 Philipp Grubwieser1,2†
Philipp Grubwieser1,2† Nina Böck3†
Nina Böck3† Erika Kvalem Soto3
Erika Kvalem Soto3 Richard Hilbe1Patrizia Moser4
Richard Hilbe1Patrizia Moser4 Markus Seifert1
Markus Seifert1 Stefanie Dichtl2Miriam Alisa Govrins2
Stefanie Dichtl2Miriam Alisa Govrins2 Wilfried Posch2
Wilfried Posch2 Thomas Sonnweber1
Thomas Sonnweber1 Manfred Nairz1
Manfred Nairz1 Igor Theurl1
Igor Theurl1 Zlatko Trajanoski3*
Zlatko Trajanoski3* Günter Weiss1,5*
Günter Weiss1,5*- 1Department of Internal Medicine II, Infectious Diseases, Immunology, Rheumatology, Medical University of Innsbruck, Innsbruck, Austria
- 2Institute of Hygiene and Medical Microbiology, Medical University of Innsbruck, Innsbruck, Austria
- 3Biocenter, Institute of Bioinformatics, Medical University of Innsbruck, Innsbruck, Austria
- 4INNPATH, Innsbruck Medical University Hospital, Innsbruck, Austria
- 5Christian Doppler Laboratory for Iron Metabolism and Anemia Research, Medical University of Innsbruck, Innsbruck, Austria
Introduction: Airway epithelial cells play a central role in the innate immune response to invading bacteria, yet adequate human infection models are lacking.
Methods: We utilized mucociliary-differentiated human airway organoids with direct access to the apical side of epithelial cells to model the initial phase of Pseudomonas aeruginosa respiratory tract infection.
Results: Immunofluorescence of infected organoids revealed that Pseudomonas aeruginosa invades the epithelial barrier and subsequently proliferates within the epithelial space. RNA sequencing analysis demonstrated that Pseudomonas infection stimulated innate antimicrobial immune responses, but specifically enhanced the expression of genes of the nitric oxide metabolic pathway. We demonstrated that activation of inducible nitric oxide synthase (iNOS) in airway organoids exposed bacteria to nitrosative stress, effectively inhibiting intra-epithelial pathogen proliferation. Pharmacological inhibition of iNOS resulted in expansion of bacterial proliferation whereas a NO producing drug reduced bacterial numbers. iNOS expression was mainly localized to ciliated epithelial cells of infected airway organoids, which was confirmed in primary human lung tissue during Pseudomonas pneumonia.
Discussion: Our findings highlight the critical role of epithelial-derived iNOS in host defence against Pseudomonas aeruginosa infection. Furthermore, we describe a human tissue model that accurately mimics the airway epithelium, providing a valuable framework for systemically studying host-pathogen interactions in respiratory infections.
1 Introduction
Bacterial lower respiratory tract infections remain a major public health concern, causing over 2 million deaths worldwide annually (1). Substantial progress has been made in understanding host responses to these infections. Animal and in-vitro models have underscored the importance of innate immune cells, such as macrophages, in frontline defence against pulmonary infections (2, 3). However, the role of the respiratory epithelium in early defence against bacterial pathogens is increasingly recognized (4). Positioned at the interface between the external environment and the internal milieu, the respiratory epithelium is the primary site of contact with invading bacteria, suggesting its active participation in early innate immune defence (5).
Pseudomonas aeruginosa (PA) is a gram-negative opportunistic bacterium and a growing health concern due to its pathogenicity and antibiotic resistance (6). PA lung infections are common and associated with high mortality rates, greatly impacting patients with cystic fibrosis (CF),chronic obstructive pulmonary disease (COPD) as well as patients with nosocomial pneumonia (7, 8).
During infection of the respiratory tract, PA closely interacts with epithelial cells and can adopt an intracellular lifecycle (9). PA can invade and actively replicate inside various epithelial cell types, including bronchial epithelia (10–12). Consequently, it has been hypothesized that epithelial internalization is the prerequisite for invasion, facilitating dissemination of the bacterium to the bloodstream and distant organs, while allowing the pathogen to evade innate immune cells, thus promoting bacterial persistence (13–15).
Although the airway epithelium initiates an inflammatory response and secretes antimicrobial effector molecules after pathogen detection, little is known about its direct antimicrobial responses. Only recently, we showed that airway epithelial cells differentially regulate nutrient trafficking in response to intra- or extracellular bacteria, thereby affecting pathogen multiplication (16).
It has been hypothesized that the production of reactive oxygen- and nitrogen species (ROS/RNS) by airway epithelial cells facilitates intracellular pathogen killing (5). Indeed, the human respiratory epithelium expresses nitric oxide synthases (NOS) (17, 18). In mice, the expression of the inducible isoform iNOS, mainly by monocytic cells including macrophages, is induced by inflammatory stimuli, such as bacterial lipopolysaccharide (LPS) or cytokines, such as interferon-gamma, tumor-necrosis factor-alpha, and interleukin- (IL-) 17 (19, 20). However, limited information is available on iNOS function in human infection models, particularly regarding its potential role within the human respiratory epithelium (21).
Despite significant progress, the multistage and cell specific mechanisms of host responses to bacterial invasion in the lung remain poorly understood. Herein we apply a novel infection model, in which differentiated human airway organoids (AOs) are challenged with the opportunistic pathogen Pseudomonas aeruginosa. In direct suspension culture, apical-out organoids are exposed to viable bacteria, closely mimicking the in-vivo situation as bacteria interact with the epithelial layer at the correct spatial localization. This enables monitoring of the bacterial entry and intracellular fate, and in parallel allows for comprehensive analysis of host epithelial cell responses to infection.
2 Material and methods
2.1 Organoid and bacteria culture
Patient derived human airway organoids were acquired from Foundation Hubrecht Organoid Biobank (www.hubrechtorganoidbiobank.org) and cultured as described by Sachs et al. (22). Apical-out polarity switch and mucociliary differentiation was performed as described by Co et al. (23) and Zhou et al. (24), respectively.
Pseudomonas aeruginosa, strain P14 as well as strain P14 stably expressing GFP (PA-GFP), were a kind gift of Dirk Bumann, Biozentrum Basel, Switzerland. This reference strain is characterized by its similarity to patient isolates in terms of virulence and thus is the preferred model for infection studies with virulent PA (25). Detailed information is provided in the Supplementary Methods.
2.2 Organoid infection
Fully differentiated, apical-out human airway organoids were washed two times with PBS, and seeded in equal density in proximal differentiation medium without antibiotics and hydrocortisone into 12-well plates treated with anti-adherence solution. On the next day, PA from the mid-logarithmic growth phase was added to organoids at a final concentration of 25*106/ml for 3h. Bacterial outgrowth in the medium is then prevented by washing thrice in PBS containing gentamicin (25µg/ml) and adding fresh medium containing gentamicin (8µg/ml) for further incubation. To minimize the use of gentamicin and thus off-target effects, we determined the minimal inhibitory concentration (MIC) of gentamicin with conventional microbiological methods (Etest, MIC: 1µg/ml, Supplementary Figure 2A) and in experiment-specific conditions (growth in ALI or LB medium; Supplementary Figure 2B). This antibiotic is not able to penetrate cell membranes, and thus only acts in the extracellular space (26). Thus, bacteria, which have invaded the epithelial formation, are not exposed to this antibiotic and remain viable inside epithelial cells. During this gentamicin-protected phase, organoids and intra-organoid bacteria further interact for up to 24 hours, enabling analysis of intra-organoid bacterial numbers and organoid innate immune responses at various time intervals. To quantify intra-organoid bacteria, AOs were washed thrice in PBS, and subsequently lysed in 0.5% sodium deoxycholic acid (Sigma-Aldrich). Organoid lysates, containing viable bacteria, were plated immediately on LB plates, and colony-forming units (CFUs) were quantified after overnight incubation. Where indicated, Organoids were treated with 100µM of the NO-donor NOC-18 (MedchemExpress, HY-136278) or 50µM of the iNOS inhibitor L-NIL (MedchemExpress, HY- 12116) during the active and gentamicin-protected infection phase.
2.3 Statistical analysis
RNA sequencing analysis is described in detail in the Supplementary Methods section. Statistical analysis of CFU and qPCR data was performed in GraphPad Prism (9.4.1). For pairwise comparisons, an unpaired student t-test was used. ANOVA with Sidaks post-hoc test was used for multiple group comparisons. Data was log-transformed as appropriate. A p-value of <0.05 was used as the significance threshold. Additional methods can be found in the Supplementary Material.
3 Results
3.1 Pseudomonas aeruginosa infects differentiated human airway organoids and resides in the intracellular space
To investigate the epithelium-pathogen interaction, we first generated apical-out airway organoids. To this end, we treated organoids with EDTA for complete matrix degradation and cultured them in suspension, resulting in apical-out airway organoids (AOAOs, Figure 1A). After 16 days of culture in the differentiation medium, AOAOs present an abundance of ciliated cells at the apical side, considered to be the hallmark of terminal mucociliary differentiation. During the differentiation phase, markers of ciliated (FOXJ1) and goblet (MUC5AC) cells increased significantly, whereas club cells (SCGB1A1) decreased, and basal cells (P63) remained constant (Supplementary Figure 1).
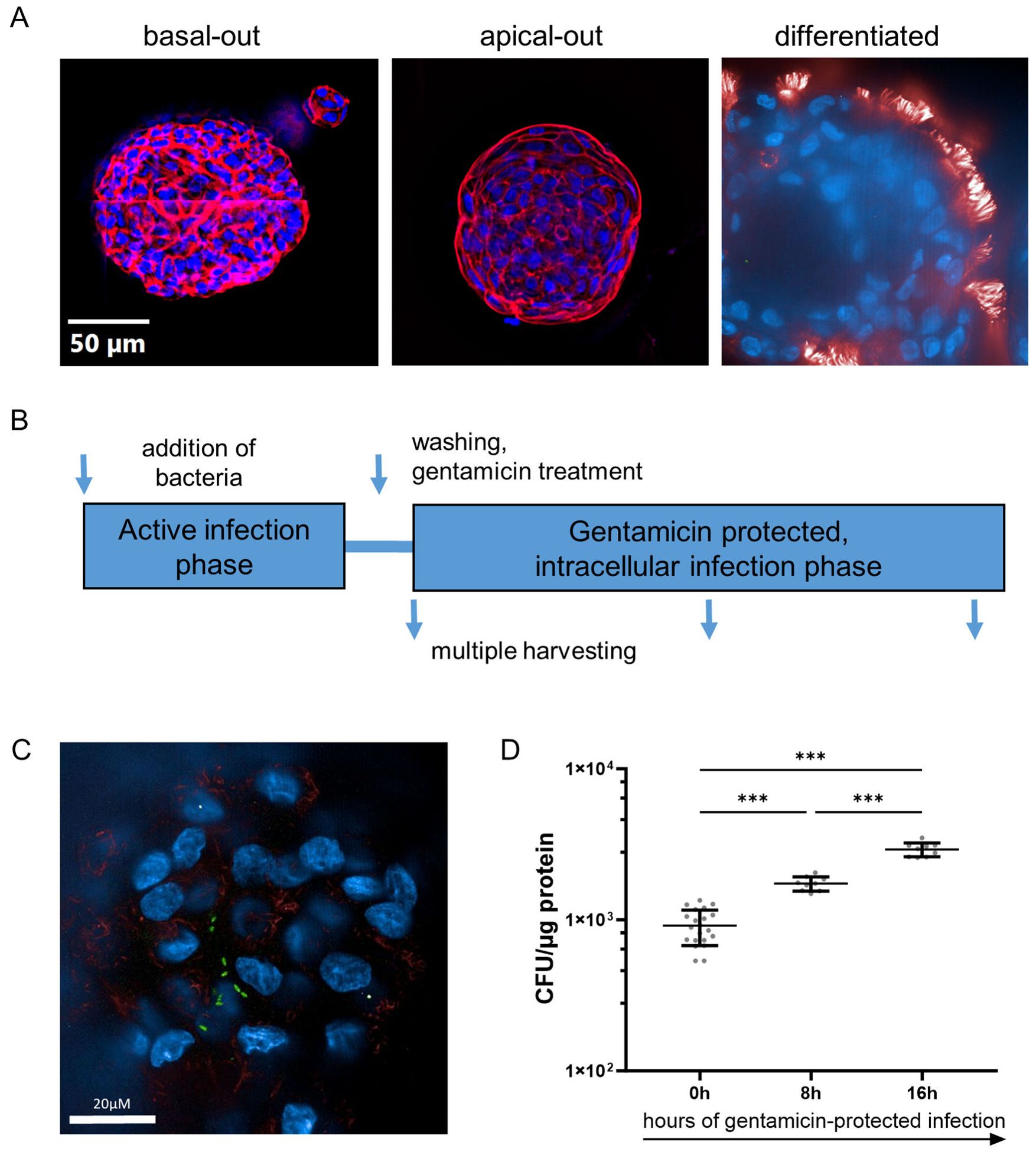
Figure 1. Pseudomonas aeruginosa infects differentiated human airway organoids and resides in the intracellular space. (A) Immune fluorescence imaging of human airway organoids in basal-out configuration (left panel) and apical-out configuration (middle panel). Representative epifluorescence images of organoids stained for actin (red) and nuclei (blue), mounted on slides. Terminally differentiated organoid (right panel). Representative confocal image, stained for apically located cilia (red) and nuclei (blue). (B) Schematic representation of the organoid infection model used herein. (C) Confocal immune fluorescence imaging revealing intra-organoid bacteria (PA-GFP, green rods) in infected organoids after 3h of active infection and removal of extracellular bacteria. (D) Quantification of intracellular bacteria during the gentamicin-protected course of infection. Infected organoids were lysed at indicated time intervals, and lysates were plated onto LB-agar plates for CFU quantification. Data from three independent experiments are shown as a scatter plot with mean ± SD. *** denotes p < 0.001 for ANOVA with post hoc statistical testing.
AOAOs were then challenged with the bacterial pathogen PA in a gentamicin-protected infection model (Figure 1B): viable bacteria are directly added to the antibiotic-free cell culture medium, after which AOs and bacteria directly interact for 3h. Subsequently, to prevent bacterial overgrowth, AOs are washed and treated with gentamicin, which eliminates bacteria in the extracellular space but does not affect intracellular bacteria which have invaded the organoid. Infected AOs are then further incubated in a gentamicin-containing medium and harvested at multiple time intervals for analysis.
We first applied immunofluorescence of organoids infected with PA expressing GFP (PA-GFP), harvested after the active infection phase and after the removal of extracellular bacteria. Interestingly, confocal imaging revealed bacteria (green, rod-shaped) inside the epithelial tissue (Figure 1C), showing that PA has invaded the epithelial barrier. Subsequently, to quantify the number of viable intracellular bacteria, the organoid lysate was plated at multiple time intervals of the gentamicin-protected phase (Figure 1D). Viable PA bacteria were recovered directly after the active infection phase (0h), indicating the number of bacteria that have invaded AOs. The number of intra-organoid bacteria subsequently increased, with peak bacterial burden at 16h, suggesting intra-organoid bacterial multiplication.
3.2 Pro-inflammatory signaling shapes organoid responses to infection
To shed light on organoid responses to infection, bulk RNA sequencing of infected and uninfected organoids was performed. Direct comparison revealed major differences in the transcriptome of infected organoids compared to uninfected controls (Figures 2A, B). Principal-component analysis (PCA) demonstrated close similarities among samples belonging to the same treatment group after batch effect correction (Supplementary Figure 3). Infection with PA induced the upregulation of 343 genes and downregulation of 182 genes in AOs, with a significant increase in the expression of genes associated with the inflammatory response, such as the pro-inflammatory cytokine IL-17C (Volcano plot, Figure 2A; Supplementary Table 2). The ORA revealed, that these changes reflect pathways involved in cellular responses to bacteria and lipopolysaccharides (LPS), mainly including a pro-inflammatory cytokine signature (IL1-B, TNFA, IL-8) and production of antimicrobial peptides (DEFB, S100A8, S100A9). To confirm if those alterations are also found by sequential analysis of mRNA and protein expression of such genes, we studied IL-6 and IL-8 expression via qPCR and ELISA over time (Figures 2C, D). The transcriptional induction of both cytokines was immediately increased after the initiation of the infection phase and significantly elevated throughout the gentamicin-protected infection phase, being in line with the RNA-sequencing data. Consistent with previous findings, uninfected AOs exhibited basal secretion of IL-8 (27). Interestingly, aside from the expected inflammatory response, differential regulation of metabolic pathways, specifically the nitric oxide (NO) metabolic pathway (GO:0046209) was evident (Figure 2E). This pathway contains several genes associated with nitric oxide production, including iNOS (NOS2), which was significantly induced in infected organoids compared to uninfected controls. Together, these results indicate that human airway organoids exposed to viable bacteria induce multiple inflammatory and anti-microbial pathways, secrete inflammatory cytokines and upregulate genes that are associated with NO production.
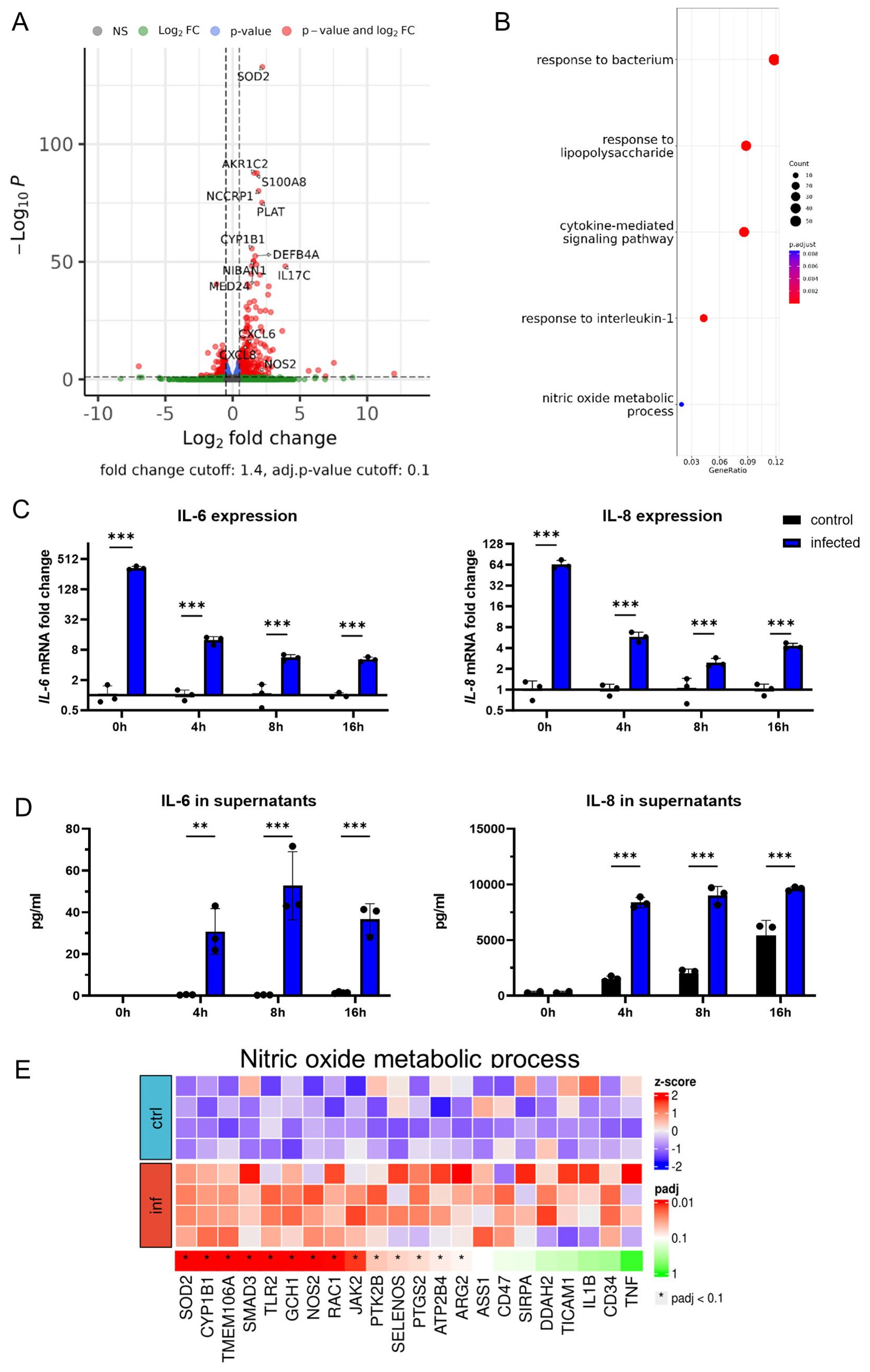
Figure 2. Pro-inflammatory signaling shapes organoid responses to infection. (A) Volcano plot showing differentially expressed genes of infected human airway organoids in comparison to uninfected controls after 4h of gentamicin-protected infection. (B) Gene-set analysis with over-representation test (ORA) showing Gene ontology biological processes. (C) Differential IL-6 (left panel) and IL-8 (right panel) mRNA expression in infected organoids. Organoids were harvested at indicated time intervals of gentamicin-protected infection. Data is shown as mean ± SD of a triplicate experiment. (D) Levels of the inflammatory cytokines IL-6 (left panel) and IL-8 (right panel) in supernatants of infected organoids. Supernatants were collected at indicated time intervals of gentamicin-protected infection. Data is shown as mean ± SD of a triplicate experiment. (E) Heat map graphical representation of differentially expressed genes in infected versus uninfected organoids. The genes (X axis) are derived from Gene ontology nitric oxide metabolic process (GO:0046209) set of genes. The gene expression is indicated by z-score. ** denotes p < 0.01, *** denotes p < 0.001 for ANOVA with post-hoc statistical testing.
3.3 Human airway organoids induce iNOS in response to infection with Pseudomonas aeruginosa
Next, to further examine organoid iNOS induction in response to infection, we evaluated the involved regulatory networks. To accomplish this, bulk RNA sequencing data was analysed for pathogen detection and subsequent activation of inflammatory signalling pathways, which finally led to antimicrobial effector induction (Figure 3A). In all entities, a significant positive regulation pattern in infected organoids was evident, including a prominent induction of iNOS. We confirmed the transcriptional upregulation of iNOS by qPCR at multiple time-intervals of the gentamicin-protected infection phase (Figure 3B). A striking increase in NOS2 mRNA levels is visible directly after the end of the active infection phase (3h of bacterial exposure), with a consecutive upregulation thereafter. Interestingly, not only infection with viable PA but also treatment with heat-inactivated bacteria (HI PA) led to a significant increase of NOS2 mRNA levels (Figure 3C). In contrast, treatment with the sterile bacterial supernatant (SN), which contains soluble bacterial products including toxins but no viable bacteria, did not result in iNOS induction. Notably, the induction of iNOS in infected organoids was evident at the protein level (Figures 3D, E). Induction of iNOS was paralleled by increased phosphorylation of p38, indicating increased mitogen-activated protein kinase (MAPK) activation. This data suggests, that the host-pathogen interaction during active PA infection leads to the activation of pro-inflammatory pathways in airway epithelial cells including iNOS formation at the transcriptional and translational level.
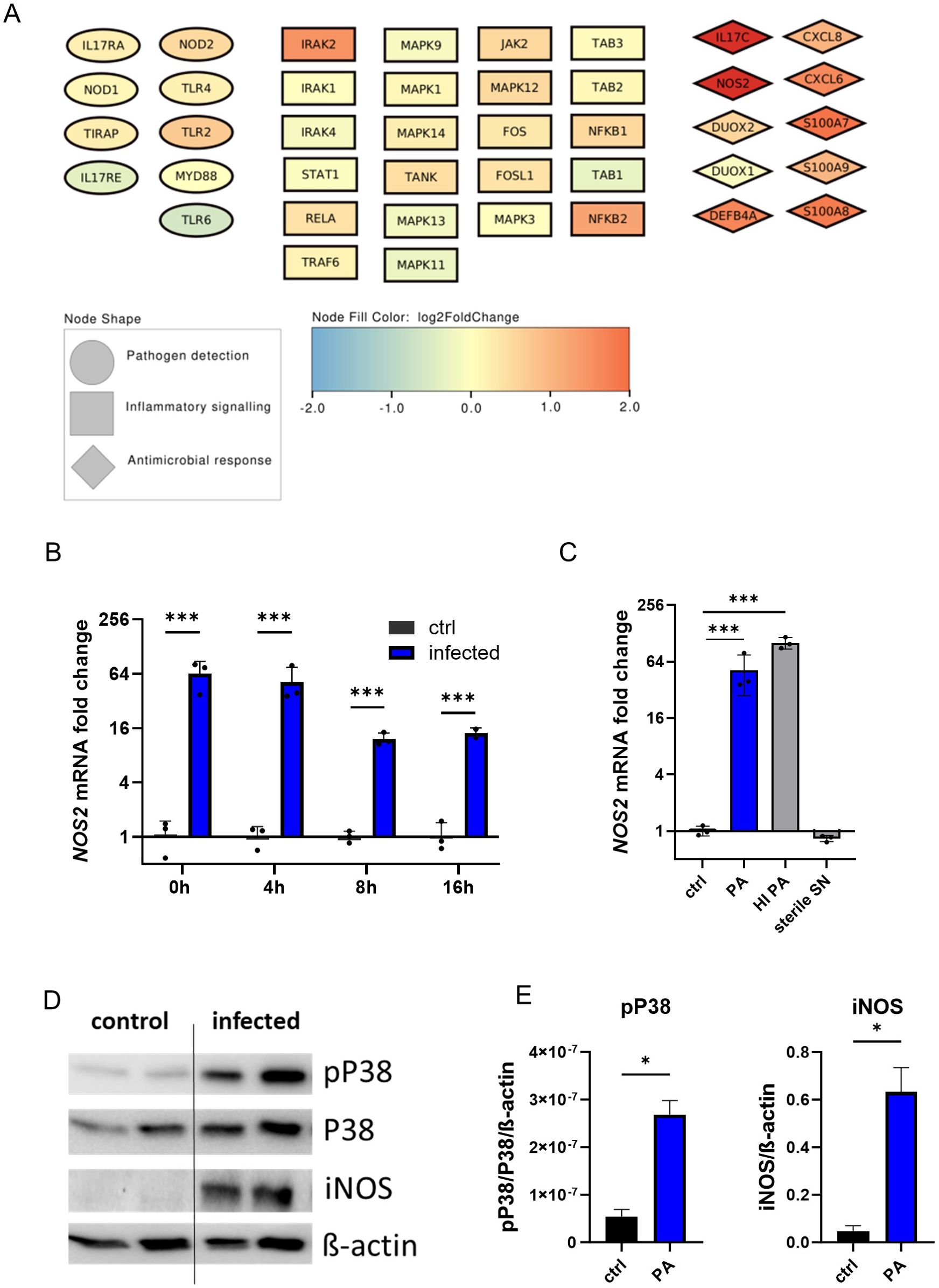
Figure 3. Human airway organoids induce iNOS in response to infection with Pseudomonas aeruginosa. (A) Graphical representation of infection-associated induced genes as nodes. Pathogen-detection-associated genes are shown as ellipses, intracellular inflammatory signalling genes are shown as squares, and antimicrobial effectors are depicted as diamonds. The colour of the nodes indicates the log2-fold change from the differential expression analysis of infected human airway organoids compared to uninfected controls (B) Differential NOS2 mRNA expression in infected organoids. Organoids were harvested at indicated time intervals of gentamicin-protected infection. Data is shown as mean ± SD of three independent experiments. (C) Differential NOS2 mRNA expression at the 4h time interval in infected organoids and organoids exposed to heat-inactivated (HI) bacteria or treated with sterile-filtrated bacterial supernatant (SN). Organoids were harvested at indicated time intervals of gentamicin protected infection. Data is shown as mean ± SD of three independent experiments. (D) Western blot of the MAPK subunit p38, phosphorylated p38, and iNOS protein of infected organoids and uninfected controls harvested directly after the active infection phase (0h-time interval), shown in duplicates. (E) Densitometric quantification of the Western blots targets iNOS and phosphorylated p38. * denotes p < 0.05 for statistical testing with a two-sided unpaired t-test, *** denotes p < 0.001 for ANOVA with post-hoc statistical testing.
3.4 iNOS is induced in ciliated cells of infected airway organoids and human bronchial epithelia during Pseudomonas aeruginosa pneumonia
Next, we applied immunofluorescence imaging to reveal cellular localization of iNOS-protein expression in infected organoids. After the active infection, organoids were stained for ciliated cells (acetylated tubulin) and iNOS protein. Imaging revealed induction of iNOS protein in infected organoids but not in uninfected controls (Figure 4A). Furthermore, regions with high iNOS expression in infected AOs structurally resembled ciliated cells. Indeed, co-staining with acetylated tubulin, a specific marker for ciliated cells, revealed near congruent expression of iNOS protein (Figure 4A). Finally, iNOS expression was investigated in human lung tissue specimen (Figure 4B). Immunohistochemistry staining confirmed iNOS protein presence in the bronchial epithelium in a patient infected with PA, specifically in ciliated cells of the bronchial region. Compared to this, no obvious iNOS expression was found in a non-infected control lung sample. These results indicate, that iNOS is induced in human respiratory epithelium upon pathogen contact, specifically in ciliated cells. Corroborating our findings of the organoid infection model, the iNOS protein expression showed a similar pattern in histological analysis of human lung specimens.
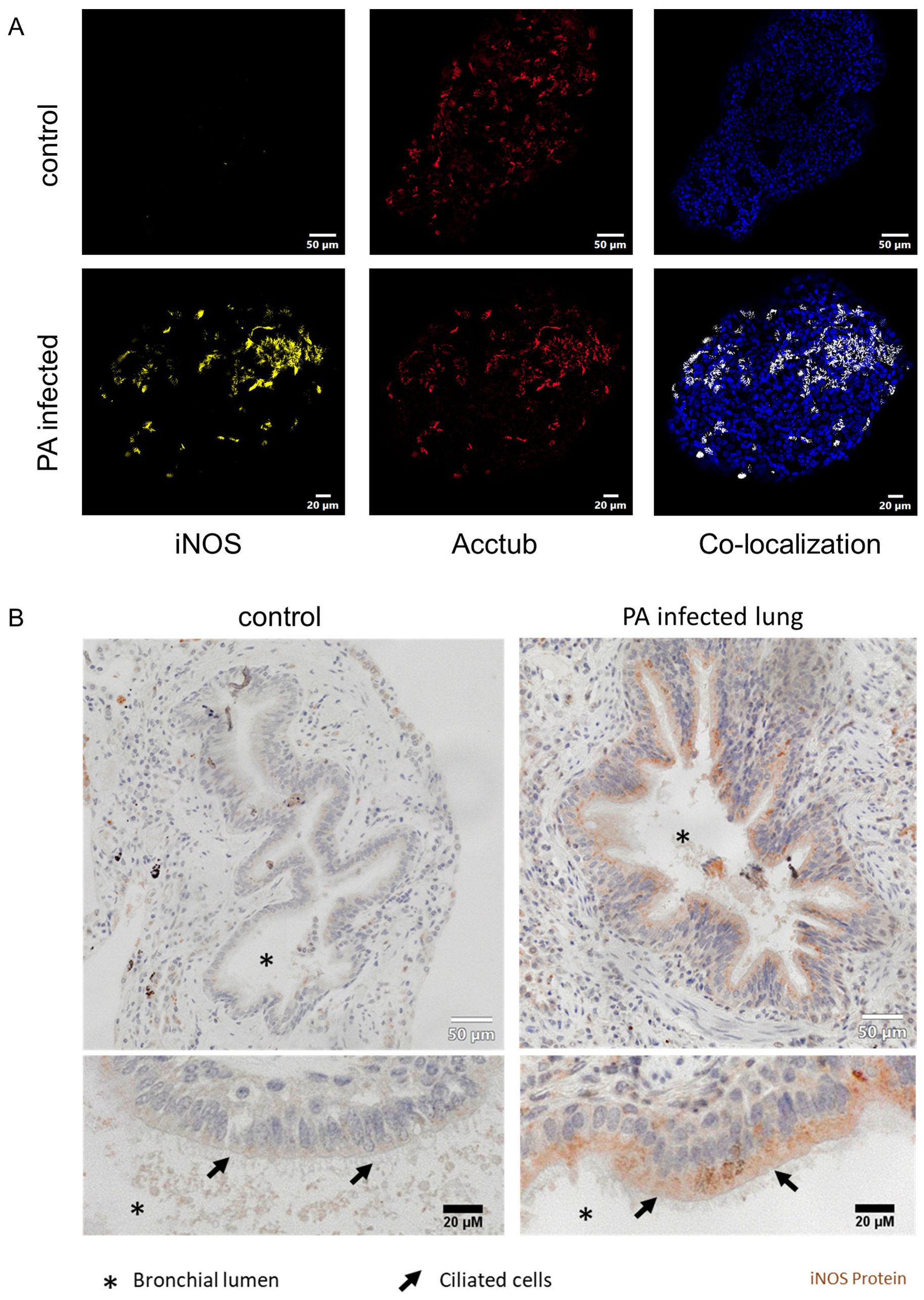
Figure 4. iNOS is induced in ciliated cells of infected airway organoids and human bronchial epithelia during Pseudomonas aeruginosa pneumonia. (A) Immune fluorescence imaging of differentiated human airway organoids, infected with PA14 (lower panels). Representative epifluorescence images of organoids stained for iNOS (yellow), ciliated cells (red) and nuclei (blue). Co-localization of iNOS and ciliated cells is depicted in white (right panel). (B) Histological analysis of human lung samples stained for iNOS protein obtained from a non-infected lung (left panel) and a lung with confirmed Pseudomonas aeruginosa pneumonia (right panel).
3.5 Human airway organoids expose Pseudomonas aeruginosa to nitrosative stress
After confirming the expression of iNOS protein in infected AOs, we next assessed the impact of iNOS expression on bacterial genes associated with response to nitrosative stress exerted by host formation of NO. For this, PA bacteria were either incubated in AO medium alone or in the presence of AOs (graphical representation Figure 5A). After 3h of incubation, mRNA was extracted and expression of bacterial genes associated with NO detoxification, namely nitrite reductase (NirS), nitric oxide reductase (NorCB), nitrous oxide reductase (NosZ), flavohemoglobin (fhp)) were analysed (Figure 5B) (28). In bacteria exposed to AOs, several genes for detoxification of nitric-oxide stress were significantly elevated, indicating that direct interaction with human AOs leads to the induction of nitrosative stress in bacteria and subsequently to expression of bacterial NO detoxification enzymes.
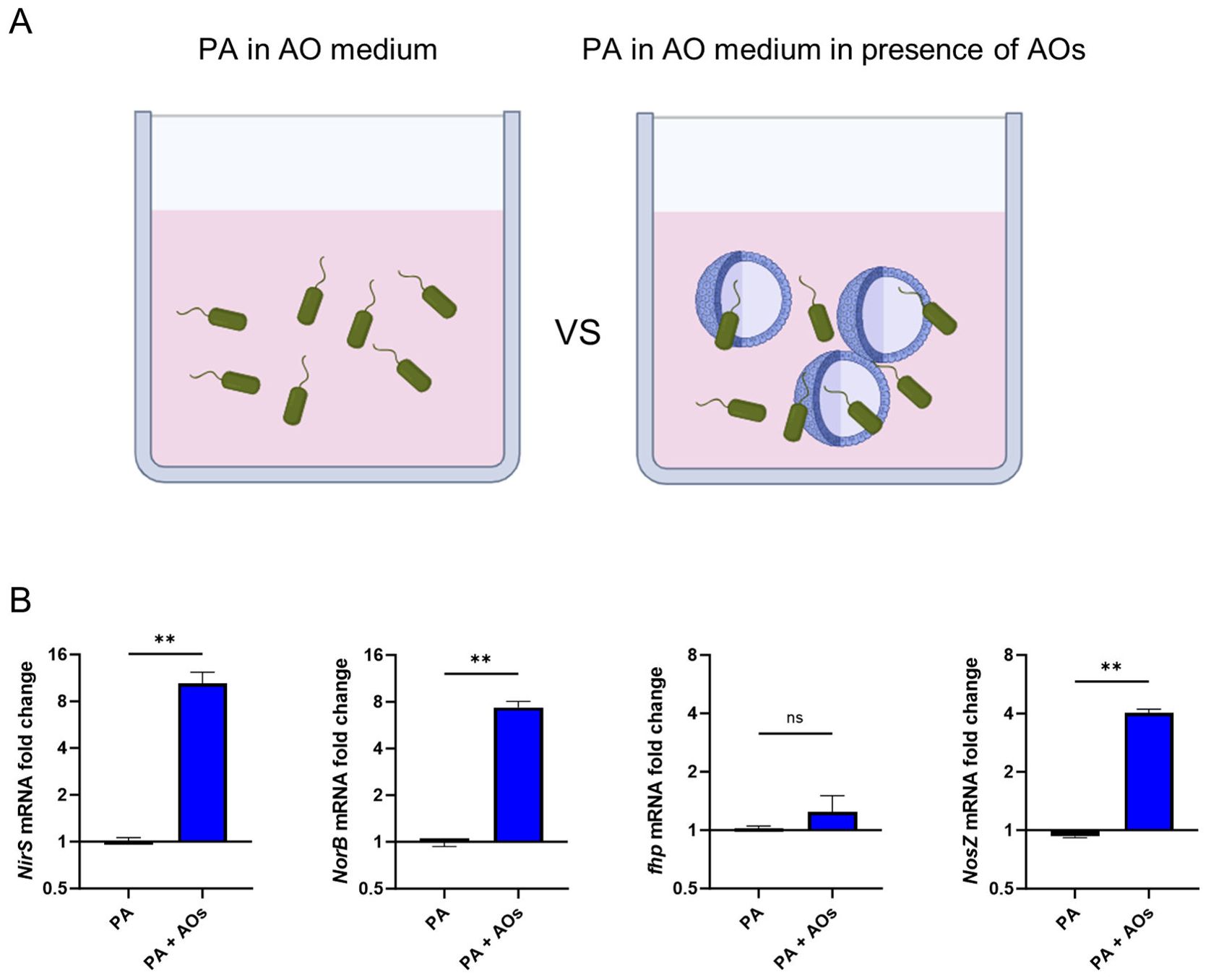
Figure 5. Human airway organoids expose Pseudomonas aeruginosa to nitrosative stress. (A) Graphical illustration of the experimental setup: PA were either incubated for 3h in antibiotic-free AO medium alone or in the presence of AOs. After this, mRNA was extracted. (B) Differential mRNA expression of bacterial genes associated with denitrification, in PA versus PA exposed to human AOs. Data is shown as mean ± SD of three independent experiments. ns denotes not significant, ** denotes p < 0.01 for a two-sided, unpaired t-test.
3.6 Human airway organoids control intracellular Pseudomonas aeruginosa growth via iNOS
To determine the functional relevance of epithelial iNOS expression for the control of intraepithelial bacterial multiplication, we applied the specific iNOS inhibitor L-NIL in our AO infection model (illustration Figure 6A). To this aim, AOs were infected with PA and treated with 50µM L-NIL during the active infection and gentamicin-protected infection phase, or left untreated. At the peak of bacterial intra-organoid pathogen burden (16h), total RNA was extracted and expression of bacterial genes associated with denitrification (NirS, NorB, fhp, NosZ) were analysed (Figure 6B). In intra-organoid bacteria from AOs treated with the iNOS inhibitor L-NIL, expression of denitrification genes (NirS, NorB, fhp, NosZ) was generally lower compared to bacteria exposed to AOs under standard conditions. A statistically significant reduction was only observed for the nitrous oxide reductase (NosZ). Next, intra-organoid bacteria were quantified after the gentamicin-protected infection phase in AOs treated with either the iNOS inhibitor L-NIL or the NO-donor NOC-18 (Figure 6C). Underlining the role of iNOS activation in pathogen control, recovered CFUs from AOs treated with L-NIL were significantly increased. Accordingly, bacterial numbers recovered from AOs treated with the NO forming drug NOC-18 were significantly decreased. In sum, this data demonstrates that human airway organoids infected with PA control intra-epithelial bacterial growth at least in part by induction of the iNOS pathway in epithelial cells.
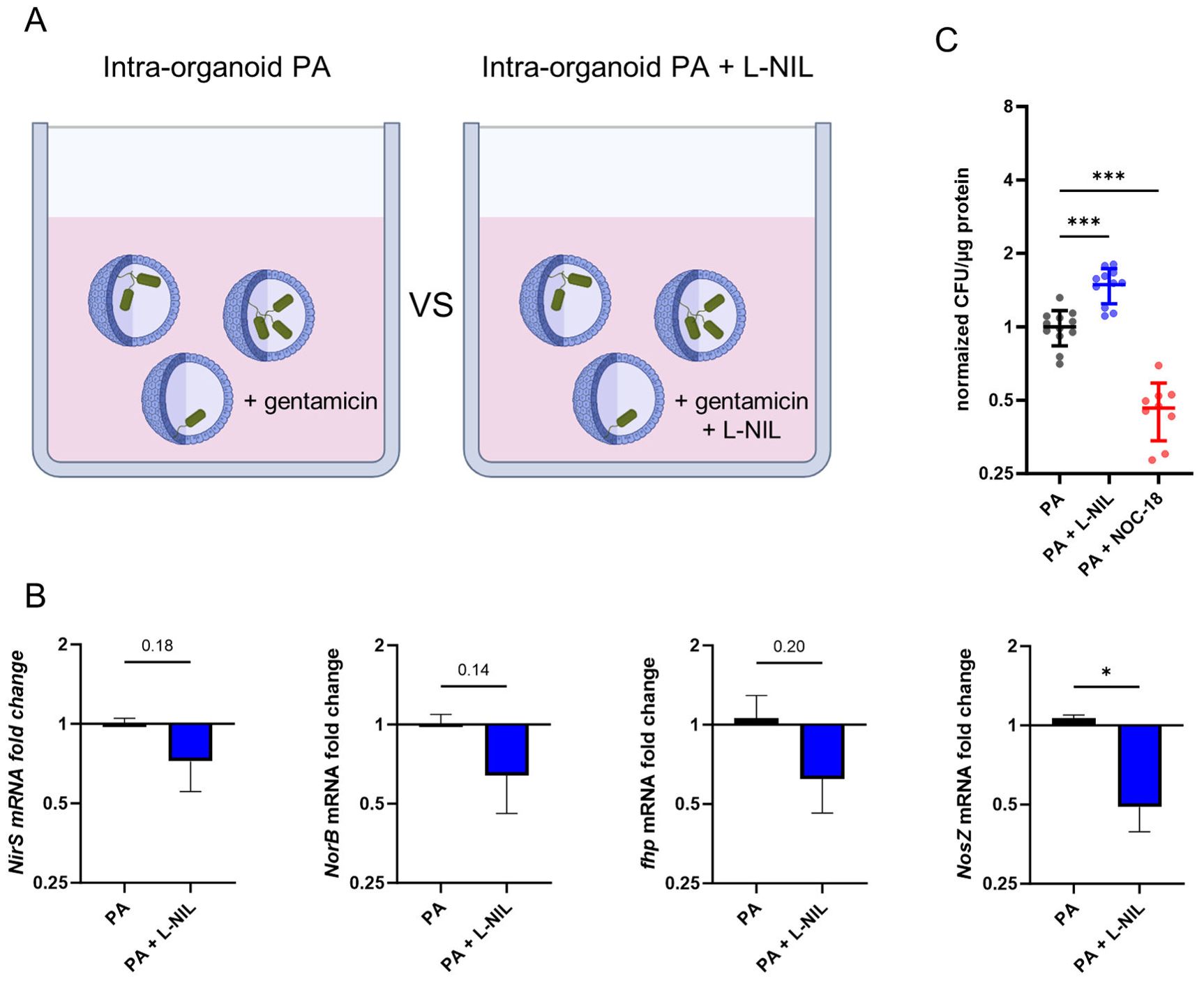
Figure 6. Human airway organoids control intracellular Pseudomonas aeruginosa growth via iNOS. (A) Graphical illustration of the experimental setup: Infected AOs were incubated with or without the specific iNOS inhibitor L-NIL during the 16h gentamicin-protected infection phase. Afterwards, mRNA was extracted. (B) Differential mRNA expression of bacterial genes associated with denitrification, in PA infecting AOs versus PA infection of AOs in the presence of the iNOS inhibitor L-NIL after 16h of gentamicin protected infection. Data is shown as mean ± SD of three independent experiments. (C) Quantification of intracellular bacteria after 24h of gentamicin-protected infection of AOs treated with 50µM iNOS inhibitor L-NIL, 100µM NO-donor NOC-18 or left untreated. Infected organoids were lysed, and lysates were plated onto LB-agar plates for CFU quantification. Data from three independent experiments performed in triplicates are shown as a scatter plot with mean ± SD, normalized to infection control (PA). * denotes p < 0.05 for unpaired t-test, *** denotes p < 0.001 for ANOVA with post-hoc statistical testing.
4 Discussion
To date, a bottleneck in respiratory infection research is the sparse offering of physiologically relevant in-vitro or ex-vivo models to study bacterial interactions with human airway epithelial cells. Here, we developed an ex-vivo human model and demonstrate that human mucociliary differentiated bronchiolar airway organoids recapitulate the early steps of Pseudomonas aeruginosa respiratory tract infection. AOs recognize invading pathogens and initiate innate immune responses, as indicated by induction of multiple inflammatory and anti-microbial pathways, which is consistent with previous studies using Pseudomonas-epithelial infection models (29, 30). Furthermore, suppressive regulators of innate immunity, like TRIM29 or IL-19, showed a positive regulation pattern in infected AOs. TRIM29 has been reported to be highly expressed by airway epithelial cells and is modulating innate immune responses (31, 32). Interestingly, increased presence of TRIM29 promotes endoplasmic reticulum stress and production of reactive species, potentially exposing an intracellular pathogen to oxidative stress (33). Considering this dual role in both innate immune modulation and stress response, epithelial-derived TRIM29 potentially impacts PA respiratory tract infection.
In this work, we provide the first evidence of the airway epithelial iNOS axis in controlling intra-organoid bacterial growth. Although iNOS activity is a well-established antimicrobial effector molecule in murine infection models, the role of iNOS-derived antimicrobial RNS in humans remained less clear (20, 34). Most human ex-vivo infection models employing innate immune cells, such as macrophages, failed to show induction of iNOS during infection or sufficient production of antimicrobial iNOS-derived RNS species. Several reasons have been postulated, including epigenetic silencing of the respective promotor or the artificial environment during ex-vivo experiments (35, 36). Nevertheless, induction of iNOS following infection has been demonstrated for specific infections in-vivo, including tuberculosis (37).
In respiratory infections, it has been proposed that iNOS may not originate from classical innate immune cells, such as macrophages, but rather from airway epithelial cells (17, 18). Accordingly, dysfunctional airway epithelium, for instance in CF or COPD patients, critically alters host defence functions (38). In a cellular co-culture model, CF bronchial epithelial cells failed to induce iNOS in response to neutrophilic infiltration (39). In line with this observation, lower epithelial iNOS expression was shown in CF subjects as compared to controls (40). Nonetheless, NO formation in a CF epithelial cell-line model resulted in decreased PA adherence and improved the elimination of internalized bacteria (21). Together, these findings suggest that the reduced functionality of epithelial iNOS in CF patients significantly contributes in PA colonization and infection. Additionally, the use of inhaled corticosteroids in COPD patients is associated with an increased pneumonia rate, potentially due to decreased epithelial NO production (41).
To determine the role of epithelial iNOS production in immune defence against PA in our airway organoid model, we used the specific pharmacological iNOS inhibitor L-NIL or the NO-forming drug NOC-18. We observed enhanced clearance of PA upon addition of NOC-18, whereas treatment with L-NIL increased bacterial numbers in our organoid infection model. Together, these findings confirmed that upregulation of iNOS activity upon PA infection plays critical part in epithelial infection control. The upregulation of iNOS in epithelial cells by both viable and heat-inactivated PA suggests that bacterial cell wall components, as well as interaction with intracellular PAMPs induce iNOS expression.
An improved understanding of innate immune responses mediated by airway epithelial cells is pivotal to effectively combat early infection and impede PA persistence, especially given the rising burden of anti-microbial resistance. This may contribute to the development of adjunct therapeutic concepts by modulating specific innate immune pathways, including the stimulation of iNOS-mediated antimicrobial effector mechanisms, in order to improve infection outcomes.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: European Nucleotide Archive, accession number: PRJEB82833.
Ethics statement
The studies involving humans were approved by Ethics committee, Medical University Innsbruck, Austria (study number 1046/2024). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by- product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
PG: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. NB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Visualization, Writing – original draft, Writing – review & editing. ES: Data curation, Formal analysis, Software, Visualization, Writing – original draft, Writing – review & editing. RH: Methodology, Software, Writing – original draft, Writing – review & editing. PM: Methodology, Validation, Writing – original draft, Writing – review & editing. MS: Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. SD: Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. MG: Data curation, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. WP: Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. TS: Investigation, Validation, Writing – original draft, Writing – review & editing. MN: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. IT: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. ZT: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Writing – original draft, Writing – review & editing. GW: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded in whole or in part by the Austrian Science Fund (FWF, DOC 82 doc.fund; doi: 10.55776/DOC82). For open access purposes, the author has applied a CC BY public copyright license to any author accepted manuscript version arising from this submission. Furthermore, this work was supported by the Medical University of Innsbruck (project CONNECT) and by the European Research Council (project EPIC grant agreement no. 786895 to ZT). Financial support by the Christian Doppler Society (Laboratory of Iron Metabolism and Anemia Research) and the “Verein zur Förderung von Forschung und Weiterbildung in Infektiologie und Immunologie an der Medizinischen Universität Innsbruck” is gratefully acknowledged.
Acknowledgments
The authors want to thank Beatrice Claudi and Prof. Dr. Dirk Bumann (Biozentrum, University of Basel) for providing the PA14 and PA14-GFP bacterial strains. Furthermore, the authors thank Werner Klotz (Laboratory infectious disease diagnostics, Medical University of Innsbruck) for performing the Luminex ELISA essay.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1508727/full#supplementary-material
References
1. GBD Collaborators LRI. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990-2016: A systematic analysis for the global burden of disease study 2016. Lancet Infect Dis. (2018) 18:1191–210. doi: 10.1016/s1473-3099(18)30310-4
2. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. (2020) 11:1722. doi: 10.3389/fimmu.2020.01722
3. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
4. Mettelman RC, Allen EK, Thomas PG. Mucosal immune responses to infection and vaccination in the respiratory tract. Immunity. (2022) 55:749–80. doi: 10.1016/j.immuni.2022.04.013
5. Hiemstra PS, McCray PB Jr., Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J. (2015) 45:1150–62. doi: 10.1183/09031936.00141514
6. Reyes J, Komarow L, Chen L, Ge L, Hanson BM, Cober E, et al. Global epidemiology and clinical outcomes of carbapenem-resistant pseudomonas aeruginosa and associated carbapenemases (Pop): A prospective cohort study. Lancet Microbe. (2023) 4:e159–e70. doi: 10.1016/S2666-5247(22)00329-9
7. Collaborators GAR. Global mortality associated with 33 bacterial pathogens in 2019: A systematic analysis for the global burden of disease study 2019. Lancet. (2022) 400:2221–48. doi: 10.1016/s0140-6736(22)02185-7
8. Martinez-García MA, Rigau D, Barrecheguren M, García-Ortega A, Nuñez A, Oscullo Yepez G, et al. Long-term risk of mortality associated with isolation of pseudomonas aeruginosa in copd: A systematic review and meta-analysis. Int J Chron Obstruct Pulmon Dis. (2022) 17:371–82. doi: 10.2147/copd.S346294
9. Angus AA, Lee AA, Augustin DK, Lee EJ, Evans DJ, Fleiszig SM. Pseudomonas aeruginosa induces membrane blebs in epithelial cells, which are utilized as a niche for intracellular replication and motility. Infect Immun. (2008) 76:1992–2001. doi: 10.1128/iai.01221-07
10. Kroken AR, Gajenthra Kumar N, Yahr TL, Smith BE, Nieto V, Horneman H, et al. Exotoxin S secreted by internalized pseudomonas aeruginosa delays lytic host cell death. PLoS Pathog. (2022) 18:e1010306. doi: 10.1371/journal.ppat.1010306
11. Jolly AL, Takawira D, Oke OO, Whiteside SA, Chang SW, Wen ER, et al. Pseudomonas aeruginosa-induced bleb-niche formation in epithelial cells is independent of actinomyosin contraction and enhanced by loss of cystic fibrosis transmembrane-conductance regulator osmoregulatory function. mBio. (2015) 6:e02533. doi: 10.1128/mBio.02533-14
12. Kumar NG, Nieto V, Kroken AR, Jedel E, Grosser MR, Hallsten ME, et al. Pseudomonas aeruginosa can diversify after host cell invasion to establish multiple intracellular niches. mBio. (2022) 13:e02742–22. doi: 10.1128/mbio.02742-22
13. Lepanto P, Bryant DM, Rossello J, Datta A, Mostov KE, Kierbel A. Pseudomonas aeruginosa interacts with epithelial cells rapidly forming aggregates that are internalized by a lyn-dependent mechanism. Cell Microbiol. (2011) 13:1212–22. doi: 10.1111/j.1462-5822.2011.01611.x
14. Lovewell RR, Patankar YR, Berwin B. Mechanisms of phagocytosis and host clearance of pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L591–603. doi: 10.1152/ajplung.00335.2013
15. Muggeo A, Coraux C, Guillard T. Current concepts on pseudomonas aeruginosa interaction with human airway epithelium. PLoS Pathog. (2023) 19:e1011221. doi: 10.1371/journal.ppat.1011221
16. Grubwieser P, Hoffmann A, Hilbe R, Seifert M, Sonnweber T, Böck N, et al. Airway epithelial cells differentially adapt their iron metabolism to infection with klebsiella pneumoniae and escherichia coli in vitro. Front Cell Infect Microbiol. (2022) 12:875543. doi: 10.3389/fcimb.2022.875543
17. Guo FH, De Raeve HR, Rice TW, Stuehr DJ, Thunnissen FB, Erzurum SC. Continuous nitric oxide synthesis by inducible nitric oxide synthase in normal human airway epithelium in vivo. Proc Natl Acad Sci U.S.A. (1995) 92:7809–13. doi: 10.1073/pnas.92.17.7809
18. Asano K, Chee CB, Gaston B, Lilly CM, Gerard C, Drazen JM, et al. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc Natl Acad Sci U.S.A. (1994) 91:10089–93. doi: 10.1073/pnas.91.21.10089
19. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. (2015) 264:182–203. doi: 10.1111/imr.12266
20. Bogdan C. Nitric oxide and the immune response. Nat Immunol. (2001) 2:907–16. doi: 10.1038/ni1001-907
21. Darling KE, Evans TJ. Effects of nitric oxide on pseudomonas aeruginosa infection of epithelial cells from a human respiratory cell line derived from a patient with cystic fibrosis. Infect Immun. (2003) 71:2341–9. doi: 10.1128/iai.71.5.2341-2349.2003
22. Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Böttinger L, Klay D, et al. Long-term expanding human airway organoids for disease modeling. EMBO J. (2019) 38:e100300. doi: 10.15252/embj.2018100300
23. Co JY, Margalef-Català M, Monack DM, Amieva MR. Controlling the polarity of human gastrointestinal organoids to investigate epithelial biology and infectious diseases. Nat Protoc. (2021) 16:5171–92. doi: 10.1038/s41596-021-00607-0
24. Zhou J, Li C, Sachs N, Chiu MC, Wong BH, Chu H, et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc Natl Acad Sci U.S.A. (2018) 115:6822–7. doi: 10.1073/pnas.1806308115
25. Grace A, Sahu R, Owen DR, Dennis VA. Pseudomonas aeruginosa reference strains pao1 and pa14: A genomic, phenotypic, and therapeutic review. Front Microbiol. (2022) 13:1023523. doi: 10.3389/fmicb.2022.1023523
26. Elsinghorst EA. Measurement of invasion by gentamicin resistance. Methods Enzymol. (1994) 236:405–20. doi: 10.1016/0076-6879(94)36030-8
27. Iakobachvili N, Leon-Icaza SA, Knoops K, Sachs N, Mazères S, Simeone R, et al. Mycobacteria–host interactions in human bronchiolar airway organoids. Mol Microbiol. (2022) 117:682–92. doi: 10.1111/mmi.14824
28. Line L, Alhede M, Kolpen M, Kühl M, Ciofu O, Bjarnsholt T, et al. Physiological levels of nitrate support anoxic growth by denitrification of pseudomonas aeruginosa at growth rates reported in cystic fibrosis lungs and sputum. Front Microbiol. (2014) 5:554. doi: 10.3389/fmicb.2014.00554
29. Tang M, Liao S, Qu J, Liu Y, Han S, Cai Z, et al. Evaluating bacterial pathogenesis using a model of human airway organoids infected with pseudomonas aeruginosa biofilms. Microbiol Spectr. (2022) 10:e02408–22. doi: 10.1128/spectrum.02408-22
30. Cholon DM, Greenwald MA, Higgs MG, Quinney NL, Boyles SE, Meinig SL, et al. A novel co-culture model reveals enhanced cftr rescue in primary cystic fibrosis airway epithelial cultures with persistent pseudomonas aeruginosa infection. Cells. (2023) 12:2618. doi: 10.3390/cells12222618
31. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. Trim29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8:945. doi: 10.1038/s41467-017-00101-w
32. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for trim29 in the control of innate immunity in the respiratory tract. Nat Immunol. (2016) 17:1373–80. doi: 10.1038/ni.3580
33. Wang J, Lu W, Zhang J, Du Y, Fang M, Zhang A, et al. Loss of trim29 mitigates viral myocarditis by attenuating perk-driven er stress response in male mice. Nat Commun. (2024) 15:3481. doi: 10.1038/s41467-024-44745-x
34. Chakravortty D, Hensel M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infection. (2003) 5:621–7. doi: 10.1016/S1286-4579(03)00096-0
35. Gross TJ, Kremens K, Powers LS, Brink B, Knutson T, Domann FE, et al. Epigenetic silencing of the human nos2 gene: rethinking the role of nitric oxide in human macrophage inflammatory responses. J Immunol. (2014) 192:2326–38. doi: 10.4049/jimmunol.1301758
36. Palmieri EM, McGinity C, Wink DA, McVicar DW. Nitric oxide in macrophage immunometabolism: hiding in plain sight. Metabolites. (2020) 10. doi: 10.3390/metabo10110429
37. Choi H-S, Rai PR, Chu HW, Cool C, Chan ED. Analysis of nitric oxide synthase and nitrotyrosine expression in human pulmonary tuberculosis. Am J Respir Crit Care Med. (2002) 166:178–86. doi: 10.1164/rccm.2201023
38. De Rose V, Molloy K, Gohy S, Pilette C, Greene CM. Airway epithelium dysfunction in cystic fibrosis and copd. Mediators Inflammation. (2018) 2018:1309746. doi: 10.1155/2018/1309746
39. Meng QH, Polak JM, Edgar AJ, Chacon MR, Evans TJ, Gruenert DC, et al. Neutrophils enhance expression of inducible nitric oxide synthase in human normal but not cystic fibrosis bronchial epithelial cells. J Pathol. (2000) 190:126–32. doi: 10.1002/(sici)1096-9896(200002)190:2<126::Aid-path500>3.0.Co;2-#
40. Moeller A, Horak F Jr., Lane C, Knight D, Kicic A, Brennan S, et al. Inducible no synthase expression is low in airway epithelium from young children with cystic fibrosis. Thorax. (2006) 61:514–20. doi: 10.1136/thx.2005.054643
Keywords: airway organoids, Pseudomonas aeruginosa, iNOS, airway epithelia, innate immunity
Citation: Grubwieser P, Böck N, Soto EK, Hilbe R, Moser P, Seifert M, Dichtl S, Govrins MA, Posch W, Sonnweber T, Nairz M, Theurl I, Trajanoski Z and Weiss G (2024) Human airway epithelium controls Pseudomonas aeruginosa infection via inducible nitric oxide synthase. Front. Immunol. 15:1508727. doi: 10.3389/fimmu.2024.1508727
Received: 09 October 2024; Accepted: 05 November 2024;
Published: 03 December 2024.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Catherine Chaput, NEC Laboratories Europe, GermanyCuncai Guo, Washington University in St. Louis, United States
Copyright © 2024 Grubwieser, Böck, Soto, Hilbe, Moser, Seifert, Dichtl, Govrins, Posch, Sonnweber, Nairz, Theurl, Trajanoski and Weiss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Günter Weiss, Z3VlbnRlci53ZWlzc0BpLW1lZC5hYy5hdA==; Zlatko Trajanoski, emxhdGtvLnRyYWphbm9za2lAaS1tZWQuYWMuYXQ=
†These authors have contributed equally to this work