 Vedanjali Gogineni
Vedanjali Gogineni Joonseok Oh1
Joonseok Oh1 Amanda L. Waters
Amanda L. Waters Michelle Kelly
Michelle Kelly Robert Stone
Robert Stone Mark T. Hamann
Mark T. Hamann- 1Department of BioMolecular Sciences, Division of Pharmacognosy, Research Institute of Pharmaceutical Sciences, School of Pharmacy, The University of Mississippi, University, MS, United States
- 2Department of Chemistry, University of Central Oklahoma, Edmond, OK, United States
- 3Coasts and Oceans, National Institute of Water and Atmospheric Research Ltd., Auckland, New Zealand
- 4Auke Bay Laboratories, Alaska Fisheries Science Center, NOAA Fisheries, Juneau, AK, United States
- 5Department of Drug Discovery and Biomedical Sciences, Medical University of South Carolina, Charleston, SC, United States
Marine sources have long been known for their potential to produce unique skeletons and various biological activities. Fractionation of the ethanol extracts of an undescribed species of Monanchora Carter, 1883 and a specimen closely comparable to Monanchora pulchra (Lambe, 1894/1895) (Class Demospongiae, Order Poecilosclerida, Family Crambeidae), yielded a known compound, monanchocidin A. Monanchocidin A, a secondary metabolite, showed very modest antibacterial, antifungal, and antiprotozoal activities with IC50 values ranging between 255.75 and 7288.92 μM. Monanchocidin A also exhibited potent selective activity for the melanoma panel in the NCI cancer cell screening panel.
Introduction
Marine invertebrates, particularly sponges, have been considered among the richest sources of pharmacologically active secondary metabolites with anticancer, antibacterial, antifungal, anti-inflammatory, antiviral, and antimalarial activities (Sipkema et al., 2005). A continuing hypothesis is that this richness is related to the metabolic contribution of symbiotic microorganisms; sponges are a reservoir of microbial biomass of up to 60% (Mehbub et al., 2014). Research involving marine sources has generated significant discoveries of pharmacologically relevant tools for identifying distinctive cellular targets; some of which have turned out to be essential tools in biochemical research and played fundamental roles in current advancement of life-sciences (Glaser and Mayer, 2009). Natural products have the advantage of possessing unique and stereodense pharmacophores along with a high degree of stereochemistry. Such compounds can provide hits against more challenging screening targets such as protein-protein interactions. Additionally, natural products being natural metabolites have the advantage of “metabolite-likeness” making them not only biologically active but also being substrates for many transporter systems delivering the compounds to their intracellular sites of action (Harvey et al., 2015). Following the food and drug administration (FDA) approval of two marine natural products (MNPs), cytarabine (Ara-C), and vidarabine (Ara-A) in 1969 and 1976, respectively, it was not until 2004 that Prialt® (ω-conotoxin, ziconotide), another synthetic equivalent of a marine conopeptide was approved (Glaser and Mayer, 2009). Yondelis® (trabectedin) is the latest FDA approved drug that was approved on October, 2015 which was originally isolated from a sea squirt, and now chemically synthesized for the treatment of ovarian and other cancers1. Hence, there is the need for the isolation and structural elucidation of chemical constituents from various marine sources that could possess unique skeleton and biological activities.
Pentacyclic guanidine alkaloids are frequently found in sponge species in genera Ptilocaulis (Kashman et al., 1989), Hemimycale (Kashman et al., 1989), Crambe (Jares-Erijman et al., 1991), Neofolitispa (Venkateswarlu et al., 1999), and Monanchora (Carter, 1883; Braekman et al., 2000; Guzii et al., 2010; Makarieva et al., 2011) as well as in starfishes of the Fromia and Celerina genera (Palagiano et al., 1995). These molecules are highly complex structures with diverse bioactivities that has stimulated interest in the scientific research groups resulting in several synthetic approaches (Nagasawa et al., 2002; Ma et al., 2015). An isolated pentacyclic guanidine alkaloid, Monanchocidin A (Figure 1) has aroused several questions regarding the role of spermidine “anchor” in its biological activities (Dyshlovoy et al., 2015) and recent synthetic efforts have been made to understand its structural requirements (Shi and Pierce, 2015).
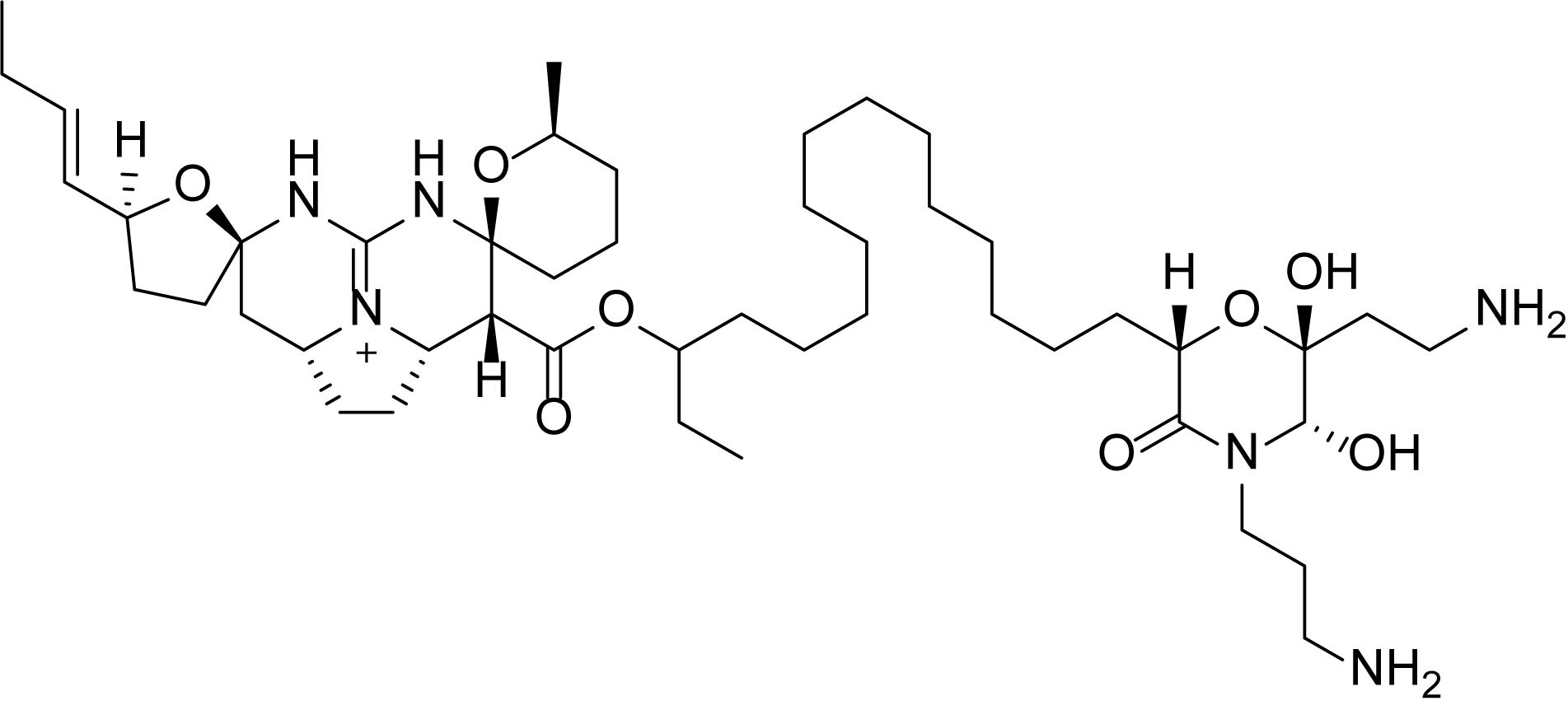
Figure 1. Structure of monanchocidin A.
During our exploration for bioactive constituents from marine organisms (Zou and Hamann, 2013), we began to explore the deep ocean and remote regions of the North Pacific Ocean with remotely operated vehicles with a focus on sponge extracts (about 50 g of each sponge) collected from various locations along the Aleutian Island Archipelago for screening against antibacterial, antifungal, and antiprotozoal activities along with anticancer activities. The ethanolic extracts of an undescribed species of Monanchora Carter, 1883 (Figure 2A) and a specimen closely comparable to Monanchora pulchra (Lambe, 1894/1895) (Class Demospongiae, Order Poecilosclerida, Family Crambeidae) (Figure 2B), showed unique selectivity for melanoma cells in the NCI 60 cell panel. Simultaneous bioassay-guided fractionation and isolation resulted in the identification of monanchocidin A (Guzii et al., 2010) as the active component of both sponges.

Figure 2. Comparison of Monanchora pulchra and Monanchora sp. (Order Poecilosclerida, Family Crambeidae): (A) DDDBS voucher 2010-AK-49 (NIWA 92956; NHMUK2011.2.11.3), Monanchora sp., ex situ image showing pale orange-yellow coloration and deep cracks in the ectosomal membrane upon collection; (B) M. pulchra (Lambe, 1894/1895) sensu stricto, DDDBS voucher AB13-0142 (NIWA 92966): Southwest of Atka Island, Central Aleutian Islands, 150 m, collected by Robert Stone, NOAA Fisheries, Alaska on the submarine DELTA, image reprinted from Stone et al. (2011) with permission.
Materials and Methods
Sample Collection
Department of Drug Discovery and Biomedical Sciences (DDDBS) voucher, 2010-AK-49 (Figure 2A), was collected on 25 July 2010, with a research otter trawl, during biannual fish stock assessment surveys, conducted by the FV Sea Storm south of Kanaga Island in the central Aleutian Islands of Alaska (51.648° N, 177.450° W). The collection site was in rough, rocky bottom, moderate current, a depth of 133 m, and water temperature at the time of collection of 4.9°C. In life, the sponge formed a divided fan, with incised, slightly foliose margins (other specimens collected had compressed branches, broadly expanded, emerging, and anastomosing from a short stem). In life, the surface was smooth, appearing almost inflated, with abundant, raised, membranous oscules on both surfaces and margins. In life, the surface appears inflated but on deck and on drying, deep cracks appeared where the ectosomal membrane had split. The sponge was supported on a short, tough, “cored” stem, that extends into the lamellae as dense “veins,” not clearly visible in life. The texture of the fan in life was firm, flabby at the margins, elastic and easily torn, but the lower stem was tough and fibrous. The color in life was yellow with an orange tinge. The sponge is an undescribed species of Monanchora (Order Poecilosclerida, Family Crambeidae). A voucher of 2010-AK-49 has been deposited at the NIWA Invertebrate Collection (NIC), Wellington, New Zealand (NIWA 92956) and the Natural History Museum, London (NHMUK2011.2.11.3).
Another DDDBS voucher, 2011-AK-001, was also dredged from the Aleutian Islands, but on 28 July 2011 (52.070° N, 177.239° E). In life, the sponge appeared almost identical in morphology to 2010-AK-49 (Monanchora sp.) (Figure 2A), forming a thin, multi-lamellate fan (Figure 3A). Voucher 2011-AK-001 has slightly smaller styles and smaller, thinner anchorate chelae than in 2010-AK-49 (Monanchora sp.), and has occasional unguiferous, arcuate chelae, with numerous sharp alae, in the microsclere complement. The voucher, 2011-AK-001, is closely comparable to M. pulchra, originally described by Lambe (1894/1895), and subsequently by Abbas et al. (2011), and Stone et al. (2011), but M. pulchra sensu stricto forms a very thin, yellowish fan, whereas the voucher is much more robust, and the coloration is darker. The voucher, 2011-AK-001, is hereafter referred to as M. cf. pulchra. A voucher of 2011-AK-001 has been deposited at the NIWA Invertebrate Collection (NIC), Wellington, New Zealand (NIWA 92955).

Figure 3. Monanchora cf. pulchra (Lambe, 1894/1895) (Order Poecilosclerida, Family Crambeidae): (A) DDDBS voucher 2011-AK-001 (NIWA 92955), ex situ image showing preserved multi-lamellate form; (B) DDDBS voucher AB13-0177 (NIWA 92958), in situ image showing planar form with inflated surface and brick-red coloration; (C) DDDBS voucher AB13-0177 (NIWA 92958), ex situ image.
The undescribed species of Monanchora [exemplified by voucher 2010-AK-49 (Figure 2A)] occurs in sympatry with M. pulchra sensu stricto [exemplified by voucher AB13-0142 (Figure 2B)] and M. cf. pulchra [exemplified by 2011-AK-001 (Figure 3A) and others (Figures 3B,C)], and can be differentiated in the following ways: M. pulchra sensu stricto and Monanchora sp. form light orange yellow fans, but the former is always much thinner. Monanchora cf. pulchra also forms thin fans but is a brighter orange-yellow to brick red coloration. M. pulchra sensu stricto has megasclere styles up to 1100 μm (Lambe, 1894/1895), while Monanchora sp. is characterized by two size categories of megascleres: a choanosomal style up to 1200 μm long, and ectosomal subtylostyles that range from 200 to 550 μm long. Monanchora cf. pulchra has slightly shorter choanosomal styles (900 μm) with noticeably bulbous heads. M. pulchra sensu stricto has sigmas, and small, robust, unguiferous, arcuate chelae, with longish teeth-like alae (Lambe, 1894/1895). Monanchora sp. has two forms of microscleres: an anchorate isochelae, 30–55 μm and sigmas, about 20 μm long. Both forms of microsclere are also much larger than those in M. cf. pulchra in which the anchorate isochelae are only about 19 μm long and the sigmas only about 13 μm long. Monanchora sp. has large, robust anchorate/arcuate chelae with multiple very short alae, not found in M. cf. pulchra or M. pulchra sensu stricto.
Extraction and Isolation
Both frozen sponges [2010-AK-49 and 2011-AK-001] were homogenized and extracted with ethanol (190 proof) at room-temperature. The crude extracts (14.35 and 9.99 g both wet) were subjected to liquid partition yielding ethyl acetate and water fractions. The water fraction is further subjected to liquid partition with butanol to yield 12 fractions that were eluted with a stepwise gradient solvent of water-methanol (MeOH). The fractions that yielded monanchocidin A are from 1:1 water-MeOH and 100% MeOH followed by analytical HPLC [Phenomenex C18 analytical column; flow rate 0.5 mL/min using 50:50 TFA 0.1–100% ethyl acetate].
General Experimental Procedures
1H and 13C NMR spectra were obtained from Bruker Avance NMR spectrometer functioning at 400 MHz and 100 MHz, respectively. CD3OD was used as solvent and TMS was used as the internal standard. HPLC and LC-MS analyses were used to investigate monanchocidin A. HPLC was carried out using a Waters System. The UV spectrum was obtained using a Hewlett Packard 8452A diode array spectrometer. ESIMS and HRESIMS were obtained using Bruker MicroTOF mass spectrometer. All the organic solvents and water used were of HPLC grade or better and procured from Sigma-Aldrich.
NMR Processing Procedures
Comprehensive NMR data set was collected for monanchocidin A. Individual experiments were compared to the known monanchocidin A standard data set (Supplementary Table S1). Both 1D and 2D data sets were overlaid utilizing the NMR processing software, MestReNova v.7.1.0-9185. The experimental data matched that of the standard data of monanchocidin A.
Antibacterial and Antifungal Screening
The antimicrobial or opportunistic infections (OI) tests were conducted on samples to test their ability to inhibit a panel of five bacteria and five fungi which are pathogenic to humans. The bacterial panel included Staphylococcus aureus ATCC 29213, Methicillin-resistant S. aureus (MRSA) ATCC 33591, Escherichia coli ATCC 35218, Pseudomonas aeruginosa ATCC 27853, and Mycobacterium intracellulare ATCC 23068, while the fungal panel included Candida albicans ATCC 90028, Candida glabrata ATCC 90030, Candida krusei ATCC 6258, Aspergillus fumigatus ATCC 204305, and Cryptococcus neoformans ATCC 90113. All the bacterial and fungal organisms that were used for the evaluation were attained from the American-Type Culture Collections (Manassas, VA, United States). Susceptibility testing was achieved via a modified version of CLSI (previously NCCLS) methods (Samoylenko et al., 2009 and referenced herein). A modified method was used to test M. intracellulare (Franzblau et al., 1998). Samples were consecutively diluted in 20% DMSO/saline and placed in duplicate into a 96-well flat bottom microplates. In the incubation broth, the OD630 of the microbe suspensions was corrected in order to prepare the microbial inocula. Ciprofloxacin (ICN Biomedicals, Dublin, OH, United States), the antibacterial drug control and amphotericin B (ICN Biomedicals, Dublin, OH, United States), the antifungal drug control were used as the positive controls in each corresponding assay. All organisms were read at either 630 nm by means of the BioTek Powerwave XS plate reader (BioTek Instruments, VT) or 544ex/590em (M. intracellulare, A. fumigatus) utilizing the Polarstar Galaxy plate reader (BMG LabTechnologies, Germany) prior to and after the incubation. The percent growth was plotted against the test concentration to calculate the IC50.
In the Primary Screen, crude extracts were initially tested at a concentration of 50 μg/mL in duplicate and the percent inhibitions were calculated with respect to positive and negative controls. Extracts that exhibit greater than or equal to 50% inhibition proceed to Secondary Assay.
During the Secondary Assay, samples (crude extracts/column fractions) that were dissolved to 20 mg/mL were tested at 50, 10, and 2 μg/mL, while samples that were dissolved to 2 mg/mL (pure compounds/column fractions) were tested at 20, 4, 0.8 μg/mL and the values of IC50 against all 10 microbial strains were reported. Pure compounds with an IC50 value of less than or equal to 7 μg/mL in the secondary OI assay proceed to Tertiary Assay.
In the Tertiary Assay, the pure compounds were tested against all 10 microbes at concentrations of 20, 10, 5.0, …. 0.02 μg/mL and the IC50’s were calculated. In addition to the IC50, the minimum inhibitory concentration (MIC) and either the minimum bactericidal or minimum fungicidal concentration (MBC or MFC, respectively), were also reported. MIC is the lowest test concentration in μg/mL that can inhibit the organism 100%. MBC or MFC is the lowest test concentration in μg/mL that kills the organism. A pure compound may have an MIC where the cells might still be alive and not growing. MBC and MFC is a way to monitor the cidality or killing ability of the test sample.
Anti-malarial Screening
The anti-malarial [or anti-protozoal (AP)] screening was performed on samples to test their ability to inhibit the chloroquine-sensitive (D6, Sierra Leone) and/or chloroquine-resistant (W2, Indo China) strains of Plasmodium falciparum protozoan by determining the plasmodial LDH activity as mentioned earlier (Makler and Hinrichs, 1993). Compounds that needed testing were dissolved at 2 mg/mL in DMSO. A 200 μL P. falciparum culture suspension (2% hematocrit and 2% parasitemia in a RPMI 1640 medium supplemented with 60 μg/mL amikacin and 10% human serum) was added to the plates of the 96-well plate comprising 10 μL of serially diluted samples. The plate was then flushed with a gas mixture of 90% N2, 5% CO2, and 5% O2 and incubated for 72 h at 37°C in a modular incubation chamber. Plasmodial LDH activity was established using the MalstatTM reagent (Flow Inc., Portland, OR, United States). In summary, 20 μL of the incubation mixture was combined with 100 μL of Malstat reagent and incubated for 30 min. To this, 20 μL of 1:1 NBT/PES mixture (Sigma, St. Louis, MO) was added and the plate was further incubated in dark for 1 h. The reaction was ceased by adding 100 μL of 5% acetic acid solution. The plate was read using the EL-340 Biokinetics Reader (BioTek Instruments, Winooski, VT, United States) at 650 nm. IC50 values were determined from the dose-response curves that were generated by plotting the percent growth against the drug concentration. Chloroquine was used as the positive control in each assay. 0.25% DMSO was used as the vehicle control.
Crude extracts were initially tested in the Primary Screen at a concentration of 15867 ng/mL in duplicate against the D6 P. falciparum strain and the percent inhibitions were calculated relative to positive and negative controls. Extracts with ≥50% inhibition proceeded to Secondary Assay.
In the Secondary Assay, samples dissolved to 20 mg/mL (crude extracts/column fractions) were tested at 47600, 15867, and 5289 mg/mL and the IC50 values against both the D6 and W2 strains were reported. Samples dissolved to 2 mg/mL (pure compounds/column fractions) were tested at 4760, 1587, and 529 ng/mL and the IC50 values against both D6 and W2 strains were reported.
In vitro Cytotoxicity
Samples were also tested in the mammalian kidney fibroblasts (VERO cells) for general cytotoxicity. The assay was performed in a 96-well tissue culture treated plates as outlined earlier (Mustafa et al., 2004). Cells were seeded into the wells of a 96-well plate (25000 cells/well) and incubated for 24 h. Samples were then added, and the plates were incubated again for 48 h. The number of viable cells was determined using neutral red assay. IC50 values were then calculated from the dose-response curves as described above. Doxorubicin was used as the positive control, while DMSO was used as the vehicle control. The selectivity indices (SI) ratio of the IC50 of VERO to the IC50 of D6 or W2 strains were calculated.
Anti-leishmanial Screening (LEM)
The LEM screening was conducted on samples to test their ability to inhibit the culture of Leishmania donovani promastigotes (Ma et al., 2004), a fly-borne protozoan responsible for visceral leishmaniasis. The promastigotes were grown in an RPMI 1640 medium enhanced with 10% fetal calf serum (Gibco Chem. Co.) at 26°C. A 3-day old culture was diluted to 5 × 105 promastigotes/mL. Sample dilutions were prepared directly in cell suspension in the 96-well plates. Plates were then incubated for 48 h at 26°C and the growth of leishmania promastigotes was determined using Alamar blue assay as described earlier. Standard fluorescence was measured at an excitation wavelength of 544 nm and an emission wavelength of 590 nm using Fluostar Galaxy plate reader (BMG Lab Technologies). Amphotericin B and pentamidine were used as the positive controls. IC50 values were computed as above from the dose-response curves.
Crude extracts were initially tested in a Primary Screen at a concentration of 80 μg/mL in duplicate and the percent inhibitions were calculated with respect to positive and negative controls. Extracts with ≥50% inhibition exceed to Secondary Assay. In the Secondary LEM Assay, the samples (2 and 20 mg/mL) were tested at 40, 8.0, and 1.6 μg/mL and the IC50 values along with the IC90 values (test concentration exhibiting 90% inhibition of the protozoan relative to controls) were reported. Samples with an IC50 value of <1.6 μg/mL in the Secondary Assay proceeded to Tertiary Assay where the samples were tested at 40, 8, 1.6, 0.32, 0.064, 0.0128 μg/mL and the values of IC50 and IC90 were reported.
National Cancer Institute (NCI)-60 Human Tumor Cell Line Screen
Testing of monanchocidin A (NSC763554) in the NCI-60 cell panel was performed by the NIH NCI developmental therapeutics program (DTP). The specific procedures and techniques used were published in a series of articles (Shoemaker, 2006; NCI-60 Screening Methodology, 2015). Monanchocidin A was first tested at a single dose of 10 μM in the NCI-60 cell panel and then moved onto a five-dose testing for the determination of the IC50 value for each cell line. Cancer cell lines were grown in an RPMI 1640 medium containing 2 mM L-glutamine and 5% fetal bovine serum. Cells were inoculated at 5000–40,000 cells per well based on the doubling time of the cell-line in a 96 well microtiter plate in 100 μL. Plates were then incubated at 37°C, 100% relative humidity, 95% air, and 5% CO2 for 24 h. Before the addition of the compound, a measurement of the cell population was taken in situ with trichloroacetic acid (TCA) (Tz). Monanchocidin A was dissolved at 400-fold the maximum test concentration in DMSO for stock storage solution. Monanchocidin A was then diluted to two times the final maximum concentration with 50 μg/mL gentamicin in the complete medium. Additionally, four 10-fold serial dilutions were prepared. To the growing cells in the microtiter plates, 100 μL of each dilution was added. Under the aforementioned conditions, plates were then incubated for an added 48 h. Assays were then terminated and the cells were fixed in situ by adding 50 μL of 80% TCA for suspension cells or 50 μL of cold 50% (w/v) TCA and incubated at 4°C for 60 min for adherent cells. The supernatant was discarded, and the plates were washed with water five times and air dried. To each well, 100 μL of sulforhodamine B (SRB) solution 0.4% (w/v) in 1% acetic acid was added and the plates were incubated for 10 min at room temperature. Using 1% acetic acid, the plates were washed five times and air dried to remove the unbound dye. The bound dye is solubilized using 10 mM trizma® base and the absorbance reading was recorded at 515 nm. Taxol was used as the control. The growth inhibition percentages were calculated as below:
where Ti = test growth at five concentration levels in the presence of monanchocidin A, Tz = time zero, and C = control growth.
In addition, 50% growth inhibition (compound concentration resulting in 50% reduction of the net-protein increase in control cells measured by SRB staining, GI50), total growth-inhibition concentration (TGI), and 50% lethal concentration (compound concentration ensuing 50% reduction of the protein at the end of treatment compared to the beginning, LC50) were calculated using the growth inhibition percentage equations (Alley et al., 1988).
Results
In order to provide an insight into the development of novel biological agents from natural products with high activity, a panel of antifungal, antibacterial, and antiprotozoal assays were performed. The ethanol extracts of Monanchora cf. pulchra and an undescribed Monanchora sp., both collected off the central Aleutian Islands of Alaska, were found to have the highest inhibition against these assays.
The antibacterial activities were evaluated against S. aureus ATCC 29213, E. coli ATCC 35218, methicillin-resistant S. aureus ATCC 33591 (MRS), M. intracellulare ATCC 23068, and P. aeruginosa ATCC 27853. Ciprofloxacin was included as a positive control for antibacterial activity. The antifungal activities were evaluated against a panel of pathogenic fungi (C. albicans ATCC 90028, Candida glabrata ATCC 90030, C. krusei ATCC 6258, C. neoformans ATCC 90113, and A. fumigatus ATCC 90906) associated with OI. Amphotericin B was included as a standard antifungal drug for comparison. In vitro antimalarial activity was seen against chloroquine sensitive (D6, Sierra Leone) and chloroquine resistant (W2, Indo China) strains of P. falciparum by measuring the activities of plasmodial LDH and anti-leishmanial against L. donovani promastigotes using Alamar BlueTM assay.
The ethanol extracts of M. cf. pulchra and the undescribed species of Monanchora showed antibacterial activity against S. aureus, MRS, and E. coli with IC50 values of 6.03, 5.49, and 17.75 μg/mL, respectively. The extracts also exhibited in vitro antifungal activity against C. albicans, C. glabrata, C. krusei, A. fumigatus, and C. neoformans with IC50 values of 23.07, 4.51, 3.42, 21.06, and 2.77 μg/mL, respectively, and <90% inhibition on antiprotozoal assays (Table 1).

Table 1. IC50 Values of the Crude Ethanol Extracts of Monanchocidin A in μg/mL.
Following a bioassay-guided fractionation strategy, the ethanol extracts were subjected to a liquid- liquid partition in ethyl acetate (EtOAc) and water. The water partition was further fractionated with butanol to obtain three main fractions: EtOAc, butanol, and water fractions, which were tested for their antibacterial and antifungal activities. The butanol portion exhibited antibacterial and antifungal activities with IC50 values of 1.52, 1.40, 7.08, 2.51, 1.63, 1.96, 9.06, and <0.8 μg/mL against S. aureus, MRS, E. coli, C. albicans, C. glabrata, C. krusei, A. fumigatus, and C. neoformans, respectively. The EtOAc and water portions did not show in vitro antibacterial or antifungal activities (Table 2). The butanol fractions were subjected to Sephadex LH-20 column chromatography and subsequent purification using HPLC C18 yielded the pentacyclic guanidine, monanchocidin A, as the major and active compound. Monanchocidin A showed significant IC50 values against S. aureus, MRS, E. coli, and P. aeruginosa at 348.75, 372.00, 778.88, and 941.63 μM, respectively, in the tertiary assay. No activity was seen against M. intracellulare in the antibacterial panel of assays (Table 3). Monanchocidin A showed antifungal activity against C. albicans, C. glabrata, C. krusei, C. neoformans, and A. fumigatus with IC50 values of 581.25, 837.00, 651.00, 127.88, and 395.25 μM, respectively (Table 4). Antiprotozoal activity of monanchocidin A against L. donovani was moderate with IC50 values of 7288.92 and IC90 of 8160.80 μM and good IC50 values of 360.38 and 255.75 μM were seen against P. falciparum D6 and W2, respectively (Table 5).
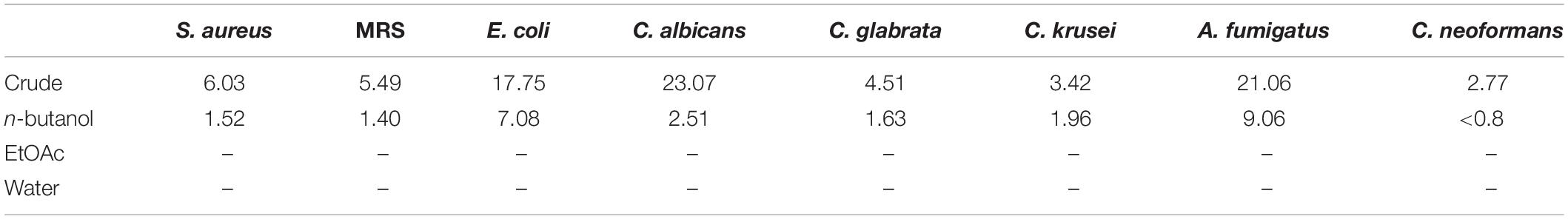
Table 2. IC50 values for the fractions resulting from the ethanol extracts in μg/mL.
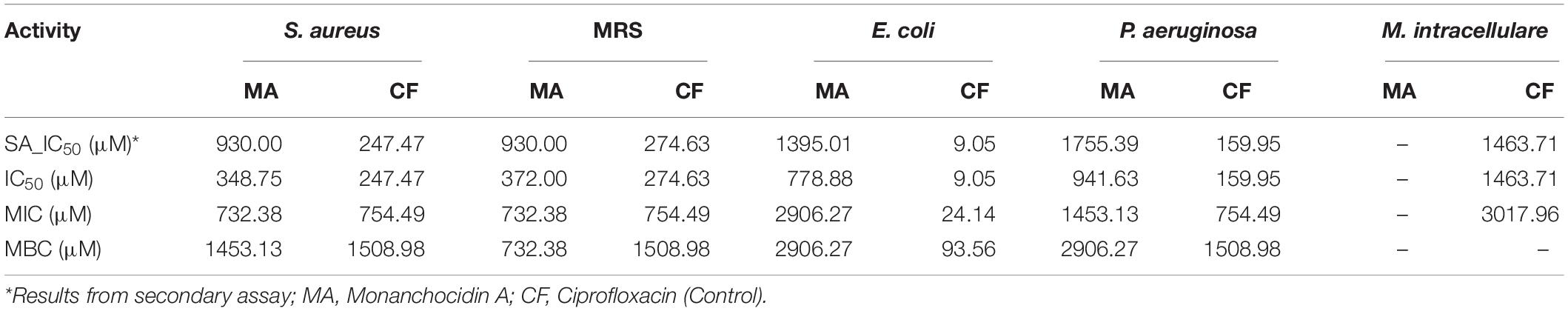
Table 3. Antibacterial activity of monanchocidin A in μM.
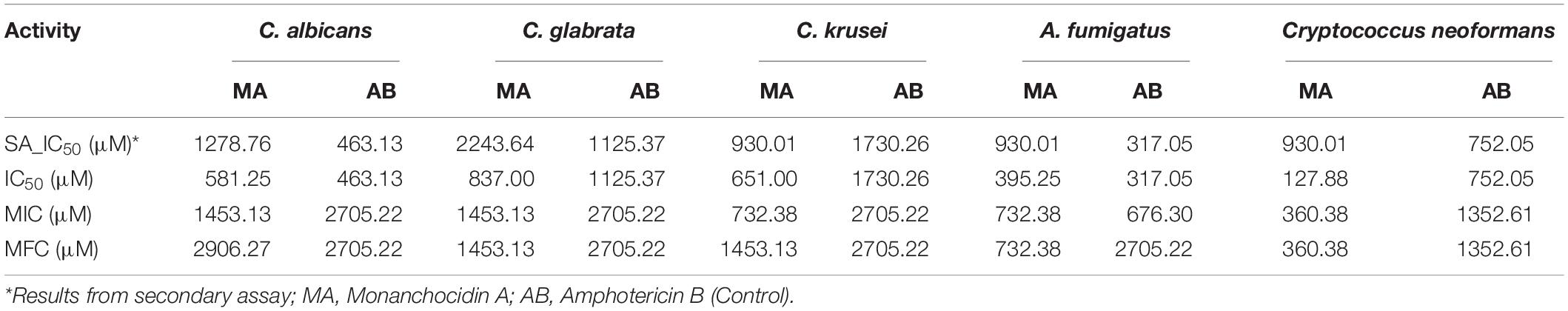
Table 4. Antifungal activity of monanchocidin A in μM.
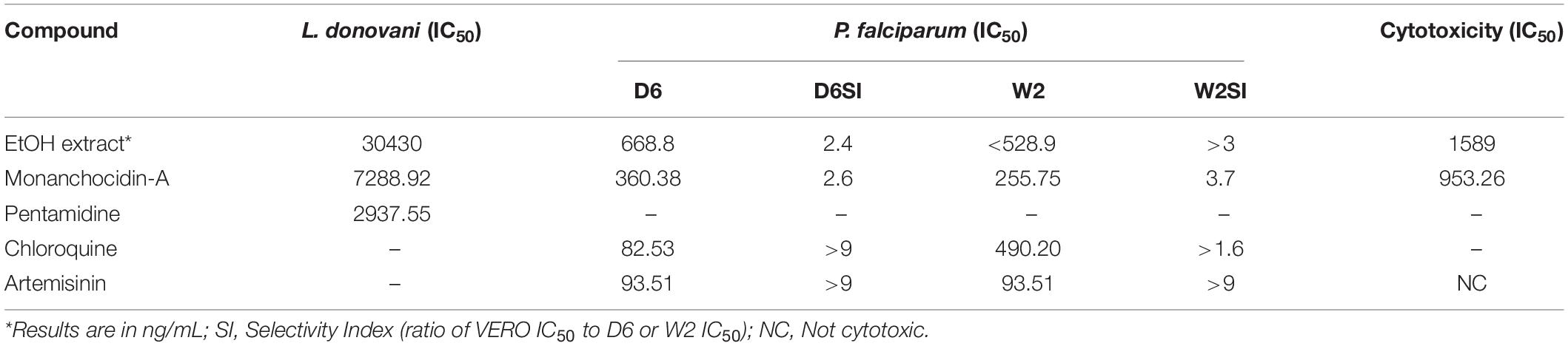
Table 5. Antiprotozoal activity of monanchocidin A in μM.
The 1H (Supplementary Figure S1) and 13C (Supplementary Figure S2) NMR spectroscopic and high-resolution mass spectrometry data (Supplementary Figures S3, S4) of (1) were compared with the isolates in both cases with those reported in literature and found to be consistent with monanchocidin A (1) (Guzii et al., 2010). However, monanchocidin A showed cytotoxicity against mammalian kidney fibroblasts (vero cells) with an IC50 value of 953.26 μM. The broad spectrum of biological activities exhibited by monanchocidin A, as well as the recent report about the cytotoxic efficacy of compound 1 against human prostate and bladder cancer cells (Nagasawa et al., 2002), revealed that compound 1 should have a strong potential in drug discovery. Hence, monanchocidin A was evaluated in NCI 60 tumor cell line assay including characteristic cell lines of melanoma, leukemia, non-small cell lung, colon, CNS, ovarian, renal, prostate, and breast cancer. Figure 4 shows the dose response curves in different cancer cell lines. In the NCI-60 cell panel, monanchocidin A was most sensitive for leukemia cell lines K-562 (GI50 = 0.081 μM), MOLT-4 (GI50 = 0.045 μM), RPMI-8226 (GI50 = 0.110 μM), and SR (GI50 = 0.038 μM); non-small cell lung cancer lines A549/ATCC (GI50 = 0.048 μM), EKVX (GI50 = 0.062 μM), HOP-62 (GI50 = 0.135 μM), NCI-H226 (GI50 = 0.138 μM), NCI-H460 (GI50 = 0.068 μM), and NCI-H522 (GI50 = 0.083 μM); colon cancer cell lines COLO 205 (GI50 = 0.102 μM), HCT-116 (GI50 = 0.042 μM), and HT29 (GI50 = 0.051 μM); CNS cancer cell lines SF-268 (GI50 = 0.081 μM), and U251 (GI50 = 0.100 μM); melanoma cell lines LOX IMVI (GI50 = 0.022 μM), MALME-3M (GI50 = 0.095 μM), M14 (GI50 = 0.018 μM), MDA-MB-435 (GI50 = 0.023 μM), SK-MEL-2 (GI50 = 0.138 μM), SK-MEL-28 (GI50 = 0.063 μM), SK-MEL-5 (GI50 = 0.034 μM), UACC-257 (GI50 = 0.035 μM), and UACC-62 (GI50 = 0.024 μM); renal cancer cell line SN12C (GI50 = 0.107 μM); prostate cancer cell line PC-3 (GI50 = 0.141 μM); and breast cancer cell lines MDA-MB-231/ATCC (GI50 = 0.068 μM), and MDA-MB-468 (GI50 = 0.095 μM). The results showed an overall GI50 of −6.78 μM, with greater sensitivity toward melanoma cancer cell groups (Figure 5). Within the melanoma cancer cell panel, NSC763554 caused 50% or more growth inhibition of nine melanoma cell lines at concentrations between 1 and 10 μM. Figure 6 illustrates how monanchocidin A compares to the FDA-approved cancer drug taxol (NSC 125973). All the melanoma cell lines were highly sensitive to monanchocidin A exhibiting greater activity than taxol in vitro (Figure 5).
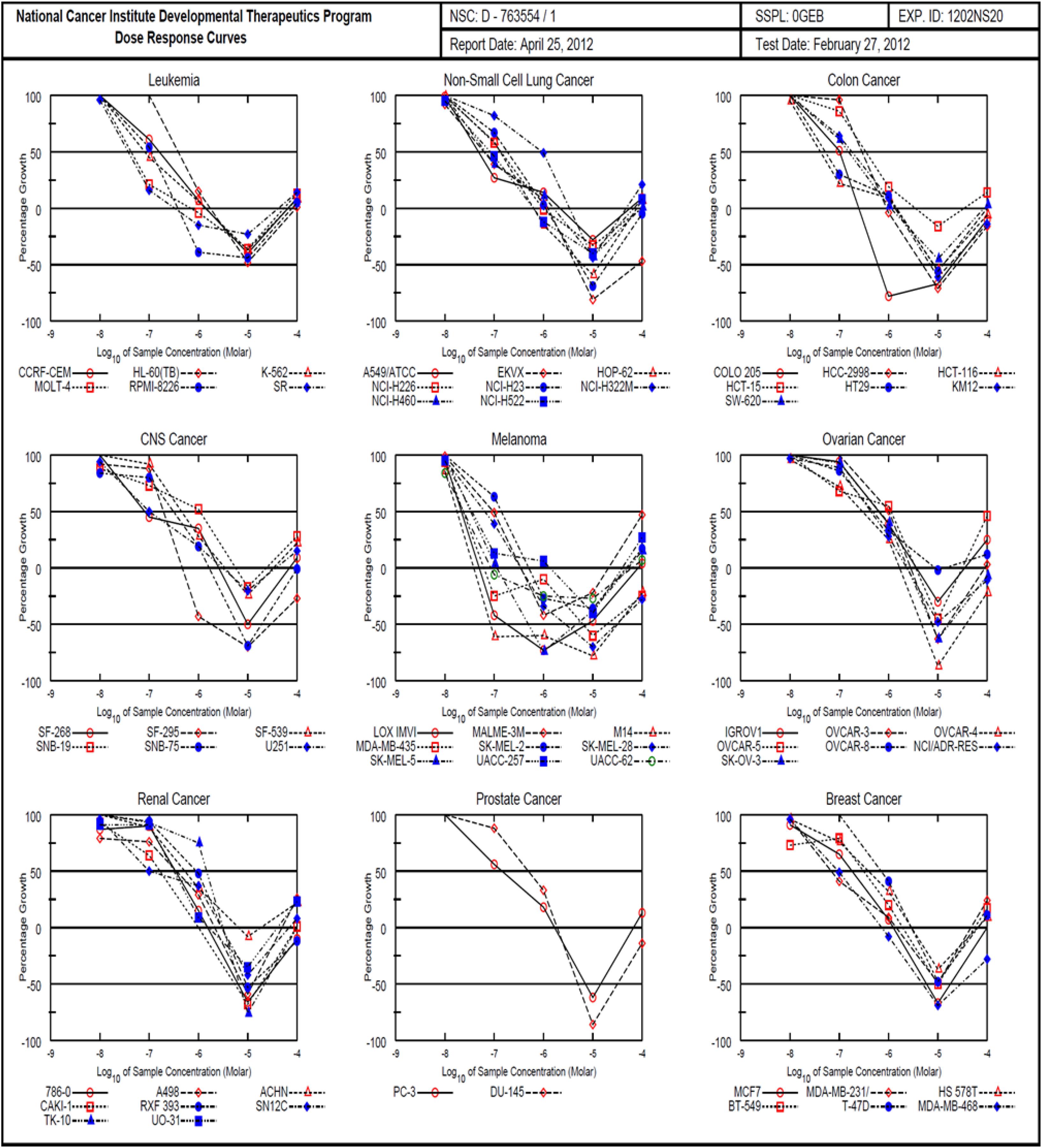
Figure 4. Comparison of NCI-60 cell panel for monanchocidin A. Differential toxicity of monanchocidin A toward NCI 60 cancer cell lines: Growth inhibition of NCI 60 cancer cell lines after exposure to compound 1. The cell lines were plated 24 h prior to adding the compound and then incubated for additional 48 h. The cell numbers were then estimated using Sulforhodamine B as staining.
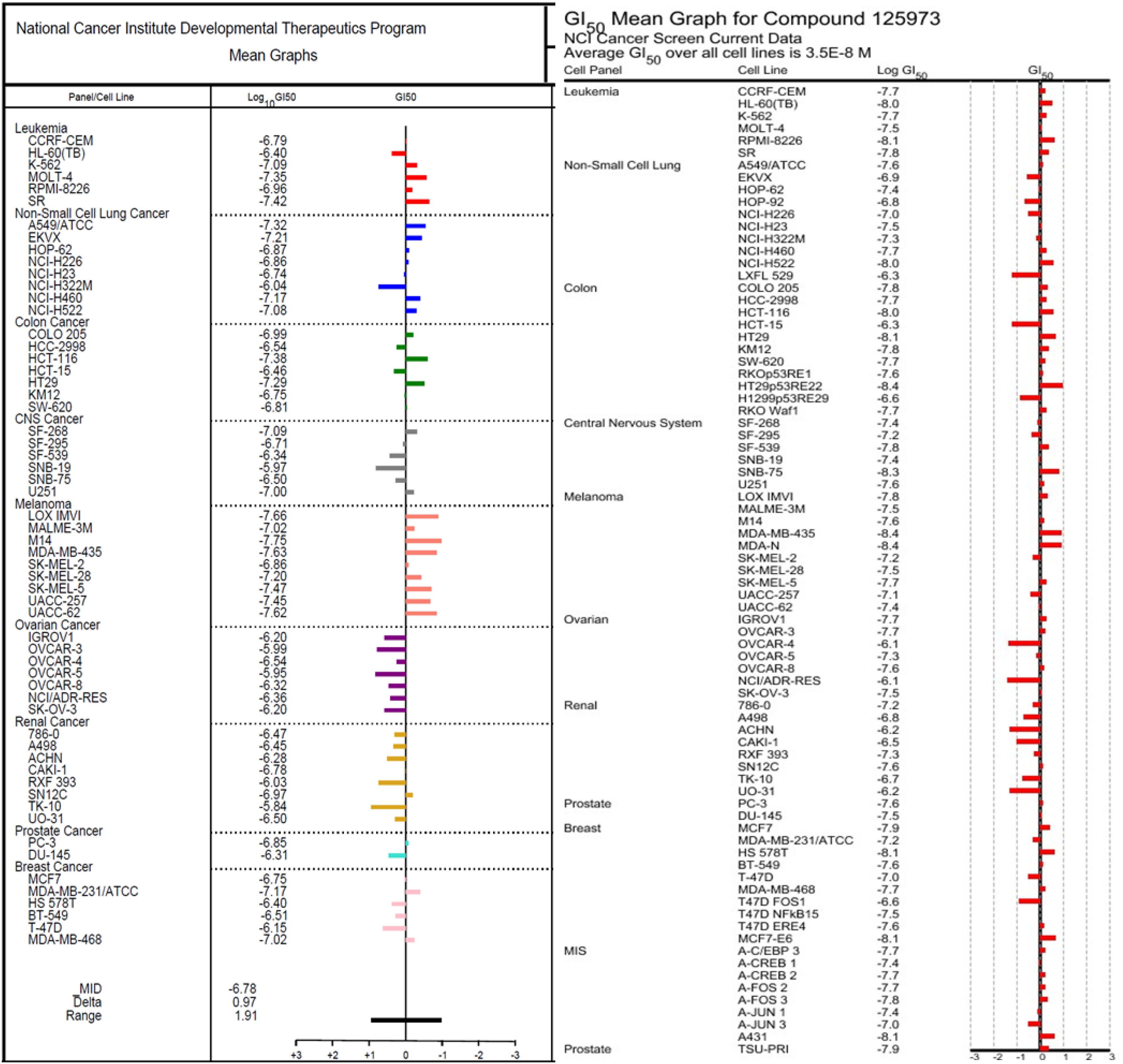
Figure 5. Comparison of the GI50’s between NSC763554 (Monanchocidin A) and Taxol. GI50 is the calculated micro-molar concentration that results in 50% reduction of the measured protein at the end of drug-treatment compared to the beginning. The median log GI50 for NSC763554 over all cell lines was –6.78.
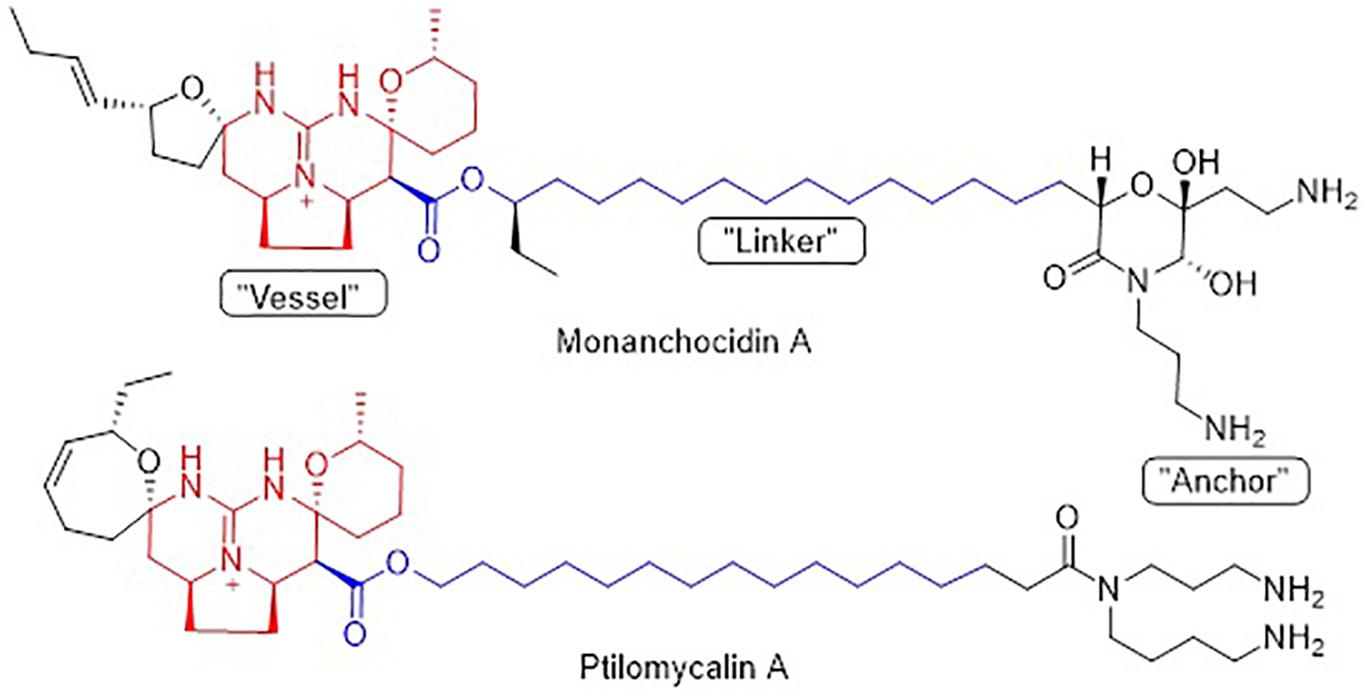
Figure 6. Structural similarity between monanchocidin A and ptilomycalin A.
Discussion
Monanchocidin A was evaluated for antibacterial, antifungal, and antiprotozoal activities, for which there are no previous reports. Monanchocidin A showed weak antibacterial activity against S. aureus and MRS at IC50 values of 349 and 372 μM, respectively, and antifungal activity against A. fumigatus and C. neoformans at IC50 values of 395 and 128 μM, respectively. Monanchocidin A also presented antimalarial activity against P. falciparum D6 and W2 cells at IC50 values of 360 and 256 μM, respectively. Monanchocidin A exhibited good biological spectrum against all the assays showing its potential in drug discovery for the treatment of diseases caused by bacteria, fungi, and protozoa.
Monanchocidin A was compared against all publicly available NCI-60 data using the CellMiner Database Version 1.4 (Reinhold et al., 2012). Utilizing the Compare (Zhou et al., 2000) algorithm and Pearson correlation coefficients, it was found that monanchocidin A shared a 0.638 Pearson correlation with ptilomycalin A (Figure 6). A Pearson correlation value above 0.6 is considered somewhat statistically significant. Upon further examination of the ptilomycalin A, the structure was found to be a similar polycyclic guanidine structure (Kashman et al., 1989). Pentacyclic guanidine alkaloids like monanchocidin A and ptilomycalin A includes two types of “anchor” fragments namely morpholinone and spermidine types (Tabakmakher et al., 2015). The NCI-60 data for ptilomycalin A was first reported in 2000 as being particularly selective for non-small cell lung cancer (HOP-62) and melanoma (M14) (Coffey et al., 2000). Crambescidin 816, a hydroxylated form of ptilomycalin A, is reported to be a powerful in vitro Ca2+ channel blocker (Coffey et al., 2000). Ptilomycalin A like compounds are known to activate the ERK1/2 and JNK1/2, ensuing AP-1 activation and resulting in p53-independent programmed cell-death and the arrest of S-phase cell cycle (Dyshlovoy et al., 2016). While there are several hypotheses for the mechanism of action (MOA) of this class of molecules, the function at a molecular level remains largely unresolved (Shi et al., 2017). This data shows the potential of monanchocidin A in anticancer activity and hence, further studies should be done to modulate the activity and to further explore the mechanism of action in these kinds of cancer cells.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Material. Efforts to resupply the drug lead for further investigation are currently underway.
Author Contributions
VG, JO, and MH designed the experiments and wrote the manuscript. AW performed the NCI60 human tumor cell-line screening on Monanchocidin A. RS helped with sponge collection while MK identified the sponges and provided taxonomic information and comparisons.
Funding
Funding for this research was provided by the Abney Foundation, the Charles and Carol Cooper Endowment, and the South Carolina SmartState Programs.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Alaska Fisheries Science Center (National Oceanic and Atmospheric Administration) for supporting the sample collections and Dr. James Sims, for Invertebrate Collections, ETH, Zürich, who helped in the collection of the sponges. We also thank the NCI in vitro evaluation using the NCI 60 cell panel and the NCNPR for antifungal and antibacterial assays. The findings and conclusions in this manuscript are those of the authors and do not necessarily represent the views of the National Marine Fisheries Service.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00058/full#supplementary-material
Footnotes
References
Abbas, S., Kelly, M., Bowling, J., Sims, J., Waters, A., and Hamann, M. (2011). Advancement into the arctic region for bioactive sponge secondary metabolites. Mar. Drugs 9, 2423–2437. doi: 10.3390/md9112423
Alley, M. C., Scudiero, D. A., Monks, A., Hursey, M. L., Czerwinski, M. J., Fine, D. L., et al. (1988). Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 48, 589–601.
Braekman, J. C., Daloze, D., Tavares, R., Hajdu, E., and Van Soest, R. W. M. (2000). Novel polycyclic guanidine alkaloids from two marine sponges of the genus Monanchora. J. Nat. Prod. 63, 193–196. doi: 10.1021/np990403g
Carter, H. J. (1883). XLVII.–new genus of sponges. J. Nat. Hist. 11, 369–370. doi: 10.1080/00222938309459164
Coffey, D. S., McDonald, A. I., Overman, L. E., Rabinowitz, M. H., and Renhowe, P. A. (2000). A practical entry to the crambescidin family of guanidine alkaloids. Enantioselective total syntheses of ptilomycalin A, crambescidin 657 and its methyl ester (neofolitispates 2), and crambescidin 800. J. Am. Chem. Soc. 122, 4893–4903. doi: 10.1021/ja000234i
Dyshlovoy, S. A., Hauschild, J., Amann, K., Tabakmakher, K. M., Venz, S., Walther, R., et al. (2015). Marine alkaloid monanchocidin A overcomes drug resistance by induction of autophagy and lysosomal membrane permeabilization. Oncotarget 6, 17328–17341. doi: 10.18632/oncotarget.4175
Dyshlovoy, S. A., Tabakmakher, K. M., Hauschild, J., Shchekaleva, R. K., Otte, K., Guzii, A. G., et al. (2016). Guanidine Alkaloids from the marine sponge Monanchora pulchra Show cytotoxic properties and prevent EGF-Induced neoplastic transformation in Vitro. Mar. Drugs 14:133. doi: 10.3390/md14070133
Franzblau, S. G., Witzig, R. S., McLaughlin, J. C., Torres, P., Madico, G., Hernandez, A., et al. (1998). Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 36, 362–366.
Glaser, K. B., and Mayer, A. M. S. (2009). A renaissance in marine pharmacology: from preclinical curiosity to clinical reality. Biochem. Pharmacol. 78, 440–448. doi: 10.1016/j.bcp.2009.04.015
Guzii, A. G., Makarieva, T. N., Denisenko, V. A., Dmitrenok, P. S., Kuzmich, A. S., Dyshlovoy, S. A., et al. (2010). Monanchocidin: a new apoptosis-inducing polycyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 12, 4292–4295. doi: 10.1021/ol101716x
Harvey, A. L., Edrada-Ebel, R., and Quinn, R. J. (2015). The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 14, 111–129. doi: 10.1038/nrd4510
Jares-Erijman, E. A., Sakai, R., and Rinehart, K. L. (1991). Crambescidins: new antiviral and cytotoxic compounds from the Sponge Crambe crambe. J. Org. Chem. 56, 5712–5715. doi: 10.1021/jo00019a049
Kashman, Y., Hirsh, S., McConell, O. J., Ohtani, I., Kusumi, T., and Kakisawa, H. (1989). Ptilomycalin A: a novel polycyclic guanidine alkaloid of marine origin. J. Am. Chem. Soc. 111, 8925–8926. doi: 10.1021/ja00206a029
Lambe, L. M. (1894/1895). Sponges from the western coast of North America. Trans. R. Soc. Can. 12, 113–138
Ma, G., Khan, S. I., Jacob, M. R., Tekwani, B. L., Li, Z., Pasco, D. S., et al. (2004). Antimicrobial and antileishmanial activities of hypocrellins A and B. Antimicrob. Agents Chemother. 48, 4450–4452. doi: 10.1128/AAC.48.11.4450-4452.2004
Ma, Y., De, S., and Chen, C. (2015). Syntheses of cyclic guanidine-containing natural products. Tetrahedron 71, 1145–1173. doi: 10.1016/j.tet.2014.11.056
Makarieva, T. N., Tabakmaher, K. M., Guzii, A. G., Denisenko, V. A., Dmitrenok, P. S., Shubina, L. K., et al. (2011). Monanchocidins B–E: polycyclic guanidine alkaloids with potent antileukemic activities from the sponge Monanchora pulchra. J. Nat. Prod. 74, 1952–1958. doi: 10.1021/np200452m
Makler, M. T., and Hinrichs, D. J. (1993). Measurement of the lactate-dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 48, 205–210. doi: 10.4269/ajtmh.1993.48.205
Mehbub, M. F., Lei, J., Franco, C., and Zhang, W. (2014). Marine sponge derived natural products between 2001 and 2010: trends and opportunities for discovery of bioactives. Mar. Drugs 12, 4539–4577. doi: 10.3390/md12084539
Mustafa, J., Khan, S. I., Ma, G., Walker, L. A., and Khan, I. A. (2004). Synthesis and anticancer activities of fatty acid analogs of podophyllotoxin. Lipids 39, 167–172. doi: 10.1007/s11745-004-1215-5
Nagasawa, K., Georgieva, A., Koshino, H., Nakata, T., Kita, T., and Hashimoto, Y. (2002). Total synthesis of crambescidin 359. Org. Lett. 4, 177–180. doi: 10.1021/ol0168263
NCI-60 Screening Methodology, (2015). DTP Human Cancer Cell Line Screen. Avaliable at: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed August 26, 2015).
Palagiano, E., De Marino, S., Minale, L., Riccio, R., Zollo, F., Lorizzi, M., et al. (1995). Ptilomycalin A, crambescidin 800 and related new highly cytotoxic guanidine alkaloids from the starfishes Fromia monilis and Celerina heffernani. Tetrahedron 51, 3675–3682. doi: 10.1016/0040-4020(95)00082-J
Reinhold, W. C., Sunshine, M., Liu, H., Varma, S., Kohn, K. W., Morris, J., et al. (2012). CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 72, 3499–3511. doi: 10.1158/0008-5472
Samoylenko, V., Jacob, M. R., Khan, S. I., Zhao, J., Tekwani, B. L., Midiwo, J. O., et al. (2009). Antimicrobial, antiparasitic and cytotoxic spermine alkaloids from Albizia schimperiana. Nat. Prod. Commun. 4, 791–796. doi: 10.1177/1934578X0900400611
Shi, Y., Moazami, Y., and Pierce, J. G. (2017). Structure, synthesis and biological properties of the pentacyclic guanidinium alkaloids. Bioorg. Med. Chem. 25, 2817–2824. doi: 10.1016/j.bmc.2017.03.015
Shi, Y., and Pierce, J. G. (2015). Synthesis of the 5,6-dihydroxymorpholin-3-one fragment of monanchocidin A. Org. Lett. 17, 968–971. doi: 10.1021/acs.orglett.5b00069
Shoemaker, R. H. (2006). The NCI60 human tumor cell line anticancer drug screen. Nat. Rev. Cancer 6, 813–823. doi: 10.1038/nrc1951
Sipkema, D., Franssen, M. C. R., Osinga, R., Tramper, J., and Wijffels, R. H. (2005). Marine sponges as pharmacy. Mar. Biotechnol. 7, 142–162. doi: 10.1007/s10126-004-0405-5
Stone, R. P., Lehnert, H., and Reiswig, H. (2011). “A guide to the deep-water sponges of the Aleutian Island Archipelago,” in NOAA Professional Paper NMFS 12, (Silver Spring, MA: NOAA), 187.
Tabakmakher, K. M., Makarieva, T. N., Denisenko, V. A., Guzii, A. G., Dmitrenok, P. S., Kuzmich, A. S., et al. (2015). Normonanchocidins A, B and D, new pentacyclic guanidine alkaloids from the Far-Eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 10, 913–916.
Venkateswarlu, Y., Reddy, M. V. R., Ramesh, P., and Rao, J. V. (1999). Neofolitispates, pentacyclic guanidine alkaloids from the sponge Neofolitispa dianchora. Indian J. Chem. 38B, 254–256.
Zhou, B.-N., Hoch, J. M., Johnson, R. K., Mattern, M. R., Eng, W.-K., Ma, J., et al. (2000). Use of COMPARE analysis to discover new natural product drugs: isolation of camptothecin and 9-methoxycamptothecin from a new source. J. Nat. Prod. 63, 1273–1276. doi: 10.1021/np000058r
Keywords: marine natural products, sponges, guanidine alkaloids, monanchocidin A, anticancer
Citation: Gogineni V, Oh J, Waters AL, Kelly M, Stone R and Hamann MT (2020) Monanchocidin A From Subarctic Sponges of the Genus Monanchora and Their Promising Selectivity Against Melanoma in vitro. Front. Mar. Sci. 7:58. doi: 10.3389/fmars.2020.00058
Received: 11 September 2019; Accepted: 27 January 2020;
Published: 18 February 2020.
Edited by:
Chiara Lauritano, Stazione Zoologica Anton Dohrn, ItalyReviewed by:
Bin Wu, Zhejiang University, ChinaGiovanna Romano, Stazione Zoologica Anton Dohrn, Italy
Copyright © 2020 Gogineni, Oh, Waters, Kelly, Stone and Hamann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark T. Hamann, aGFtYW5ubUBtdXNjLmVkdQ==
†Vedanjali Gogineni, Analytical Development Department, Cambrex Pharmaceuticals, Charles City, IA, United States; Affiliate Instructor, Drug Discovery and Biomedical Sciences, College of Pharmacy, Medical University of South Carolina, Charleston, SC, United States