 Saravanan Subramaniam1,2*
Saravanan Subramaniam1,2* Asha Jose2
Asha Jose2 Devin Kenney3,4
Devin Kenney3,4 Aoife K. O’Connell4
Aoife K. O’Connell4 Markus Bosmann5,6
Markus Bosmann5,6 Florian Douam3,4Nicholas Crossland3,4,6
Florian Douam3,4Nicholas Crossland3,4,6- 1Department of Pharmacology and Toxicology, Massachusetts College of Pharmacy and Health Sciences, Boston, MA, United States
- 2Renal Section, Department of Medicine, Chobanian & Avedisian School of Medicine, Boston University, Boston, MA, United States
- 3Department of Virology, Immunology and Microbiology, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, United States
- 4National Emerging Infectious Diseases Laboratories (NEIDL), Boston University, Boston, MA, United States
- 5Department of Medicine, Pulmonary Center, Chobanian & Avedisian School of Medicine, Boston University, Boston, MA, United States
- 6Department of Pathology and Laboratory Medicine, Chobanian & Avedisian School of Medicine, Boston University, Boston, MA, United States
Introduction
Coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2, is a public health emergency with phenotypes ranging from asymptomatic to severe sequelae that can lead to multiple organ failure and death (1, 2). SARS-CoV-2 efficiently infects airway epithelial cells and alveolar pneumocytes, causing in high viral loads and inflammatory responses, including the interferon response (3). In hospitalized patients, COVID-19 increases the risk of venous and arterial thromboembolic events due to vascular barrier failure, edema, endotheliitis, thrombosis, and inflammatory cell infiltration (4, 5). Hypercoagulation and micro- and macro-circulatory thrombosis are major causes of multiple organ failure in COVID-19 (6). Although many people have survived COVID-19 without long-term symptoms, a considerable portion of COVID-19 survivors reportedly have continuing cardiovascular issues such as coagulopathy or bleeding disorders (7). This suggests that, in addition to the respiratory epithelium, the endothelium lining of blood vessels may also be impacted by SARS-CoV-2 infection. The pathophysiology of COVID-19 has been explored in recent reviews [reviewed in (8, 9)]. Due to the conflicting data, there is ongoing controversy about the endothelial tropism (refers to the ability of SARS-CoV-2 to interact with endothelial cells) and productive endothelial infection (viral replication within the ECs) of SARS-CoV-2. Here, we share our perspective on challenging the notion of endothelial tropism and productive endothelial infection, drawing insights from the current scientific evidence.
Endothelial dysfunction and hypercoagulation in COVID-19
The endothelium, which lines the inside of arteries, is crucial for controlling vascular tone and preserving vascular homeostasis (10). Disseminated intravascular coagulation (DIC), vasculitis, and thrombosis can all be attributable to endothelial damage (11, 12). Numerous prevalent viruses and bacteria have been found to directly infect ECs, causing necrosis, apoptosis and/or damage to the vessel wall (13–15). Upon infection by viruses such as Dengue, Hantaan, Marburg, Lassa, and Ebola, both immune and non-immune cells (including endothelial cells, monocytes, and macrophages) express tissue factor (TF), leading to hypercoagulation and often culminating in disseminated intravascular coagulation (DIC) (16–20). Studies have demonstrated that Dengue, Ebola, and Marburg viruses can directly infect endothelial cells (ECs) and replicate within them, as reviewed in detail elsewhere (13, 21, 22). However, it remains unclear whether SARS-CoV-2 exhibits a similar phenomenon due to conflicting observations.
SARS-CoV-2 infection of endothelium is less studied than airway epithelium and alveolar pneumocytes (10, 23–25). Despite thromboprophylaxis, 31-49% of COVID-19 emergency care patients had arterial and venous thromboembolism (26–30). This shows that endothelial impairment must be addressed aggressively to prevent thrombosis. However, it is unclear whether the hypercoagulation is driven by lung-induced systemic inflammation upon infection, or by endothelial injury or dysfunction due to direct SARS-CoV-2 infection. Several pro-inflammatory cytokines including TNF-α, IL-1α, IL-1β, IL-6, IL-8, MCP-1, IFN-γ that are responsible for the cytokine storm in COVID-19 (31, 32) may induce COVID-19-associated coagulopathy (CAC) via expression of TF on ECs, monocytes, macrophages and T cells (33–40). The IL-6 signaling complex damages liver sinusoidal ECs and produces liver injury, suggesting that endothelial dysfunction and hypercoagulation may cause severe COVID-19 (41). SARS-CoV-2 infection in the Syrian hamster model showed inflammation and type I interferon dysregulation in respiratory and non-respiratory tissues like the heart and kidney, shedding light on COVID-19 as a multiorgan disease and possible post-acute sequelae (42). SARS-CoV-2 Spike and Nucleocapsid protein directly activate ECs, inducing mitochondrial dysfunction, vasculopathy, and coagulopathy (43, 44) (Figure 1). Furthermore, our recent study showed that the early host response of the endothelium to SARS-CoV-2 infection declines with aging, potentially contributing to increased disease severity (45).
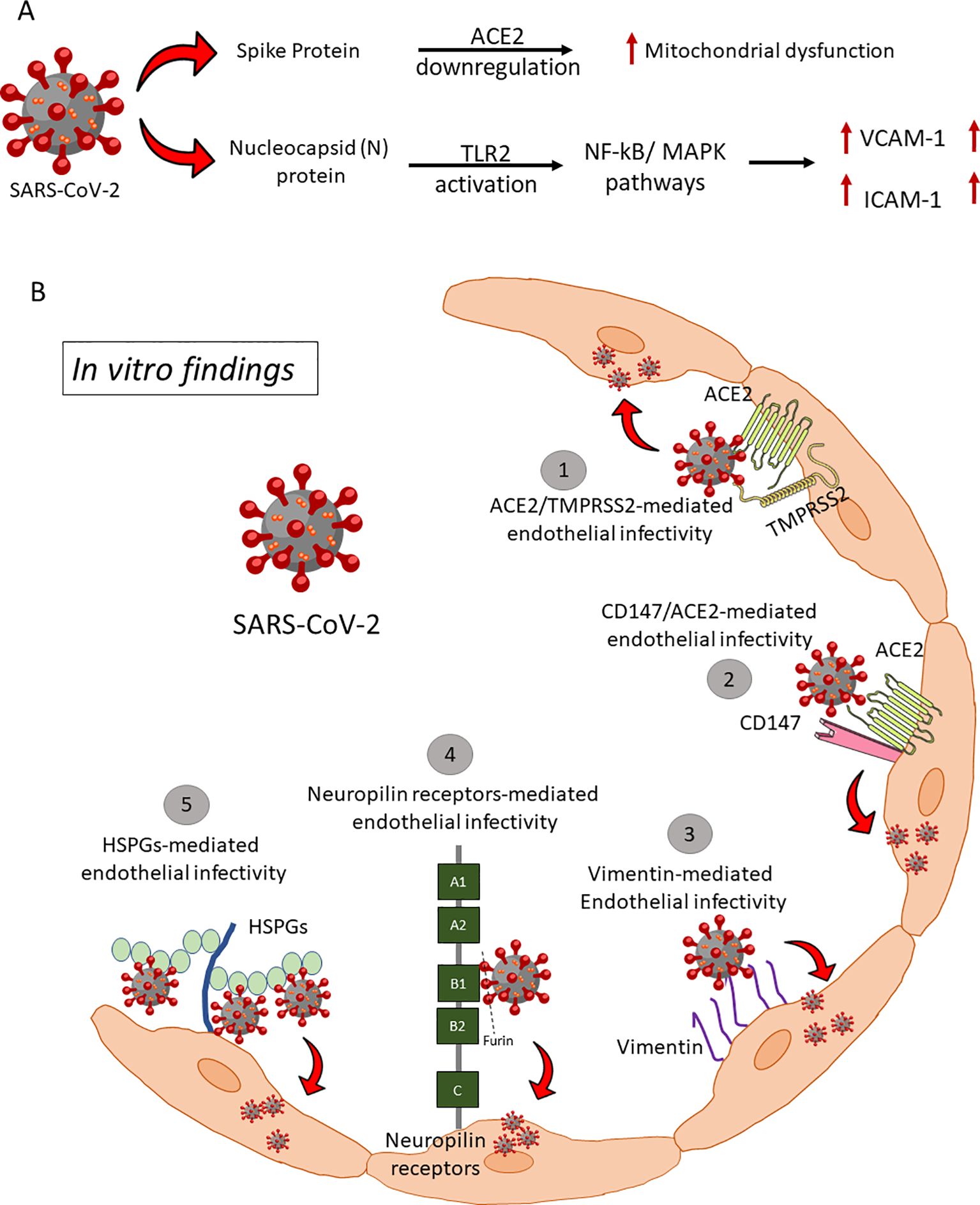
Figure 1. Endothelial infectivity and downstream signaling. (A) SARS-COV-2 Spike and Nucleocapsid proteins mediated endotheliitis and endothelial dysfunction: There are two mechanisms reported for the direct interaction of SARS-CoV-2 viral proteins with ECs and subsequent endothelial activation and dysfunction. (i) S protein of SARS-CoV-2 directly interacts with ACE2 and impairs mitochondrial function and increases redox stress which may lead to AMPK activation, MDM2 upregulation, and ultimately ACE2 destabilization. (ii) Nucleocapsid Protein (NP) of SARS-CoV-2 significantly activates human ECs through Toll-like receptor 2 (TLR2)/NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, which lead to expression of inflammatory markers (TNF-α, IL-1β) and adhesive molecules (ICAM-1, VCAM-1, and E-SELE). HSPGs, Heparan sulfate proteoglycans; ACE2, Angiotensin-Converting Enzyme 2; TMPRSS2, Transmembrane serine protease 2. (B) Proposed alternative (co)-receptors causing endothelial infectivity by in vitro assays. SARS-CoV-2 binds to Heparan sulfate and sialic acid-containing glycoproteins and gangliosides on ECs. Proteolytic cleavage of polybasic furin-type cleavage sites in the SARS-CoV-2 Spike protein was shown to expose a conserved C-terminal motif that binds cell surface Neuropilin-1 and Neuropilin-2 receptors, thus significantly potentiating SARS-CoV-2 entry, and infectivity in vitro. Vimentin on the endothelial surface acts as a co-receptor for SARS-CoV-2 entrance, according to two independent studies. CD147, a transmembrane glycoprotein of the immunoglobulin superfamily, has been identified as a potential receptor to bind spike protein and mediate virus entry.
Proposed novel endothelial (co)-receptors for SARS-CoV-2 entry
SARS-CoV-2 and SARS-CoV utilize human ACE2 as an entry receptor and TMPRSS2, primarily expressed by ECs in the respiratory and digestive tracts, as a co-factor to degrade extracellular matrix proteins for viral entry (46). ACE2 is variably expressed on arterial and venous ECs, smooth muscle cells, and pericytes across organs, facilitating systemic viral dissemination upon entry into the circulatory system. However, studies, including in-house immunohistochemistry, showed that humanized ACE2 mice express hACE2 in brain blood vessels but not in lung, gastrointestinal, or renal vessels, suggesting SARS-CoV-2 may employ both hACE2-dependent and independent entry mechanisms (47) (Figure 1B). Low expression of ACE2 and TMPRSS2 in human ECs limits the ability of SARS-CoV-2 to infect ECs (48). Thus, the variations in ACE2 expression across different microvascular beds, or alternative receptors on ECs may facilitate the entry of infectious particles. Supporting this concept, numerous additional receptors have been identified over the past three years as potentially relevant to viral particle entry in ECs. Endosomal cysteine peptidases like cathepsins B and L activate the spike (S) protein, enhancing viral entry (49–51). SARS-CoV-2 also binds heparan sulfate, sialic acid-containing glycoproteins, and gangliosides on ECs (52, 53). Proteolytic cleavage at furin-type cleavage sites in the S protein exposes a conserved motif that interacts with Neuropilin-1/2 receptors, significantly increasing infectivity (54, 55). Vimentin, CD147, and TMEM106B have been identified as co-receptors or alternative receptors, though role of TMEM106B in COVID-19 pathology lacks experimental validation (56–59). Further research is needed to confirm these mechanisms and in vivo relevance.
Controversies in COVID-19 and direct infection of ECs
Endotheliitis is regarded as a host immune-inflammatory response of the endothelium forming the inner surface of blood vessels in association with a direct consequence of infectious pathogen invasion. Systemic endotheliitis causes organ damage (60). Human autopsies, non-human primates (NHPs) and mice models showed sporadic endothelial infection and consistently observed in Syrian hamsters (61, 62). Compared to healthy individuals, circulating markers of endothelial and platelet activation are elevated in severe COVID-19 (63). Evidence of myeloid polarization, such as elevated levels of shed CD16 and CD163, have been linked to the expression of TF by proinflammatory macrophages and are related with poor clinical outcomes (64).
Elevated D-dimer and thrombocytopenia in severe COVID-19 could be explained by dysregulated inflammation and microthrombus formation that are complicated by endothelial dysfunction (65). Consecutively, in patients with severe COVID-19, hypoxia due to pulmonary microvascular dysfunction might cause the classic acute respiratory distress syndrome (ARDS) (66). Furthermore, compared to controls, human pulmonary microvascular ECs isolated from human lungs challenged with lipopolysaccharide and tumor necrosis factor alpha showed increased pro-coagulant activity and PAI-1, and decreased fibrinolytic potential, emphasizing the pro-coagulant features of the pulmonary endothelium in ARDS (67).
Earlier studies support that SARS-CoV-2 viral particles were detected in highly vascularized organs in which endothelial dysfunction plays a fundamental role (24, 68–76). Initial transmission electron microscopy (TEM) studies revealed the presence of viral particles in kidney ECs, venous ECs, and liver sinusoidal ECs in autopsy samples from COVID-19 patients (76–78). However, owing to the challenges of interpreting TEM and the high variability in experience of those interpreting images, the presence of viral particle in the endothelium remains debatable (76, 79, 80). Irrespective of these controversies, there has been increasing evidence to indicate that coated vesicles and multivesicular bodies closely mimic viral particles even in the lung epithelium by TEM and are not uncommonly misinterpreted (81). Despite the thrombo-inflammatory phenotype, no definitive animal models or human biopsies have yet shown direct SARS-CoV-2 infection of ECs or the presence of viral particles (82–85). Human micro- and macrovascular ECs are resistant to SARS-CoV-2 infection, and ACE2 overexpression is necessary for endothelial infection (86–89). Montezano et al. demonstrated that recombinant Spike protein-1 induced endothelial inflammation via ACE2 independent of ACE2 enzymatic activity and viral replication in vitro (90). On ex vivo lung cultures from a patient who had SARS-CoV-2 infection found no signs of the virus in the vascular endothelium following immunohistochemical labeling of SARS-CoV-2 Spike protein (91). ECs primed with (IL-1) produced more pro-inflammatory cytokines, such as IL-6 and IL-8, and were resistant to direct SARS-CoV-2 infection (92). Furthermore, two separate investigations found that human pulmonary microvascular ECs were resistant to SARS-CoV-2 infection (MOI=0.5-3 after 2 hours of adsorption) (93, 94).
Based on these findings, it is possible to hypothesize that prior reports of endothelial viral presence in patients are not a universal hallmark of illness and may be restricted to certain patient groups or isolated episodes. In light of this, we carefully evaluated the immunoreactivity of the SARS-CoV-2 Nucleocapsid (N) Protein in lung slices from translational preclinical animal models (transgenic K18-hACE2 mice (expression of hACE2 in lung epithelial cells), hACE2-KI (global hACE2 knock-in by replacing mouse ACE2), Syrian hamsters, and African green monkeys (AGM) and human postmortem lung samples. A board-certified veterinary pathologist (N.A.C.) immunohistochemically analyzed hundreds of organ slices from previous studies in each species (45, 61, 62, 69, 95–99). A PCR-positive human autopsy samples with clear hyaline membrane formation and AGMs at 7 days post-infection (dpi) showed no N Protein, suggesting viral antigen is only present during the acute phase of disease (Figures 2A, B). The lung epithelium of K18-hACE2, Syrian hamsters, NHPs, and ACE2-KI mice showed varying and decreasing levels of SARS-CoV-2 N protein, with airway tropism only present in ACE2-KI mice and Syrian hamsters. No tissues had viral antigens in ECs (Figures 2C–E). To further support the absence of direct endothelial infection, we performed duplex fluorescent IHC targeting the pulmonary endothelium (CD34 or CD31) and SARS-CoV-2 N protein. In both the AGM and K18-hACE2 mouse, luminal alveolar pneumocytes exclusively displayed SARS-CoV-2 N protein as evidenced by absence of colocalization with vascular endothelium at 4DPI (Figures 2F, G).
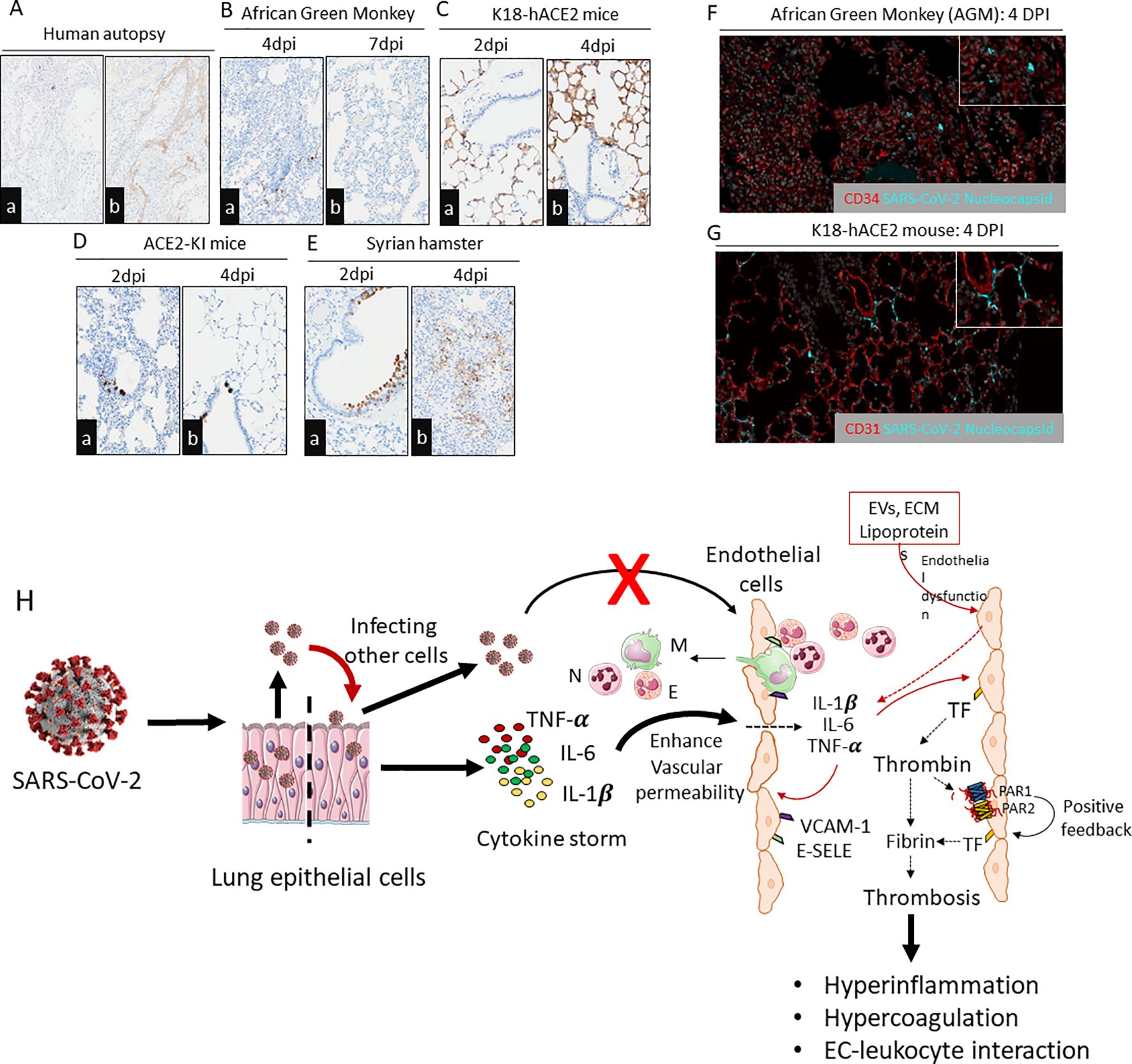
Figure 2. Immunohistochemistry staining of SARS-CoV-2 N protein in lung slices from different species exposed to SARS-CoV-2. (Aa,b) Examination of a human autopsy illustrated absence of SARS-CoV-2 N protein in the ECs from a PCR positive patient with histologic features of diffuse alveolar damage including hyaline membranes evident by fibrinogen gamma IHC. Scale bar=200µm. (Ba,b) NHPs with infected alveolar pneumocytes after 4dpi and 7dpi with no endothelial infectivity. Scale bar=200µm. (Ca,b) K18-hACE2 mice infected with SARS-CoV-2 (USA/WA1/2020) illustrating N protein within alveolar pneumocytes and no endothelial infectivity. (Da,b) Global ACE2-KI mice infected with SARS-CoV-2 and N protein detectable in airways but not in ECs. Scale bar=200µm. (Ea,b) Syrian hamster infected with SARS-CoV-2 with lung sections immunostained for N protein at 2dpi and 4dpi. Presence of N protein in airways was more prominent at 2dpi and alveolar pneumocytes at 4 dpi. Scale bar=200µm. (F, G) Absence of SARS-CoV-2 N protein localization within pulmonary endothelium of two translationally relevant preclinical models of COVID-19. (E) African Green Monkey, 4DPI; SARS-CoV-2 N protein localizes to alveolar pneumocytes with absence of immunoreactivity in neighboring endothelium. Red-CD34 vascular endothelial marker, Grey-DAPI, Teal-SARS-CoV-2 N protein. (F) K18-hACE2 mouse, 4DPI; SARS-CoV-2 N protein localizes to alveolar pneumocytes with absence of immunoreactivity of neighboring endothelium. Red-CD31 vascular endothelial marker, Grey-DAPI, Teal-SARS-CoV-2 N protein. All SARS-CoV-2 infections were from parental viruses originating from the beginning of the pandemic. All infections were from parental viruses originating from the beginning of the pandemic. Images are representative of hundreds of samples examined by a board-certified veterinary pathologist (N.A.C). (H) Endothelial dysfunction and endotheliitis in COVID-19- associated coagulopathy: SARS-CoV-2 infects lung epithelium and propagates within the cells for further infection. During infection, lung epithelium releases cytokines that act on endothelium and promote vascular permeability. Increased vascular permeability leads to infiltration of leukocytes into the alveolar space as well as leaking inflammatory cytokines (e.g., TNF-α, IL-6, and IL-1β) into the systemic circulation. These cytokines further stimulate luminal ECs to release more inflammatory cytokines and expression of adhesive receptors (e.g., VCAM-1, ICAM-1, E-SELE) and procoagulant receptors (e.g., Tissue factor). Elevated thrombin levels during viral infection activate the PAR1/2-dependent positive feedback mechanism, which further aggravates the expression of inflammatory cytokines, adhesive receptors, and tissue factor. These factors propagate EC-leukocyte interaction, hyperinflammation, and hypercoagulation. Other mediators like EVs, ECM, and lipoproteins trigger endothelial dysfunction which leads to expression of adhesive molecules and release of cytokines and chemokines. Direct infection of ECs by SARS-CoV-2 is an unlikely event. N, Neutrophil; M, Monocyte/macrophage; E, Eosinophil; TF, Tissue factor; PAR1, Protease-activated receptor-1; PAR2, Protease activated receptor-2; E-SELE, E-selectin; VCAM-1, Vascular cell adhesion protein 1.
EC and inflammatory mediators in COVID-19
Findings from our group and others collectively question the universality of direct endothelial infection by SARS-CoV-2. Because active viral infection of lung epithelial cells is associated with a systemic pro-inflammation, it is possible that inflammatory mediators can facilitate endothelial injury by activating immunothrombosis mechanisms such as complement activation, antiphospholipid antibodies, and so on. ECs treated with human sera from COVID-19 hospitalized patients (n=118) demonstrated anti-cardiolipin IgG/IgM and anti-phosphatidlyserine/prothrombin (anti-PS/PT) IgG/IgM-driven elevation of surface adhesion markers E-selectin, VCAM-1, and ICAM-1 (100–102).
Infection-induced proinflammatory cytokines, such as IL1β, TNFα, can stimulate coagulation, which may influence thrombin generation, fibrin formation, and TF-dependent thrombo-inflammatory responses via protease-activated receptors (PARs) (6, 22, 103–106) (Figure 2H). SARS-CoV-2 infection induces increased production of superoxide anion and release of mitochondrial DNA (mtDNA), activating Toll-like receptor 9 (TLR9) and NFκ-B. Consequently, this activation orchestrates the expression of inflammatory genes, contributing to the pathological processes associated with COVID-19 (107).
Numerous studies have highlighted changes in lipid profiles linked with COVID-19. Among the most commonly observed changes are reductions in serum cholesterol and ApoA1 levels, coupled with elevated triglycerides (108). Lipidomic analysis in COVID-19 patients showed high levels of eicosanoids in the lungs which might be a potential contributor for endothelial dysfunction.
A systemic inflammatory response and aberrant expression of the extracellular matrix (ECM) during COVID-19 controls the balance and repair of ECs (109). A study on human lung autopsy confirmed that Hyaluronan is an important compound ECM of all vital organ systems (110). COVID-19 is characterized by significantly increased levels of MMP-1 and vascular endothelial growth factor (VEGF)-A, which are directly correlated with the severity of the disease (111). More than 50% of patients who had experienced moderate or severe cases of COVID-19 had reduced pulmonary diffusion and early fibrotic changes, which were correlated with elevated levels of MMP-1 (112). A detailed dysregulation of the ECM in COVID-19 reviewed elsewhere (113).
The endothelial glycocalyx is crucial for maintaining vascular homeostasis, and it is linked to vascular endothelial dysfunction (114). Numerous studies indicate that severe COVID-19 patients experience endothelial glycocalyx damage on the endothelial cell surface, evidenced by elevated plasma levels of glycocalyx components like syndecan-1, heparan sulfate, and hyaluronan. These biomarkers, along with high levels of IL-1β, IL-6, TNF-α, hsCRP, and procalcitonin, are associated with increased severity and mortality in COVID-19 cases (115–117). Furthermore, several drugs, including heparin and tocilizumab, are already being used in COVID-19 treatment to protect the endothelial glycocalyx damage (118–121). However, despite numerous reports of glycocalyx injury in COVID-19, the underlying mechanisms are still not fully understood (117, 122–125). Vascular endothelial glycocalyx damage and potential targeted therapy in COVID-19 has been explored in recent reviews (reviewed in (84, 126, 127).
Most extracellularvesicles (EVs) found in the blood originate from platelets and erythrocytes (128). Under physiological conditions, the proportion of circulating EVs secreted by ECs is relatively low, but notably increases in pathological conditions marked by endothelial dysfunction. EVs released by ECs contain numerous endothelial markers, including endoglin/CD105, E-selectinCD62E, S-endo/CD146, vascular endothelial cadherin/CD144, platelet endothelial cell adhesion molecule 1/CD31, and intercellular adhesion molecule 1/CD54 (129). SARS-CoV-2 infection associated with the release of EVs carrying TF into the bloodstream which activate platelets and ECs, thereby contributing to COVID-19-related thrombosis in patients (130, 131).
Given the critical role of ECs in vascular homeostasis and the prospective association of COVID-19 with endothelial injury or dysfunction, it appears that patients with preexisting endothelial dysfunction in various disease states (e.g., diabetes, atherosclerosis, and hypertension) are vulnerable to a more severe disease course (132). For instance, among the comorbidities, diabetes mellitus (DM) was the most frequently reported (10.9% of cases) condition (133). Studies from China, Europe, UK and the US have also found that when people with DM acquire COVID-19, they are more likely to develop COVID-19-related complications, require ICU hospitalization, or die from the disease (134–136). The possible root cause might be chronic endothelial dysfunction due to DM together with the direct damage of ECs by SARS-CoV-2-mediated inflammatory responses result in further impairment of the microcirculation contributing to pathophysiology of acute respiratory syndrome and multi-organ failure.
Conclusion
Experimental and clinical evidence from our group and others suggests that SARS-CoV-2 is unlikely to productively infect endothelial cells. Instead, elevated circulatory mediators, such as cytokines, extracellular matrix components, extracellular vesicles, lipids/lipoproteins, and thrombin, are likely the primary drivers of endothelial dysfunction during SARS-CoV-2 infection. Additionally, accumulating evidence indicates that SARS-CoV-2-mediated endothelial glycocalyx damage disrupts vascular homeostasis by altering vascular permeability, cell adhesion, mechanosensing, and antithrombotic and anti-inflammatory functions.
Given the mixed findings on endothelial infectivity and the involvement of multiple (co)-receptors, further research is needed. First, developing better animal models that demonstrate endothelial infectivity of SARS-CoV-2 could help identify potential co-receptors and validate direct infection. Second, there is a possibility that SARS-CoV-2 enters endothelial cells without effective replication. Initial entry may trigger significant interferon and inflammatory signaling, but rapid RNA degradation could render the infection undetectable. These hypotheses warrant further investigation.
Besides, the variation in detection of SARS-CoV-2 or viral particles might be due to the methodological differences, such as tissue sampling techniques, sensitivity of detection methods, and variations in experimental models. In addition, biological variability, including differences in patient population and disease severity, could lay a role in the observed variability across studies. Thus, further systematic review is required to identify the methodological approaches contributes to discrepancies.
Alternatively, infected ECs can be cleared from the system by various immune mechanisms, such as phagocytosis by immune cells like macrophages, or through apoptosis induced by immune responses. This can make it challenging to detect infections or inflammation in the endothelial layer, as the infected cells may be removed before they can be adequately studied or identified. However, considering that studies typically sample at different time points, the likelihood of this occurring is low. Nonetheless, it is important to carefully investigate this possibility to draw a well-informed conclusion, ensuring that immune clearance mechanisms are accounted for in the analysis.Nonetheless, current clinical and experimental evidence confirms that endothelial dysfunction is a hallmark of COVID-19, making it a critical therapeutic target. Addressing this dysfunction may help protect vulnerable individuals from severe hyperinflammation and hypercoagulation. To mitigate pro-thrombotic complications, the International Society on Thrombosis and Haemostasis (ISTH) recommends universal standard thromboprophylaxis with low molecular weight heparin or unfractionated heparin (LMWH/UFH) for all hospitalized patients, unless contraindicated (137).
Author contributions
SS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AJ: Conceptualization, Formal analysis, Visualization, Writing – review & editing. DK: Writing – review & editing. AO: Resources, Writing – review & editing, Methodology. MB: Funding acquisition, Resources, Writing – review & editing. FD: Resources, Writing – review & editing, Funding acquisition. NC: Data curation, Formal analysis, Investigation, Resources, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Aniara Diagnostica (Coagulation Research Grant 2020-2021) and the National Institutes of Health (1UL1TR001430 to S.S., R01 HL141513 and R01 HL139641 to MB). MCPHS faculty development grant (to SS). Research in the Douam Lab related to this work is partly funded by a Boston University Startup funds and a Peter Paul Career Development Professorship (to FD). We thank the Evans Center for Interdisciplinary Biomedical Research at Boston University School of Medicine for their support of the Affinity Research Collaborative on ‘Respiratory Viruses: A Focus on COVID-19’. This work used NIH S10 Shared Instrumentation Grants S10-OD026983 and S10-OD030269.
Acknowledgments
We are grateful to our collaborators who provided the FFPE samples utilized in Figures 2A–G preparation. Specifically, our external collaborators Paul Duprex, Chad Mire and Thomas Beach.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mokhtari T, Hassani F, Ghaffari N, Ebrahimi B, Yarahmadi A, Hassanzadeh G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J Mol Histol. (2020) 51:613–28. doi: 10.1007/s10735-020-09915-3
2. Zaim S, Chong JH, Sankaranarayanan V, Harky A. COVID-19 and multiorgan response. Curr Probl Cardiol. (2020) 45:100618. doi: 10.1016/j.cpcardiol.2020.100618
3. Deinhardt-Emmer S, Bottcher S, Haring C, Giebeler L, Henke A, Zell R, et al. SARS-CoV-2 causes severe epithelial inflammation and barrier dysfunction. J Virol. (2021) 95:e00110-21. doi: 10.1128/JVI.00110-21
4. Bilaloglu S, Aphinyanaphongs Y, Jones S, Iturrate E, Hochman J, Berger JS. Thrombosis in hospitalized patients with COVID-19 in a New York City health system. JAMA. (2020) 324:799–801. doi: 10.1001/jama.2020.13372
5. Al-Ani F, Chehade S, Lazo-Langner A. Thrombosis risk associated with COVID-19 infection. A scoping review. Thromb Res. (2020) 192:152–60. doi: 10.1016/j.thromres.2020.05.039
6. Subramaniam S, Ruf W, Bosmann M. Advocacy of targeting protease-activated receptors in severe coronavirus disease 2019. Br J Pharmacol. (2021) 179:2086–99. doi: 10.1111/bph.15587
7. Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med. (2021) 27:601–15. doi: 10.1038/s41591-021-01283-z
8. Yuki K, Fujiogi M, Koutsogiannaki S. COVID-19 pathophysiology: A review. Clin Immunol. (2020) 215:108427. doi: 10.1016/j.clim.2020.108427
9. Chen B, Tian EK, He B, Tian L, Han R, Wang S, et al. Overview of lethal human coronaviruses. Signal Transduct Target Ther. (2020) 5:89. doi: 10.1038/s41392-020-0190-2
10. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
11. Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. (2015) 15:130. doi: 10.1186/s12872-015-0124-z
12. Poredos P, Jezovnik MK. Endothelial dysfunction and venous thrombosis. Angiology. (2018) 69:564–7. doi: 10.1177/0003319717732238
13. Goodrum F, Bughio F. Viral infection at the endothelium. Oncotarget. (2015) 6:26541–2. doi: 10.18632/oncotarget.v6i29
14. Valbuena G, Walker DH. The endothelium as a target for infections. Annu Rev Pathol. (2006) 1:171–98. doi: 10.1146/annurev.pathol.1.110304.100031
15. van Gorp EC, Suharti C, ten Cate H, Dolmans WM, van der Meer JW, ten Cate JW, et al. Review: infectious diseases and coagulation disorders. J Infect Dis. (1999) 180:176–86. doi: 10.1086/jid.1999.180.issue-1
16. Goeijenbier M, Meijers JC, Anfasa F, Roose JM, van de Weg CA, Bakhtiari K, et al. Effect of Puumala hantavirus infection on human umbilical vein endothelial cell hemostatic function: platelet interactions, increased tissue factor expression and fibrinolysis regulator release. Front Microbiol. (2015) 6:220. doi: 10.3389/fmicb.2015.00220
17. Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. (2003) 163:2371–82. doi: 10.1016/S0002-9440(10)63592-4
18. Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis. (2003) 188:1618–29. doi: 10.1086/jid.2003.188.issue-11
19. Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. (2010) 115:161–7. doi: 10.1182/blood-2009-03-210179
20. Huerta-Zepeda A, Cabello-Gutierrez C, Cime-Castillo J, Monroy-Martinez V, Manjarrez-Zavala ME, Gutierrez-Rodriguez M, et al. Crosstalk between coagulation and inflammation during Dengue virus infection. Thromb Haemost. (2008) 99:936–43. doi: 10.1160/TH07-08-0483
21. Fosse JH, Haraldsen G, Falk K, Edelmann R. Endothelial cells in emerging viral infections. Front Cardiovasc Med. (2021) 8:619690. doi: 10.3389/fcvm.2021.619690
22. Subramaniam S, Scharrer I. Procoagulant activity during viral infections. Front Biosci (Landmark Ed). (2018) 23:1060–81. doi: 10.2741/4633
23. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
24. Colmenero I, Santonja C, Alonso-Riano M, Noguera-Morel L, Hernandez-Martin A, Andina D, et al. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br J Dermatol. (2020) 183:729–37. doi: 10.1111/bjd.v183.4
25. Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol. (2022) 20:270–84. doi: 10.1038/s41579-022-00713-0
26. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb Res. (2020) 191:148–50. doi: 10.1016/j.thromres.2020.04.041
27. Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. (2020) 18:1023–6. doi: 10.1111/jth.14810
28. Barrett CD, Moore HB, Yaffe MB, Moore EE. ISTH interim guidance on recognition and management of coagulopathy in COVID-19: A comment. J Thromb Haemost. (2020) 18:2060–3. doi: 10.1111/jth.14860
29. Akima S, McLintock C, Hunt BJ. RE: ISTH interim guidance to recognition and management of coagulopathy in COVID-19. J Thromb Haemost. (2020) 18:2057–8. doi: 10.1111/jth.14853
30. Iba T, Connors JM, Levy JH. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflammation Res. (2020) 69:1181–9. doi: 10.1007/s00011-020-01401-6
31. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
32. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. (2020) 130:2620–9. doi: 10.1172/JCI137244
33. Nawroth PP, Stern DM. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. (1986) 163:740–5. doi: 10.1084/jem.163.3.740
34. Nawroth PP, Handley DA, Esmon CT, Stern DM. Interleukin 1 induces endothelial cell procoagulant while suppressing cell-surface anticoagulant activity. Proc Natl Acad Sci U S A. (1986) 83:3460–4. doi: 10.1073/pnas.83.10.3460
35. Herbert JM, Savi P, Laplace MC, Lale A. IL-4 inhibits LPS-, IL-1 beta- and TNF alpha-induced expression of tissue factor in endothelial cells and monocytes. FEBS Lett. (1992) 310:31–3. doi: 10.1016/0014-5793(92)81139-D
36. Schwager I, Jungi TW. Effect of human recombinant cytokines on the induction of macrophage procoagulant activity. Blood. (1994) 83:152–60. doi: 10.1182/blood.V83.1.152.152
37. Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, et al. Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J Biol Chem. (1997) 272:28568–73. doi: 10.1074/jbc.272.45.28568
38. Neumann FJ, Ott I, Marx N, Luther T, Kenngott S, Gawaz M, et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb Vasc Biol. (1997) 17:3399–405. doi: 10.1161/01.ATV.17.12.3399
39. Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res. (2005) 96:1233–9. doi: 10.1161/01.RES.0000171805.24799.fa
40. De Palma R, Cirillo P, Ciccarelli G, Barra G, Conte S, Pellegrino G, et al. Expression of functional tissue factor in activated T-lymphocytes in vitro and in vivo: A possible contribution of immunity to thrombosis? Int J Cardiol. (2016) 218:188–95. doi: 10.1016/j.ijcard.2016.04.177
41. McConnell MJ, Kawaguchi N, Kondo R, Sonzogni A, Licini L, Valle C, et al. Liver injury in COVID-19 and IL-6 trans-signaling-induced endotheliopathy. J Hepatol. (2021) 75(3):647–58. doi: 10.1016/j.jhep.2021.04.050
42. Francis ME, Goncin U, Kroeker A, Swan C, Ralph R, Lu Y, et al. SARS-CoV-2 infection in the Syrian hamster model causes inflammation as well as type I interferon dysregulation in both respiratory and non-respiratory tissues including the heart and kidney. PLoS Pathog. (2021) 17:e1009705. doi: 10.1371/journal.ppat.1009705
43. Qian Y, Lei T, Patel PS, Lee CH, Monaghan-Nichols P, Xin HB, et al. Direct activation of endothelial cells by SARS-CoV-2 nucleocapsid protein is blocked by simvastatin. J Virol. (2021) 95:e0139621. doi: 10.1128/JVI.01396-21
44. Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, et al. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res. (2021) 128:1323–6. doi: 10.1161/CIRCRESAHA.121.318902
45. Subramaniam S, Kenney D, Jayaraman A, O’Connell AK, Walachowski S, Montanaro P, et al. Aging is associated with an insufficient early inflammatory response of lung endothelial cells in SARS-CoV-2 infection. Front Immunol. (2024) 15:1397990. doi: 10.3389/fimmu.2024.1397990
46. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A. (2020) 117:11727–34. doi: 10.1073/pnas.2003138117
47. McCray PB Jr., Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. (2007) 81:813–21. doi: 10.1128/JVI.02012-06
48. Schimmel L, Chew KY, Stocks CJ, Yordanov TE, Essebier P, Kulasinghe A, et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin Transl Immunol. (2021) 10:e1350. doi: 10.1002/cti2.v10.10
49. Milan Bonotto R, Mitrovic A, Sosic I, Martinez-Orellana P, Dattola F, Gobec S, et al. Cathepsin inhibitors nitroxoline and its derivatives inhibit SARS-CoV-2 infection. Antiviral Res. (2023) 216:105655. doi: 10.1016/j.antiviral.2023.105655
50. Zhao MM, Yang WL, Yang FY, Zhang L, Huang WJ, Hou W, et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther. (2021) 6:134. doi: 10.1038/s41392-021-00558-8
51. Pislar A, Mitrovic A, Sabotic J, Pecar Fonovic U, Perisic Nanut M, Jakos T, et al. The role of cysteine peptidases in coronavirus cell entry and replication: The therapeutic potential of cathepsin inhibitors. PLoS Pathog. (2020) 16:e1009013. doi: 10.1371/journal.ppat.1009013
52. Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. (2020) 183:1043–57.e15. doi: 10.1016/j.cell.2020.09.033
53. Robson B. Bioinformatics studies on a function of the SARS-CoV-2 spike glycoprotein as the binding of host sialic acid glycans. Comput Biol Med. (2020) 122:103849. doi: 10.1016/j.compbiomed.2020.103849
54. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. (2020) 370:856–60. doi: 10.1126/science.abd2985
55. Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Anton-Plagaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. (2020) 370:861–5. doi: 10.1126/science.abd3072
56. Suprewicz L, Swoger M, Gupta S, Piktel E, Byfield FJ, Iwamoto DV, et al. Extracellular vimentin as a target against SARS-CoV-2 host cell invasion. Small. (2021) 18(6):e2105640. doi: doi: 10.1002/smll.202105640
57. Amraei R, Xia C, Olejnik J, White MR, Napoleon MA, Lotfollahzadeh S, et al. Extracellular vimentin is an attachment factor that facilitates SARS-CoV-2 entry into human endothelial cells. Proc Natl Acad Sci. (2022) 119:e2113874119. doi: 10.1073/pnas.2113874119
58. Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. (2020) 5:283. doi: 10.1038/s41392-020-00426-x
59. Baggen J, Jacquemyn M, Persoons L, Vanstreels E, Pye VE, Wrobel AG, et al. TMEM106B is a receptor mediating ACE2-independent SARS-CoV-2 cell entry. Cell. (2023) 186(16):3427–42.e22. doi: 10.1016/j.cell.2023.06.005
60. Hattori Y, Hattori K, Machida T, Matsuda N. Vascular endotheliitis associated with infections: Its pathogenetic role and therapeutic implication. Biochem Pharmacol. (2022) 197:114909. doi: 10.1016/j.bcp.2022.114909
61. Yen HL, Valkenburg S, Sia SF, Choy KT, Peiris JSM, Wong KHM, et al. Cellular tropism of SARS-CoV-2 in the respiratory tract of Syrian hamsters and B6.Cg-Tg(K18-ACE2)2Prlmn/J transgenic mice. Vet Pathol. (2022) 59:639–47. doi: 10.1177/03009858211043084
62. Kenney DJ, O’Connell AK, Turcinovic J, Montanaro P, Hekman RM, Tamura T, et al. Humanized mice reveal a macrophage-enriched gene signature defining human lung tissue protection during SARS-CoV-2 infection. Cell Rep. (2022) 39:110714. doi: 10.1016/j.celrep.2022.110714
63. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. (2020) 7:e575–e82. doi: 10.1016/S2352-3026(20)30216-7
64. Schulte-Schrepping J, Reusch N, Paclik D, Bassler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. (2020) 182:1419–40.e23. doi: 10.1016/j.cell.2020.08.001
65. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
66. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. COVID-19 does not lead to a “Typical” Acute respiratory distress syndrome. Am J Respir Crit Care Med. (2020) 201:1299–300. doi: 10.1164/rccm.202003-0817LE
67. Grau GE, de Moerloose P, Bulla O, Lou J, Lei Z, Reber G, et al. Haemostatic properties of human pulmonary and cerebral microvascular endothelial cells. Thromb Haemost. (1997) 77:585–90. doi: 10.1055/s-0038-1656009
68. Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. (2020) 383:590–2. doi: 10.1056/NEJMc2011400
69. Carossino M, Kenney D, O’Connell AK, Montanaro P, Tseng AE, Gertje HP, et al. Fatal neurodissemination and SARS-CoV-2 tropism in K18-hACE2 mice is only partially dependent on hACE2 expression. Viruses. (2022) 14(3):535. doi: 10.3390/v14030535
70. Libby P, Luscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. (2020) 41:3038–44. doi: 10.1093/eurheartj/ehaa623
71. Ambrosino P, Calcaterra IL, Mosella M, Formisano R, D’Anna SE, Bachetti T, et al. Endothelial dysfunction in COVID-19: A unifying mechanism and a potential therapeutic target. Biomedicines. (2022) 10(4):812. doi: 10.3390/biomedicines10040812
72. Lowenstein CJ, Solomon SD. Severe COVID-19 is a microvascular disease. Circulation. (2020) 142:1609–11. doi: 10.1161/CIRCULATIONAHA.120.050354
73. Ma Z, Yang KY, Huang Y, Lui KO. Endothelial contribution to COVID-19: an update on mechanisms and therapeutic implications. J Mol Cell Cardiol. (2022) 164:69–82. doi: 10.1016/j.yjmcc.2021.11.010
74. Flaumenhaft R, Enjyoji K, Schmaier AA. Vasculopathy in COVID-19. Blood. (2022) 140:222–35. doi: 10.1182/blood.2021012250
75. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. (2020) 20:389–91. doi: 10.1038/s41577-020-0343-0
76. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel A, et al. Electron microscopy of SARS-CoV-2: a challenging task - Authors’ reply. Lancet. (2020) 395:e100. doi: 10.1016/S0140-6736(20)31185-5
77. Stahl K, Brasen JH, Hoeper MM, David S. Direct evidence of SARS-CoV-2 in gut endothelium. Intensive Care Med. (2020) 46:2081–2. doi: 10.1007/s00134-020-06237-6
78. Kondo Y, Larabee JL, Gao L, Shi H, Shao B, Hoover CM, et al. L-SIGN is a receptor on liver sinusoidal endothelial cells for SARS-CoV-2 virus. JCI Insight. (2021) 6(14):e148999. doi: 10.1172/jci.insight.148999
79. Goldsmith CS, Miller SE, Martines RB, Bullock HA, Zaki SR. Electron microscopy of SARS-CoV-2: a challenging task. Lancet. (2020) 395:e99. doi: 10.1016/S0140-6736(20)31188-0
80. Miller SE, Brealey JK. Visualization of putative coronavirus in kidney. Kidney Int. (2020) 98:231–2. doi: 10.1016/j.kint.2020.05.004
81. Frelih M, Erman A, Wechtersbach K, Plesko J, Avsic-Zupanc T, Kojc N. SARS-CoV-2 virions or ubiquitous cell structures? Actual dilemma in COVID-19 era. Kidney Int Rep. (2020) 5:1608–10. doi: 10.1016/j.ekir.2020.07.003
82. Zheng J, Wong LR, Li K, Verma AK, Ortiz ME, Wohlford-Lenane C, et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature. (2021) 589:603–7. doi: 10.1038/s41586-020-2943-z
83. Qin Z, Liu F, Blair R, Wang C, Yang H, Mudd J, et al. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics. (2021) 11:8076–91. doi: 10.7150/thno.61810
84. Xu SW, Ilyas I, Weng JP. Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol Sin. (2023) 44:695–709. doi: 10.1038/s41401-022-00998-0
85. Gao S, Tang AT, Wang M, Buchholz DW, Imbiakha B, Yang J, et al. Endothelial SARS-CoV-2 infection is not the underlying cause of COVID-19-associated vascular pathology in mice. Front Cardiovasc Med. (2023) 10:1266276. doi: 10.3389/fcvm.2023.1266276
86. Schimmel L, Chew KY, Stocks C, Yordanov T, Essebier T, Kulasinghe A, et al. Endothelial cells are not productively infected by SARS-CoV-2. Transl Immunology. (2021) 10(10):e1350. doi: 10.1002/cti2.1350
87. Nascimento Conde J, Schutt WR, Gorbunova EE, Mackow ER. Recombinant ACE2 expression is required for SARS-CoV-2 to infect primary human endothelial cells and induce inflammatory and procoagulative responses. mBio. (2020) 11(6):e03185-20. doi: 10.1128/mBio.03185-20
88. Liu F, Han K, Blair R, Kenst K, Qin Z, Upcin B, et al. SARS-CoV-2 infects endothelial cells in vivo and in vitro. Front Cell Infect Microbiol. (2021) 11:701278. doi: 10.3389/fcimb.2021.701278
89. McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S, et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation. (2021) 143:865–8. doi: 10.1161/CIRCULATIONAHA.120.052824
90. Montezano AC, Camargo LL, Mary S, Neves KB, Rios FJ, Stein R, et al. SARS-CoV-2 spike protein induces endothelial inflammation via ACE2 independently of viral replication. Sci Rep. (2023) 13:14086. doi: 10.1038/s41598-023-41115-3
91. Hui KPY, Cheung MC, Perera R, Ng KC, Bui CHT, Ho JCW, et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir Med. (2020) 8:687–95. doi: 10.1016/S2213-2600(20)30193-4
92. Ahmetaj-Shala B, Peacock TP, Baillon L, Swann OC, Gashaw H, Barclay WS, et al. Resistance of endothelial cells to SARS-CoV-2 infection in vitro. bioRxiv. (2020). doi: 10.1101/2020.11.08.372581
93. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. (2020) 182:429–46.e14. doi: 10.1016/j.cell.2020.05.042
94. Bernard I, Limonta D, Mahal LK, Hobman TC. Endothelium infection and dysregulation by SARS-CoV-2: evidence and caveats in COVID-19. Viruses. (2020) 13(1):29. doi: 10.3390/v13010029
95. Chen DY, Chin CV, Kenney D, Tavares AH, Khan N, Conway HL, et al. Spike and nsp6 are key determinants of SARS-CoV-2 Omicron BA.1 attenuation. Nature. (2023) 615:143–50. doi: 10.1038/s41586-023-05697-2
96. Hartmann JA, Cardoso MR, Talarico MCR, Kenney DJ, Leone MR, Reese DC, et al. Evasion of NKG2D-mediated cytotoxic immunity by sarbecoviruses. Cell. (2024) 187:2393–410.e14. doi: 10.1016/j.cell.2024.03.026
97. Kehrer T, Cupic A, Ye C, Yildiz S, Bouhaddou M, Crossland NA, et al. Impact of SARS-CoV-2 ORF6 and its variant polymorphisms on host responses and viral pathogenesis. Cell Host Microbe. (2023) 31:1668–84 e12. doi: 10.1016/j.chom.2023.08.003
98. Nambulli S, Escriou N, Rennick LJ, Demers MJ, Tilston-Lunel NL, McElroy AK, et al. A measles-vectored vaccine candidate expressing prefusion-stabilized SARS-CoV-2 spike protein brought to phase I/II clinical trials: protection of African green monkeys from COVID-19 disease. J Virol. (2024) 98:e0176223. doi: 10.1128/jvi.01762-23
99. Thieulent CJ, Dittmar W, Balasuriya UBR, Crossland NA, Wen X, Richt JA, et al. Mouse-adapted SARS-CoV-2 MA10 strain displays differential pulmonary tropism and accelerated viral replication, neurodissemination, and pulmonary host responses in K18-hACE2 mice. mSphere. (2023) 8:e0055822. doi: 10.1128/msphere.00558-22
100. Shi H, Zuo Y, Gandhi AA, Sule G, Yalavarthi S, Gockman K, et al. Endothelial cell-activating antibodies in COVID-19. medRxiv. (2021). doi: 10.1101/2021.01.18.21250041
101. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol. (2021) 17:46–64. doi: 10.1038/s41581-020-00357-4
102. Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. (2017) 129:2291–302. doi: 10.1182/blood-2016-11-749879
103. Subramaniam S, Ogoti Y, Hernandez I, Zogg M, Botros F, Burns R, et al. A thrombin-PAR1/2 feedback loop amplifies thromboinflammatory endothelial responses to the viral RNA analogue poly(I:C). Blood Adv. (2021) 5:2760–74. doi: 10.1182/bloodadvances.2021004360
104. Kirchhofer D, Tschopp TB, Hadvary P, Baumgartner HR. Endothelial cells stimulated with tumor necrosis factor-alpha express varying amounts of tissue factor resulting in inhomogenous fibrin deposition in a native blood flow system. Effects of thrombin inhibitors. J Clin Invest. (1994) 93:2073–83. doi: 10.1172/JCI117202
105. Bode M, Mackman N. Regulation of tissue factor gene expression in monocytes and endothelial cells: Thromboxane A2 as a new player. Vascul Pharmacol. (2014) 62:57–62. doi: 10.1016/j.vph.2014.05.005
106. Bevilacqua MP, Pober JS, Majeau GR, Cotran RS, Gimbrone MA Jr. Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J Exp Med. (1984) 160:618–23. doi: 10.1084/jem.160.2.618
107. Costa TJ, Potje SR, Fraga-Silva TFC, da Silva-Neto JA, Barros PR, Rodrigues D, et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vascul Pharmacol. (2022) 142:106946. doi: 10.1016/j.vph.2021.106946
108. Rezaei A, Neshat S, Heshmat-Ghahdarijani K. Alterations of lipid profile in COVID-19: A narrative review. Curr Probl Cardiol. (2022) 47:100907. doi: 10.1016/j.cpcardiol.2021.100907
109. Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol. (2010) 341:126–40. doi: 10.1016/j.ydbio.2009.10.026
110. Hellman U, Karlsson MG, Engstrom-Laurent A, Cajander S, Dorofte L, Ahlm C, et al. Presence of hyaluronan in lung alveoli in severe Covid-19: An opening for new treatment options? J Biol Chem. (2020) 295:15418–22. doi: 10.1074/jbc.AC120.015967
111. Syed F, Li W, Relich RF, Russell PM, Zhang S, Zimmerman MK, et al. Excessive matrix metalloproteinase-1 and hyperactivation of endothelial cells occurred in COVID-19 patients and were associated with the severity of COVID-19. J Infect Dis. (2021) 224:60–9. doi: 10.1093/infdis/jiab167
112. Safont B, Tarraso J, Rodriguez-Borja E, Fernandez-Fabrellas E, Sancho-Chust JN, Molina V, et al. Lung function, radiological findings and biomarkers of fibrogenesis in a cohort of COVID-19 patients six months after hospital discharge. Arch Bronconeumol. (2022) 58:142–9. doi: 10.1016/j.arbres.2021.08.014
113. Huang JJ, Wang CW, Liu Y, Zhang YY, Yang NB, Yu YC, et al. Role of the extracellular matrix in COVID-19. World J Clin Cases. (2023) 11:73–83. doi: 10.12998/wjcc.v11.i1.73
114. Yamaoka-Tojo M. Vascular endothelial glycocalyx damage in COVID-19. Int J Mol Sci. (2020) 21(24):9712. doi: 10.3390/ijms21249712
115. Karampoor S, Zahednasab H, Farahmand M, Mirzaei R, Zamani F, Tabibzadeh A, et al. A possible pathogenic role of Syndecan-1 in the pathogenesis of coronavirus disease 2019 (COVID-19). Int Immunopharmacol. (2021) 97:107684. doi: 10.1016/j.intimp.2021.107684
116. Zhang D, Li L, Chen Y, Ma J, Yang Y, Aodeng S, et al. Syndecan-1, an indicator of endothelial glycocalyx degradation, predicts outcome of patients admitted to an ICU with COVID-19. Mol Med. (2021) 27:151. doi: 10.1186/s10020-021-00412-1
117. Goonewardena SN, Grushko OG, Wells J, Herty L, Rosenson RS, Haus JM, et al. Immune-mediated glycocalyx remodeling in hospitalized COVID-19 patients. Cardiovasc Drugs Ther. (2023) 37:307–13. doi: 10.1007/s10557-021-07288-7
118. Ikonomidis I, Pavlidis G, Katsimbri P, Lambadiari V, Parissis J, Andreadou I, et al. Tocilizumab improves oxidative stress and endothelial glycocalyx: A mechanism that may explain the effects of biological treatment on COVID-19. Food Chem Toxicol. (2020) 145:111694. doi: 10.1016/j.fct.2020.111694
119. Kinaneh S, Khamaysi I, Karram T, Hamoud S. Heparanase as a potential player in SARS-CoV-2 infection and induced coagulopathy. Biosci Rep. (2021) 41(7):BSR20210290. doi: 10.1042/BSR20210290
120. Yuan L, Cheng S, Sol W, van der Velden AIM, Vink H, Rabelink TJ, et al. Heparan sulfate mimetic fucoidan restores the endothelial glycocalyx and protects against dysfunction induced by serum of COVID-19 patients in the intensive care unit. ERJ Open Res. (2022) 8:00652–2021. doi: 10.1183/23120541.00652-2021
121. Drost CC, Rovas A, Osiaevi I, Rauen M, van der Vlag J, Buijsers B, et al. Heparanase is a putative mediator of endothelial glycocalyx damage in COVID-19 - A proof-of-concept study. Front Immunol. (2022) 13:916512. doi: 10.3389/fimmu.2022.916512
122. Drost CC, Rovas A, Osiaevi I, Schughart K, Lukasz A, Linke WA, et al. Interleukin-6 drives endothelial glycocalyx damage in COVID-19 and bacterial sepsis. Angiogenesis. (2024) 27:411–22. doi: 10.1007/s10456-024-09916-w
123. Lambadiari V, Mitrakou A, Kountouri A, Thymis J, Katogiannis K, Korakas E, et al. Association of COVID-19 with impaired endothelial glycocalyx, vascular function and myocardial deformation 4 months after infection. Eur J Heart Fail. (2021) 23:1916–26. doi: 10.1002/ejhf.v23.11
124. Vollenberg R, Tepasse PR, Ochs K, Floer M, Strauss M, Rennebaum F, et al. Indications of persistent glycocalyx damage in convalescent COVID-19 patients: A prospective multicenter study and hypothesis. Viruses. (2021) 13:2324. doi: 10.3390/v13112324
125. Veraldi N, Vives RR, Blanchard-Rohner G, L’Huillier AG, Wagner N, Rohr M, et al. Endothelial glycocalyx degradation in multisystem inflammatory syndrome in children related to COVID-19. J Mol Med (Berl). (2022) 100:735–46. doi: 10.1007/s00109-022-02190-7
126. Zha D, Fu M, Qian Y. Vascular endothelial glycocalyx damage and potential targeted therapy in COVID-19. Cells. (2022) 11:1972. doi: 10.3390/cells11121972
127. Lv B, Huang S, Huang H, Niu N, Liu J. Endothelial glycocalyx injury in SARS-CoV-2 infection: molecular mechanisms and potential targeted therapy. Mediators Inflamm. (2023) 2023:6685251. doi: 10.1155/2023/6685251
128. Alberro A, Iparraguirre L, Fernandes A, Otaegui D. Extracellular vesicles in blood: sources, effects, and applications. Int J Mol Sci. (2021) 22:8163. doi: 10.3390/ijms22158163
129. Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol. (2011) 31:27–33. doi: 10.1161/ATVBAHA.110.218123
130. Hernandez-Diazcouder A, Diaz-Godinez C, Carrero JC. Extracellular vesicles in COVID-19 prognosis, treatment, and vaccination: an update. Appl Microbiol Biotechnol. (2023) 107:2131–41. doi: 10.1007/s00253-023-12468-6
131. Mackman N, Antoniak S, Wolberg AS, Kasthuri R, Key NS. Coagulation abnormalities and thrombosis in patients infected with SARS-CoV-2 and other pandemic viruses. Arterioscler Thromb Vasc Biol. (2020) 40:2033–44. doi: 10.1161/ATVBAHA.120.314514
132. Russell CD, Lone NI, Baillie JK. Comorbidities, multimorbidity and COVID-19. Nat Med. (2023) 29:334–43. doi: 10.1038/s41591-022-02156-9
133. Team CC-R. Preliminary estimates of the prevalence of selected underlying health conditions among patients with coronavirus disease 2019 - United States, February 12-March 28, 2020. MMWR Morb Mortal Wkly Rep. (2020) 69:382–6. doi: 10.15585/mmwr.mm6913e2
134. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
135. Barron E, Bakhai C, Kar P, Weaver A, Bradley D, Ismail H, et al. Associations of type 1 and type 2 diabetes with COVID-19-related mortality in England: a whole-population study. Lancet Diabetes Endocrinol. (2020) 8:813–22. doi: 10.1016/S2213-8587(20)30272-2
136. Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, Morton CE, et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature. (2020) 584:430–6. doi: 10.1038/s41586-020-2521-4
Keywords: COVID-19, endothelial cells (EC), SARS-CoV-2, coreceptors, endothelial infection
Citation: Subramaniam S, Jose A, Kenney D, O’Connell AK, Bosmann M, Douam F and Crossland N (2025) Challenging the notion of endothelial infection by SARS-CoV-2: insights from the current scientific evidence. Front. Immunol. 16:1443932. doi: 10.3389/fimmu.2025.1443932
Received: 04 June 2024; Accepted: 14 January 2025;
Published: 04 February 2025.
Edited by:
Aleksandra Piechota-Polanczyk, Medical University of Lodz, PolandReviewed by:
Nhat Tu Le, Houston Methodist Research Institute, United StatesLaura Michalick, Charité University Medicine Berlin, Germany
Copyright © 2025 Subramaniam, Jose, Kenney, O’Connell, Bosmann, Douam and Crossland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saravanan Subramaniam, c2FyYXZhbmFuLnN1YnJhbWFuaWFtQG1jcGhzLmVkdQ==