 Samuel N. Bogan
Samuel N. Bogan Kevin M. Johnson
Kevin M. Johnson Gretchen E. Hofmann
Gretchen E. Hofmann- 1Department of Ecology, Evolution and Marine Biology, University of California, Santa Barbara, Santa Barbara, CA, United States
- 2Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, United States
Epigenetic processes such as variation in DNA methylation may promote phenotypic plasticity and the rapid acclimatization of species to environmental change. The extent to which an organism can mount an epigenetic response to current and future climate extremes may influence its capacity to acclimatize or adapt to global change on ecological rather than evolutionary time scales. The thecosome pteropod Limacina helicina antarctica is an abundant macrozooplankton endemic to the Southern Ocean and is considered a bellwether of ocean acidification as it is highly sensitive to variation in carbonate chemistry. In this study, we quantified variation in DNA methylation and gene expression over time across different ocean acidification regimes. We exposed L. helicina antarctica to pCO2 levels mimicking present-day norms in the coastal Southern Ocean of 255 μatm pCO2, present-day extremes of 530 μatm pCO2, and projected extremes of 918 μatm pCO2 for up to 7 days before measuring global DNA methylation and sequencing transcriptomes in animals from each treatment across time. L. helicina antarctica significantly reduced DNA methylation by 29–56% after 1 day of exposure to 918 μatm pCO2 before DNA methylation returned to control levels after 6 days. In addition, L. helicina antarctica exposed to 918 μatm pCO2 exhibited drastically more differential expression compared to cultures replicating present-day pCO2 extremes. Differentially expressed transcripts were predominantly downregulated. Furthermore, downregulated genes were enriched with signatures of gene body methylation. These findings support the potential role of DNA methylation in regulating transcriptomic responses by L. helicina antarctica to future ocean acidification and in situ variation in pCO2 experienced seasonally or during vertical migration. More broadly, L. helicina antarctica was capable of mounting a substantial epigenetic response to ocean acidification despite little evidence of metabolic compensation or recovery of the cellular stress response in this species at future pCO2 levels.
Introduction
Marine ecosystems are already reaching extremes of environmental change on par with projected global climate change over the next century (Hoegh-Guldberg and Bruno, 2010; Harris et al., 2013; Chan et al., 2017; Oliver et al., 2018). Species inhabiting regions such as polar oceans are currently faced with a necessity to acclimatize or adapt to stressors that will only intensify as climate change progresses. Driven by the rapid advancement of extreme physical conditions in ecosystems today and in modeled projections, global change biologists have begun to direct attention to the mechanisms and consequences of species’ abilities to rapidly respond to anthropogenic stress via acquired, adaptive traits (Chown et al., 2007; Calosi et al., 2008; Chevin et al., 2010; Nicotra et al., 2010; Beldade et al., 2011; Donelson et al., 2018; Kelly, 2019) and how mechanisms of acclimatization vary within a system between current and projected environmental change (Hennige et al., 2010; Duarte et al., 2018). We examined such a rapidly-acting process – namely, changes in DNA methylation and gene expression in the Antarctic pteropod Limacina helicina antarctica during exposure to present and future ocean acidification (OA) levels for the Southern Ocean.
In recent studies of marine metazoans, epigenetic processes have been demonstrated to be associated with the phenotypic plasticity of fitness-related traits in species experiencing climate change-driven stressors (Zhang et al., 2013; Putnam et al., 2016; Clark et al., 2018; Strader et al., 2019; Wong et al., 2019), a relationship that can be explained in part by the link between epigenetic mechanisms, gene expression, translation, and the traits underpinned by these processes (True et al., 2004). For example, experimental manipulation of DNA methylation levels in Arabidopsis thaliana has been shown to substantially alter the phenotypic plasticity of key developmental and physiological traits in low- and high-nutrient environments (Bossdorf et al., 2010). In humans, quantitative trait loci at methylated bases are linked to differential gene expression underpinning multiple physiological traits across a diversity of tissue types (Taylor et al., 2019). Overall, understanding how epigenetic processes may promote adaptive responses to environmental stressors will require investigations in diverse, ecologically critical taxa and experimentation across gradients of stress eliciting adaptive and pathological responses.
Among a suite of epigenetic modifications, DNA methylation, the addition of a -CH3 methyl group to either cytosine or adenine bases, has received considerable focus for its role in regulating gene expression, particularly in vertebrate lineages and plants (Schubeler, 2015; Zhang et al., 2018). Investigations into the role of DNA methylation in invertebrates have uncovered marked differences in methylation’s influence on gene expression and performance between phylogenetic lineages and across different environmental contexts (Sarda et al., 2012; Dimond and Roberts, 2016; Hofmann, 2017; Eirin-Lopez and Putnam, 2019). However, studies of DNA methylation and its role in environment-organism interactions in marine invertebrates remain sparse and confined to a small number of species for any given phyla.
Antarctic pteropods offer a valuable system for examining the role of epigenetics in shaping organismal responses to global change in the marine environment. Shelled pteropods or thecosomes have been proposed as a bellwether species for OA (Manno et al., 2017). The Antarctic thecosome L. helicina antarctica is a widely distributed and abundant pteropod endemic to the Southern Ocean (Boysen-Ennen et al., 1991; Steinberg et al., 2015; Thibodeau et al., 2019) where it can make up > 50% of total zooplankton measured as individuals per unit volume (Hunt et al., 2008). Limacina sp. is also a globally distributed genus spanning both polar and temperate environments (Bernard and Froneman, 2009; Hunt et al., 2010). L. helicina antarctica presently experiences a large degree of seasonal variability in seawater pCO2 and pH, with pH varying by 0.6 units in regions like Prydz Bay (Gibson and Trull, 1999) and the Ross Sea (McNeil et al., 2010). This seasonal variability currently exceeds modeled predictions of an increase in annual mean pH of 0.4 units within the Southern Ocean by 2100 (IPCC, 2013). L. helicina antarctica also experiences present-day undersaturation in Ωaragonite in some regions of the Southern Ocean during the austral winter (Bednaršek et al., 2012b). The frequency and duration of undersaturation events are expected to increase as OA advances in the Southern Ocean (McNeil and Matear, 2008; Hauri et al., 2015; Negrete-García et al., 2019). The onset of month long undersaturation events are predicted in the Southern Ocean within 14–29 years, along with annual mean Ωaragonite undersaturation as soon as 10–20 years from today (IPCC, 2019).
In the face of future increases in pCO2, current evidence suggests that L. helicina antarctica is poorly poised to acclimatize to predicted OA over the next century (Bednaršek et al., 2012a, 2014b; Gardner et al., 2018). Juvenile L. helicina antarctica collected from the McMurdo Sound, Ross Sea, do not appear to alter their metabolic rate over the course of 2-week acclimations to a regional, present-day pCO2 extreme of 427–513 μatm (Hoshijima et al., 2017). By contrast, acclimation to future pCO2 extremes of 901–1000 μatm has been shown to significantly alter metabolic rate under near-ambient temperatures (Seibel et al., 2012; Hoshijima et al., 2017). Similarly, gene expression by L. helicina antarctica has shown little differential expression in response to intermediate pCO2 levels of 432 μatm when compared to differential expression under 902 μatm at which pervasive downregulation is apparent among transcripts even after 21 days of acclimation (Johnson and Hofmann, 2017). The dynamic shifts in metabolism and gene expression that arise when L. helicina antarctica are exposed to present-day vs. future pCO2 extremes may be driven or regulated by epigenetic mechanisms such as DNA methylation.
Determining (i) whether DNA methylation in L. helicina antarctica varies across exposures to current and future pCO2 extremes and (ii) whether changes in DNA methylation are associated with differential gene expression may provide valuable insight into molecular mechanisms underpinning this species’ performance under OA and its ability to rapidly acclimatize to changing ocean conditions. Findings regarding epigenetic responses by L. helicina antarctica to OA may provide translational information for similar studies in other members of Limacina sp. and thecosome pteropods, a clade of shelled pteropods considered to be a bellwether for the severity of global OA (Bednaršek et al., 2014a). Furthermore, epigenetic studies revealing the importance of DNA methylation for biological functions in molluscs (Gavery and Roberts, 2013; Riviere et al., 2013, 2017; Diaz-Freije et al., 2014; Lian et al., 2015; Garcia-Fernandez et al., 2017; Suarez-Ulloa et al., 2019) thus far have been heavily skewed toward bivalves and in particular, oysters, necessitating a broader assessment of diversity in environmental epigenetics across the speciose molluscan phylum.
To this end, we conditioned juvenile L. helicina antarctica collected from McMurdo Sound under present-day pCO2 levels of 255 μatm, present-day pCO2 extremes of 530 μatm, and future pCO2 extremes of 918 μatm, resulting in Ωaragnoite undersaturation, for up to 7 days (Figure 1). Following this experiment, we quantified the proportion of 5-methylcytosine in L. helicina limacina DNA following 1, 3, and 6 days of exposure to each pCO2 level. Additionally, we sequenced and analyzed the transcriptomes of pteropods sampled from each pCO2 level after 0.5 and 7 days of conditioning in order to (i) quantify differential expression in response to current and future pCO2 extremes across time and (ii) test for correlations between differential expression and signatures of DNA methylation among transcribed genes.
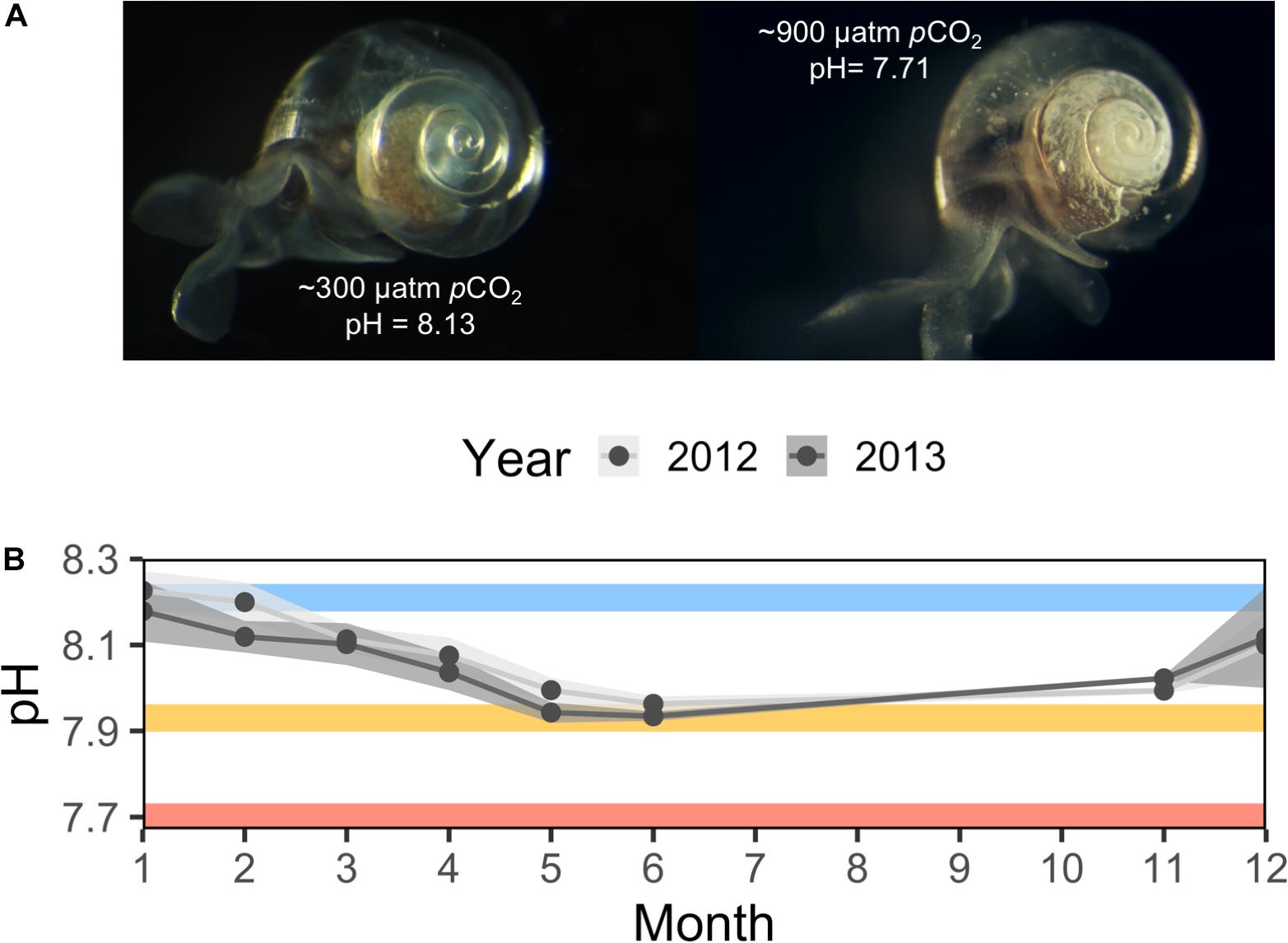
Figure 1. In situ and experimental ocean acidification experienced by Limacina helicina antarctica. (A) Representative images of shell dissolution following exposure to ∼300 and ∼900 μatm pCO2 for 14 days. (B) Mean surface ocean pH recordings per month from McMurdo Sound taken at McMurdo Station circa 2012 and Cape Evans circa 2013 recorded by Kapsenberg et al., 2015 and Hoshijima et al. (unpublished). Error windows represent ± SD. Mean pH from 255, 530, and 918 μatm pCO2 experimental conditions are subimposed as blue, yellow, and red lines, respectively.
Materials and Methods
Collection and Conditioning of L. helicina antarctica
Juvenile L. helicina antarctica were collected in October of 2014 from Cape Evans, McMurdo Sound, via plankton net as previously described (Hoshijima et al., 2017; Johnson and Hofmann, 2017) and transported to the Crary Laboratory, McMurdo Station, where they were held under near-ambient temperature (−0.59°C) in filtered seawater for 24 h before conditioning at different pCO2 levels. During conditioning, L. helicina antarctica were held in 1 L tanks with a flow rate of 2 L h–1 at a starting density of 200 animals L–1. L. helicina antarctica were not fed at any point during the experiment as the winter-summer transition in the McMurdo Sound is characterized by low phytoplankton abundance (Foster et al., 1987).
Seawater pCO2 was manipulated using three reservoir containers held at different pCO2 levels, each connected to three replicate culturing tanks of the volume and flow rate described above. pCO2 was controlled inside of reservoir tanks by injecting mixtures of filtered, CO2-scrubbed, dry air and pure CO2 with seawater. Pure air and CO2 were mixed using SmartTrak® 100 Series Mass Flow Controllers and Micro-Trak© 101 Series Mass Flow Controllers (Sierra Instruments, United States), respectively. Reservoirs were held in a 1240 L seawater table filled with near-ambient seawater. Culture tanks held a mean temperature of −0.59°C ± 0.11 SD over the course of the 7-day experiment. Seawater temperature, pCO2, pH, and carbonate chemistry were measured as previously described (Johnson and Hofmann, 2017). Low, intermediate, and high pCO2 levels averaged 255 μatm ± 3.52 SD, 530 μatm ± 12.03 SD, and 918 μatm ± 19.53 SD. A full report of temperature and seawater chemistry in each culture tank over time is available in Supplementary Figure S1.
Quantification and Analysis of Genomic 5-Methylcytosine
DNA was extracted from pooled replicates containing 10 juvenile L. helicina antarctica sampled from each pCO2 treatment at 1, 3, and 6 days using a CTAB DNA extraction modified from Worden (2009). The step-by-step DNA extraction protocol is detailed under Supplementary Data S2. DNA purity, quality, and concentration were respectively assessed via Nanodrop, agarose gel, and Qubit Broad Range DNA quantification. DNA extractions were performed on five biological replicates per pCO2 level x timepoint by sampling two pooled replicates from two of the triplicate cultures and one pooled replicate from the third.
The proportion of 5-methylcytosine (5-mC) present in L. helicina antarctica DNA was quantified colorimetrically using the ELISA-based MethylFlash DNA 5-mC Quantification Kit (Epigentek, Farmingdale, NY, United States) according to manufacturer guidelines. Contrary to manufacturer instructions, standards and samples were measured in triplicate rather than duplicate in order to improve accuracy. Additionally, the 45 samples were randomly split between two different plates such that 3/5 or 2/5 of replicates for each pCO2 level x timepoint group would be present on a given plate. Within a single plate, the arrangement of standards, negative controls, and samples was randomized. In keeping with manufacturer instructions, technical replicates remained grouped together within each plate. OD450 was measured using an Epoch Microplate Spectrophotometer (Biotek Instruments, Winooski, VT, United States).
Variation in 5-mC content was modeled as a function of pCO2 level, exposure time, and their interaction by fitting a linear regression to the data using the lm() function of the R ‘stats’ package v3.5.1. A full report describing the linear model used to assess changes in 5-mC content is available in Supplementary Table S1. Reported F-statistics from the fitted model were generated using the anova() function of the R ‘stats’ package. Cliff’s delta estimates, mean effect size estimates, and post hoc Mann–Whitney U-tests comparing 5-mC content between groups were performed with the ‘dabest’ package for estimation statistics (Ho et al., 2019).
The presence of outliers within the data was assessed by measuring the non-normality, residual, and leverage of each individual replicate fitted to the linear model using Q–Q plots, residual vs. leverage plots, and Cook’s distance estimates executed with the R ‘stats’ base package. Any sample exhibiting non-normality, high leverage, and high residual was excluded from the linear model, resulting in the removal of a single data point.
RNA Sequencing and Transcriptomic Analyses
RNA was extracted and sequenced from three pooled L. helicina antarctica replicates (10 individuals/pool) per treatment for each of the two sampling time points (n = 18 libraries; 2 μg library–1) as previously described (Johnson and Hofmann, 2017). Sequencing was performed at the UC Davis Genome Center on an Illumina HiSeq4000 sequencer using paired-end 100 bp reads. Raw sequencing reads were trimmed using Trimmomatic to remove adapter sequences, low quality base pairs (PHRED < 20), and sequences smaller then 75 bp (Bolger et al., 2014). The quality of trimmed reads was assessed using FastQC v0.11.8 (Andrews, 2018). Trimmed reads were mapped to the L. helicina antarctica reference transcriptome (Johnson and Hofmann, 2016) and counted using RSEM v1.3.1. RSEM was chosen for its accurate quantification of read counts mapped to de novo reference assemblies (Li and Dewey, 2011). Read processing, alignment, and counting was performed with support from the Indiana University Carbonate computing cluster (Stewart et al., 2017). A differential expression analysis was performed using edgeR v3.24.3 (Robinson et al., 2010) set to ‘robust’ calculation of dispersion and ‘robust’ fitting of general linear models using the glmQLFit() function of edgeR. Differentially expressed genes (DEGs) were identified using an FDR cutoff < 0.05 and an absolute log2 foldchange cutoff of > 1.5 (Chen et al., 2016). A principal coordinates analysis was also performed in order to cluster RNAseq samples by inputting log2-adjusted counts per million (CPM) read counts to the pcoa() function of the R package ‘ape’ (Paradis et al., 2004). R scripts used to analyze differential gene expression are included in Supplementary Data S3. Enriched gene ontologies (GO) were identified among up- and down-regulated genes using Mann–Whitney U-tests input with signed, −log p-values using the ‘Rank Based Gene Ontology Analysis with Adaptive Clustering’ method: https://github.com/z0on/GO_MWU (Wright et al., 2015).
Germline gene body methylation levels were estimated within exons by calculating observed over expected CpG frequencies (CpGOE) across each transcript within the L. helicina antarctica reference transcriptome (Johnson and Hofmann, 2016) using python scripts written by Dimond and colleagues https://github.com/jldimond/Coral-CpG (Dimond and Roberts, 2016). Means and distributions of transcript CpGOE values were then compared between up- and down-regulated DEGs within treatment groups using permutation tests and Kolmogorov–Smirnov (KS) tests, respectively. A linear model was used to assess variation in transcript CpGOE as a function of foldchange direction, duration of exposure to high pCO2, and their interaction using the R package ‘lmPerm’ v2.1.0, which generates p-values for linear models using permutation tests (Wheeler and Torchiano, 2016). Residuals within this linear model were bimodal, necessitating a statistical approach such as a permutation test that does not assume a normal distribution. Two-sided KS tests were performed using the ks.test() function of the R package ‘dgof’ v1.2 (Arnold and Emerson, 2013). The influence of categorical CpGOE bins (bin size = 0.5) on transcriptional variation represented as the CV of logCPM was assessed using the lm() function of R ‘stats.’
Results
Genomic Methylation in L. helicina antarctica Under Current and Future pCO2 Extremes
ELISA-based quantification of 5-methylcytosine in L. helicina antarctica revealed significant effects of pCO2 (F1,40 = 10.24; p = 0.0027), and the interaction of pCO2 and time (F1,40 = 7.74; p = 0.0082) on DNA methylation. Specifically, acute exposure to future pCO2 extremes induced hypomethylation of the L. helicina antarctica genome followed by a return to normal levels over time. Exposure to current pCO2 extremes did not induce detectable changes in genomic methylation relative to ambient pCO2 (Figure 2).
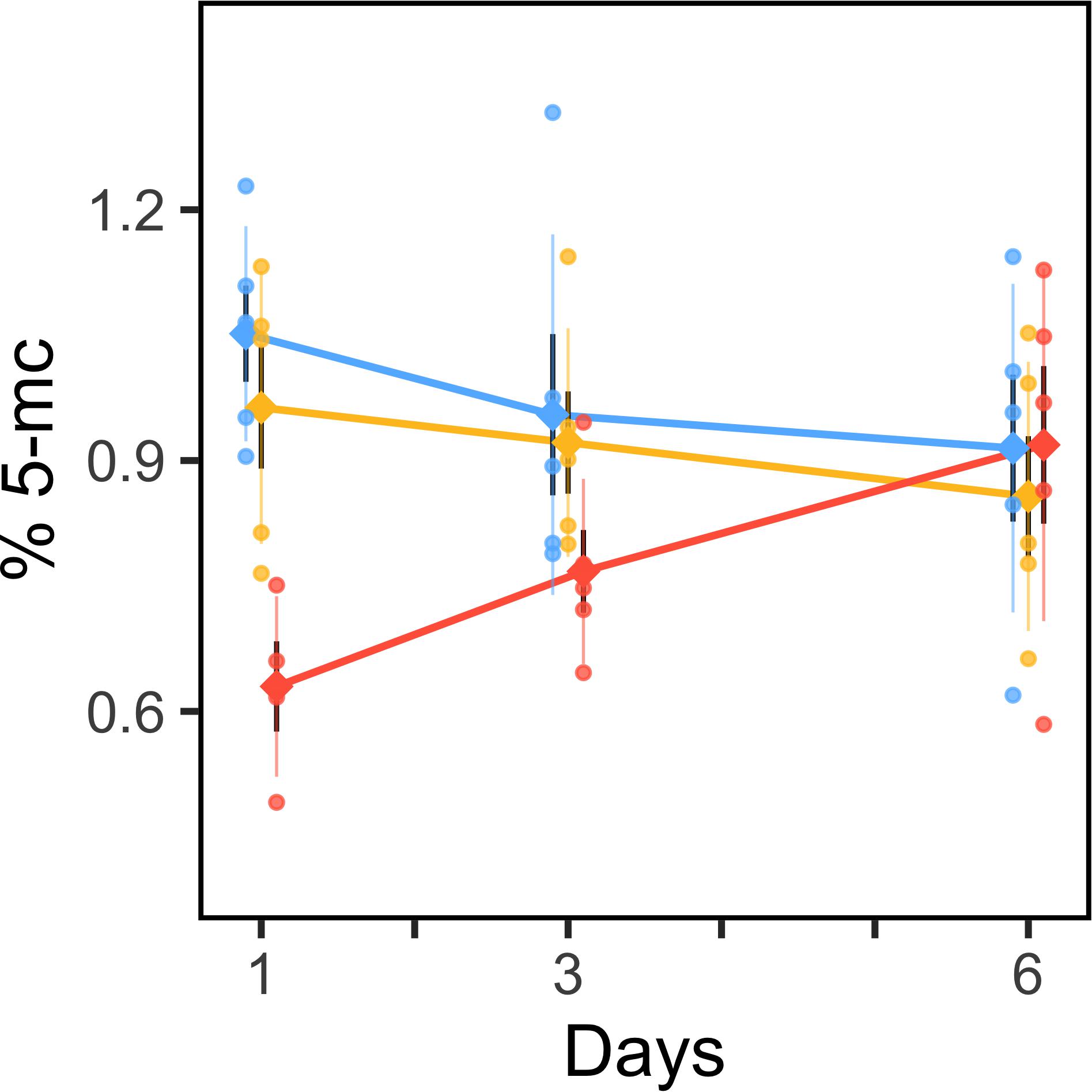
Figure 2. Differential methylation of the Limacina helicina antarctica genome during acute exposure to future pCO2 extremes. Mean values (diamonds) and individual replicates (circles) of percent 5-methylcytosine quantified using ELISA-based methods are plotted for 255 μatm (blue), 530 μatm (yellow), and 918 μatm (red) pCO2 treatments level across time. ± SE is represented by wide, black error bars. ± SD is represented by narrow, colored error bars.
Individuals exposed to 918 μatm pCO2 (future extremes) for 1 day exhibited a 0.42% (0.29–0.56 95% CI) reduction in 5-mC content relative to 255 μatm (Mann–Whitney p = 0.020). For context, the mean 5-mC content of the L. helicina antarctica genome equaled 0.944 ± 0.17% when animals exposed to 918 μatm pCO2 were excluded from the mean. 5-mC in 918 μatm cultures remained 0.19% lower than the 255 μatm treatment after 3 days of exposure (Mann–Whitney p = 0.060) before converging at a difference of 0.0039% after 6 days. L. helicina antarctica showed no differences in 5-mC content between 530 and 255 μatm pCO2 treatments at any timepoint (Figure 2).
One replicate exhibited 2.14-fold greater 5-mC content than the overall mean. The non-normality, residual, and leverage of this replicate were examined in order to determine its status as an outlier using (i) Cook’s distance, (ii) quantile–quantile plots to assess deviation from normality and (iii) residual-leverage plots. The replicate exhibited a substantially high residual and leverage and was thus removed from all analyses (Supplementary Figure S2). A full report describing the linear model used to assess changes in 5-mC content (Supplementary Table S1) and CV values per replicate (Supplementary Figure S3) are available in Supplementary Data S1.
Transcriptomic Responses to Current and Future pCO2 Extremes
As genomic methylation influences both magnitude and variation in gene expression in multiple invertebrate lineages, we sought to pair genome-wide methylation data with transcriptomes sequenced for early and late timepoints under low, medium, and high pCO2. Overall, we found that gene expression in L. helicina antarctica varied as a function of pCO2 and exposure time. A principal coordinates analysis of CPM read counts demonstrated that a coordinate axis associated with pCO2 level and time explained 32.53% of transcriptional variation. A second axis associated with time alone explained 8.4% (Figure 3A).
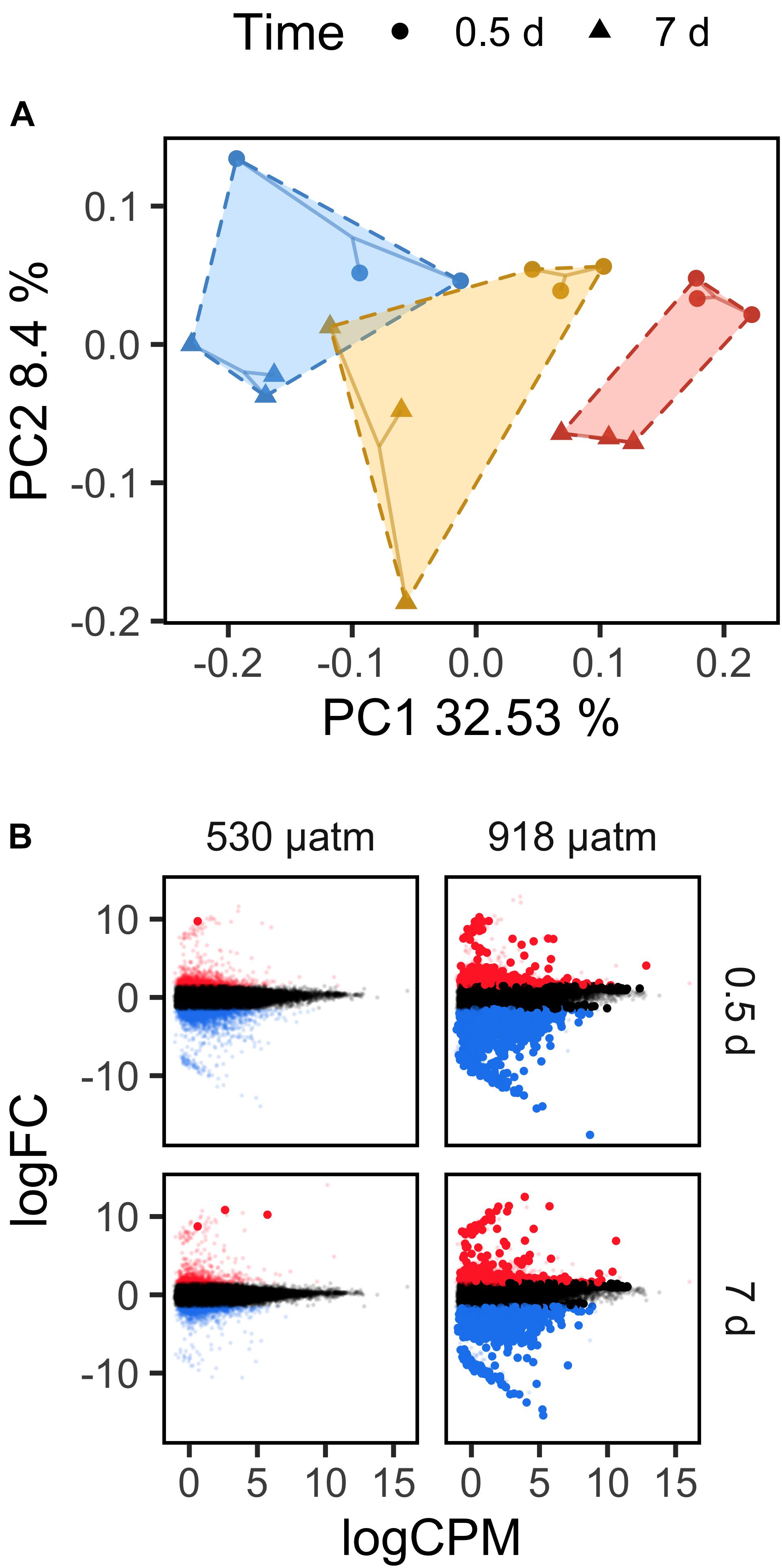
Figure 3. Transcriptomic responses by Limacina helicina antarctica to current and future pCO2 extremes. (A) Loading of RNAseq samples to latent variables derived from a principal coordinates analysis of CPM-normalized read counts. Polygons are drawn around pCO2 255 μatm (blue), 530 μatm (yellow), and 918 μatm (red) pCO2 treatments. Replicates sampled at different times are denoted by different shapes. (B) Differential expression under 530 and 918 μatm pCO2 relative to 255 μatm. Transcript log2 foldchanges are plotted against log2 CPM read counts. Red and blue points denote up- and down-regulated transcripts, respectively, above an absolute log2 foldchange of 1.5. Large, solid points denote differentially expressed transcripts with an FDR p-value < 0.05.
Limacina helicina antarctica exhibited substantially more differential expression under 918 μatm pCO2 than under 530 μatm. After 0.5 day of exposure, pteropods in the 530 μatm treatment exhibited only one significant DEG relative to 255 μatm (FDR < 0.05; absolute log2FC > 1.5). After 7 days at 530 μatm, 3 transcripts were differentially expressed. Additionally, each DEG in the 530 μatm treatment group was upregulated. By contrast, L. helicina antarctica differentially expressed 6,649 transcripts after 0.5 day of exposure to 918 μatm and 6,815 transcripts after 7 days. 69.91% of DEGs detected at 0.5 day under 918 μatm pCO2 were downregulated. Downregulated transcripts composed 61.20% of DEGs after 7 days of exposure to 918 μatm (Figure 3B). 3,607 transcripts were differentially expressed at both 0.5 and 7 days under 918 μatm pCO2, leaving 3,042 DEGs unique to 0.5 day of exposure and 3,773 DEGs unique to 7 days.
Transcripts that were up- or down-regulated at 0.5 day under 918 μatm pCO2 were collectively enriched for 88 GO terms while 111 GO terms were enriched among up- and down-regulated transcripts from 7 days of 918 μatm exposure (FDR < 0.01). 81 of these GO terms were commonly enriched among both time points. 56.81% of GO terms enriched at 0.5 day were associated with downregulated transcripts while 54.95% of GO terms enriched at 7 days were associated with downregulation. Ontologies uniquely enriched at 0.5 day pertained to glucose metabolism, protein metabolism, and the oxidative stress response. Ontologies unique to the 7 days timepoint were diverse but included multiple GO terms pertaining to singular functions such as fatty-acid metabolism, antioxidant activity, transcription, trans-membrane transport, and transferase activity for glycosyl and hexosyl groups. Enriched ontologies that were shared between 0.5 and 7 days included functions relating to protein degradation, protein synthesis, cytoskeletal structure, ATP synthesis, and methyltransferase activity (Supplementary Figures S4, S5).
Since methyltransferases are of broad interest in an epigenetic context, we queried enriched GO terms for these subsets of genes. We found that 182 and 174 transcripts associated with methyltransferase activity were significantly enriched among down or upregulated genes at 0.5 and 7 days, respectively, under 918 μatm pCO2. On average, enriched methyltransferases were upregulated at both time points. Sub-ontologies of “methyltransferase activity” (GO:0008168) that were enriched among DEGs at both time points included RNA methyltransferases, tRNA methyltransferases, and SAM-dependent methyltransferases: a class that includes enzymes targeting DNA and/or RNA (Supplementary Figures S4, S5). Histone methyltransferases were not enriched among down or upregulated genes, but some were differentially expressed under future pCO2 extremes. 1 EZH2 histone-lysine N-methyltransferase was upregulated after 0.5 day exposure while four histone-arginine methyltransferases, 1 UTY histone demethylase, and 4 histone-lysine N-methyltransferases showed downregulation. 1 SETD8 histone-lysine N-methyltransferase was downregulated at both 0.5 and 7 days of exposure to 918 μatm (Supplementary Figures S6, S7).
Full lists of transcripts differentially expressed at 918 μatm pCO2 relative to 255 μatm are available in Supplementary Data S4, S5. GO terms significantly enriched among sets of up- and down-regulated genes are visualized in Supplementary Figures S4, S5.
Signatures of Gene Body Methylation Among Differentially Expressed Genes
Transcripts that were differentially expressed in response to 918 μatm pCO2 varied in mean CpGOE ratio depending on fold change direction (F1,13459 = 85.05; p < 2e-16). CpGOE is inversely related to germline methylation and, when it is applied to transcripts, demonstrates a signature of gene body methylation. Specifically, downregulated genes were enriched with low CpGOE genes at both 0.5 and 7 days (Figure 4A). A significant interaction between foldchange direction and time was also observed (F1,13459 = 71.45; p < 2e-16) in which the mean difference in CpGOE between up- and down-regulated DEGs was greater at 0.5 day (Cliff’s delta = 0.1409 ± 0.0330) than 7 days (Cliff’s delta = 0.0292 ± 0.0296).
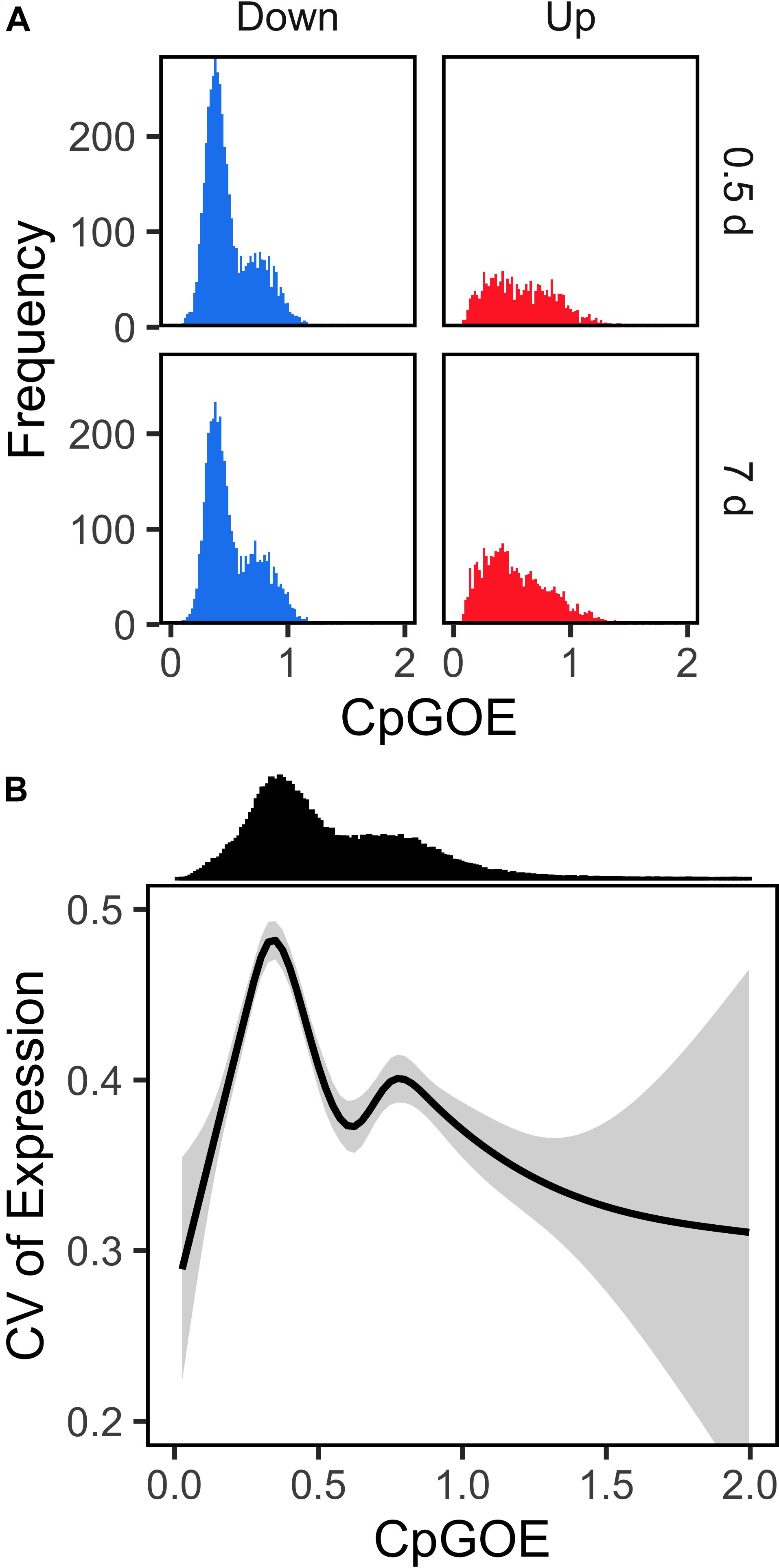
Figure 4. Transcripts downregulated by Limacina helicina antarctica in response to future pCO2 extremes show enriched signatures of gene body methylation. (A) CpGOE histograms are plotted for transcripts exhibiting significant downregulation (blue) or upregulation (red) under 918 μatm pCO2. Bin widths = 0.02 CpGOE. (B) Mean CV of expression is plotted across CpGOE. Shaded regions depict ± 95% CI.
As (i) the distribution of CpGOE in the L. helicina antarctica transcriptome is bimodal and (ii) the maximum and minimum percentiles of CpGOE distributions were equivalent between up- and down-regulated transcripts, changes in the densities of CpGOE distributions among DEGs served as a more accurate measurement of the frequency of high and low CpGOE values. A Kolmogorov–Smirnov (KS) test demonstrated significant differences in the modality of CpGOE values between up- and down-regulated DEGs at 0.5 day (D = 0.19075; p < 1.0e-15) and 7 days (D = 0.15198; p < 1.0e-15) under 918 μatm pCO2. Density beneath the low CpGOE mode was 2.44-fold greater among downregulated DEGs compared to upregulated DEGs at 0.5 day and 1.84-fold greater after 7 days (Figure 4A). Differences in the modality of CpGOE among DEGs that were downregulated either at 0.5 and 7 days relative to the whole L. helicina antarctica transcriptome were significant but less pronounced: KS test D values equaled 0.09318–0.06263 for 0.5 and 7 days, respectively (p < 1.0e-15). Cumulative distributions visualizing the modalities of CpGOE in each of the groups are available in Supplementary Figure S8. In contrast to the relationship between genes’ CpGOE and differential expression in L. helicina antarctica, a negligible association between CpGOE and transcriptional variation was observed. A linear model reported a significant effect of categorical, binned CpGOE (bin size = 0.5) on the CV of expression of transcripts in which the low CpGOE mode exhibited a higher mean CV (Figure 4B), albeit with a large degree of variation surrounding the mean.
Discussion
Our analyses of DNA methylation and gene expression in L. helicina antarctica showed dynamic changes in both processes following experimental conditioning under future pCO2 levels predicted for Antarctic coastal waters. Juvenile L. helicina antarctica appeared to mount a strong epigenetic response to future pCO2 extremes, reducing DNA methylation by 29–56% after brief exposure to high pCO2 before steadily increasing DNA methylation levels over time. In addition, differential gene expression showed an association with DNA methylation as evidenced by an enrichment of low CpGOE transcripts among genes that were significantly downregulated in response to future pCO2 extremes. Changes in global DNA methylation and differential expression were absent or negligible following exposure to present-day pCO2 extremes. Taken together, these observations suggest that DNA methylation plays a role in regulating the cellular responses of L. helicina antarctica to future OA.
Below, we discuss the functional significance of DNA methylation in the context of marine molecular ecology, compare our findings to studies of DNA methylation in other Antarctic organisms, and discuss the available data on epigenetic responses by other species to OA. Lastly, we point to next steps for the study of epigenetics in polar systems, including future work on L. helicina antarctica and other calcifying marine invertebrates.
Hypomethylation Following Acute Exposure to High pCO2
While L. helicina antarctica has exhibited a poor capacity to acclimate to experimental OA as evidenced by a lack of metabolic compensation and upregulation of stress response pathways following 21 days of acclimation to future pCO2 levels (Hoshijima et al., 2017; Johnson and Hofmann, 2017), in this study, it appears that juvenile L. helicina antarctica were capable of mounting a significant epigenetic response to high pCO2. Our preliminary analyses in L. helicina antarctica have identified dynamic change in levels of DNA methylation in response to high pCO2. This outcome might not be expected if we were to stereotype this species as an Antarctic, cold-adapted invertebrate with diminished cellular responses to environmental change. In fact, studies of environmental epigenetics in two Antarctic marine invertebrates, a benthic polychete and an intertidal gastropod, have both documented differential methylation in response to variation in temperature (Marsh and Pasqualone, 2014; Clark et al., 2018).
It is worth noting that one limit to our approach toward quantifying global patterns of DNA methylation is the non-specific nature of the acquired data. While global levels of methylation provide insight into changes across the whole genome, these measurements are limited in that they (i) provide no indication of functional consequences and (ii) are less sensitive than NGS methods such as bisulfite sequencing. For example, if X methylated CpGs become demethylated in response to a given treatment and are proceeded by methylation at X CpGs that were previously unmethylated, global 5-mC quantification would not detect a change in methylation. For this reason, our results do not rule out the possibility that L. helicina antarctica may differentially methylate its genome in response to present-day pCO2 extremes. Additionally, the large effect strength of 918 μatm pCO2 on genomic methylation in L. helicina antarctica is not unprecedented in the context of acute exposure to stress (Rodrigues et al., 2015; Li et al., 2016; Robinson et al., 2019), but the quantity of this effect should be further evaluated with replicate experiments or similar studies.
Although we did detect changes in total methylation and gene expression, our results are presently not sufficient to directly link differential methylation with the expression of specific genes. However, other studies on invertebrates indicate a relationship between gene expression and genomic methylation in response to variation in multiple abiotic factors. Many invertebrate phyla including molluscs predominantly exhibit DNA methylation within gene coding regions (Zemach et al., 2010; Wang et al., 2014; Jeong et al., 2018; Li et al., 2018; Liew et al., 2018). Hypomethylation at intragenic regions is generally associated with reduced gene expression and increased transcriptional variability and/or spuriousness in both vertebrates (Kobayashi et al., 2012; Neri et al., 2017) and invertebrates (Zemach et al., 2010; Dixon et al., 2014, 2018; Gavery and Roberts, 2014; Li et al., 2018). Many studies have also noted associations between hypomethylation and environmentally-induced or disease-induced pathologies spanning a wide range of taxa including plants, vertebrates, and invertebrates for which hypomethylation was demonstrated in both promoters and/or gene bodies (Aina et al., 2004; Pavet et al., 2006; Pogribny et al., 2006; Rusiecki et al., 2008; Luttmer et al., 2013; Xiu et al., 2019). While mechanistic links between hypomethylation and pathology are unclear, there is strong evidence in support of its role in cellular responses to infection and stress.
Despite extensive research on organismal responses to OA, the cellular mechanisms that contribute to pathological vs. adaptive responses to pCO2 stress remain poorly understood (Melzner et al., 2020). It is plausible that hypomethylation of the L. helicina antarctica genome in response to high pCO2 is not adaptive. Rather, it may be related to pathological processes associated with chronic stress seen in L. helicina antarctica under OA. The 29–56% reduction in mean 5-mC by L. helicina antarctica in response to pCO2 stress contrasts observations of differential methylation under OA that have been documented in more pCO2-tolerant calcifying invertebrates. Specifically, scleractinian corals and purple sea urchins of differing life history stages respectively exhibited global hypermethylation or a combination of hypo- and hypermethylation at CpGs in response to elevated levels of pCO2 (Putnam et al., 2016; Liew et al., 2018; Strader et al., 2019; Strader et al., in review). It is unclear to what extent such variation between these taxa is due to phylogenetic differences, life history or to pCO2 sensitivity, but they remain noteworthy. Further comparative work exploring differences in the variation of DNA methylation under OA in thecosome pteropods relative to more pCO2-tolerant calcifying invertebrates could help elucidate mutually exclusive aspects of genomic methylation that contribute to acclimatization and pathology.
Evidence of a Role for DNA Methylation in Differential Expression Under High pCO2
Limacina helicina antarctica mounted a large transcriptomic response to future pCO2 extremes that predominantly consisted of downregulated DEGs. This response corroborates our past reporting on differential expression in response to OA among L. helicina antarctica collected from the McMurdo Sound (Johnson and Hofmann, 2017). Since (i) gene expression in molluscs and other invertebrates positively correlates with gene body methylation (Zemach et al., 2010; Gavery and Roberts, 2013; Dixon et al., 2014, 2018; Gavery and Roberts, 2014; Li et al., 2018), and (ii) DNA methylation occurs primarily within gene bodies among molluscs and other invertebrates, it is possible that hypomethylation of L. helicina antarctica DNA observed in response to high pCO2 contributed to the differential expression of downregulated genes. Indeed, downregulated DEGs were enriched with low CpGOE transcripts relative to upregulated genes. This demonstrated that downregulated DEGs were likely to have a greater degree of methylation at exons within germline cells, and further, suggested a link between hypomethylation and downregulation across the L. helicina antarctica genome.
Despite evidence of DNA methylation’s influence on downregulated transcripts, the timing of changes in 5 m-C and differential expression were not synchronized throughout the duration of the experiment. Mean 5-mC levels in L. helicina antarctica exposed to 918 μatm became comparable to 530 μatm and 255 μatm cultures by 6 days of exposure while the quantity of downregulation in 918 μatm cultures remained high and relatively unchanged even by 7 days. Thus, DNA methylation and differential expression did not correlate over time despite evidence of their association revealed by CpGOE values. DNA methylation and differential expression do not necessarily correlate throughout time when a causal relationship is present (Secco et al., 2015; Pacis et al., 2019), and have been documented to be unpaired during invertebrate development under high pCO2 (Strader et al., in review). The relationship between DNA methylation and gene expression in molluscs has proved tenuous in some cases. For example, the offspring of diuron-exposed oysters have exhibited differential expression and DNA methylation relative to offspring from control parents, but only a small number of DEGs were correlated with differentially methylated CpGs (Rondon et al., 2017). Other reports in oysters have documented stronger associations between gene expression and DNA methylation (Olson and Roberts, 2014). The lack of synchronicity in the timing of differential methylation and differential expression in this experiment does not entirely rule out their mutual influence. Rather, it may suggest a more complex relationship that includes other epigenetic factors.
We also observed interesting patterns of differential expression in genes with various functions related to other epigenetic modifications to DNA. For example, the differential expression of histone modifying enzymes by L. helicina antarctica correlated strongly with changes in global 5-mC over time under high pCO2: 1 EZH2 histone-lysine N-methyltransferase was upregulated by L. helicina antarctica after 0.5 day exposure to 918 μatm while 4 histone-arginine methyltransferases, 1 UTY histone demethylase, and 4 histone-lysine N-methyltransferases were downregulated. Only one of these genes remained differentially expressed by 7 days of exposure to 918 μatm, demonstrating an early signature of downregulation in histone methyltransferases. In at least some invertebrates and mammals, gene bodies that exhibit higher levels of baseline DNA methylation are also enriched with H3K36me3, histones with lysine 36 trimethylation (Nanty et al., 2011; Baubec et al., 2015). H3K36me3 can chaperone de novo methyltransferases to gene bodies (Baubec et al., 2015). Inversely, the absence of H3K36me3 at gene bodies has been documented to be a predictor of hypomethylation (Hahn et al., 2011). Arginine methylation at H4R3me2 has a similar relationship with de novo methyltransferases but ultimately has a more repressive effect on gene expression (Zhao et al., 2009). Therefore, differential methylation of intragenic regions is associated with chromatin accessibility in certain contexts and both of these processes mediate the influence of intragenic regions on differential expression. Overall, our results suggest that differential methylation of DNA and histones may jointly influence persistent transcriptomic responses by L. helicina antarctica to future pCO2 extremes. Histone modifications (e.g., histone methylation) and subsequent changes in transcription have been shown to regulate development and responses to environmental stress in molluscs (Fellous et al., 2015, 2019; Gonzalez-Romero et al., 2017) and other calcifying marine invertebrates (Rodriguez-Casariego et al., 2018). Interactions between chromatin structure and DNA methylation should be explored in greater functional detail in this system in order to substantiate these observations and understand their influence on pervasive and persistent downregulation by L. helicina antarctica under OA.
Pervasive downregulation or “dampening” transcriptomic and/or proteomic responses are commonly observed under high pCO2 across marine molluscs and other calcifying invertebrates (Todgham and Hofmann, 2009; O’Donnell et al., 2010; Dineshram et al., 2012; Kenkel et al., 2017; De Wit et al., 2018; Kriefall et al., 2018), including some species of Limacina pteropods (Koh et al., 2015; Johnson and Hofmann, 2017). Other genera of shelled pteropods have not exhibited dampening transcriptomic responses to OA (Maas et al., 2015; Moya et al., 2016) and strong time-dependent effects on the proportion of upregulated vs. downregulated genes have been documented in Limacina retroversa in response to high pCO2 (Maas et al., 2018). Thus, there does not appear to be a generalizable transcriptional response to OA within Pteropoda or the Limacina genus. Studying the potential for phylogenetic variation in epigenetic processes among Pteropods may further explain the contribution of mechanisms such as DNA methylation to cellular responses to OA. Similarly, expanding epigenetic studies of cellular responses to OA across a broader diversity of taxa may reveal mechanistic similarities between evolutionarily distant groups that exhibit similar transcriptional responses to high pCO2.
Conclusion
Perhaps the most important conclusion we can draw from this experiment is that variation in DNA methylation across the L. helicina antarctica genome was responsive to ocean acidification and likely yielded consequences for cellular functions due to (i) the magnitude of variation, and (ii) evidence of its influence on gene expression. From a polar biology perspective, our results demonstrate that DNA methylation is dynamic in an otherwise stenothermic Antarctic macrozooplankton. Many Antarctic ectotherms have classically been considered to be specialized to an invariable environment, subsequently lacking phenotypic and physiological plasticity necessary for acclimatizing to environmental shifts on par with future climate change (Peck et al., 2004, 2010, 2014; Beers and Jayasundara, 2015). A growing body of work is beginning to demonstrate, however, that physiological plasticity is retained and sufficient for metabolic recovery under a warmer and/or more acidic ocean in at least some Antarctic ectotherms (Seebacher et al., 2005; Peck et al., 2010; Enzor and Place, 2014; Reed and Thatje, 2015; Huth and Place, 2016a, b; Morley et al., 2016; Enzor et al., 2017; Davis et al., 2018; Hawkins et al., 2018). The responsiveness of DNA methylation in L. helicina antarctica to variation in pCO2 may be linked to this species’ environmental experience in situ. Recent oceanographic observations suggest that seasonal variation in pCO2 in coastal waters of the Southern Ocean is substantial (Gibson and Trull, 1999; McNeil et al., 2010; Kapsenberg et al., 2015; Negrete-García et al., 2019). Thus, it is likely that L. helicina antarctica is presently faced with low pH conditions that are undersaturated for aragonite calcification during vertical migration.
Lastly, recent modeling efforts have indicated that Ωaragonite undersaturation in the Southern Ocean will appear at quite shallow depths (∼400 m) by the end of the century (Negrete-García et al., 2019) creating a habitat compression or “squeeze” event for calcifying macrozooplankton such as L. helicina antarctica. Thus, quantifying the epigenetic, transcriptional, and physiological plasticity of L. helicina antarctica populations is important in assessing their capacity to respond to future changes in ocean carbonate chemistry. With that said, it remains unclear whether or not dynamic changes in DNA methylation are adaptive for L. helicina populations experiencing high pCO2. Future research concerning this ecologically critical species that aims to expand on our findings and past OA experiments will benefit from (i) executing long-term cultures of L. helicina antarctica under future pCO2 extremes for at least 3–6 weeks and (ii) conducting integrated analyses of metabolic rate, calcification, gene expression, and epigenetic profiling. This 3–6 week range has proven to be a metabolic and transcriptional tipping point for other Antarctic ectotherms acclimating to increases in temperature and pCO2 (Peck et al., 2010; Enzor and Place, 2014; Enzor et al., 2017; Davis et al., 2018). Quantifying DNA methylation at whole-genome or base-by-base levels across long-term acclimations will expand on dynamic epigenetic changes documented in this study and reveal whether or not they associate with or initiate plastic responses to global change. If DNA methylation or other epigenetic mechanisms ultimately do not drive plastic responses to environmental variation in L. helicina antarctica, this species still represents a valuable comparative system for studying the contribution of epigenetic mechanisms to adaptive vs. pathological responses to abiotic stress and future OA.
Data Availability Statement
The datasets generated for this study can be found in the Sequence Read Archive (SRA) BioProject PRJNA576909.
Author Contributions
GH conceived the aims, scope, and design of the study. GH and KJ collected, cultured, sampled animals for the study, and edited the manuscript. KJ extracted and prepared RNA for sequencing. SB extracted DNA, performed 5-mC quantification, executed downstream 5-mC analyses, and wrote the manuscript. SB and KJ performed transcriptomic analyses.
Funding
This work was funded in part by a grant from the U.S. National Science Foundation (NSF) through the U.S. Antarctic Program (PLR-1246202) to GH. KJ was supported by an NSF Graduate Research Fellowship during the time of the experiment. SB was supported by a Graduate Student Fellowship from the Department of Ecology, Evolution, and Marine Biology at UC Santa Barbara throughout his contribution to the study.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the late Dr. Umi Hoshijima for his invaluable assistance throughout the experiment, for collecting in situ pH data reported in this study, and for his consultation regarding these data. The authors acknowledge both Maddie Housh for her assistance in extracting DNA as well as the Crary Laboratory staff at McMurdo Station, Antarctica, for their support. Additionally, other members of the U.S. Antarctic Program, and of Lockheed’s Antarctic Support Corporation (ASC) supported field work for this study. The authors also acknowledge the Indiana University Pervasive Technology Institute for providing computational resources on the Carbonate cluster that have contributed to the results reported within this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00788/full#supplementary-material
References
Aina, R., Sgorbati, S., Santagostino, A., Labra, M., Ghiani, A., and Citterio, S. (2004). Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp. Physiol. Plant. 121, 472–480. doi: 10.1111/j.1399-3054.2004.00343.x
Andrews, S. (2018). FastQC: A Quality Control Tool for High Throughput Sequence Data. (Babraham Bioinformatics). Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed August 15, 2019).
Arnold, T. B., and Emerson, J. W. (2013). Discrete Goodness-of-Fit Tests. London: Comprehensive R Archive Network.
Baubec, T., Colombo, D. F., Wirbelauer, C., Schmidt, J., Burger, L., Krebs, A. R., et al. (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247. doi: 10.1038/nature14176
Bednaršek, N., Feely, R. A., Reum, J. C., Peterson, B., Menkel, J., Alin, S. R., et al. (2014a). Limacina helicina shell dissolution as an indicator of declining habitat suitability owing to ocean acidification in the California Current Ecosystem. Proc. R. Soc. B Biol. Sci. 281:20140123. doi: 10.1098/rspb.2014.0123
Bednaršek, N., Tarling, G. A., Bakker, D. C., Fielding, S., and Feely, R. A. (2014b). Dissolution dominating calcification process in polar pteropods close to the point of aragonite undersaturation. PLoS One 9:e109183. doi: 10.1371/journal.pone.0109183
Bednaršek, N., Tarling, G. A., Bakker, D. C. E., Fielding, S., Cohen, A., Kuzirian, A., et al. (2012a). Description and quantification of pteropod shell dissolution: a sensitive bioindicator of ocean acidification. Glob. Change Biol. 18, 2378–2388. doi: 10.1111/j.1365-2486.2012.02668.x
Bednaršek, N., Tarling, G. A., Bakker, D. C. E., Fielding, S., Jones, E. M., Venables, H. J., et al. (2012b). Extensive dissolution of live pteropods in the Southern Ocean. Nat. Geosci. 5, 881–885. doi: 10.1007/s00227-017-3261-3
Beers, J. M., and Jayasundara, N. (2015). Antarctic notothenioid fish: what are the future consequences of ‘losses’ and ‘gains’ acquired during long-term evolution at cold and stable temperatures? J. Exp. Biol. 218, 1834–1845. doi: 10.1242/jeb.116129
Beldade, P., Mateus, A. R., and Keller, R. A. (2011). Evolution and molecular mechanisms of adaptive developmental plasticity. Mol. Ecol. 20, 1347–1363. doi: 10.1111/j.1365-294X.2011.05016.x
Bernard, K. S., and Froneman, P. W. (2009). The sub-Antarctic euthecosome pteropod, Limacina retroversa: distribution patterns and trophic role. Deep Sea Res. Part I Oceanogr. Res. Pap. 56, 582–598. doi: 10.1016/j.dsr.2008.11.007
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bossdorf, O., Arcuri, D., Richards, C. L., and Pigliucci, M. (2010). Experimental alteration of DNA methylation affects the phenotypic plasticity of ecologically relevant traits in Arabidopsis thaliana. Evol. Ecol. 24, 541–553. doi: 10.1007/s10682-010-9372-7
Boysen-Ennen, E., Hagen, W., Hubold, G., and Piatowski, U. (1991). Zooplankton biomass in the ice-covered Weddell Sea, Antarctica. Mar. Biol. 111, 227–235. doi: 10.1007/bf01319704
Calosi, P., Bilton, D. T., and Spicer, J. I. (2008). Thermal tolerance, acclimatory capacity and vulnerability to global climate change. Biol. Lett. 4, 99–102. doi: 10.1098/rsbl.2007.0408
Chan, F., Barth, J. A., Blanchette, C. A., Byrne, R. H., Chavez, F., Cheriton, O., et al. (2017). Persistent spatial structuring of coastal ocean acidification in the California Current System. Sci. Rep. 7:2526. doi: 10.1038/s41598-017-02777-y
Chen, Y., Lun, A. T., and Smyth, G. K. (2016). From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 5:1438. doi: 10.12688/f1000research.8987.2
Chevin, L. M., Lande, R., and Mace, G. M. (2010). Adaptation, plasticity, and extinction in a changing environment: towards a predictive theory. PLoS Biol. 8:e1000357. doi: 10.1371/journal.pbio.1000357
Chown, S. L., Slabber, S., McGeouch, M., Janion, C., and Leinaas, H. P. (2007). Phenotypic plasticity mediates climate change responses among invasive and indigenous arthropods. Proc. R. Soc. B Biol. Sci. 274, 2531–2537. doi: 10.1098/rspb.2007.0772
Clark, M. S., Thorne, M. A. S., King, M., Hipperson, H., Hoffman, J. I., Peck, L. S., et al. (2018). Life in the intertidal: cellular responses, methylation and epigenetics. Funct. Ecol. 32, 1982–1994. doi: 10.1111/1365-2435.13077
Davis, B. E., Flynn, E. E., Miller, N. A., Nelson, F. A., Fangue, N. A., and Todgham, A. E. (2018). Antarctic emerald rockcod have the capacity to compensate for warming when uncoupled from CO2-acidification. Glob. Change Biol. 24, e655–e670. doi: 10.1111/gcb.13987
De Wit, P., Durland, E., Ventura, A., and Langdon, C. J. (2018). Gene expression correlated with delay in shell formation in larval Pacific oysters (Crassostrea gigas) exposed to experimental ocean acidification provides insights into shell formation mechanisms. BMC Genomics 19:160. doi: 10.1186/s12864-018-4519-y
Diaz-Freije, E., Gestal, C., Castellanos-Martinez, S., and Moran, P. (2014). The role of DNA methylation on Octopus vulgaris development and their perspectives. Front. Physiol. 5:62. doi: 10.3389/fphys.2014.00062
Dimond, J. L., and Roberts, S. B. (2016). Germline DNA methylation in reef corals: patterns and potential roles in response to environmental change. Mol. Ecol. 25, 1895–1904. doi: 10.1111/mec.13414
Dineshram, R., Wong, K. K. W., Xiao, S., Yu, Z., Qian, P. Y., and Thiyagarajan, V. (2012). Analysis of Pacific oyster larval proteome and its response to high-CO2. Mar. Pollut. Bull. 64, 2160–2167. doi: 10.1016/j.marpolbul.2012.07.043
Dixon, G., Bay, L. K., and Matz, M. V. (2014). Bimodal signatures of germline methylation are linked with gene expression plasticity in the coral Acropora millepora. BMC Genomics 15:1109. doi: 10.1186/1471-2164-15-1109
Dixon, G., Liao, Y., Bay, L. K., and Matz, M. V. (2018). Role of gene body methylation in acclimatization and adaptation in a basal metazoan. Proc. Natl. Acad. Sci. U.S.A. 115, 13342–13346. doi: 10.1073/pnas.1813749115
Donelson, J. M., Salinas, S., Munday, P. L., and Shama, L. N. S. (2018). Transgenerational plasticity and climate change experiments: where do we go from here? Glob. Change Biol. 24, 13–34. doi: 10.1111/gcb.13903
Duarte, B., Martins, I., Rosa, R., Matos, A. R., Roleda, M. Y., Reusch, T. B. H., et al. (2018). Climate change impacts on seagrass meadows and macroalgal forests: an integrative perspective on acclimation and adaptation potential. Front. Mar. Sci. 5:190. doi: 10.3389/fmars.2018.00190
Eirin-Lopez, J. M., and Putnam, H. M. (2019). Marine environmental epigenetics. Annu. Rev. Mar. Sci. 11, 335–368. doi: 10.1146/annurev-marine-010318-095114
Enzor, L. A., Hunter, E. M., and Place, S. P. (2017). The effects of elevated temperature and ocean acidification on the metabolic pathways of notothenioid fish. Conserv. Physiol. 5:cox019. doi: 10.1093/conphys/cox019
Enzor, L. A., and Place, S. P. (2014). Is warmer better? Decreased oxidative damage in notothenioid fish after long-term acclimation to multiple stressors. J. Exp. Biol. 217, 3301–3310. doi: 10.1242/jeb.108431
Fellous, A., Favrel, P., and Riviere, G. (2015). Temperature influences histone methylation and mRNA expression of the Jmj-C histone-demethylase orthologues during the early development of the oyster Crassostrea gigas. Mar. Genomics 19, 23–30. doi: 10.1016/j.margen.2014.09.002
Fellous, A., Lefranc, L., Jouaux, A., Goux, D., Favrel, P., and Riviere, G. (2019). Histone methylation participates in gene expression control during the early development of the Pacific oyster Crassostrea gigas. Genes 10:E695. doi: 10.3390/genes10090695
Foster, B. A., Cargill, J. M., and Montgomery, J. C. (1987). Planktivory in Pagothenia borchgrevinki (Pisces: Nototheniidae) in McMurdo Sound, Antarctica. Polar Biol. 8, 49–54. doi: 10.1007/bf00297164
Garcia-Fernandez, P., Garcia-Souto, D., Almansa, E., Moran, P., and Gestal, C. (2017). Epigenetic DNA methylation mediating Octopus vulgaris early development: effect of essential fatty acids enriched diet. Front. Physiol. 8:292. doi: 10.3389/fphys.2017.00292
Gardner, J., Manno, C., Bakker, D. C. E., Peck, V. L., and Tarling, G. A. (2018). Southern Ocean pteropods at risk from ocean warming and acidification. Mar. Biol. 165:8. doi: 10.1007/s00227-017-3261-3
Gavery, M. R., and Roberts, S. B. (2013). Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 1:e215. doi: 10.7717/peerj.215
Gavery, M. R., and Roberts, S. B. (2014). A context dependent role for DNA methylation in bivalves. Brief. Funct. Genomics 13, 217–222. doi: 10.1093/bfgp/elt054
Gibson, J. A. E., and Trull, T. W. (1999). Annual cycle of fCO2 under sea-ice and in open water in Prydz Bay, East Antarctica. Mar. Chem. 66, 187–200. doi: 10.1016/s0304-4203(99)00040-7
Gonzalez-Romero, R., Suarez-Ulloa, V., Rodriguez-Casariego, J., Garcia-Souto, D., Diaz, G., Smith, A., et al. (2017). Effects of Florida Red Tides on histone variant expression and DNA methylation in the Eastern oyster Crassostrea virginica. Aquat. Toxicol. 186, 196–204. doi: 10.1016/j.aquatox.2017.03.006
Hahn, M. A., Wu, X., Li, A. X., Hahn, T., and Pfeifer, G. P. (2011). Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One 6:e18844. doi: 10.1371/journal.pone.0018844
Harris, K. E., DeGrandpre, M. D., and Hales, B. (2013). Aragonite saturation state dynamics in a coastal upwelling zone. Geophys. Res. Lett. 40, 2720–2725. doi: 10.1002/grl.50460
Hauri, C., Friedrich, T., and Timmermann, A. (2015). Abrupt onset and prolongation of aragonite undersaturation events in the Southern Ocean. Nat. Clim. Change 6, 172–176. doi: 10.1038/nclimate2844
Hawkins, S. J., Evans, A. J., Dale, A. C., Firth, L. B., and Smith, I. P. (2018). Antarctic marine biodiversity: adaptations, environments and responses to change. Oceanogr. Mar. Biol. 56, 105–236. doi: 10.1201/9780429454455-3
Hennige, S. J., Smith, D. J., Walsh, S.-J., McGinley, M. P., Warner, M. E., and Suggett, D. J. (2010). Acclimation and adaptation of scleractinian coral communities along environmental gradients within an Indonesian reef system. J. Exp. Mar. Biol. Ecol. 391, 143–152. doi: 10.1016/j.jembe.2010.06.019
Ho, J., Tumkaya, T., Aryal, S., Choi, H., and Claridge-Chang, A. (2019). Moving beyond P values: data analysis with estimation graphics. Nat. Methods 16, 565–566. doi: 10.1038/s41592-019-0470-3
Hoegh-Guldberg, O., and Bruno, J. F. (2010). The impact of climate change on the world’s marine ecosystems. Science 328, 1523–1528. doi: 10.1126/science.1189930
Hofmann, G. E. (2017). Ecological epigenetics in marine metazoans. Front. Mar. Sci. 4:4. doi: 10.3389/fmars.2017.00004
Hoshijima, U., Wong, J. M., and Hofmann, G. E. (2017). Additive effects of pCO2 and temperature on respiration rates of the Antarctic pteropod Limacina helicina antarctica. Conserv. Physiol. 5:cox064. doi: 10.1093/conphys/cox064
Hunt, B., Strugnell, J., Bednarsek, N., Linse, K., Nelson, R. J., Pakhomov, E., et al. (2010). Poles apart: the “bipolar” pteropod species Limacina helicina is genetically distinct between the Arctic and Antarctic oceans. PLoS One 5:e9835. doi: 10.1371/journal.pone.0009835
Hunt, B. P. V., Pakhomov, E. A., Hosie, G. W., Siegel, V., Ward, P., and Bernard, K. (2008). Pteropods in Southern Ocean ecosystems. Prog. Oceanogr. 78, 193–221. doi: 10.1016/j.pocean.2008.06.001
Huth, T. J., and Place, S. P. (2016a). RNA-seq reveals a diminished acclimation response to the combined effects of ocean acidification and elevated seawater temperature in Pagothenia borchgrevinki. Mar. Genomics 28, 87–97. doi: 10.1016/j.margen.2016.02.004
Huth, T. J., and Place, S. P. (2016b). Transcriptome wide analyses reveal a sustained cellular stress response in the gill tissue of Trematomus bernacchii after acclimation to multiple stressors. BMC Genomics 17:127. doi: 10.1186/s12864-016-2454-3
IPCC (2013). Climate Change 2013: the Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, eds T. F. Stocker, D. Qin, G. K. Plattner, M. Tignor, S. K. Allen, J. Boschung, et al. (Cambridge: Cambridge University Press).
IPCC (2019). IPCC Special Report on the Ocean and Cryosphere in a Changing Climate, eds H. O. Pörtner, D. C. Roberts, V. Masson-Delmotte, P. Zhai, M. Tignor, E. Poloczanska, et al. (Geneva: IPCC).
Jeong, H., Wu, X., Smith, B., and Yi, S. V. (2018). Genomic landscape of methylation islands in hymenopteran insects. Genome Biol. Evol. 10, 2766–2776. doi: 10.1093/gbe/evy203
Johnson, K. M., and Hofmann, G. E. (2016). A transcriptome resource for the Antarctic pteropod Limacina helicina antarctica. Mar. Genomics 28, 25–28. doi: 10.1016/j.margen.2016.04.002
Johnson, K. M., and Hofmann, G. E. (2017). Transcriptomic response of the Antarctic pteropod Limacina helicina antarctica to ocean acidification. BMC Genomics 18:812. doi: 10.1186/s12864-017-4161-0
Kapsenberg, L., Kelley, A. L., Shaw, E. C., Martz, T. R., and Hofmann, G. E. (2015). Near-shore Antarctic pH variability has implications for the design of ocean acidification experiments. Sci. Rep. 5:9638.
Kelly, M. (2019). Adaptation to climate change through genetic accommodation and assimilation of plastic phenotypes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374:20180176. doi: 10.1098/rstb.2018.0176
Kenkel, C. D., Moya, A., Strahl, J., Humphrey, C., and Bay, L. K. (2017). Functional genomic analysis of corals from natural CO2-seeps reveals core molecular responses involved in acclimatization to ocean acidification. Glob. Change Biol. 24, 158–171. doi: 10.1111/gcb.13833
Kobayashi, H., Sakurai, T., Imai, M., Takahashi, N., Fukuda, A., Yayoi, O., et al. (2012). Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 8:e1002440. doi: 10.1371/journal.pgen.1002440
Koh, H. Y., Lee, J. H., Han, S. J., Park, H., Shin, S. C., and Lee, S. G. (2015). A transcriptomic analysis of the response of the arctic pteropod Limacina helicina to carbon dioxide-driven seawater acidification. Polar Biol. 38, 1727–1740. doi: 10.1007/s00300-015-1738-4
Kriefall, N. G., Pechenik, J. A., Pires, A., and Davies, S. W. (2018). Resilience of Atlantic slippersnail Crepidula fornicata larvae in the face of severe coastal acidification. Front. Mar. Sci. 5:312. doi: 10.3389/fmars.2018.00312
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, S., Papale, L. A., Zhang, Q., Madrid, A., Chen, L., Chopra, P., et al. (2016). Genome-wide alterations in hippocampal 5-hydroxymethylcytosine links plasticity genes to acute stress. Neurobiol. Dis. 86, 99–108. doi: 10.1016/j.nbd.2015.11.010
Li, Y., Liew, Y. J., Cui, G., Cziesielski, M. J., Zahran, N., Michell, C. T., et al. (2018). DNA methylation regulates transcriptional homeostasis of algal endosymbiosis in the coral model Aiptasia. Sci. Adv. 4:eaat2142. doi: 10.1126/sciadv.aat2142
Lian, S., He, Y., Li, X., Zhao, B., Hou, R., Hu, X., et al. (2015). Changes in global DNA methylation intensity and DNMT1 transcription during the aging process of scallop Chlamys farreri. J. Ocean Univ. China 14, 685–690. doi: 10.1007/s11802-015-2507-2
Liew, Y. J., Didier, Z., Li, Y., Tamubtté, E., Venn, A. A., Michell, C. T., et al. (2018). Epigenome-associated phenotypic acclimatization to ocean acidification in a reef-building coral. Sci. Adv. 4:eaar8028. doi: 10.1126/sciadv.aar8028
Luttmer, R., Spijkerman, A. M., Kok, R. M., Jakobs, C., Blom, H. J., Serne, E. H., et al. (2013). Metabolic syndrome components are associated with DNA hypomethylation. Obes. Res. Clin. Pract. 7, 106–115. doi: 10.1016/j.orcp.2012.06.001
Maas, A. E., Lawson, G. L., Bergan, A. J., and Tarrant, A. M. (2018). Exposure to CO2 influences metabolism, calcification and gene expression of the thecosome pteropod Limacina retroversa. J. Exp. Biol. 221:jeb164400. doi: 10.1242/jeb.164400
Maas, A. E., Lawson, G. L., and Tarrant, A. M. (2015). Transcriptome-wide analysis of the response of the thecosome pteropod Clio pyramidata to short-term CO2 exposure. Comp. Biochem. Physiol. Part D Genomics Proteomics 16, 1–9. doi: 10.1016/j.cbd.2015.06.002
Manno, C., Bednaršek, N., Tarling, G. A., Peck, V. L., Comeau, S., Adhikari, D., et al. (2017). Shelled pteropods in peril: assessing vulnerability in a high CO2 ocean. Earth Sci. Rev. 169, 132–145. doi: 10.1016/j.earscirev.2017.04.005
Marsh, A. G., and Pasqualone, A. A. (2014). DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai. Front. Physiol. 5:173. doi: 10.3389/fphys.2014.00173
McNeil, B. I., and Matear, R. J. (2008). Southern Ocean acidification: a tipping point at 450-ppm atmospheric CO2. Proc. Natl. Acad. Sci. U.S.A. 105, 18860–18864. doi: 10.1073/pnas.0806318105
McNeil, B. I., Tagliabue, A., and Sweeney, C. (2010). A multi-decadal delay in the onset of corrosive ‘acidified’ waters in the Ross Sea of Antarctica due to strong air-sea CO2 disequilibrium. Geophys. Res. Lett. 37:L19607.
Melzner, F., Mark, F. C., Seibel, B. A., and Tomanek, L. (2020). Ocean acidification and coastal marine invertebrates: tracking CO2 effects from seawater to the cell. Annu. Rev. Mar. Sci. 12, 1–25.
Morley, S. A., Berman, J., Barnes, D. K. A., de Juan Carbonell, C., Downey, R. V., and Peck, L. S. (2016). Extreme phenotypic plasticity in metabolic physiology of Antarctic demosponges. Front. Ecol. Evol. 3:157. doi: 10.3389/fevo.2015.00157
Moya, A., Howes, E. L., Lacoue-Labarthe, T., Foret, S., Hanna, B., Medina, M., et al. (2016). Near-future pH conditions severely impact calcification, metabolism and the nervous system in the pteropod Heliconoides inflatus. Glob. Change Biol. 22, 3888–3900. doi: 10.1111/gcb.13350
Nanty, L., Carbajosa, G., Heap, G. A., Ratnieks, F., van Heel, D. A., Down, T. A., et al. (2011). Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 21, 1841–1850. doi: 10.1101/gr.121640.111
Negrete-García, G., Lovenduski, N. S., Hauri, C., Krumhardt, K. M., and Lauvset, S. K. (2019). Sudden emergence of a shallow aragonite saturation horizon in the Southern Ocean. Nat. Clim. Change 9, 313–317. doi: 10.1038/s41558-019-0418-8
Neri, F., Rapelli, S., Krepelova, A., Incarnato, D., Parlato, C., Basile, G., et al. (2017). Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77. doi: 10.1038/nature21373
Nicotra, A. B., Atkin, O. K., Bonser, S. P., Davidson, A. M., Finnegan, E. J., Mathesius, U., et al. (2010). Plant phenotypic plasticity in a changing climate. Trends Plant Sci. 15, 684–692. doi: 10.1016/j.tplants.2010.09.008
O’Donnell, M. J., Todgham, A. E., Sewell, M. A., Hammond, L. M., Ruggiero, K., Fangue, N. A., et al. (2010). Ocean acidification alters skeletogenesis and gene expression in larval sea urchins. Mar. Ecol. Prog. Ser. 398, 157–171. doi: 10.3354/meps08346
Oliver, E. C. J., Donat, M. G., Burrows, M. T., Moore, P. J., Smale, D. A., Alexander, L. V., et al. (2018). Longer and more frequent marine heatwaves over the past century. Nat. Commun. 9:1324. doi: 10.1038/s41467-018-03732-9
Olson, C. E., and Roberts, S. B. (2014). Genome-wide profiling of DNA methylation and gene expression in Crassostrea gigas male gametes. Front. Physiol. 5:224. doi: 10.3389/fphys.2014.00224
Pacis, A., Mailhot-Leonard, F., Tailleux, L., Randolph, H. E., Yotova, V., Dumaine, A., et al. (2019). Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 116, 6938–6943. doi: 10.1073/pnas.1814700116
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Pavet, V., Quintero, C., Cecchini, N. M., Rosa, A. L., and Alvarez, M. E. (2006). Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by Pseudomonas syringae. Phytopathology 19, 577–587. doi: 10.1094/mpmi-19-0577
Peck, L. S., Morley, S. A., and Clark, M. S. (2010). Poor acclimation capacities in Antarctic marine ectotherms. Mar. Biol. 157, 2051–2059. doi: 10.1007/s00227-010-1473-x
Peck, L. S., Morley, S. A., Richard, J., and Clark, M. S. (2014). Acclimation and thermal tolerance in Antarctic marine ectotherms. J. Exp. Biol. 217, 16–22. doi: 10.1242/jeb.089946
Peck, L. S., Webb, K. E., and Bailey, D. M. (2004). Extreme sensitivity of biological function to temperature in Antarctic marine species. Funct. Ecol. 18, 625–630. doi: 10.1111/j.0269-8463.2004.00903.x
Pogribny, I. P., Ross, S. A., Wise, C., Pogribna, M., Jones, E. A., Tryndyak, V. P., et al. (2006). Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat. Res. 593, 80–87. doi: 10.1016/j.mrfmmm.2005.06.028
Putnam, H. M., Davidson, J. M., and Gates, R. D. (2016). Ocean acidification influences host DNA methylation and phenotypic plasticity in environmentally susceptible corals. Evol. Appl. 9, 1165–1178. doi: 10.1111/eva.12408
Reed, A. J., and Thatje, S. (2015). Long-term acclimation and potential scope for thermal resilience in Southern Ocean bivalves. Mar. Biol. 162, 2217–2224. doi: 10.1007/s00227-015-2752-3
Riviere, G., He, Y., Tecchio, S., Crowell, E., Gras, M., Sourdaine, P., et al. (2017). Dynamics of DNA methylomes underlie oyster development. PLoS Genet. 13:e1006807. doi: 10.1371/journal.pgen.1006807
Riviere, G., Wu, G. C., Fellous, A., Goux, D., Sourdaine, P., and Favrel, P. (2013). DNA methylation is crucial for the early development in the oyster C. gigas. Mar. Biotechnol. 15, 739–753. doi: 10.1007/s10126-013-9523-2
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Robinson, N. A., Johnsen, H., Moghadam, H., Andersen, O., and Tveiten, H. (2019). Early developmental stress affects subsequent gene expression response to an acute stress in Atlantic salmon: an approach for creating robust fish for aquaculture? G3 9, 1597–1611. doi: 10.1534/g3.119.400152
Rodrigues, G. M. Jr., Toffoli, L. V., Manfredo, M. H., Francis-Oliveira, J., Silva, A. S., Raquel, H. A., et al. (2015). Acute stress affects the global DNA methylation profile in rat brain: modulation by physical exercise. Behav. Brain Res. 279, 123–128. doi: 10.1016/j.bbr.2014.11.023
Rodriguez-Casariego, J. A., Ladd, M. C., Shantz, A. A., Lopes, C., Cheema, M. S., Kim, B., et al. (2018). Coral epigenetic responses to nutrient stress: histone H2A. X phosphorylation dynamics and DNA methylation in the staghorn coral Acropora cervicornis. Ecol. Evol. 8, 12193–12207. doi: 10.1002/ece3.4678
Rondon, R., Grunau, C., Fallet, M., Charlemagne, N., Sussarellu, R., Chaparro, C., et al. (2017). Effects of a parental exposure to diuron on Pacific oyster spat methylome. Environ. Epigenet. 3:dvx004. doi: 10.1093/eep/dvx004
Rusiecki, J. A., Baccarelli, A., Bollati, V., Tarantini, L., Moore, L. E., and Bonefeld-Jorgensen, E. C. (2008). Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ. Health Perspect. 116, 1547–1552. doi: 10.1289/ehp.11338
Sarda, S., Zeng, J., Hunt, B. G., and Yi, S. V. (2012). The evolution of invertebrate gene body methylation. Mol. Biol. Evol. 29, 1907–1916. doi: 10.1093/molbev/mss062
Schubeler, D. (2015). Function and information content of DNA methylation. Nature 517, 321–326. doi: 10.1038/nature14192
Secco, D., Wang, C., Shou, H., Schultz, M. D., Chiarenza, S., Nussaume, L., et al. (2015). Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 4:e09343. doi: 10.7554/eLife.09343
Seebacher, F., Davison, W., Lowe, C. J., and Franklin, C. E. (2005). A falsification of the thermal specialization paradigm: compensation for elevated temperatures in Antarctic fishes. Biol. Lett. 1, 151–154. doi: 10.1098/rsbl.2004.0280
Seibel, B. A., Maas, A. E., and Dierssen, H. M. (2012). Energetic plasticity underlies a variable response to ocean acidification in the pteropod, Limacina helicina antarctica. PLoS One 7:e30464. doi: 10.1371/journal.pone.0030464
Steinberg, D. K., Ruck, K. E., Gleiber, M. R., Garzio, L. M., Cope, J. S., Bernard, K. S., et al. (2015). Long-term (1993–2013) changes in macrozooplankton off the Western Antarctic Peninsula. Deep Sea Res. Part I Oceanogr. Res. Pap. 101, 54–70. doi: 10.1016/j.dsr.2015.02.009
Stewart, C. A., Welch, V., Plale, B., Fox, G., Pierce, M., and Sterling, T. (2017). Indiana University Pervasive Technology Institute. Bloomington: Indiana University.
Strader, M. E., Wong, J. M., Kozal, L. C., Leach, T. S., and Hofmann, G. E. (2019). Parental environments alter DNA methylation in offspring of the purple sea urchin, Strongylocentrotus purpuratus. J. Exp. Mar. Biol. Ecol. 517, 54–64. doi: 10.1016/j.jembe.2019.03.002
Suarez-Ulloa, V., Rivera-Casas, C., and Michel, M. (2019). Seasonal DNA methylation variation in the flat tree oyster Isognomon alatus from a mangrove ecosystem in North Biscayne Bay, Florida. J. Shellfish Res. 38, 79–88.
Taylor, D. L., Jackson, A. U., Narisu, N., Hemani, G., Erdos, M. R., Chines, P. S., et al. (2019). Integrative analysis of gene expression, DNA methylation, physiological traits, and genetic variation in human skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 116, 10883–10888. doi: 10.1073/pnas.1814263116
Thibodeau, P. S., Steinberg, D. K., Stammerjohn, S. E., and Hauri, C. (2019). Environmental controls on pteropod biogeography along the Western Antarctic Peninsula. Limnol. Oceanogr. 64, S240–S256.
Todgham, A. E., and Hofmann, G. E. (2009). Transcriptomic response of sea urchin larvae Strongylocentrotus purpuratus to CO2-driven seawater acidification. J. Exp. Biol. 212, 2579–2594. doi: 10.1242/jeb.032540
True, H. L., Berlin, I., and Lindquist, S. L. (2004). Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 431, 184–187. doi: 10.1038/nature02885
Wang, X., Li, Q., Lian, J., Li, L., Jin, L., Cai, H., et al. (2014). Genome-wide and single-base resolution DNA methylomes of the Pacific oyster Crassostrea gigas provide insight into the evolution of invertebrate CpG methylation. BMC Genomics 15:1119. doi: 10.1186/1471-2164-15-1119
Wheeler, R., and Torchiano, M. (2016). Permutation Tests for Linear Models. London: Comprehensive R Archive Network.
Wong, J. M., Kozal, L. C., Leach, T. S., Hoshijima, U., and Hofmann, G. E. (2019). Transgenerational effects in an ecological context: conditioning of adult sea urchins to upwelling conditions alters maternal provisioning and progeny phenotype. J. Exp. Mar. Biol. Ecol. 517, 65–77. doi: 10.1016/j.jembe.2019.04.006
Worden, A. (2009). DNA Extraction - CTAB Method. Moss Landing, CA: Monterey Bay Aquarium Research Institute.
Wright, R. M., Aglyamova, G. V., Meyer, E., and Matz, M. V. (2015). Gene expression associated with white syndromes in a reef building coral, Acropora hyacinthus. BMC Genomics 16:371. doi: 10.1186/s12864-015-1540-2
Xiu, Y., Shao, C., Zhu, Y., Li, Y., Gan, T., Xu, W., et al. (2019). Differences in DNA methylation between disease-resistant and disease-susceptible Chinese tongue sole (Cynoglossus semilaevis) families. Front. Genet. 10:847. doi: 10.3389/fgene.2019.00847
Zemach, A., McDaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Zhang, H., Lang, Z., and Zhu, J. K. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506. doi: 10.1038/s41580-018-0016-z
Zhang, Y. Y., Fischer, M., Colot, V., and Bossdorf, O. (2013). Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol. 197, 314–322. doi: 10.1111/nph.12010
Keywords: epigenetics, DNA methylation, pteropod, ocean acidification, gene expression, Limacina helicina antarctica
Citation: Bogan SN, Johnson KM and Hofmann GE (2020) Changes in Genome-Wide Methylation and Gene Expression in Response to Future pCO2 Extremes in the Antarctic Pteropod Limacina helicina antarctica. Front. Mar. Sci. 6:788. doi: 10.3389/fmars.2019.00788
Received: 11 October 2019; Accepted: 06 December 2019;
Published: 22 January 2020.
Edited by:
Jose M. Eirin-Lopez, Florida International University, United StatesReviewed by:
Yi Jin Liew, Commonwealth Scientific and Industrial Research Organisation (CSIRO), AustraliaAlexandre Fellous, Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research (AWI), Germany
Copyright © 2020 Bogan, Johnson and Hofmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gretchen E. Hofmann, aG9mbWFubkB1Y3NiLmVkdQ==