 Guowang Zhao1†
Guowang Zhao1† Xinghai Li2†Ying Zhang3,4†Xingzi Wang5Li Deng6Juan Xu3Shumei Jin7
Xinghai Li2†Ying Zhang3,4†Xingzi Wang5Li Deng6Juan Xu3Shumei Jin7 Zan Zuo3,4Linting Xun3,4Mei Luo3,4
Zan Zuo3,4Linting Xun3,4Mei Luo3,4 Fan Yang8*
Fan Yang8* Jialong Qi3,4,9,10*Ping Fu1*
Jialong Qi3,4,9,10*Ping Fu1*- 1Department of Rheumatology and Clinical Immunology, The Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, China
- 2Department of Minimal Invasive Intervention Radiology, Ganzhou People’s Hospital, Ganzhou, Jiangxi, China
- 3Yunnan Digestive Endoscopy Clinical Medical Center, Department of Gastroenterology, The First People’s Hospital of Yunnan Province, Affiliated by Kunming University of Science and Technology, Kunming, Yunnan, China
- 4School of Medicine, The First People’s Hospital of Yunnan Province, Kunming University of Science and Technology, Kunming, Yunnan, China
- 5Department of Nephrology, Yueyang Central Hospital, Yueyang, Hunan, China
- 6Department of Internal Medicine, Community Health Service Station of Dian Mian Avenue, Kunming, Yunnan, China
- 7Yunnan Institute of Food and Drug Supervision and Control, Medical Products Administration of Yunnan Province, Kunming, Yunnan, China
- 8School of Medicine, Kunming University of Science and Technology, Kunming, Yunnan, China
- 9Yunnan Provincial Key Laboratory of Clinical Virology, The First People’s Hospital of Yunnan Province, Kunming, Yunnan, China
- 10Yunnan Provincial Key Laboratory of Birth Defects and Genetic Diseases, First People’s Hospital of Yunnan Province, Kunming, Yunnan, China
Systemic lupus erythematosus (SLE) is a chronic inflammatory and autoimmune disease with multiple tissue damage. However, the pathology remains elusive, and effective treatments are lacking. Multiple types of programmed cell death (PCD) implicated in SLE progression have recently been identified. Although ferroptosis, an iron-dependent form of cell death, has numerous pathophysiological features similar to those of SLE, such as intracellular iron accumulation, mitochondrial dysfunction, lipid metabolism disorders and concentration of damage associated-molecular patterns (DAMPs), only a few reports have demonstrated that ferroptosis is involved in SLE progression and that the role of ferroptosis in SLE pathogenesis continues to be neglected. Therefore, this review elucidates the potential intricate relationship between SLE and ferroptosis to provide a reliable theoretical basis for further research on ferroptosis in the pathogenesis of SLE.
1 Introduction
Systemic lupus erythematosus (SLE) is a chronic systemic autoimmune disease characterized by inflammation and immune-mediated injury to various organs and systems. Approximately 3.4 million people are affected by SLE, with approximately 400,000 people diagnosed with SLE each year worldwide (1, 2). Despite the diversity of clinical manifestations and the extremely complex pathogenesis in SLE, high levels of interferons (IFNs) and autoantibodies are common features (3), which also are primarily the result of cell death. In SLE, excessive cell death and impaired clearance machinery contribute to the continuous release of intracellular nucleic acids and their complexes into the extracellular space, thereby promoting responses by autoreactive B cells and IFN-responsive mechanisms (4). Interestingly, some prevalent features of SLE, including iron overload, mitochondrial dysfunction, lipid metabolism disorders and damage associated molecular pattern (DAMP) concentrations, are complexly associated with cell death, especially ferroptosis (5, 6).
Ferroptosis is an iron-dependent immunogenic form of cell death that is distinct from cell death mediated by perforin rupture (7). The occurrence of ferroptosis is attributed mainly to an imbalance in intracellular oxidation and antioxidant mechanisms, which triggers a cascade of hydrogen peroxide-lipid reactions and mediates the rupture of the cell membrane. Its mechanism mainly includes lipid peroxidation (mainly involving iron homeostasis and mitochondrial dysfunction) and antioxidant system disorders [mainly involving the cystine/glutamate transport system (xCT), glutathione (GSH)-glutathione peroxidase 4 (GPX4)] (5). Ferroptosis is induced by many factors, such as excessive DAMPs [IFN-α, IFN-γ, high-mobility group box 1 (HMGB1)] (8), ionizing radiation (9), the accumulation of iron, and increases in free radicals and oxidized lipids (10). Although the physiological role of ferroptosis is still unclear, its pathological role in a variety of acute and chronic diseases has been widely reported (11, 12), and it is potentially closely related to the pathogenesis of SLE (13, 14).
To date, the pathogenesis of SLE has not been classified. PCD has been shown outstanding role in SLE pathogenesis. Ferroptosis is a novel form of PCD, which exhibits several pathophysiological features reminiscent of those seen in SLE, including intracellular iron accumulation, mitochondrial dysfunction, lipid metabolism disorders, and regulation by DAMPs (Table 1). These similarities warrant further exploration of the potential interplay between ferroptosis and SLE pathogenesis. Although ferroptosis has been reported to play a role in SLE, the relationship between ferroptosis and SLE remains unclear. Therefore, this review discusses those parallel features between ferroptosis and SLE promote further research on ferroptosis in the pathogenesis of SLE.
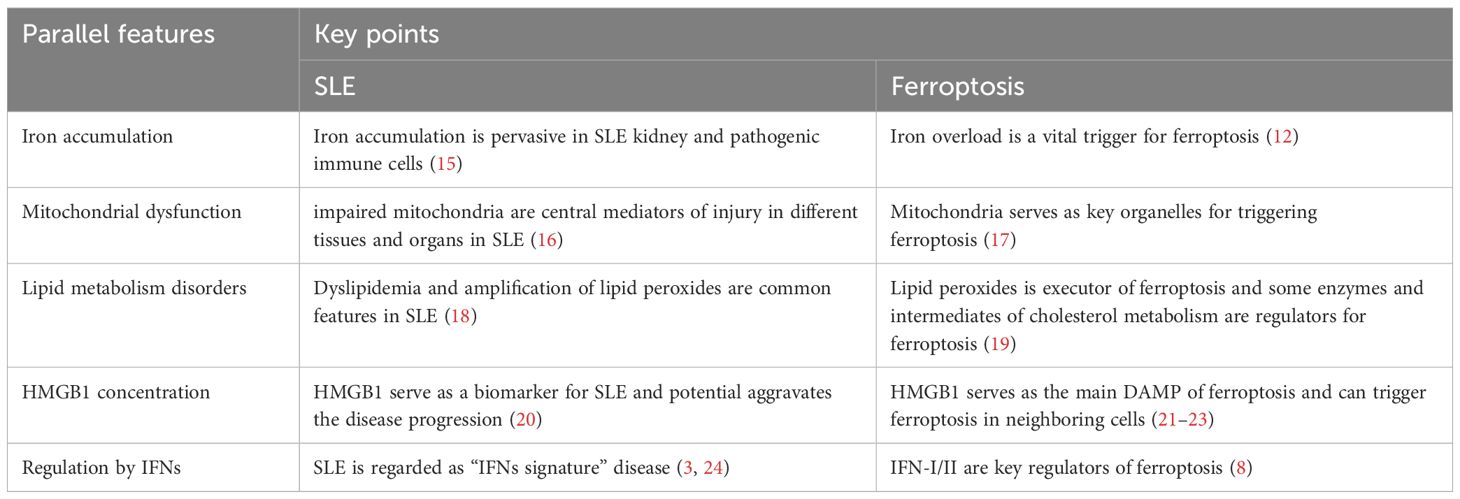
Table 1. Parallel features in SLE and ferroptosis.
2 Primary regulation in ferroptosis
Current evidence demonstrated intracellular oxidation primary related to some intracellular alterations, encompassing iron accumulation, mitochondrial dysfunction and lipid metabolism disorder (5). And antioxidant systems related to defend ferroptosis mainly including the xCT-GPX4, ferroptosis suppressor protein 1 (FSP1)-coenzyme Q10 (CoQ10), Guanosine triphosphate cyclohydrolase 1 (GCH1)-tetrahydrobiopterin (BH4) and dihydroorotate dehydrogenase (DHODH)-dihydroubiquione (CoQH2) systems (5, 25). Conceivably, biological processes related to the homeostasis of oxidation-reduction are peculiarly prone to regulate ferroptosis, such as lipogenesis (26, 27), autophagy (mainly including ferritinophagy, lipophagy and mitophagy) (28, 29), and the tricarboxylic acid (TCA) cycle (17). Moreover, plenty of and metabolic molecules are crucial for primary ferroptostic suppressors, such as: isopentenylation is required for the synthesis of selenoenzymes including GPX4 and ubiquinone (also known as coenzyme Q) is a major substrate of FSP1, both them is metabolites of mevalonate pathway (30). Additionally, multiple DAMPs can regulate ferroptosis, such as: IFNs can regulate solute carrier family 7a member 11 (SLC7A11, subunit of xCT) and GPX4 expression by activating JAK-STAT1 signaling (8); HMGB1 can induce ferroptosis by promoting autophagy and iron accumulation (31, 32); Mitochondrial DNA (mtDNA) performs ability to trigger ferroptosis by activating the cyclic GMP-AMP receptor stimulator of interferon genes (STING) (33).
3 Similar features in SLE and ferroptosis
3.1 Iron accumulation
Iron is one of the essential trace elements in the human body and is crucial for maintaining various biological functions, such as transporting oxygen, participating in the production of energy in the respiratory chain, and being involved in key biological reactions as a key component of various enzymes (34). However, iron accumulation can also mediate a series of pathophysiological processes, such as reactive oxygen species (ROS) production, lipid peroxide production and mitochondrial dysfunction, thereby promoting disease progression, such as SLE (35). Iron accumulation has been detected in the kidneys of both SLE patients and lupus mice (36, 37), and multiple molecules related to iron metabolism [such as ferritin, transferrin, ceruloplasmin, and neutrophil gelatinase-associated lipid carrier protein (NGAL)] have been identified as biomarkers of SLE and/or LN, which are associated with disease activity and/or renal involvement (38–43). Additionally, the excessive intake of iron exacerbates SLE progression (44–46), whereas iron chelation can alleviate the disease (37, 47).
Kidney is major damaged organ in SLE and main organ attacked by iron overload because it is site of iron filtration and reabsorption. Over transferrin bound (TBI) and non-transferrin bound iron (NTBI) can overwhelm the heavy chain ferritin (FtH) capacity, which lead to release of labile iron and render proximal tubular epithelial cells (PTEC) susceptible to iron oxidant damage, especial in distal tubular epithelial cells (DTEC) with lack iron storage (light and heavy chain ferritin) and export protein (ferroportin) (15). Moreover, this damage can be worse under some pathological condition like LN with glomerular injury resulting in an increased leakage of TBI and NTBI (48). Additionally, SLE is a typical autoimmune disease, and the overactivation of CD4+ T lymphocytes is a feature of SLE (28). In SLE, CD4+ T cells are over differentiated into pathogenic Th1 and Th17 cells, whereas Treg cells are suppressed (49, 50). Interestingly, iron is crucial for regulating the activation, proliferation and differentiation of CD4+ T lymphocytes (51). Some reports have demonstrated that the iron concentration in CD4+ T cells is significantly greater in SLE patients than in healthy controls (52, 53). CD71 (also referred to as ferritin receptor TfR1, which mediates iron endocytosis) on the surface of CD4+ T cells is essential for their activation and proliferation and promotes their differentiation into Th17 cells (54–56). Moreover, Th17 cells in SLE patients express high levels of CD71, which is positively correlated with disease activity (56). Similarly, follicular helper T (Tfh) cells, as a specific subset of CD4+ T cells, contribute to the pathogenesis of SLE by promoting the maturation of germinal center B cells and the production of antibodies (57, 58). However, the accumulation of iron in CD4+ T cells in SLE can promote their differentiation into Tfh cells and aggravate the progression of SLE (53). Moreover, Treg cells can restrict CD4+ T cell overactivation and its dysfunction is involved in SLE pathogenesis (59–61). Gao XF et al. demonstrated iron deficiency could alleviate pristane-induced lupus progression by promoting Treg cell expansion (62). And Feng P et al. verified iron overload leaded to systemic autoimmune disorders by aggravating Treg cell death (63). These findings verify that iron overload is involved in the pathogenesis of SLE, but whether it occurs through ferroptosis is not clear.
Iron overload is also conducive to ferroptosis. Too much iron intake, too little iron excretion, or impaired iron metabolism can contribute to iron overload. Physiologically, there are several ways to reduce the level of intracellular iron when the level of intracellular iron is excessive: i. iron-responsive proteins/iron-responsive elements (IRPs/IREs) hamper the transfer of iron by disrupting the stability of transferrin receptor mRNA while promoting the transcription and synthesis of ferritin to enhance the binding of free iron (64–66); ii. increased expression of hepcidin promotes the expression of iron transfer protein (ferroportin), which promotes iron outward transport (67–69); and iii. ferritin is targeted to lysosomes by nuclear receptor coactivator 4 (NCOA4)-mediated autophagosomes for degradation (often referred to as ferritinophagy) (70, 71). Iron overload is a vital trigger for ferroptosis. i.Iron-mediated Fenton reactions are necessary for ferroptosis and can facilitate the production of phospholipid hydroperoxides (PLOOH, a biomarker of ferroptosis). ii.Key enzymes that trigger lipid peroxidation [lipoxygenases (LOXs) and cytochrome P450 oxidoreductases (PORs)] require iron catalysis. iii.Iron is required for numerous redox-based metabolic processes and is a major source of intracellular ROS (11, 34). iv.Iron chelating agents not only prevent ferroptosis but also reduce the production of lipid peroxides (72–75). Feng P et al. reported that iron overload in Treg cells can promote iron-dependent cell death (63). Liu Y et al. shown iron overload in the joint cavity of rheumatoid arthritis patients and in vitro can promote ferroptosis in macrophages (76). Additionally, Alli, A. A. et al. demonstrated iron sequestration within the proximal tubules promoted LN progression by exacerbating ferroptosis (77). This evidence demonstrated that iron accumulation can trigger ferroptosis not only in the extracellular space but also in the intracellular space.
Thus, iron overload is a shared feature of SLE and ferroptosis and multiple evidences showed iron overload was involved in disease progression by triggering ferroptosis (Figure 1). Although, numerous reports demonstrated iron overload can aggravate the progression of SLE, whether the effect is ascribed to promoting ferroptosis. Anyhow, preventing iron accumulation in tissues and cells by mediating the key molecules of iron metabolism (transferrin receptor, ferroportin, hepcidin, etc.) may be a novel direction for the treatment of SLE.
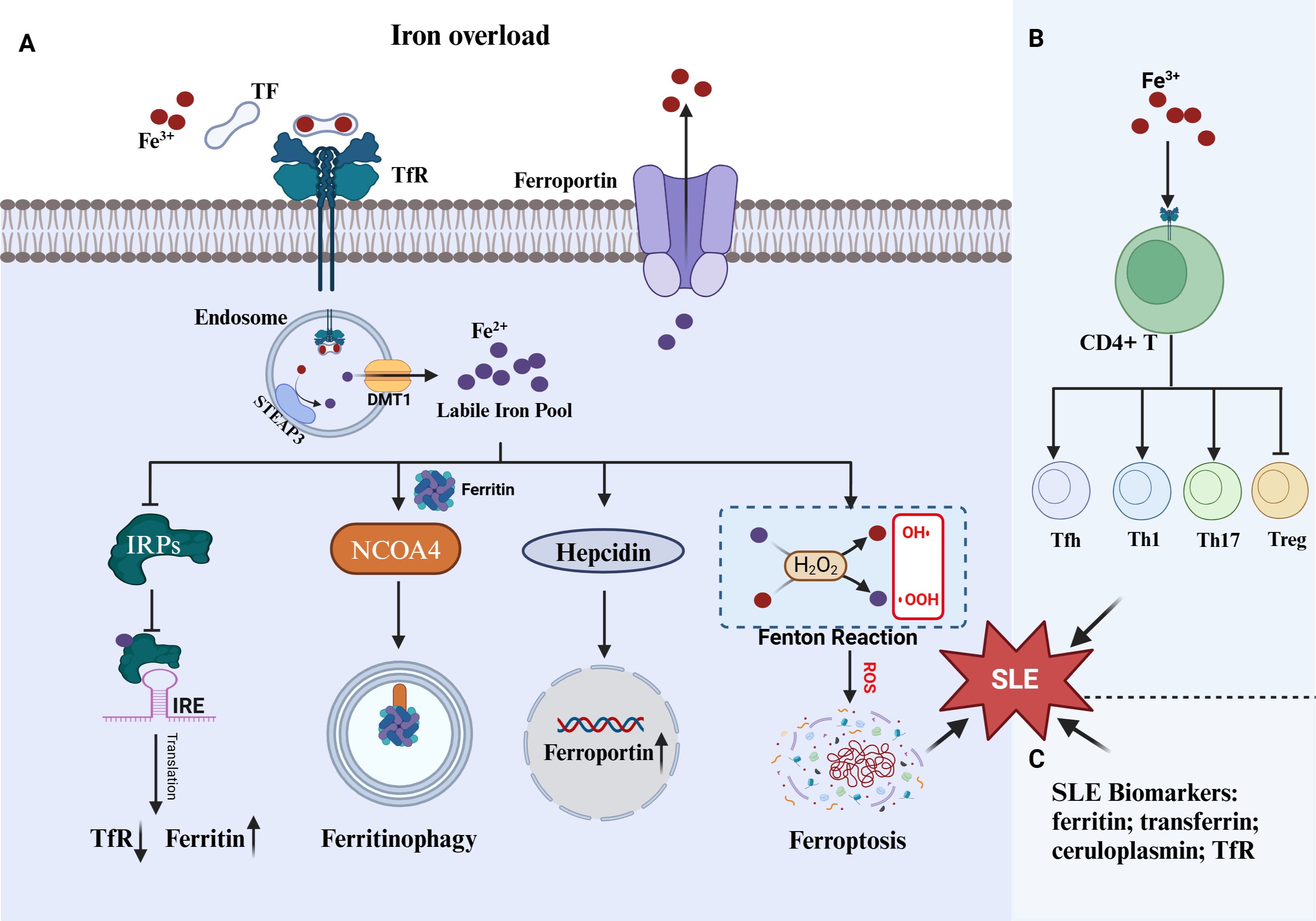
Figure 1. Iron overload in ferroptosis and SLE. (A) Three mechanism including IRPs/IRE system, ferritinophagy and hepcidin-ferroportin roadways against iron overload. The cell undergoes ferroptosis when those mechanism are impaired. (B) Iron overload promote CD4+T abnormal proliferation and differentiation, which can promote SLE progress. (C) Numerous serum protein closed to iron regulation were identified as SLE biomarkers. TF, transferrin; TfR, transferrin receptor; IRPs, iron-responsive proteins; IREs, iron-responsive elements; STEAP3, six-transmembrane epithelial antigen of the prostate 3; DMT1, Divalent metal transporter 1; NOCA4, nuclear receptor coactivator 4.
3.2 Mitochondrial dysfunction
Mitochondria are essential organelles for maintaining the normal physiological function of the cell and play a crucial role in physiological processes such as energy production, calcium homeostasis, and iron metabolism. It is also a key site for ROS elevation, self-nucleic acid antigen production, and the promotion of various forms of cell death (such as ferroptosis), which are closely related to a variety of diseases, such as SLE (26, 78, 79). Overactivation and immune intolerance of immune cells is the most pivotal link in the pathogenesis of SLE (28). The rapid proliferation and differentiation of immune cells require mitochondria to supply a large amount of energy. Oxidative phosphorylation (OXPHOS) of the electron transport chain (ETC) on the inner mitochondrial membrane is the process of ATP production, which is also accompanied by ROS production (80). Dysfunction of mitochondria limits ATP production but aggravates the release of ROS, which are closely related to immune disorders, autoantibody production, excessive cell death and ineffective clearance mechanisms (81). Previous studies have shown that mitochondria in CD4+ T cells from SLE patients exhibit numerous abnormalities, such as large size (mitochondrial fusion), membrane hyperpolarization (elevated mitochondrial transmembrane potential), ATP depletion, fragility, increased mitochondrial ROS (mtROS) and decreased antioxidant (GSH) levels (82–84). Moreover, massive ROS production in SLE patients is the main contributor to mitochondrial dysfunction (85). Second, mitochondrial DNA (mtDNA) oxidized by ROS (ox-mtDNA) has been identified as an important DAMP. Under normal physiological conditions, it can be dissociated from mitochondrial transcription factor A (TFAM) and degraded in lysosomes. However, in SLE, the restriction of TFAM function promotes the continuous accumulation of Ox-mtDNA. In addition, mtDNA that is released into the cytoplasm can not only activate cyclic GMP-AMP synthase (cGAS) or Toll-like receptors 9 (TLR9) to promote the production of IFNs (86, 87) but also serve as an antigen to directly activate the immune system to exacerbate autoantibody production (81). Since the kidney is one of the most involved organs in lupus and iron consumption is prevalent in it, mitochondrial are particularly susceptible to oxidative attack in glomerular and tubular cells. Tian Y at al. Demonstrated the molecular mechanism of iron-induced injury in these cells was contributed to mtROS overproduction (88). Remarkably, recent evidence demonstrated impairment of mitochondrial degradation in glomerular and tubular cells was implicated in proteinuria and renal failure in LN (16, 89, 90). Moreover, both mtROS and impair mitochondrial degradation promote Ox-mtDNA accumulation (91). On the other side, Ox-mtDNA can exacerbate mtROS production in a vicious cycle, which further aggravate the disease progression.
Moreover, mitochondria serve as central hubs for multiple forms of cell death, such as apoptosis, necroptosis, and pyroptosis, and are also key organelles that trigger ferroptosis (5, 78, 92). It is still debated whether mitochondria are necessary for ferroptosis. For example, some cells become insensitive to ferroptosis-inducing agents after the removal of mitochondria, whereas others do not (93, 94). Moreover, it has also been shown that mitochondria are required for erastin-induced ferroptosis but not for RSL3-induced ferroptosis (17). However, deficiency of optic atrophy protein 1 (OPA1), which maintains mitochondrial homeostasis and function, leads to resistance to both erastin- and RSL3-induced ferroptosis (95). In ferroptosis, significant changes in mitochondrial function and morphology are observed, including a decrease in volume, increase in membrane density, and decrease in ridge and ATP production (7). The role of mitochondria in triggering ferroptosis is explained as follows: First, mitochondria are one of the main sources of ROS and the key site of energy production and iron metabolism (65, 96). Mitochondrial dysfunction not only reduces ATP production but also contributes ETC to leaking large amounts of ROS (such as O2·- and H2O2) and disturb iron homeostasis. H2O2 combines with the Fe2+-mediated Fenton cascade and further exacerbates the massive release of ROS, which promotes lipid peroxidation (97). Depletion of mtROS can alleviate mitochondrial ferroptosis (98). Second, the tricarboxylic acid cycle (TCA) cycle is located in the mitochondria. The mitochondrial TCA cycle can increase ferroptosis sensitivity (17). Glutamate metabolism is an important complementary part of the TCA cycle. In mitochondria, glutamine can be converted to glutamate and TCA intermediate metabolites (α-ketoglutarate, αKG), both of which can promote ferroptosis (99–101). Moreover, the inactive αKG dehydrogenase complex can prevent ferroptosis induced by cystine depletion (102). Additionally, ferroptosis has been identified as a form of autophagy-dependent cell death (103). And interesting, some reports demonstrated mtDNA could trigger autophagy-dependent ferroptosis by activating STING (33) and peroxisome proliferator activated receptor alpha (PPARα) (104). Moreover, mtDNA is prone to transform B- to Z-DNA structures under mtROS stress (105), which could promote ferroptosis by activating Z-DNA binding protein 1 (ZBP1) (106). These results suggest that mitochondrial dysfunction can promote ferroptosis.
In summary, in SLE patients, mtROS accumulation and mtDNA release may promote ferroptosis and subsequent release of intracellular antigens and DAMPs, thereby mediating the progression of SLE (Figure 2). Therefore, limiting mtROS production and mtDNA release to maintain mitochondrial function may be a promising treatment strategy for SLE.
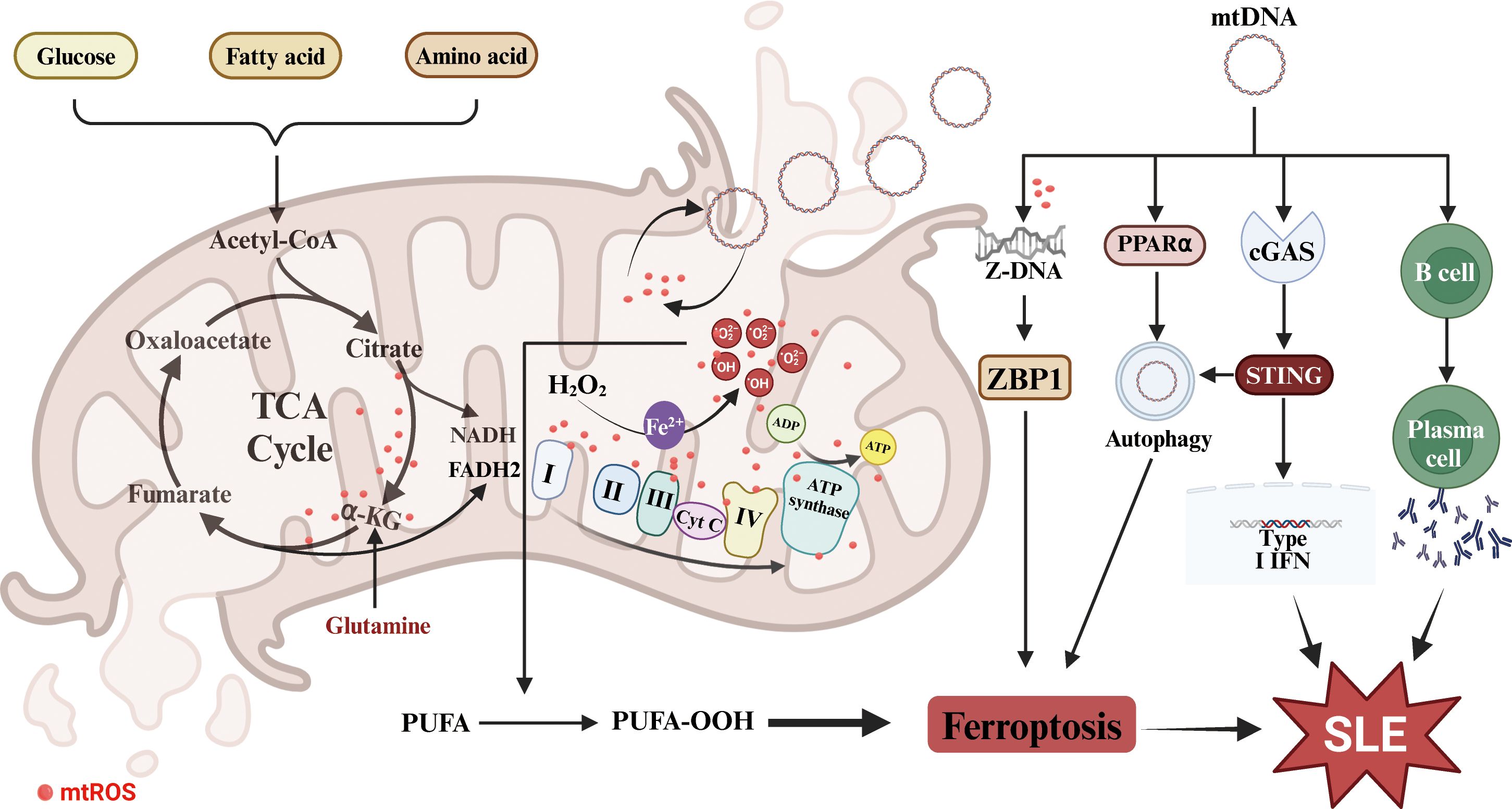
Figure 2. Mitochondrial dysfunction in SLE and ferroptosis. The dysfunction promotes mtROS accumulation and mtDNA release, which triggers cell ferroptosis and enhances the production of autoantibodies and IFNs. PUFA, polyunsaturated fatty acids; TCA, the tricarboxylic acid cycle; α-KG, α-ketoglutarate; mtROS, mitochondrial reactive oxygen species; mtDNA, mitochondrial DNA; ZBP1, Z-DNA binding protein 1; PPARα, peroxisome proliferator activated receptor alpha.
3.3 Lipid metabolism disorder
Lipid metabolism is a major physiological process that includes catabolism [fatty acid oxidation (FAO)], anabolism (de novo lipogenesis) and storage [lipid droplets (LDs)], and lipid metabolism disorders are closely related to multiple pathological processes and diseases, including immune dysfunction and SLE (107). Dyslipidemia, a major type of lipid metabolic disruption, is most common in SLE patients (68%-100%) and is characterized primarily by increased plasma levels of very low-density lipoprotein (VLDL), triglyceride (TG) and total cholesterol (TC) but decreased high-density lipoprotein (HDL) levels (108). This disorder not only is closely related to SLE activity and organ involvement (including the cardiovascular system and kidney) but is also related to high mortality (109, 110). Among them, low HDL-cholesterol (HDL-c) levels are the most common sign of dyslipidemia in SLE patients (18, 111, 112). Under normal conditions, HDL performs anti-inflammatory and antioxidative functions despite its cholesterol efflux capacity. However, in SLE, it functions in an opposite manner with an uncertain mechanism (113, 114). Interestingly, a previous study demonstrated that limiting cholesterol flux could aggravate the IFN and ISG response in a STING-dependent manner in macrophages (115), and its precursor concentration, 7-dehydrocholesterol (7-DHC), could also significantly increase I-IFN production via the phosphatidylinositol 3-kinase (PI3K)-protein kinase B 3 (AKT3)-interferon regulatory factor 3 (IRF3) pathway (116). However, 7-DHC, as a precursor to form Vitamin D3 in the skin by solar ultraviolet B radiation, is the main source of Vitamin D. The deficiency Vitamin D is prevailed in SLE population and its supplementation seems to ameliorated the diseases (117), which may attribute that adequate level of Vitamin D prevents the synthesis of 7-DHC. Additionally, the metabolite of cholesterol, 7α, 25-dihydroxycholesterol (7α, 25-OHC), is dramatically elevated in the plasma of SLE patients and binds to the G protein-coupled receptor (GPCR) Epstein–Barr virus-induced gene 2 (EBI2) in macrophages to alleviate the IFN and ISG response for protecting against SLE progress (118). Consequently, homeostasis lipidemia, especially cholesterol metabolism, is crucial for SLE pathogenesis.
Additionally, lipids are highly vulnerable to oxidation, and many lipid peroxides, including oxidized HDL (ox-HDL), ox-LDL, malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), have been identified as potential biomarkers for SLE and are related to disease activity (111, 118–121). Because oxidative stress is involved in the pathogenesis of SLE, aberrant oxylipins have been revealed in SLE patients by lipidomic analysis (122, 123). Oxylipin is a series of oxidative metabolites produced by polyunsaturated fatty acids (PUFAs), such as arachidonic acid (AA), linoleic acid (LA), and alpha-linolenic acid (ALA), that undergo non-enzymatic or specific enzymatic [lipoxygenase (LOX) and cytochrome P450 (CYP450)] oxidations. Multiple oxylipins, such as 12-hydroxy-heptecotrienoic acid (12-HHTrE) and prostaglandin E1 (PGE1), are critical for SLE pathogenesis and have been identified as biomarkers for SLE pathogenesis (124, 125). Interestingly, the concentration of serum PUFAs is significantly decreased in SLE patients, which demonstrates that the antioxidant system is impaired and that lipid peroxidation is prevalent (107, 124). Moreover, lysophospholipids containing PUFA chains and lipid peroxidation substances (i.e., 4-hydroxyalkenals) strongly accumulate in the peripheral blood mononuclear cells of SLE patients (126). In summary, lipid metabolic disorder is one of the outstanding features in SLE, especially pertaining to oxidized-lipid metabolism.
Moreover, lipid metabolic disorder is essential for ferroptosis, because the executor of this cell death is lipid peroxidation, which triggers ion flux disturbances and ultimately plasma membrane permeabilization and fragmentation (127–129). Lipid metabolism is crucial for the initiation, propagation and termination of lipid peroxidation (130). PUFAs [especially AA (C20:4) and LA (C18:2)] are regarded as primary inducers of ferroptosis because whose bis-allylic hydrogens have relatively low bond dissociation energies and are more sensitive to lipid peroxidation (19, 131). In brief, the contributions of PUFAs to ferroptosis can be divided into two main categories: extension into the membrane and peroxidation. First, PUFAs are taken into the cell or converted by other lipids inside the cell. Then, extension can occur into membrane phospholipids (PUFA-PLs) via acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), which are very vulnerable to peroxidation. Second, PUFA-PLs are oxidated by lipoxygenases or via nonenzymatic autoxidation reactions [such as the Fenton reaction). The lipid peroxidation chain reaction is subsequently maintained by lipid radicals (such as the phospholipid peroxyl radical (PLOO·)] derived from lipid peroxidation. When antioxidative agents, including GPX4 and coenzyme Q10 (CoQ10), are depleted, the cell ultimately undergoes ferroptosis (132).
Although the direct relationship between SLE and ferroptosis related to lipid metabolism has not been reported, there are numerous potential connections as follows: i.The dyslipidemia feature of SLE further promotes cholesterol metabolic dysfunction, which can affect ferroptosis. The intermediate, isopentenyl pyrophosphate (IPP), is required for the synthesis of GPX4 (133, 134) and CoQ10 (135–137), which are known to prevent ferroptosis. Similarly, squalene (138, 139) and 7-DHC (140, 141) effectively prevent lipids from autoxidation and subsequent fragmentation (142). Furthermore, some enzymes related to cholesterol metabolism serve as potential suppressors of ferroptosis, such as 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) in mitochondria (143) and sterol C5-desaturase (SCD5), whereas 7-dehydrocholesterol reductase (DHCR7) (140) and squalene monooxygenase (SQLE) (138) function as pro-ferroptostic molecules (3). Elevation of the cellular cholesterol content specifically restrains elevated lipid peroxidation and reduces susceptibility to ferroptosis by decreasing membrane fluidity (138). ii.PUFA, an inducer of ferroptosis, is significantly decreased in the extracellular space but increased in the intracellular space in SLE patients (Figure 3). Moreover, the levels of lipid peroxidation substances, especially MDA and 4-HNE (ferroptostic biomarkers), are markedly increased in SLE patients. Moreover, elevated MDA was related with multiple lupus manifestations (such as vasculitis, musculoskeletal, cutaneous and nephritis) (144–146). And also, the serum levels of anti-MDA IgG antibody positively were related with disease activity, active nephritis, inflammatory indictors and the consumption of complement factors (147). This phenomenon may be explained by the fact that PUFAs are taken into cells to be involved in ferroptosis in SLE patients.
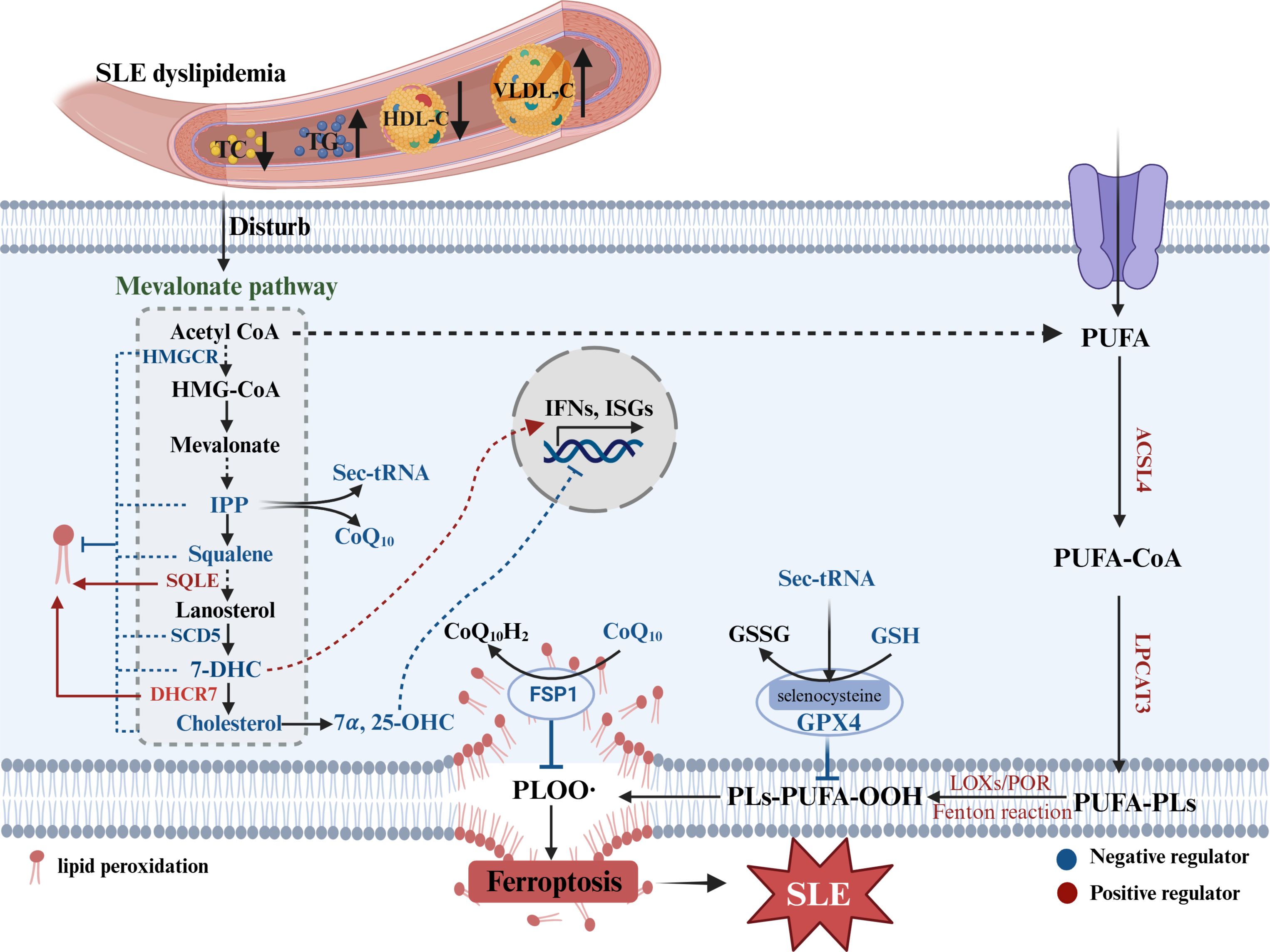
Figure 3. Lipid metabolism disorder in SLE and ferroptosis. SLE dyslipidemia can disturb cholesterol metabolism, which intermediates or enzymes not only affect IFNs response but mediate ferroptosis. Sec-tRNA, selenocysteine tRNA required for GPX4 synthesis; TC, total cholesterol; TG, triglyceride; HDL-C, high density lipoprotein-cholesterol; VLDL-C, very low density lipoprotein-cholesterol; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; IPP, isopentenyl pyrophosphate; SQLE, squalene monooxygenase; CoQ10, coenzyme Q10; 7-DHC, 7-dehydrocholesterol; DHCR7, 7-dehydrocholesterol reductase; SCD5, sterol C5-desaturase; 7α,25-OHC, 7α, 25-dihydroxycholesterol; FSP1, ferroptosis suppressor protein 1; ISGs, interferon-stimulated genes; LOXs, lipoxygenases; POR, cytochrome P450 reductase; PLs, phospholipids; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3.
3.4 Function of DAMPs
3.4.1 HMGB1
DAMPs are endogenous molecules that are released into the extracellular space under some pathologic conditions, mainly during cell death. Although numerous DAMPs released from cell death or necrosis, including TNF-α (148, 149), IL-1β, IL-18 (150, 151) and IL-33 (152), have been identified as biomarkers for SLE, their potential role in SLE has not been well defined.
However, extracellular HMGB1, a ubiquitous DAMP, belongs to the HMG family and performs well in SLE pathogenesis. HMGB1 was originally thought to be located only in the nucleus as a DNA chaperone and participated in regulating chromatin structure and function; however, recently, it was found that it can also be located in the cytoplasm and cell membrane. In addition, HMGB1 can be secreted actively or passively into the extracellular space to perform various biological functions (153, 154). In recent years, much more attention has been given to its extracellular function. A growing number of reports have suggested that extracellular (both urine and plasma) HMGB1 can serve as a biomarker for SLE, which could predict disease activity and renal involvement (152–157). Moreover, anti-HMGB1 antibodies in plasma have also been identified as biomarkers of SLE (158, 159). Extracellular HMGB1 can bind to numerous receptors, such as toll-like receptors (TLRs), and advanced glycosylation end-product-specific receptor (AGER), triggering receptor expression on myeloid cells-1 (TREM1), which promotes the release of pro-inflammatory factors. In addition, when released outside the cell, HMGB1 usually binds to intranuclear material (e.g., RNA, DNA), which can activate multiple intracellular nucleic acid sensors, including TLR7, TLR9 and cGAS (160, 161). These sensors are tightly correlated with increased IFN production and SLE pathogenesis (162, 163). In addition, both I-IFN and II-IFN can promote HMGB1 release, resulting in a vicious cycle (24, 164), which may aggravate the progression of SLE. Overall, numerous results suggest that extracellular HMGB1 is involved in the pathogenesis of SLE. Similarly, treatment with an HMGB1 monoclonal neutralizing antibody can alleviate the phenotype of MRL/lpr and BXSB Lupus mice, but it has also been shown to be ineffective in MRL/lpr mice (20).
Extracellular HMGB1 is derived mainly from cell death, including apoptosis, necroptosis and ferroptosis (165). Various ferroptostic agonists (including erastin and RSL3) can increase HMGB1 release, and this process is blocked by ferroptostic antagonists (such as ferrostatin-1 and liproxstatin-1) or pro-ferroptostic genetic deficiency (e.g., Acsl4 shRNA) in cancer and noncancer cells (21). Wiernicki B et al. recently demonstrated that ferroptosis can be clearly distinguished into three stages: (1) At the beginning, there is intracellular lipid peroxide accumulation; (2) In the intermediate stage, cell membrane permeability increases, resulting in ATP release; and (3) In the final stage, there is complete disintegration of the cell membrane, and HMGB1, LDH and inflammatory factors are released (166). HMGB1 serves as the main DAMP of ferroptosis and mediates the pathological effects of ferroptosis. It is released during ferroptosis of M2 macrophages and can promote the inflammatory response of M1 cells through the TLR4/STAT3 pathway. Moreover, the inhibition of ferroptosis rescues this effect (167). Wang CB et al. revealed that the hepatotoxic effect of methotrexate (MTX) was mainly mediated by HMGB1 released from ferroptostic cells (31). In addition, Zhang DF et al. reported that renal dysfunction related to imidacloprid was primarily attributed to HMGB1 release from ferroptosis, which promoted the activation of the nucleotide-binding domain (NBD), leucine-rich repeat (LRR), and pyrin domain (PYD)-containing protein 3 (NLRP3) inflammasome in neighboring cells through the RAGE/TLR4-NF-κB signaling pathway, further aggravating pyroptosis (22). Interestingly, numerous recent reports have shown that HMGB1 released from ferroptosis can also trigger ferroptosis in neighboring cells, thus forming a vicious cycle and aggravating the progression of the disease. Wei Q et al. revealed that ultraviolet B radiation induces autophagy-dependent ferroptosis and that subsequently released HMGB1 can promote ferroptosis in neighboring cells through TfR1 (32). Davaanyam D et al. demonstrated that ferroptosis occurs in neurons at the early stage of cerebral ischemia and that HMGB1 can upregulate the expression of hepcidin through TLR4/C-X-C chemokine receptor type 4 (CXCR4) to promote ferroptosis and aggravate cerebral ischemia. Moreover, inhibition of HMGB1 can intercept ferroptosis and alleviate cerebral ischemia (23). In addition, Deng YL et al. reported that the overexpression of casepase-6 in THP-1 cells promoted ferroptosis in HTR8/SVneo cells. However, HMGB1-neutralizing antibodies can prevent this process (168). Taken together, these findings indicate that HMGB1 is an important effector of ferroptosis that can mediate the involvement of ferroptosis in various pathological mechanisms and disease progression.
In summary, ferroptosis can increase the release of HMGB1, which can intensify the release of inflammatory factors through its sensors. Additionally, binding to nuclear substances can activate related nucleic acid receptors (TLR7/9, cGAS, etc.) to mediate the mass production of IFNs, and subsequently, IFNs can promote ferroptosis and accelerate the release of HMGB1. Moreover, HMGB1 can further promote ferroptosis in neighboring cells. Overall, these events form a vicious cycle, which may exacerbate the progression of SLE (Figure 4).
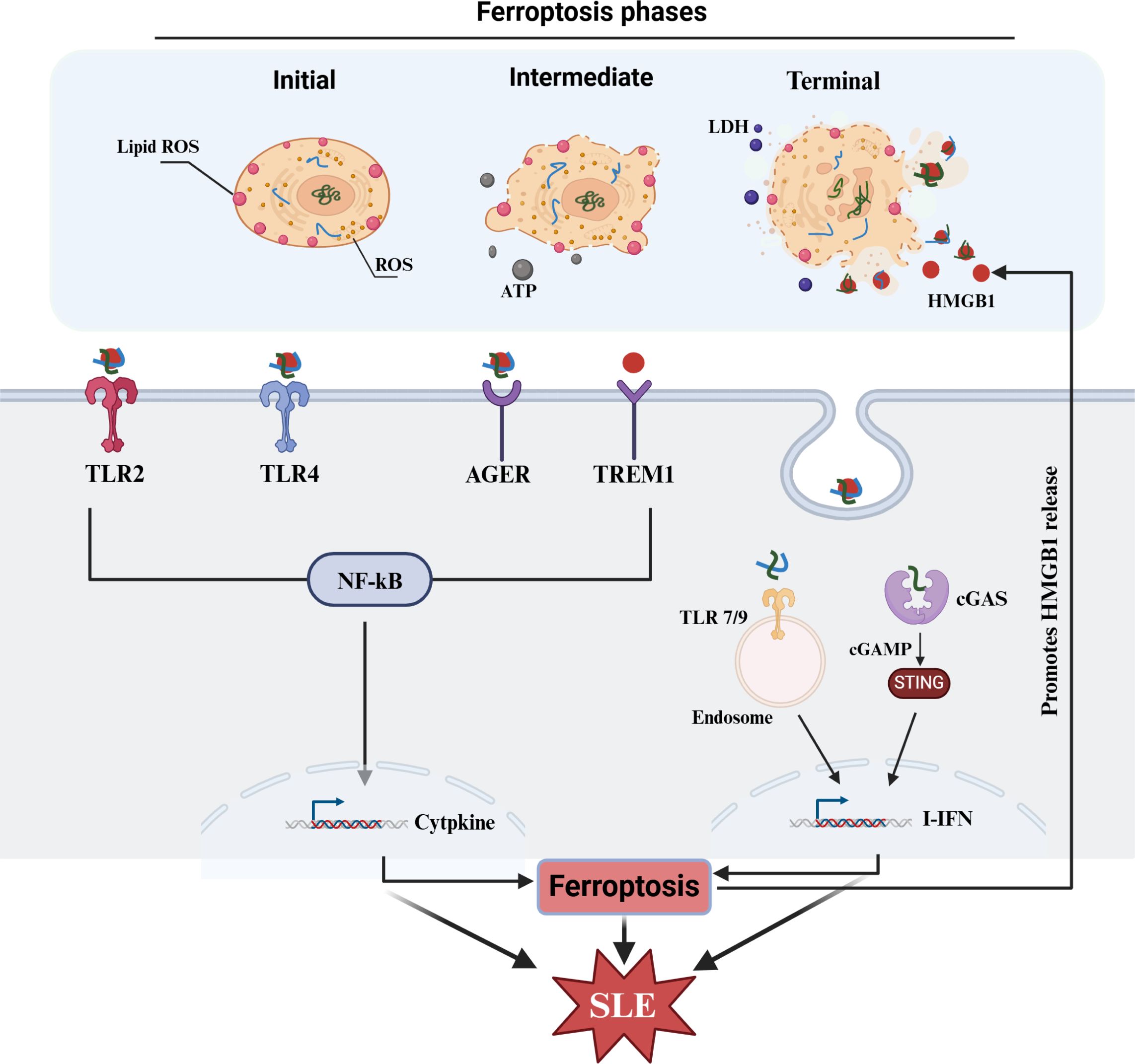
Figure 4. HMGB1 in SLE and ferroptosis. HMGB1 as a major DAMPs released from ferroptosis can intensify the production of IFNs and other cytokines, which subsequently contribute ferroptosis and SLE progress. HMGB1, high-mobility group box 1; TLR, toll-like receptors; AGER, advanced glycosylation end-product-specific receptor; TREM1, triggering receptor expressed on myeloid cells-1; cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon genes; cGAMP, cyclic GMP-AMP; ISGs, interferon-stimulated genes.
3.4.2 IFNs
SLE is known as an “interferon signature” disease and the overactivation of the interferon system is closely related to SLE pathogenesis, especially I-IFN, which is elevated before the onset of SLE and closely associated with disease activity and organ involvement (169–171). Nucleic acid substances bind to the endosomal or intracellular nucleic acid receptors (TLRs, cGAS, Nucleotide-binding and leucine-rich repeat receptors (NLRs), etc.) and promote the massive production of interferon-stimulated genes (ISGs) and Type I IFNs (I-IFNs), subsequently contributing to SLE progression. In the past, type I IFNs (II-IFNs) were considered to play the most crucial role in the pathogenesis of SLE (169). Recently, other types of IFNs, especially II IFNs, have also been confirmed to play an important role in the pathogenesis of SLE (170, 171). Moreover, in recent years, IFNs have been shown to be crucial for the regulation of ferroptosis (8). Li PC et al. reported that, in SLE, IFN-α or IgG can restrain GPX4 transcription through CaMKIV/CREMα, which promotes an increase in the level of intracellular lipid ROS and ultimately facilitates neutrophil ferroptosis. Moreover, they confirmed that neutrophil ferroptosis is a key form of cell death involved in the pathogenesis of SLE (172). In addition, evidence that IFN-α can induce ferroptosis was confirmed by Zhang SL et al. (173). They reported that manganese could disturb the expression of DHODH by activating cGAS-STING pathway, promoting an increase in mitochondrial lipid peroxides and ROS, ultimately leading to ferroptosis of tumor cells. Zhang SL et al. also found that blocking IFNAR could rescue this effect of manganese. As the downstream pathway of IFNs, the JAK-STAT pathway is considered one of the most important pathways involved in the pathogenesis of SLE (mainly JAK-STAT1) (174, 175). In addition, many JAK inhibitors (such as tofacitinib, baricitinib, upadacitinib and filgotinib) are being investigated in clinical trials (176). Recent studies have confirmed that spermine can effectively alleviate the progression of SLE by inhibiting JAK1 (177).
Moreover, a close correlation between the IFNs-JAK-STAT pathway and ferroptosis has been demonstrated in recent years. In another autoimmune disease, Sjogren’s syndrome, high expression of IFN-γ could decrease the expression of solute carrier family 3 member 2 (SLC3A2, a subunit of xCT), glutathione, and GPX4 in salivary gland epithelial cells and then trigger ferroptosis through activation of the JAK/STAT1 pathway, which can be inhibited by JAK1/2 or STAT1 antagonists (178). In addition, in retinal pigment epithelial cells, IFN-γ can downregulate the expression of solute carrier family 40 member 1 (SLC40A1, iron export protein) and SLC7A11 through the JAK1-2/STAT1 pathway, which promotes the accumulation of intracellular Fe2+ and the exhaustion of glutathione and ultimately induces ferroptosis (179). In tumor cells, IFN-γ can also prevent the transcription of both SLC3A2 and SLC7A11 and then accelerate ferroptosis via JAK1/2-STAT1-IRF1 (180). Furthermore, a chromatin immunoprecipitation assay confirmed that IFN-γ promoted the binding of STAT1 to the SLC7A11 promoter to intercept its transcription (181). However, it has also been reported that inhibition of STAT1 can downregulate GPX4 and SLC7A11 expression to promote ferroptosis (182). Fan Li also confirmed that STAT1 can bind to the promoter region of SLC7A11 and promote its transcription using a dual luciferase reporter assay (183). These results suggest that STAT1 can regulate ferroptosis, but the regulatory mechanism is contradictory and may be related to differences in cells and their environment.
In conclusion, JAK-STAT1 not only play important roles in the pathogenesis of SLE but also regulate ferroptosis. Since the regulation of IFNs-JAK-STAT signaling is inconsistent in ferroptosis, whether this signaling is implicated in SLE pathogenesis by regulating ferroptosis needs more evidences (Figure 5). Although many inhibitors targeting the JAK-STAT pathway have been investigated in clinical trials, their actual effects are not satisfactory (176). Notably, whether the inhibition of the JAK-STAT pathway potentially aggravates ferroptosis and promotes the progression of SLE deserves further explore.
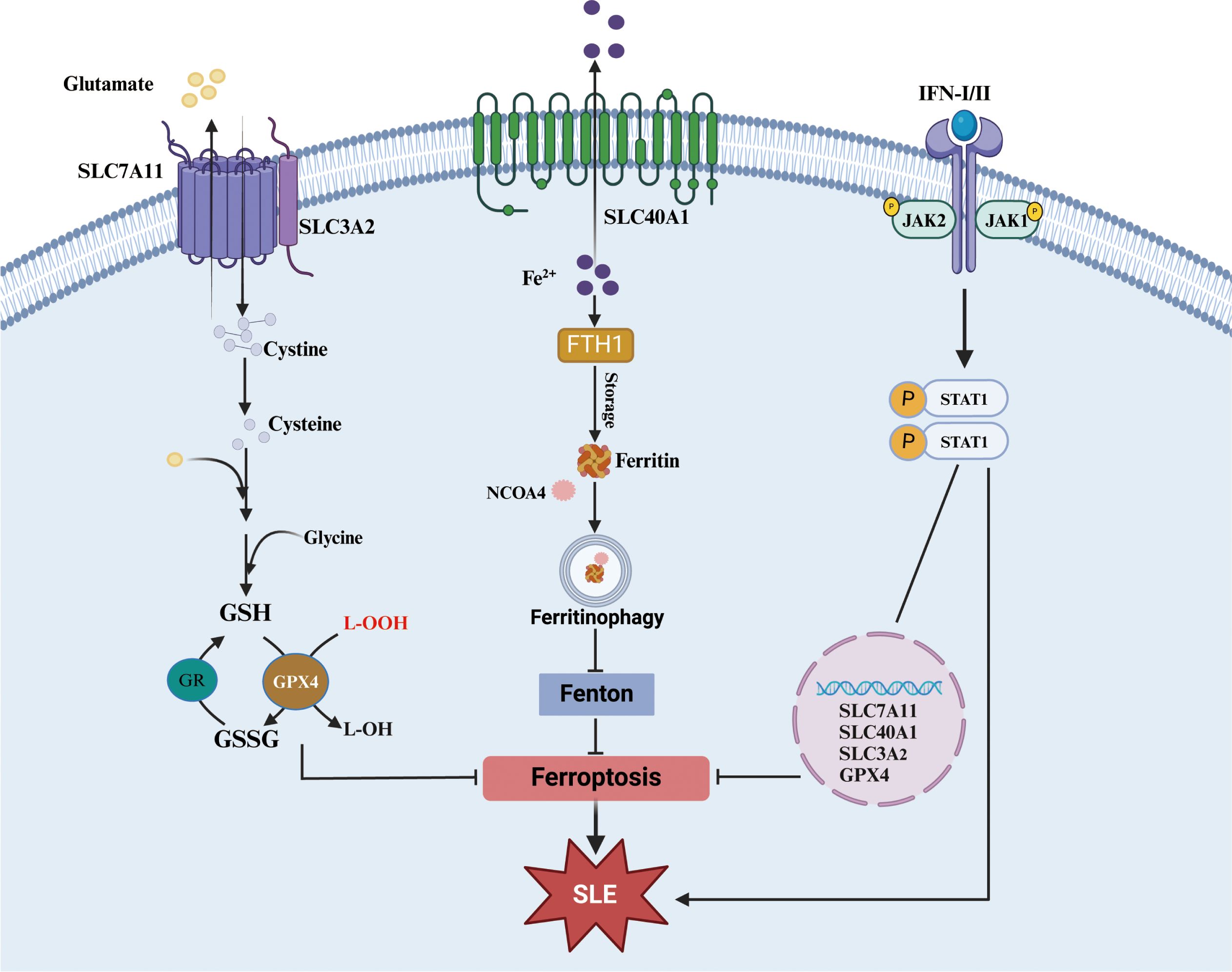
Figure 5. JAK-STAT1 pathway in ferroptosis. Both xCT-GPX4 and FTH1-Ferritinophagy are important roadways to against ferroptosis. And the accumulation of some cytokines (especial I/II-IFN) could significantly affect pivotal molecules of the two roadways although JAK-STAT1 pathway. xCT, cystine/glutamate transport system; SLC7A11, solute carrier family 7a member 11, SLC3A2, solute carrier family 3 member 2; SLC40A1, solute carrier family 40 member 1; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, glutathione disulfide; FTH1, ferritin heavy chain 1; NCOA4, nuclear receptor coactivator 4.
4 Promising therapeutical reagents for SLE by inhibiting ferroptosis
Given that several pathophysiological features of ferroptosis, including iron accumulation, mitochondrial dysfunction, overproduction of lipid peroxidation and suppression xCT-GPX4 pathway are potentially implicated in SLE progression, reversing those features may be beneficial to SLE disease. Below, we summarize some the bioactive pharmacological agents that can against those ferroptostic process (Table 2).
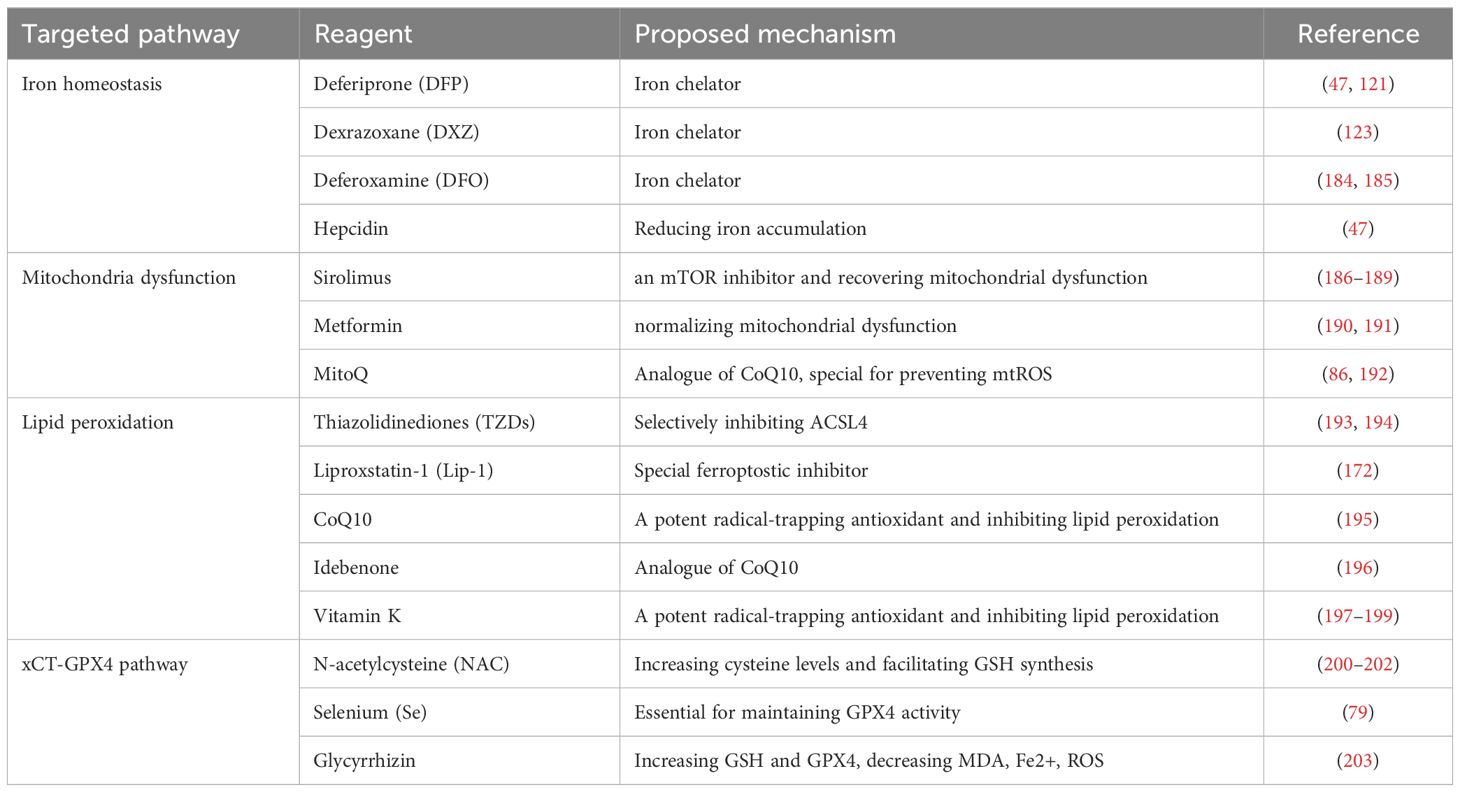
Table 2. The promising therapeutical reagents for SLE by inhibiting ferroptosis.
4.1 Inhibiting iron accumulation
Intracellular iron accumulation is the chief culprit of ferroptosis and can promote SLE progression, the therapeutical reagents to reduce iron concentration may be beneficial to treat SLE. Iron chelators, including deferiprone (DFP), dexrazoxane (DXZ) and deferoxamine (DFO), are commonly used in clinical. Although only few reports demonstrated DFO could alleviated SLE progression, multiple evidences confirmed those chelators performed an effectivity on some chronical diseases by preventing ferroptosis, such as osteoarthritis (184), I/R-induced injury (185), non-alcoholic steatohepatitis (204), neurodegeneration (205) and so on. Additionally, hepcidin, the iron-regulatory hormone, has been verified it could reduce progression and severity of LN by decreasing renal iron accumulation (47).
4.2 Recovering mitochondrial dysfunction
Mitochondrial dysfunction not only drivers ferroptostic process but promotes SLE progression. Recovering mitochondrial dysfunction to prevent ferroptosis may improve prognosis of SLE disease. Recent evidences demonstrated mTOR inhibition suppress ferroptosis by mediating mitochondrial dysfunction (186). Sirolimus is an mTOR inhibitor that server as an antifungal or immunosuppressive agent in clinical, which also effectively alleviates lupus mice manifestation (187, 188). Moreover, Lai ZW et al. demonstrated sirolimus improve SLE patient activity by recovering mitochondrial dysfunction (189). Metformin, commonly used in clinical to treat type II diabetes, can reduces oxidants by normalizing mitochondrial dysfunction (190, 191). Remarkably, it has been verified to improve SLE patients by recovering mitochondrial dysfunction (206–208). Additionally, MitoQ, as an analogue of CoQ10, special for preventing mtROS production, was shown to alleviate lupus mice (86, 192).
4.3 Reducing lipid peroxidation
Lipid peroxidation is essential to ferroptosis and prevent the process was shown to improve numerous ferroptosis-related diseases. Thiazolidinediones (TZDs), including rosiglitazone, pioglitazone and troglitazone, are applied to treat type II diabetes. Remarkably, TZDs performed an ability to suppress ferroptosis though selectively inhibiting ACSL4 and reduce mortality in kidney-specific Gpx4 knockout mice (193, 194). Liproxstain-1 (Lip-1), a spiroquinoxalinamine derivative, was identified as a special ferroptostic inhibitor by high-throughput screening. Recent evidence shown it could alleviate MRL/lpr lupus mice symptom (172). Additionally, some lipid antioxidants also display potential therapeutical prospect as follow: i. CoQ10 is a major substrate of FSP1 that known as ferroptostic suppressor. The dietary supplement of it as a potential candidate for the treatment of various noncommunicable diseases (195). Moreover, its analogues, idebenone with better bioavailability and efficacy, was shown to attenuate murine lupus (196). ii. Vitamin K, resembled CoQ10 structure, was used to overcome warfarin poisoning in clinical practice and its supplementation could positively affect multiple chronical diseases (197). Interesting, it also was identified as a lipid radical-trapping antioxidant to prevent ferroptosis via FSP1-mediated pathway (198, 199).
4.4 Activating xCT-GPX4 pathway
As described above, the xCT and GSH-GPX4 pathway are identified as primary defense mechanism to ferroptosis. N-acetylcysteine (NAC), a precursor to cysteine, has been shown to prevent ferroptosis by increasing cysteine levels and facilitating the synthesis of γ-glutamyl-cysteine and GSH (200). And its supplement is potentially beneficial for comorbid disorders associated with heavy alcohol consumption (201) and Parkinson’s disease symptoms (202). Micronutrient selenium (Se) is essential for maintaining GPX4 activity. Optimal Se supplement could improve hemorrhagic and ischemic stroke prognosis via inhibiting ferroptosis in mice (79). Additionally, glycyrrhizin, extracted from the glycyrrhiza and commonly used in clinical, could alleviate acute liver failure by increasing GSH and GPX4, decreasing MDA, Fe2+, ROS to inhibit ferroptosis (203).
5 Conclusion
In summary, SLE is a complex chronic disease for which its pathophysiology and effective treatments remain speculative. Ferroptosis is a prevalent type of pathogenic cell death in chronic disease. Although the studies on ferroptosis in SLE pathogenesis are still in early stages, there are intricate and close connections among them, which provides opportunities for understanding SLE pathophysiology and development of novel therapeutics.
Author contributions
GZ: Writing – original draft, Formal analysis, Writing – review & editing. XL: Writing – review & editing, Conceptualization, Writing – original draft. YZ: Writing – original draft, Conceptualization, Writing – review & editing. XW: Visualization, Writing – review & editing. LD: Visualization, Writing – review & editing. JX: Visualization, Writing – review & editing. SJ: Resources, Writing – review & editing. ZZ: Resources, Writing – review & editing. LX: Resources, Writing – review & editing. ML: Resources, Writing – review & editing. FY: Supervision, Writing – review & editing. JQ: Funding acquisition, Writing – review & editing. PF: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No.82460111,81860287), and Foreign Cooperative research project of the Second Affiliated Hospital of Kunming Medical University (No. 2022dwhz11), and the Kunming University of Science and Technology Joint Special Project on medical project (KUST-KH2023002Z), and The First People’s Hospital of Yunnan Province Talent Project program (No.KHBS-2022013, KHYJ-2023-05-01, 2022-KHRCBZ-B03), and the Yunnan Provincial Key Laboratory of Birth Defects and Genetic Diseases (No.2022ZDKFKT001), and the Yunnan Provincial Key Laboratory of Clinical Virology (No. 202205AG070053), and the Kunming Medical University Joint Special Project on Applied Basic Research (202301AY070001-210), and Yunnan Fundamental basic research project (202301AT070034).
Acknowledgments
We are grateful to Biorender (BioRender.com) for their assistance in graphic illustration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel CH, Sammaritano LR. Systemic lupus erythematosus: A review. JAMA. (2024) 331:1480–91. doi: 10.1001/jama.2024.2315
2. Tian J, Zhang D, Yao X, Huang Y, Lu Q. Global epidemiology of systemic lupus erythematosus: a comprehensive systematic analysis and modelling study. Ann Rheum Dis. (2023) 82:351–6. doi: 10.1136/ard-2022-223035
3. Crow MK. Pathogenesis of systemic lupus erythematosus: risks, mechanisms and therapeutic targets. Ann Rheum Dis. (2023) 82:999–1014. doi: 10.1136/ard-2022-223741
4. Xu Y, Li P, Li K, Li N, Liu H, Zhang X, et al. Pathological mechanisms and crosstalk among different forms of cell death in systemic lupus erythematosus. J Autoimmun. (2022) 132:102890. doi: 10.1016/j.jaut.2022.102890
5. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. (2024) 25:424–42. doi: 10.1038/s41580-024-00703-5
6. Dai E, Chen X, Linkermann A, Jiang X, Kang R, Kagan VE, et al. A guideline on the molecular ecosystem regulating ferroptosis. Nat Cell Biol. (2024) 26:1447–57. doi: 10.1038/s41556-024-01360-8
7. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
8. Chen Y, Fang ZM, Yi X, Wei X, Jiang DS. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. (2023) 14:205. doi: 10.1038/s41419-023-05716-0
9. Yang P, Li J, Zhang T, Ren Y, Zhang Q, Liu R, et al. Ionizing radiation-induced mitophagy promotes ferroptosis by increasing intracellular free fatty acids. Cell Death Differ. (2023) 30:2432–45. doi: 10.1038/s41418-023-01230-0
10. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
11. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
12. Ru Q, Li Y, Chen L, Wu Y, Min J, Wang F. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct Target Ther. (2024) 9:271. doi: 10.1038/s41392-024-01969-z
13. Chen Q, Wang J, Xiang M, Wang Y, Zhang Z, Liang J, et al. The potential role of ferroptosis in systemic lupus erythematosus. Front Immunol. (2022) 13:855622. doi: 10.3389/fimmu.2022.855622
14. Lai B, Wu CH, Wu CY, Luo SF, Lai JH. Ferroptosis and autoimmune diseases. Front Immunol. (2022) 13:916664. doi: 10.3389/fimmu.2022.916664
15. van Swelm RPL, Wetzels JFM, Swinkels DW. The multifaceted role of iron in renal health and disease. Nat Rev Nephrol. (2020) 16:77–98. doi: 10.1038/s41581-019-0197-5
16. Zhao L, Hu X, Xiao F, Zhang X, Zhao L, Wang M. Mitochondrial impairment and repair in the pathogenesis of systemic lupus erythematosus. Front Immunol. (2022) 13:929520. doi: 10.3389/fimmu.2022.929520
17. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–363.e3. doi: 10.1016/j.molcel.2018.10.042
18. Yuan J, Li LI, Wang Z, Song W, Zhang Z. Dyslipidemia in patients with systemic lupus erythematosus: Association with disease activity and B-type natriuretic peptide levels. BioMed Rep. (2016) 4:68–72. doi: 10.3892/br.2015.544
19. Pope LE, Dixon SJ. Regulation of ferroptosis by lipid metabolism. Trends Cell Biol. (2023) 33:1077–87. doi: 10.1016/j.tcb.2023.05.003
20. Liu T, Son M, Diamond B. HMGB1 in systemic lupus erythematosus. Front Immunol. (2020) 11:1057. doi: 10.3389/fimmu.2020.01057
21. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
22. Zhang D, Wu C, Ba D, Wang N, Wang Y, Li X, et al. Ferroptosis contribute to neonicotinoid imidacloprid-evoked pyroptosis by activating the HMGB1-RAGE/TLR4-NF-κB signaling pathway. Ecotoxicol Environ Saf. (2023) 253:114655. doi: 10.1016/j.ecoenv.2023.114655
23. Davaanyam D, Lee H, Seol SI, Oh SA, Kim SW, Lee JK. HMGB1 induces hepcidin upregulation in astrocytes and causes an acute iron surge and subsequent ferroptosis in the postischemic brain. Exp Mol Med. (2023) 55:2402–16. doi: 10.1038/s12276-023-01111-z
24. Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. (2003) 170:3890–7. doi: 10.4049/jimmunol.170.7.3890
25. Dong W, Xu H, Wei W, Ning R, Chang Y. Advances in the study of ferroptosis and its relationship to autoimmune diseases. Int Immunopharmacol. (2024) 140:112819. doi: 10.1016/j.intimp.2024.112819
26. Chen H, Qi Q, Wu N, Wang Y, Feng Q, Jin R, et al. Aspirin promotes RSL3-induced ferroptosis by suppressing mTOR/SREBP-1/SCD1-mediated lipogenesis in PIK3CA-mutant colorectal cancer. Redox Biol. (2022) 55:102426. doi: 10.1016/j.redox.2022.102426
27. Huang G, Cai Y, Ren M, Zhang X, Fu Y, Cheng R, et al. Salidroside sensitizes Triple-negative breast cancer to ferroptosis by SCD1-mediated lipogenesis and NCOA4-mediated ferritinophagy. J Adv Res. (2024) 29:S2090–1232(24)00429-6. doi: 10.1016/j.jare.2024.09.027
28. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. (2020) 27:420–35. doi: 10.1016/j.chembiol.2020.02.005
29. Chen X, Yu C, Kang R, Kroemer G, Tang D. Cellular degradation systems in ferroptosis. Cell Death Differ. (2021) 28:1135–48. doi: 10.1038/s41418-020-00728-1
30. Conrad M, Proneth B. Selenium: tracing another essential element of ferroptotic cell death. Cell Chem Biol. (2020) 27:409–19. doi: 10.1016/j.chembiol.2020.03.012
31. Wang C, Leng M, Ding C, Zhu X, Zhang Y, Sun C, et al. Ferritinophagy-mediated ferroptosis facilitates methotrexate-induced hepatotoxicity by high-mobility group box 1 (HMGB1). Liver Int. (2024) 44:691–705. doi: 10.1111/liv.15811
32. Wei Q, He F, Rao J, Xiang X, Li L, Qi H. Targeting non-classical autophagy-dependent ferroptosis and the subsequent HMGB1/TfR1 feedback loop accounts for alleviating solar dermatitis by senkyunolide I. Free Radic Biol Med. (2024) 223:263–80. doi: 10.1016/j.freeradbiomed.2024.08.004
33. Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. (2021) 17:948–60. doi: 10.1080/15548627.2020.1739447
34. Zhang DD. Ironing out the details of ferroptosis. Nat Cell Biol. (2024) 26:1386–93. doi: 10.1038/s41556-024-01361-7
35. Roemhild K, von Maltzahn F, Weiskirchen R, Knüchel R, von Stillfried S, Lammers T. Iron metabolism: pathophysiology and pharmacology. Trends Pharmacol Sci. (2021) 42:640–56. doi: 10.1016/j.tips.2021.05.001
36. Van Raaij S, Van Swelm R, Bouman K, Cliteur M, Van Den Heuvel MC, Pertijs J, et al. Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease. Sci Rep. (2018) 8:9353. doi: 10.1038/s41598-018-31457-8
37. Marks ES, Bonnemaison ML, Brusnahan SK, Zhang W, Fan W, Garrison JC, et al. Renal iron accumulation occurs in lupus nephritis and iron chelation delays the onset of albuminuria. Sci Rep. (2017) 7:12821. doi: 10.1038/s41598-017-13029-4
38. Vanarsa K, Ye Y, Han J, Xie C, Mohan C, Wu T. Inflammation associated anemia and ferritin as disease markers in SLE. Arthritis Res Ther. (2012) 14:R182. doi: 10.1186/ar4012
39. Hinze CH, Suzuki M, Klein-Gitelman M, Passo MH, Olson J, Singer NG, et al. Neutrophil gelatinase-associated lipocalin is a predictor of the course of global and renal childhood-onset systemic lupus erythematosus disease activity. Arthritis Rheum. (2009) 60:2772–81. doi: 10.1002/art.24751
40. Urrego T, Ortiz-Reyes B, Vanegas-Garcia AL, Munoz CH, Gonzalez LA, Vasquez G, et al. Utility of urinary transferrin and ceruloplasmin in patients with systemic lupus erythematosus for differentiating patients with lupus nephritis. Reumatol Clin. (2020) 16:17–23. doi: 10.1016/j.reuma.2018.02.002
41. Suzuki M, Wiers K, Brooks EB, Greis KD, Haines K, Klein-Gitelman MS, et al. Initial validation of a novel protein biomarker panel for active pediatric lupus nephritis. Pediatr Res. (2009) 65:530–6. doi: 10.1203/PDR.0b013e31819e4305
42. Suzuki M, Wiers KM, Klein-Gitelman MS, Haines KA, Olson J, Onel KB, et al. Neutrophil gelatinase-associated lipocalin as a biomarker of disease activity in pediatric lupus nephritis. Pediatr Nephrol. (2008) 23:403–12. doi: 10.1007/s00467-007-0685-x
43. Abulaban KM, Song H, Zhang X, Kimmel PL, Kusek JW, Nelson RG, et al. Predicting decline of kidney function in lupus nephritis using urine biomarkers. Lupus. (2016) 25:1012–8. doi: 10.1177/0961203316631629
44. Brown AC. Lupus erythematosus and nutrition: a review of the literature. J Ren Nutr. (2000) 10:170–83. doi: 10.1053/jren.2000.16323
45. Oh VM. Iron dextran and systemic lupus erythematosus. BMJ. (1992) 305:1000. doi: 10.1136/bmj.305.6860.1000-a
46. Leiter LM, Reuhl KR, Racis SP Jr, Sherman AR. Iron status alters murine systemic lupus erythematosus. J Nutr. (1995) 125:474–84. doi: 10.1093/jn/125.3.474
47. ScIndia Y, Wlazlo E, Ghias E, Cechova S, Loi V, Leeds J, et al. Modulation of iron homeostasis with hepcidin ameliorates spontaneous murine lupus nephritis. Kidney Int. (2020) 98:100–15. doi: 10.1016/j.kint.2020.01.025
48. Wlazlo E, Mehrad B, Morel L, ScIndia Y. Iron metabolism: an under investigated driver of renal pathology in lupus nephritis. Front Med (Lausanne). (2021) 8:643686. doi: 10.3389/fmed.2021.643686
49. Akiela B, Kosałka J, Plutecka H, Bazan-Socha S, Sanak M, Musiał J. Facilitated expansion of Th17 cells in lupus nephritis patients. Clin Exp Immunol. (2018) 194:283–94. doi: 10.1111/cei.13196
50. Tsanaktsi A, Solomou EE, Liossis SC. Th1/17 cells, a subset of Th17 cells, are expanded in patients with active systemic lupus erythematosus. Clin Immunol. (2018) 195:101–6. doi: 10.1016/j.clim.2018.08.005
51. Ni S, Yuan Y, Kuang Y, Li X. Iron metabolism and immune regulation. Front Immunol. (2022) 13:816282. doi: 10.3389/fimmu.2022.816282
52. Zhao M, Li MY, Gao XF, Jia SJ, Gao KQ, Zhou Y, et al. Downregulation of BDH2 modulates iron homeostasis and promotes DNA demethylation in CD4+ T cells of systemic lupus erythematosus. Clin Immunol. (2018) 187:113–21. doi: 10.1016/j.clim.2017.11.002
53. Gao X, Song Y, Wu J, Lu S, Min X, Liu L, et al. Iron-dependent epigenetic modulation promotes pathogenic T cell differentiation in lupus. J Clin Invest. (2022) 132:e152345. doi: 10.1172/JCI152345
54. Jabara HH, Boyden SE, Chou J, Ramesh N, Massaad MJ, Benson H, et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat Genet. (2016) 48:74–8. doi: 10.1038/ng.3465
55. Aljohani AH, Al-Mousa H, Arnaout R, Al-Dhekri H, Mohammed R, Alsum Z, et al. Clinical and immunological characterization of combined immunodeficiency due to TFRC mutation in eight patients. J Clin Immunol. (2020) 40:1103–10. doi: 10.1007/s10875-020-00851-1
56. Voss K, Sewell AE, Krystofiak ES, Gibson-Corley KN, Young AC, Basham JH, et al. Elevated transferrin receptor impairs T cell metabolism and function in systemic lupus erythematosus. Sci Immunol. (2023) 8:eabq0178. doi: 10.1126/sciimmunol.abq0178
57. Su C, Johnson ME, Torres A, Thomas RM, Manduchi E, Sharma P, et al. Mapping effector genes at lupus GWAS loci using promoter Capture-C in follicular helper T cells. Nat Commun. (2020) 11:3294. doi: 10.1038/s41467-020-17089-5
58. Toellner KM, Luther SA, Sze DM, Choy RK, Taylor DR, MacLennan IC, et al. T helper 1 (Th1) and Th2 characteristics start to develop during T cell priming and are associated with an immediate ability to induce immunoglobulin class switching. J Exp Med. (1998) 187:1193–204. doi: 10.1084/jem.187.8.1193
59. Jacquemin C, Augusto JF, Scherlinger M, Gensous N, Forcade E, Douchet I, et al. OX40L/OX40 axis impairs follicular and natural Treg function in human SLE. JCI Insight. (2018) 3:e122167. doi: 10.1172/jci.insight.122167
60. Álvarez-Rodríguez L, Martínez-Taboada V, Calvo-Alén J, Beares I, Villa I, López-Hoyos M. Altered th17/treg ratio in peripheral blood of systemic lupus erythematosus but not primary antiphospholipid syndrome. Front Immunol. (2019) 10:391. doi: 10.3389/fimmu.2019.00391
61. Zhao C, Chu Y, Liang Z, Zhang B, Wang X, Jing X, et al. Low dose of IL-2 combined with rapamycin restores and maintains the long-term balance of Th17/Treg cells in refractory SLE patients. BMC Immunol. (2019) 20:32. doi: 10.1186/s12865-019-0305-0
62. Gao X, Song Y, Lu S, Hu L, Zheng M, Jia S, et al. Insufficient iron improves pristane-induced lupus by promoting treg cell expansion. Front Immunol. (2022) 13:799331. doi: 10.3389/fimmu.2022.799331
63. Feng P, Yang Q, Luo L, Sun Y, Lv W, Wan S, et al. The kinase PDK1 regulates regulatory T cell survival via controlling redox homeostasis. Theranostics. (2021) 11:9503–18. doi: 10.7150/thno.63992
64. Billesbølle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S, et al. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature. (2020) 586:807–11. doi: 10.1038/s41586-020-2668-z
65. Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. (2024) 25:133–55. doi: 10.1038/s41580-023-00648-1
66. Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. (2008) 28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521
67. Nemeth E, Ganz T. Hepcidin and iron in health and disease. Annu Rev Med. (2023) 74:261–77. doi: 10.1146/annurev-med-043021-032816
68. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306:2090–3. doi: 10.1126/science.1104742
69. Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. (2000) 403:776–81. doi: 10.1038/35001596
70. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. (2014) 509:105–9. doi: 10.1038/nature13148
71. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. (2014) 16:1069–79. doi: 10.1038/ncb3053
72. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
73. Doll S, Conrad M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life. (2017) 69:423–34. doi: 10.1002/iub.1616
74. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523
75. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
76. Liu Y, Luo X, Chen Y, Dang J, Zeng D, Guo X, et al. Heterogeneous ferroptosis susceptibility of macrophages caused by focal iron overload exacerbates rheumatoid arthritis. Redox Biol. (2024) 69:103008. doi: 10.1016/j.redox.2023.103008
77. Alli AA, Desai D, Elshika A, Conrad M, Proneth B, Clapp W, et al. Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis. Clin Immunol. (2023) 248:109213. doi: 10.1016/j.clim.2022.109213
78. Glover HL, Schreiner A, Dewson G, Tait SWG. Mitochondria and cell death. Nat Cell Biol. (2024) 26:1434–46. doi: 10.1038/s41556-024-01429-4
79. Lin L, Ren R, Xiong Q, Zheng C, Yang B, Wang H. Remodeling of T-cell mitochondrial metabolism to treat autoimmune diseases. Autoimmun Rev. (2024) 23:103583. doi: 10.1016/j.autrev.2024.103583
80. Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin Immunol. (2012) 144:200–13. doi: 10.1016/j.clim.2012.07.001
81. Cao S, Jiang J, Yin H, Wang L, Lu Q. Abnormal energy metabolism in the pathogenesis of systemic lupus erythematosus. Int Immunopharmacol. (2024) 134:112149. doi: 10.1016/j.intimp.2024.112149
82. Morel L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol. (2017) 13:280–90. doi: 10.1038/nrrheum.2017.43
83. Gergely P Jr, Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. (2002) 46:175–90. doi: 10.1002/1529-0131(200201)46:1
84. Doherty E, Oaks Z, Perl A. Increased mitochondrial electron transport chain activity at complex I is regulated by N-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid Redox Signal. (2014) 21:56–65. doi: 10.1089/ars.2013.5702
85. Yang Z, Matteson EL, Goronzy JJ, Weyand CM. T-cell metabolism in autoimmune disease. Arthritis Res Ther. (2015) 17:29. doi: 10.1186/s13075-015-0542-4
86. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
87. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
88. Chun J, Chung H, Wang X, Barry R, Taheri ZM, Platnich JM, et al. NLRP3 localizes to the tubular epithelium in human kidney and correlates with outcome in IgA nephropathy. Sci Rep. (2016) 6:24667. doi: 10.1038/srep24667
89. Qi YY, Zhou XJ, Cheng FJ, Hou P, Ren YL, Wang SX, et al. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Ann Rheum Dis. (2018) 77:1799–809. doi: 10.1136/annrheumdis-2018-213028
90. Tian Y, Guo H, Miao X, Xu J, Yang R, Zhao L, et al. Nestin protects podocyte from injury in lupus nephritis by mitophagy and oxidative stress. Cell Death Dis. (2020) 11:319. doi: 10.1038/s41419-020-2547-4
91. Halfon M, Tankeu AT, Ribi C. Mitochondrial dysfunction in systemic lupus erythematosus with a focus on lupus nephritis. Int J Mol Sci. (2024) 25:6162. doi: 10.3390/ijms25116162
92. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. (2021) 220:e202105043. doi: 10.1083/jcb.202105043
93. Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. (2018) 13:1013–20. doi: 10.1021/acschembio.8b00199
94. Bogacz M, Krauth-Siegel RL. Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death. Elife. (2018) 7:e37503. doi: 10.7554/eLife.37503
95. Liang FG, Zandkarimi F, Lee J, Axelrod JL, Pekson R, Yoon Y, et al. OPA1 promotes ferroptosis by augmenting mitochondrial ROS and suppressing an integrated stress response. Mol Cell. (2024) 84:3098–3114.e6. doi: 10.1016/j.molcel.2024.07.020
96. Borcherding N, Brestoff JR. The power and potential of mitochondria transfer. Nature. (2023) 623:283–91. doi: 10.1038/s41586-023-06537-z
97. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. (2020) 32:920–37. doi: 10.1016/j.cmet.2020.10.011
98. Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med. (2018) 117:45–57. doi: 10.1016/j.freeradbiomed.2018.01.019
99. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
100. Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. (2021) 33:174–189.e7. doi: 10.1016/j.cmet.2020.12.007
101. Conlon M, Poltorack CD, Forcina GC, Armenta DA, Mallais M, Perez MA, et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat Chem Biol. (2021) 17:665–74. doi: 10.1038/s41589-021-00751-4
102. Shin D, Lee J, You JH, Kim D, Roh JL. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. (2020) 30:101418. doi: 10.1016/j.redox.2019.101418
103. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
104. Chen Z, Sun X, Li X, Liu N. Oleoylethanolamide alleviates hyperlipidaemia-mediated vascular calcification via attenuating mitochondrial DNA stress triggered autophagy-dependent ferroptosis by activating PPARα. Biochem Pharmacol. (2023) 208:115379. doi: 10.1016/j.bcp.2022.115379
105. Carvalho G, Repolês BM, Mendes I, Wanrooij PH. Mitochondrial DNA instability in mammalian cells. Antioxid Redox Signal. (2022) 36:885–905. doi: 10.1089/ars.2021.0091
106. Lai K, Wang J, Lin S, Chen Z, Lin G, Ye K, et al. Sensing of mitochondrial DNA by ZBP1 promotes RIPK3-mediated necroptosis and ferroptosis in response to diquat poisoning. Cell Death Differ. (2024) 31:635–50. doi: 10.1038/s41418-024-01279-5
107. Ferreira HB, Pereira AM, Melo T, Paiva A, Domingues MR. Lipidomics in autoimmune diseases with main focus on systemic lupus erythematosus. J Pharm BioMed Anal. (2019) 174:386–95. doi: 10.1016/j.jpba.2019.06.005
108. Sun W, Li P, Cai J, Ma J, Zhang X, Song Y, et al. Lipid metabolism: immune regulation and therapeutic prospectives in systemic lupus erythematosus. Front Immunol. (2022) 13:860586. doi: 10.3389/fimmu.2022.860586
109. Zhou B, Xia Y, She J. Dysregulated serum lipid profile and its correlation to disease activity in young female adults diagnosed with systemic lupus erythematosus: a cross-sectional study. Lipids Health Dis. (2020) 19:40. doi: 10.1186/s12944-020-01232-8
110. Ganjali S, Shirmohammadi L, Read MI, Sahebkar A. High-density lipoprotein functionality in systemic lupus erythematosus. Semin Arthritis Rheum. (2020) 50:769–75. doi: 10.1016/j.semarthrit.2020.05.011
111. Kim SY, Yu M, Morin EE, Kang J, Kaplan MJ, Schwendeman A. High-density lipoprotein in lupus: disease biomarkers and potential therapeutic strategy. Arthritis Rheumatol. (2020) 72:20–30. doi: 10.1002/art.41059
112. Gaál K, Tarr T, Lőrincz H, Borbás V, Seres I, Harangi M, et al. High-density lipopoprotein antioxidant capacity, subpopulation distribution and paraoxonase-1 activity in patients with systemic lupus erythematosus. Lipids Health Dis. (2016) 15:60. doi: 10.1186/s12944-016-0229-0
113. Smith CK, Seto NL, Vivekanandan-Giri A, Yuan W, Playford MP, Manna Z, et al. Lupus high-density lipoprotein induces proinflammatory responses in macrophages by binding lectin-like oxidised low-density lipoprotein receptor 1 and failing to promote activating transcription factor 3 activity. Ann Rheum Dis. (2017) 76:602–11. doi: 10.1136/annrheumdis-2016-209683
114. Ronda N, Favari E, Borghi MO, Ingegnoli F, Gerosa M, Chighizola C, et al. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. (2014) 73:609–15. doi: 10.1136/annrheumdis-2012-202914
115. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. (2015) 163:1716–29. doi: 10.1016/j.cell.2015.11.045
116. Xiao J, Li W, Zheng X, Qi L, Wang H, Zhang C, et al. Targeting 7-dehydrocholesterol reductase integrates cholesterol metabolism and IRF3 activation to eliminate infection. Immunity. (2020) 52:109–122.e6. doi: 10.1016/j.immuni.2019.11.015
117. Ho LJ, Wu CH, Luo SF, Lai JH. Vitamin D and systemic lupus erythematosus: Causality and association with disease activity and therapeutics. Biochem Pharmacol. (2024) 227:116417. doi: 10.1016/j.bcp.2024.116417
118. Zhang F, Zhang B, Ding H, Li X, Wang X, Zhang X, et al. The oxysterol receptor EBI2 links innate and adaptive immunity to limit IFN response and systemic lupus erythematosus. Adv Sci (Weinh). (2023) 10:e2207108. doi: 10.1002/advs.202207108
119. Frostegård J, Svenungsson E, Wu R, Gunnarsson I, Lundberg IE, Klareskog L, et al. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. (2005) 52:192–200. doi: 10.1002/art.20780
120. Khan F, Moinuddin, Mir AR, Islam S, Alam K, Ali A. Immunochemical studies on HNE-modified HSA: Anti-HNE-HSA antibodies as a probe for HNE damaged albumin in SLE. Int J Biol Macromol. (2016) 86:145–54. doi: 10.1016/j.ijbiomac.2016.01.053
121. Tanhapour M, Miri A, Vaisi-Raygani A, Bahrehmand F, Kiani A, Rahimi Z, et al. Synergism between apolipoprotein E Ɛ4 allele and paraoxonase (PON1) 55-M allele is associated with risk of systemic lupus erythematosus. Clin Rheumatol. (2018) 37:971–7. doi: 10.1007/s10067-017-3859-3
122. Wójcik P, Gęgotek A, Žarković N, Skrzydlewska E. Oxidative stress and lipid mediators modulate immune cell functions in autoimmune diseases. Int J Mol Sci. (2021) 22:723. doi: 10.3390/ijms22020723
123. Danieli MG, Antonelli E, Longhi E, Gangemi S, Allegra A. The role of microbiota and oxidative stress axis and the impact of intravenous immunoglobulin in systemic lupus erythematosus. Autoimmun Rev. (2024) 23:103607. doi: 10.1016/j.autrev.2024.103607
124. He J, Ma C, Tang D, Zhong S, Yuan X, Zheng F, et al. Absolute quantification and characterization of oxylipins in lupus nephritis and systemic lupus erythematosus. Front Immunol. (2022) 13:964901. doi: 10.3389/fimmu.2022.964901
125. Pellefigues C, Dema B, Lamri Y, Saidoune F, Chavarot N, Lohéac C, et al. Prostaglandin D (2) amplifies lupus disease through basophil accumulation in lymphoid organs. Nat Commun. (2018) 9:725. doi: 10.1038/s41467-018-03129-8
126. Hu C, Zhang J, Hong S, Li H, Lu L, Xie G, et al. Oxidative stress-induced aberrant lipid metabolism is an important causal factor for dysfunction of immunocytes from patients with systemic lupus erythematosus. Free Radic Biol Med. (2021) 163:210–9. doi: 10.1016/j.freeradbiomed.2020.12.006
127. Hirata Y, Cai R, Volchuk A, Steinberg BE, Saito Y, Matsuzawa A, et al. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr Biol. (2023) 33:1282–1294.e5. doi: 10.1016/j.cub.2023.02.060
128. Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J, et al. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. (2021) 28:1644–57. doi: 10.1038/s41418-020-00691-x
129. Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. (2020) 11:922. doi: 10.1038/s41419-020-03118-0
130. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. (2022) 82:2215–27. doi: 10.1016/j.molcel.2022.03.022
131. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U.S.A. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
132. Kim JW, Lee JY, Oh M, Lee EW. An integrated view of lipid metabolism in ferroptosis revisited via lipidomic analysis. Exp Mol Med. (2023) 55:1620–31. doi: 10.1038/s12276-023-01077-y
133. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. (2017) 551:247–50. doi: 10.1038/nature24297
134. Warner GJ, Berry MJ, Moustafa ME, Carlson BA, Hatfield DL, Faust JR. Inhibition of selenoprotein synthesis by selenocysteine tRNA [Ser]Sec lacking isopentenyladenosine. J Biol Chem. (2000) 275:28110–9. doi: 10.1074/jbc.M001280200
135. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
136. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
137. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
138. Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature. (2019) 567:118–22. doi: 10.1038/s41586-019-0945-5
139. Sun Q, Liu D, Cui W, Cheng H, Huang L, Zhang R, et al. Cholesterol mediated ferroptosis suppression reveals essential roles of Coenzyme Q and squalene. Commun Biol. (2023) 6:1108. doi: 10.1038/s42003-023-05477-8
140. Freitas FP, Alborzinia H, Dos Santos AF, Nepachalovich P, Pedrera L, Zilka O, et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. (2024) 626:401–10. doi: 10.1038/s41586-023-06878-9
141. Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L, et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature. (2024) 626:411–8. doi: 10.1038/s41586-023-06983-9
142. Lee J, Roh JL. Cholesterol-ferroptosis nexus: Unveiling novel cancer therapeutic avenues. Cancer Lett. (2024) 597:217046. doi: 10.1016/j.canlet.2024.217046
143. Wang H, Shu L, Lv C, Liu N, Long Y, Peng X, et al. BRCC36 deubiquitinates HMGCR to regulate the interplay between ferroptosis and pyroptosis. Adv Sci (Weinh). (2024) 11:e2304263. doi: 10.1002/advs.202304263
144. Merino de Paz N, García-González M, Gómez-Bernal F, Quevedo-Abeledo JC, de Vera-González A, López-Mejias R, et al. Relationship between malondialdehyde serum levels and disease features in a full characterized series of 284 patients with systemic lupus erythematosus. Antioxidants (Basel). (2023) 12:1535. doi: 10.3390/antiox12081535
145. Liu L, de Leeuw K, Arends S, Doornbos-van-der-Meer B, Bulthuis MLC, van Goor H, et al. Biomarkers of oxidative stress in systemic lupus erythematosus patients with active nephritis. Antioxidants (Basel). (2023) 12:1627. doi: 10.3390/antiox12081627
146. Shruthi S, Thabah MM, Zachariah B, Negi VS. Association of oxidative stress with disease activity and damage in systemic lupus erythematosus: A cross sectional study from a tertiary care centre in southern India. Indian J Clin Biochem. (2021) 36:185–93. doi: 10.1007/s12291-020-00879-5
147. Hardt U, Larsson A, Gunnarsson I, Clancy RM, Petri M, Buyon JP, et al. Autoimmune reactivity to malondialdehyde adducts in systemic lupus erythematosus is associated with disease activity and nephritis. Arthritis Res Ther. (2018) 20:36. doi: 10.1186/s13075-018-1530-2
148. Ghorbaninezhad F, Leone P, Alemohammad H, Najafzadeh B, Nourbakhsh NS, Prete M, et al. Tumor necrosis factor-α in systemic lupus erythematosus: Structure, function and therapeutic implications (Review). Int J Mol Med. (2022) 49:43. doi: 10.3892/ijmm.2022.5098
149. Yang M, Shen Z, Zhang X, Song Z, Zhang Y, Lin Z, et al. Ferroptosis of macrophages facilitates bone loss in apical periodontitis via NRF2/FSP1/ROS pathway. Free Radic Biol Med. (2023) 208:334–47. doi: 10.1016/j.freeradbiomed.2023.08.020
150. Aringer M. Inflammatory markers in systemic lupus erythematosus. J Autoimmun. (2020) 110:102374. doi: 10.1016/j.jaut.2019.102374
151. Galozzi P, Bindoli S, Doria A, Sfriso P. The revisited role of interleukin-1 alpha and beta in autoimmune and inflammatory disorders and in comorbidities. Autoimmun Rev. (2021) 20:102785. doi: 10.1016/j.autrev.2021.102785
152. Sarrand J, Soyfoo M. Involvement of IL-33 in the pathophysiology of systemic lupus erythematosus: review. Int J Mol Sci. (2022) 23:3138. doi: 10.3390/ijms23063138
153. Chen R, Zou J, Kang R, Tang D. The redox protein high-mobility group box 1 in cell death and cancer. Antioxid Redox Signal. (2023) 39:569–90. doi: 10.1089/ars.2023.0236
154. Tang D, Kang R, Zeh HJ, Lotze MT. The multifunctional protein HMGB1: 50 years of discovery. Nat Rev Immunol. (2023) 23:824–41. doi: 10.1038/s41577-023-00894-6
155. Tanaka A, Ito T, Kibata K, Inagaki-Katashiba N, Amuro H, Nishizawa T, et al. Serum high-mobility group box 1 is correlated with interferon-α and may predict disease activity in patients with systemic lupus erythematosus. Lupus. (2019) 28:1120–7. doi: 10.1177/0961203319862865
156. Burbano C, Gómez-Puerta JA, Muñoz-Vahos C, Vanegas-García A, Rojas M, Vásquez G, et al. HMGB1+ microparticles present in urine are hallmarks of nephritis in patients with systemic lupus erythematosus. Eur J Immunol. (2019) 49:323–35. doi: 10.1002/eji.201847747
157. Zhao G, Wang X, Lei H, Ruan N, Yuan B, Tang S, et al. Serum HMGB-1 released by ferroptosis and necroptosis as a novel potential biomarker for systemic lupus erythematosus. Int Immunopharmacol. (2024) 140:112886. doi: 10.1016/j.intimp.2024.112886
158. Abdulahad DA, Westra J, Bijzet J, Limburg PC, Kallenberg CG, Bijl M. High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Arthritis Res Ther. (2011) 13:R71. doi: 10.1186/ar3332
159. Wirestam L, Schierbeck H, Skogh T, Gunnarsson I, Ottosson L, Erlandsson-Harris H, et al. Antibodies against High Mobility Group Box protein-1 (HMGB1) versus other anti-nuclear antibody fine-specificities and disease activity in systemic lupus erythematosus. Arthritis Res Ther. (2015) 17:338. doi: 10.1186/s13075-015-0856-2
160. Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. (2009) 462:99–103. doi: 10.1038/nature08512
161. Andreeva L, Hiller B, Kostrewa D, Lässig C, de Oliveira Mann CC, Jan Drexler D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. (2017) 549:394–8. doi: 10.1038/nature23890
162. Wen L, Zhang B, Wu X, Liu R, Fan H, Han L, et al. Toll-like receptors 7 and 9 regulate the proliferation and differentiation of B cells in systemic lupus erythematosus. Front Immunol. (2023) 14:1093208. doi: 10.3389/fimmu.2023.1093208
163. Hu Y, Chen B, Yang F, Su Y, Yang D, Yao Y, et al. Emerging role of the cGAS-STING signaling pathway in autoimmune diseases: Biologic function, mechanisms and clinical prospection. Autoimmun Rev. (2022) 21:103155. doi: 10.1016/j.autrev.2022.103155
164. Lu B, Antoine DJ, Kwan K, Lundbäck P, Wähämaa H, Schierbeck H, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U.S.A. (2014) 111:3068–73. doi: 10.1073/pnas.1316925111
165. Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. (2022) 54:91–102. doi: 10.1038/s12276-022-00736-w
166. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. (2022) 13:3676. doi: 10.1038/s41467-022-31218-2
167. Feng Z, Meng F, Huo F, Zhu Y, Qin Y, Gui Y, et al. Inhibition of ferroptosis rescues M2 macrophages and alleviates arthritis by suppressing the HMGB1/TLR4/STAT3 axis in M1 macrophages. Redox Biol. (2024) 75:103255. doi: 10.1016/j.redox.2024.103255
168. Deng Y, Yu L, Lai W, Xiao S, Zhang W. Knocking down macrophages Caspase-6 through HMGB1 coordinates macrophage trophoblast crosstalk to suppress ferroptosis and alleviate preeclampsia. Int Immunopharmacol. (2024) 140:112859. doi: 10.1016/j.intimp.2024.112859
169. Psarras A, Wittmann M, Vital EM. Emerging concepts of type I interferons in SLE pathogenesis and therapy. Nat Rev Rheumatol. (2022) 18:575–90. doi: 10.1038/s41584-022-00826-z
170. Caielli S, Wan Z, Pascual V. Systemic lupus erythematosus pathogenesis: interferon and beyond. Annu Rev Immunol. (2023) 41:533–60. doi: 10.1146/annurev-immunol-101921-042422
171. Liu W, Zhang S, Wang J. IFN-g, should not be ignored in SLE. Front Immunol. (2022) 13:954706. doi: 10.3389/fimmu.2022.954706
172. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol. (2021) 22:1107–17. doi: 10.1038/s41590-021-00993-3
173. Zhang S, Kang L, Dai X, Chen J, Chen Z, Wang M, et al. Manganese induces tumor cell ferroptosis through type-I IFN dependent inhibition of mitochondrial dihydroorotate dehydrogenase. Free Radic Biol Med. (2022) 193:202–12. doi: 10.1016/j.freeradbiomed.2022.10.004
174. Philips RL, Wang Y, Cheon H, Kanno Y, Gadina M, Sartorelli V, et al. The JAK-STAT pathway at 30: Much learned, much more to do. Cell. (2022) 185:3857–76. doi: 10.1016/j.cell.2022.09.023
175. Yiu G, Rasmussen TK, Tsai BL, Diep VK, Haddon DJ, Tsoi J, et al. High interferon signature leads to increased STAT1/3/5 phosphorylation in PBMCs from SLE patients by single cell mass cytometry. Front Immunol. (2022) 13:833636. doi: 10.3389/fimmu.2022.833636
176. Richter P, Cardoneanu A, Burlui AM, Macovei LA, Bratoiu I, Buliga-Finis ON, et al. Why do we need JAK inhibitors in systemic lupus erythematosus? Int J Mol Sci. (2022) 23:11788. doi: 10.3390/ijms231911788
177. Xu H, Zhang X, Wang X, Li B, Yu H, Quan Y, et al. Cellular spermine targets JAK signaling to restrain cytokine-mediated autoimmunity. Immunity. (2024) 57:1796–1811.e8. doi: 10.1016/j.immuni.2024.05.025
178. Cao T, Zhou J, Liu Q, Mao T, Chen B, Wu Q, et al. Interferon-γ induces salivary gland epithelial cell ferroptosis in Sjogren’s syndrome via JAK/STAT1-mediated inhibition of system Xc. Free Radic Biol Med. (2023) 205:116–28. doi: 10.1016/j.freeradbiomed.2023.05.027
179. Wei TT, Zhang MY, Zheng XH, Xie TH, Wang W, Zou J, et al. Interferon-γ induces retinal pigment epithelial cell Ferroptosis by a JAK1-2/STAT1/SLC7A11 signaling pathway in Age-related Macular Degeneration. FEBS J. (2022) 289:1968–83. doi: 10.1111/febs.16272
180. Kong R, Wang N, Han W, Bao W, Lu J. IFNγ-mediated repression of system xc- drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J Leukoc Biol. (2021) 110:301–14. doi: 10.1002/JLB.3MA1220-815RRR
181. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
182. Long Q, Tao H, Wang P, Wu B, Zhu Q, Chen H, et al. Fludarabine enhances radiosensitivity by promoting ferroptosis in B-cell lymphoma. Radiat Res. (2024) 201:224–39. doi: 10.1667/RADE-23-00018.1
183. Li F, Hao S, Gao J, Jiang P. EGCG alleviates obesity-exacerbated lung cancer progression by STAT1/SLC7A11 pathway and gut microbiota. J Nutr Biochem. (2023) 120:109416. doi: 10.1016/j.jnutbio.2023.109416
184. Cao S, Wei Y, Xu H, Weng J, Qi T, Yu F, et al. Crosstalk between ferroptosis and chondrocytes in osteoarthritis: a systematic review of in vivo and in vitro studies. Front Immunol. (2023) 14:1202436. doi: 10.3389/fimmu.2023.1202436
185. Yamada N, Karasawa T, Wakiya T, Sadatomo A, Ito H, Kamata R, et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am J Transplant. (2020) 20:1606–18. doi: 10.1111/ajt.15773
186. Cosialls E, Pacreau E, Duruel C, Ceccacci S, Elhage R, Desterke C, et al. mTOR inhibition suppresses salinomycin-induced ferroptosis in breast cancer stem cells by ironing out mitochondrial dysfunctions. Cell Death Dis. (2023) 14:744. doi: 10.1038/s41419-023-06262-5
187. Ji L, Xie W, Zhang Z. Efficacy and safety of sirolimus in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Semin Arthritis Rheumatol. (2020) 50:1073–80. doi: 10.1016/j.semarthrit.2020.07.006
188. Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol. (2016) 68:2728–39. doi: 10.1002/art.39791
189. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet. (2018) 391:1186–96. doi: 10.1016/S0140-6736(18)30485-9
190. Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. (2020) 32:44–55.e6. doi: 10.1016/j.cmet.2020.04.015
191. Wang M, Tian T, Zhou H, Jiang SY, Jiao YY, Zhu Z, et al. Metformin normalizes mitochondrial function to delay astrocyte senescence in a mouse model of Parkinson’s disease through Mfn2-cGAS signaling. J Neuroinflamm. (2024) 21:81. doi: 10.1186/s12974-024-03072-0
192. Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal. (2016) 9:ra115. doi: 10.1126/scisignal.aaf1933
193. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
194. Askari B, Kanter JE, Sherrid AM, Golej DL, Bender AT, Liu J, et al. Rosiglitazone inhibits acyl-CoA synthetase activity and fatty acid partitioning to diacylglycerol and triacylglycerol via a peroxisome proliferator-activated receptor-gamma-independent mechanism in human arterial smooth muscle cells and macrophages. Diabetes. (2007) 56:1143–52. doi: 10.2337/db06-0267
195. Arenas-Jal M, Suñé-Negre JM, García-Montoya E. Coenzyme Q10 supplementation: Efficacy, safety, and formulation challenges. Compr Rev Food Sci Food Saf. (2020) 19:574–94. doi: 10.1111/1541-4337.12539
196. Blanco LP, Pedersen HL, Wang X, Lightfoot YL, Seto N, Carmona-Rivera C, et al. Improved mitochondrial metabolism and reduced inflammation following attenuation of murine lupus with coenzyme Q10 analog idebenone. Arthritis Rheumatol. (2020) 72:454–64. doi: 10.1002/art.41128
197. Mladěnka P, Macáková K, Kujovská Krčmová L, Javorská L, Mrštná K, OEMONOM researchers and collaborators. Vitamin K - sources, physiological role, kinetics, deficiency, detection, therapeutic use, and toxicity. Nutr Rev. (2022) 80:677–98. doi: 10.1093/nutrit/nuab061
198. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. (2022) 608:778–83. doi: 10.1038/s41586-022-05022-3
199. Mishima E, Wahida A, Seibt T, Conrad M. Diverse biological functions of vitamin K: from coagulation to ferroptosis. Nat Metab. (2023) 5:924–32. doi: 10.1038/s42255-023-00821-y
200. Monti DA, Zabrecky G, Kremens D, Liang TW, Wintering NA, Cai J, et al. N-acetyl cysteine may support dopamine neurons in parkinson’s disease: preliminary clinical and cell line data. PloS One. (2016) 11:e0157602. doi: 10.1371/journal.pone.0157602
201. Morley KC, Baillie A, Van Den Brink W, Chitty KE, Brady K, Back SE, et al. N-acetyl cysteine in the treatment of alcohol use disorder in patients with liver disease: Rationale for further research. Expert Opin Investig Drugs. (2018) 27:667–75. doi: 10.1080/13543784.2018.1501471
202. Monti DA, Zabrecky G, Kremens D, Liang TW, Wintering NA, Bazzan AJ, et al. N-acetyl cysteine is associated with dopaminergic improvement in parkinson’s disease. Clin Pharmacol Ther. (2019) 106:884–90. doi: 10.1002/cpt.1548
203. Wang Y, Chen Q, Shi C, Jiao F, Gong Z. Mechanism of glycyrrhizin on ferroptosis during acute liver failure by inhibiting oxidative stress. Mol Med Rep. (2019) 20:4081–90. doi: 10.3892/mmr.2019.10660
204. Qi J, Kim JW, Zhou Z, Lim CW, Kim B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am J Pathol. (2020) 190:68–81. doi: 10.1016/j.ajpath.2019.09.011
205. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U.S.A. (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
206. He L. Metformin and systemic metabolism. Trends Pharmacol Sci. (2020) 41:868–81. doi: 10.1016/j.tips.2020.09.001
207. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. (2015) 7:274ra18. doi: 10.1126/scitranslmed.aaa0835
Keywords: systemic lupus erythematosus (SLE), ferroptosis, interferons (IFNs), mitochondrial, damage-associated molecular patterns (DAMPs)
Citation: Zhao G, Li X, Zhang Y, Wang X, Deng L, Xu J, Jin S, Zuo Z, Xun L, Luo M, Yang F, Qi J and Fu P (2025) Intricating connections: the role of ferroptosis in systemic lupus erythematosus. Front. Immunol. 16:1534926. doi: 10.3389/fimmu.2025.1534926
Received: 26 November 2024; Accepted: 17 January 2025;
Published: 04 February 2025.
Edited by:
Yu’e Liu, Boston Children’s Hospital and Harvard Medical School, United StatesReviewed by:
Jin-Ming Zhang, University of Texas Health Science Center at Houston, United StatesZurong Wan, Cornell University, United States
Copyright © 2025 Zhao, Li, Zhang, Wang, Deng, Xu, Jin, Zuo, Xun, Luo, Yang, Qi and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Yang, NjQ1NjE2NTFAcXEuY29t; Jialong Qi, OTYyOTkyNTk4QHFxLmNvbQ==; Ping Fu, MTc1NjA0NzI2N0BxcS5jb20=
†These authors share first authorship