 R. Hal Scofield
R. Hal Scofield Jonathan D. Wren
Jonathan D. Wren Valerie M. Lewis
Valerie M. Lewis
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 20 February 2025
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1479814
This article is part of the Research Topic Risk and Protective Factors in the Natural History of Autoimmunity View all 6 articles
Systemic lupus erythematosus (SLE) predominately affects women with a ratio of females-to-males of about 9:1. The complement of sex chromosomes may play and important role in the mechanism of the sex bias. Previous work has shown that men with Klinefleter’s syndrome (47,XXY) as well as women with 47,XXX are found in excess among SLE patients well as among Sjogren’s disease, systemic sclerosis and idiopathic inflammatory myositis. in cells with more than one X chromosome, all but one is inactivated. However, X chromosome inactivation, as mediated by the long noncoding RNA X-inactive specific transcript, or XIST, is not complete with approximately 10% of genes in the non-recombining region of the X chromosome escaping X inactivation. In the TLR7 signaling pathway, both the TLR7 and TLR adaptor interacting with endolysosomal SLC15A4 (TASL) escape X inactivation. Comparing male and female immune cells, there is increased TLR7 signaling related to increased expression of these genes in cells with more than one X chromosome. Cells with more than one X chromosome also express XIST, while cells with one X chromosome do not. XIST, as a source of ligand for TLR7, has also been shown to increase TLR7 signaling. Thus, we propose that both these mechanisms operating in immune cells with more than one X chromosome may act in a mutual way to mediate an X chromosome dose effect for the sex bias of autoimmune disease.
Systemic illness among patients with the rash of lupus erythematosus was first noted by Moriz Kaposi in Vienna during the late 19th century (1). During the remainder of the 19th century and through the middle of the 20th century, the entity of systemic lupus erythematosus was established (2). The bias of this disease to affect women was also noted during this period, with assembled cohorts comprised by ~90% of women (3). This ratio of ~9:1 women to men in cohorts of SLE has continued to be true into the 21st century with modern epidemiological methods (4). This relationship holds true in all racial and ethnic groups studied.
While there are sex hormone differences between SLE patients and matched controls, be they men or women (reviewed in (5)), a fundamental biological explanation for these findings and their relationship to the gender-bias of SLE has not been forthcoming (6). Clearly, some men with SLE have primary hypogonadism. For instance, Mok, et al, found that 5 of 35 men with SLE had low serum testosterone and high luteinizing hormone (LH) while none of 33 control men did (7). The etiology of the hypogonadism in these men was not determined. Higher serum prolactin is also found in both men and women with SLE compared to controls (8, 9). However, men with SLE have the same degree of hypogonadism and low testosterone as do men with other non-female biased chronic illnesses (10), suggesting chronic illness causes hypogonadism in SLE rather than vice versa. Furthermore, at the onset of disease, prior to treatment, there are no sex hormone differences between SLE patients and a matched control population (11).
Seeking another explanation to the sex bias of SLE, we examined the complement of sex chromosomes, initially among men with SLE. We found that these SLE-affected men were much more likely than matched control men to have Klinefelter’s syndrome, that is, 47,XXY (12). Subsequent work found that 47,XXX was found in excess among women with SLE (13). We have also found the rare mosaic, 45XO/46XX/47XXX, is associated with SLE (14), while Turner’s syndrome (female 45,XO) was not found in excess among SLE patients (15). We have now extended these findings to other female-biased autoimmune diseases (16, 17), and others have replicated the findings in SLE (18, 19). Thus, this work established that the number of X chromosomes was a risk factor for SLE, and that the number of X chromosomes might underly the female predominance of the disease.
Discussing the potential mechanisms by which an X chromosome dose effect might operate requires a brief review of the biology of the sex chromosomes, which are in mammals, of course, are the X and Y. The X and Y chromosomes pair in meiosis and mitosis by virtue of short regions at the distal ends of both chromosomes known as the pseudoautosomal regions (PAR); namely PAR1 and PAR2. Each PAR contains a handful of genes, which behave identically to autosomal genes. That is, there is expression of one copy on X and one copy on Y with genetic crossover occurring within PAR1 and PAR2 of the X and Y chromosomes. Meanwhile, on the X chromosome, centromeric to the two PARs are about 2000 genes that are X-linked. Similarly, on the Y chromosome centromeric to the two PARs are about 40 genes in the non-recombining region of Y. Almost all these Y genes are expressed in male gonadal tissue and function in spermatogenesis. In contrast, X-linked genes, like other chromosomes, are not functionally organized; and, generally do not have a Y homologue (although there are exceptions).
In cells with 2 or more X chromosomes, all but one is inactivated by methylation through the action of the X inactive-specific transcript (Xist) gene (Figure 1), which encodes a long non-coding RNA (20). That is, since women have two X-chromosomes and men have one, the imbalance in X chromosome gene expression is equalized by each cell with 2 or more X chromosomes randomly undergoing inactivation (which is mediated by methylation of CpG) of all but one X chromosome. However, despite the fact that the inactive X chromosome makes up the cytoplasmic Barr body, X inactivation is not an all-or-none phenomenon. On the inactivated X chromosome (Xi), about 15% of the genes escape methylation partially or completely giving women (and Klinefelter men) more phenotypic variability compared to normal (i.e., 46XY) men (21).
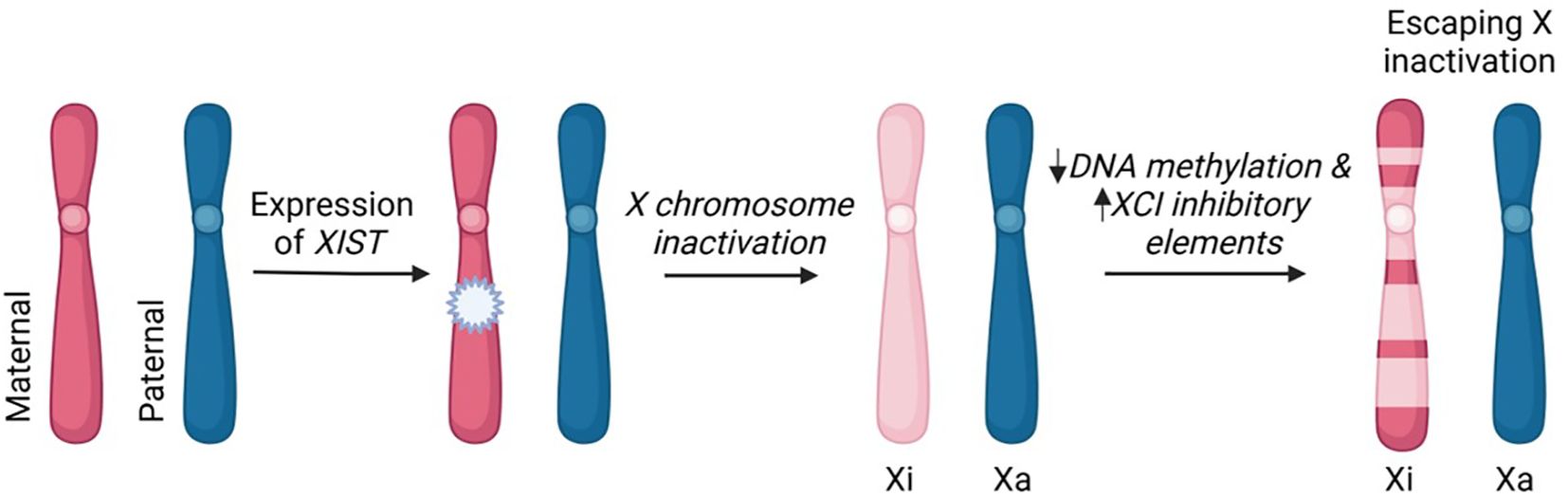
Figure 1. X chromosome inactivation and escaping X inactivation. The process of XCI occurs in mammalian cells that have two or more X chromosomes. In early stages of embryonic development, the maternal or paternal X chromosome is randomly silenced. This X-inactivation is initiated by long non-coding RNA, XIST, and subsequent DNA methylation and histone modifications. The incomplete inactivation of the X chromosome (pseudoautosomal region and variable genes throughout the X chromosome) results in approximately 15% of X-linked genes remaining transcriptionally active. These "escapee" genes contribute to differential expression of X-linked genes between men and women. Xi-inactive; Xa-active.This image was created in Biorender.com.
Continued presence of Xist transcripts were not thought to be needed for maintenance of X inactivation (22). However, recent data demonstrate that this may not be the case in immune cells. Yu and colleague showed that deletion of Xist in CD11c-positive atypical memory B lymphocytes along with TLR7 activation induced isotype switching. In addition, Xist down regulation was found among B cells from women with SLE (23). Also, Anguera has found different localization patterns of the Xist non-coding RNA in B cells with upregulation of 20 X chromosome genes in female cells (24, 25). In a published preprint, conditional knock of Xist in female mice (BALB/c and C57BL/6) produced a spontaneous lupus phenotype (26). Thus, there may be differences in the physiology of this long non-coding RNA in B cells that change X chromosome inactivation in such a way that predisposes to a SLE.
The idea that immune genes are enriched on the X chromosome is frequently evoked. However, we find this is not the case. All human genes and Gene Ontology (GO) categories were downloaded from NCBI’s FTP server (ftp.ncbi.nlm.nih.gov/Gene/DATA/) on August 6th, 2024. Only protein-coding and RNA-producing (eg, ncRNA) transcripts with at least one GO category annotation were selected for analysis. GO categories associated with all transcripts on each human chromosome were then identified, summed, and hypergeometric tests performed to determine relative chromosomal enrichments or depletions in each GO category. False Discovery Rate (FDR) corrections for the most significant p-value (enriched or depleted) were performed to correct for multiple testing. As a positive control, we find the Y chromosome highly enriched (p-value = 0) in the GO categories “spermatogenesis” and “gonadal mesoderm development”. We find that, although there are many immune-related genes on the X chromosome, it is not particularly enriched for immune-related genes more than any other chromosome. This was true for all genes related to immune function with 50 of 1,482 (3.4%) on the X chromosome. Furthermore, no individual category of immune function had enrichment on the X chromosome (Table 1). In fact, we found significant immune-related transcript enrichment on other chromosomes, particularly chromosome 9 (Table 2), and we found other GO categories enriched on the X chromosome (Supplementary Table S1). Some of the categories in Supplementary Table S1 might impact immune processes (eg, miRNA-mediated gene silencing), but none are not immune-specific.
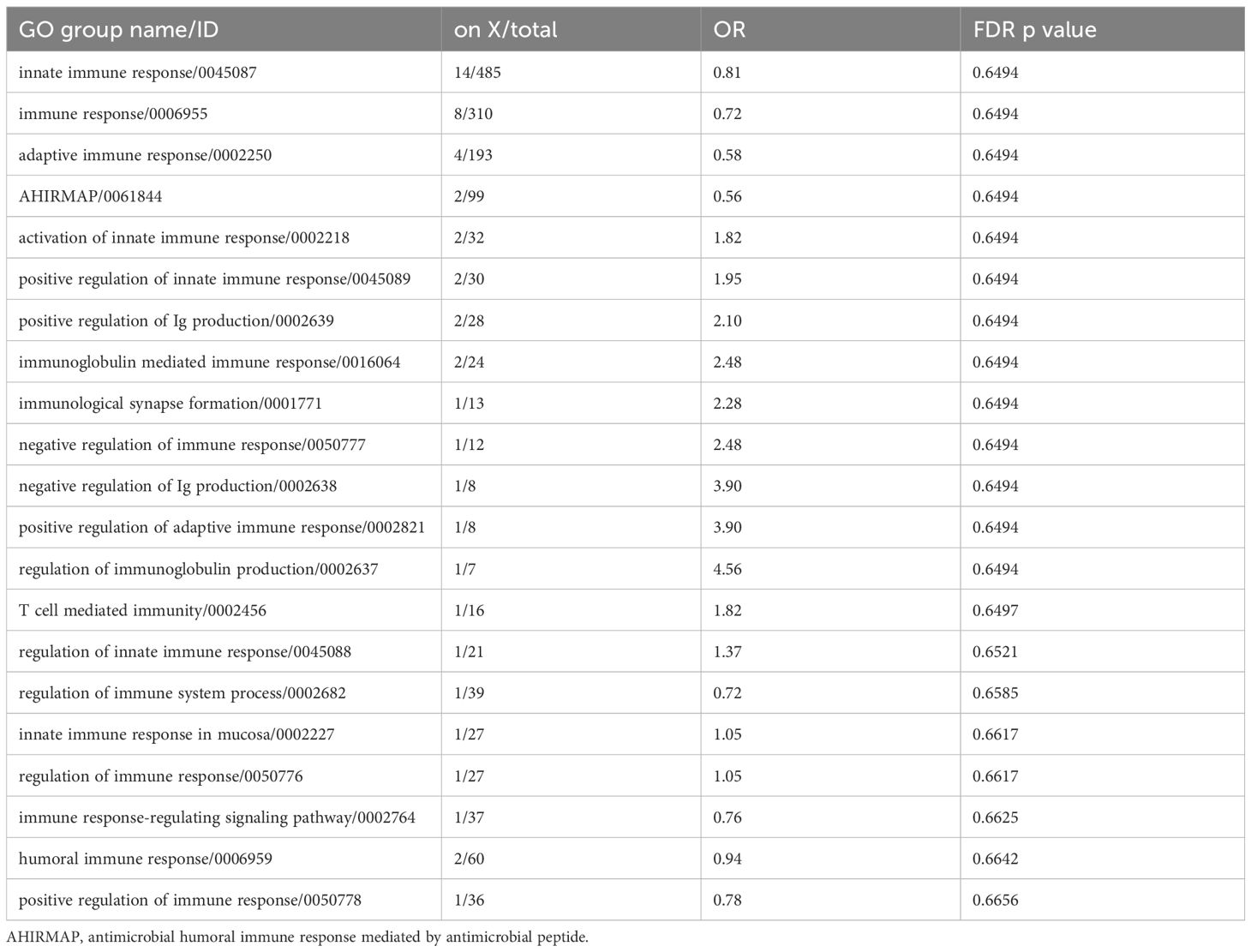
Table 1. Immune related gene categories for the X chromosome.
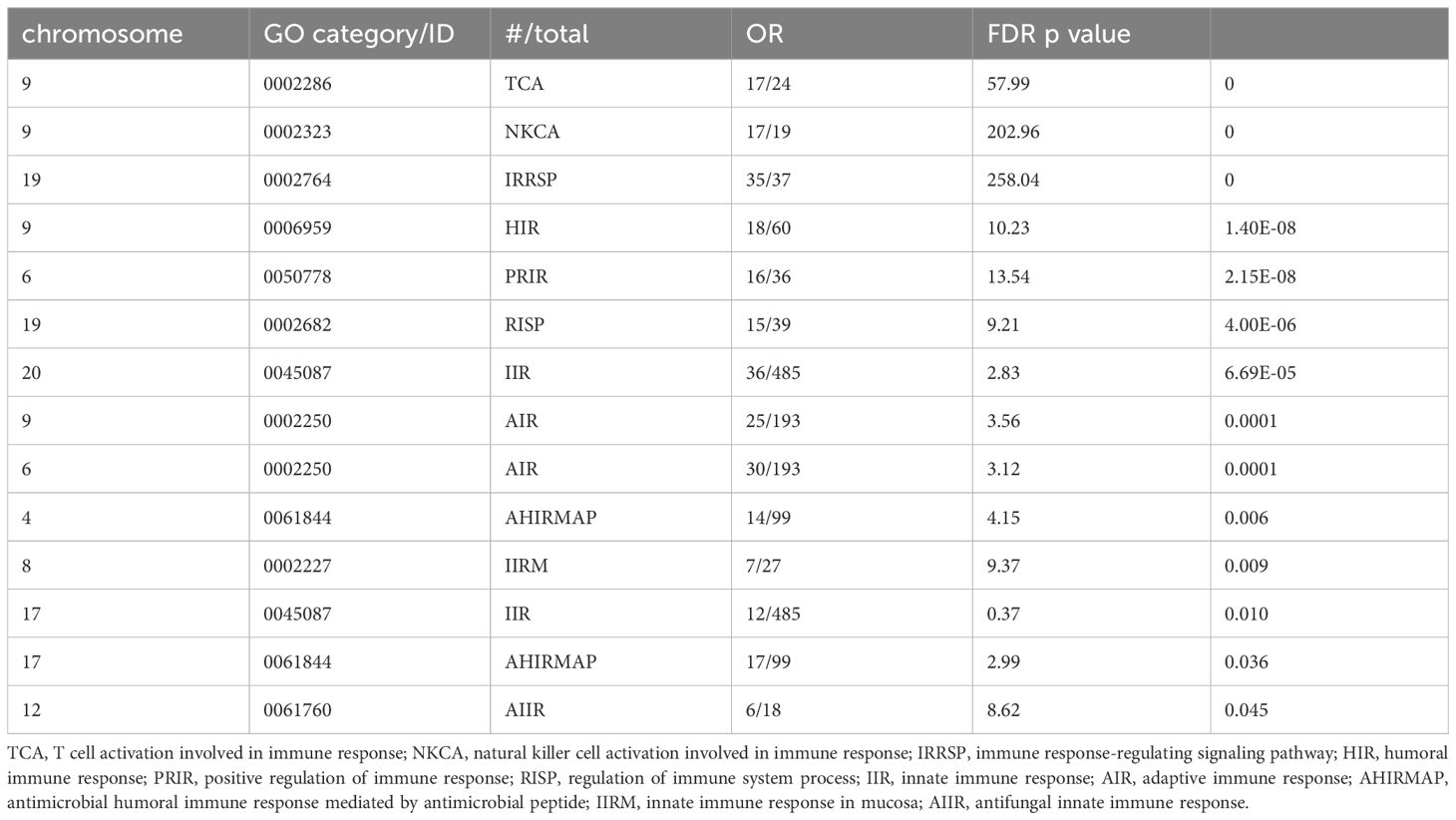
Table 2. Gene ontology categories that are significantly found increased on a given chromosome.
X chromosome genes that escape X inactivation; and, thus have expression of the gene from each of X chromosome, are candidates to mediate the X chromosome dose effect. Our attention was drawn to two genes in the toll like receptor 7 (TLR7) pathway that routinely escape X inactivation; namely, TLR7 itself and TASL (TLR Adaptor Interacting With Endolysosomal SLC15A4). The TLR7 pathway is critical for the pathogenesis of SLE, both in murine models and humans. For instance, rare gain-of-function TLR7 mutations can cause monogenic pediatric SLE (27–29) and mice with TLR7 over-expression due to a translocation between the X and Y chromosome develop a lupus-like illness (30, 31). The TLR7 protein is localized to the endosome and is critical for recognition of viruses and subsequent activation of the innate immune system. TLR7 binds single-stranded RNA or metabolites thereof, which activates the pathway, leading to production of interferon as well as other cytokines (32). Furthermore, common population variants of genes encoding protein that function in the TLR7 pathway show genetic association to the SLE phenotype. These include TLR7, TASL, SLC15a4 (a binding partner of TASL (33)), and UNC93B1, a regulator of TLR7 movement into the endosome (34–37). Many functional studies also implicate the TLR7 pathway in SLE pathogenesis in both human and murine lupus models (30, 31, 38–43).
Given the critical nature of the TLR7 pathway in SLE and the association of X chromosome number with the sex bias of the disease, we elected to study the role of TASL in the TLR7 pathway. As described above, the TASL gene routinely escapes X inactivation and TASL is expressed in several immune cells, including B lymphocytes and monocytes, contains an SLE risk allele (19, 35) and binds SLC15A4 on the lysosomal surface (44). SLC15a4 regulates lysosomal pH, to which TLR7 signaling is highly sensitive (45, 46). In addition, knockout of the gene is known to abrogate TLR7 signaling (47).
Given these data, we undertook studies to examine the role of TASL in the TLR7 pathway (48). In particular, since TASL and SLC15a4 are binding partners and SLC15a4, at least in part, determines lysosomal pH, we studied lysosomal pH. First, we examined expression of the TASL protein in human primary monocytes, B cells and lymphoblastoid cells lines. In each case, TASL was expressed more highly in female cells compared to male cells (49). Additional studies from Odham et al, also found TASL was more highly expressed in female cells and this sexual dimorphism was magnified when stimulated with type I interferons (50). Using a ratiometric measurement of lysosomal pH via fluorescence in unstimulated female monocytes, we found lysosomal pH averaged 4.9 versus 5.6 in male cells (p=0.0001) (48). A similar difference in lysosomal pH was also found between male and female B cells and dendritic cells, while we did not find a female:male dichotomy for lysosomal pH in NK or T cells, neither of which express TASL (48). Thus, the sex difference in lysosomal pH is likely to be associated with increased TLR7 signaling, and may be dependent upon increased expression of TASL in female cells.
In order to determine if, in fact, TASL participates in lysosomal pH regulation and TLR7 signaling, we undertook a series of knockdown experiments using CRISPR-Cas9 and primary human monocytes (CD14+/CD16−). In female cells treated with a TLR7 agonist, TASL knockdown abrogated interferon-alpha, IL-6 and TNF production (49). Thus, TASL is critical for TLR7 pathway signaling. Furthermore, knockdown of TASL expression resulted in a rise in lysosomal pH in female monocytes to the pH we found in male monocytes. And, intracellular transport of NOD1 antigens, a function of SLC15a4, was also abrogated by TASL knockdown (49). However, it should be noted that these results have not been independently replicated; and, thus, are not confirmed.
Several other lines of evidence support a sex-biased function of the TRL7 pathway (51–53). Our studies in primary monocytes and LCLs suggest TASL is involved in the TLR7 in a sexually dimorphic manner such that lysosomal pH is lower and TLR7 signaling greater in female versus male cells. As of late, studies on TASL have shown that the once uncharacterized protein functions as enzyme that regulates interferon regulatory factor 5 (IRF5), colocalizes with TLR7 and is interferon inducible. TASL ability to increase interferon production (our work and others) and its own protein level to be subsequently amplified by interferon stimulations suggest a positive feedforward response that would result in increased production that is often found in SLE affected subjects. Thus, increased expression of both TLR7 (54) and TASL (48, 49) may underlie not only improved outcome of women compared to men in some infections (55) but also female disposition to autoimmunity mediated via TLR7 (56).
Other investigators have taken a different tack in studying the role of the X chromosome in the sex bias of SLE (57, 58); however, the data generated also concern the TLR7 pathway. As mentioned above, XIST long non-coding RNA mediates X chromosome inactivation (Figure 1); and, thus, is expressed only in cells with more than one X chromosome. Dou and colleagues preformed a series of experiments that indicate XIST is a source of ligand for TLR7; and, of course, this is a sex specific source of ligand (57, 58).
First, these investigators noted that XIST is rich in potential TLR7 ligands. A putative TLR7 stimulatory motif, the UU dinucleotide, was found 2,140 times in XIST RNA. XIST was the sex-biased transcript with the highest degree of UU dinucleotide gene expression; and, further, was the only sex-biased expression source of the extended TLR7 motif 5′-GUCCUUCAA-3′ (57, 58). Overall, XIST was the strongest sex biased source of self TLR7 ligand.
Next, these investigators turned to stimulation of TLR7 by XIST nucleotides using HEK-hTLR7 cells as a reporter. The extended TLR7 motif found in XIST as well as a longer sequence of XIST (containing the A-repeat, UU dinucleotide rich region) were also found to stimulate TLR7 signaling as indicated by production of interferon-alpha. Further, not only was the response due to specific binding of XIST nucleotide and dose-dependent, the TLR7 response was inhibited by depletion of XIST as well by hydroxychloroquine (57). Additional studies found that XIST levels were higher in peripheral leukocytes among women with SLE compared to non-SLE affected matched controls, and that levels of XIST correlated with disease activity. The investigators concluded, and we certainly agree, that the XIST long non-coding RNA is the most potent source of sex biased TLR7 ligands in female cells.
We further conclude that these two sets of data suggest synergism for a female biased expansion of the TLR7 signaling pathway that could underlie the X chromosome dose effect found in various autoimmune diseases, including SLE (12–15), Sjögren’s disease (13, 16), polymyositis/dermatomyositis (17), and systemic sclerosis (17). The idea, we think, is straight forward. XIST RNA supplies TLR7 ligand in female cells. In addition, female B lymphocytes, dendritic cells, and monocytes have enhanced TLR7 pathway signaling by virtue of the over-expression (compared to male cells) of not only TLR7 but also TASL. Enhanced TLR7 signaling activity deploys a feed forward loop in the TLR7 pathway that leads to increased expression and activity of the pathway (59). Thus, both increased ligand and enhanced activity support further enhancement of TLR7 signaling in female cells. Of course, these phenomena are universal in cells with more than one X chromosome; that is, from women or Klinefelter men. So, other factors must be in play such as other genetics or environmental exposure.
What environmental exposure might interact with this sex-biased enhancement of TLR7 signaling induced by Xist and genes in the TLR7 pathway that escape X inactivation? One candidate is Epstein Barr virus (EBV). Epidemiological evidence supports the idea that this near ubiquitous infection is necessary but not sufficient for the expression of SLE as well as multiple sclerosis, and there some evidence in Sjögren’s disease (60–65). Recent studies have found that single nucleotide polymorphisms demonstrating genetic association with SLE or Sjögren’s disease are more likely to be found in promoter regions bound by the EBV transcription factor EBV nuclear antigen 2 (EBNA2) (66, 67). Overall, the preponderance of evidence indicates that EBV infection is likely one of the environmental triggers for disease. Furthermore, EBV infects B lymphocytes, a cell type with expression of TASL, engaging and increasing expression of TLR7 (68). B cell hyperplasia is one of the hallmarks of systemic autoimmune disease (69). Thus, these data concerning enhanced expressed XIST, TLR7 and TASL in female cells impacting TLR7 signaling may interact with data concerning a role of EBV in promoting SLE and other autoimmune diseases (62, 63, 70). Of course, estrogen and differential expression of estrogen-regulated genes remain a potential biological trigger of the disease. The sex bias of SLE is present in prepubescent children at about 5 to 1, but of course is less pronounced than after puberty (71). These data suggest an effect of estrogen. Further, there are clear effects of estrogen on B lymphocytes and humeral immunity (72, 73) with effects on development, immune tolerance, immunoglobulin somatic hypermutation, and class switching. In addition, some estrogen effects in B cells may be mediated through cell surface (as opposed to nuclear) estrogen receptors (74).
The evidence is strong that the number of X chromosomes is important for the female bias of some, but not all, autoimmune diseases. The mechanism by which a dose effect for the X chromosome is not understood. Available evidence suggests that multiple factors may play roles that are complementary. These include expression of XIST, which provides TRL7 ligand, and escape of X inactivation by genes whose protein products are critical for TLR7 signaling (see Figure 2).
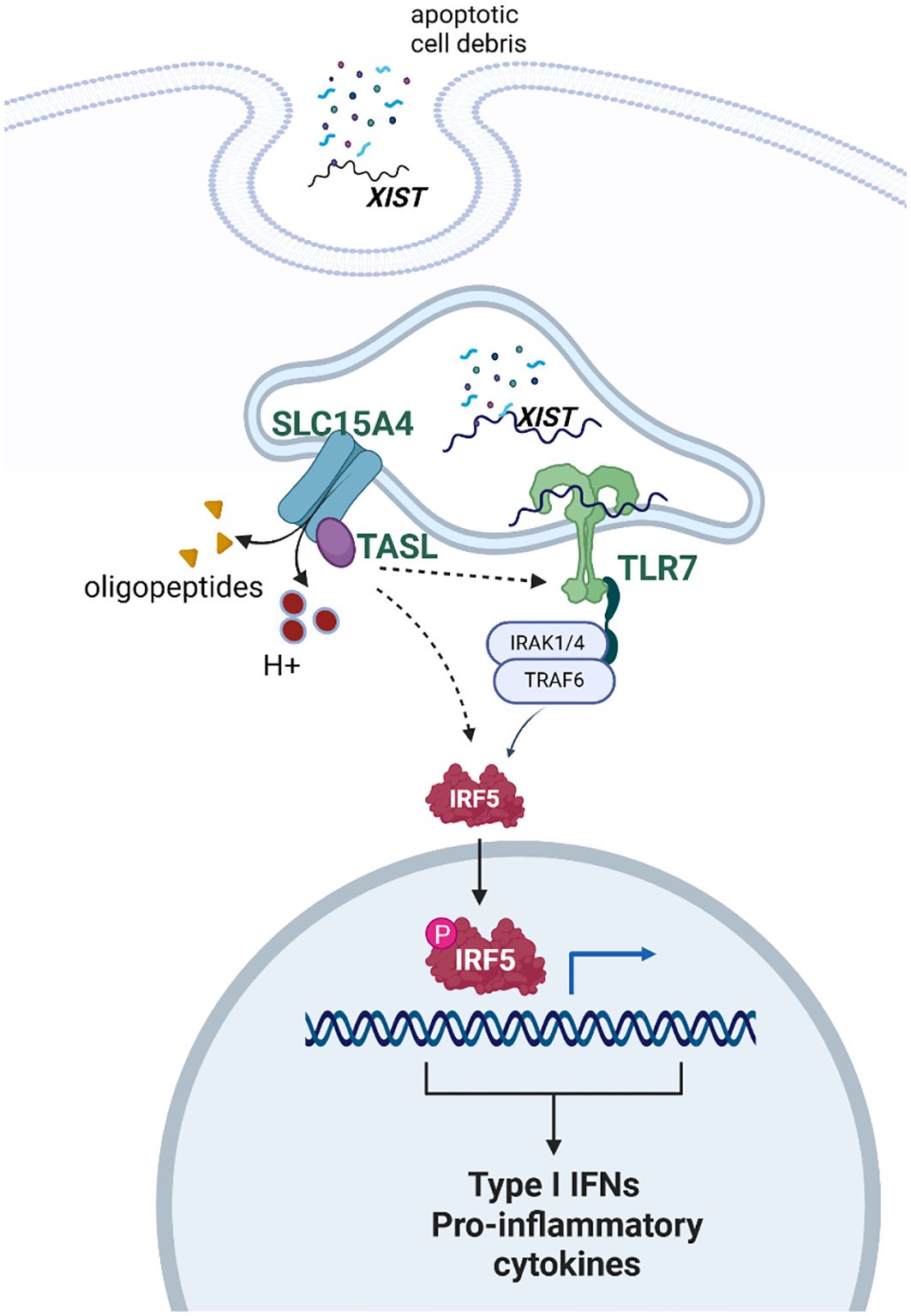
Figure 2. Schematic depicting the proposed interaction of XIST, TLR7, and TASL in response to self-antigen. XIST provides ligand for TLR7. Once TLR7 signaling is activated, there is a feed forward stimulation of the pathway. The genes for both TLR7 and TASL are on the X chromosome and escape X inactivation. Thus, some data suggest that TRL7 signaling is more robust in female cells, compared to male cells, on this basis. Created with Biorender.com.
RHS: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. JW: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. VL: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by research award BX001451 to RS from the US Department of Veterans Affairs as well as NIH grants AR053483, AI082717, and GM104938. The funders had no role in the concept, the data gathering, the data analysis, the writing, or the decision to publish this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1479814/full#supplementary-material
1. Kaposi M. Lupus eythematosus. In: Hebra F, editor. On Diseases of the Skin including the Exanthemata. The New syndenham society, London (1875). p. 14–35.
2. Scofield RH, Oates J. The place of William Osler in the description of systemic lupus erythematosus. Am J Med Sci. (2009) 338:409–12. doi: 10.1097/MAJ.0b013e3181acbd71
3. Harvey AM, Shulman LE, Tumulty PA, Conley CL, Schoenrich EH. Systemic lupus erythematosus: review of the literature and clinical analysis of 138 cases. Med (Baltimore). (1954) 33:291–437. doi: 10.1097/00005792-195412000-00001
4. Dall’Era M, Cisternas MG, Snipes K, Herrinton LJ, Gordon C, Helmick CG. The incidence and prevalence of systemic lupus erythematosus in San Francisco County, California: the California lupus surveillance project. Arthritis Rheumatol. (2017) 69:1996–2005. doi: 10.1002/art.v69.10
5. Lu LJ, Wallace DJ, Ishimori ML, Scofield RH, Weisman MH. Review: Male systemic lupus erythematosus: a review of sex disparities in this disease. Lupus. (2010) 19:119–29. doi: 10.1177/0961203309350755
6. Lahita RG, Bradlow HL. Klinefelter’s syndrome: hormone metabolism in hypogonadal males with systemic lupus erythematosus. J Rheumatol. (1987) 14 Suppl 13:154–7.
7. Mok CC, Lau CS. Profile of sex hormones in male patients with systemic lupus erythematosus. Lupus. (2000) 9:252–7. doi: 10.1191/096120300680198926
8. Jara LJ, Benitez G, Medina G. Prolactin, dendritic cells, and systemic lupus erythematosus. Autoimmun Rev. (2008) 7:251–5. doi: 10.1016/j.autrev.2007.11.018
9. McMurray RW. Prolactin in murine systemic lupus erythematosus. Lupus. (2001) 10:742–7. doi: 10/1191/096120301717164985
10. Mackworth-Young CG, Parke AL, Morley KD, Fotherby K, Hughes GR. Sex hormones in male patients with systemic lupus erythematosus: a comparison with other disease groups. Eur J Rheumatol Inflammation. (1983) 6:228–32.
11. Chang DM, Chang CC, Kuo SY, Chu SJ, Chang ML. Hormonal profiles and immunological studies of male lupus in Taiwan. Clin Rheumatol. (1999) 18:158–62. doi: 10.1007/s100670050075
12. Scofield RH, Bruner GR, Namjou B, Kimberly RP, Ramsey-Goldman R, Petri M, et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheumatism. (2008) 58:2511–7. doi: 10.1002/art.23701
13. Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, et al. X chromosome dose and sex bias in autoimmune diseases: increased prevalence of 47,XXX in systemic lupus erythematosus and sjogren’s syndrome. Arthritis Rheumatol. (2016) 68:1290–300. doi: 10.1002/art.39560
14. Sharma R, Harris VM, Cavett J, Kurien BT, Liu K, Koelsch KA, et al. Rare X chromosome abnormalities in systemic lupus erythematosus and sjogren’s syndrome. Arthritis Rheumatol. (2017) 69:2187–92. doi: 10.1002/art.v69.11
15. Cooney CM, Bruner GR, Aberle T, Namjou-Khales B, Myers LK, Feo L, et al. 46,X,del(X)(q13) Turner’s syndrome women with systemic lupus erythematosus in a pedigree multiplex for SLE. Genes Immunity. (2009) 10:478–81. doi: 10.1038/gene.2009.37
16. Harris VM, Sharma R, Cavett J, Kurien BT, Liu K, Koelsch KA, et al. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjogren’s syndrome. Clin Immunol. (2016) 187:137–8. doi: 10.1016/J.clim.2016.01.002
17. Scofield RH, Lewis VM, Cavitt J, Kurien BT, Assassi S, Martin J, et al. 47XXY and 47XXX in scleroderma and myositis. ACR Open Rheumatol. (2022) 4:528–33. doi: 10.1002/acr2.11413
18. Tangtanatakul P, Lei Y, Jaiwan K, Yang W, Boonbangyang M, Kunhapan P, et al. Association of genetic variation on X chromosome with systemic lupus erythematosus in both Thai and Chinese populations. Lupus Sci Med. (2024) 11(1):e001061. doi: 10.1136/lupus-2023-001061
19. Bentham J, Morris DL, Cunninghame Graham DS, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. (2015) 47(12):1457–65. doi: 10.1038/ng.3434
20. Kanata E, Duffié R, Schulz EG. Establishment and maintenance of random monoallelic expression. Development. (2024) 151(10):dev201741. doi: 10.1242/dev.201741
21. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. (2005) 434:400–4. doi: 10.1038/nature03479
22. Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet. (1999) 22:323–4. doi: 10.1038/11887
23. Yu B, Qi Y, Li R, Shi Q, Satpathy AT, Chang HY. B cell-specific XIST complex enforces X-inactivation and restrains atypical B cells. Cell. (2021) 184:1790–803.e17. doi: 10.1016/j.cell.2021.02.015
24. Wang J, Syrett CM, Kramer MC, Basu A, Atchison ML, Anguera MC. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc Natl Acad Sci USA. (2016) 113(14):E2029-38. doi: 10.1073/pnas.1520113113
25. Syrett CM, Sierra I, Beethem ZT, Dubin AH, Anguera MC. Loss of epigenetic modifications on the inactive X chromosome and sex-biased gene expression profiles in B cells from NZB/W F1 mice with lupus-like disease. J Autoimmun. (2020) 107:102357. doi: 10.1016/j.jaut.2019.102357
26. Lovell CD, Jiwrajka N, Amerman HK, Cancro MP, Anguera MC. Xist deletion in B cells results in systemic lupus erythematosus phenotypes. bioRxiv. (2024). doi: 10.1101/2024.05.15.594175
27. Stremenova Spegarova J, Sinnappurajar P, Al Julandani D, Navickas R, Griffin H, Ahuja M, et al. A de novo TLR7 gain-of-function mutation causing severe monogenic lupus in an infant. J Clin Invest. (2024) 134(13):e179193. doi: 10.1172/jc1179193
28. Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. (2022) 605:349–56. doi: 10.1038/s41586-022-04642-z
29. David C, Arango-Franco CA, Badonyi M, Fouchet J, Rice GI, Didry-Barca B, et al. Gain-of-function human UNC93B1 variants cause systemic lupus erythematosus and chilblain lupus. J Exp Med. (2024) 221(8):e20232066. doi: 10.1084/jem.20232066
30. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. (2006) 312:1669–72. doi: 10.1126/science.1124978
31. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci United States America. (2006) 103:9970–5. doi: 10.1073/pnas.0603912103
32. Li F, Song B, Zhou WF, Chu LJ. Toll-like receptors 7/8: A paradigm for the manipulation of immunologic reactions for immunotherapy. Viral Immunol. (2023) 36:564–78. doi: 10.1089/vim.2023.0077
33. Zhang H, Bernaleau L, Delacrétaz M, Hasanovic E, Drobek A, Eibel H, et al. SLC15A4 controls endolysosomal TLR7-9 responses by recruiting the innate immune adaptor TASL. Cell Rep. (2023) 42:112916. doi: 10.1016/j.celrep.2023.112916
34. Al-Azab M, Idiiatullina E, Liu Z, Lin M, Hrovat-Schaale K, Xian H, et al. Genetic variants in UNC93B1 predispose to childhood-onset systemic lupus erythematosus. Nat Immunol. (2024) 25:969–80. doi: 10.1038/s41590-024-01846-5
35. Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet. (2016) 48:940–6. doi: 10.1038/ng.3603
36. Baccala R, Gonzalez-Quintial R, Blasius AL, Rimann I, Ozato K, Kono DH, et al. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci U S A. (2013) 110:2940–5. doi: 10.1073/pnas.1222798110
37. Blasius AL, Krebs P, Sullivan BM, Oldstone MB, Popkin DL. Slc15a4, a gene required for pDC sensing of TLR ligands, is required to control persistent viral infection. PloS Pathog. (2012) 8:e1002915. doi: 10.1371/journal.ppat.1002915
38. von Hofsten S, Fenton KA, Pedersen HL. Human and murine toll-like receptor-driven disease in systemic lupus erythematosus. Int J Mol Sci. (2024) 24(10):5351. doi: 10.3390/ijms25105351
39. Sakata K, Nakayamada S, Miyazaki Y, Kubo S, Ishii A, Nakano K, et al. Up-regulation of TLR7-mediated IFN-alpha production by plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Front Immunol. (2018) 9:1957. doi: 10.3389/fimmu.2018.01957
40. Clancy RM, Markham AJ, Buyon JP. Endosomal Toll-like receptors in clinically overt and silent autoimmunity. Immunol Rev. (2016) 269:76–84. doi: 10.1111/imr.2016.269.issue-1
41. Miquel CH, Faz-Lopez B, Guéry JC. Influence of X chromosome in sex-biased autoimmune diseases. J Autoimmun. (2023) 137:102992. doi: 10.1016/j.jaut.2023.102992
42. Wolf C, Lim EL, Mokhtari M, Kind B, Odainic A, Lara-Villacanas E, et al. UNC93B1 variants underlie TLR7-dependent autoimmunity. Sci Immunol. (2024) 9:eadi9769. doi: 10.1126/sciimmunol.adi9769
43. Mishra H, Schlack-Leigers C, Lim EL, Thieck O, Magg T, Raedler J, et al. Disrupted degradative sorting of TLR7 is associated with human lupus. Sci Immunol. (2024) 9:eadi9575. doi: 10.1126/sciimmunol.adi9575
44. Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, et al. The bioPlex network: A systematic exploration of the human interactome. Cell. (2015) 162:425–40. doi: 10.1016/j.cell.2015.06.043
45. Kobayashi T, Tanaka T, Toyama-Sorimachi N. How do cells optimize luminal environments of endosomes/lysosomes for efficient inflammatory responses? J Biochem. (2013) 154:491–9. doi: 10.1093/jb/mvt099
46. Kobayashi T, Tsutsui H, Shimabukuro-Demoto S, Sugitani-Yoshida R, Karyu H, Furuyama-Tanaka K, et al. Lysosome biogenesis regulated by the amino-acid transporter SLC15A4 is critical for functional integrity of mast cells. Int Immunol. (2017) 29(12):551–6. doi: 10.1093/intimm/dxx063
47. Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, Furuyama-Tanaka K, Karyu H, Sugiura Y, et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity. (2014) 41:375–88. doi: 10.1016/j.immuni.2014.08.011
48. Harris VM, Harley ITW, Kurien BT, Koelsch KA, Scofield RH. Lysosomal pH is regulated in a sex dependent manner in immune cells expressing CXorf21. Front Immunol. (2019) 10:578. doi: 10.3389/fimmu.2019.00578
49. Harris VM, Koelsch KA, Kurien BT, Harley ITW, Wren JD, Harley JB, et al. Characterization of cxorf21 provides molecular insight into female-bias immune response in SLE pathogenesis. Front Immunol. (2019) 10:2160. doi: 10.3389/fimmu.2019.02160
50. Odhams CA, Roberts AL, Vester SK. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus. Nat. Commun. (2019) 10:2164. doi: 10.1038/s41467-019-10106-2
51. Guéry JC. Sex differences in primary HIV infection: revisiting the role of TLR7-driven type 1 IFN production by plasmacytoid dendritic cells in women. Front Immunol. (2021) 12:729233. doi: 10.3389/fimmu.2021.729233
52. Laffont S, Guéry JC. Deconstructing the sex bias in allergy and autoimmunity: From sex hormones and beyond. Adv Immunol. (2019) 142:35–64. doi: 10.1016/bs.ai.2019.04.001
53. Rubtsova K, Marrack P, Rubtsov AV. TLR7, IFNγ, and T-bet: their roles in the development of ABCs in female-biased autoimmunity. Cell Immunol. (2015) 294:80–3. doi: 10.1016/j.cellimm.2014.12.002
54. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol. (2018) 3(19):eaap8855. doi: 10.1126/sciimmunol.aap8855
55. Spiering AE, de Vries TJ. Why females do better: the X chromosomal TLR7 gene-dose effect in COVID-19. Front Immunol. (2021) 12:756262. doi: 10.3389/fimmu.2021.756262
56. Souyris M, Mejia JE, Chaumeil J, Guery JC. Female predisposition to TLR7-driven autoimmunity: gene dosage and the escape from X chromosome inactivation. Semin. Immunopathol. (2019) 41:153–64. doi: 10.1007/s00281-018-0712-y
57. Crawford JD, Wang H, Trejo-Zambrano D, Cimbro R, Talbot CC Jr., Thomas MA, et al. The XIST lncRNA is a sex-specific reservoir of TLR7 ligands in SLE. JCI Insight. (2023) 8(20):e169344. doi: 10.1172/jci.insight.169344
58. Dou DR, Zhao Y, Belk JA, Zhao Y, Casey KM, Chen DC, et al. Xist ribonucleoproteins promote female sex-biased autoimmunity. Cell. (2024) 187:733–49.e16. doi: 10.1016/j.cell.2023.12.037
59. Assil S, Coléon S, Dong C, Décembre E, Sherry L, Allatif O, et al. Plasmacytoid dendritic cells and infected cells form an interferogenic synapse required for antiviral responses. Cell Host Microbe. (2019) 25:730–45.e6. doi: 10.1016/j.chom.2019.03.005
60. Almohmeed YH, Avenell A, Aucott L, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein Barr virus and multiple sclerosis. PloS One. (2013) 8:e61110. doi: 10.1371/journal.pone.0061110
61. Draborg A, Izarzugaza JM, Houen G. How compelling are the data for Epstein-Barr virus being a trigger for systemic lupus and other autoimmune diseases? Curr Opin Rheumatol. (2016) 28:398–404. doi: 10.1097/BOR0000000000000289
62. Jog NR, James JA. Epstein barr virus and autoimmune responses in systemic lupus erythematosus. Front Immunol. (2020) 11:623944. doi: 10.3389/fimmu.2020.623944
63. Laurynenka V, Ding L, Kaufman KM, James JA, Harley JB. A high prevalence of anti-EBNA1 heteroantibodies in systemic lupus erythematosus (SLE) supports anti-EBNA1 as an origin for SLE autoantibodies. Front Immunol. (2022) 13:830993. doi: 10.3389/fimmu.2022.830993
64. Singh D, Oudit O, Hajtovic S, Sarbaugh D, Salis R, Adebowale T, et al. Antibodies to an Epstein Barr Virus protein that cross-react with dsDNA have pathogenic potential. Mol Immunol. (2021) 132:41–52. doi: 10.1016/j.molimm.2021.01.013
65. Wood RA, Guthridge L, Thurmond E, Guthridge CJ, Kheir JM, Bourn RL, et al. Serologic markers of Epstein-Barr virus reactivation are associated with increased disease activity, inflammation, and interferon pathway activation in patients with systemic lupus erythematosus. J Transl Autoimmun. (2021) 4:100117. doi: 10.1016/j.jtauto.2021.100117
66. Harley JB, Chen X, Pujato M, Miller D, Maddox A, Forney C, et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. (2018) 50:699–707. doi: 10.1038/s41588-018-0102-3
67. Hong T, Parameswaran S, Donmez OA, Miller D, Forney C, Lape M, et al. Epstein-Barr virus nuclear antigen 2 extensively rewires the human chromatin landscape at autoimmune risk loci. Genome Res. (2021) 31:2185–98. doi: 10.1101/gr.264705.120
68. Liu L, Wang Y, Wang W, Ying W, Sun B, Wang X, et al. Increased expression of the TLR7/9 signaling pathways in chronic active EBV infection. Front Pediatr. (2022) 10:1091571. doi: 10.3389/fped.2022.1091571
69. Brauner S, Folkersen L, Kvarnstrom M, Meisgen S, Petersen S, Franzen-Malmros M, et al. H1N1 vaccination in Sjogren’s syndrome triggers polyclonal B cell activation and promotes autoantibody production. Ann Rheumatic Dis. (2017) 76:1755–63. doi: 10.1136/annrheumdis-2016-210509
70. Poole BD, Templeton AK, Guthridge JM, Brown EJ, Harley JB, James JA. Aberrant Epstein-Barr viral infection in systemic lupus erythematosus. Autoimmun Rev. (2009) 8:337–42. doi: 10.1016/j.autrev.2008.12.008
71. Smith EMD, Lythgoe H, Midgley A, Beresford MW, Hedrich CM. Juvenile-onset systemic lupus erythematosus: Update on clinical presentation, pathophysiology and treatment options. Clin Immunol. (2019) 209:108274. doi: 10.1016/j.clim.2019.108274
72. Hughes GC, Choubey D. Modulation of autoimmune rheumatic diseases by oestrogen and progesterone. Nat Rev Rheumatol. (2014) 10:740–51. doi: 10.1038/nrrheum.2014.144
73. Sakiani S, Olsen NJ, Kovacs WJ. Gonadal steroids and humoral immunity. Nat Rev Endocrinol. (2013) 9:56–62. doi: 10.1038/nrendo.2012.206
Keywords: systemic lupus erythematosus, sex bias, TLR7, TASL, XIST
Citation: Scofield RH, Wren JD and Lewis VM (2025) The toll like receptor 7 pathway and the sex bias of systemic lupus erythematosus. Front. Immunol. 16:1479814. doi: 10.3389/fimmu.2025.1479814
Received: 12 August 2024; Accepted: 30 January 2025;
Published: 20 February 2025.
Edited by:
Esther Erdei, University of New Mexico Health Sciences Center, United StatesReviewed by:
Anne Satterthwaite, University of Texas Southwestern Medical Center, United StatesCopyright © 2025 Scofield, Wren and Lewis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valerie M. Lewis, dmFsZXJpZS1sZXdpc0BvbXJmLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.