ‘Yes’ and ‘Obligatory’. Species were labeled ‘Unknown’ whenever the literature provided no evidence of pathogenicity whatsoever. ‘Rarely’ is a term used in the literature almost solely in reference to human pathogens. The commonly used term ‘Facultative’ was split into ‘Pathobiont’ and ‘Opportunistic’, differentiating them by defining the former as a mutualistic symbiont becoming virulent under certain conditions, while the latter refers to a commensal symbiont of pathogenic capabilities, a ‘hitchhiker’ that usually does not provide useful services to the host – nor causes harm – but turns virulent when conditions are favorable of it. ‘Yes’ marks an uncertainty whether the pathogen should be categorized as ‘Rarely’, ‘Pathobiont’ or ‘Opportunistic’. ‘Obligatory’ refers to obligatory pathogens, meaning they always express virulence. This is a rare attribute found in bacteria and no obligatory pathogens were found in this study.
Phylogenetic trees
A detailed account of the parameters used for creating trees is given in the Supplementary Information File. Briefly, sequences identified as belonging to the several genera chosen for deeper enquiry were uploaded to Silva (https://www.arb-silva.de/aligner/) for preparing phylogenetic files (Quast et al., 2013; Yilmaz et al., 2014; Oliver et al., 2017). The ACT (Alignment, Classification and Tree Service) tool was used (SINA v1.2.11) (Pruesse et al., 2012). Output TREE format files were extracted for visualization with the FigTree v1.4.3 software (http://tree.bio.ed.ac.uk/software/figtree/).
Results
All of the 89 fish collected appeared healthy both externally and internally upon inspection and necropsy. The community structure of the gill samples differed between fish species (Figure 1). Atlantic chub mackerel (ACM; Scomber colias) exhibited a higher and richer composition (Simpson index average: 0.9) than the Lessepsian lizardfish (LLF; Saurida lessepsianus) and Bluespotted seabream (BSSB; Pagrus caeruleostictus), which displayed indexes of 0.75 and 0.81, respectively. The LLF had the highest variance between samples. In all three fish species, low-richness outliers did not display an unusual increase in pathogenic agents’ presence. A comparison of compositions (Figure 2) shows a clustering of microbiomes among species, with ACM displaying a community structure least similar to the others, and BSSB sharing most of its microbiome with the two other species.
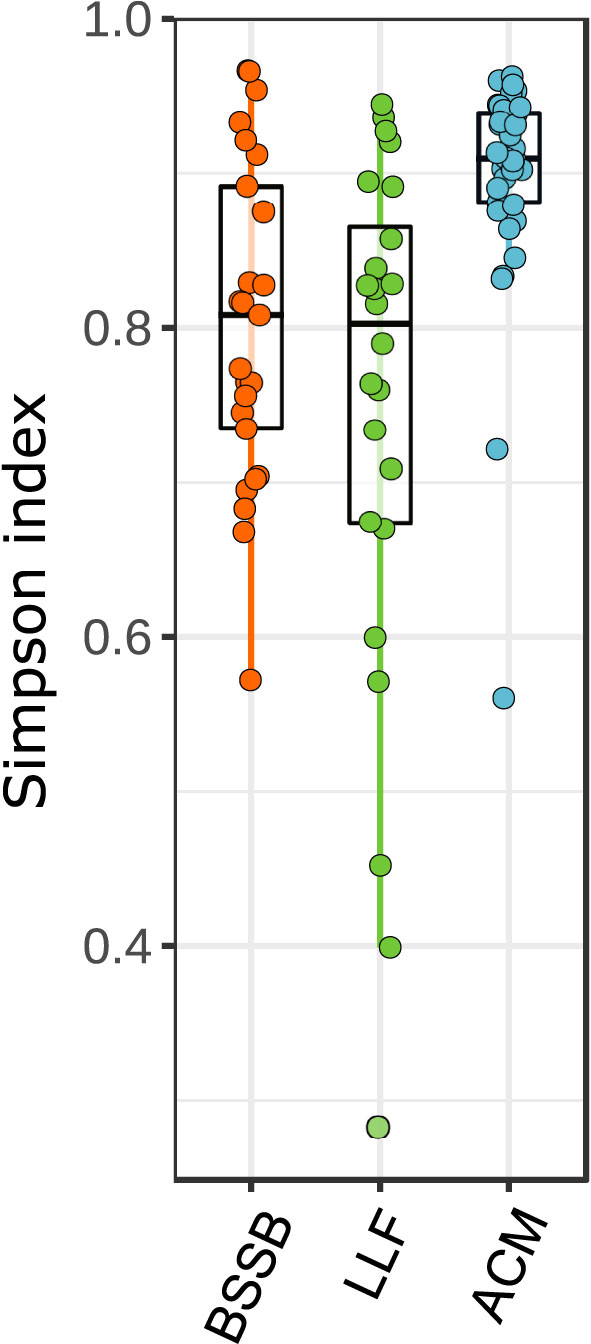
Figure 1 Community structure of fish gills samples. Data clustered by fish species. Kruskal-Wallis statistic: 24.928, P<0.001.
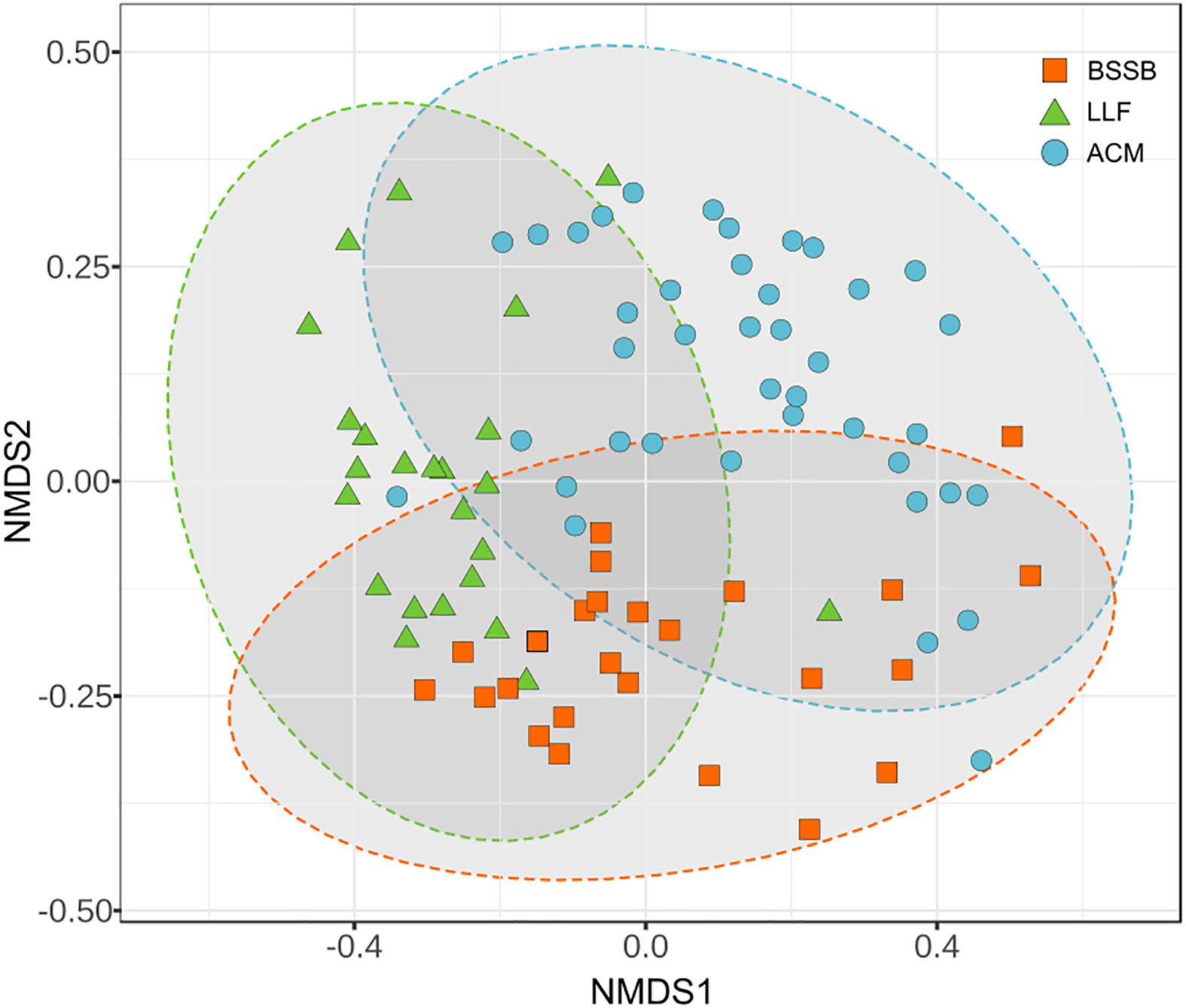
Figure 2 Comparison of compositions of the gills’ microbiome between the sampled fish species. PERMANOVA: F: 7.7605, R²: 0.1529, P<0.001, NMDS stress = 0.1740.
An interaction network (Figure 3), expressing the strength of ties between bacteria genera to each other and their tendency to be hosted by the different fish species, highlights three main cohorts: the ‘Psychrobacter cohort’, the ‘Photobacterium cohort’ and the ‘Staphylococcus-Streptococcus cohort’. The Psychrobacter cohort was the most diverse and contained few genera associated with pathogenicity. It was mostly associated with ACM. The ‘Photobacterium cohort’ expressed correlation especially to BSSB samples, and presented ties between the Gram-negative Gammaproteobacteria class members Vibrio, Aliivibrio, Photobacterium and Shewanella, together with Cetobacterium (class Fusobacteriia). The ‘Staphylococcus-Streptococcus cohort’ was most strongly associated with LLF, and exhibited correlations between Gram-positive bacteria Staphylococcus, Streptococcus and Gemella (Bacilli) with Actinomyces, Cutibacterium, Micrococcus and Rothia (Actinobacteria). It also exhibited correlations with the Gram-negative Cloacibacteria (Bacteroidia) and Enhydrobacter (Gammaproteobacteria).
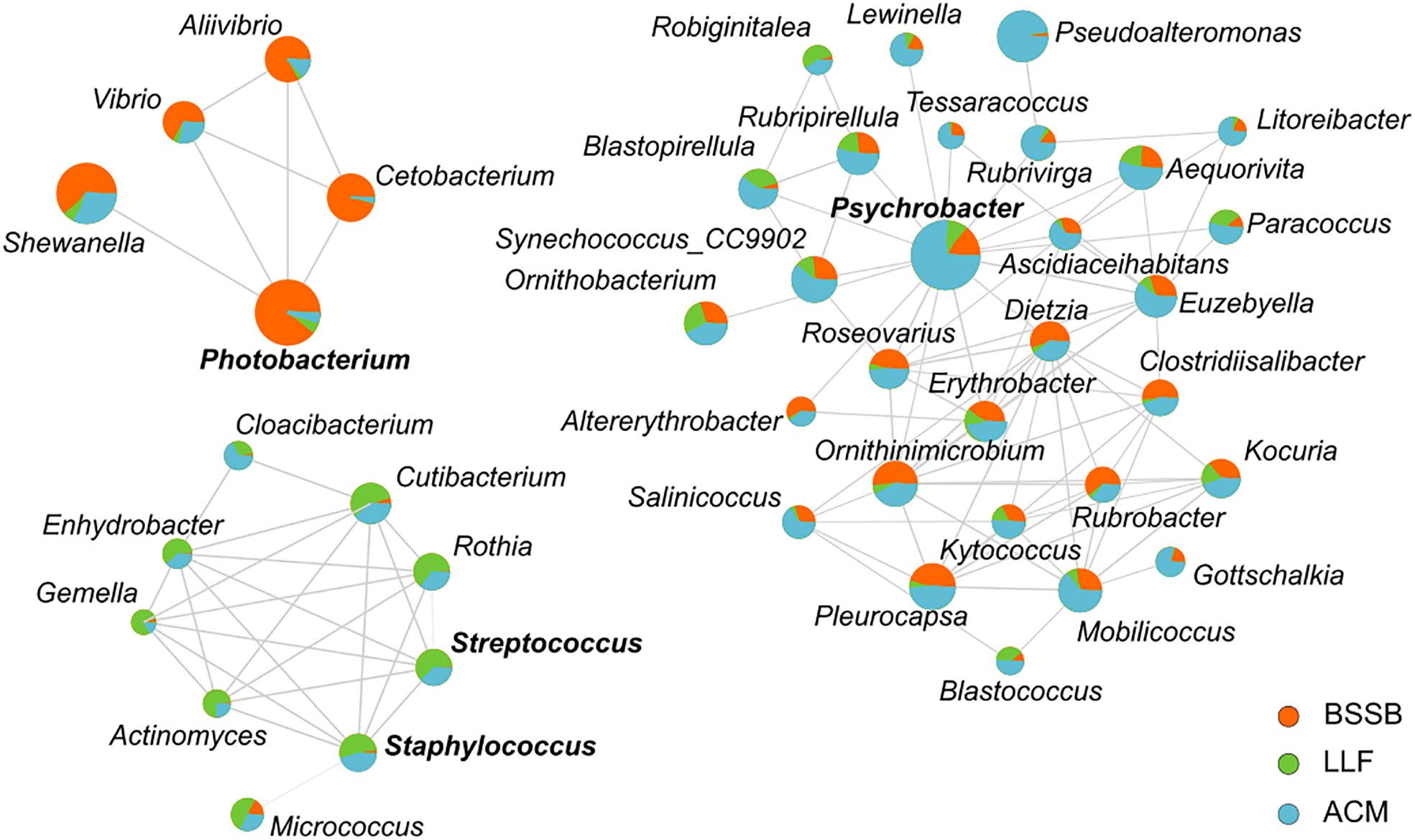
Figure 3 An interaction network between bacterial genera, in reference to fish species. Network calculation used Spearman’s rank correlation coefficient and the threshold settings placed at: correlation > 0.5; P<0.05. Size of circles represents abundance and the coloring associates bacteria with host in regards to mean abundance. Names of 4 genera are emphasized by bold type, in order to identify them as those after which each cohort was named (i.e., Photobacterium cohort, Psychrobacter cohort and Staphylococcus-Streptococcus cohort).
The relative abundance of each genus by fish type further supports species-specific microbial composition (Figure S2). The ACM gill microbiome was predominated by Psychrobacter (28.3%), and BSSB by Photobacterium (30.2%). These two genera were notably present in all three species, alongside Shewanella. The Cetobacterium, Aliivibrio, Cutibacterium, Vibrio, Rothia, Staphylococcus and Streptococcus genera were distributed in lower abundance between two or three species. Over 65% of the LLF, 27% of the ACM and 18% of the BSSB microbiome reads were classified as “Not-assigned” with an additional set of reads in each returned as “others”, a summary of low count genera. The genera known to include many potential pathogenic species – Photobacterium, Shewanella, Staphylococcus, Streptococcus, and Vibrio – were also unevenly distributed between fish types (Figure 4). From this figure there are several observations to be made: (i) except for Staphylococcus, BSSB is host to a larger percentage of potentially pathogenic bacterial species than the other two fish species; (ii) error bars across all three fish species indicate that large variances occur amongst samples from each fish species, especially in the top quarter percentile of samples.
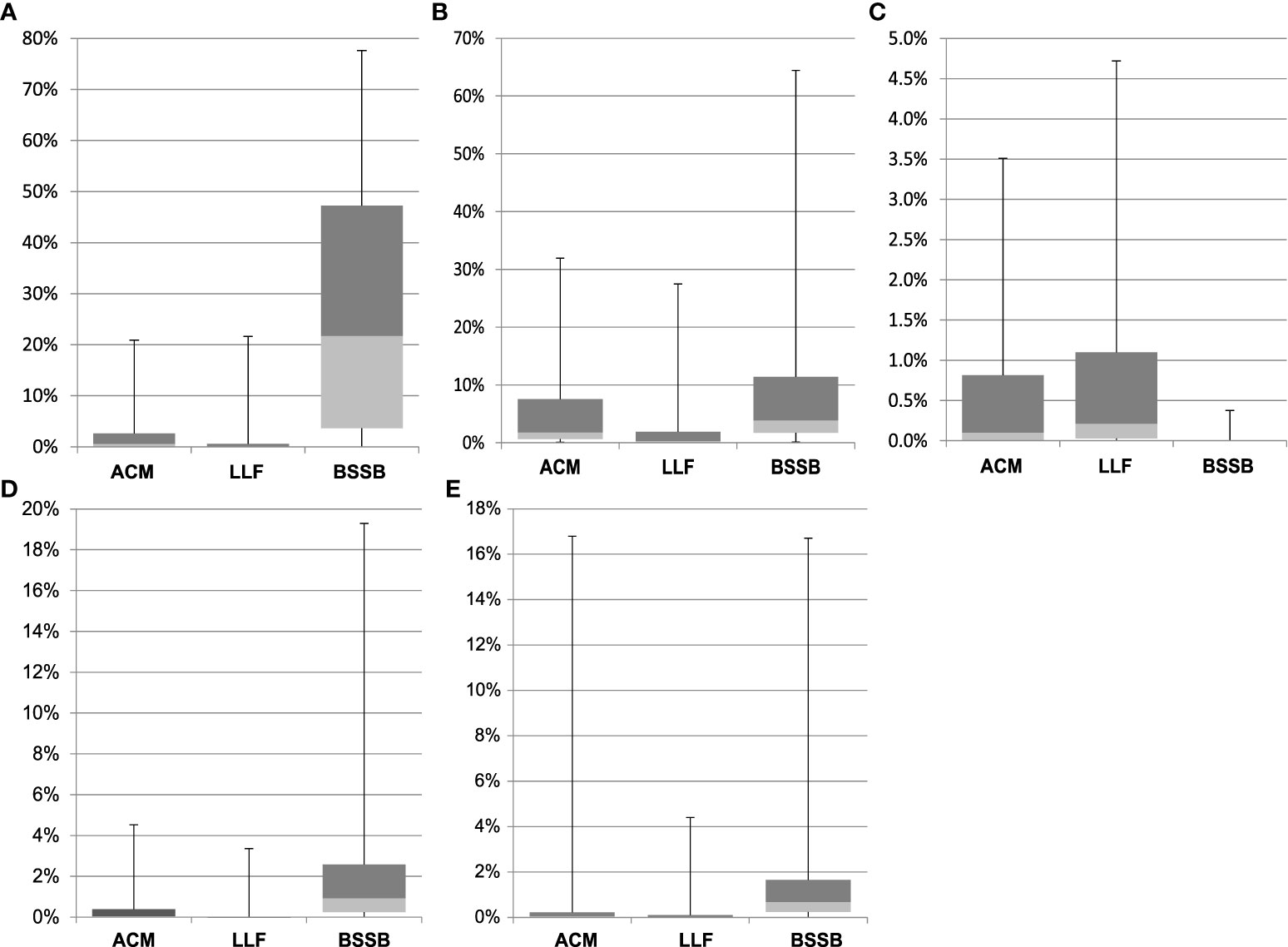
Figure 4 Relative abundance of five genera, clustered by fish species. Genera represented: Photobacterium (A), Shewanella (B), Staphylococcus (C), Streptococcus (D), and Vibrio (E). Error bars depict quarterly percentiles, i.e., 25%, median, 75%, 100%.
The NGS data analyses resulted in an output of 5,798 unique amplicon sequence variants (ASVs) of which 5,717 were identified as bacteria, 15 as archaea, five as eukaryotes and the rest unidentified. None of the ASVs appeared in 100% of the samples, nor in 100% of the samples of any specific fish species. Of those bacterial ASVs, 189 were initially identified to the taxonomic level of species, with 177 unique values (i.e., species). Of this list, 41 species were identified as bearing some pathogenic potential to humans and/or marine animals (Table S1): 36 had varying human clinical relevance or zoonotic potential. These were divided into the following taxonomic classes: Gammaproteobacteria (n = 15); Actinobacteria (n = 10); Bacilli (n = 8); Campylobacteria (n = 1); Alphaproteobacteria (n = 1); and Fusobacteriia (n = 1). The literature also revealed that of the 41 potentially pathogenic species, a total of 14 were known marine animal pathogens (meaning, some of those potentially pathogenic to humans may also cause disease in marine wildlife). These 14 species were of the taxonomic classes Gammaproteobacteria (n = 9); Bacilli (n = 3); and Bacteroidia (n = 2). Thirteen of the fish pathogens appeared in samples belonging to ACM, six in LLF and four in BSSB. The results are visualized in Figure 5. It shows S. baltica to be highly prevalent in these fish species (found in 95% of ACM, 46% of LLF and 92% of BSSB), and that P. damselae was also prevalent (73%, 38% and 84% of the ACM, LLF and BSSB samples, respectively). In contrast, pathogenic Streptococcus, Staphylococcus and Vibrio species were found less frequently.
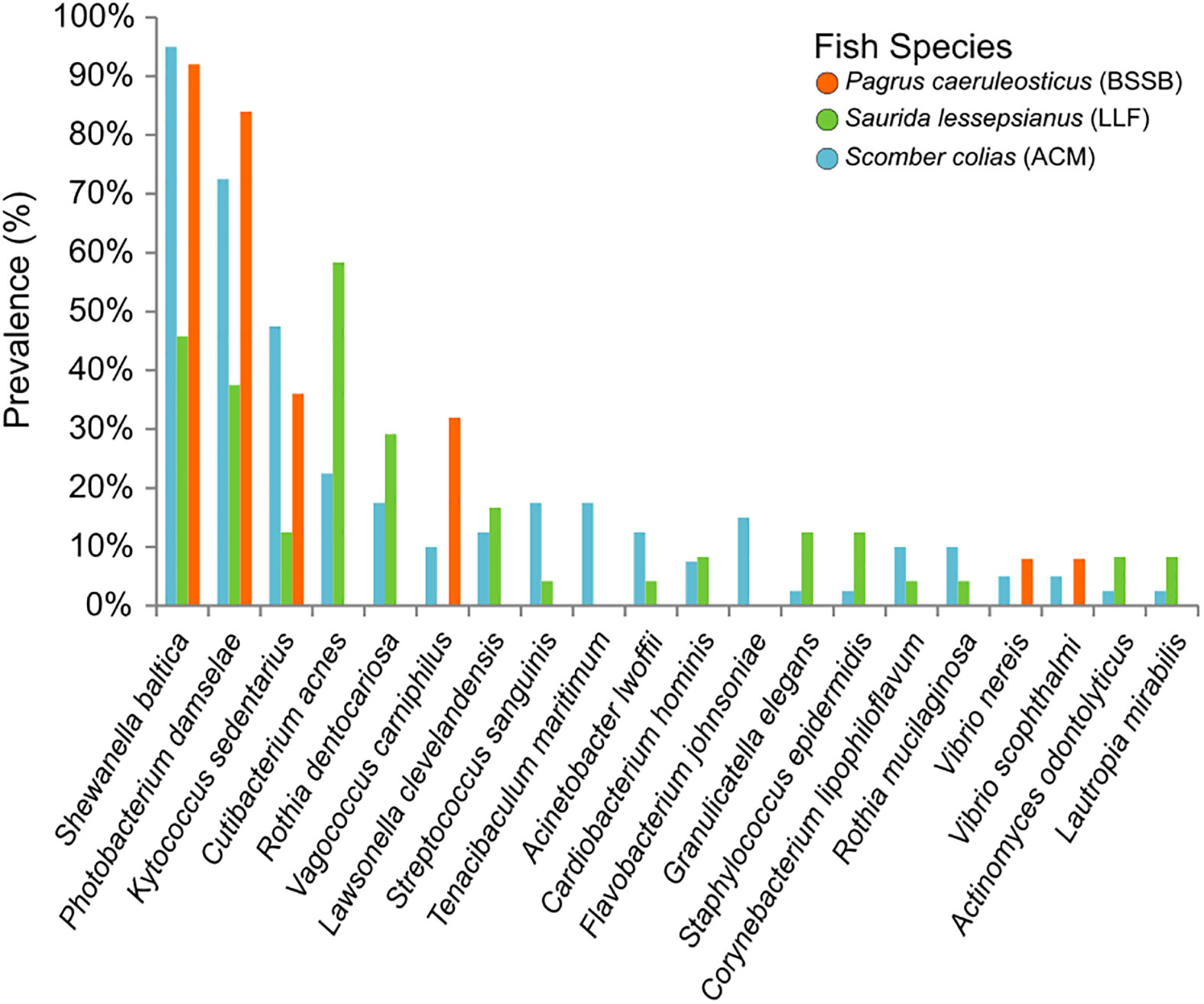
Figure 5 Prevalence of the 20 most commonly found pathogens in the gills of the fish sampled. The prevalence (y axis) refers to amount of samples per species found to contain these specific potential pathogens. Pathogens arranged (on the x axis) from the most (overall) prevalent (left) to the least (right).
Phylogenetic analysis of these five genera of interest demonstrated the most closely related reference species to the ASVs with pathogenic potential. The P. damselae clade (Figure S4) appears divided between ASVs associated with P. damselae subsp. damselae and P. damselae subsp. piscicida, in which the former is predominant – both in number of ASVs and total number of reads. In total, P. damselae makes up >30% of the genus’ ASV reads. The Shewanella phylogenetic tree (Figure S5) is comprised of numerous ASVs, as this genus is the most prevalent (though not most abundant) of all genera analyzed. The S. baltica associated ASVs were responsible for >70% of all Shewanella reads. The trees of Staphylococcus, Streptococcus and Vibrio (Figures S6−S8, respectively) are similar in terms of the pathogenic/non-pathogenic ratios they exhibit: between 1−4%. This data is summarized in Figure 6.
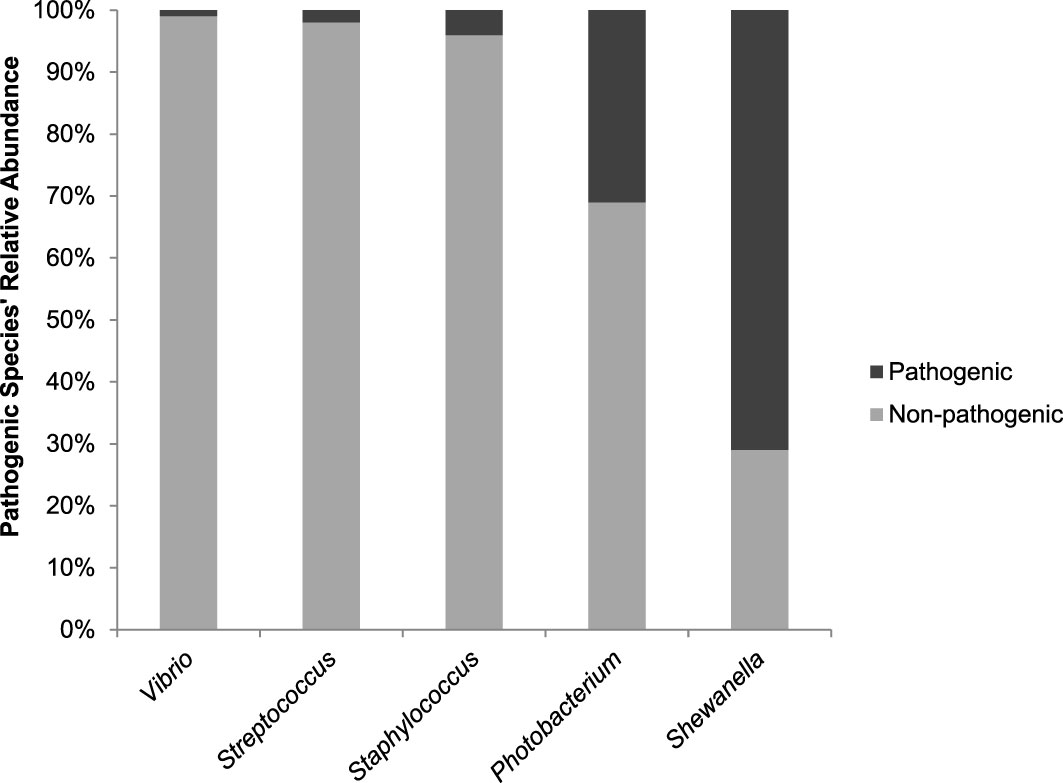
Figure 6 A comparison of the Pathogenic/Non-pathogenic ratios per genera of interest. Values represent relative abundance of potential pathogenic species identified out of all ASVs associated with a certain genus.
Discussion
The use of fish gill microbiomes to assess pathogen prevalence is gaining importance and becoming increasingly routine (Hess et al., 2015; Pratte et al., 2018; Brown et al., 2019; Rosado et al., 2019; Minich et al., 2020). However, to the best of our knowledge, this is the first study to be carried out in the Eastern Mediterranean. In previous studies, the presence of pathogens was inferred from taxonomic levels higher than species (Pratte et al., 2018; Rosado et al., 2019), sometimes supported by data on shifts in microbiome composition between treatment and control groups (Hess et al., 2015; Mohammed and Arias, 2015; Brown et al., 2019; Minich et al., 2020) or spatio-temporal differences (Minich et al., 2020). In the current study, we based our findings regarding pathogens only on ASVs that we could identify to the species level at a high degree of certainty. The data show that Atlantic chub mackerel (ACM), which was found to host the richest microbial community of the three fish species, also shows the largest total number of bacterial species with pathogenic potential. The microbiome of ACM included 35 out of the total of 41 pathogenic bacterial species found (and 13 of the 14 fish pathogens), while Lessepsian lizardfish (LLF) had 26 (6/14) and Bluespotted seabream (BSSB) just 6 (5/14 fish pathogens). A possible explanation for this result may be the pelagic-migratory nature of ACM, which means that it passes through diverse geographic zones, where it may accumulate a variety of bacteria on its gills. BSSB has a relatively high abundance of Photobacterium, and within the Photobacterium genus, ~30% of the reads belong to a specific and well-known pathogenic species. This entails that ~10% of BSSB gill microbiome is P. damselae. Had this putative pathogen been obligatory, many of the samples would have shown signs of infection. Since this was not the case, it raises the question whether the two subspecies of P. damselae (a multi-gene PCR array for sub-speciation was not performed in this study) are opportunistic or pathobionts. Previous studies suggest that a genetic diversity within P. damselae subsp. damselae strains means disease outbreaks in fish are most likely caused by multiclonal populations, containing several complementing virulence factors (Terceti et al., 2016). Cases in which P. damselae is highly prevalent in fish without causing disease may reflect the need for a few variants of this pathogen to ‘join forces’ to create effective infection (Terceti et al., 2016). This is supported by a study conducted on P. damselae subsp. piscicida, which found variability of virulence in different strains, governed by a protein exotoxin, AIP56 which is secreted by virulent P. damselae subsp. piscicida in large quantities but not by avirulent strains. This is a key factor responsible for apoptogenic activity targeting fish macrophages and neutrophils (do Vale et al., 2005).
The observed high correlations between Gammaproteobacteria of the Photobacterium cohort, and especially the Vibrionaceae members, Vibrio, Aliivibrio and Photobacterium, suggest that the ecological niche existing in the form of BSSB gills provides these genera with preferable conditions. A similar assumption can be made regarding the Streptococcus-Staphylococcus cohort and the gills of LLF. In reference to potentially pathogenic species, within the Streptococcus-Staphylococcus genera-related sequences, pathogenic species make up only a small percentage of the total reads, and these two genera are comparatively less dominant in terms of relative abundance. On the other hand, S. baltica, a bacterium that is not known to be pathogenic to marine animals, but is rather a major cause of food spoilage (Zhang et al., 2020), dominates Shewanella-associated ASVs. A comparison of the pathogenic/non-pathogenic ratios per genus (Figure 1) summarizes these differences. According to our analyses, Vibrio, a genus found relatively much less abundant than Photobacterium, also comprised almost entirely of non-pathogenic species.
Nevertheless, it is important to remember that separating Vibrionaceae members by means of their 16S genes is problematic (Machado and Gram, 2015), therefore better identification may require lengthening the amplicon sequences by using a different set of primers (Morales and Holben, 2009; Martínez-porchas et al., 2016), or by complementing the identification using Multilocus Sequence Analysis (MLSA) – targeting protein-encoding genes such as toxR and rpoD (Pascual et al., 2010).
While using rRNA amplicon sequencing for the study of microbiomes has great advantages and is extremely useful, its limitations must be considered. In a highly diverse microbiome, different bacteria can display functional redundancy. Hence, a ‘function over phylogeny’ approach studying the functional characteristics of the microbial community – in addition to the species composition – may be advantageous and more informative (Moya and Ferrer, 2016; Gibbons, 2017; Louca et al., 2017). It was suggested by de Bruijn et al. (2018) (Louca et al., 2017) that the functional characteristics of greatest importance would be the protection of the fish against pathogens, and that this could be achieved by research of gene clusters involved in biosynthesis and production of proteins and metabolites, and studying these genes’ frequency, diversity and transcription (Lynn and De Leenheer, 2019). Also, parallel investigation of both phylogeny and function is likely to help cut to a minimum the over- or underrepresentation of certain bacterial families, due to bias resulting from DNA extraction methods (Kashinskaya et al., 2017) and PCR, when relying solely on rRNA amplicon sequencing (Chakravorty et al., 2007; Sinclair et al., 2015; Stoddard et al., 2015).
The formation and function of the gill microbiome can be better understood when considering the much more investigated gut microbiome. During evolution, as the fish body structure developed and became increasingly complex, the adapting mucosa epithelia would have likely required additional resources, higher metabolic rates and improved metabolic capacities. In the ancient ocean, where microorganisms presumably flourished, interactions with fish inevitably formed the basis for creating early microbiomes (Gomez et al., 2013). According to Maynard et al. (2012), in the intestine and microbiome relationship, one counterpart receives a protected gut environment while the other benefits from the microbial highly adaptive metabolic engine that provides essential factors (e.g., vitamins) as well as an improved host ability to obtain nutrients from food. Gill et al., (2006) suggested that the human metabolism is the result of an amalgamation of microbial and human attributes. The microbiome can therefore be viewed as representing an evolutionary collection of species which is integral to the host’s constant strive to augment the utilization of its food components (Maynard et al., 2012). In line with this hypothesis, i.e., the acceptance of commensal presence on the mucosal barriers, likely evolved in early vertebrates (e.g., fish), to profit from the ready-made microbial genetic material rather than independently, creating novel metabolic capabilities. It follows, that the greater the microbiome diversity, the better the chances of improving performance. The gill mucosa, as a semipermeable barrier between the fish and external milieu, would be faced with similar challenges (Koppang et al., 2015).
In the gills, similarly to the gut, the fish immune system must be able to identify commensal bacteria to avoid unnecessary inflammatory reactions triggered when pathogens threaten the intactness of the mucosal barrier (Gomez et al., 2013). The development of such immune tolerance to bacteria has been demonstrated in carp, where repeated anal administration of allogeneic cells has led to loss of allospecific cell-mediated immune response (Sato et al., 2005). Certainly, “false-alarm” inflammatory responses to innocuous bacteria, e.g., resulting in hyperplasia of the branchial epithelium, would not only place an unnecessary burden on the fish vital resources but also compromise the gas exchange processes of the respiratory epithelium.
Apart from host-pathogen relations, bacterial social interactions are another factor affecting virulence expression – both in terms of creating the conditions favoring virulence, and also in the ability to control virulence expression. Bacterial populations experience pressures of conflict and cooperation, which become a major factor in the organization and function of microbial communities (Asfahl and Schuster, 2017). Such interactions are found to be a stabilizing element widespread in Vibrio species, creating a protocooperation-based community, which shows increasing growth yield, while creating incentives to prevent a “Tragedy of the Commons” (Bruger and Waters, 2018). It was further shown that cooperative communities include members responsible for “policing”, so that when challenged by cheaters, cooperative behavior can persist, provided that four conditions hold: (i) toxin-producers are present; (ii) the cost of toxin production surpasses that of public good production, meaning, policing becomes more expensive than cooperation; (iii) the toxin’s harmful effects on the cooperator has to be sufficiently high – in order to counterweigh that policing is more costly than cooperation; and finally, (iv) the toxin’s effects on the cheater must be even higher (Lynn and De Leenheer, 2019). An example coinciding with this theory is the pathogenic habits of Vibrio harveyi. In one study (Montánchez et al., 2019), this species was found to express virulent genes under heat stress, while cells of its community as a whole suffered extensive fatalities, demonstrating that disease outbreaks due to elevated sea surface temperatures, is but an escape route taken to avoid mortality. This means that pathogenicity in this species is not a display of offensive behavior, but rather a defensive mechanism. Some pathogens can survive for substantial periods in the open water, which explains how fish-cage originated secretions may travel many kilometers with the currents and still affect wildlife (or other cage farm stocks) downstream (Viau et al., 2011; Shapiro et al., 2013). Yet, pathogens are mostly prevalent in hotspots (e.g., immunosuppressed animals, naïve hosts, or those associated with anthropogenic pollution-hit areas) (Lyons et al., 2010; Lobelle and Cunliffe, 2011). Therefore, perhaps Pathogenicity, being defined as “the quality of producing or the ability to produce pathologic changes or disease” (Shapiro-Ilan et al., 2005), is inherently a trait ‘designed’ to affect a host. Being host-oriented, the presence of the ‘right’ host cells is an essential (though not sufficient) prerequisite for expressing pathogenicity. Yet, variances in genotypic attributes between different fish species (even those sharing diets, trophic levels and habitats), will create different ‘environmental’ conditions within gills (and other fish organs, for that matter), meaning pathogens in these organs will face different microbial communities (Pratte et al., 2018), which in turn may have an effect on the expression of pathogenicity of a given pathogen. Such a mechanism may help explain why asserting whether some less known bacteria are pathogenic (and to which degree), may prove tricky: some pathogens are not necessarily the cause of a certain disease, but are rather secondary agents of it, helping progress it, or just gaining benefit from the change in conditions within the infected organ (Brink et al., 2019). It is known (Laanto et al., 2012) that some pathogens may develop into symbionts with opportunistic pathogenic capabilities, using this ability as a survival tool, which may hint why co-infection is common – an arrival of a non-symbiotic pathogen (due to injury), may cause the host to become immunosuppressed and create conditions favorable of pathogenesis for the symbiotic pathogen.
As a general rule, bacteria of all phyla are known to have certain ‘preferable’ physio-biochemical conditions in which they thrive, and a range of conditions that can be regarded as ‘tolerable’. The Eastern Mediterranean water column displays stratification, with different physiochemical properties existing in different layers that affect the local microbial communities (Techtmann et al., 2015). It was also shown that Gorgonians (Octocorallia, Anthozoa, Cnidaria), which are at the heart of extremely biodiverse ecosystems second only to tropical coral reefs, exhibit great differences in their microbiomes relative to their surrounding waters (van de Water et al., 2017). It is therefore obvious that fish gills offer a niche with a unique set of conditions, inducing the formation of certain microbial communities that are different than those in the water the fishes swim in. However, pathogenesis expressed in fish gills may also be affected by: (i) changes in the gills’ microbiome throughout the life-time of the fish (Nagelkerken and van der Velde, 2002; Wilson et al., 2010; Mercier et al., 2012); and (ii) the possible presence of protistan parasites (e.g., ciliates, flagellates, etc.), which frequently facilitate the establishment of secondary microbial species (Gibbons, 2017), as well as many other pathogenic agents – viruses, fungi (Bui et al., 2019), macro and micro-parasites such as helminths and myxosporea (Molnár, 2002; Liyanage et al., 2003; Nguyen et al., 2021).
Fish gills harbor species-specific microbiomes, exhibiting strong correlations between certain taxonomic groups. In the present study, we demonstrated some overlap between the three host species sampled, perhaps expressing a form of core microbiome. The genera which were the focus of the study, Vibrio, Photobacterium, Shewanella, Staphylococcus and Streptococcus are important members within these fish species gills microbiomes, and at least in the case of Bluespotted seabream, a substantial percent of its gills microbiome is populated by generalist pathogenic species, which are notorious marine pathogens. In conclusion, given that all fish sampled appeared healthy, and based on the notion that pathogenicity is also influenced by environmental pressure against virulence (coming from microbial community interactions, carrying a strong preference for cooperation over cheating strategies), it can be inferred that pathogenesis is but one of many tools for survival and reproduction that bacteria are equipped with. This in turn explains the fact that pathogens are very rarely obligatory. What it also means is that healthy wildlife populations are not necessarily devoid of pathogens, but have a mix they coevolved with and which protect them from invasions of novel types.
Data availability statement
The datasets presented in this study can be found in online repositories. The name of the repository and link to the data can be found below: DRYAD; https://doi.org/10.5061/dryad.wh70rxwr3.
Ethics statement
Ethical review and approval was not required for the animal study because the subjects of research were dead fish provided by another institute conducting an approved ecological study.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was financially supported by the Kahn Foundation and by the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou, China) (SMSEGL20SC02).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1008103/full#supplementary-material
References
Abraham T. J., Paul P., Adikesavalu H., Patra A., Banerjee S. (2016). Stenotrophomonas maltophilia as an opportunistic pathogen in cultured African catfish Clarias gariepinus (Burchell, 1822). Aquaculture 450, 168–172. doi: 10.1016/j.aquaculture.2015.07.015
Abu Fanas S., Brigi C., Varma S. R., Desai V., Senok A., D’Souza J. (2021). The prevalence of novel periodontal pathogens and bacterial complexes in stage II generalized periodontitis based on 16S rRNA next generation sequencing abstract. J. Appl. Oral. Sci. 29, e20200787. doi: 10.1590/1678-7757-2020-0787
Alberti M. O., Hindler J. A., Humphries R. M. (2016). Antimicrobial susceptibilities of Abiotrophia defectiva, Granulicatella adiacens, and Granulicatella elegans. Antimicrob. Agents Chemother. 60 (3), 1411–1421. doi: 10.1128/AAC.02645-15
Al-Hebshi N. N., Nasher A. T., Idris A. M., Chen T. (2015). Robust species taxonomy assignment algorithm for 16S rRNA NGS reads: application to oral carcinoma samples. J. Oral. Microbiol. 7 (1), 28934. doi: 10.3402/jom.v7.28934
Andrade T. S., Henriques J. F., Almeida A. R., Soares A. M. V. M., Scholz S., Domingues I. (2017). Zebrafish embryo tolerance to environmental stress factors — concentration–dose response analysis of oxygen limitation, ph, and uv-light irradiation. Environ. Toxicol. Chem. 36 (3), 682–690. doi: 10.1002/etc.3579
Apprill A. (2017). Marine animal microbiomes: Toward understanding host – microbiome interactions in a changing ocean. Front. Mar. Sci. 4, 222. doi: 10.3389/fmars.2017.00222
Arechavala-Lopez P., Sanchez-Jerez P., Bayle-Sempere J. T., Uglem I., Mladineo I. (2013). Reared fish, farmed escapees and wild fish stocks — a triangle of pathogen transmission of concern to Mediterranean aquaculture management. Aquac Environ. Interact. 3, 153–161. doi: 10.3354/aei00060
Aschenbrenner I. A., Cernava T., Berg G., Grube M. (2016). Understanding microbial multi-species symbioses. Front. Microbiol. 7, 180. doi: 10.3389/fmicb.2016.00180
Asfahl K. L., Schuster M. (2017). Social interactions in bacterial cell – cell signaling. FEMS Microbiol. Rev. 41 (1), 92–107. doi: 10.1093/femsre/fuw038
Atalah J., Sanchez-jerez P. (2020). Global assessment of ecological risks associated with farmed fish escapes. Glob Ecol. Conserv. 21, e00842. doi: 10.1016/j.gecco.2019.e00842
Barash A., Pickholtz R., Nativ H., Malamud S., Scheinin A., Tchernov D. (2018). Seasonal arrival and feeding of injured coastal sharks at fish farms in the Eastern Mediterranean. J. Black Sea/Mediterr Environ. 24 (1), 86–90.
Barrett L. T., Swearer S. E., Dempster T. (2019). Impacts of marine and freshwater aquaculture on wildlife: A global meta-analysis. Rev. Aquac. 11, 1022–1044. doi: 10.1111/raq.12277
Bernard K. (2005). Corynebacterium species and coryneforms: an update on taxonomy and diseases attributed to these taxa. Clin. Microbiol. Newsl. 27 (2), 9–18. doi: 10.1016/j.clinmicnews.2005.01.002
Brassil B., Mores C. R., Wolfe A. J., Putonti C. (2020). Characterization and spontaneous induction of urinary tract Streptococcus anginosus prophages. J. Gen. Virol. 101, 685–691. doi: 10.1099/jgv.0.001407
Brennan C. A., Garrett W. S. (2019). Fusobacterium nucleatum — symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 17, 156–166. doi: 10.1038/s41579-018-0129-6
Brink M., Rhode C., Macey B. M., Christison K. W., Roodt-wilding R. (2019). Metagenomic assessment of body surface bacterial communities of the sea urchin, Tripneustes gratilla. Mar. Genomics 47, 100675. doi: 10.1016/j.margen.2019.03.010
Brooke J. S., Bonaventura G., Berg G., Martinez J. (2017). Editorial: A multidisciplinary look at Stenotrophomonas maltophilia: An emerging multi-Drug-Resistant global opportunistic pathogen. Front. Microbiol. 8, 1511. doi: 10.3389/fmicb.2017.01511
Brown R. M., Wiens G. D., Salinas I. (2019). Analysis of the gut and gill microbiome of resistant and susceptible lines of rainbow trout (Oncorhynchus mykiss). Fish Shellfish Immunol. 86, 497–506. doi: 10.1016/j.fsi.2018.11.079
Bruger E. L., Waters M. (2018). Maximizing growth yield and dispersal via quorum sensing promotes cooperation in Vibrio bacteria. Appl. Environ. Microbiol. 84 (14), e00402–e00418. doi: 10.1128/AEM.00402-18
Bui S., Oppedal F., Sievers M., Dempster T. (2019). Behaviour in the toolbox to outsmart parasites and improve fish welfare in aquaculture. Rev. Aquac. 11, 168–186. doi: 10.1111/raq.12232
Callahan B. J., Mcmurdie P. J., Rosen M. J., Han A. W., Johnson A. J., Holmes S. P. (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 13, 581–583. doi: 10.1038/nmeth.3869.
Cao S., Geng Y., Yu Z., Deng L., Gan W., Wang K., et al. (2018). Acinetobacter lwoffii, an emerging pathogen for fish in Schizothorax genus in China. Transbound Emerg. Dis. 65, 1816–1822. doi: 10.1111/tbed.12957
Caporaso J. G., Lauber C. L., Walters W. A., Berg-lyons D., Huntley J., Fierer N., et al. (2012). Ultra-high-throughput microbial community analysis on the illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Caporaso J. G., Lauber C. L., Walters W. A., Berg-lyons D., Lozupone C. A., Turnbaugh P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Caspar Y., Recule C., Pouzol P., Lafeuillade B., Mallaret M., Maurin M., et al. (2013). Psychrobacter arenosus bacteremia after blood transfusion, France. Emerg. Infect. Dis. 19 (7), 1118–1120. doi: 10.3201/eid1907.121599
Castillo D. E., Nanda S., Keri J. E. (2019). Propionibacterium (Cutibacterium) acnes bacteriophage therapy in acne: Current evidence and future perspectives. Dermatol. Ther. 9, 19–31. doi: 10.1007/s13555-018-0275-9
Chakravorty S., Helb D., Burday M., Connell N., Alland D. (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69 (2), 330–339. doi: 10.1016/j.mimet.2007.02.005
Chang H., Dai F., Duan B., Duan G., Zu F., Yang Z., et al. (2018). Isolation and characterization of Vagococcus carniphilus from diseased crucian carp. Biotechnol. Biotechnol. Equip. 32 (4), 936–941. doi: 10.1080/13102818.2017.1413420
Chen C. (1996). Distribution of a newly described species, Kingella oralis, in the human oral cavily. Oral. Microbiol. Immunol. 11, 425–427. doi: 10.1111/j.1399-302X.1996.tb00206.x
Chong J., Liu P., Zhou G., Xia J. (2020). Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821. doi: 10.1038/s41596-019-0264-1
Colombo A. P. V., Bennet S., Cotton S. L., Goodson J. M., Kent R., Haffajee A. D., et al. (2012). Impact of periodontal therapy on the subgingival microbiota of severe periodontitis: Comparison between good responders and individuals with refractory periodontitis using the human oral microbe identification microarray. J. Periodontol. 83 (10), 1279–1287. doi: 10.1902/jop.2012.110566
Conte F. S. (2004). Stress and the welfare of cultured fish. Appl. Anim. Behav. Sci. 86, 205–223. doi: 10.1016/j.applanim.2004.02.003
de Bruijn I., Liu Y., Wiegertjes G. F., Raaijmakers J. M. (2018). Exploring fish microbial communities to mitigate emerging diseases in aquaculture. FEMS Microbiol. Ecol. 94 (1), fix161. doi: 10.1093/femsec/fix161
Dhariwal A., Chong J., Habib S., King I. L., Agellon L. B., Xia J. (2017). MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, 180–188. doi: 10.1093/nar/gkx295
do Vale A., Silva M. T., dos Santos N. M. S., Nascimento D. S., Reis-Rodrigues P., Costa-Ramos C., et al. (2005). AIP56, a novel plasmid-encoded virulence factor of Photobacterium damselae subsp. piscicida with apoptogenic activity against sea bass macrophages and neutrophils. Mol. Microbiol. 58 (4), 1025–1038. doi: 10.1111/j.1365-2958.2005.04893.x.
Egerton S., Culloty S., Whooley J., Stanton C., Ross R. P. (2018). The gut microbiota of marine fish. Front. Microbiol. 9, 873. doi: 10.3389/fmicb.2018.00873
Elarabany N., Bahnasawy M., Edrees G., Alkazagli R. (2017). Effects of salinity on some haematological and biochemical parameters in Nile tilapia, Oreochromus niloticus. Agric. For Fish. 6 (6), 200–205. doi: 10.11648/j.aff.20170606.13
Esteve C., Merchán R., Alcaide E. (2017). An outbreak of Shewanella putrefaciens group in wild eels Anguilla anguilla l. favoured by hypoxic aquatic environments. J. Fish Dis. 40, 929–939. doi: 10.1111/jfd.12574
Evans D. H., Piermarini P. M., Choe K. P. (2005). The multifunctional fish gill: Dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol. Rev. 85, 97–177. doi: 10.1152/physrev.00050.2003
Funke G., Hutson R. A., Hilleringmann M., Heizmann W. R., Collins M. D. (1997). Corynebacterium lipophiloflavum sp. nov. isolated from a patient with bacterial vaginosis. FEMS Microbiol. Lett. 150, 219–224. doi: 10.1016/S0378-1097(97)00118-3
Genin A., Levy L., Sharon G., Raitsos D. E., Diamant A. (2020). Rapid onsets of warming events trigger mass mortality of coral reef fish. PNAS 117 (41), 25378–25385. doi: 10.1073/pnas.2009748117
Ghyselinck J., Pfeiffer S., Heylen K., Sessitsch A., De Vos P. (2013). The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PloS One 8 (8), e71360. doi: 10.1371/journal.pone.0071360
Gibbons S. M. (2017). Function over phylogeny. Nat. Ecol. Evol. 1 (1), 0032. doi: 10.1038/s41559-016-0032.
Gill S. R., Pop M., DeBoy R. T., Eckburg P. B., Turnbaugh P. J., Samuel B. S., et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science (80) 312 (5778), 1355–1359. doi: 10.1126/science.1124234.
Gomez D., Sunyer J. O., Salinas I. (2013). The mucosal immune system of fish: The evolution of tolerating commensals while fighting pathogens. Fish Shellfish Immunol. 35 (6), 1729–1739. doi: 10.1016/j.fsi.2013.09.032
Greay T. L., Evasco K. L., Evans M. L., Oskam C. L., Magni P. A., Ryan U. M., et al. (2021). Illuminating the bacterial microbiome of Australian ticks with 16S and Rickettsia-specific next-generation sequencing. Curr. Res. Parasitol. Vector-Borne Dis. 1, 100037. doi: 10.1016/j.crpvbd.2021.100037
Halpern B. S., Walbridge S., Selkoe K. A., Kappel C. V., Micheli F., D’Agrosa C., et al. (2008). A global map of human impact on marine ecosystems. Sci. (80-). 319 (5865), 948–952. doi: 10.1126/science.1149345
Harvell C. D., Kim K., Burkholder J. M., Colwell R. R., Epstein P. R., Grimes D. J., et al. (1999). Emerging marine diseases — climate links and anthropogenic factors. Science (80-) 285, 1505–1510. doi: 10.1126/science.285.5433.1505
Hess S., Wenger A. S., Ainsworth T. D., Rummer J. L. (2015). Exposure of clownfish larvae to suspended sediment levels found on the great barrier reef: Impacts on gill structure and microbiome. Sci. Rep. 5, 10561. doi: 10.1038/srep10561
Johnson E. O., Kamilaris T. C., Chrousos G. P., Gold P. W. (1992). Mechanisms of stress: A dynamic overview of hormonal and behavioral homeostasis. Neurosci. Biobehav. Rev. 16, 115–130. doi: 10.1016/S0149-7634(05)80175-7
Johnston D., Earley B., Cormican P., Murray G., Kenny D. A., Waters S. M., et al. (2017). Illumina MiSeq 16S amplicon sequence analysis of bovine respiratory disease associated bacteria in lung and mediastinal lymph node tissue. BMC Vet. Res. 13, 118. doi: 10.1186/s12917-017-1035-2
Kashinskaya E. N., Andree K. B., Simonov E. P., Solovyev M. M. (2017). DNA Extraction protocols may influence biodiversity detected in the intestinal microbiome: A case study from wild Prussian carp, Carassius gibelio. FEMS Microbiol. Ecol. 93 (2), fiw240. doi: 10.1093/femsec/fiw240
Khajanchi B. K., Fadl A. A., Borchardt M. A., Berg R. L., Horneman A. J., Stemper M. E., et al. (2010). Distribution of virulence factors and molecular fingerprinting of Aeromonas species isolates from water and clinical samples: Suggestive evidence of water-to-Human transmission. Appl. Environ. Microbiol. 76 (7), 2313–2325. doi: 10.1128/AEM.02535-09
Kiernan T. J., Flaherty N. O., Gilmore R., Ho E., Hickey M., Tolan M., et al. (2008). Abiotrophia defectiva endocarditis and associated hemophagocytic syndrome – a first case report and review of the literature. Int. J. Infect. Dis. 12, 478–482. doi: 10.1016/j.ijid.2008.01.014
King W. L., Jenkins C., Seymour J. R., Labbate M. (2019). Oyster disease in a changing environment: Decrypting the link between pathogen, microbiome and environment. Mar. Environ. Res. 143, 124–140. doi: 10.1016/j.marenvres.2018.11.007
Koppang E. O., Kvellestad A., Fischer U. (2015). “Fish mucosal immunity: gill,” in Mucosal health in aquaculture. Eds. Beck B. H., Peatman E. (San Diego: Academic Press), 93–133 p.
Krotman Y., Yergaliyev T. M., Shani R. A., Avrahami Y., Szitenberg A. (2020). Dissecting the factors shaping fish skin microbiomes in a heterogeneous inland water system. Microbiome. 8, 9. doi: 10.1186/s40168-020-0784-5
Krzyściak W., Jurczak A., Kościelniak D., Bystrowska B., Skalniak A. (2014). The virulence of Streptococcus mutans and the ability to form biofilms. Eur. J. Clin. Microbiol. Infect. Dis. 33, 499–515. doi: 10.1007/s10096-013-1993-7
Kuang T., He A., Lin Y., Huang X., Liu L., Zhou L. (2020). Comparative analysis of microbial communities associated with the gill, gut, and habitat of two filter-feeding fish. Aquac Rep. 18, 100501. doi: 10.1016/j.aqrep.2020.100501
Kubilay A., Uluköy G. (2004). First isolation of Staphylococcus epidermidis from cultured gilthead sea bream (Sparus aurata) in Turkey. Bull. Eur. Assoc. Fish Pathol. 24 (3), 137–143.
Kusejko K., Auñón Á, Jost B., Natividad B., Strahm C., Thurnheer C., et al. (2021). The impact of surgical strategy and rifampin on treatment outcome in Cutibacterium periprosthetic joint infections. Clin. Infect. Dis. XX (XX), 1–10. doi: 10.1093/cid/ciaa1839
Laanto E., Bamford J. K. H., Laakso J., Sundberg L.-R. (2012). Phage-driven loss of virulence in a fish pathogenic bacterium. PloS One 7 (12), e53157. doi: 10.1371/journal.pone.0053157
Lejeusne C., Chevaldonné P., Pergent-Martini C., Boudouresque C. F., Pérez T. (2010). Climate change effects on a miniature ocean: the highly diverse, highly impacted Mediterranean Sea. Trends Ecol. Evol. 25 (4), 250–260. doi: 10.1016/j.tree.2009.10.009
Levenga H., Donnelly P., Blijelevens N., Verweij P., de Pauw B. (2004). Fatal hemorrhagic pneumonia caused by infection due to Kytococcus sedentarius — a pathogen or passenger? Ann. Hematol. 83, 447–449. doi: 10.1007/s00277-003-0831-x.
Li N., Lin Q., Fu X., Guo H., Liu L., Wu S. (2015). Development and efficacy of a novel streptomycin-resistant Flavobacterium johnsoniae vaccine in grass carp (Ctenopharyngodon idella). Aquaculture 448, 93–97. doi: 10.1016/j.aquaculture.2015.05.047
Lim Y. K., Kweon O. J., Kim H. R., Kim T. H., Lee M.-K. (2017). First case of bacteremia caused by Janibacter hoylei. APMIS. 125, 665–668. doi: 10.1111/apm.12693
Liu H., Guo X., Gooneratne R., Lai R., Zeng C., Zhan F., et al. (2016). The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 6, 24340. doi: 10.1038/srep24340
Liyanage Y. S., Yokoyama H., Wakabayashi H. (2003). Dynamics of experimental production of Thelohanellus hovorkai (Myxozoa: Myxosporea) in fish and oligochaete alternate hosts. J. Fish Dis. 26, 575–582. doi: 10.1046/j.1365-2761.2003.00492.x
Llewellyn M. S., Boutin S., Hoseinifar S. H., Derome N., Biron D. G., Romero J., et al. (2014). Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 5, 207. doi: 10.3389/fmicb.2014.00207
Lobelle D., Cunliffe M. (2011). Early microbial biofilm formation on marine plastic debris. Mar. pollut. Bull. 62, 197–200. doi: 10.1016/j.marpolbul.2010.10.013
Louca S., Jacques S. M. S., Pires A. P. F., Leal J. S., Srivastava D. S., Parfrey L. W., et al. (2017). High taxonomic variability despite stable functional structure across microbial communities. Nat. Ecol. Evol. 1 (1), 15. doi: 10.1038/s41559-016-0015
Lynn B. K., De Leenheer P. (2019). Division of labor in bacterial populations. Math Biosci. 316, 108257. doi: 10.1016/j.mbs.2019.108257
Lyons M. M., Ward J. E., Gaff H., Hicks R. E., Drake J. M., Dobbs F. C. (2010). Theory of island biogeography on a microscopic scale: organic aggregates as islands for aquatic pathogens. Aquat Microb. Ecol. 60, 1–13. doi: 10.3354/ame01417
Machado H., Gram L. (2015). The fur gene as a new phylogenetic marker for Vibrionaceae species. Appl. Environ. Microbiol. 81 (8), 2745–2752. doi: 10.1128/AEM.00058-15
MacKinnon M. M., Amezaga M. R., MacKinnon J. R. (2001). A case of Rothia dentocariosa Endophthalmitis. Eur. J. Clin. Microbiol. Infect. Dis. 20, 756–757. doi: 10.1007/s100960100589
Malani A. N., Aronoff D. M., Bradley S. F., Kauffman C. A. (2006). Cardiobacterium hominis endocarditis: two cases and a review of the literature. Eur. J. Clin. Microbiol. Infect. Dis. 25, 587–595. doi: 10.1007/s10096-006-0189-9
Maraki S., Papadakis I. S. (2015). Rothia mucilaginosa pneumonia: a literature review. Infect. Dis. (Auckl). 47, 125–129. doi: 10.3109/00365548.2014.980843
Marbjerg L. H., Gaini S., Justesen S. (2015). First report of Sphingomonas koreensis as a human pathogen in a patient with meningitis. J. Clin. Microbiol. 53 (3), 1028–1030. doi: 10.1128/JCM.03069-14
Marcel G., Sabri M. Y., Siti-Zahrah A., Emikpe B. O. (2013). Water condition and identification of potential pathogenic bacteria from red tilapia reared in cage-cultured system in two different water bodies in Malaysia. Afr. J. Microbiol. Res. 7 (47), 5330–5337. doi: 10.5897/AJMR12.1468.
Martínez-porchas M., Villalpando-canchola E., Vargas-albores F. (2016). Significant loss of sensitivity and specificity in the taxonomic classification occurs when short 16S rRNA gene sequences are used. Heliyon 2, e0017. doi: 10.1016/j.heliyon.2016.e00170
Matanza X. M., Osorio C. R. (2020). Exposure of the opportunistic marine pathogen Photobacterium damselae subsp. damselae to human body temperature is a stressful condition that shapes the transcriptome, viability, cell morphology, and virulence. Front. Microbiol. 11, 1771. doi: 10.3389/fmicb.2020.01771.
Maynard C. L., Elson C. O., Hatton R. D., Weaver C. T. (2012). Reciprocal interactions of the intestinal microbiota and immune system. Nature 489 (7415), 231–241. doi: 10.1038/nature11551.
Menezes M. F., Sousa M. J., Paixão P., Atouguia J., Negreiros I., Simões M. J. (2018). Lawsonella clevelandensis as the causative agent of a breast abscess. IDCases. 12, 95–96. doi: 10.1016/j.idcr.2018.03.014
Mercier L., Mouillot D., Bruguier O., Vigliola L., Darnaude A. M. (2012). Multi-element otolith fingerprints unravel sea–lagoon lifetime migrations of gilthead sea bream Sparus aurata. Mar. Ecol. Prog. Ser. 444, 175–194. doi: 10.3354/meps09444
Meron D., Davidovich N., Ofek-Lalzar M., Berzak R., Scheinin A., Regev Y., et al. (2020). Specific pathogens and microbial abundance within liver and kidney tissues of wild marine fish from the Eastern Mediterranean Sea. Microb. Biotechnol., 1–11. doi: 10.1111/1751-7915.13537
Merrifield D. L., Rodiles A. (2015). “The fish microbiome and its interactions with mucosal tissues,” in Mucosal health in aquaculture, vol. p . Eds. Beck B. H., Peatman E. (Plymouth: Academic Press), 273–289. Available at: https://www.sciencedirect.com/science/article/pii/B9780124171862000108.
Minich J. J., Petrus S., Michael J. D., Michael T. P., Knight R., Allen E. E. (2020). Temporal, environmental, and biological drivers of the mucosal microbiome in a wild marine fish, Scomber japonicus. mSphere. 5, e00401–e00420. doi: 10.1128/mSphere.00401-20
Minich J. J., Power C., Melanson M., Knight R., Webber C., Rough K., et al. (2020). The southern bluefin tuna mucosal microbiome is influenced by husbandry method, net pen location, and anti-parasite treatment. Front. Microbiol. 11, 2015. doi: 10.3389/fmicb.2020.02015
Mohammed H. H., Arias C. R. (2015). Potassium permanganate elicits a shift of the external fish microbiome and increases host susceptibility to columnaris disease. Vet. Res. 46, 82. doi: 10.1186/s13567-015-0215-y
Molnár K. (2002). Site preference of fish myxosporeans in the gill. Dis. Aquat Organ. 48, 197–207. doi: 10.3354/dao048197
Mondal S. K., Lijon B., Reza R., Ishika T. (2016). Isolation and identification of Vibrio nereis and Vibrio harveyi in farm raised penaeus monodon marine shrimp. Int. J. Biosci. 8 (4), 55–61. doi: 10.12692/ijb/8.4.55-61.
Montaña S., Palombarani S., Carulla M., Kunst A., Rodriguez C. H., Nastro M., et al. (2018). First case of bacteraemia due to Acinetobacter schindleri harbouring bla NDM-1 in an immunocompromised patient. New Microbes New Infect. 21, 28–30. doi: 10.1016/j.nmni.2017.10.004
Montánchez I., Ogayar E., Plágaro A. H., Esteve-codina A., Gómez-Garrido J., Orruño M., et al. (2019). Analysis of Vibrio harveyi adaptation in sea water microcosms at elevated temperature provides insights into the putative mechanisms of its persistence and spread in the time of global warming. Sci. Rep. 9, 289. doi: 10.1038/s41598-018-36483-0
Morales S. E., Holben W. E. (2009). Empirical testing of 16S rRNA gene PCR primer pairs reveals variance in target specificity and efficacy not suggested by in silico analysis. Appl. Environ. Microbiol. 75 (9), 2677–2683. doi: 10.1128/AEM.02166-08
Moya A., Ferrer M. (2016). Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 24 (5), 402–413. doi: 10.1016/j.tim.2016.02.002
Nagelkerken I., van der Velde G. (2002). Do non-estuarine mangroves harbour higher densities of juvenile fish than adjacent shallow-water and coral reef habitats in Curaçao (Netherlands Antilles)? Mar. Ecol. Prog. Ser. 245, 191–204. doi: 10.3354/meps245191
Nakasaki K., Tran L. T. H., Idemoto Y., Abe M., Palenzuela Rollon A. (2009). Comparison of organic matter degradation and microbial community during thermophilic composting of two different types of anaerobic sludge. Bioresour Technol. 100, 676–682. doi: 10.1016/j.biortech.2008.07.046
Naqib A., Poggi S., Wang W., Hyde M., Kunstman K., Green S. J. (2018). “Making and sequencing heavily multiplexed, high-throughput 16S ribosomal RNA gene amplicon libraries using a flexible, two-stage PCR protocol,” in Gene expression analysis: Methods and protocols, vol. p . Eds. Raghavachari N., Garcia-Reyero N.l. (New York, NY: Humana Press), 149–169.
Nguyen T. H., Dorny P., Nguyen T. T. G., Dermauw V. (2021). Helminth infections in fish in Vietnam: A systematic review. Int. J. Parasitol. Parasites Wildl. 14, 13–32. doi: 10.1016/j.ijppaw.2020.12.001
Nguyen K., Liou Y. (2019). Global mapping of eco-environmental vulnerability from human and nature disturbances. Sci. Total Environ. 664, 995–1004. doi: 10.1016/j.scitotenv.2019.01.407
Ni J., Yan Q., Yu Y., Zhang T. (2013). Factors influencing the grass carp gut microbiome and its effect on metabolism. FEMS Microbiol. Ecol. 87, 704–714. doi: 10.1111/1574-6941.12256
Oliver F., Yilmaz P., Quast C., Gerken J., Beccati A., Ciuprina A., et al. (2017). 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 261, 169–176. doi: 10.1016/j.jbiotec.2017.06.1198
Otto M. (2009). Staphylococcus epidermidis — the a’ccidental’ pathogen. Nat. Rev. Microbiol. 7, 555–567. doi: 10.1038/nrmicro2182
Papastamatiou Y. P., Itano D. G., Dale J. J., Meyer C. G., Holland K. N. (2010). Site fidelity and movements of sharks associated with ocean-farming cages in Hawaii. Mar. Freshw. Res. 61 (12), 1366–1375. doi: 10.1071/MF10056
Pascual J., Macián M. C., Arahal D. R., Garay E., Pujalte M. (2010). Multilocus sequence analysis of the central clade of the genus Vibrio by using the 16S rRNA, recA, pyrH, rpoD, gyrB, rctB and toxR genes. Int. J. Syst. Evol. Microbiol. 60, 154–165. doi: 10.1099/ijs.0.010702-0
Paździor E. (2016). Shewanella putrefaciens – a new opportunistic pathogen of freshwater fish. J. Vet. Res. 60 (4), 429–434. doi: 10.1515/jvetres-2016-0064
Pérez-Ruzafa A., Pérez-Marcos M., Marcos C. (2018). From fish physiology to ecosystems management: Keys for moving through biological levels of organization in detecting environmental changes and anticipate their consequences. Ecol. Indic. 90, 334–345. doi: 10.1016/j.ecolind.2018.03.019
Petrova M. I., Reid G., Vaneechoutte M., Lebeer S. (2017). Lactobacillus iners: Friend or foe? Trends Microbiol. 25 (3), 182–191. doi: 10.1016/j.tim.2016.11.007.
Pham T. H., Cheng P. T., Wang P., Chen S. (2020). Genotypic diversity, and molecular and pathogenic characterization of Photobacterium damselae subsp. piscicida isolated from different fish species in Taiwan. J. Fish Dis. 43, 757–774. doi: 10.1111/jfd.13173
Piroddi C., Bearzi G., Christensen V. (2011). Marine open cage aquaculture in the eastern Mediterranean Sea: a new trophic resource for bottlenose dolphins. Mar. Ecol. Prog. Ser. 440, 255–266. doi: 10.3354/meps09319
Powell M. D., Harris J. O., Carson J., Hill J. V. (2005). Effects of gill abrasion and experimental infection with Tenacibaculum maritimum on the respiratory physiology of Atlantic salmon Salmo salar affected by amoebic gill disease. Dis. Aquat Organ. 63, 169–174. doi: 10.3354/dao063169
Pratte Z. A., Besson M., Hollman R. D., Stewart J. (2018). The gills of reef fish support a distinct microbiome influenced by host-specific factors. Appl. Environ. Microbiol. 84 (9), e00063–e00018. doi: 10.1128/AEM.00063-18
Pruesse E., Peplies J., Glöckner F. O. (2012). SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 28 (14), 1823–1829. doi: 10.1093/bioinformatics/bts252
Qiao G., Lee D. C., Sung H. W., Li H., Xu D.-H., Il P. S. (2012). Microbiological characteristics of Vibrio scophthalmi isolates from diseased olive flounder Paralichthys olivaceus. Fish Sci. 78, 853–863. doi: 10.1007/s12562-012-0502-8
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Ramsey M. M., Freire M. O., Gabrilska R. A., Rumbaugh K. P., Lemon K. P. (2016). Staphylococcus aureus shifts toward commensalism in response to Corynebacterium species. Front. Microbiol. 7, 1230. doi: 10.3389/fmicb.2016.01230
Rasmussen-ivey C. R., Figueras M. J., Mcgarey D., Liles M. R. (2016). Virulence factors of Aeromonas hydrophila: In the wake of reclassification. Front. Microbiol. 7, 1337. doi: 10.3389/fmicb.2016.01337
Reddy M. R. K., Mastan S. A. (2013). Emerging Acinetobacter schindleri in red eye infection of Pangasius sutchi. Afr. J. Biotechnol. Full. 12 (50), 6992–6996. doi: 10.5897/AJB2013.12342.
Regalado N. G., Martin G., Antony S. J. (2009). Acinetobacter lwoffii: Bacteremia associated with acute gastroenteritis. Travel Med. Infect. Dis. 7, 316–317. doi: 10.1016/j.tmaid.2009.06.001
Reilly G. D., Reilly C. A., Smith E. G., Baker-Austin C. (2011). Vibrio alginolyticus-associated wound infection acquired in British waters, Guernsey, July 2011. Eurosurveillance 16 (42), pii=19994. doi: 10.2807/ese.16.42.19994-en
Rivas A. J., Lemos M. L., Osorio C. R. (2013). Photobacterium damselae subsp. damselae, a bacterium pathogenic for marine animals and humans. Front. Microbiol. 4, 283. doi: 10.3389/fmicb.2013.00283.
Rosado D., Pérez-losada M., Severino R., Cable J., Xavier R. (2019). Characterization of the skin and gill microbiomes of the farmed seabass (Dicentrarchus labrax) and seabream (Sparus aurata). Aquaculture. 500, 57–64. doi: 10.1016/j.aquaculture.2018.09.063
Salinas I. (2015). The mucosal immune system of teleost fish. Biol. (Basel). 4, 525–539. doi: 10.3390/biology4030525
Sato A., Somamoto T., Yokooka H., Okamoto N. (2005). Systemic priming of alloreactive cytotoxic cells in carp, following anal administration of allogeneic cell antigens. Fish Shellfish Immunol. 19 (1), 43–52. doi: 10.1016/j.fsi.2004.11.010
Selvin J., Lipton A. P. (2003). Vibrio alginolyticus associated with white spot disease of Penaeus monodon. Dis. Aquat Organ. 57, 147–150. doi: 10.3354/dao057147
Sevellec M., Pavey S. A., Boutin S., Filteau M., Derome N., Bernatchez L. (2014). Microbiome investigation in the ecological speciation context of lake whitefish (Coregonus clupeaformis) using next-generation sequencing. J. Evol. Biol. 27, 1029–1046. doi: 10.1111/jeb.12374
Shapiro-Ilan D. I., Fuxa J. R., Lacey L. A., Onstad D. W., Kaya H. K. (2005). Definitions of pathogenicity and virulence in invertebrate pathology. J. Invertebr Pathol. 88 (1), 1–7. doi: 10.1016/j.jip.2004.10.003
Shapiro K., Miller W. A., Silver M. W., Odagiri M., Largier J. L., Conrad P. A., et al. (2013). Research commentary: Association of zoonotic pathogens with fresh, estuarine, and marine macroaggregates. Microb. Ecol. 65, 928–933. doi: 10.1007/s00248-012-0147-2
Shea D., Bateman A., Li S., Tabata A., Schulze A., Mordecai G., et al. (2020). Environmental DNA from multiple pathogens is elevated near active Atlantic salmon farms. Proc. R Soc. B. 287, 20202010. doi: 10.1098/rspb.2020.2010
Sinclair L., Osman O. A., Bertilsson S., Eiler A. (2015). Microbial community composition and diversity via 16S rRNA gene amplicons: Evaluating the illumina platform. PloS One 10 (2), e0116955. doi: 10.1371/journal.pone.0116955
Siqueira J. F. Jr., Rôças I. N. (2003). Campylobacter gracilis and Campylobacter rectus in primary endodontic infections. Int. Endodotnic J. 36, 174–180. doi: 10.1046/j.1365-2591.2003.00636.x
Stoddard S. F., Smith B. J., Hein R., Roller B. R. K., Schmidt T. M. (2015). rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 43 (D1), D593–D598. doi: 10.1093/nar/gku1201
Takiguchi Y., Terano T., Hirai A. (2003). Lung abscess caused by Actinomyces odontolyticus. Intern. Med. 42, 723–725. doi: 10.2169/internalmedicine.42.723
Tarnecki A. M., Brennan N. P., Schloesser R. W., Rhody N. R. (2019). Shifts in the skin-associated microbiota of hatchery-reared common snook Centropomus undecimalis during acclimation to the wild. Microb. Ecol. 77, 770–781. doi: 10.1007/s00248-018-1252-7
Techtmann S. M., Fortney J. L., Ayers K. A., Joyner D. C., Linley T. D., Pfiffner S. M., et al. (2015). The unique chemistry of Eastern Mediterranean water masses selects for distinct microbial communities by depth. PloS One 10 (3), e0120605. doi: 10.1371/journal.pone.0120605
Terceti M. S., Ogut H., Osorio C. R. (2016). Photobacterium damselae subsp. damselae, an emerging fish pathogen in the black Sea: Evidence of a multiclonal origin. Appl. Environ. Microbiol. 82 (13), 3736–3745. doi: 10.1128/AEM.00781-16.
Terceti M. S., Rivas A. J., Alvarez L., Noia M., Cava F., Osorio C. R. (2017). rstB regulates expression of the Photobacterium damselae subsp. damselae major virulence factors damselysin, phobalysin P and phobalysin C. Front. Microbiol. 8, 582. doi: 10.3389/fmicb.2017.00582.
Tevell S., Hellmark B., Nilsdotter-Augustinsson Ä, Söderquist B. (2017). Staphylococcus capitis isolated from prosthetic joint infections. Eur. J. Clin. Microbiol. Infect. Dis. 36, 115–122. doi: 10.1007/s10096-016-2777-7
van de Water J. A. J. M., Melkonian R., Voolstra C. R., Junca H., Beraud E., Allemand D., et al. (2017). Comparative assessment of Mediterranean gorgonian-associated microbial communities reveals conserved core and locally variant bacteria. Microb. Ecol. 73, 466–478. doi: 10.1007/s00248-016-0858-x
Vayssier-Taussat M., Moutailler S., Michelet L., Devillers E., Bonnet S., Cheval J., et al. (2013). Next generation sequencing uncovers unexpected bacterial pathogens in ticks in Western Europe. PloS One 8 (11), e81439. doi: 10.1371/journal.pone.0081439
Viau E. J., Goodwin K. D., Yamahara K. M., Layton B. A., Sassoubre L. M., Burns S. L., et al. (2011). Bacterial pathogens in Hawaiian coastal streams-associations with fecal indicators, land cover, and water quality. Water Res. 45, 3279–3290. doi: 10.1016/j.watres.2011.03.033
Vignier N., Barreau M., Olive C., Baubion E., Théodose R., Hochedez P., et al. (2013). Human infection with Shewanella putrefaciens and S. algae: Report of 16 cases in Martinique and review of the literature. Am. Soc. Trop. Med. Hyg. 89 (1), 151–156. doi: 10.4269/ajtmh.13-0055.
Vorburger C., Perlman S. J. (2018). The role of defensive symbionts in host – parasite coevolution. Biol. Rev. 93, 1747–1764. doi: 10.1111/brv.12417
Walters W., Hyde E. R., Berg-lyons D., Ackermann G., Humphrey G., Parada A., et al. (2015). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems. 1 (1), e00009–e00015. doi: 10.1128/mSystems.00009-15.
Wang Z., Cai C., Cao X., Zhu J., He J., Wu P., et al. (2018). Supplementation of dietary astaxanthin alleviated oxidative damage induced by chronic high pH stress, and enhanced carapace astaxanthin concentration of Chinese mitten crab Eriocheir sinensis. Aquaculture. 483, 230–237. doi: 10.1016/j.aquaculture.2017.10.006
Wang T., Costa V., Jenkins S. G., Hartman B. J., Westblade L. F. (2019). Acinetobacter radioresistens infection with bacteremia and pneumonia. IDCases. 16, e00495. doi: 10.1016/j.idcr.2019.e00495
Wilson S. K., Depczynski M., Fisher R., Holmes T. H., O’Leary R. A., Tinkler P. (2010). Habitat associations of juvenile fish at Ningaloo Reef, Western Australia: The importance of coral and algae. PloS One 5 (12), e15185. doi: 10.1371/journal.pone.0015185
Woo P. C. Y., Tse H., Lau S. K. P., Leung K., Woo K. S., Wong M. K. M., et al. (2005). Alkanindiges hongkongensis sp. nov. a novel Alkanindiges species isolated from a patient with parotid abscess. Syst. Appl. Microbiol. 28, 316–322. doi: 10.1016/j.syapm.2005.01.003
Xie Z.-Y., Hu C.-Q., Chen C., Zhang L.-P., Ren C.-H. (2005). Investigation of seven Vibrio virulence genes among Vibrio alginolyticus and Vibrio parahaemolyticus strains from the coastal mariculture systems in guangdong, China. pdf. Lett. Appl. Microbiol. 41, 202–207. doi: 10.1111/j.1472-765X.2005.01688.x
Yanong R. P. E. (2003). Necropsy techniques for fish. Semin. Avian Exot Pet Med. 12 (2), 89–105. doi: 10.1053/saep.2003.127885
Yildirimer C. C., Brown K. H. (2018). Intestinal microbiota lipid metabolism varies across rainbow trout (Oncorhynchus mykiss) phylogeographic divide. J. Appl. Microbiol. 125 (6), 1614–1625. doi: 10.1111/jam.14059
Yilmaz P., Parfrey L. W., Yarza P., Gerken J., Pruesse E., Quast C., et al. (2014). The SILVA and “All-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, 643–648. doi: 10.1093/nar/gkt1209
Zarantoniello M., Bortoletti M., Olivotto I., Ratti S., Poltronieri C., Negrato E., et al. (2021). Salinity, temperature and ammonia acute stress response in seabream (Sparus aurata) juveniles: A multidisciplinary study. Animals. 11, 97. doi: 10.3390/ani11010097
Zhang L., Liu Y., Zheng H. J., Zhang C. P. (2020). The oral microbiota may have influence on oral cancer. Front. Cell Infect. Microbiol. 9, 476. doi: 10.3389/fcimb.2019.00476
Zhang C., Wang C., Jatt A.-N., Liu H., Liu Y. (2020). Role of rpoS in stress resistance, biofilm formation and quorum sensing of Shewanella baltica. Lett. Appl. Microbiol. 72, 307–315. doi: 10.1111/lam.13424
Zhang Z., Yu Y., Wang Y., Liu X., Wang L., Zhang H., et al. (2020). Complete genome analysis of a virulent Vibrio scophthalmi strain VSc190401 isolated from diseased marine fish half-smooth tongue sole, Cynoglossus semilaevis. BMC Microbiol. 20, 341. doi: 10.1186/s12866-020-02028-7
Zhu B., Macleod L. C., Kitten T., Xu P. (2018). Streptococcus sanguinis biofilm formation & interaction with oral pathogens. Future Microbiol. 13 (8), 915–932. doi: 10.2217/fmb-2018-0043
Keywords: Photobacterium, Shewanella, Staphylococcus, Streptococcus, Vibrio, Marine fish, Wild fish pathogens, Gill microbiome
Citation: Itay P, Shemesh E, Ofek-Lalzar M, Davidovich N, Kroin Y, Zrihan S, Stern N, Diamant A, Wosnick N, Meron D, Tchernov D and Morick D (2022) An insight into gill microbiome of Eastern Mediterranean wild fish by applying next generation sequencing. Front. Mar. Sci. 9:1008103. doi: 10.3389/fmars.2022.1008103
Received: 31 July 2022; Accepted: 01 September 2022;
Published: 29 September 2022.
Edited by:
Dusan Palic, Chair for Fish Diseases and Fisheries Biology, Ludwig-Maximilians-University Munich, GermanyReviewed by:
Mateus De Souza Terceti, University of Santiago de Compostela, SpainTa Chih Chneg, National Pingtung University of Science and Technology, Taiwan
Copyright © 2022 Itay, Shemesh, Ofek-Lalzar, Davidovich, Kroin, Zrihan, Stern, Diamant, Wosnick, Meron, Tchernov and Morick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danny Morick, dmorick@univ.haifa.ac.il