 Zhi-Xun Guo1
Zhi-Xun Guo1 Jia-Li Ma1Jin-Qiu Zhang2Ling-Ling Yan2Ying Zhou2Xin-li Mao2,3,4*
Jia-Li Ma1Jin-Qiu Zhang2Ling-Ling Yan2Ying Zhou2Xin-li Mao2,3,4* Shao-Wei Li2,3,4*Xian-Bin Zhou2,3,4*
Shao-Wei Li2,3,4*Xian-Bin Zhou2,3,4*- 1Taizhou Hospital of Zhejiang Province affiliated to Wenzhou Medical University, Linhai, Zhejiang, China
- 2Department of Gastroenterology, Taizhou Hospital of Zhejiang Province affiliated to Wenzhou Medical University, Linhai, Zhejiang, China
- 3Key Laboratory of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor of Zhejiang Province, Linhai, Zhejiang, China
- 4Institute of Digestive Disease, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
Background: Esophageal cancer (EC) is the seventh-most prevalent cancer worldwide and is a significant contributor to cancer-related mortality. Metabolic reprogramming in tumors frequently coincides with aberrant immune function alterations, and extensive research has demonstrated that perturbations in energy metabolism within the tumor microenvironment influence the occurrence and progression of esophageal cancer. Current treatment modalities for esophageal cancer primarily include encompass chemotherapy and a limited array of targeted therapies, which are hampered by toxicity and drug resistance issues. Immunotherapy, particularly immune checkpoint inhibitors (ICIs) targeting the PD-1/PD-L1 pathway, has exhibited promising results; however, a substantial proportion of patients remain unresponsive. The optimization of these immunotherapies requires further investigation. Mounting evidence underscores the importance of modulating metabolic traits within the tumor microenvironment (TME) to augment anti-tumor immunotherapy.
Methods: We selected relevant studies on the metabolism of the esophageal cancer tumor microenvironment and immune cells based on our searches of MEDLINE and PubMed, focusing on screening experimental articles and reviews related to glucose metabolism, amino acid metabolism, and lipid metabolism, as well their interactions with tumor cells and immune cells, published within the last five years. We analyzed and discussed these studies, while also expressing our own insights and opinions.
Results: A total of 137 articles were included in the review: 21 articles focused on the tumor microenvironment of esophageal cancer, 33 delved into research related to glucose metabolism and tumor immunology, 30 introduced amino acid metabolism and immune responses, and 17 focused on the relationship between lipid metabolism in the tumor microenvironment and both tumor cells and immune cells.
Conclusion: This article delves into metabolic reprogramming and immune alterations within the TME of EC, systematically synthesizes the metabolic characteristics of the TME, dissects the interactions between tumor and immune cells, and consolidates and harnesses pertinent immunotherapy targets, with the goal of enhancing anti-tumor immunotherapy for esophageal cancer and thereby offering insights into the development of novel therapeutic strategies.
1 Introduction
Esophageal cancer (EC) is the seventh-most common cancer and sixth leading cause of cancer-related mortality worldwide. It is a complex multifactorial disease with varying distribution worldwide (1). Among 456,000 annual cases of esophageal cancer, esophageal squamous cell carcinoma (ESCC) comprises approximately 90% of all cases. High-incidence regions include East Asia to Central Asia, the East African Rift Valley, and South Africa, with East Asia having the highest incidence (2). Statistics indicate that China bears over 50% of the global burden of EC cases, with ESCC serving as the primary histological subtype (3).
The emergence, progression, and dissemination of EC are closely related to its cellular microenvironment, also known as the tumor microenvironment (TME). A refractory TME is a key issues that clinically impede effective cancer treatment. Increasing evidence highlights the significance of TME in driving cancer heterogeneity and treatment resistance (4–7). Tumor cells, immune cells, stromal cells, and various cytokines collectively constitute the TME (8), forming a precisely structured ecological environment that favors the expansion, proliferation and dissemination of cancer cells. Immunocompetent cells and supporting stromal elements acting as essential components of the TME, exhibit high specialization and heterogeneity in the phenotype and function, participating throughout the entire process of tumor development and treatment response (9, 10). Metabolic reprogramming plays a crucial role in cancer progression, and the TME is a significant factor influencing this process (11) (Table 1).
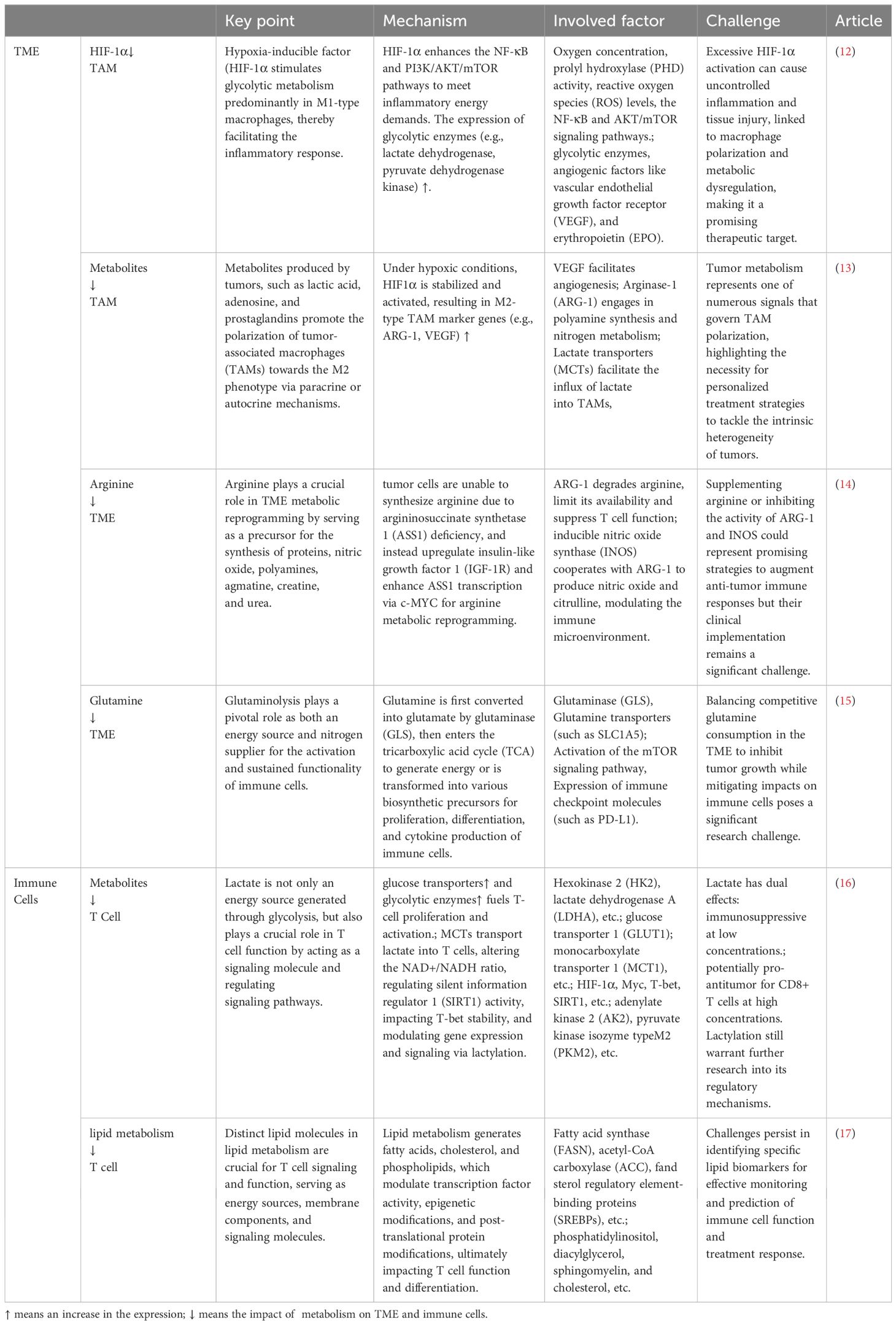
Table 1. Details, key insights, and shortcomings concerning metabolic reprogramming of esophageal cancer and immune cells.
Tumors activate immune and stromal cells through immune metabolic reprogramming, such as tumor-associated macrophages (TAMs) (18), tumor-associated neutrophils (TANs) (19), regulatory T cells (Tregs) (20), myeloid-derived suppressor cells (MDSCs) (21), endothelial cells (ECs) (22), and cancer-associated fibroblasts (CAFs) (23). Tumors promote their occurrence and development by increasing aerobic glycolysis (24), affecting protein palmitoylation of oncogenes and tumor suppressor genes (25), and altering microenvironmental cellular metabolism such as abnormal fatty acid synthesis and oxidation, as well as competing for oxygen and glucose (26), and essential or non-essential amino acids (27, 28). Investigating the impact of TME metabolism on stromal and immune cells can significantly enhance our understanding of EC progression and facilitate the development of more effective EC treatment methods.
Chemotherapy remains the primary treatment method for most patients with EC, although it is associated with dose-limiting toxicity (29). Significant advancements in targeted therapy have led to better treatment strategies. However, the National Comprehensive Cancer Network (NCCN) advocates only for the administration of trastuzumab, which specifically targets human epidermal growth factor receptor 2 (HER2), and ramucirumab, an inhibitor of vascular endothelial growth factor receptor 2 (VEGFR-2), as therapeutic options for patients diagnosed with EC (30). In recent years, findings concerning the efficacy of immunotherapy for the treatment of diverse types of cancer have been exciting. For instance, immune checkpoint inhibitors (ICIs), immune-modulating compounds, monospecific antibodies and other immunotherapies represent new methods for treating EC (31, 32). This approach harnesses the patient’s own immune system to fight malignant cells by blocking the immune checkpoint pathways. Inhibiting programmed death 1 (PD-1) and programmed death ligand 1 (PD-L1), classic ICIs, have demonstrated compelling clinical benefits in various malignancies, including ESCC (33, 34). However, despite the availability of numerous treatment options for EC, there is a dearth of effective treatments.
Given the above, we attempted to augment anti-tumor immunotherapy for EC through a comprehensive summation of the metabolic characteristics within the TME, and identify and utilize relevant immunotherapy targets.
2 Microenvironment of EC
Within the TME, anti-tumor cells and tumor cells counteract each other. Various molecules released by stromal cells facilitate tumor growth by directly activating cancer cell growth signals or by remodeling the microenvironment (35). Upon activation, immune cells, such as dendritic cells (DCs) (36), effector T cells (T effs) (37), memory T cells (T mems) (38), and natural killer cells (NKs) (39), mount an immune response against tumor cells in the TME, thereby controlling tumor progression and preventing evasion of immune surveillance.
In EC, vascular endothelial growth factor receptor (VEGF) plays a crucial role in tumor progression. Shimada et al. identified a positive correlation between serum VEGF levels and the tumor stage and prognosis by comparing the serum VEGF content in patients diagnosed with ESCC (40). Cancer-associated fibroblasts (CAFs) also constitute a vital component of the EC TME, playing significant roles in the disease progression and prognosis. During disease progression, inhibition of the transcription factor KLF4 in epithelial cells results in a marked reduction in the expression of ANXA1, which serves as a ligand for formyl peptide receptor type 2 (FPR2). This decrease subsequently triggers the unregulated conversion of normal fibroblasts into CAFs, ultimately facilitating crosstalk (41). Cancer-derived S100A8 engages with CD147 receptors on CAFs, triggering their polarization and subsequently fostering chemoresistance via the activation of the intracellular RhoA-ROCK-MLC2-MRTF-A signaling pathway (42). Chen et al. discovered that precancerous esophageal epithelial cells could reprogram normal resident fibroblasts into CAFs by downregulating the ANXA1-FRP2 signaling pathway (43). Qiu et al. emphasized the future prospects and clinical trends of targeting CAFs for the ESCC treatment. A deeper understanding of the molecular biology of CAFs could potentially contribute to the development of novel anti-ESCC strategies (44). Concurrently, prolonged exposure of the esophagus to a gastric acid environment fosters a chronic inflammatory microenvironment. Evidence supports the crucial role of CAFs in the progression of Barrett’s esophagus (BE) and esophageal adenocarcinoma (EAC) (45, 46). In EAC, the distal esophageal epithelium undergoes metaplasia as a consequence of reflux disease, and inflammatory cytokines are significantly upregulated, affecting the cancer prognosis. The infiltration of M2-macrophages, especially the predominance of M2-like cells over M1-like cells, correlates with an unfavorable prognosis (47, 48). In EAC and ESCC, tumor-infiltrating macrophages appear to play a crucial role in the aggressive progression of malignancy and resistance to therapy (49). Some studies have indicated that MDSCs play a role in fostering the growth of esophageal tumors in experimental settings, and elevated infiltration levels of these cells in patients with EC correlate with adverse prognostic indicators (50, 51). This suggests that MDSCs may serve as a potential oncogenic factor within the TME of EC.
Some bacteria also influence the TME in EC. Certain bacterial infections in the TME activate immune responses that directly eliminate tumor cells, while other bacteria induce immune evasion of tumor cells by inhibiting inflammatory pathways (52, 53). Examples of bacteria that influence the TME in EC include Helicobacter pylori (54) and Streptococcus (55).
The interaction between neoplastic cells and tumor-suppressing immune cells in the local microenvironment of esophageal tumors reveals meaningful therapeutic strategies. By selectively upregulating or downregulating anti-tumor substances, specifically inhibiting tumor cells, and activating or restoring immune cell functions, an anti-TME is created. Therefore, we have summarized the characteristics of metabolic reprogramming in the esophageal cancer EC TME, providing strategies for the treatment of EC (Figure 1).
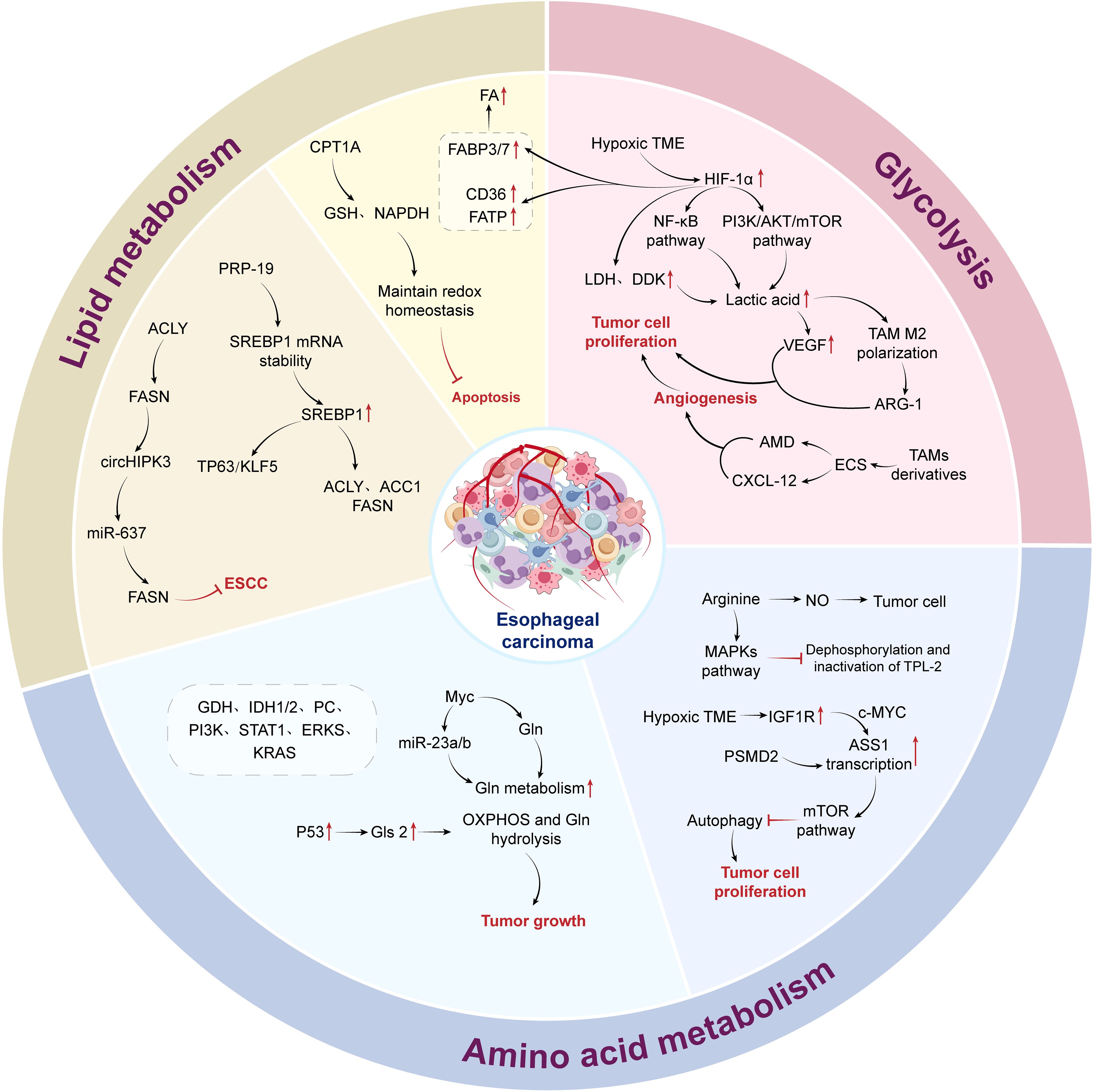
Figure 1. Metabolic reprogramming in the tumor microenvironment of esophageal cancer.
In this section, we have summarized 21 articles, while 10 discussed immune cells in the TME of EC, 7 explored the roles of VEGF and CAFs in the progression of EC, and 4 investigated the impact of microflora on the esophageal TME.
3 Glucose metabolism
In 1956, Otto Warburg noted that, unlike normal cells, cells predominantly utilize glycolysis for energy production, even under aerobic conditions, rather than oxidative phosphorylation (OXPHOS). This metabolic phenomenon is referred to as aerobic glycolysis, also known as the “Warburg effect (56, 57). The increase in aerobic glycolysis results in glucose deprivation and lactate accumulation of lactate within tumor cells. Meanwhile, numerous dysfunctional tumor blood vessels and rapidly proliferating tumor cells create a hypoxic TME, forcing cells to upregulate the expression of glucose transporter 1 (GLUT-1) to counteract glucose consumption by tumor cells and further resulting in an acidic and hypoxic TME (58–60). High glycolytic activity and poor blood exchange reduce glucose availability (61), exacerbating a vicious cycle. This indicates that the improvement of hypoxia and excessive glucose levels can be used as a target to inhibit tumor progression.
Hypoxia-inducible factor (HIF-1α) is expressed in the hypoxic TME and acts as a transcription factor involved in the transcription of glycolytic or glucose transporter genes (59, 62, 63). The transcription of HIF-1α significantly enhances the two major pathways of NF-κB and PI3K/AKT/mTOR, allowing cells to increase glucose metabolism under oxygen-independent conditions (12). Pyruvate kinase isozyme typeM2 (PKM2) is highly expressed in both tumor cells and tumor-associated fibroblasts (64) and is upregulated in patients as well (65). Some studies have revealed that the interplay between PKM2, heat shock protein 90 (HSP90), and HIF-1α leads to the stabilization of PKM2, which in turn facilitates aerobic glycolysis and inhibits the process of cell apoptosis (66). The observed correlation between an elevated PKM2 expression and an adverse prognosis further supports the crucial role of glycolysis in the progression of ESCC.
Lactate dehydrogenase (LDH), pyruvate dehydrogenase (PDH), and pyruvate dehydrogenase kinase (PDK) are key enzymes in pyruvate metabolism. HIF-1α upregulates the expression of LDH and PDK, catalyzing the conversion of pyruvate to lactate and leading to further lactate accumulation (12, 59, 67, 68). Lactate further mediates the expression of VEGF and M2 polarization of TAMs through HIF-1α (69), creating immunosuppressive interactions within TAMs. The accumulation of lactate caused by a hypoxic microenvironment is a crucial factor in reducing anti-tumor immunity.
In some case of tumor progression, the TME can promote the transformation of macrophages into TAMs (70). In a TME characterized by low glucose levels, TAMs modulate their functions through glycolysis activation, while lactate promotes the polarization of M2-type TAMs, leading to elevated expression of VEGF and arginase-1 (ARG-1) and thereby stimulating cancer cell proliferation (71). TAMs indirectly increase the bioavailability of targeted nutrients in the TME, providing nutrition for malignant cells, and their immunosuppressive effects also promote tumor progression (72). The nutritional support mechanism involves the recruitment of TAM-secreted bioactive molecules or activation of the ECs, leading to the generation of TAM-derived adrenomedullin (AMD), chemokine (C-X-C motif) ligand 12 (CXCL12), and other factors, which promote new blood vessel formation (73, 74). Therefore, the role of TAMs in oncogenesis promotion and immunodepression makes them potential targets for cancer therapies.
Glucose is a crucial energy source for activating T-cells, and a low-glucose TME environment can inhibit the T-cell function and reduce their persistence during the initiation of adaptive immune responses (75). As glycolysis increases in tumor cells and TAMs, the abundance of glucose in the TME decreases, eliciting impairment of the T-cell function and further exacerbating the immunosuppressive capacity of the TME. Lactate produced by cancer cells stabilizes extracellular signal-regulated kinases 1/2 (ERK1/2), signal transducer and activator of transcription (STAT) 3, and HIF-1α to induce ARG-1, thereby inhibiting the T-cell function and promoting tumor growth (69, 76). In addition, enhanced glycolysis in tumor cells increases the secretion of colony-stimulating factor (CSF) and macrophage colony-stimulating factor (M-CSF), further inhibiting the function of T cells (77). At this point, low glucose induces increased expression of FoxP3, promoting the transition from T eff cells to T reg cells (78), while also reprogramming T-lymphocyte metabolic processes to adapt to the glucose-deprived environment, characterized by the suppression of glycolysis and the augmentation of OXPHOS (58).
EC cells secrete transforming growth factor-beta (TGF-β), thereby enhancing the immune tolerance of tumors. Clinical studies have established a significant correlation between TGF-β expression levels and the prognosis of patients with EC (79). The TGF-β signaling pathway is abnormally high in patients (80). TGF-β activates regulatory T cells (Tregs) directly, while simultaneously inhibiting the cytotoxicity of T cells and natural killer cells (NKs), impairing DCs antigen-presenting function, and blocking the differentiation of naïve T cells towards effector T cells (81). A study found that TGF-β is highly expressed in patients treated with conventional chemotherapy regimens, suggesting that chemotherapy may upregulate TGF-β levels, leading to the development of immune resistance (80). Furthermore, IL-6 upregulation is evident in both ESCC and EAC (82), originating from the TME and expressed or implicated in related pathways across diverse EC phenotypes (83). This upregulation promotes epithelial-to-mesenchymal transition (EMT), clonogenicity, and chemoresistance in EC (83).
In this section, we have reviewed 33 articles on glucose metabolism in the TME of EC, 11 of which were related to the mechanisms of glucose metabolism, 11 discussed the roles of factors such as HIF-1α and TGF-β in EC, and 13 explored the impact of changes in TME glucose metabolism on immune cells.
4 Amino acid metabolism
Amino acids, as essential resources and metabolites for the sustenance of cellular life, participate in the TME, and their increased demand supports the rapid proliferation of cancer cells (84). Among amino acids, arginine is a precursor for the synthesis of proteins, polyamines, nitric oxide, creatine, agmatine, and urea (85), and is also a source of NO, an important substance in tumor regulation (86). Nutritional arginine in the TME activates the MAPK pathway by inhibiting dephosphorylation and subsequent inactivation of TPL-2, a tumor-promoting locus of MAPK kinase (87). This finding highlights the important role of arginine metabolism in cancer progression. In the hypoxic TME of ESCC, the receptor tyrosine kinase insulin-like growth factor 1 (IGF-1R) is upregulated and increases the transcription of argininosuccinate synthetase 1 (ASS1) regulated by c-MYC, achieving reprogramming of arginine metabolism (14). In the rate-controlling steps of arginine metabolism, the differences in the levels of ASS1 and argininosuccinate lyase (ASL) between ESCC tissues and their metastatic derivatives suggest that blocking ASS1 or ASL may hinder the proliferation of ESCC at the original site and potentiate distant metastatic dissemination (88).
Glutamine is the predominantly consumed amino acid in tumors, and glutamine addiction is a typical characteristic of cancer (89). Research has shown that tumor cells have a stronger dependence on glutamine (90), which is widely present in EC cells, as well as other tumor cells. Glutamine serves as a precursor for the biosynthesis of arginine or other non-essential amino acids, purines, pyridines, and glucose, and provides nutrition for rapidly proliferating malignant cells. Its redox reaction removes reactive oxygen species, making it indispensable for the survival of cancer cell. Indeed cancer cells that lack glutamine undergo rapid cell death, making glutamine a crucial target for cancer therapy (91–93).
The metabolic reprogramming of glutamine is orchestrated by various oncogenic genes (94). Specifically, Myc can directly bind to the promoters of glutamine metabolism genes to enhance glutamine metabolism (95), or indirectly stimulate it by inhibiting the expression of miR-23a/b (96). In contrast, p53 promotes OXPHOS and glutamine hydrolysis, thereby inhibiting tumor growth by upregulating the expression of glutaminase isoenzyme Gls2 (97). Other genes, such as glutamate dehydrogenase (GDH) (98), IDH1/2 (99), pyruvate carboxylase (PC) (98), phosphatidylinositol 3-kinase (PI3K) (100), signal transducer and activator of transcription 1 (STAT1) (101, 102), extracellular signal-regulated kinases (ERKs) (103), and KRAS (104), are associated with metabolic reprogramming of glutamine. In experiments with porcine intestinal ECs, the mTOR and MAPK/ERK signaling pathways were inactivated in a low-glutamine environment and reactivated after glutamine supplementation (105). Glutamine is also essential for T-cell function. Indeed, recent studies have shown that glutamine is a key in-vivo fuel for CD8 T-cells (106). Glutamine depletion inhibits T-cell proliferation and cytokines production, and this inhibitory effect on T-cells is irreversible (107).
The competition for glutamine between cancer cells and T cells results in a relative deficiency of glutamine in T cells (108, 109). Therefore, we can combat tumor proliferation by specifically modulating glutamine levels in the TME of EC. For example, the glutaminase (GLS) inhibitor CB839 not only delay tumor proliferation (110), but also enhances the therapeutic effect of chimeric antigen receptor T-cells (CAR-T) (111). The glutamine transporter inhibitor V-9302 has been demonstrated in animal experiments to reduce the uptake of glutamine by malignant cells in mice (112). Edwards et al. further showed that V-9302 not only diminishes the uptake by malignant cells but also augments the infiltration of CD8+ T cells, elevates the count of Th1 cells secreting the anti-tumor effector molecule IFNγ within the TME, and mitigates tumor-induced glutamine deprivation in lymphocytes, ultimately attenuating the impairment of anti-tumor immunity (109). Therefore, we anticipate that new targeted glutaminase drugs will contribute to the management of EC.
In summary, the amino acids in the TME are indispensable for cell metabolism. The proliferative activity and expansion of cancer cells, along with the activation and differentiation of immune cells, demand a high-energy microenvironment. Amino acids, which serve as secondary major energy providers, exert a profound influence on cellular metabolism. In this section, 30 articles were included, while 6 focused on arginine metabolic reprogramming, 19 focused on glutamine metabolic reprogramming, and 5 discussed therapeutic strategies based on amino acid metabolic reprogramming. A deeper understanding of amino acid metabolism in the TME can aid in identifying novel anti-tumor targets and offer new perspectives for targeted therapies.
5 Lipid metabolism
Alterations in lipid metabolism represent a significant aspect of cellular reprogramming in cancer and serve as a crucial mode of interaction between tumor cells and the TME. Their synthetic and catabolic processes can be effective targets for lipid metabolism.
ESCC progression requires the enhanced synthesis and uptake of lipids (113). Lipid synthesis metabolism is associated with several enzymes, including ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), and sterol regulatory element binding protein 1 (SREBP1). ATP citrate lyase (ACLY) catalyzes a key rate-limiting step in the biosynthesis of fatty acids, cholesterol, and other lipids during de novo fat synthesis. Research has demonstrated that overexpression of ACLY stimulates the proliferation of tumor cells, whereas suppression of ACLY expression hinders their growth (114–116). FASN serves as the catalyst for the final step of fatty acid production, forming palmitate, which involves the conversion of intermediates into saturated fatty acids in the presence of NADPH. FASN exhibits low expression in quiescent normal cells (117), but is overexpressed in many types of cancer. Elevated expression and activity of FASN contributes to the survival of cancer cells (118) and has implicated in the progression of ESCC. For example, the competitive endogenous RNA circHIPK3 upregulates FASN expression in ESCC cells by sponging miR-637, increasing fatty acid biosynthesis, and promoting tumor progression (118). In hypoxic tumor cells, the upregulation of the hypoxia-inducible factor HIF-1α leads to a subsequent increase in FABP3/7 expression (119). Concurrently, there is a significant elevation in the gene and protein levels of CD36 and FATP in these cells (26). This coordinated upregulation facilitates the influx and accumulation of fatty acids (FAs), ultimately improving the survival of hypoxic tumor cells. Preclinical investigations have revealed that the internalization of exogenous fatty acids by CD36 is contingent on CD36 expression, and notably, the combined application of CD36 inhibitors with FASN inhibitors and anti-PD-1 therapy exhibits marked synergistic efficacy (120).
SREBP1 is an important transcriptional regulator of lipid synthesis that regulates the fat generation process by activating ACLY, ACC1, and FASN (121). It can also synergize with TP63/kruppel-like factor 5 (KLF5) in the regulation of fatty acid biosynthesis (122). SREBP1 overexpression correlates with an unfavorable prognosis in ESCC patients and facilitates ESCC progression through the stimulation of fatty acid biosynthesis. In ESCC, pre-mRNA processing factor 19 (PRP19) enhances the stability of SREBP1 mRNA in an n6-methyladenosine-dependent manner, mediating SREBP-dependent fatty acid synthesis and ESCC progression (122). Therefore, we may be able to reduce fatty acid synthesis in TME and disrupt the energy intake of ESCC by targeting these key enzymes, ultimately achieving the goal of delaying tumor progression.
Fatty acid oxidation (FAO) is a process in which fatty acids are shortened to produce acetyl-CoA, NADH, and FADH2 (123). Carnitine palmitoyl transferase I (CPT1A) is a key rate-limiting enzyme in FAO, facilitating the transport of long-chain fatty acids into the mitochondria for oxidation. Its upregulation in ESCC is associated with low survival rates in patients (124). The concentrations of medium- and long-chain acylcarnitines, which serve as the primary substrates for energy generation through FAO in the mitochondria, are markedly reduced in the peripheral blood of patients with ESCC compared to healthy controls. This reduction suggests alterations in β-oxidation activity and the tricarboxylic acid (TCA) cycle in the ESCC cells (125). In ESCC, CPT1A maintains redox homeostasis by providing GSH and NADPH, thereby inhibiting apoptosis. Inhibiting CPT1A, which leads to a decrease in NADPH supply, can thus inhibit anchorage-independent growth of ESCC cells both in vitro and in vivo (124). Overexpression of CPT1A activates FAO and is closely correlated with grading, metastasis, clinical staging, and a poor prognosis in EC patients.
At the same time, FAO is also one of the sources of OXPHOS for some immune cells. For example, compared to T eff cells, the sustained upregulation of CPT1A expression in T cells and studies using CPT1A inhibitors indicate that FAO is an important metabolic pathway in T cells (126). The differentiation and activation of TAM are related to FAO (127). By inhibiting mitochondrial OXPHOS, particularly FAO, the tendency for M2 polarization of TAMs is diminished, resulting in reduced tumor proliferation, angiogenesis, and immunosuppression. These observations underscore the potential of CPT1A as a promising target for clinical intervention in ESCC treatment strategies.
The high energy demands of malignant cells necessitate increased intake, resynthesis, and oxidative breakdown of exogenous lipids through additional fat metabolism to obtain energy. This process of energy alteration also exerts an influence on immune cells. In the TME, lipid metabolism synergizes with glucose and amino acid metabolism to alter the normal functions of immune cells, leading to immunosuppression (Figure 2). This provides tumors with an opportunity to evade immune defense mechanisms, thereby promoting the development of escape mechanisms and cancer progression. We have selected 17 articles that reference research on the relationship between lipid metabolism in the tumor microenvironment and the interaction between tumor cells and immune cells, 12 of which focused on the anabolic metabolism of lipids, and 5 focused on the oxidative catabolism of lipids, hoping to better integrate the relevant targets of lipid metabolism and provide new directions for the development of new drugs.
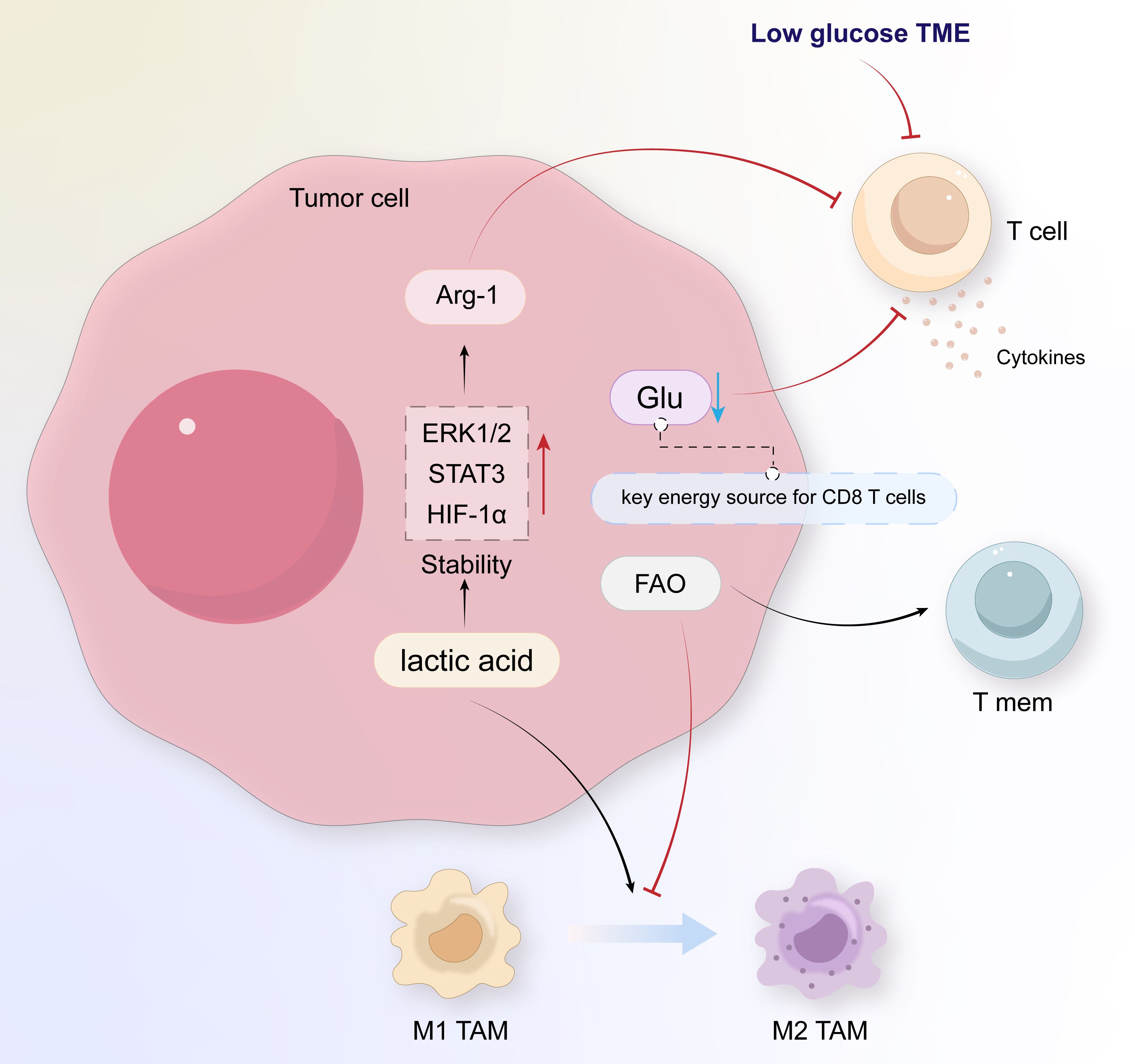
Figure 2. Immunological changes in the tumor microenvironment of esophageal cancer.
6 Conclusion and outlook
EC is a complex and malignant disease that involves tumor cells, creating a nutritious environment conducive to tumor growth by activating various pathways. Tumor metabolites and metabolic regulation affect the function of immune cells, thereby resulting in local immunosuppression that enables tumors to evade the host’s immune surveillance mechanisms. Therefore, addressing metabolic abnormalities in the TME is crucial for the initiation and progression of esophageal cancer.
Currently, effective treatment options for EC are limited, and the treatment response and overall survival rates of patients undergoing tumor immunotherapy remain suboptimal. The essential roles of glucose, glutamine, and other nutrients in cell proliferation and immune defense make it challenging to avoid the toxic and side effects associated with targeted modulation of their levels within the TME. However, targeted therapies continue to face the challenge of drug resistance. For instance, an elevated GLS expression in tumor cells enables glutamine synthesis from glutamate, thereby permitting malignant cells to sustain proliferation even during glutamine deprivation (128).
Metabolic reprogramming is a distinctive feature of both tumor cells and immune responses. The immunosuppressive microenvironment in EC tumors, marked by elevated lactate levels, reduced amino acid availability, and increased fatty acid accumulation along with heightened energy consumption, represents a key focus for metabolic interventions in targeted therapies. Several microenvironment-targeted drugs, including PD-1/PD-L1 inhibitors and anti-angiogenic agents, have progressed to clinical trials and shown promising efficacy. Nevertheless, the therapeutic mechanisms underlying metabolic reprogramming in the EC microenvironment are in the preclinical stage. Further exploration of its immunological and biological facets is pivotal for the development of novel drugs and the refinement of existing immunotherapies (Table 2).
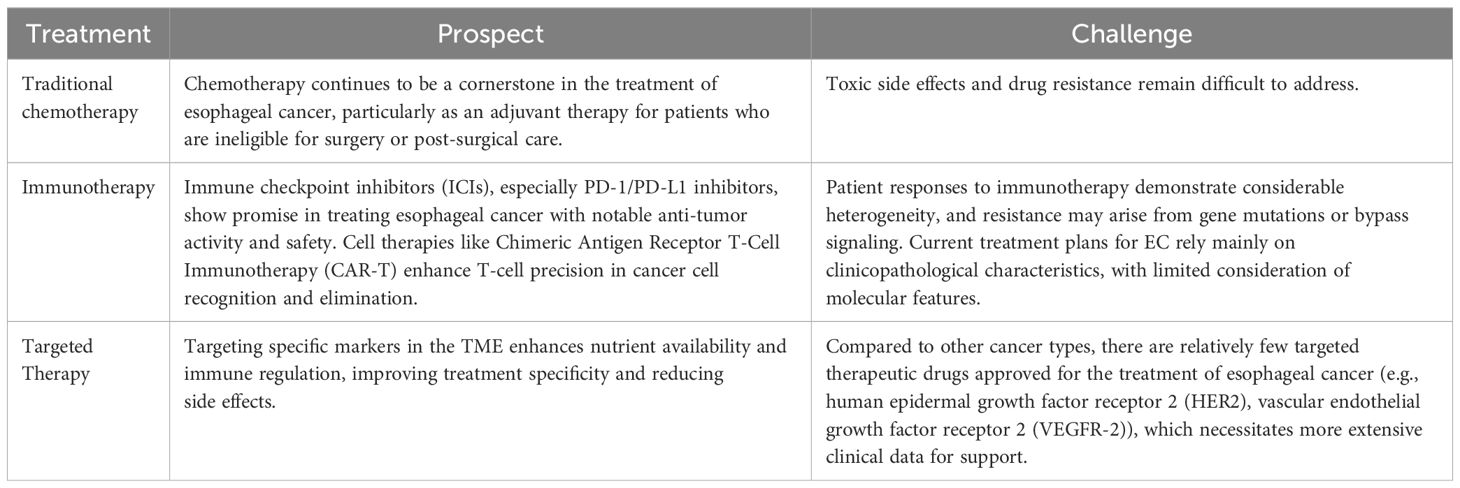
Table 2. Prospects and challenges of EC therapies.
Consequently, we anticipate utilizing more advanced sequencing techniques to delve deeper into the interactions between immune cells and tumor cells in EC metabolism. For instance, the application of spatial tri-omic sequencing technologies enables the delineation of spatial dynamic remodeling within EC (129). Perturb-DBiT has the capacity to elucidate clonal dynamics and cooperative interactions, while also identifying differential and synergistic perturbations that promote or inhibit immune infiltration in tumors (130). In addition, spatial CITE sequencing can reveal spatially distinct germinal center reactions in EC (131). Our goal was to identify suitable biomarkers that can reveal more characteristic targets, ultimately leading to the discovery of more effective novel therapies that can enhance nutrient availability and improve immune regulation within the TME. We should also consider the differences in sensitivity to targeted therapy between EAC and ESCC, as well as the potential risks associated with novel targeted therapies. Furthermore, it is important to consider potential risks associated with novel targeted therapies. By collecting and analyzing effective clinical data, we can explore more effective combinations of different targeted therapies, immunotherapy in conjunction with targeted therapy, and targeted therapy combined with conventional chemotherapy (132). These efforts will aid in the development of new anti-EC drugs and enhance the overall efficacy of EC treatments.
Author contributions
Z-XG: Writing – original draft, Writing – review & editing. J-LM: Conceptualization, Writing – review & editing. J-QZ: Conceptualization, Writing – review & editing. L-LY: Conceptualization, Funding acquisition, Writing – review & editing. YZ: Conceptualization, Funding acquisition, Writing – review & editing. X-lM: Funding acquisition, Supervision, Writing – review & editing. X-BZ: Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by the Medical Science and Technology Project of Zhejiang Province (2024KY1788), Medical Science and Technology Project of Zhejiang Province (2024KY1792), Program of Taizhou Science and Technology Grant (23ywa33&23ywa35), Program of Taizhou Science and Technology Grant (23ywb03), and the Program of Taizhou Enze Medical Center Grant (24EZCG02) and the Program of Zhejiang Province "Pioneer and Leading Goose" Science and Technology Program (2025C02139).
Acknowledgments
We thank all authors who participated in this study.
Conflict of interest
The authors declare that this study was conducted in the absence of any commercial or financial relationships that could be interpreted as potential conflicts of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Abnet CC, Arnold M, Wei W-Q. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. (2018) 154:360–73. doi: 10.1053/j.gastro.2017.08.023
3. Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen W, et al. Mortality, morbidity, and risk factors in China and its provinces, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Lond Engl. (2019) 394:1145–58. doi: 10.1016/S0140-6736(19)30427-1
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
6. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
7. Croft W, Evans RPT, Pearce H, Elshafie M, Griffiths EA, Moss P. The single cell transcriptional landscape of esophageal adenocarcinoma and its modulation by neoadjuvant chemotherapy. Mol Cancer. (2022) 21:200. doi: 10.1186/s12943-022-01666-x
8. Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. (2015) 368:7–13. doi: 10.1016/j.canlet.2015.07.039
9. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. (2018) 20:1349–60. doi: 10.1038/s41556-018-0236-7
10. Hirata E, Sahai E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med. (2017) 7:a026781. doi: 10.1101/cshperspect.a026781
11. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
12. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
13. Zhang J, Dong Y, Di S, Xie S, Fan B, Gong T. Tumor associated macrophages in esophageal squamous carcinoma: Promising therapeutic implications. BioMed Pharmacother Biomed Pharmacother. (2023) 167:115610. doi: 10.1016/j.biopha.2023.115610
14. Liu L-X, Heng J-H, Deng D-X, Zhao H, Zheng Z-Y, Liao L-D, et al. Sulconazole induces PANoptosis by triggering oxidative stress and inhibiting glycolysis to increase radiosensitivity in esophageal cancer. Mol Cell Proteomics MCP. (2023) 22:100551. doi: 10.1016/j.mcpro.2023.100551
15. Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal CCS. (2022) 20:114. doi: 10.1186/s12964-022-00909-0
16. Wu H, Huang H, Zhao Y. Interplay between metabolic reprogramming and post-translational modifications: from glycolysis to lactylation. Front Immunol. (2023) 14:1211221. doi: 10.3389/fimmu.2023.1211221
17. Lim SA, Su W, Chapman NM, Chi H. Lipid metabolism in T cell signaling and function. Nat Chem Biol. (2022) 18:470–81. doi: 10.1038/s41589-022-01017-3
18. Netea-Maier RT, Smit JWA, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. (2018) 413:102–9. doi: 10.1016/j.canlet.2017.10.037
19. Shaul ME, Fridlender ZG. Cancer-related circulating and tumor-associated neutrophils - subtypes, sources and function. FEBS J. (2018) 285:4316–42. doi: 10.1111/febs.14524
20. Wolf D, Sopper S, Pircher A, Gastl G, Wolf AM. Treg(s) in cancer: friends or foe? J Cell Physiol. (2015) 230:2598–605. doi: 10.1002/jcp.25016
21. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
22. Hida K, Maishi N, Torii C, Hida Y. Tumor angiogenesis–characteristics of tumor endothelial cells. Int J Clin Oncol. (2016) 21:206–12. doi: 10.1007/s10147-016-0957-1
23. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. (2006) 6:392–401. doi: 10.1038/nrc1877
24. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
25. Ko P-J, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep. (2018) 19:e46666. doi: 10.15252/embr.201846666
26. Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci CMLS. (2016) 73:377–92. doi: 10.1007/s00018-015-2070-4
27. Geck RC, Toker A. Nonessential amino acid metabolism in breast cancer. Adv Biol Regul. (2016) 62:11–7. doi: 10.1016/j.jbior.2016.01.001
28. Salisbury TB, Arthur S. The regulation and function of the L-type amino acid transporter 1 (LAT1) in cancer. Int J Mol Sci. (2018) 19:2373. doi: 10.3390/ijms19082373
29. Liu H, Zhao J, Fu R, Zhu C, Fan D. The ginsenoside Rk3 exerts anti-esophageal cancer activity in vitro and in vivo by mediating apoptosis and autophagy through regulation of the PI3K/Akt/mTOR pathway. PloS One. (2019) 14:e0216759. doi: 10.1371/journal.pone.0216759
30. Samson P, Lockhart AC. Biologic therapy in esophageal and gastric Malignancies: current therapies and future directions. J Gastrointest Oncol. (2017) 8:418–29. doi: 10.21037/jgo.2016.11.13
31. Tanaka T, Nakamura J, Noshiro H. Promising immunotherapies for esophageal cancer. Expert Opin Biol Ther. (2017) 17:723–33. doi: 10.1080/14712598.2017.1315404
32. Zhang Y, Zhang Y, Zhang L. Expression of cancer-testis antigens in esophageal cancer and their progress in immunotherapy. J Cancer Res Clin Oncol. (2019) 145:281–91. doi: 10.1007/s00432-019-02840-3
33. Janjigian YY, Bendell J, Calvo E, Kim JW, Ascierto PA, Sharma P, et al. CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol Off J Am Soc Clin Oncol. (2018) 36:2836–44. doi: 10.1200/JCO.2017.76.6212
34. Kudo T, Hamamoto Y, Kato K, Ura T, Kojima T, Tsushima T, et al. Nivolumab treatment for oesophageal squamous-cell carcinoma: an open-label, multicentre, phase 2 trial. Lancet Oncol. (2017) 18:631–9. doi: 10.1016/S1470-2045(17)30181-X
35. Oya Y, Hayakawa Y, Koike K. Tumor microenvironment in gastric cancers. Cancer Sci. (2020) 111:2696–707. doi: 10.1111/cas.14521
36. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
37. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
38. Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T cell, exhaustion, and tumor immunity. Immunol Med. (2020) 43:1–9. doi: 10.1080/25785826.2019.1698261
39. O’Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. (2019) 19:282–90. doi: 10.1038/s41577-019-0139-2
40. Shimada H, Takeda A, Nabeya Y, Okazumi SI, Matsubara H, Funami Y, et al. Clinical significance of serum vascular endothelial growth factor in esophageal squamous cell carcinoma. Cancer. (2001) 92:663–9. doi: 10.1002/1097-0142(20010801)92:3<663::aid-cncr1368>3.0.co;2-l
41. Chen Y, Zhu S, Liu T, Zhang S, Lu J, Fan W, et al. Epithelial cells activate fibroblasts to promote esophageal cancer development. Cancer Cell. (2023) 41:903–918.e8. doi: 10.1016/j.ccell.2023.03.001
42. Chen X, Cheng G, Zhu L, Liu T, Yang X, Liu R, et al. Alarmin S100A8 imparts chemoresistance of esophageal cancer by reprogramming cancer-associated fibroblasts. Cell Rep Med. (2024) 5:101576. doi: 10.1016/j.xcrm.2024.101576
43. Lavon H, Scherz-Shouval R. Insights into the co-evolution of epithelial cells and fibroblasts in the esophageal tumor microenvironment. Cancer Cell. (2023) 41:826–8. doi: 10.1016/j.ccell.2023.03.020
44. Qiu L, Yue J, Ding L, Yin Z, Zhang K, Zhang H. Cancer-associated fibroblasts: An emerging target against esophageal squamous cell carcinoma. Cancer Lett. (2022) 546:215860. doi: 10.1016/j.canlet.2022.215860
45. Wang J, Zhang G, Wang J, Wang L, Huang X, Cheng Y. The role of cancer-associated fibroblasts in esophageal cancer. J Transl Med. (2016) 14:30. doi: 10.1186/s12967-016-0788-x
46. Fujiya T, Asanuma K, Koike T, Okata T, Saito M, Asano N, et al. Nitric oxide could promote development of Barrett’s esophagus by S-nitrosylation-induced inhibition of Rho-ROCK signaling in esophageal fibroblasts. Am J Physiol Gastrointest Liver Physiol. (2022) 322:G107–16. doi: 10.1152/ajpgi.00124.2021
47. Blank S, Nienhüser H, Dreikhausen L, Sisic L, Heger U, Ott K, et al. Inflammatory cytokines are associated with response and prognosis in patients with esophageal cancer. Oncotarget. (2017) 8:47518–32. doi: 10.18632/oncotarget.17671
48. Cao W, Peters JH, Nieman D, Sharma M, Watson T, Yu J. Macrophage subtype predicts lymph node metastasis in oesophageal adenocarcinoma and promotes cancer cell invasion in vitro. Br J Cancer. (2015) 113:738–46. doi: 10.1038/bjc.2015.292
49. Miyashita T, Tajima H, Shah FA, Oshima M, Makino I, Nakagawara H, et al. Impact of inflammation-metaplasia-adenocarcinoma sequence and inflammatory microenvironment in esophageal carcinogenesis using surgical rat models. Ann Surg Oncol. (2014) 21:2012–9. doi: 10.1245/s10434-014-3537-5
50. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother CII. (2011) 60:1419–30. doi: 10.1007/s00262-011-1028-0
51. Karakasheva TA, Waldron TJ, Eruslanov E, Kim S-B, Lee J-S, O’Brien S, et al. CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. (2015) 75:4074–85. doi: 10.1158/0008-5472.CAN-14-3639
52. Ma Q, Xing C, Long W, Wang HY, Liu Q, Wang R-F. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflamm. (2019) 16:53. doi: 10.1186/s12974-019-1434-3
53. Yang L, Li A, Wang Y, Zhang Y. Intratumoral microbiota: roles in cancer initiation, development and therapeutic efficacy. Signal Transduct Target Ther. (2023) 8:35. doi: 10.1038/s41392-022-01304-4
54. Matsuda H, Iwahori K, Takeoka T, Kato R, Urakawa S, Saito T, et al. Helicobacter pylori infection affects the tumor immune microenvironment of esophageal cancer patients. Anticancer Res. (2024) 44:3799–805. doi: 10.21873/anticanres.17205
55. Wu H, Leng X, Liu Q, Mao T, Jiang T, Liu Y, et al. Intratumoral microbiota composition regulates chemoimmunotherapy response in esophageal squamous cell carcinoma. Cancer Res. (2023) 83:3131–44. doi: 10.1158/0008-5472.CAN-22-2593
56. Vaupel P, Schmidberger H, Mayer A. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol. (2019) 95:912–9. doi: 10.1080/09553002.2019.1589653
57. Hochwald JS, Zhang J. Glucose oncometabolism of esophageal cancer. Anticancer Agents Med Chem. (2017) 17:385–94. doi: 10.2174/1871520616666160627092716
58. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
59. Fang H-Y, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. (2009) 114:844–59. doi: 10.1182/blood-2008-12-195941
60. Wang T, Liu H, Lian G, Zhang S-Y, Wang X, Jiang C. HIF1α-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflammation. (2017) 2017:9029327. doi: 10.1155/2017/9029327
61. Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. (2020) 78:1019–33. doi: 10.1016/j.molcel.2020.05.034
62. Lu Z-N, Song J, Sun T-H, Sun G. UBE2C affects breast cancer proliferation through the AKT/mTOR signaling pathway. Chin Med J (Engl). (2021) 134:2465–74. doi: 10.1097/CM9.0000000000001708
63. Buckley AM, Lynam-Lennon N, O’Neill H, O’Sullivan J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat Rev Gastroenterol Hepatol. (2020) 17:298–313. doi: 10.1038/s41575-019-0247-2
64. Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab TEM. (2012) 23:560–6. doi: 10.1016/j.tem.2012.06.010
65. Ma R, Liu Q, Zheng S, Liu T, Tan D, Lu X. PKM2-regulated STAT3 promotes esophageal squamous cell carcinoma progression via TGF-β1-induced EMT. J Cell Biochem. (2019) 120:11539–50. doi: 10.1002/jcb.28434
66. Feng J, Wu L, Ji J, Chen K, Yu Q, Zhang J, et al. PKM2 is the target of proanthocyanidin B2 during the inhibition of hepatocellular carcinoma. J Exp Clin Cancer Res CR. (2019) 38:204. doi: 10.1186/s13046-019-1194-z
67. Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
68. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
69. Mazzone M, Menga A, Castegna A. Metabolism and TAM functions-it takes two to tango. FEBS J. (2018) 285:700–16. doi: 10.1111/febs.14295
70. Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjärvi S, Oliynyk G, et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci U.S.A. (2020) 117:32005–16. doi: 10.1073/pnas.2015343117
71. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
72. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
73. Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, et al. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res Off J Am Assoc Cancer Res. (2011) 17:7230–9. doi: 10.1158/1078-0432.CCR-11-1354
74. Hughes R, Qian B-Z, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. (2015) 75:3479–91. doi: 10.1158/0008-5472.CAN-14-3587
75. Voss K, Larsen SE, Snow AL. Metabolic reprogramming and apoptosis sensitivity: Defining the contours of a T cell response. Cancer Lett. (2017) 408:190–6. doi: 10.1016/j.canlet.2017.08.033
76. Mojsilovic SS, Mojsilovic S, Villar VH, Santibanez JF. The metabolic features of tumor-associated macrophages: opportunities for immunotherapy? Anal Cell Pathol Amst. (2021) 2021:5523055. doi: 10.1155/2021/5523055
77. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. (2018) 28:87–103.e6. doi: 10.1016/j.cmet.2018.04.022
78. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
79. Zhang H, Xie C, Yue J, Jiang Z, Zhou R, Xie R, et al. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol Carcinog. (2017) 56:1150–63. doi: 10.1002/mc.22581
80. Blum AE, Venkitachalam S, Ravillah D, Chelluboyina AK, Kieber-Emmons AM, Ravi L, et al. Systems biology analyses show hyperactivation of transforming growth factor-β and JNK signaling pathways in esophageal cancer. Gastroenterology. (2019) 156:1761–74. doi: 10.1053/j.gastro.2019.01.263
81. Chen J, Gingold JA, Su X. Immunomodulatory TGF-β Signaling in hepatocellular carcinoma. Trends Mol Med. (2019) 25:1010–23. doi: 10.1016/j.molmed.2019.06.007
82. Vinocha A, Grover RK, Deepak R. Clinical significance of interleukin-6 in diagnosis of lung, oral, esophageal, and gall bladder carcinomas. J Cancer Res Ther. (2018) 14:S758–60. doi: 10.4103/0973-1482.183217
83. Ebbing EA, van der Zalm AP, Steins A, Creemers A, Hermsen S, Rentenaar R, et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc Natl Acad Sci U.S.A. (2019) 116:2237–42. doi: 10.1073/pnas.1820459116
84. Sivanand S, Vander Heiden MG. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell. (2020) 37:147–56. doi: 10.1016/j.ccell.2019.12.011
85. Chen C-L, Hsu S-C, Ann DK, Yen Y, Kung H-J. Arginine signaling and cancer metabolism. Cancers. (2021) 13:3541. doi: 10.3390/cancers13143541
86. Keshet R, Erez A. Arginine and the metabolic regulation of nitric oxide synthesis in cancer. Dis Model Mech. (2018) 11:dmm033332. doi: 10.1242/dmm.033332
87. Mieulet V, Yan L, Choisy C, Sully K, Procter J, Kouroumalis A, et al. TPL-2-mediated activation of MAPK downstream of TLR4 signaling is coupled to arginine availability. Sci Signal. (2010) 3:ra61. doi: 10.1126/scisignal.2000934
88. Sun W, Kou H, Fang Y, Xu F, Xu Z, Wang X, et al. FOXO3a-regulated arginine metabolic plasticity adaptively promotes esophageal cancer proliferation and metastasis. Oncogene. (2024) 43:216–23. doi: 10.1038/s41388-023-02906-0
89. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) 16:619–34. doi: 10.1038/nrc.2016.71
90. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. (2017) 3:169–80. doi: 10.1016/j.trecan.2017.01.005
91. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. (2007) 178:93–105. doi: 10.1083/jcb.200703099
92. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. (2013) 123:3678–84. doi: 10.1172/JCI69600
93. Yang L, Venneti S, Nagrath D. Glutaminolysis: A hallmark of cancer metabolism. Annu Rev BioMed Eng. (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
94. Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol. (2012) 23:362–9. doi: 10.1016/j.semcdb.2012.02.002
95. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U.S.A. (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
96. Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. (2009) 458:762–5. doi: 10.1038/nature07823
97. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U.S.A. (2010) 107:7455–60. doi: 10.1073/pnas.1001006107
98. Takeuchi Y, Nakayama Y, Fukusaki E, Irino Y. Glutamate production from ammonia via glutamate dehydrogenase 2 activity supports cancer cell proliferation under glutamine depletion. Biochem Biophys Res Commun. (2018) 495:761–7. doi: 10.1016/j.bbrc.2017.11.088
99. Borodovsky A, Seltzer MJ, Riggins GJ. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr Opin Oncol. (2012) 24:83–9. doi: 10.1097/CCO.0b013e32834d816a
100. Liu J, Zhang C, Lin M, Zhu W, Liang Y, Hong X, et al. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget. (2014) 5:2635–47. doi: 10.18632/oncotarget.1862
101. Zhao L, Huang Y, Zheng J. STAT1 regulates human glutaminase 1 promoter activity through multiple binding sites in HIV-1 infected macrophages. PloS One. (2013) 8:e76581. doi: 10.1371/journal.pone.0076581
102. Zhao L, Huang Y, Tian C, Taylor L, Curthoys N, Wang Y, et al. Interferon-α regulates glutaminase 1 promoter through STAT1 phosphorylation: relevance to HIV-1 associated neurocognitive disorders. PloS One. (2012) 7:e32995. doi: 10.1371/journal.pone.0032995
103. Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V, et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U.S.A. (2012) 109:7705–10. doi: 10.1073/pnas.1116573109
104. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. (2013) 496:101–5. doi: 10.1038/nature12040
105. Zhu Y, Lin G, Dai Z, Zhou T, Li T, Yuan T, et al. L-Glutamine deprivation induces autophagy and alters the mTOR and MAPK signaling pathways in porcine intestinal epithelial cells. Amino Acids. (2015) 47:2185–97. doi: 10.1007/s00726-014-1785-0
106. Ma EH, Dahabieh MS, DeCamp LM, Kaymak I, Kitchen-Goosen SM, Oswald BM, et al. 13C metabolite tracing reveals glutamine and acetate as critical in vivo fuels for CD8 T cells. Sci Adv. (2024) 10:eadj1431. doi: 10.1126/sciadv.adj1431
107. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol Baltim Md 1950. (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
108. Pallett LJ, Dimeloe S, Sinclair LV, Byrne AJ, Schurich A. A glutamine “tug-of-war”: targets to manipulate glutamine metabolism for cancer immunotherapy. Immunother Adv. (2021) 1:ltab010. doi: 10.1093/immadv/ltab010
109. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest. (2021) 131:e140100, 140100. doi: 10.1172/JCI140100
110. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) 13:890–901. doi: 10.1158/1535-7163.MCT-13-0870
111. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of th17 and th1 cell differentiation by glutaminase-dependent metabolism. Cell. (2018) 175:1780–1795.e19. doi: 10.1016/j.cell.2018.10.001
112. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. (2018) 24:194–202. doi: 10.1038/nm.4464
113. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun Lond Engl. (2018) 38:27. doi: 10.1186/s40880-018-0301-4
114. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. (2012) 72:3709–14. doi: 10.1158/0008-5472.CAN-11-4112
115. Hanai J, Doro N, Sasaki AT, Kobayashi S, Cantley LC, Seth P, et al. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol. (2012) 227:1709–20. doi: 10.1002/jcp.22895
116. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem. (2018) 157:1276–91. doi: 10.1016/j.ejmech.2018.09.001
117. Hopperton KE, Duncan RE, Bazinet RP, Archer MC. Fatty acid synthase plays a role in cancer metabolism beyond providing fatty acids for phospholipid synthesis or sustaining elevations in glycolytic activity. Exp Cell Res. (2014) 320:302–10. doi: 10.1016/j.yexcr.2013.10.016
118. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
119. Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. (2014) 9:349–65. doi: 10.1016/j.celrep.2014.08.056
120. Wang H, Franco F, Tsui Y-C, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. (2020) 21:298–308. doi: 10.1038/s41590-019-0589-5
121. Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol J Int Soc Oncodevelopmental Biol Med. (2015) 36:4133–41. doi: 10.1007/s13277-015-3047-5
122. Zhang G-C, Yu X-N, Guo H-Y, Sun J-L, Liu Z-Y, Zhu J-M, et al. PRP19 enhances esophageal squamous cell carcinoma progression by reprogramming SREBF1-dependent fatty acid metabolism. Cancer Res. (2023) 83:521–37. doi: 10.1158/0008-5472.CAN-22-2156
123. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang X-Y, et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
124. Tian T, Lu Y, Lin J, Chen M, Qiu H, Zhu W, et al. CPT1A promotes anoikis resistance in esophageal squamous cell carcinoma via redox homeostasis. Redox Biol. (2022) 58:102544. doi: 10.1016/j.redox.2022.102544
125. Wang PP, Song X, Zhao XK, Wei MX, Gao SG, Zhou FY, et al. Serum metabolomic profiling reveals biomarkers for early detection and prognosis of esophageal squamous cell carcinoma. Front Oncol. (2022) 12:790933. doi: 10.3389/fonc.2022.790933
126. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir actions on regulatory and memory T cells are independent of cpt1a-mediated fatty acid oxidation. Cell Metab. (2018) 28:504–515.e7. doi: 10.1016/j.cmet.2018.06.002
127. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. (2020) 80:1438–50. doi: 10.1158/0008-5472.CAN-19-2994
128. Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. (2015) 17:1556–68. doi: 10.1038/ncb3272
129. Zhang D, Rubio Rodríguez-Kirby LA, Lin Y, Song M, Wang L, Wang L, et al. Spatial dynamics of mammalian brain development and neuroinflammation by multimodal tri-omics mapping. BioRxiv Prepr Serv Biol. (2024), 2024.07.28.605493. doi: 10.1101/2024.07.28.605493
130. Baysoy A, Tian X, Zhang F, Renauer P, Bai Z, Shi H, et al. Spatially Resolved in vivo CRISPR Screen Sequencing via Perturb-DBiT. BioRxiv Prepr Serv Biol. (2024), 2024.11.18.624106. doi: 10.1101/2024.11.18.624106
131. Liu Y, DiStasio M, Su G, Asashima H, Enninful A, Qin X, et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat Biotechnol. (2023) 41:1405–9. doi: 10.1038/s41587-023-01676-0
Keywords: esophageal cancer, tumor microenvironment, metabolic reprogramming, immune cells, glycolysis, targeted therapy
Citation: Guo Z-X, Ma J-L, Zhang J-Q, Yan L-L, Zhou Y, Mao X-l, Li S-W and Zhou X-B (2025) Metabolic reprogramming and immunological changes in the microenvironment of esophageal cancer: future directions and prospects. Front. Immunol. 16:1524801. doi: 10.3389/fimmu.2025.1524801
Received: 08 November 2024; Accepted: 06 January 2025;
Published: 24 January 2025.
Edited by:
Lisha Mou, Shenzhen Second People’s Hospital, ChinaReviewed by:
Fu Gao, Yale University, United StatesXinmei Dong, BeiGene, China
Ping Han, Baylor College of Medicine, United States
Copyright © 2025 Guo, Ma, Zhang, Yan, Zhou, Mao, Li and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin-li Mao, bWFveGxAZW56ZW1lZC5jb20=; Shao-Wei Li, bGlfc2hhb3dlaTgxQGhvdG1haWwuY29t; Xian-Bin Zhou, MTEyMDkzMTY4QHFxLmNvbQ==