 Bingqian Huang
Bingqian Huang Yulin Yang
Yulin Yang Jinming Liu
Jinming Liu Biao Zhang
Biao Zhang Nengming Lin
Nengming Lin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 11 March 2025
Sec. Cancer Immunity and Immunotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1533007
This article is part of the Research Topic The Role of Ubiquitination in Disease Development, Progression, and Prognosis View all 10 articles
Mitochondrial homeostasis (MH) refers to the dynamic balance of mitochondrial number, function, and quality within cells. Maintaining MH is significant in the occurrence, development, and clinical treatment of Gastrointestinal (GI) tumors. Ubiquitination, as an important post-translational modification mechanism of proteins, plays a central role in the regulation of MH. Over the past decade, research on the regulation of MH by ubiquitination has focused on mitochondrial biogenesis, mitochondrial dynamics, Mitophagy, and mitochondrial metabolism during these processes. This review summarizes the mechanism and potential therapeutic targets of ubiquitin (Ub)-regulated MH intervention in GI tumors.
Gastrointestinal (GI) tumors, including hepatocellular carcinoma (HCC), esophageal cancer (ESCA), gastric cancer (GC), colorectal cancer (CRC), and pancreatic cancer (PAAD) (1), are the leading causes of cancer-related deaths worldwide. Highlighting its severe public health burden, there are an estimated 4.9 million new cases and 3.9 million deaths annually, according to the latest data (2). Mitochondria (Mt) plays a crucial role in the occurrence and development of GI tumors (3). As a key organelle in the metabolic reprogramming of cancer cells, Mt dysfunction is one of the main drivers of cancer initiation and progression (4). Mt stress releases Mitochondrial DNA (mtDNA) into the cytoplasm and extracellular space, activating multiple innate immune signals (5). In GI tumors, the mtDNA mutation rate is higher, mainly in the D-loop region, which is the hypervariable region of mtDNA and is responsible for the regulation of mtDNA transcription and replication, which may be related to the special physiological environment of gastrointestinal cells (such as acidic environment, frequent cell turnover, etc.) (6, 7). Concurrently, the increased electron leakage from Mt stress generates a high concentration of reactive oxygen species (ROS), which further aggravates tissue damage and inflammation and suppresses the signal presentation between dendritic cells and T cells, leading to immune cell dysfunction and further promoting the infiltration of tumor-associated macrophages and the formation of an immunosuppressive tumor microenvironment (8, 9). Additionally, DNA and ROS released by Mt can change the balance of intestinal microbiota, leading to damage to the intestinal barrier and the occurrence of inflammatory bowel disease (IBD) (10). IBD is one of the risk factors for CRC (11). In the late GI tumors stage, Mitochondrial metabolism (MM) may instead increase, promoting cancer growth (10). This may be related to the B-cell lymphoma/leukemia-2 (BCL-2) protein family promoting the increase of mitochondrial permeability transition and mitochondrial permeability transition resistance, affecting the release of cytochrome C, thereby promoting the malignant transformation and progression of tumors (12). Studies have shown that Mt can also act as strategic molecular intermediaries or transport media for targeted therapies involved in antitumor activities (13). In summary, Mt dysfunction and structural damage further complicate the mechanism of GI tumorigenesis and development. Therefore, maintaining the normal function of Mt and regulating Mitochondrial homeostasis (MH) is key to intervening in the progression of GI tumors. MH refers to a state of balance in which Mt maintains normal function and structure within cells, including processes such as Mitochondrial biogenesis (MB), Mitochondrial dynamics (MK), Mitophagy, and MM (14–16).
Ubiquitination is involved in regulating these processes to maintain normal cellular functions. Ubiquitination is one of the main post-translational modifications of proteins, involving the addition of Ub molecules to target proteins, and plays a role in regulating protein degradation, signal transduction, DNA repair, cell cycle, and apoptosis within organisms (17–19). The process of ubiquitination is primarily catalyzed by E1 Ub-activating enzymes, E2 Ub-conjugating enzymes, and E3 Ub ligases, which play roles at different stages of the enzymatic ubiquitination cascade. Deubiquitinating enzymes (DUBs) can reverse ubiquitination by removing Ub. Under normal conditions, the ubiquitination process balances MH through the action of ubiquitinating enzymes and DUBs (20). However, when the ubiquitination process is disrupted, and MH is imbalanced, the progression of GI tumors is affected (Figure 1). This study to elucidate the molecular mechanisms and therapeutic targets of ubiquitination-regulated MH in the intervention of gastrointestinal tumors by reviewing the literature on Mt, ubiquitination, and GI tumors published in the past decade from databases such as PubMed and Web of Science, thereby providing theoretical support for the clinical diagnosis and treatment of GI tumors.
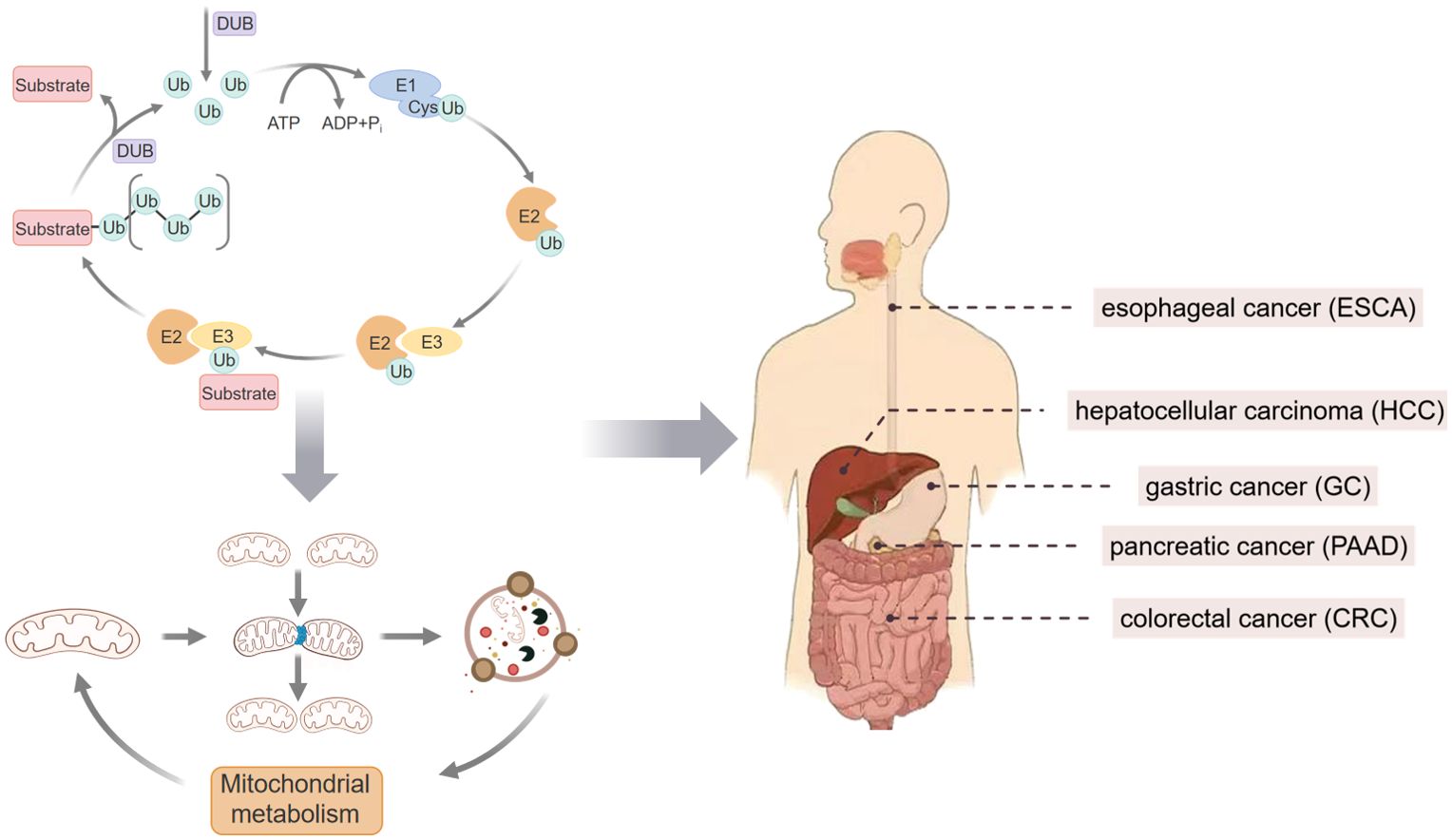
Figure 1. Schematic representation of MH targeted by ubiquitination affecting GI tumors.
MB refers to the synthesis of new Mt within cells, a process activated by various physiological and environmental signals such as cellular stress, increased energy demands, exercise training, and hormonal changes (21). This process primarily involves the transcriptional activation of nuclear-encoded mitochondrial genes, the translocation of corresponding proteins to Mt, the replication of mtDNA, and the synthesis of mitochondrial phospholipids (22). We found that ubiquitination targets peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) to participate in the transcriptional activation of nuclear-encoded mitochondrial genes, Mitochondrial ubiquitin ligase/the E3 ligase Membrane-associated ring-gh-type finger 5 (MITOL/MARCH5) ubiquitinates DNA polymerase gamma catalytic subunit (PolγA) to mediate mtDNA replication, and the tumor necrosis factor receptor-associated aactor 6 (TRAF6) E3 ligase restricts protein translocation, while mitochondrial phospholipid synthesis and membrane changes also reciprocally affect ubiquitination (23, 24).
PGC-1α is a primary regulatory factor in MB and energy metabolism, controlling both the nuclear and mitochondrial genomes (25, 26). PGC-1α regulates mtDNA replication, transcription, and translation, as well as the assembly of oxidative phosphorylation (OXPHOS), through co-activating nuclear respiratory factors (NRFs) and estrogen-related receptor alpha, thus maintaining mitochondrial quantity and function (27). Additionally, as a cellular energy responder, PGC-1α is activated via the AMP-activated protein kinase/silent information regulator 1 (SIRT1) pathway, enhancing MB (28). The heme oxygenase 1/PGC-1α pathway responds to oxidative stress by directly activating antioxidant enzymes such as superoxide dismutase, catalase, and glutathione peroxidase, thereby improving the cell’s antioxidant capacity, reducing ROS production, maintaining mitochondrial adenosine-triphosphate (ATP) levels and membrane potential (MMP), and protecting Mt from oxidative stress-induced damage (29).
The mechanisms of ubiquitination regulating PGC-1α mainly include promoting PGC-1α degradation, modulating its stability, and controlling its activity. Research has shown that radiation-induced DNA-dependent protein kinase can phosphorylate serine 636 of PGC-1α, thereby enhancing the binding of the E3 Ub ligase Ring finger protein 34 to PGC-1α and accelerating its ubiquitination and degradation (25). However, this binding can be competitively blocked by lysine methyltransferase 5C, which reduces the ubiquitination level of PGC-1α and extends its half-life (30). In addition, neural precursor cell expressed, developmentally downregulated protein 4-1 (NEDD4-1) is also an E3 ligase that can mediate the ubiquitination and degradation of PGC-1α by enhancing the recognition of the “TPPTTPP” sequence in PGC-1α through phosphorylation mediated by glycogen synthase kinase 3β (GSK-3β) (31).
The mitochondrial membrane-integrated Ub ligase MITOL is a key regulator of mitochondrial membrane fission, fusion, and mitophagy (32). PolγA is the only DNA polymerase in Mt, and it replicates mtDNA through a process known as “D-loop replication,” where the heavy strand is replicated first, followed by the light strand (33). Recent studies have shown that MITOL ubiquitinates PolγA, negatively regulating its interaction with the translocase of the outer mitochondrial membrane 20 (Tom20), thereby inhibiting its entry into Mt (34).
TRAF6 limits the translocation of tumor protein 53(p53) toMt by promoting the ubiquitination of p53 at lysine 24 (K24) in the cytoplasm through K63-linked Ub chains. This modification restricts the interaction of p53 with myeloid cell leukemia-1 (MCL-1)/BCL-2 Antagonist 1 (BAK), thereby inhibiting p53 mitochondrial translocation (35).
Cardiolipin (CL) is a characteristic phospholipid of the mitochondrial inner membrane, playing a crucial role in cellular energy metabolism, MK, and the initiation of apoptotic pathways (36). The precursor for CL synthesis is phosphatidic acid (PA), which is synthesized in the endoplasmic reticulum and then transported to the outer mitochondrial membrane (OMM) (37). From there, it is transferred to the inner membrane for CL synthesis by the uncharacterized protein with a phosphatidic acid transfer protein-like domain1-mitochondrial distribution and morphology Protein 35 (MDM35) protein complex in the intermembrane space (38). Studies have shown that common lipids in the mitochondrial membrane interact with MITOL and influence its activity and stability depending on the in vitro lipid composition (39). Notably, the binding of CL to purified MITOL significantly reduces its thermal stability, whereas the presence of PA enhances its stability most strongly (32), which further confirms that lipids can directly affect the activity of Ub ligases and may control the ubiquitination-dependent mechanisms regulating MK and turnover. Phosphatidylcholine is a lipid essential for mitochondrial membrane construction, primarily synthesized through the Kennedy pathway using choline as a substrate. During phosphatidylcholine synthesis, choline kinase accelerates mitochondrial damage. The mitochondrial kinase PTEN-induced putative kinase 1 (PINK1) accumulates on the membrane of damaged Mt, activates the Parkin rbr E3 ub protein Ligase (Parkin), and promotes substrate ubiquitination to initiate mitophagy (40).
Mt are highly dynamic organelles capable of undergoing continuous cycles of fusion and fission, thereby altering their morphology, size, and spatial distribution (41). This physiological process is referred to as MK. MK refers to the state in which Mt maintains homeostasis through fusion and fission processes in cells. This dynamic change causes Mt to assume a variety of morphologies in the cytoplasm, such as punctate, fragmented, strip, or linear. MK is closely related to the functions of Mt, such as cell proliferation, cell metabolism, and cell migration (42). This balance is maintained by various dynamin-related guanosine triphosphatase (GTPases), which play critical roles in these processes (43). Key proteins involved include dynamin-related protein 1 (Drp1), mitofusin 1 (MFN1), mitofusin 2 (MFN2), and aptic atrophy 1 (OPA1), which mediate membrane dynamics and structural changes (44).
Drp1 is an essential protein in the mitochondrial fission process, containing a GTPase domain that enables it to bind and hydrolyze GTP (45). Drp1 primarily resides in the cytoplasm and, in response to mitochondrial fission signals, is recruited to the mitochondrial surface. There, it assembles into ring-like structures and utilizes the energy from GTP hydrolysis to divide Mt into two separate units (46). Identified Drp1 receptors include mitochondrial fission factor (Fis1), mitochondrial fission factor, and MK proteins of 49 kDa and 51 kDa (47). These receptor proteins facilitate the localization of Drp1 toMt through protein-protein interactions. Recent studies have revealed that the clustered Mt homolog gene and its drosophila homolog clueless promote the recruitment of Drp1 to the mitochondrial surface via its receptors in both human cell lines and Drosophila models, thereby enhancing mitochondrial fission (48). Current studies have shown that Drp1 can be targeted for ubiquitination and degradation by E3 Ub ligases, such as MITOL and Parkin, thereby influencing the normal mitochondrial fission-fusion process (49, 50). Knockout of either enzyme results in increased Drp1 activity and uncontrolled mitochondrial hyper-fragmentation (49). In cells lacking MITOL, re-expression of MITOL reverses the suppression of stress-induced apoptosis (51). Furthermore, emerging evidence indicates that Drp1 is not only a substrate of MITOL but also a regulatory factor of MITOL activity. Drp1 may modulate MK by influencing the functional activity of MITOL (52).
The mitochondrial fusion proteins MFN1, MFN2, and OPA1 are essential GTPases responsible for the structural fusion of mitochondrial membranes. MFN1 and MFN2 regulate the fusion of the OMM, while OPA1 mediates the fusion of the inner mitochondrial membrane (IMM) (53). Studies have shown that a family with sequence similarity 73 member A/B promotes mitochondrial fusion through the regulation of phospholipid metabolism, particularly via mitochondrial phospholipase D, in collaboration with MFN1/2 on the OMM (54). Additionally, the active domain of G-protein β2 (Gβ2) undergoes structural remodeling with MFN1’s binding domain, regulating MFN1 migration on the mitochondrial membrane and facilitating mitochondrial fusion. However, Gβ2 does not interact with MFN2 (55). Sudeshna Nag et al. (56) reported that under stress conditions induced by carbonyl cyanide 3-chlorophenylhydrazone (CCCP), the interaction between mitochondrial phosphatase phosphoglycerate mutase 5 (PGAM5) and MFN2 is weakened. Instead, PGAM5 shifts to interact with Drp1. Concurrently, MFN2 undergoes phosphorylation and ubiquitination by kinases and E3 Ub ligases, leading to proteasomal degradation. This results in an increased proportion of Mt failing to undergo fusion, as well as mitochondrial fragmentation and degradation. This process can be reversed by deubiquitinating enzymes such as Ub-specific protease 30 (USP30). USP30, a deubiquitinase, inhibits mitochondrial fusion by reducing the non-degradative ubiquitination levels of MFN1/2 (57). The SCFMdm30 complex, an intracellular E3 Ub ligase complex composed of S phase kinase-associated protein 1, Cullin1 (CUL1), F-box protein (FBX), and Ring-box 1, promotes the K48-linked ubiquitination of fusion of Mt protein 1 (Fzo1) (the yeast homolog of MFN1/2), leading to its degradation and subsequently impairing mitochondrial fusion (58). In contrast, USP2 specifically binds to the ubiquitinated form of Fzo1, removing the Ub modification to regulate Fzo1 stability and enhance mitochondrial fusion efficiency (59).
Compared to MFN1/2, OPA1 exhibits a broader range of functions, including maintaining the respiratory chain, MMP, cristae structure, regulation of apoptosis, and mitochondrial DNA stability (60). OPA1 exists in multiple isoforms, primarily categorized into long isoforms (L-OPA1, including a and b) and short isoforms (S-OPA1, including c, d, and e). L-OPA1 predominantly regulates the fusion of IMM, while S-OPA1 is involved in IMM fission (61). Studies have shown that treatment of SH-SY5Y cells with 6-hydroxydopamine (6-OHDA) results in a decrease in the protein levels of MFN2 and OPA1. Conversely, 6-OHDA treatment increases the expression of Fis1 and Drp1, leading to excessive mitochondrial fission, thereby affecting mitochondrial morphology and function (62). Moreover, the study found that carnosic acid, a rosemary extract, enhances the ubiquitination of inhibitor of nuclear factor kappa-B kinase subunit gamma (IKKγ), activating the Parkin/IKKγ/p65 signaling pathway to upregulate OPA1 expression and maintain MK homeostasis (62). Optic atrophy 1 (OMA1) is a metalloprotease located in the IMM, with OPA1 being one of its primary substrates (63). Under physiological conditions, OMA1 remains inactive but is rapidly activated during mitochondrial stress, such as MMP loss or excessive ROS production, negatively regulating OPA1 (64). It was demonstrated that leptin increased OPA1 expression by promoting the ubiquitin-mediated degradation of OMA1 via the GSK3 pathway, thereby enhancing the anti-apoptotic capacity of these cells (65).
Similar to MK and MB, Mitophagy is another key process in the maintenance of line MH. Mitophagy is a cellular autophagic process that involves the selective sequestration and degradation of damaged or dysfunctional Mt, thereby maintaining the integrity of the mitochondrial network and cellular homeostasis (66, 67). Ubiquitination participates in mitophagy primarily through the PINK1/Parkin pathway, the PINK1/SYNPHILIN1/SIAH1 complex, as well as the interactions of Mitochondrial E3 Ub protein ligase 1 (MUL1) (68, 69).
The PINK1/Parkin pathway is the most widely studied mitophagy pathway and is a classic Ub-dependent pathway (70). PINK1, as a sensor of mitochondrial health, accumulates on the outer membrane of damaged Mt when they are compromised and activates Parkin, which in turn promotes the recruitment of Parkin to the Mt. PINK1 phosphorylates Ub on the Ser65 site of substrates on the mitochondrial outer membrane, a process that activates Parkin, allowing it to ubiquitinate numerous substrates on the mitochondrial outer membrane, thereby triggering selective autophagy (71). In healthy Mt, PINK1 is imported into the inner membrane, where its membrane-binding portion is cleaved by the protease presenilin-associated rhomboid-like (PARL) (72). The cleaved catalytic portion exposes unstable amino acid residues at the N-terminus and is rapidly degraded by the Ub-proteasome system (UPS) (72). This process is a crucial step in mitochondrial quality control, ensuring that only functionally intact Mt remains in the cell (73). When the function of PINK1 or Parkin is impaired, the removal of damaged Mt is hindered, leading to mitochondrial dysfunction and pathological changes in the organism (74). The loss of DJ-1 inhibits the recruitment of the selective autophagy receptor, synphilin, to depolarized Mt, further blocking PINK1/Parkin-mediated mitophagy (75). In addition to removing damaged mt, the PINK1/Parkin pathway also contributes to MB development. It has been shown that loss of PINK1/Parkin in nerve cells inhibits MB and ubiquitinates Parkin-interacting substrate, thereby relieving the inhibitory effect on PGC-1α (76).
Similar to Parkin, seven in absentia homolog 1 (SIAH1) is also an E3 Ub ligase (77). Through forming a complex with PINK1 and synphiln1, SIAH1 promotes the recruitment of autophagic markers, Microtubule-associated protein one light chain 3 (LC3), and the lysosome-associated membrane protein 1 (Lamp1), thereby facilitating mitochondrial autophagy (78). LC3 is a key marker in the autophagy process. During autophagosome formation, the cytosolic form of LC3-I participates in a Ub-like reaction involving Autophagy-related (Atg) 7 and Atg3 (E1-like Ub-activating enzyme and E2-like Ub-conjugating enzyme), binding to phosphatidyl ethanolamine to form the lipidated form, LC3-II, which attaches to the autophagosomal membrane and serves as a structural protein of the autophagosome (79). LAMP1 is commonly used as a marker for lysosomes, and LAMP1-positive organelles are often referred to as lysosomal compartments. After the fusion of the autophagosome with the lysosome, lysosomal hydrolases can degrade the autophagosome’s contents, and LAMP1-labeled organelles participate in this degradation process (80).
MUL1 participates in mitochondrial autophagy and mediates MM and MK (81). MUL1 mediates sodium selenite-induced mitochondrial autophagy and the stability of the autophagy-related protein Unc-51, Like autophagy activating kinase 1 (ULK1), during this process (82). Interacting with the ULK1/ATG13 complex, MUL1 promotes the formation of K48 polyubiquitin chains on ULK1, leading to its degradation via the UPS. In muscle cells, MUL1 mediates the degradation of MFN2 via the UPS while also inducing mitochondrial autophagy (82). Additionally, by promoting the Small Ub-like modifier conjugation (SUMOylation) of the highly dynamic protein Drp1, which is recruited to Mt, MUL1 enhances the stability of Drp1 on the mitochondrial surface, playing a critical role in the dynamic regulation of mitochondrial morphology (83). The regulatory effect of MUL1 on mitochondrial energy metabolism has also been observed. MUL1 regulates the protein levels of protein kinase Bβ and hypoxia-inducible factor 1-alpha (HIF-1α) through K48-specific polyubiquitination, and the loss of MUL1 leads to the accumulation and activation of these substrates, affecting mitochondrial respiration and resulting in a shift to a new metabolic and lipidomic state (84). This is evident in the fact that, compared to wild-type cells, MUL1(-/-) cells show impaired mitochondrial respiration and increased ATP production through glycolysis, indicating a metabolic shift from oxidative phosphorylation to glycolysis (84).
Under physiological conditions, mitochondrial energy metabolism includes the tricarboxylic acid cycle, OXPHOS, and fatty acid oxidation (85). During these processes, ubiquitination plays a key role in regulating mitochondrial homeostasis through various metabolic products such as glucose, fatty acids, amino acids, and the electron transport chain (ETC) (86, 87).
Currently, the mechanisms by which ubiquitination participates in mitochondrial glucose metabolism under physiological conditions are not well understood. However, in certain pathological states, ubiquitination may play a role in reshaping mitochondrial glycolysis (88). In a study using a heart-specific promoter cTnT, the deletion of NEDD8-Activating enzyme E1 impaired cardiac oxidative metabolism and mitochondrial function, leading to the down-regulation of genes related to fatty acid utilization, while genes associated with glucose utilization were significantly up-regulated (89). Another study found that inhibition of polycomb repressive complex 1 reduced histone H2A ubiquitination (H2Aub) occupancy and, by suppressing ubiquitination, promoted the expression of Hsp27 (heat shock protein 27). Hsp27 enhances glycolysis during myocardial ischemia by activating the NF-κB/PFKFB3 signaling pathway, and it also reduces mitochondrial ROS production by interacting with Coenzyme Q9, inhibiting ferroptosis during reperfusion (90). Research progress shows that E3 ligases and deubiquitinating enzymes influence the Warburg effect in tumors by regulating glycolysis-related signaling pathways and transcription factors (91, 92). Phosphofructokinase platelet (PFKP) is a gene encoding a rate-limiting enzyme of glycolysis, and its role in mediating glycolytic regulation of tumor progression has been well-established in lung cancer and advanced prostate cancer (93, 94). HMG-CoA Reductase Degradation 1 (HRD1), as a metabolic enzyme, catalyzes the ubiquitination of PFKP and promotes its degradation, thereby inhibiting the expression and activity of PFKP in cancer cells and obstructing cell invasion and proliferation (95). The gut microbiota and its derivative metabolite taurocholic acid can epigenetically promote the glycolysis of Myeloid-derived suppressor cells by enhancing the monomethylation of the target gene H3K4 and inhibiting C-terminus of Hsc70-interacting protein-mediated PDL1 ubiquitination, which in turn suppresses the proliferation and function of effector T cells (96).
Under energy stress conditions, tumor cells mobilize lipids stored in lipid droplets and generate energy through mitochondrial fatty acid oxidation (β-oxidation) (97). The direct contact between lipid droplets promotes the hydrolysis of triglycerides in the lipid droplets into fatty acids and glycerol, which are then transported into theMt (98). In this process, nicotinamide adenine dinucleotide kinase (NADK) regulates fatty acid synthesis by maintaining the intracellular coenzyme nicotinamide adenine dinucleotide phosphate levels, thereby controlling lipid storage and metabolic homeostasis in lipid droplets (98). The knockdown of NADK affects the levels of acetyl-CoA, thereby regulating the acetylation modification of the key transcription factor PGC-1α and MB and influencing mitochondrial function and number by affecting CL synthesis (99). A Study found that mitochondrial Signal Transducer and Activator of Transcription 3 (STAT3) could reduce the ubiquitination and degradation of carnitine palmitoyltransferase 1a, thereby inhibiting fatty acid oxidation metabolism and reducing oxidative stress formation (100). In stem cells, the fatty acid synthesis regulated by the lipid synthesis enzyme Acetyl-CoA Carboxylase 1 can influence acetylation-mediated Fis1 Ub-proteasomal degradation by consuming Acetyl-Coenzyme A. At the same time, it can produce lipid products that drive the shift of Mt from a dynamic equilibrium to fission, thereby enhancing mitochondrial fission (101).
Almost all amino acids are synthesized or degraded in the Mt (102). Impaired amino acid metabolism is associated with primary mitochondrial diseases and mitochondrial dysfunction disorders. For example, abnormalities in branched-chain amino acid metabolism are closely related to the progression of diseases such as diabetes (103), atherosclerosis (104), and cancer (105). Wang T et al. (106) conducted experiments on amino acid starvation in tumor cells, assessing the levels of K48-polyubiquitinated proteins in cultured cells after starvation. They found that short-term starvation promoted protein ubiquitination, but after prolonged treatment, there was a significant decrease. This suggests that early amino acid depletion promotes protein ubiquitination, while later stages lead to the degradation of these polyubiquitinated proteins. This phenomenon may be related to the energy depletion induced by amino acid starvation, which in turn triggers mitochondrial autophagy. Current research generally considers the UPS and autophagy-lysosome systems to have distinct functions, but the two systems can interact and influence each other. Amino acid starvation may simultaneously affect the UPS (107), protease activity in mTOR-inhibited Human Embryonic Kidney 293 cells, and polyubiquitination of the 26S proteasome (108). These pathways can all lead to autophagy to varying extents, with amino acid starvation being an important ubiquitination-mediated mechanism for regulating mitochondrial homeostasis (109).
The mechanistic target of rapamycin complex 1 (mTORC1) is a serine/threonine kinase that integrates various environmental signals to regulate cell growth and metabolism. Activation of mTORC1 requires binding to the lysosome through the Ragulator-Rag complex. One essential component of Ragulator, mTOR Activator 1 (LAMTOR1), undergoes dynamic ubiquitination modifications in response to the abundance of amino acids. The E3 ligase TRAF4 directly interacts with Late Endosomal/Lysosomal Adaptor and MAPK and LAMTOR1 and catalyzes polyubiquitination at the K151 site with K63 linkages. This ubiquitination promotes the binding of LAMTOR1 to Rag GTPases and enhances the activation of mTORC1 (110).
ETC in Mt consists of a series of protein complexes (NADH dehydrogenase, succinate dehydrogenase, cytochrome c reductase, and cytochrome c oxidase) located on the IMM. These complexes are responsible for transferring electrons from one complex to another, ultimately transferring electrons to oxygen, resulting in the formation of water (111). Ubiquitination plays a role in the ETC mechanism, primarily through the targeting of specific proteins. The epigenetic regulator Unfolded Protein Response Factor 1 modulates K27 ubiquitination through NLRP14, thereby maintaining the stability of the mitochondrial Na+/Ca2+ exchanger protein Mitochondrial Sodium/Calcium/Lithium Exchanger, which ensures the stability of mitochondrial morphology and function (112). The contact sites between the endoplasmic reticulum membrane and the mitochondrial membrane, known as mut-associated membranes, represent multifunctional microdomains involved in mitochondrial homeostasis (113). Mt-associated membrane-specific E3 Ub ligases can ubiquitinate nascent proteins, thereby activating TANK binding kinase 1 on the Mt-associated membrane (114), leading to the degradation of ribosomal proteins and disrupting the overall mitochondrial homeostasis. Meanwhile, the Mt-associated membrane is closely linked to Ca2+ homeostasis. Upon ubiquitination of inositol 1,4,5-trisphosphate receptor type 2, the mitochondrial outer membrane protein Fundc1 loses its binding site, promoting increased mitochondrial Ca2+, mitochondrial fragmentation, and apoptosis (112).
In addition, RNA molecules also have a significant association with ETC. BDNF-AS is a natural antisense long non-coding RNA of brain-derived neurotrophic factor (BDNF) (115). The expression of BDNF-AS is significantly positively correlated with Voltage dependent anion channel 3 (VDAC3) expression. Previous studies have shown that VDAC3 influences cellular ferroptosis by regulating mitochondrial iron ion flux (116), but the mechanism by which BDNF-AS regulates VDAC3 expression is still unclear. Circular RNA Microtubule crosslinking factor 1 can promote its development by inhibiting the Ub-mediated degradation of the mitochondrial protein complement C1q binding protein and mediating β-catenin activation (117). Under various stress-induced cellular senescence conditions, the expression of SIRT1 protein is down-regulated through Ub-mediated proteasomal degradation (118), a process typically involving Ub-dependent proteasomal degradation. However, some studies suggest that DNA damage-induced cellular senescence follows the autophagosome-lysosome pathway, which may be linked to mitochondrial homeostasis imbalance caused by DNA damage, leading to a decline in the NAD+-dependent biological function of SIRT1, affecting the ubiquitination binding process (119).
The mechanism of interaction between ubiquitination and deubiquitination targeting MH is shown in Figure 2 and Table 1.
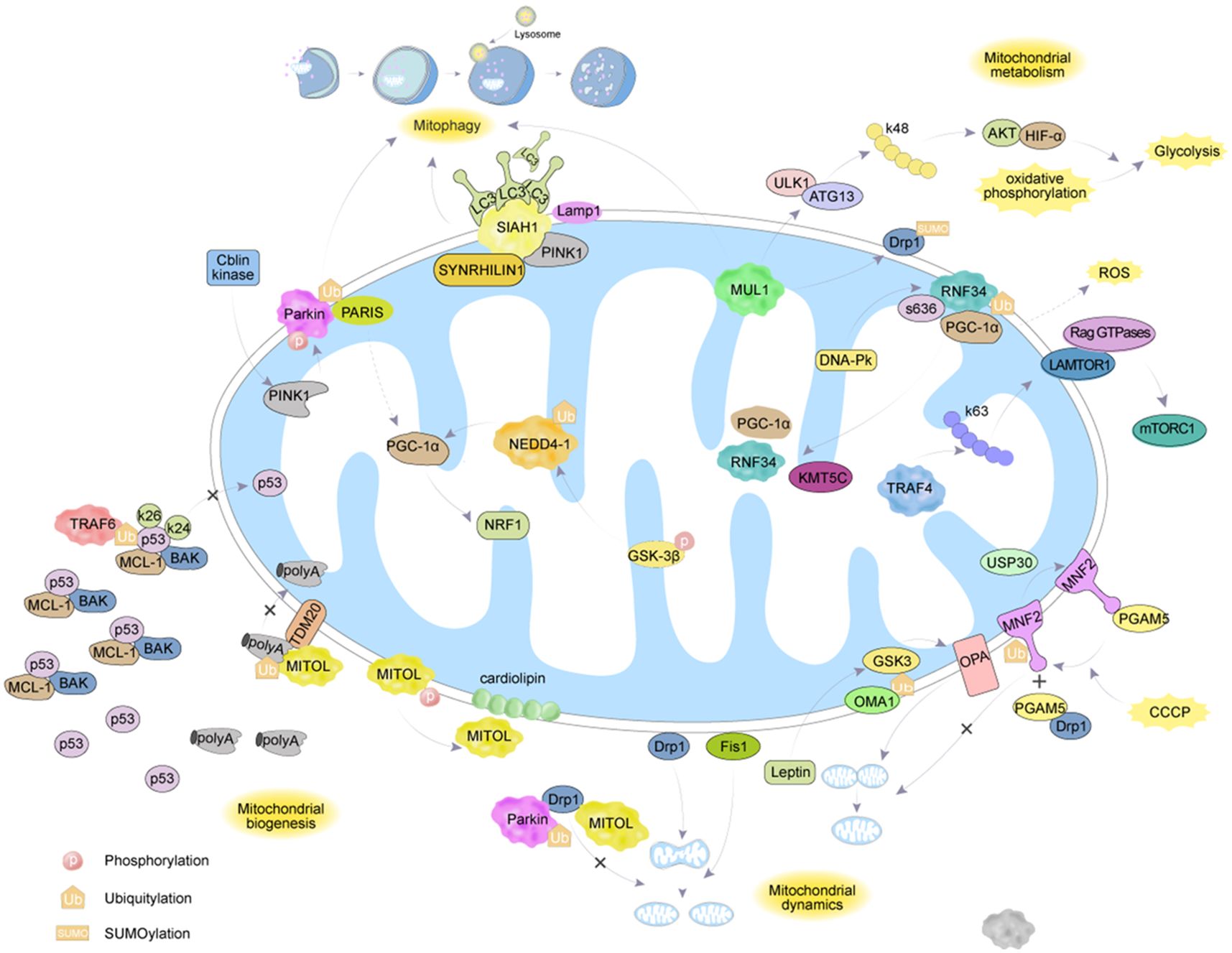
Figure 2. The regulatory mechanisms of ubiquitination enzymes and deubiquitination enzymes on MH.
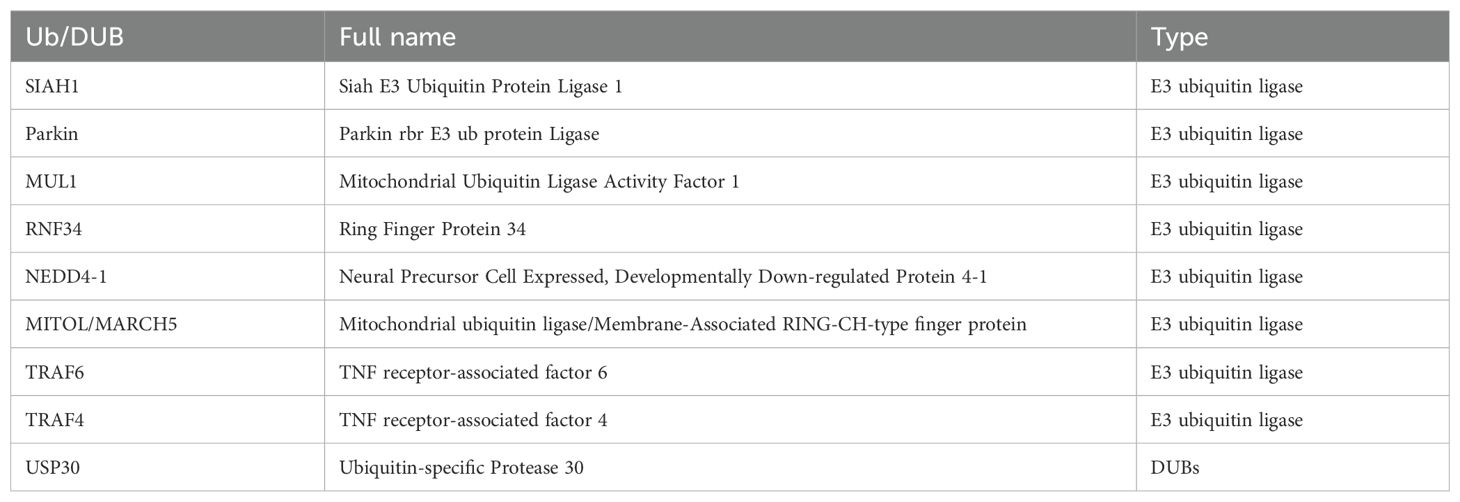
Table 1. Ubiquitinating enzymes targeting MH.
In HCC, In HCC, ubiquitination targeting the MH mechanism with several therapeutic agents was found (Table 2). PINK1/Parkin-mediated mitophagy plays a crucial role. Cyclin-dependent kinase 9 (CDK9) activates SIRT1 and promotes the stabilization of PINK1 protein through its mediated deacetylation, but this effect is blocked by Wogonin (122). Matrine, a traditional Chinese medicine (TCM), triggers mitochondrial dysfunction and induces the upregulation of PINK1/Parkin through phosphatase and PTEN. The elevation of the p62 and LC3-II/I ratio suggests that Matrine acts as both an inducer of autophagy and an inhibitor of autophagosome-lysosome formation, while the blockade of autophagy promotes Matrine-induced cell death (124). The TCM Quercetin (123) upregulates the expression of PINK1/Parkin in Huh7 and Hep3B cells, thereby exerting its anti-cancer effects in HCC. The hepatitis B virus (HBV)-encoded X protein (HBx) plays a key role in inducing HCC (129). Studies have found that thyroid hormone (TH) induces the ubiquitination of mitochondrial-related HBx through PINK1/Parkin and triggers selective mitophagy, thereby inhibiting HBx-promoted ROS and carcinogenesis (125). Lipid metabolism disorder is one of the important characteristics of HCC. Ubiquitin conjugating enzyme E2 O (UBE2O), as an E2 enzyme, has been found to promote HCC progression with high expression. Meanwhile, UBE2O interacts with the mitochondrial β-oxidation enzyme and mediates its ubiquitination and degradation, thereby regulating lipid metabolism reprogramming under the action of E2 and E3 enzyme activities (126). Studies have shown that the VDAC1 inhibitor Novobiocin can reduce the mono-ubiquitination level of VDAC1 K274, and subsequent mutation of this site weakens the interaction between Hsp90α-VDAC1, increases the oligomerization of VDAC1, and thus affects the progression of HCC (120). Crosstalk between some forms of cell death has also been found in HCC. During ferroptosis, 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) is primarily localized to the Mt, but after treatment with pyroptosis inducers, HMGCR translocates to the endoplasmic reticulum. BRCA1/BRCA2-containing complex subunit 36 deubiquitinates HMGCR through DUB activity and inhibits ferroptosis while promoting pyroptosis (127). Transarterial chemoembolization (TACE) is the main treatment method for advanced liver cancer, but postoperative hypoxia can easily worsen the patient’s condition. The hypoxic environment caused by TACE leads to overexpression of S100 calcium-binding protein A9, which forms trimers as a penta-glycine motif protein, triggers mitochondrial fission and ROS production through the deubiquitination and stabilization of PGAM5, and ultimately promotes the growth and metastasis of HCC (121).
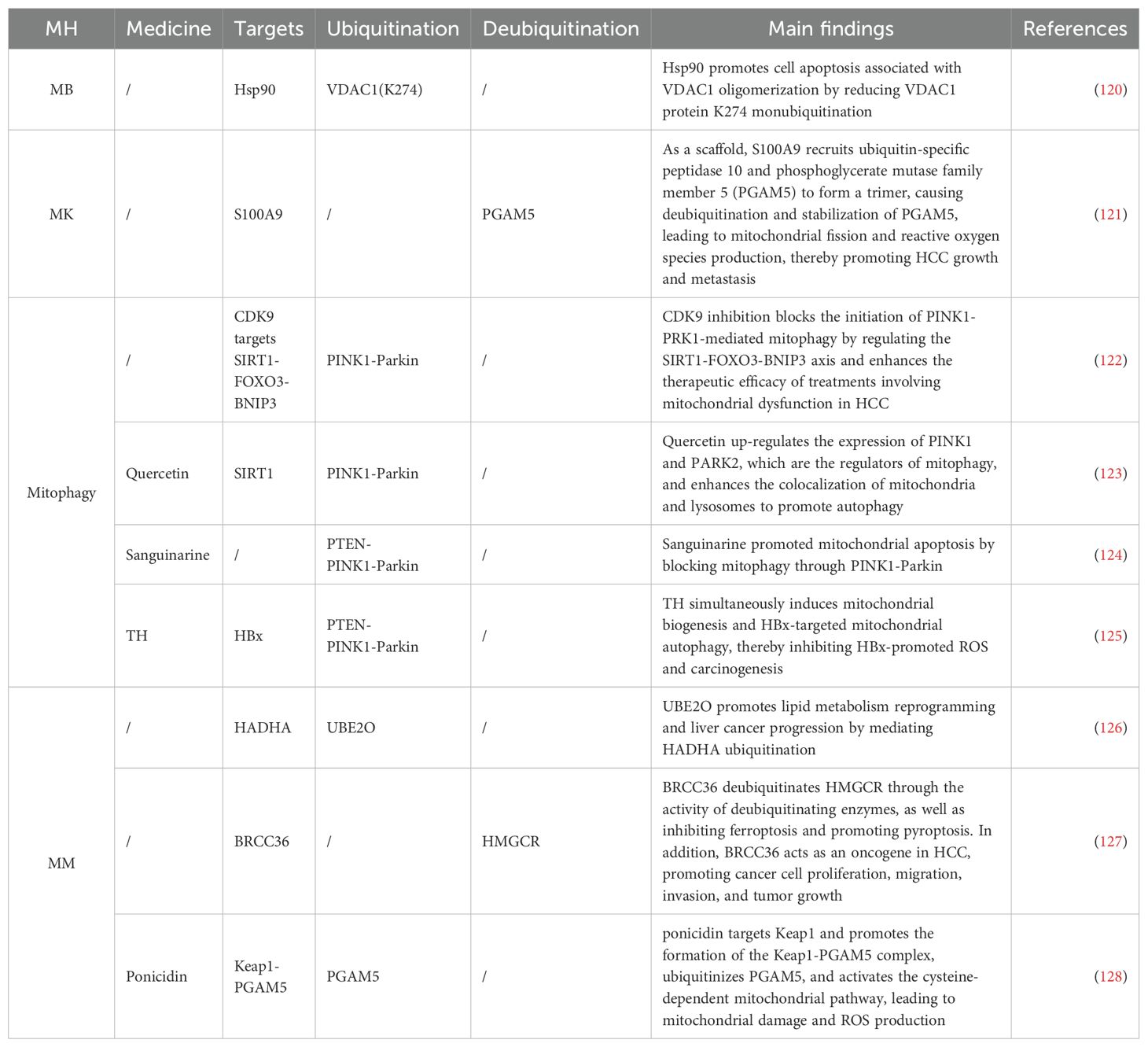
Table 2. Ubiquitination targeting the MH mechanism in HCC.
In PAAD, the enhancement of ferroptosis or drug sensitivity through ubiquitination has garnered attention (Table 3). Due to the dense stroma of PAAD tumors, most tumor-targeting drugs are not sensitive to PAAD treatment (136). As a first-line oncology drug, gemcitabine exhibits a significant chemoresistance phenomenon in pancreatic cancer (137). Stomatin like 2 (STOML2), also known as SLP-2, is a protein located in the mitochondrial inner membrane that participates in maintaining mitochondrial stability (138). Studies have found (113) that increased expression levels of STOML2 can stabilize PARL, thereby preventing PINK1-dependent mitophagy induced by gemcitabine (131). In mammals, ULK1 is a key component of the autophagy initiation complex (139). In PAAD, the E3 ligase NEDD4 like E3 ubiquitin protein ligase NEDD4L binds to ULK1 and is involved in its ubiquitination regulation. In NEDD4L knockout cells, genetic or pharmacological inhibition of ULK1 or Solute Carrier Family 1 Member 5 (SLC1A5/ASCT2) can sensitize PAAD cells, especially under nutrient deprivation conditions (135). Ferroptosis activator imidazole erastin (IKE) upregulates its E3 Ub ligase RANBP2-type and C3HC4-type zinc finger containing 1 (RBCK1), and the knockdown of RBCK1 enhances the cytotoxic effect of IKE on PAAD cells (134). This is attributed to the interaction between RBCK1 and MFN2, leading to polyubiquitination and promoting proteasomal degradation under ferroptotic stress, which results in reduced ROS and lipid peroxidation. Another study indicates that the PINK1-PARK2 pathway-mediated degradation of SLC25A37 and SLC25A28 increases mitochondrial iron accumulation, leading to HIF-1α dependent Warburg effect and AIM2 inflammasome activation in tumor cells, promoting the release of high mobility group box 1 and further inducing the expression of CD274/PD-L1 (133). Galactan RN0D, isolated from the TCM Sanqi, has been identified as an activator of the PINK1/Parkin pathway, ultimately activating cytotoxicity in tumor cells (132). Ubiquitination-mediated Mt gene transcription has also been observed in PAAD. As a core component of endoplasmic reticulum-associated protein degradation, the HRD1-SEL1L complex, when increased, reduces the stability of the mitochondrial protein AlkB homolog 1, leading to impaired transcription of mitochondrial DNA-encoded genes (140). The SIRT4 agonist entinostat reverses this process (130), possibly by deacetylating lysine 547 of SEL1L and increasing the protein levels of HRD1.
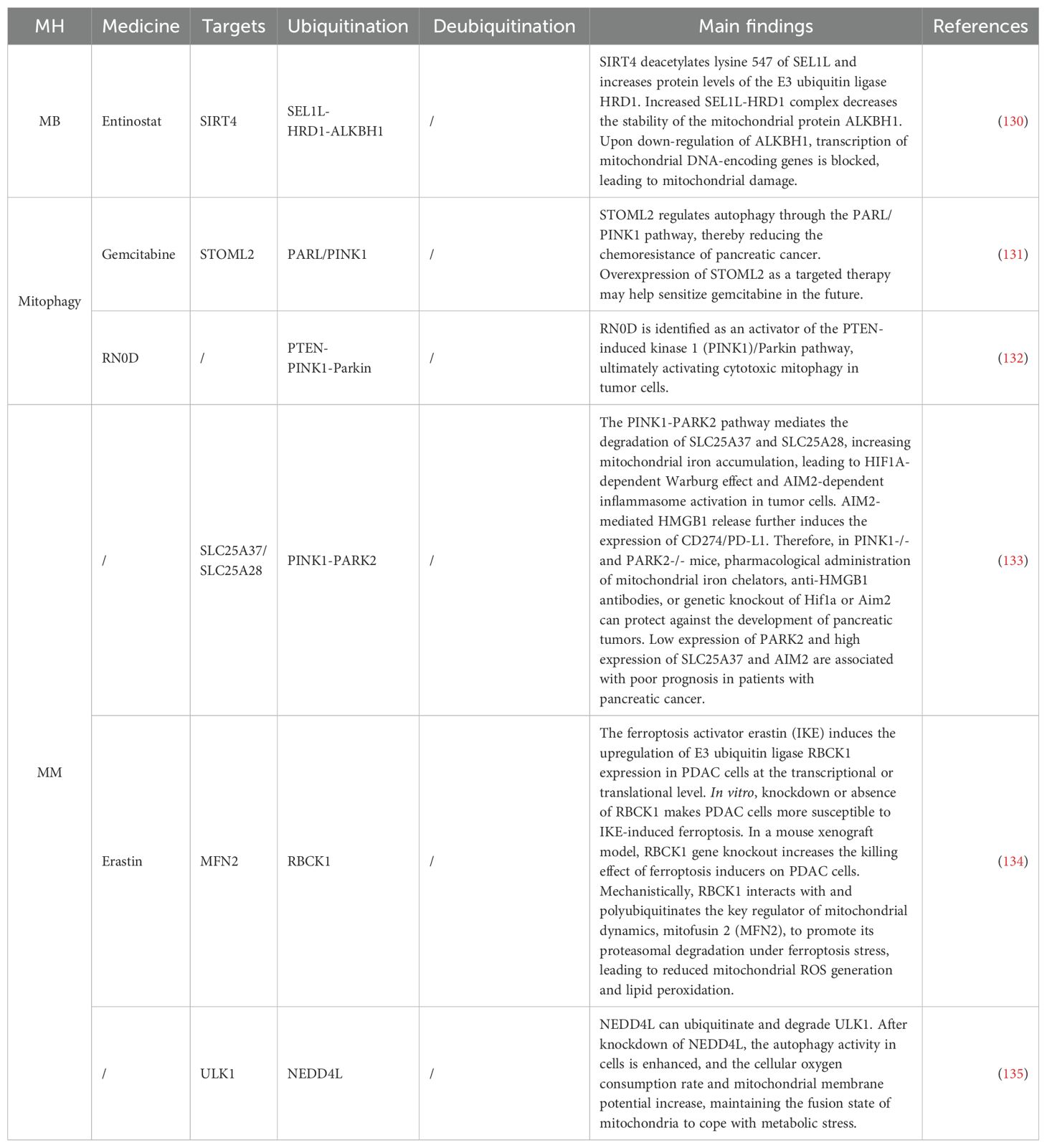
Table 3. Ubiquitination targeting the MH mechanism in PAAD.
In CRC, several ubiquitination mechanisms have also been identified (Table 4). The p53 is one of the most important tumor suppressors, which inhibits the formation and development of tumors by regulating the expression of various genes, including those that promote cell cycle arrest and apoptosis (151). In CRC, p53 ubiquitination plays a significant role. A protein named UBX domain-containing protein 2A promotes the carboxy-terminal ubiquitination of Mortalin-2 in an Hsp70 interaction-dependent manner, reducing Mortalin-2 levels, which not only inactivates p53 but also directly promotes tumor cell invasion and migration (144). The mitochondrial antiviral signaling (MAVS) protein promotes p53-dependent cell death in response to DNA damage (143). MAVS is underexpressed in CRC and inhibits p53 ubiquitination by blocking the formation of the p53-MDM2 complex (143). Lipoic acid (LA) is a dithiol compound with redox activity and is an essential cofactor for mitochondrial oxidative decarboxylation (152). The p53 is found to be ubiquitinated and degraded by the proteasome mechanism after LA treatment, a process that does not involve the MDM2. Interestingly, the combined application of LA and anticancer drugs (doxorubicin, 5-fluorouracil) attenuates the stabilization of p53-mediated p21 and exerts a synergistic cytotoxic effect on CRC cells in a p53-dependent manner (153). Dihydroartemisinin downregulates the expression of the mitochondrial inner membrane scaffold protein anti-proliferative protein 2 in a Ub-dependent manner and blocks the downregulation of p53 and p21, thereby enhancing the cytotoxicity of oxaliplatin in CRC (154). TRAF6 promotes the K63-linked ubiquitination of p53 at K24 in the cytoplasm to limit the interaction between p53 and MCL-1/BAK, thereby restricting the mitochondrial translocation of p53 and spontaneous apoptosis. Additionally, TRAF6 promotes the K63-linked ubiquitination and transactivation of nuclear p53 by recruiting p300 to acetylate p53 (35). The PINK1/Parkin pathway has also been identified in CRC. SIRT3 is highly expressed in CRC with mitochondrial dysfunction, leading to PINK1/Parkin-mediated mitophagy. Targeting histone H2Aub ubiquitination at K119 reduces it, thereby enhancing DNA damage repair induced by radiation (147). Delta-valentine, as an emerging dietary metabolite, targets SIRT3 to participate in the process (146), while Aloe Gel Polysaccharides mediate the PINK1/parkin pathway in a ROS-dependent manner (148). Non-coding RNA targeting ubiquitination to regulate mitochondrial homeostasis mechanisms in CRC has been discovered for the first time. Non-coding RNA piR-823 interacts with PINK1, promoting its ubiquitination and proteasome-dependent degradation, thereby alleviating mitophagy, a mechanism reversed by Ant-823, which promotes Parkin activation (145). Regulation of the ETC and MM has also been identified in CRC. Receptor-interacting protein kinase 1 (RIPK1) interacts with the mitochondrial calcium uniporter (MCU), promoting cell proliferation by increasing mitochondrial calcium uptake and energy metabolism. The ubiquitination site of RIPK1 (RIPK1-K377) is a key site for interaction with MCU and the promotion of cell proliferation (150). As a molecular chaperone of Hsp90, Tumor necrosis factor receptor-associated protein 1 regulates the glycolytic enzyme phosphofructokinase-1 (PFK1) to maximize lactate production, balancing low OXPHOS. This depends on the interaction between TRAP1 and PFK1, which favors the glycolytic activity of PFK1 and prevents its ubiquitination/degradation (155). The interaction between membrane glycoprotein CD36 and glypican 4 (GPC4) induces proteasome-dependent ubiquitination and degradation of GPC4 in CRC cells, reducing the high addiction of CRC cells to glucose or glycolytic inhibition through mitochondrial reduction, i.e., reducing the expression of glycolytic target genes GLUT1, HK2, PKM2, and LDHA, thereby inhibiting the energy metabolism and growth of tumor cells (149). Deubiquitination is the reverse process of ubiquitination (142). USP36 exerts its pro-apoptotic function by targeting cIAP1 and survivin, and it has been found that USP36 can be degraded through polyubiquitination, although the E3 ligase responsible for this process remains unidentified (156).
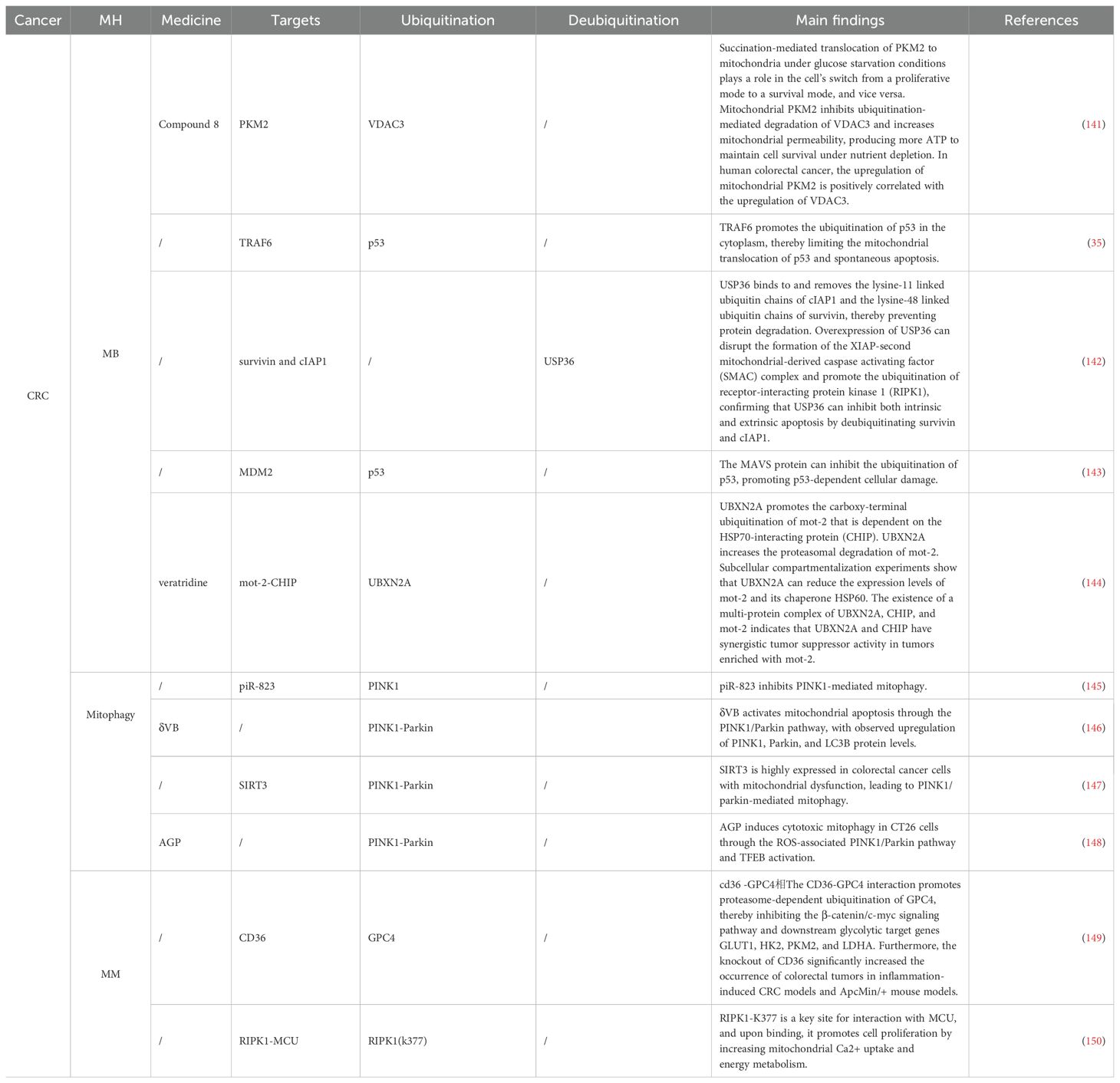
Table 4. Ubiquitination targeting the MH mechanism in CRC and IBD.
IBD is a chronic relapsing inflammatory disorder associated with an increased risk of developing CRC (157). Compared with sporadic CRC, IBD-related CRC typically occurs at a younger age, progresses more rapidly, and has a worse prognosis (158). Therefore, focusing on the pathogenesis of IBD can help prevent the development of CRC in advance. Studies have shown that mitochondrial dysfunction activates pyroptosis through multiple pathways, thereby exacerbating the occurrence of IBD (159). Mitochondrial dysfunction, combined with impaired autophagy, leads to increased ROS levels, which activate the Nucleotide-binding oligomerization domain, Leucine-rich repeat, and Pyrin domain-containing protein 3 (NLRP3) inflammasome and subsequently induce pyroptosis (160). Meanwhile, defects in mitochondrial respiration lead to cellular energy metabolism disorders, making cells more prone to pyroptosis (160). Pyroptosis of intestinal epithelial cells disrupts the intestinal barrier, rendering the gut more susceptible to attacks from pathogens and inflammatory factors. Additionally, the pore-forming action of Gasdermin proteins, which causes cell membrane rupture and the release of large amounts of inflammatory factors (such as IL-1β and IL-18), further intensifies the intestinal inflammatory state (159). Research has found that the E3 Ub ligase gp78 mediates mixed ubiquitination of NLRP3, inhibiting its activity by preventing the oligomerization and subcellular translocation of the nucleotide-binding and oligomerization domain of NLRP3, thereby reducing inflammasome activation and its detrimental effects (161). Parkin-driven mitophagy and inhibition of NLRP3 inflammasome activation in the colon exert a protective effect against DSS-induced colitis in mice (162). Moreover, in IBD, the epigenetic modifier SET and MYND Domain containing protein 5 (SMYD5) regulates toll-like receptor 4 target genes in macrophages at the K20 site (163). This regulation increases the risk of IBD progression to CRC by mediating PGC-1α ubiquitination and degradation through methylation, thereby inhibiting MB.
For GC patients, clinical drug resistance has always been a limitation in late-stage treatment. X-ray repair cross-complementing 1 (XRCC1) is a key regulator of cisplatin-induced DNA damage and apoptosis (164). Thioredoxin-like 1 mediates cisplatin resistance by negatively regulating the expression of XRCC1 through the UPS (165). Myocyte enhancer factor 2A activates PGC1α transcription and inhibits Kelch-like ECH-associated protein 1, reducing the ubiquitination and degradation of NRF2, thereby regulating ROS levels and mediating GC cisplatin resistance (166). Ferroptosis resistance is one of the key factors leading to GC drug resistance. Studies have shown that SRY-box transcription factor 13 (SOX13) promotes the protein reshaping of ETC complexes by directly transactivating Supercomplex assembly factor 1, leading to the assembly of supercomplexes, mitochondrial respiration, mitochondrial energetics, and increased chemo- and immuno-resistance (167). Zanamivir restores the ferroptosis resistance phenotype by directly targeting SOX13 and promoting the ubiquitination and degradation of SOX13 mediated by a tripartite motif containing 25 (TRIM25) (167). Research has found that LncRNA BDNF-AS can affect the ubiquitination modification of VDAC3 by FBXW7 by recruiting WD repeat-containing protein 5 (168). The USP7 can stabilize Heterogeneous nuclear ribonucleoprotein A1 in cancer-associated fibroblasts through deubiquitination, leading to increased secretion of exosomal miR-522, thereby inhibiting ferroptosis and promoting acquired drug resistance in GC (169), primarily by targeting arachidonate 15-lipoxygenase and blocking the accumulation of lipid ROS in Mt. In TCM treatment, the compound herbal medicine Huachansu induces apoptosis in GC cells by increasing ROS levels and inhibiting USP activity (170). Mechanisms and targets are shown in Table 5.
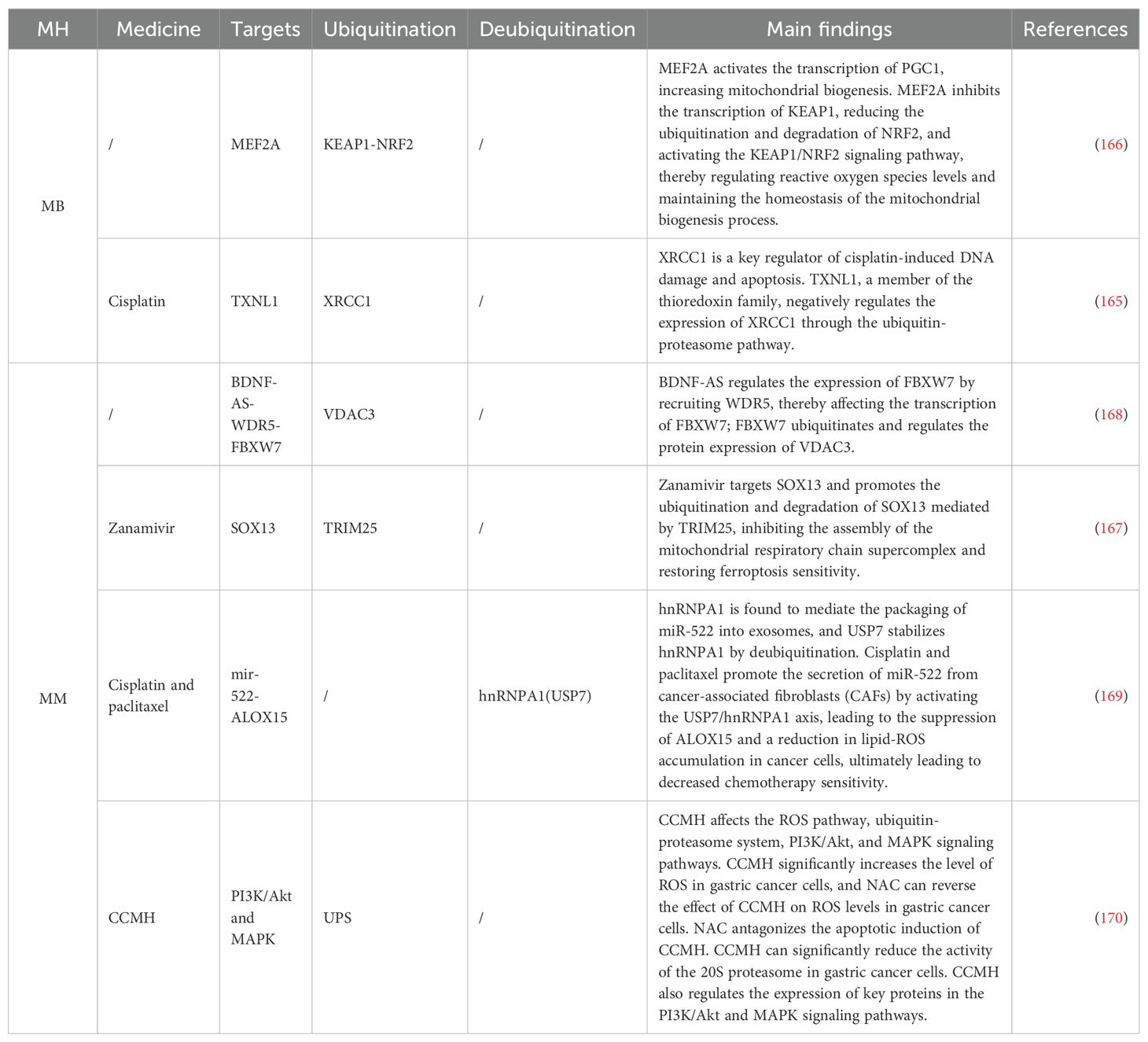
Table 5. Ubiquitination targeting the MH mechanism in GC.
In ESCA, OTU deubiquitinase 1 is a deubiquitinating enzyme that regulates the apoptosis-inducing factor (AIF), capable of ubiquitinating AIF at K244, impairing mitochondrial oxidative phosphorylation, and reducing cell viability. Additionally, its deubiquitination at K255 enhances AIF’s binding capacity to DNA, promoting the occurrence of parthanatos (171). On the other hand, the E3 Ub ligase Itch plays a significant role in TNF-related apoptosis-inducing ligand-mediated apoptosis in ESCA. Knockdown of Itch leads to resistance to TNF-related apoptosis-inducing ligand-mediated apoptosis and significantly alters mitochondrial morphology, increasing mitochondrial cholesterol content. High cholesterol levels reduce membrane fluidity, further intervening in mitochondrial dynamic homeostasis (172). Apart from cholesterol, proteins are also an important factor affecting mitochondrial dynamic homeostasis. Syntaphilin (SNPH) is a static mitochondrial anchor protein primarily expressed in the brain, playing a crucial role in neurotransmitter release and MK. In various tumor cells, SNPH is downregulated or even silenced, leading to the redistribution of Mt from the perinuclear area to the cell periphery, resulting in increased tumor cell migration and invasion (173). Studies have found that CUL1 can ubiquitinate SNPH, disrupting mitochondrial dynamic homeostasis and promoting tumor metastasis and radioresistance (174). In the transformation of ESCA keratinocytes, cells with high CD44 expression exhibit a series of mitochondrial autophagy characteristics: mitochondrial fragmentation, reduced mitochondrial content, and Parkin mitochondrial translocation (175). Mechanisms and targets are shown in Table 6.
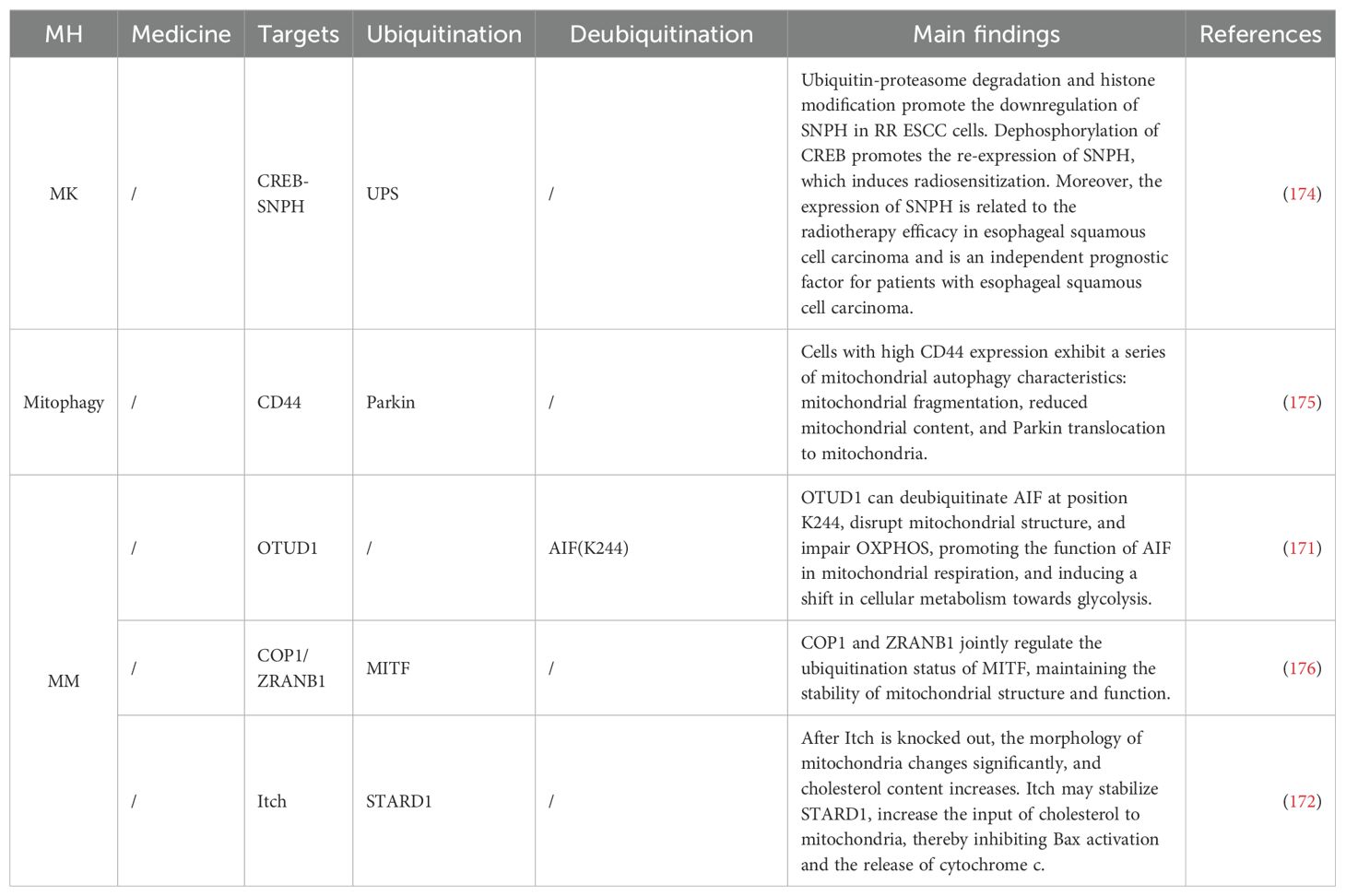
Table 6. Ubiquitination targeting the MH mechanism in ESCA.
In summary, we have summarized the various aspects of MH regulation by different ubiquitinases and deubiquitinases in current scientific research, including MB, mitophagy, and the MM involved in these processes. In gastrointestinal tumors, through the ubiquitination regulation of MH, we have discovered different cell death crosstalk mechanisms, such as mitophagy, ferroptosis, and apoptosis, tumor drug resistance mechanisms, metabolic reprogramming, and new targets for TCM treatment. However, the specific roles of these mechanisms and the potential crosstalk between signaling pathways in different tumor types have not yet been fully elucidated. Clinical treatment results suggest that single-target therapies may not be sufficient for gastrointestinal tumors, and the development of new drugs is needed. Proton beam therapy (PBT) has been shown to inhibit colon cancer metastasis by stimulating mitochondrial biogenesis through the upregulation of PGC-1α and its co-transcription factors (NRF1α/ERRα). Additionally, compounds like hydroxytyrosol (HTyr) can promote mitochondrial biogenesis by increasing PGC-1α expression, offering potential as adjuvant anticancer agents. Advances in gene-editing technologies, such as CRISPR/Cas9, offer the potential to directly target mitochondrial dysfunction by correcting genetic defects or modulating key regulatory pathways. Although clinical translation is still in its infancy, preclinical studies have demonstrated the feasibility of these approaches. In addition, current research has not determined the mechanisms by which ubiquitination regulation of MH is involved in the immune evasion of gastrointestinal tumors, which may require further experimental data support.
BH: Conceptualization, Writing – review & editing, Writing – original draft. YY: Writing – original draft. JL: Investigation, Writing – original draft. BZ: Investigation, Visualization, Writing – review & editing. NL: Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. We acknowledge financial support from the Key Laboratory of Clinical Cancer Pharmacology and Toxicology Research of Zhejiang Province (2020E10021), Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents (ZWB-2020-18), Zhejiang Medical Key Discipline Foundation (ZWB-2018-02-03), Hangzhou Medical Key Discipline Foundation (HWF-2021-21-16).
The authors thank all the staff from Hangzhou First People’s Hospital affiliated with Westlake University Medical School, Gansu University of Chinese Medicine and Department of General Surgery, and The First Affiliated Hospital of Dalian Medical University for their continuous support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MH: Mitochondrial Homeostasis
MB: Mitochondrial Biogenesis
MM: Mitochondrial Metabolism
MK: Mitochondrial Kinetics
GI: Gastrointestinal
Mt: Mitochondria
mtDNA: Mitochondrial DNA
HCC: Hepatocellular Carcinoma
ESCA: Esophageal Cancer
PAAD: Pancreatic Cancer
GC: Gastric Cancer
CRC: Colorectal Cancer
ROS: Reactive Oxygen Species
BCL-2: B-cell lymphoma/leukemia-2
IBD: Inflammatory Bowel Disease
Ub: Ubiquitin
DUBs: Deubiquitinating enzymes
USP: Ub-specific protease
UPS: Ubiquitin-Proteasome System
PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1 alpha
OXPHOS: Oxidative Phosphorylation
NRFs: Nuclear Respiratory Factors
MITOL1/MARCH5: Mitochondrial ubiquitin ligase/Membrane-Associated RING-CH-Type Finger 5
PolγA: DNA Polymerase Gamma Catalytic Subunit
TRAF6: Tumor Necrosis Factor Receptor-Associated Factor 6
SIRT1: Silent information regulator 1
ATP: Adenosine Triphosphate
MMP: Mitochondrial Membrane Potential
GSK-3β: Glycogen synthase kinase 3β
Tom20: Translocase of the outer mitochondrial membrane 20
MCL-1/BAK: myeloid cell leukemia-1/BCL-2 Antagonist 1
p53: Tumor Protein 53
K24: Lysine 24
CL: Cardiolipin
PA: phosphatidic acid
OMM: Outer Mitochondrial Membrane
IMM: Inner Membrane Membrane
NEDD4-1: Neural Precursor Cell Expressed, Developmentally Down-regulated Protein 4-1
MDM35: Mitochondrial Distribution and Morphology Protein 35
PINK1: PTEN induced putative kinase 1
Parkin: Parkin rbr E3 ub protein Ligase
GTPase: Guanosine Triphosphatase
Drp1: Dynamin-related protein 1
MFN1: Mitofusin 1
MFN2: Mitofusin 2
OPA1: Optic Atrophy 1
Fis1: Mitochondrial fission factor
Gβ2: G-protein β2
CCCP: Carbonyl cyanide 3-chlorophenylhydrazone
PGAM5: Phosphoglycerate mutase 5
CUL1: Cullin1
FBX: F-box protein
Fzo1: Fusion of Mitochondria Protein 1
6-OHDA: 6-hydroxydopamine
IKKγ: Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Gamma
OMA1: Optic Atrophy 1
MUL1: Mitochondrial E3 ubiquitin protein ligase 1
PARL: Presenilin-Associated Rhomboid-Like
ULK1: Unc-51 Like Autophagy Activating Kinase 1
SIAH1: Seven in Absentia Homolog 1
LC3: Microtubule-associated protein 1 light chain 3
Atg: Autophagy-related
LAMP1: Lysosomal-associated membrane protein 1
SUMOylation: Small Ubiquitin-like Modifier Conjugation
HIF-1α: Hypoxia-inducible factor 1-alpha
ETC: Electron Transport Chain
H2Aub: histone H2A ubiquitination
Hsp27: heat shock protein 27
PFKP: Phosphofructokinase Platelet
HRD1: HMG-CoA Reductase Degradation 1
NADK: Nicotinamide Adenine Dinucleotide Kinase
STAT3: Signal Transducer and Activator of Transcription 3
mTORC1: mechanistic target of rapamycin complex 1
LAMTOR1: mTOR Activator 1
BDNF: brain-derived neurotrophic factor
VDAC3: Voltage dependent anion channel 3
CDK9: Cyclin-dependent kinase 9
TCM: traditional Chinese medicine
HBx: hepatitis B virus -encoded X protein
UBE2O: Ubiquitin conjugating enzyme E2 O
HMGCR: 3-hydroxy-3-methylglutaryl-CoA reductase
TACE: Transarterial chemoembolization
STOML2: Stomatin like 2
IKE: imidazole erastin
RBCK1: RANBP2-type and C3HC4-type zinc finger containing 1
MAVS: mitochondrial antiviral signaling
LA: Lipoic acid
NLRP3: Nucleotide-binding oligomerization domain, Leucine-rich repeat, and Pyrin domain-containing protein 3
RIPK1: Receptor-interacting protein kinase 1
MCU: mitochondrial calcium uniporter
PFK1: phosphofructokinase-1
GPC4: glypican 4
USP36: Ubiquitin specific peptidase 36
XRCC1: X-ray repair cross-complementing 1
SOX13: SRY-box transcription factor 13
AIF: apoptosis-inducing factor
SNPH: Syntaphilin
1. Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology. (2020) 76:182–8. doi: 10.1111/his.13975
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
3. Kenny TC, Birsoy K. Mitochondria and cancer. Cold Spring Harb Perspect Med. (2024) 14(12):a041534. doi: 10.1101/cshperspect.a041534
4. Zhu S, Chen C, Wang M, Liu Y, Li B, Qi X, et al. Pan-cancer association of a mitochondrial function score with genomic alterations and clinical outcome. Sci Rep. (2024) 14:31430. doi: 10.1038/s41598-024-83022-1
5. Feng S, Wierzbowski MC, Hrovat-Schaale K, Dumortier A, Zhang Y, Zyulina M, et al. Mechanisms of NLRP3 activation and inhibition elucidated by functional analysis of disease-associated variants. Nat Immunol. (2025) 26(3):1–13. doi: 10.1038/s41590-025-02088-9
6. Harino T, Tanaka K, Motooka D, Masuike Y, Takahashi T, Yamashita K, et al. D-loop mutations in mitochondrial DNA are a risk factor for chemotherapy resistance in esophageal cancer. Sci Rep. (2024) 14:31653. doi: 10.1038/s41598-024-80226-3
7. Wang X, Li S, Shen Y, Cao L, Lu Y, Cao J, et al. Construction of molecular subtype and prognostic model for gastric cancer based on nucleus-encoded mitochondrial genes. Sci Rep. (2024) 14:28491. doi: 10.1038/s41598-024-78729-0
8. Wang S, Wu X, Bi W, Xu J, Hou L, Li G, et al. ROS-induced cytosolic release of mitochondrial PGAM5 promotes colorectal cancer progression by interacting with MST3. Nat Commun. (2025) 16:1406. doi: 10.1038/s41467-025-56444-2
9. Zou J, Jiang C, Hu Q, Jia X, Wang S, Wan S, et al. Tumor microenvironment-responsive engineered hybrid nanomedicine for photodynamic-immunotherapy via multi-pronged amplification of reactive oxygen species. Nat Commun. (2025) 16:424. doi: 10.1038/s41467-024-55658-0
10. Haque PS, Kapur N, Barrett TA, Theiss AL. Mitochondrial function and gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. (2024) 21:32. doi: 10.1038/s41575-024-00931-2
11. Rao Z, Brunner E, Giszas B, Iyer-Bierhoff A, Gerstmeier J, Börner F, et al. Glucocorticoids regulate lipid mediator networks by reciprocal modulation of 15-lipoxygenase isoforms affecting inflammation resolution. Proc Natl Acad Sci U.S.A. (2023) 120:e2302070120. doi: 10.1073/pnas.2302070120
12. Su L, Xu J, Lu C, Gao K, Hu Y, Xue C, et al. Nano-flow cytometry unveils mitochondrial permeability transition process and multi-pathway cell death induction for cancer therapy. Cell Death Discovery. (2024) 10:176. doi: 10.1038/s41420-024-01947-y
13. Szabo I, Zoratti M, Biasutto L. Targeting mitochondrial ion channels for cancer therapy. Redox Biol. (2021) 42:101846. doi: 10.1016/j.redox.2020.101846
14. Liu L, Li Y, Chen G, Chen Q. Crosstalk between mitochondrial biogenesis and mitophagy to maintain mitochondrial homeostasis. J BioMed Sci. (2023) 30:86. doi: 10.1186/s12929-023-00975-7
15. Ryu KW, Fung TS, Baker DC, Saoi M, Park J, Febres-Aldana CA, et al. Cellular ATP demand creates metabolically distinct subpopulations of mitochondria. Nature. (2024) 635:746–54. doi: 10.1038/s41586-024-08146-w
16. Tang Y, Zhang J, Fang Y, Zhu K, Zhu J, Huang C, et al. Correcting mitochondrial loss mitigates NOTCH1-related aortopathy in mice. Nat Cardiovasc Res. (2025) 4(2):235–47. doi: 10.1038/s44161-024-00603-z
17. Jia Y, Yuan X, Feng L, Xu Q, Fang X, Xiao D, et al. m6A-modified circCacna1c regulates necroptosis and ischemic myocardial injury by inhibiting Hnrnpf entry into the nucleus. Cell Mol Biol Lett. (2024) 29:140. doi: 10.1186/s11658-024-00649-8
18. Gao J, Liu Y, Si C, Guo R, Hou S, Liu X, et al. Aspirin inhibits proteasomal degradation and promotes α-synuclein aggregate clearance through K63 ubiquitination. Nat Commun. (2025) 16:1–17. doi: 10.1038/s41467-025-56737-6
19. Hu X, Li L, Nkwocha J, Kmieciak M, Shang S, Cowart LA, et al. Src inhibition potentiates MCL-1 antagonist activity in acute myeloid leukemia. Sig Transduct Target Ther. (2025) 10:1–14. doi: 10.1038/s41392-025-02125-x
20. Ng MYW, Wai T, Simonsen A. Quality control of the mitochondrion. Dev Cell. (2021) 56:881–905. doi: 10.1016/j.devcel.2021.02.009
21. Popov L-D. Mitochondrial biogenesis: An update. J Cell Mol Med. (2020) 24:4892–9. doi: 10.1111/jcmm.15194
22. Wu L, Wang L, Du Y, Zhang Y, Ren J. Mitochondrial quality control mechanisms as therapeutic targets in doxorubicin-induced cardiotoxicity. Trends Pharmacol Sci. (2023) 44:34–49. doi: 10.1016/j.tips.2022.10.003
23. Qi Y, Rajbanshi B, Hao R, Dang Y, Xu C, Lu W, et al. The dual role of PGAM5 in inflammation. Exp Mol Med. (2025) 57(2):1–14. doi: 10.1038/s12276-025-01391-7
24. Chen S, Zhang D, Du Y, Shi J, Gu S, Zhou X, et al. Targeting TRAF6/IRF3 axis to inhibit NF-κB-p65 nuclear translocation enhances the chemosensitivity of 5-FU and reverses the proliferation of gastric cancer. Cell Death Dis. (2024) 15:1–13. doi: 10.1038/s41419-024-07290-5
25. Zhao M, Li Y, Lu C, Ding F, Xu M, Ge X, et al. PGC1α Degradation suppresses mitochondrial biogenesis to confer radiation resistance in glioma. Cancer Res. (2023) 83:1094–110. doi: 10.1158/0008-5472.CAN-22-3083
26. S S, J W, H G, X H, B M, X C, et al. PGC-1α-coordinated hypothalamic antioxidant defense is linked to SP1-lanCL1 axis during high-fat-diet-induced obesity in male mice. Antioxid (Basel Switzerland). (2024) 13:252. doi: 10.3390/antiox13020252
27. Jannig PR, Dumesic PA, Spiegelman BM, Ruas JL. SnapShot: regulation and biology of PGC-1α. Cell. (2022) 185:1444–1444.e1. doi: 10.1016/j.cell.2022.03.027
28. Chen X, Ji Y, Liu R, Zhu X, Wang K, Yang X, et al. Mitochondrial dysfunction: roles in skeletal muscle atrophy. J Transl Med. (2023) 21:503. doi: 10.1186/s12967-023-04369-z
29. Xin G-J, Chen Y-Y, Liu Z-X, Xu S-J, Zhang H-Y, Guo F, et al. Ginsenoside Re regulates mitochondrial biogenesis through Nrf2/HO-1/PGC-1α pathway to reduce hypoxia/reoxygenation injury in H9c2 cells. Zhongguo Zhong Yao Za Zhi. (2024) 49:1064–72. doi: 10.19540/j.cnki.cjcmm.20231017.702
30. Zhao Q, Cui X, Zhu Q, Li F, Bao R, Shi T, et al. Non-catalytic mechanisms of KMT5C regulating hepatic gluconeogenesis. Nat Commun. (2025) 16:1483. doi: 10.1038/s41467-025-56696-y
31. Fan S, Yan X, Hu X, Liu X, Zhao S, Zhang Y, et al. Shikonin blocks CAF-induced TNBC metastasis by suppressing mitochondrial biogenesis through GSK-3β/NEDD4-1 mediated phosphorylation-dependent degradation of PGC-1α. J Exp Clin Cancer Res. (2024) 43:180. doi: 10.1186/s13046-024-03101-z
32. Merklinger L, Bauer J, Pedersen PA, Damgaard RB, Morth JP. Phospholipids alter activity and stability of mitochondrial membrane-bound ubiquitin ligase MARCH5. Life Sci Alliance. (2022) 5:e202101309. doi: 10.26508/lsa.202101309
33. Hussain M, Saifi S, Mohammed A, Sengupta S. Protocol to detect in vitro and in cell ubiquitylation of mitochondrial DNA polymerase gamma by mitochondrial E3 ligase MITOL. STAR Protoc. (2022) 3:101710. doi: 10.1016/j.xpro.2022.101710
34. Hussain M, Mohammed A, Saifi S, Khan A, Kaur E, Priya S, et al. MITOL-dependent ubiquitylation negatively regulates the entry of PolγA into mitochondria. PloS Biol. (2021) 19:e3001139. doi: 10.1371/journal.pbio.3001139
35. Zhang X, Li C-F, Zhang L, Wu C-Y, Han L, Jin G, et al. TRAF6 restricts p53 mitochondrial translocation, apoptosis, and tumor suppression. Mol Cell. (2016) 64:803–14. doi: 10.1016/j.molcel.2016.10.002
36. Fox CA, Ryan RO. Studies of the cardiolipin interactome. Prog Lipid Res. (2022) 88:101195. doi: 10.1016/j.plipres.2022.101195
37. Jahn H, Bartoš L, Dearden GI, Dittman JS, Holthuis JCM, Vácha R, et al. Phospholipids are imported into mitochondria by VDAC, a dimeric beta barrel scramblase. Nat Commun. (2023) 14:8115. doi: 10.1038/s41467-023-43570-y
38. Yu F, He F, Yao H, Wang C, Wang J, Li J, et al. Structural basis of intramitochondrial phosphatidic acid transport mediated by Ups1-Mdm35 complex. EMBO Rep. (2015) 16:813–23. doi: 10.15252/embr.201540137
39. Shiiba I, Ito N, Oshio H, Ishikawa Y, Nagao T, Shimura H, et al. ER-mitochondria contacts mediate lipid radical transfer via RMDN3/PTPIP51 phosphorylation to reduce mitochondrial oxidative stress. Nat Commun. (2025) 16:1508. doi: 10.1038/s41467-025-56666-4
40. Mitsuhashi S, Hatakeyama H, Karahashi M, Koumura T, Nonaka I, Hayashi YK, et al. Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy. Hum Mol Genet. (2011) 20:3841–51. doi: 10.1093/hmg/ddr305
41. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. (2020) 21:204–24. doi: 10.1038/s41580-020-0210-7
42. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
43. Rovira-Llopis S, Bañuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. (2017) 11:637–45. doi: 10.1016/j.redox.2017.01.013
44. Lee H, Lee TJ, Galloway CA, Zhi W, Xiao W, de Mesy Bentley KL, et al. The mitochondrial fusion protein OPA1 is dispensable in the liver and its absence induces mitohormesis to protect liver from drug-induced injury. Nat Commun. (2023) 14:6721. doi: 10.1038/s41467-023-42564-0
45. Schmitt K, Grimm A, Dallmann R, Oettinghaus B, Restelli LM, Witzig M, et al. Circadian control of DRP1 activity regulates mitochondrial dynamics and bioenergetics. Cell Metab. (2018) 27:657–666.e5. doi: 10.1016/j.cmet.2018.01.011
46. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Sig Transduct Target Ther. (2023) 8:1–25. doi: 10.1038/s41392-023-01547-9
47. Tokuyama T, Yanagi S. Role of mitochondrial dynamics in heart diseases. Genes (Basel). (2023) 14:1876. doi: 10.3390/genes14101876
48. Yang H, Sibilla C, Liu R, Yun J, Hay BA, Blackstone C, et al. Clueless/CLUH regulates mitochondrial fission by promoting recruitment of Drp1 to mitochondria. Nat Commun. (2022) 13:1582. doi: 10.1038/s41467-022-29071-4
49. Qi Z, Huang Z, Xie F, Chen L. Dynamin-related protein 1: A critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J Cell Physiol. (2019) 234:10032–46. doi: 10.1002/jcp.27866
50. Das P, Chakrabarti O. ISGylation of DRP1 closely balances other post-translational modifications to mediate mitochondrial fission. Cell Death Dis. (2024) 15:1–17. doi: 10.1038/s41419-024-06543-7
51. Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, et al. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell. (2016) 27:349–59. doi: 10.1091/mbc.E15-09-0678
52. Cherok E, Xu S, Li S, Das S, Meltzer WA, Zalzman M, et al. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol Biol Cell. (2017) 28:396–410. doi: 10.1091/mbc.E16-04-0208
53. Xian H, Liou Y-C. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. (2021) 28:827–42. doi: 10.1038/s41418-020-00657-z
54. Zorzano A, Liesa M, Sebastián D, Segalés J, Palacín M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin Cell Dev Biol. (2010) 21:566–74. doi: 10.1016/j.semcdb.2010.01.002
55. Zhang J, Liu W, Liu J, Xiao W, Liu L, Jiang C, et al. G-protein β2 subunit interacts with mitofusin 1 to regulate mitochondrial fusion. Nat Commun. (2010) 1:101. doi: 10.1038/ncomms1099
56. Nag S, Szederkenyi K, Gorbenko O, Tyrrell H, Yip CM, McQuibban GA. PGAM5 is an MFN2 phosphatase that plays an essential role in the regulation of mitochondrial dynamics. Cell Rep. (2023) 42:112895. doi: 10.1016/j.celrep.2023.112895
57. Escobar-Henriques M. Mitofusins: ubiquitylation promotes fusion. Cell Res. (2014) 24:387–8. doi: 10.1038/cr.2014.23
58. Cohen MM, Amiott EA, Day AR, Leboucher GP, Pryce EN, Glickman MH, et al. Sequential requirements for the GTPase domain of the mitofusin Fzo1 and the ubiquitin ligase SCFMdm30 in mitochondrial outer membrane fusion. J Cell Sci. (2011) 124:1403–10. doi: 10.1242/jcs.079293
59. Cavellini L, Meurisse J, Findinier J, Erpapazoglou Z, Belgareh-Touzé N, Weissman AM, et al. An ubiquitin-dependent balance between mitofusin turnover and fatty acids desaturation regulates mitochondrial fusion. Nat Commun. (2017) 8:15832. doi: 10.1038/ncomms15832
60. Alavi MV, Fuhrmann N. Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics. Mol Neurodegener. (2013) 8:32. doi: 10.1186/1750-1326-8-32
61. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. (2006) 126:177–89. doi: 10.1016/j.cell.2006.06.025
62. Lin C-Y, Chen W-J, Fu R-H, Tsai C-W. Upregulation of OPA1 by carnosic acid is mediated through induction of IKKγ ubiquitination by parkin and protects against neurotoxicity. Food Chem Toxicol. (2020) 136:110942. doi: 10.1016/j.fct.2019.110942
63. Ahola S, Rivera Mejías P, Hermans S, Chandragiri S, Giavalisco P, Nolte H, et al. OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab. (2022) 34:1875–1891.e7. doi: 10.1016/j.cmet.2022.08.017
64. Alavi MV. Recent advances in, and challenges of, designing OMA1 drug screens. Pharmacol Res. (2022) 176:106063. doi: 10.1016/j.phrs.2022.106063
65. Yang F, Wu R, Jiang Z, Chen J, Nan J, Su S, et al. Leptin increases mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance therapeutic effects of mesenchymal stem cell transplantation. Cell Death Dis. (2018) 9:556. doi: 10.1038/s41419-018-0579-9
66. Liu B, Xu C, Liu Y, Lu Z, Fu T, Li , et al. Mitochondrial quality control in human health and disease. Military Med Res. (2024) 11:32. doi: 10.1186/s40779-024-00536-5
67. Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y, et al. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther. (2023) 8:304. doi: 10.1038/s41392-023-01503-7
68. Han R, Liu Y, Li S, Li X-J, Yang W. PINK1-PRKN mediated mitophagy: differences between in vitro and in vivo models. Autophagy. (2023) 19:1396–405. doi: 10.1080/15548627.2022.2139080
69. Huang Q, Li J, Chen J, Zhang Z, Xu P, Qi H, et al. Ginsenoside compound K protects against cerebral ischemia/reperfusion injury via Mul1/Mfn2-mediated mitochondrial dynamics and bioenergy. J Ginseng Res. (2023) 47:408–19. doi: 10.1016/j.jgr.2022.10.004
70. Narendra DP, Youle RJ. The role of PINK1-Parkin in mitochondrial quality control. Nat Cell Biol. (2024) 26:1639–51. doi: 10.1038/s41556-024-01513-9
71. Martinez A, Sanchez-Martinez A, Pickering JT, Twyning MJ, Terriente-Felix A, Chen P-L, et al. Mitochondrial CISD1/Cisd accumulation blocks mitophagy and genetic or pharmacological inhibition rescues neurodegenerative phenotypes in Pink1/parkin models. Mol Neurodegener. (2024) 19:12. doi: 10.1186/s13024-024-00701-3
72. Chen C, Gao T-Y, Yi H-W, Zhang Y, Wang T, Lou Z-L, et al. Elevated ubiquitin phosphorylation by PINK1 contributes to proteasomal impairment and promotes neurodegeneration. eLife. (2025) 14. doi: 10.7554/eLife.103945.1
73. Zhao X, Wang Z, Wang L, Jiang T, Dong D, Sun M. The PINK1/Parkin signaling pathway-mediated mitophagy: a forgotten protagonist in myocardial ischemia/reperfusion injury. Pharmacol Res. (2024) 209:107466. doi: 10.1016/j.phrs.2024.107466
74. D’Amico AG, Maugeri G, Magrì B, Bucolo C, D’Agata V. Targeting the PINK1/Parkin pathway: A new perspective in the prevention and therapy of diabetic retinopathy. Exp Eye Res. (2024) 247:110024. doi: 10.1016/j.exer.2024.110024
75. Imberechts D, Kinnart I, Wauters F, Terbeek J, Manders L, Wierda K, et al. DJ-1 is an essential downstream mediator in PINK1/parkin-dependent mitophagy. Brain. (2022) 145:4368–84. doi: 10.1093/brain/awac313
76. Lee MJ, Cho Y, Hwang Y, Jo Y, Kim Y-G, Lee SH, et al. Kaempferol alleviates mitochondrial damage by reducing mitochondrial reactive oxygen species production in lipopolysaccharide-induced prostate organoids. Foods. (2023) 12:3836. doi: 10.3390/foods12203836
77. Weerawardhana A, Herath TUB, Gayan Chathuranga WA, Kim T-H, Ekanayaka P, Chathuranga K, et al. SIAH1 modulates antiviral immune responses by targeting deubiquitinase USP19. J Med Virol. (2024) 96:e29523. doi: 10.1002/jmv.29523
78. Choubey V, Zeb A, Kaasik A. Molecular mechanisms and regulation of mammalian mitophagy. Cells. (2021) 11:38. doi: 10.3390/cells11010038
79. Mondaca-Ruff D, Quiroga C, Norambuena-Soto I, Riquelme JA, San Martin A, Bustamante M, et al. Regulation of total LC3 levels by angiotensin II in vascular smooth muscle cells. J Cell Mol Med. (2022) 26:1710–3. doi: 10.1111/jcmm.17215
80. Chaudhry N, Sica M, Surabhi S, Hernandez DS, Mesquita A, Selimovic A, et al. Lamp1 mediates lipid transport, but is dispensable for autophagy in Drosophila. Autophagy. (2022) 18:2443–58. doi: 10.1080/15548627.2022.2038999
81. Puri R, Cheng X-T, Lin M-Y, Huang N, Sheng Z-H. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat Commun. (2019) 10:3645. doi: 10.1038/s41467-019-11636-5
82. Li J, Qi W, Chen G, Feng D, Liu J, Ma B, et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy. (2015) 11:1216–29. doi: 10.1080/15548627.2015.1017180
83. Peng J, Ren K-D, Yang J, Luo X-J. Mitochondrial E3 ubiquitin ligase 1: A key enzyme in regulation of mitochondrial dynamics and functions. Mitochondrion. (2016) 28:49–53. doi: 10.1016/j.mito.2016.03.007
84. Cilenti L, Mahar R, Gregorio JD, Ambivero CT, Merritt ME, Zervos AS. Regulation of metabolism by mitochondrial MUL1 E3 ubiquitin ligase. Front Cell Dev Biol. (2022) 10:904728. doi: 10.3389/fcell.2022.904728
85. Merry TL, Chan A, Woodhead JST, Reynolds JC, Kumagai H, Kim S-J, et al. Mitochondrial-derived peptides in energy metabolism. Am J Physiol Endocrinol Metab. (2020) 319:E659–66. doi: 10.1152/ajpendo.00249.2020
86. Mizushima N. Ubiquitin in autophagy and non-protein ubiquitination. Nat Struct Mol Biol. (2024) 31:208–9. doi: 10.1038/s41594-024-01217-6
87. Zhang N, Meng Y, Mao S, Ni H, Huang C, Shen L, et al. FBXO31-mediated ubiquitination of OGT maintains O-GlcNAcylation homeostasis to restrain endometrial Malignancy. Nat Commun. (2025) 16:1274. doi: 10.1038/s41467-025-56633-z
88. Gu J, Xiao X, Zou C, Mao Y, Jin C, Fu D, et al. Ubiquitin-specific protease 7 maintains c-Myc stability to support pancreatic cancer glycolysis and tumor growth. J Trans Med. (2024) 22:1135. doi: 10.1186/s12967-024-05962-6
89. Zou J, Wang W, Lu Y, Ayala J, Dong K, Zhou H, et al. Neddylation is required for perinatal cardiac development through stimulation of metabolic maturation. Cell Rep. (2023) 42:112018. doi: 10.1016/j.celrep.2023.112018
90. Shi P, Wu J, Li M, Cao Y, Wu J, Ren P, et al. Upregulation of Hsp27 via further inhibition of histone H2A ubiquitination confers protection against myocardial ischemia/reperfusion injury by promoting glycolysis and enhancing mitochondrial function. Cell Death Discovery. (2023) 9:466. doi: 10.1038/s41420-023-01762-x
91. Zhang R, Zhang D, Han F, Song X, Zhang Y, Zhang J, et al. The deubiquitinase USP7 and E3 ligase TRIM21 regulate vasculogenic mimicry and Malignant progression of RMS by balancing SNAI2 homeostasis. J Exp Clin Cancer Res. (2024) 43:135. doi: 10.1186/s13046-024-03056-1
92. Guo D, Tong Y, Jiang X, Meng Y, Jiang H, Du L, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IκBα. Cell Metab. (2022) 34:1312–1324.e6. doi: 10.1016/j.cmet.2022.08.002
93. Xu C, Tsai Y-H, Galbo PM, Gong W, Storey AJ, Xu Y, et al. Cistrome analysis of YY1 uncovers a regulatory axis of YY1:BRD2/4-PFKP during tumorigenesis of advanced prostate cancer. Nucleic Acids Res. (2021) 49:4971–88. doi: 10.1093/nar/gkab252
94. Shen J, Jin Z, Lv H, Jin K, Jonas K, Zhu C, et al. PFKP is highly expressed in lung cancer and regulates glucose metabolism. Cell Oncol (Dordr). (2020) 43:617–29. doi: 10.1007/s13402-020-00508-6
95. Fan Y, Wang J, Xu Y, Wang Y, Song T, Liang X, et al. Anti-Warburg effect by targeting HRD1-PFKP pathway may inhibit breast cancer progression. Cell Commun Signal. (2021) 19:18. doi: 10.1186/s12964-020-00679-7
96. Liu J-L, Xu X, Rixiati Y, Wang C-Y, Ni H-L, Chen W-S, et al. Dysfunctional circadian clock accelerates cancer metastasis by intestinal microbiota triggering accumulation of myeloid-derived suppressor cells. Cell Metab. (2024) 36:1320–1334.e9. doi: 10.1016/j.cmet.2024.04.019
97. Liu X, Sun X, Mu W, Li Y, Bu W, Yang T, et al. Autophagic flux-lipid droplet biogenesis cascade sustains mitochondrial fitness in colorectal cancer cells adapted to acidosis. Cell Death Discovery. (2025) 11:1–12. doi: 10.1038/s41420-025-02301-6
98. Meng Y, Guo D, Lin L, Zhao H, Xu W, Luo S, et al. Glycolytic enzyme PFKL governs lipolysis by promoting lipid droplet-mitochondria tethering to enhance β-oxidation and tumor cell proliferation. Nat Metab. (2024) 6:1092–107. doi: 10.1038/s42255-024-01047-2
99. Benador IY, Veliova M, Liesa M, Shirihai OS. Mitochondria bound to lipid droplets: where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. (2019) 29:827–35. doi: 10.1016/j.cmet.2019.02.011
100. Li R, Li X, Zhao J, Meng F, Yao C, Bao E, et al. Mitochondrial STAT3 exacerbates LPS-induced sepsis by driving CPT1a-mediated fatty acid oxidation. Theranostics. (2022) 12:976–98. doi: 10.7150/thno.63751
101. Wang L, Zhang T, Wang L, Cai Y, Zhong X, He X, et al. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. (2017) 36:1330–47. doi: 10.15252/embj.201695417
102. Li Q, Hoppe T. Role of amino acid metabolism in mitochondrial homeostasis. Front Cell Dev Biol. (2023) 11:1127618. doi: 10.3389/fcell.2023.1127618
103. Vanweert F, Schrauwen P, Phielix E. Role of branched-chain amino acid metabolism in the pathogenesis of obesity and type 2 diabetes-related metabolic disturbances BCAA metabolism in type 2 diabetes. Nutr Diabetes. (2022) 12:35. doi: 10.1038/s41387-022-00213-3
104. Zhao S, Zhou L, Wang Q, Cao J-H, Chen Y, Wang W, et al. Elevated branched-chain amino acid promotes atherosclerosis progression by enhancing mitochondrial-to-nuclear H2O2-disulfide HMGB1 in macrophages. Redox Biol. (2023) 62:102696. doi: 10.1016/j.redox.2023.102696
105. Qian L, Li N, Lu X-C, Xu M, Liu Y, Li K, et al. Enhanced BCAT1 activity and BCAA metabolism promotes RhoC activity in cancer progression. Nat Metab. (2023) 5:1159–73. doi: 10.1038/s42255-023-00818-7
106. Wang T, Zhang Y, Liu Y, Huang Y, Wang W. Amino acid-starved cancer cells utilize macropinocytosis and ubiquitin-proteasome system for nutrient acquisition. Adv Sci (Weinh). (2024) 11:e2304791. doi: 10.1002/advs.202304791
107. Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U.S.A. (2015) 112:15790–7. doi: 10.1073/pnas.1521919112
108. Cohen-Kaplan V, Livneh I, Avni N, Fabre B, Ziv T, Kwon YT, et al. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci U.S.A. (2016) 113:E7490–9. doi: 10.1073/pnas.1615455113
109. Poulia KA, Sarantis P, Antoniadou D, Koustas E, Papadimitropoulou A, Papavassiliou AG, et al. Pancreatic cancer and cachexia-metabolic mechanisms and novel insights. Nutrients. (2020) 12:1543. doi: 10.3390/nu12061543
110. Zhao L, Gao N, Peng X, Chen L, Meng T, Jiang C, et al. TRAF4-mediated LAMTOR1 ubiquitination promotes mTORC1 activation and inhibits the inflammation-induced colorectal cancer progression. Adv Sci (Weinh). (2024) 11:e2301164. doi: 10.1002/advs.202301164
111. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. (2020) 37:101674. doi: 10.1016/j.redox.2020.101674
112. Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, et al. Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes In Vivo. Circulation. (2019) 139:1913–36. doi: 10.1161/CIRCULATIONAHA.118.033552
113. Watanabe S, Murata Y, Oka Y, Oiwa K, Horiuchi M, Iguchi Y, et al. Mitochondria-associated membrane collapse impairs TBK1-mediated proteostatic stress response in ALS. Proc Natl Acad Sci U.S.A. (2023) 120:e2315347120. doi: 10.1073/pnas.2315347120
114. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. (2015) 24:4429–42. doi: 10.1093/hmg/ddv179
115. Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. (2012) 30:453–9. doi: 10.1038/nbt.2158
116. Zinghirino F, Pappalardo XG, Messina A, Nicosia G, De Pinto V, Guarino F. VDAC genes expression and regulation in mammals. Front Physiol. (2021) 12:708695. doi: 10.3389/fphys.2021.708695
117. Wang Z, Sun A, Yan A, Yao J, Huang H, Gao Z, et al. Circular RNA MTCL1 promotes advanced laryngeal squamous cell carcinoma progression by inhibiting C1QBP ubiquitin degradation and mediating beta-catenin activation. Mol Cancer. (2022) 21:92. doi: 10.1186/s12943-022-01570-4
118. Lee SH, Yang J-H, Park U-H, Choi H, Kim YS, Yoon B-E, et al. SIRT1 ubiquitination is regulated by opposing activities of APC/C-Cdh1 and AROS during stress-induced premature senescence. Exp Mol Med. (2023) 55:1232–46. doi: 10.1038/s12276-023-01012-1
119. Xu C, Wang L, Fozouni P, Evjen G, Chandra V, Jiang J, et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol. (2020) 22:1170–9. doi: 10.1038/s41556-020-00579-5
120. Zhang J, Liu L, Li Y, Huang Y, Xiao S, Deng Z, et al. HSP90 C-terminal domain inhibition promotes VDAC1 oligomerization via decreasing K274 mono-ubiquitination in Hepatocellular Carcinoma. Neoplasia. (2023) 44:100935. doi: 10.1016/j.neo.2023.100935
121. Zhong C, Niu Y, Liu W, Yuan Y, Li K, Shi Y, et al. S100A9 derived from chemoembolization-induced hypoxia governs mitochondrial function in hepatocellular carcinoma progression. Adv Sci (Weinh). (2022) 9:e2202206. doi: 10.1002/advs.202202206
122. Yao J, Wang J, Xu Y, Guo Q, Sun Y, Liu J, et al. CDK9 inhibition blocks the initiation of PINK1-PRKN-mediated mitophagy by regulating the SIRT1-FOXO3-BNIP3 axis and enhances the therapeutic effects involving mitochondrial dysfunction in hepatocellular carcinoma. Autophagy. (2022) 18:1879–97. doi: 10.1080/15548627.2021.2007027
123. Chen F. Inhibiting Pink1/Parkin-mediated mitophagy enhances the anticancer effects of quercetin in hepatocellular carcinomaf. Biochem Biophys Res Commun. (2024) 712–713:149899. doi: 10.1016/j.bbrc.2024.149899
124. Su Q, Wang J, Liu F, Zhang Y. Blocking Parkin/PINK1-mediated mitophagy sensitizes hepatocellular carcinoma cells to sanguinarine-induced mitochondrial apoptosis. Toxicol In Vitro. (2020) 66:104840. doi: 10.1016/j.tiv.2020.104840
125. Chi H-C, Chen S-L, Lin S-L, Tsai C-Y, Chuang W-Y, Lin Y-H, et al. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene. (2017) 36:5274–84. doi: 10.1038/onc.2017.136
126. Ma M, Zhang C, Cao R, Tang D, Sang X, Zou S, et al. UBE2O promotes lipid metabolic reprogramming and liver cancer progression by mediating HADHA ubiquitination. Oncogene. (2022) 41:5199–213. doi: 10.1038/s41388-022-02509-1
127. Wang H, Shu L, Lv C, Liu N, Long Y, Peng X, et al. BRCC36 deubiquitinates HMGCR to regulate the interplay between ferroptosis and pyroptosis. Adv Sci (Weinh). (2024) 11:e2304263. doi: 10.1002/advs.202304263
128. Zhao B, Liang Z, Zhang L, Jiang L, Xu Y, Zhang Y, et al. Ponicidin promotes hepatocellular carcinoma mitochondrial apoptosis by stabilizing keap1-PGAM5 complex. Adv Sci (Weinh). (2024) 11:e2406080. doi: 10.1002/advs.202406080
129. Chen L, Lin X, Lei Y, Xu X, Zhou Q, Chen Y, et al. Aerobic glycolysis enhances HBx-initiated hepatocellular carcinogenesis via NF-κBp65/HK2 signalling. J Exp Clin Cancer Res. (2022) 41:329. doi: 10.1186/s13046-022-02531-x
130. Ping D, Pu X, Ding G, Zhang C, Jin J, Xu C, et al. Sirtuin4 impacts mitochondrial homeostasis in pancreatic cancer cells by reducing the stability of AlkB homolog 1 via deacetylation of the HRD1-SEL1L complex. Biochim Biophys Acta Gene Regul Mech. (2023) 1866:194941. doi: 10.1016/j.bbagrm.2023.194941
131. Qin C, Wang Y, Zhao B, Li Z, Li T, Yang X, et al. STOML2 restricts mitophagy and increases chemosensitivity in pancreatic cancer through stabilizing PARL-induced PINK1 degradation. Cell Death Dis. (2023) 14:191. doi: 10.1038/s41419-023-05711-5
132. Ji M, Sun L, Zhang M, Liu Y, Zhang Z, Wang P. RN0D, a galactoglucan from Panax notoginseng flower induces cancer cell death via PINK1/Parkin mitophagy. Carbohydr Polym. (2024) 332:121889. doi: 10.1016/j.carbpol.2024.121889
133. Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev Cell. (2018) 46:441–455.e8. doi: 10.1016/j.devcel.2018.07.012
134. Su D, Ding C, Wang R, Qiu J, Liu Y, Tao J, et al. E3 ubiquitin ligase RBCK1 confers ferroptosis resistance in pancreatic cancer by facilitating MFN2 degradation. Free Radic Biol Med. (2024) 221:136–54. doi: 10.1016/j.freeradbiomed.2024.05.031
135. Lee D-E, Yoo JE, Kim J, Kim S, Kim S, Lee H, et al. NEDD4L downregulates autophagy and cell growth by modulating ULK1 and a glutamine transporter. Cell Death Dis. (2020) 11:38. doi: 10.1038/s41419-020-2242-5
136. Niu N, Lu P, Yang Y, He R, Zhang L, Shi J, et al. Loss of Setd2 promotes Kras-induced acinar-to-ductal metaplasia and epithelia-mesenchymal transition during pancreatic carcinogenesis. Gut. (2020) 69:715–26. doi: 10.1136/gutjnl-2019-318362
137. Fu Y, Ricciardiello F, Yang G, Qiu J, Huang H, Xiao J, et al. The role of mitochondria in the chemoresistance of pancreatic cancer cells. Cells. (2021) 10:497. doi: 10.3390/cells10030497
138. Hájek P, Chomyn A, Attardi G. Identification of a novel mitochondrial complex containing mitofusin 2 and stomatin-like protein 2. J Biol Chem. (2007) 282:5670–81. doi: 10.1074/jbc.M608168200
139. Zhang L, Ouyang L, Guo Y, Zhang J, Liu B. UNC-51-like kinase 1: from an autophagic initiator to multifunctional drug target. J Med Chem. (2018) 61:6491–500. doi: 10.1021/acs.jmedchem.7b01684
140. Ji Y, Luo Y, Wu Y, Sun Y, Zhao L, Xue Z, et al. SEL1L-HRD1 endoplasmic reticulum-associated degradation controls STING-mediated innate immunity by limiting the size of the activable STING pool. Nat Cell Biol. (2023) 25:726–39. doi: 10.1038/s41556-023-01138-4
141. Qi H, Ning X, Yu C, Ji X, Jin Y, McNutt MA, et al. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. (2019) 10:170. doi: 10.1038/s41419-018-1271-9
142. Gao B, Qiao Y, Zhu S, Yang N, Zou S-S, Liu Y-J, et al. USP36 inhibits apoptosis by deubiquitinating cIAP1 and survivin in colorectal cancer cells. J Biol Chem. (2024) 300:107463. doi: 10.1016/j.jbc.2024.107463
143. Zhang W, Gong J, Yang H, Wan L, Peng Y, Wang X, et al. The mitochondrial protein MAVS stabilizes p53 to suppress tumorigenesis. Cell Rep. (2020) 30:725–738.e4. doi: 10.1016/j.celrep.2019.12.051
144. Sane S, Hafner A, Srinivasan R, Masood D, Slunecka JL, Noldner CJ, et al. UBXN2A enhances CHIP-mediated proteasomal degradation of oncoprotein mortalin-2 in cancer cells. Mol Oncol. (2018) 12:1753–77. doi: 10.1002/1878-0261.12372
145. Wang S, Jiang X, Xie X, Yin J, Zhang J, Liu T, et al. piR-823 inhibits cell apoptosis via modulating mitophagy by binding to PINK1 in colorectal cancer. Cell Death Dis. (2022) 13:465. doi: 10.1038/s41419-022-04922-6
146. D’Onofrio N, Martino E, Mele L, Colloca A, Maione M, Cautela D, et al. Colorectal cancer apoptosis induced by dietary δ-valerobetaine involves PINK1/parkin dependent-mitophagy and SIRT3. Int J Mol Sci. (2021) 22:8117. doi: 10.3390/ijms22158117
147. Wei Y, Xiao G, Xu H, Sun X, Shi Y, Wang F, et al. Radiation resistance of cancer cells caused by mitochondrial dysfunction depends on SIRT3-mediated mitophagy. FEBS J. (2023) 290:3629–45. doi: 10.1111/febs.16769
148. Zhang K, Zhang D, Wang J, Wang Y, Hu J, Zhou Y, et al. Aloe gel glucomannan induced colon cancer cell death via mitochondrial damage-driven PINK1/Parkin mitophagy pathway. Carbohydr Polym. (2022) 295:119841. doi: 10.1016/j.carbpol.2022.119841
149. Fang Y, Shen Z-Y, Zhan Y-Z, Feng X-C, Chen K-L, Li Y-S, et al. CD36 inhibits β-catenin/c-myc-mediated glycolysis through ubiquitination of GPC4 to repress colorectal tumorigenesis. Nat Commun. (2019) 10:3981. doi: 10.1038/s41467-019-11662-3
150. Zeng F, Chen X, Cui W, Wen W, Lu F, Sun X, et al. RIPK1 binds MCU to mediate induction of mitochondrial ca2+ Uptake and promotes colorectal oncogenesis. Cancer Res. (2018) 78:2876–85. doi: 10.1158/0008-5472.CAN-17-3082
151. Zhang C, Liu J, Xu D, Zhang T, Hu W, Feng Z. Gain-of-function mutant p53 in cancer progression and therapy. J Mol Cell Biol. (2020) 12:674–87. doi: 10.1093/jmcb/mjaa040
152. Salehi B, Berkay Yılmaz Y, Antika G, Boyunegmez Tumer T, Fawzi Mahomoodally M, Lobine D, et al. Insights on the use of α-lipoic acid for therapeutic purposes. Biomolecules. (2019) 9:356. doi: 10.3390/biom9080356
153. Neitzel C, Seiwert N, Göder A, Diehl E, Weber C, Nagel G, et al. Lipoic acid synergizes with antineoplastic drugs in colorectal cancer by targeting p53 for proteasomal degradation. Cells. (2019) 8:794. doi: 10.3390/cells8080794
154. Wang X, Zheng Y, Chai Z, Li J, Zhu C, Peng Y, et al. Dihydroartemisinin synergistically enhances the cytotoxic effects of oxaliplatin in colon cancer by targeting the PHB2-RCHY1 mediated signaling pathway. Mol Carcinog. (2023) 62:293–302. doi: 10.1002/mc.23486
155. Maddalena F, Condelli V, Matassa DS, Pacelli C, Scrima R, Lettini G, et al. TRAP1 enhances Warburg metabolism through modulation of PFK1 expression/activity and favors resistance to EGFR inhibitors in human colorectal carcinomas. Mol Oncol. (2020) 14:3030–47. doi: 10.1002/1878-0261.12814
156. Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. (2020) 38:675–8. doi: 10.1038/s41587-020-0546-8
157. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. (2015) 12:720–7. doi: 10.1038/nrgastro.2015.150
158. Birch RJ, Burr N, Subramanian V, Tiernan JP, Hull MA, Finan P, et al. Inflammatory bowel disease-associated colorectal cancer epidemiology and outcomes: an english population-based study. Am J Gastroenterol. (2022) 117:1858–70. doi: 10.14309/ajg.0000000000001941
159. Rana N, Privitera G, Kondolf HC, Bulek K, Lechuga S, De Salvo C, et al. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell. (2022) 185:283–298.e17. doi: 10.1016/j.cell.2021.12.024
160. Gong W, Liu Z, Wang Y, Huang W, Yang K, Gao Z, et al. Reprogramming of Treg cell-derived small extracellular vesicles effectively prevents intestinal inflammation from PANoptosis by blocking mitochondrial oxidative stress. Trends Biotechnol. (2024), S0167-7799(24)00347-0. doi: 10.1016/j.tibtech.2024.11.017.
161. Xu T, Yu W, Fang H, Wang Z, Chi Z, Guo X, et al. Ubiquitination of NLRP3 by gp78/Insig-1 restrains NLRP3 inflammasome activation. Cell Death Differ. (2022) 29:1582–95. doi: 10.1038/s41418-022-00947-8
162. Mai C-T, Wu M-M, Wang C-L, Su Z-R, Cheng Y-Y, Zhang X-J. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol Immunol. (2019) 105:76–85. doi: 10.1016/j.molimm.2018.10.015
163. Hou Y, Sun X, Gheinani PT, Guan X, Sharma S, Zhou Y, et al. Epithelial SMYD5 exaggerates IBD by down-regulating mitochondrial functions via post-translational control of PGC-1α Stability. Cell Mol Gastroenterol Hepatol. (2022) 14:375–403. doi: 10.1016/j.jcmgh.2022.05.006
164. London RE. The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst). (2015) 30:90–103. doi: 10.1016/j.dnarep.2015.02.005
165. Ni P, Xu W, Zhang Y, Chen Q, Li A, Wang S, et al. TXNL1 induces apoptosis in cisplatin resistant human gastric cancer cell lines. Curr Cancer Drug Targets. (2015) 14:850–9. doi: 10.2174/1568009614666141028094612
166. Shen Y, Zhang T, Jia X, Xi F, Jing W, Wang Y, et al. MEF2A, a gene associated with mitochondrial biogenesis, promotes drug resistance in gastric cancer. Biochim Biophys Acta Mol Basis Dis. (2024) 1871:167497. doi: 10.1016/j.bbadis.2024.167497
167. Yang H, Li Q, Chen X, Weng M, Huang Y, Chen Q, et al. Targeting SOX13 inhibits assembly of respiratory chain supercomplexes to overcome ferroptosis resistance in gastric cancer. Nat Commun. (2024) 15:4296. doi: 10.1038/s41467-024-48307-z
168. Huang G, Xiang Z, Wu H, He Q, Dou R, Lin Z, et al. The lncRNA BDNF-AS/WDR5/FBXW7 axis mediates ferroptosis in gastric cancer peritoneal metastasis by regulating VDAC3 ubiquitination. Int J Biol Sci. (2022) 18:1415–33. doi: 10.7150/ijbs.69454
169. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. (2020) 19:43. doi: 10.1186/s12943-020-01168-8
170. Deng Y-Q, Gao M, Lu D, Liu Q-P, Zhang R-J, Ye J, et al. Compound-composed Chinese medicine of Huachansu triggers apoptosis of gastric cancer cells through increase of reactive oxygen species levels and suppression of proteasome activities. Phytomedicine. (2024) 123:155169. doi: 10.1016/j.phymed.2023.155169
171. Luo Q, Wu X, Zhao P, Nan Y, Chang W, Zhu X, et al. OTUD1 activates caspase-independent and caspase-dependent apoptosis by promoting AIF nuclear translocation and MCL1 degradation. Adv Sci (Weinh). (2021) 8:2002874. doi: 10.1002/advs.202002874
172. Holloway J, Seeley A, Cobbe N, Turkington RC, Longley DB, Evergren E. The E3 ubiquitin ligase Itch regulates death receptor and cholesterol trafficking to affect TRAIL-mediated apoptosis. Cell Death Dis. (2024) 15:40. doi: 10.1038/s41419-023-06417-4
173. Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun. (2016) 7:13730. doi: 10.1038/ncomms13730
174. Chen X, Xu W, Zhuo S, Chen X, Chen P, Guan S, et al. Syntaphilin downregulation facilitates radioresistance via mediating mitochondria distribution in esophageal squamous cell carcinoma. Free Radical Biol Med. (2021) 165:348–59. doi: 10.1016/j.freeradbiomed.2021.01.056
175. Whelan KA, Chandramouleeswaran PM, Tanaka K, Natsuizaka M, Guha M, Srinivasan S, et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene. (2017) 36:4843–58. doi: 10.1038/onc.2017.102
Keywords: mitochondrial homeostasis, ubiquitination, gastrointestinal tumors, mitochondrial biogenesis, mitochondrial dynamics, mitophagy, mitochondrial metabolism
Citation: Huang B, Yang Y, Liu J, Zhang B and Lin N (2025) Ubiquitination regulation of mitochondrial homeostasis: a new sight for the treatment of gastrointestinal tumors. Front. Immunol. 16:1533007. doi: 10.3389/fimmu.2025.1533007
Received: 22 November 2024; Accepted: 24 February 2025;
Published: 11 March 2025.
Edited by:
Wenyi Jin, City University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Chunying Li, Georgia State University, United StatesCopyright © 2025 Huang, Yang, Liu, Zhang and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Biao Zhang, YWZuMTk5NTA1QDE2My5jb20=; Nengming Lin, bG5tMTAxM0B6anUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.