 Qiong Wang
Qiong Wang Jianjian Lv
Jianjian Lv Xianyun Ren
Xianyun Ren Jiajia Wang
Jiajia Wang Shaoting Jia1,2
Shaoting Jia1,2 Yuying He
Yuying He- 1Key Laboratory for Sustainable Utilization of Marine Fisheries Resources, Ministry of Agriculture and Rural Affairs, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China
- 2Function Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
Fenneropenaeus chinensis is a commercially cultured shrimp in China. F. chinensis adults show significant sexual dimorphism, with larger females than males. However, sex determination (SD) of F. chinensis has not yet been elucidated. Clarification of the sex-determining system of F. chinensis could enrich our knowledge of the sex differentiation mechanism in crustaceans and facilitate the study of sex-controlling technologies. Here, we studied the sex-determining system of F. chinensis using the fixation index (FST) between the sexes to detect the genetic differentiation in resequencing data of multiple males and females. We located the candidate sex chromosome in the genome of F. chinensis and concluded the female heterogametic (ZW) SD system. We also assembled female-specific sequences, which could be used as molecular markers to identify the sex of F. chinensis. However, the differentiation of the F. chinensis Z and W chromosome is limited. RNA-seq data detected many genes with male-biased expression in the Z-specific region, which possibly could further intensify the divergency between the Z and W chromosomes.
Introduction
The evolution of chromosomes carrying the sex determination (SD) factor is an interesting form of genomic evolution (Charlesworth and Charlesworth, 2000; Bachtrog et al., 2011). In some cases, selection for close linkage may promote suppressed recombination in the SD region, because the region includes a sexually antagonistic polymorphism (Bull, 1985; Bergero and Charlesworth, 2009; Wright et al., 2016). Once recombination becomes suppressed, sex-specific evolutionary pressures act on the evolving sex chromosomes (Lahn and Page, 1997; Moghadam et al., 2012), leading to adaptive and non-adaptive processes that produce distinct differences between the X and Y (or Z and W) chromosomes (Muller, 1918; Bergero and Charlesworth, 2009; Bachtrog, 2013; Li et al., 2021). However, in many species, loss of recombination near the SD locus does not spread across the sex chromosomes, which may remain heteromorphic, and display only limited differentiation (Telgmann-Rauber et al., 2007; Spigler et al., 2008; Tennessen et al., 2016; Pucholt et al., 2017) (Figure 1A).
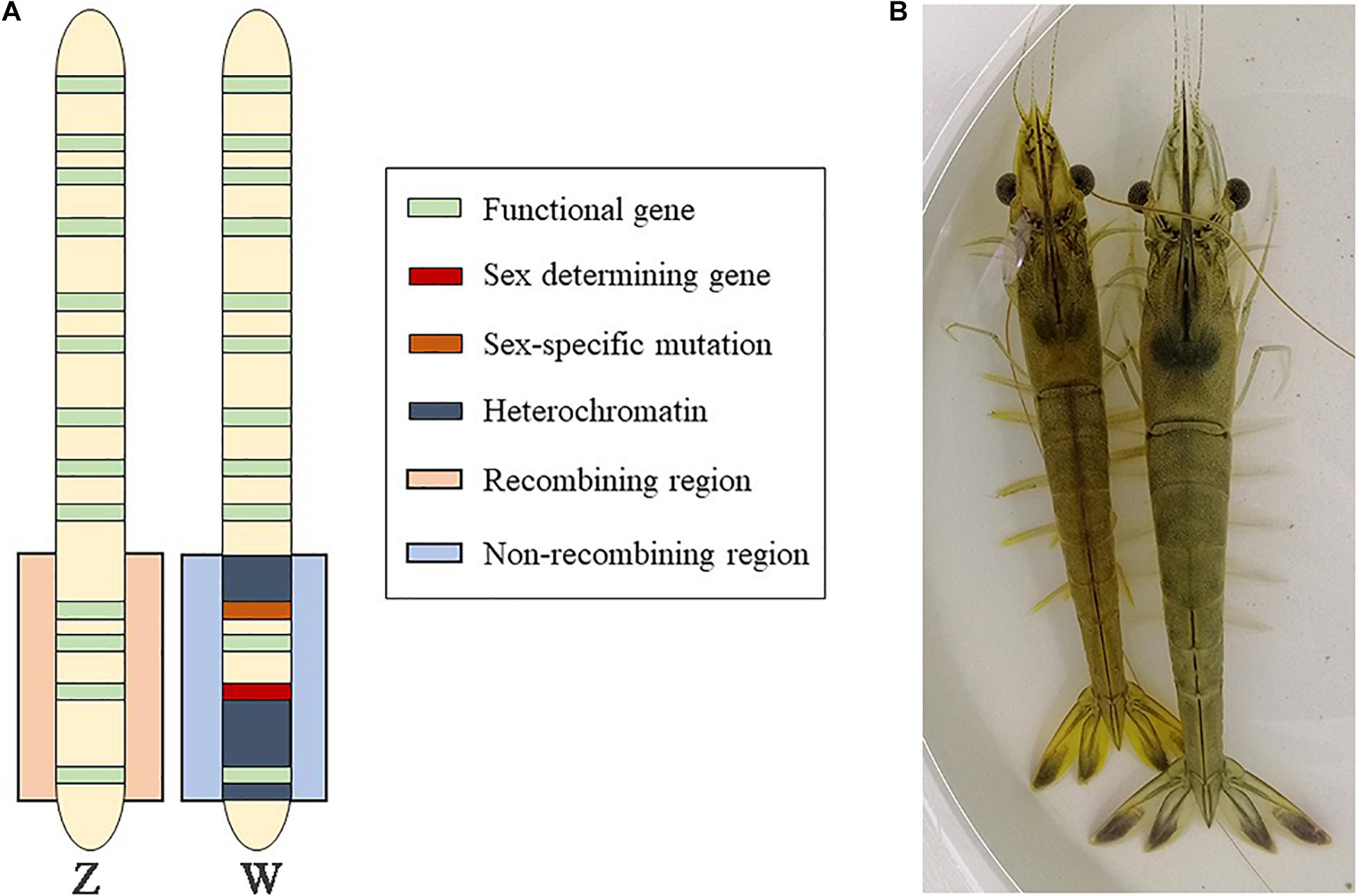
Figure 1. Background for the sex chromosome analysis of F. chinensis. (A) Model of the early-stage sex chromosome. (B) F. chinensis (male on the left and female on the right).
Fenneropenaeus chinensis is a commercially cultured shrimp in China, whose adults are sexually dimorphic. Female shrimps tend to be blue, whereas the males are yellow; the adult females are larger and heavier than the adult males (Wang et al., 2019) (Figure 1B). Studies on the sex of F. chinensis are valuable. In Crustacea, both XX/XY and ZZ/ZW system existed (Shi et al., 2018; Fang et al., 2020). The sex-determining region of a predominant aquaculture shrimp species, the Pacific white shrimp (Litopenaeus vannamei), was mapped by the integrating linkage and association analyses (Yu et al., 2017). The results supported female heterogamety. The sex-determining regions of two other Penaeus shrimps, Penaeus monodon and Penaeus japonicus, have been mapped using high-density linkage analysis (Li et al., 2003; Robinson et al., 2014), but not fine-mapped. However, even some closely related species have different SD loci (Phillip et al., 2001; Miura, 2007; Mank and Avise, 2009; Pucholt et al., 2017). For F. chinensis, several sex-related markers have been identified by QTL mapping, but no direct evidence of their connection with SD was found (Meng et al., 2021). We therefore studied the SD mechanism of F. chinensis to improve our knowledge of the sex differentiation mechanism in crustaceans and facilitate the study of sex-controlling technologies, and provide sex-linked markers to identify the sexes at early developmental stages. The genome of F. chinensis released this year (Wang et al., 2021) makes it realizable to study the sex chromosome. In this study, we used resequencing data to detect sex-linked variants, including female-specific sequences, and preliminarily conclude that the species had a female heterogametic (ZW) SD system.
Materials and Methods
Sample Collection and Sequencing
We randomly selected 10 female and 11 male F. chinensis “Huanghai No. 1” shrimps (age, 4 months) from the conservation base of Haifeng Aquaculture Co., Ltd. (Weifang, Shandong Province, China). The DNA samples of these 21 shrimps were obtained from the muscle and sequenced individually using the BGISEQ platform (BGI, Shenzhen, China), with paired ends of 150 bp. We obtained 219.9 Gb of clean DNA data (female: 104.5 Gb, male: 115.4 Gb). In addition, one female and one male were deeply sequenced, yielding 51.2 and 50.9 Gb clean data, respectively, approximately 35x cover depth.
The genome sequencing data were mapped to the improved reference genome of F. chinensis (Wang et al., 2021) using BWA (v0.7.15) (Li and Durbin, 2010) with default parameters. The bam files of 10 females and 11 males were merged into two pools by sex, and further convert to pileup file by using SAMtools (v1.9) (Li et al., 2009).
Analysis of Genetic Differentiation Between the Sexes
To evaluate the genetic differentiation between the sexes across the genome, we used PoPoolation2 software (Kofler et al., 2011) to convert the pileup file into sync file with a minimum base quality of 20. FST between the sexes and nucleotide diversity values, π, were estimated for all site types, using PoPoolation2 with the following set parameters: window size of 10 kb, step size of 5 kb, minimum allele count of 4, minimum coverage of 10, and maximum coverage of 200. FST was calculated using the merged data of all females and males, whereas π values were estimated using the two deep-sequenced individuals.
Information on mapping depth along the genome was extracted with bedtools (v2.25.0) (Quinlan and Hall, 2010) in 1 kb windows. The depth was normalized by the total read count. The ratio of the depth of male to female was log2-transformed.
Female-Specific Sequence Assembly
Besides the 21 “Huanghai No. 1” F. chinensis shrimps, we also sequenced 15 female and six male F. chinensis shrimps captured from the wild, again using the BGISEQ platform, with paired ends of 150 bp. In total, we obtained 436.3 Gb of clean resequencing data (female: 251.9 Gb, male: 184.4).
We expected a female heterogametic, or ZX system. Candidate W-linked sequences can be detected because they are female-specific. Therefore, we attempted to assemble female-specific sequences according to the method used for the snakehead (Channa argus) (Ou et al., 2017), with some adjustments. Briefly, the pooled sequencing data from the females were mapped to the reference genome, which was constructed from a male. The unmapped reads were assembled using SOAPdenovo (Luo et al., 2012), with Kmer = 31, and then aligned back to the assembled sequence to assess the accuracy of the assembly. Homozygous SNPs could reflect assembly error. The assembled contigs were mapped back to the reference genome using NCBI BLAST (v2.2.29 +) (Johnson et al., 2008) with evalue 1e-1. We deleted contigs for which more than 60% of the fragments that could be mapped to the reference genome. The resequencing data from both the males and the females were mapped to the assembled female-specific contigs. We retained only contigs with male mapping depth = 0 and female mapping depth > 30, representing W-linked candidates.
Validation With PCR Amplification
The candidate W-linked fragments based on the ‘‘Huanghai No. 1’’ strain and captured wild shrimps used for the female-specific sequence assembly were validated by PCR amplification in samples from four populations, containing two other population, ‘‘Huanghai No. 3’’ and wild shrimp bred for a generation. Primers were designed for the female-specific sequences with the web tool Primer31 and synthesized by Sangon Biotech (China). We designed a pair of reference primers that could amplify the sequence in both sexes, but with a different sequence length in the females.
Female and male DNA pools of the four populations were used as PCR templates for the preliminary primer screening. Primers that generated PCR product only in female pools were selected for further validation in the individual samples of the four populations (10 females and 10 males of “Huanghai No. 1,” 10 females and 10 males of “Huanghai No. 3,” 12 females and 12 males of wild shrimp bred for a generation, and 10 females and six males of captured wild shrimp).
RNA-Seq Data Processing
The RNA-seq data have been described in a previous study (NCBI BioProject accession number: PRJNA591354) (Wang et al., 2019). Briefly, the gonad and muscle of female and male F. chinensis shrimps at 5 months of age were collected, and total RNA was extracted. We used three biological replicates for each tissue and sex. The 12 libraries were sequenced using the Illumina NovaSeq S4 platform, with paired ends of 150 bp. Clean reads from each RNA-seq library were aligned to the reference genome using HISAT (Kim et al., 2015). The gene expression of each sample was analyzed with HTSEQ (Anders et al., 2015).
Results
Resequencing Data Identified a Candidate Sex-Linked Region
The fixation index (FST), which measures population differentiation (Weir and Cockerham, 1984; Holsinger and Weir, 2009), was calculated across the genome. Increased FST was observed on chromosome 7 (Chr7), spanning a region of approximately 3 Mb, from 34 to 37 Mb (Figures 2A,B). In the female, nucleotide diversity (π) increased in the same interval, as expected in a system of female heterogamety (Figure 2C), whereas in males, it was similar to the values in the rest of the genome. There are 60 genes located on this 3 Mb interval according to the genomic annotation file of F. chinensis.
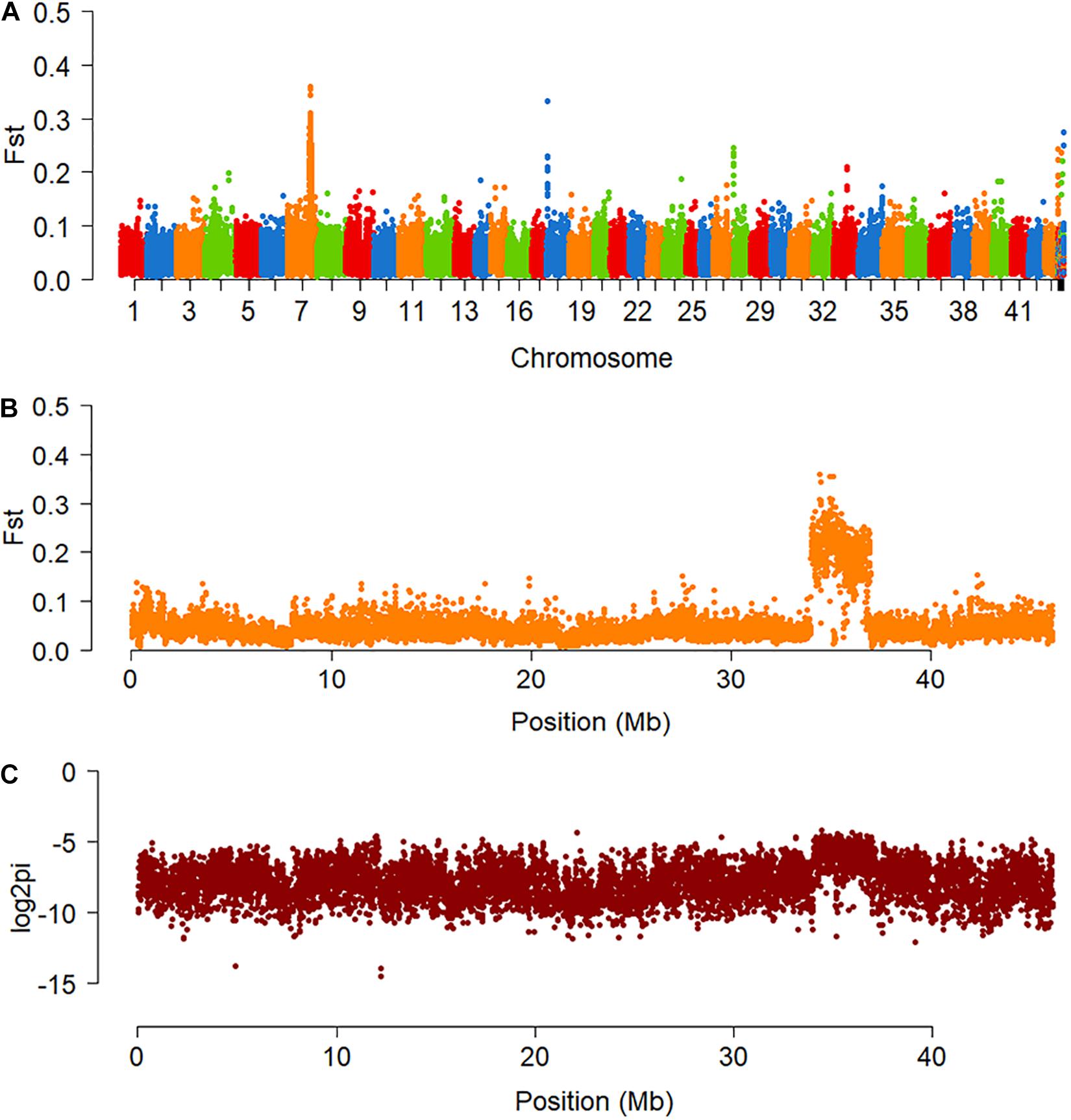
Figure 2. Genomic differentiation of the two sexes of F. chinensis. (A) Genome-wide scan of the fixation index (FST) between the two sexes of F. chinensis. (B) FST on pseudochromosome Chr7. (C) Nucleotide diversity (π) on pseudochromosome Chr7 of the female. Value of π is represented in logarithm.
The mapping depth was estimated across Chr7. More reads aligned to the 34–37 Mb interval in the males than in the females (Supplementary Figure 1), and the ratio of male to female coverage in some zones was close to 2:1 (Figure 3).
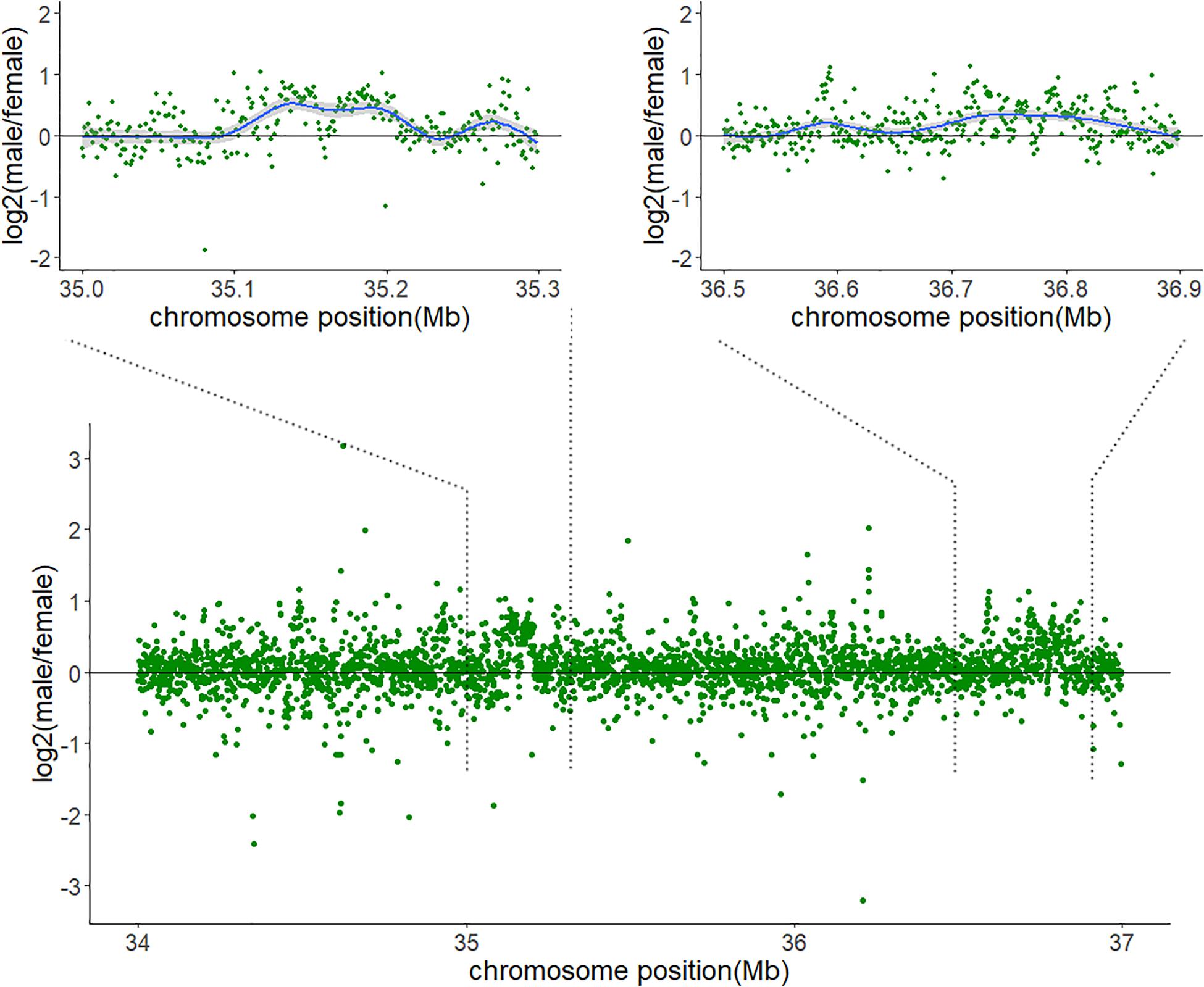
Figure 3. Sequencing depth ratios of male to female in the 34–37 Mb interval of Chr7. The values of the read depth ratios are represented in logarithm.
Assembly and Validation of Female-Specific Sequences
Among the 1.68 billion clean reads in the female pool, 92.01% mapped to the reference genome (Supplementary Table 1), and 134.12 million unmapped reads were also extracted and used in the assembly. In total, we obtained 103.25 Mb of fragments, consisting 435,866 contigs, with 70 contigs longer than 2,000 bp (Supplementary Table 2). Alignment of the unmapped reads showed a high coverage and low error rate (Supplementary Table 3). After further screening, we obtained 363 candidate female-specific contigs.
We selected 16 of the longest candidate female-specific contigs for validation. Sequences from three contigs amplified only with female DNA pools (Figure 4A). Further validation was performed using individual DNA templates (Figures 4B–D).
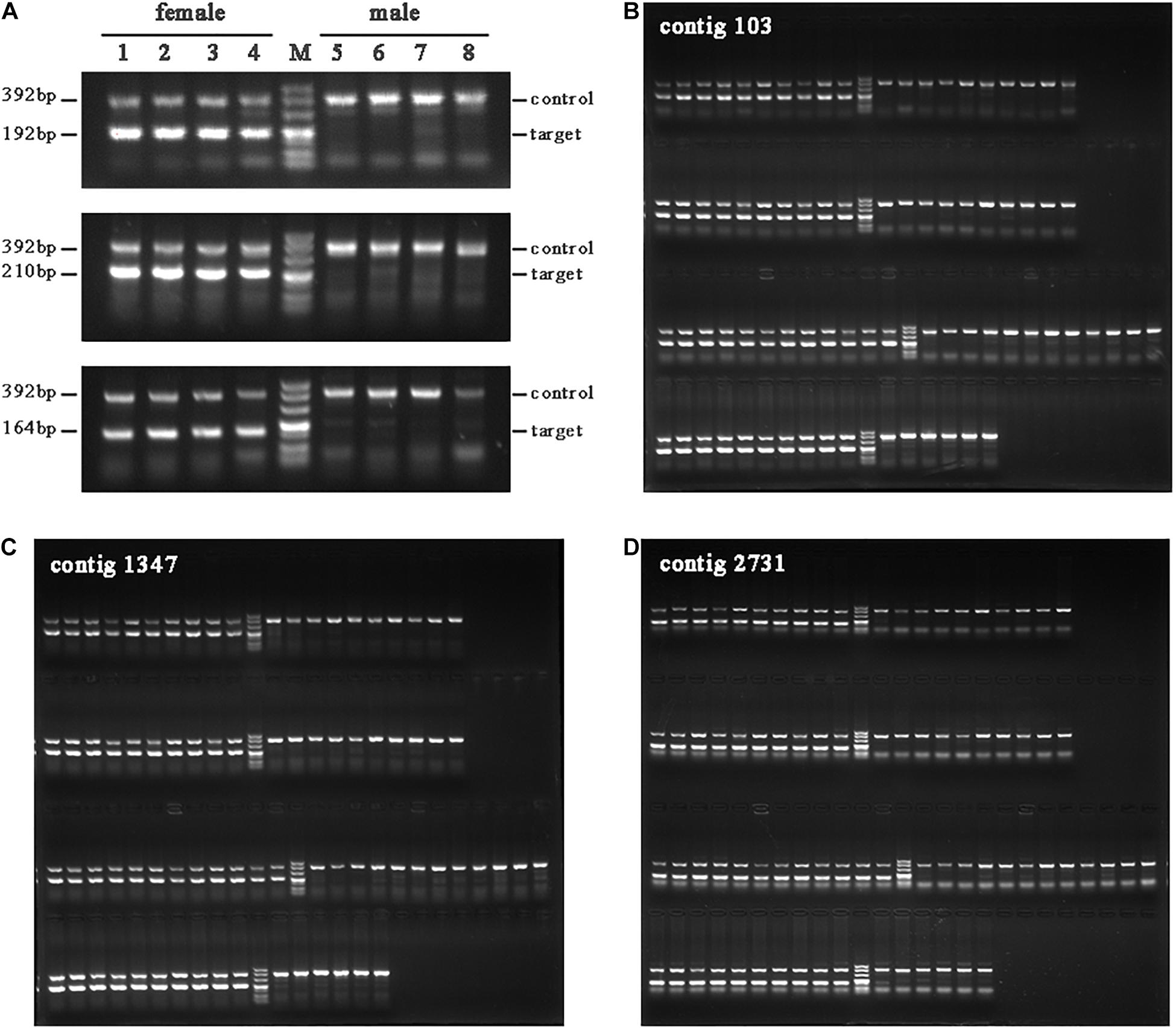
Figure 4. PCR amplification of the female-specific sequences in female and male shrimps. (A) Female-specific sequence amplification with mixed DNA templates. Lanes 1–4 refer to females of “Huanghai No. 1,” “Huanghai No. 3,” wild shrimp bred for a generation, and captured wild shrimp, respectively; lanes 5–8 refer to male shrimps in the same order. The three lines from top to bottom correspond to three contigs (contig 103, contig 1347, and contig 2731). (B–D) The three contigs were amplified with individual DNA templates.
RNA-Seq Data Detected Male-Biased Expression Genes in the Interval on Chr7
To obtain information on the expression of genes on Chr7, we re-analyzed the RNA-seq data reported in a previous study (Wang et al., 2019) (Supplementary Figure 2). In muscle and gonad tissue of 5-month-old animals, we detected abundant differentially expressed genes (DEGs). In the muscle, 743 genes had male-biased expression and 2,291 genes had female-biased expression (Supplementary Figure 3). In the gonad, there were 1,713 male-biased and 1,352 female-biased genes (Supplementary Figure 4). DEGs in the gonad showed a higher proportion (71.21%) of male-biased expression on Chr7 than other chromosomes (Supplementary Table 4).
In the 34–37 Mb interval of Chr7, we found five genes with male-biased and 11 with female-biased expression in the muscle (Figure 5A and Supplementary Table 5). In the gonad, the expression of all 13 DEGs in this region was male-biased; even expression of the adjacent genes was male-biased (Figure 5B and Supplementary Table 6).
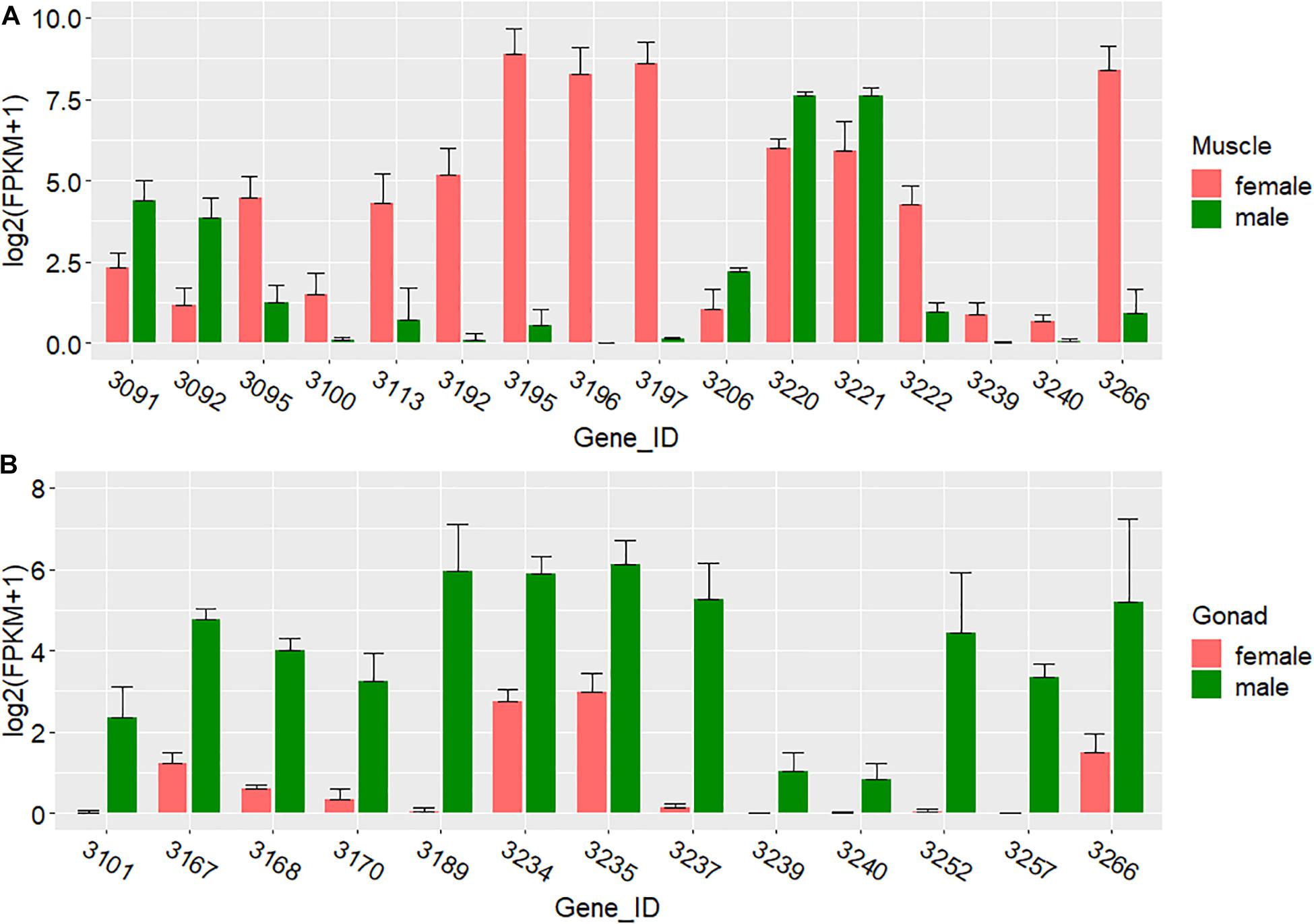
Figure 5. Expression of DEGs in the 34–37 Mb interval of Chr7. (A) Expression in the muscle. (B) Expression in the gonad.
Discussion
The FST analysis located the sex-differentiation region on Chr7, which was regarded as the candidate sex chromosome in this study. In the differentiation region, the higher read mapping depth in males than females suggests that the sex-linked region is hemizygous in females, and the increased π in the females indicating that the SD system of F. chinensis is female heterogametic (female: ZW, male: ZZ). However, this species does not have differentiated Z and W chromosomes. Most fragments of the candidate “Z chromosome” were indistinguishable from autosomal sequences, according to the FST values and gene expression analyses. The sex chromosome formation of this species may only stay a primary stage.
During sex chromosome divergence, the suppression of recombination leads to the accumulation of mutations, which can result in highly heteromorphic sex chromosomes (Wright et al., 2016). At the primary stage of the divergence, the loss of recombination near the SD locus does not spread across the sex chromosomes. The sex chromosomes display only limited levels of differentiation. There are 60 genes annotated in the interval of 34–37 Mb of Chr7; compared to the 790 genes on Chr7 (46 Mb), and 25,026 genes on the whole genome (1.45 Gb), the gene density is slightly higher, which might be caused by the intergenic region loss in the initial process of recombination suppression.
However, if recombination becomes suppressed in a large genome region carrying a sex-determining locus, transcriptional degeneration of the W (or Y) allele can occur quickly (Bachtrog et al., 2008; Papadopulos et al., 2015). The enrichment of sex-biased expression genes in such an SD region may occur before loss of genes from the sex-limited chromosome (Bergero and Charlesworth, 2011; Chibalina and Filatov, 2011; Muyle et al., 2012; Pucholt et al., 2017). All the DEGs in and near the candidate Z-linked region exhibited male-biased expression, suggesting that many mutations causing male-biased expression have accumulated. These DEGs may be related to sex development and responsible for sexual dimorphism or reproduction. However, further studies need to be performed to analyze the functions of the DEGs.
The female-specific sequences detected in our analysis may have been derived from a W chromosome-like region. However, the Z and W regions appear to carry highly similar sequences, and some of our primers were designed for both of them (Supplementary Figure 5).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA591354).
Author Contributions
QW collected the samples and carried out genetic differentiation analysis. XR extracted DNA and RNA for sequencing. JJL carried out female-specific fragment assembly. XR processed the RNA-seq data. JW and SJ performed verification experiment. JL conceived this project. YH supervised the work. QW and JJL wrote the manuscript. All authors contributed to the final manuscript.
Funding
This work was supported by the National Key R&D Program of China (No. 2019YFD0900403), China Agriculture Research System of MOF and MARA (No. CARS-48), National Natural Science Foundation of China (No. 31902367), and Central Public-interest Scientific Institution Basal Research Fund of CAFS (No. 2020TD46).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to appreciate our colleagues of the Key Laboratory for Sustainable Utilization of Marine Fisheries Resources of Yellow Sea Fisheries Research Institute, for their assistance on sample collection and helpful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.743727/full#supplementary-material
Footnotes
References
Bachtrog, D., Kirkpatrick, M., Mank, J. E., McDaniel, S. F., Pires, J. C., Rice, W., et al. (2011). Are all sex chromosomes created equal? Trends Genet 27, 350–357. doi: 10.1016/j.tig.2011.05.005
Charlesworth, B., and Charlesworth, D. (2000). The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci 355, 1563–1572. doi: 10.1098/rstb.2000.0717
Bergero, R., and Charlesworth, D. (2009). The evolution of restricted recombination in sex chromosomes. Trends Ecol Evol 24, 94–102. doi: 10.1016/j.tree.2008.09.010
Bull, J. J. (1985). Sex determining mechanisms: an evolutionary perspective. Experientia 41, 1285–1296. doi: 10.1007/bf01952071
Wright, A. E., Dean, R., Zimmer, F., and Mank, J. E. (2016). How to make a sex chromosome. Nat Commun 7, 12087. doi: 10.1038/ncomms12087
Lahn, B. T., and Page, D. C. (1997). Functional coherence of the human Y chromosome. Science 278, 675–680. doi: 10.1126/science.278.5338.675
Moghadam, H. K., Pointer, M. A., Wright, A. E., Berlin, S., and Mank, J. E. (2012). W chromosome expression responds to female-specific selection. Proc Natl Acad Sci U S A 109, 8207–8211. doi: 10.1073/pnas.1202721109
Muller, H. J. (1918). Genetic Variability, Twin Hybrids and Constant Hybrids, in a Case of Balanced Lethal Factors. Genetics 3, 422–499. doi: 10.1093/genetics/3.5.422
Li, M., Zhang, R., Fan, G., Xu, W., Zhou, Q., Wang, L., et al. (2021). Reconstruction of the origin of a neo-Y sex chromosome and its evolution in the spotted knifejaw, Oplegnathus punctatus. Mol Biol Evol 38, 2615–2626. doi: 10.1093/molbev/msab056
Bachtrog, D. (2013). Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet 14, 113–124. doi: 10.1038/nrg3366
Pucholt, P., Wright, A. E., Conze, L. L., Mank, J. E., and Berlin, S. (2017). Recent Sex Chromosome Divergence despite Ancient Dioecy in the Willow Salix viminalis. Mol Biol Evol 34, 1991–2001. doi: 10.1093/molbev/msx144
Telgmann-Rauber, A., Jamsari, A., Kinney, M. S., Pires, J. C., and Jung, C. (2007). Genetic and physical maps around the sex-determining M-locus of the dioecious plant asparagus. Mol Genet Genomics 278, 221–234. doi: 10.1007/s00438-007-0235-z
Tennessen, J. A., Govindarajulu, R., Liston, A., and Ashman, T. L. (2016). Homomorphic ZW chromosomes in a wild strawberry show distinctive recombination heterogeneity but a small sex-determining region. New Phytol 211, 1412–1423. doi: 10.1111/nph.13983
Spigler, R. B., Lewers, K. S., Main, D. S., and Ashman, T. L. (2008). Genetic mapping of sex determination in a wild strawberry, Fragaria virginiana, reveals earliest form of sex chromosome. Heredity (Edinb) 101, 507–517. doi: 10.1038/hdy.2008.100
Wang, Q., He, Y., and Li, J. (2019). Conjoint Analysis of SMRT- and Illumina-Based RNA-Sequencing Data of Fenneropenaeus chinensis Provides Insight Into Sex-Biased Expression Genes Involved in Sexual Dimorphism. Front Genet 10:1175. doi: 10.3389/fgene.2019.01175
Shi, X., Waiho, K., Li, X., Ikhwanuddin, M., Miao, G., Lin, F., et al. (2018). Female-specific SNP markers provide insights into a WZ/ZZ sex determination system for mud crabs Scylla paramamosain, S. tranquebarica and S. serrata with a rapid method for genetic sex identification. BMC Genomics 19:981. doi: 10.1186/s12864-018-5380-8
Fang, S., Zhang, Y., Shi, X., Zheng, H., Li, S., Zhang, Y., et al. (2020). Identification of male-specific SNP markers and development of PCR-based genetic sex identification technique in crucifix crab (Charybdis feriatus) with implication of an XX/XY sex determination system. Genomics 112, 404–411. doi: 10.1016/j.ygeno.2019.03.003
Yu, Y., Zhang, X., Yuan, J., Wang, Q., Li, S., Huang, H., et al. (2017). Identification of Sex-determining Loci in Pacific White Shrimp Litopeneaus vannamei Using Linkage and Association Analysis. Mar Biotechnol (NY) 19, 277–286. doi: 10.1007/s10126-017-9749-5
Robinson, N. A., Gopikrishna, G., Baranski, M., Katneni, V. K., Shekhar, M. S., Shanmugakarthik, J., et al. (2014). QTL for white spot syndrome virus resistance and the sex-determining locus in the Indian black tiger shrimp (Penaeus monodon). BMC Genomics 15:731. doi: 10.1186/1471-2164-15-731
Li, Y. T., Byrne, K., Miggiano, E., Whan, V., Moore, S., Keys, S., et al. (2003). Genetic mapping of the kuruma prawn Penaeus japonicus using AFLP markers. Aquaculture 219, 143–156. doi: 10.1016/S0044-8486(02)00355-1
Phillip, R. B., Konkol, N. R., Reed, K. M., and Stein, J. D. (2001). Chromosome painting supports lack of homology among sex chromosomes in Oncorhynchus, Salmo, and Salvelinus (Salmonidae). Genetica 111, 119–123. doi: 10.1023/a:1013743431738
Miura, I. (2007). An evolutionary witness: the frog rana rugosa underwent change of heterogametic sex from XY male to ZW female. Sex Dev 1, 323–331. doi: 10.1159/000111764
Mank, J. E., and Avise, J. C. (2009). Evolutionary diversity and turn-over of sex determination in teleost fishes. Sex Dev 3, 60–67. doi: 10.1159/000223071
Meng, X., Fu, Q., Luan, S., Luo, K., Sui, J., and Kong, J. (2021). Genome survey and high-resolution genetic map provide valuable genetic resources for Fenneropenaeus chinensis. Sci Rep 11, 7533. doi: 10.1038/s41598-021-87237-4
Wang, Q., Ren, X., Liu, P., Li, J., Lv, J., Wang, J., et al. (2021). Improved genome assembly of Chinese shrimp (Fenneropenaeus chinensis) suggests adaptation to the environment during evolution and domestication. Mol Ecol Resour doi: 10.1111/1755-0998.13463
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Kofler, R., Pandey, R. V., and Schlotterer, C. (2011). PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27, 3435–3436. doi: 10.1093/bioinformatics/btr589
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Ou, M., Yang, C., Luo, Q., Huang, R., Zhang, A. D., Liao, L. J., et al. (2017). An NGS-based approach for the identification of sex-specific markers in snakehead (Channa argus). Oncotarget 8, 98733–98744. doi: 10.18632/oncotarget.21924
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. doi: 10.1186/2047-217X-1-18
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res 36, W5–W9. doi: 10.1093/nar/gkn201
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. doi: 10.1038/nmeth.3317
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-Statistics for the Analysis of Population Structure. Evolution 38, 1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x
Holsinger, K. E., and Weir, B. S. (2009). Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat Rev Genet 10, 639–650. doi: 10.1038/nrg2611
Bachtrog, D., Hom, E., Wong, K. M., Maside, X., and de Jong, P. (2008). Genomic degradation of a young Y chromosome in Drosophila miranda. Genome Biol 9, R30. doi: 10.1186/gb-2008-9-2-r30
Papadopulos, A. S., Chester, M., Ridout, K., and Filatov, D. A. (2015). Rapid Y degeneration and dosage compensation in plant sex chromosomes. Proc Natl Acad Sci U S A 112, 13021–13026. doi: 10.1073/pnas.1508454112
Muyle, A., Zemp, N., Deschamps, C., Mousset, S., Widmer, A., and Marais, G. A. (2012). Rapid de novo evolution of X chromosome dosage compensation in Silene latifolia, a plant with young sex chromosomes. PLoS Biol 10:e1001308. doi: 10.1371/journal.pbio.1001308
Bergero, R., and Charlesworth, D. (2011). Preservation of the Y transcriptome in a 10-million-year-old plant sex chromosome system. Curr Biol 21, 1470–1474. doi: 10.1016/j.cub.2011.07.032
Keywords: Crustacea, sex differentiation, shrimp, genome, Z chromosome
Citation: Wang Q, Lv J, Ren X, Wang J, Jia S, He Y and Li J (2021) Evidences of Z- and W-Linked Regions on the Genome of Fenneropenaeus chinensis. Front. Mar. Sci. 8:743727. doi: 10.3389/fmars.2021.743727
Received: 19 July 2021; Accepted: 12 August 2021;
Published: 31 August 2021.
Edited by:
Haihui Ye, Jimei University, ChinaReviewed by:
Deborah Charlesworth, University of Edinburgh, United KingdomHongyu Ma, Shantou University, China
Copyright © 2021 Wang, Lv, Ren, Wang, Jia, He and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuying He, aGV5eUB5c2ZyaS5hYy5jbg==; Jian Li, bGlqaWFuQHlzZnJpLmFjLmNu
†These authors have contributed equally to this work