 Lin Lin
Lin Lin Yumeng Lin
Yumeng Lin Zhongyu Han
Zhongyu Han Ke Wang5†
Ke Wang5† Siyu Wang
Siyu Wang Haoran Chen
Haoran Chen- 1School of Medical and Life Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2Nanjing Tongren Hospital, School of Medicine, Southeast University, Nanjing, China
- 3School of Medicine, Southeast University, Nanjing, China
- 4Science Education Department, Chengdu Xinhua Hospital Affiliated to North Sichuan Medical College, Chengdu, China
- 5Department of Science and Education, Deyang Hospital Affiliated Hospital of Chengdu University of Traditional Chinese Medicine, Deyang, China
- 6Department of Radiology, Zhongda Hospital, Nurturing Center of Jiangsu Province for State Laboratory of AI Imaging & Interventional Radiology, School of Medicine, Southeast University, Nanjing, China
- 7Department of Respiratory and Critical Care Medicine, The First People’s Hospital of Lianyungang, Lianyungang, China
- 8Department of Preventive Medicine, Kunshan Hospital of Chinese Medicine, Kunshan, China
Lung disease development involves multiple cellular processes, including inflammation, cell death, and proliferation. Research increasingly indicates that autophagy and its regulatory proteins can influence inflammation, programmed cell death, cell proliferation, and innate immune responses. Autophagy plays a vital role in the maintenance of homeostasis and the adaptation of eukaryotic cells to stress by enabling the chelation, transport, and degradation of subcellular components, including proteins and organelles. This process is essential for sustaining cellular balance and ensuring the health of the mitochondrial population. Recent studies have begun to explore the connection between autophagy and the development of different lung diseases. This article reviews the latest findings on the molecular regulatory mechanisms of autophagy in lung diseases, with an emphasis on potential targeted therapies for autophagy.
Introduction
Pulmonary diseases, especially chronic pulmonary diseases, including chronic obstructive pulmonary disease (COPD), pulmonary tuberculosis (PTB), and lung cancer, pose significant threats to human health. Despite notable advancements in research globally in recent years, effective and precise treatments are still insufficient, leaving many lung diseases without a cure.
Autophagy is a common phenomenon in eukaryotic cells that fuses with lysosomes and hydrolyzes intramembrane components by encasing damaged or functionally degenerated organelles and certain proteins and certain macromolecules. Autophagy was first identified in the 1850s and named in 1963 by de Duve et al (1). Recent research has indicated that autophagy is important for maintaining cellular survival and homeostasis (2–4). Through the processing of metabolic precursors from cytoplasmic substrates, this process maintains homeostasis in healthy respiratory cells and ensures survival in conditions of nutrient scarcity (5). In nutrient deficiency, cells acquire nutrients through autophagy; damaged or senescent organelles can be removed by autophagy when cells are damaged or senescent; and these microorganisms or toxins can be cleared by autophagy when cells are infected by microorganisms or invaded by toxins. Eukaryotes have well-preserved degradation and recycling processes critical to maintaining cellular homeostasis and coping with stress. To some extent, autophagy is an effective mechanism to protect cells.
Autophagy is intricately associated with the clearance of organelles and, more significantly, plays a crucial part in the development and progression of various diseases. The relationship between autophagy and disease pathogenesis has not been fully confirmed. Nonetheless, a growing body of evidence indicates that autophagy may play a significant role in various human diseases (2, 6), including inflammatory diseases (7–9), cardiovascular diseases (10, 11), neurodegenerative diseases (12), and cancer (13) (Figure 1). Alterations in autophagic activities may also result from variations in the activation of proteins that regulate autophagy (2, 14). Until now, only limited studies have investigated the role of autophagy in lung disease Figure 2.
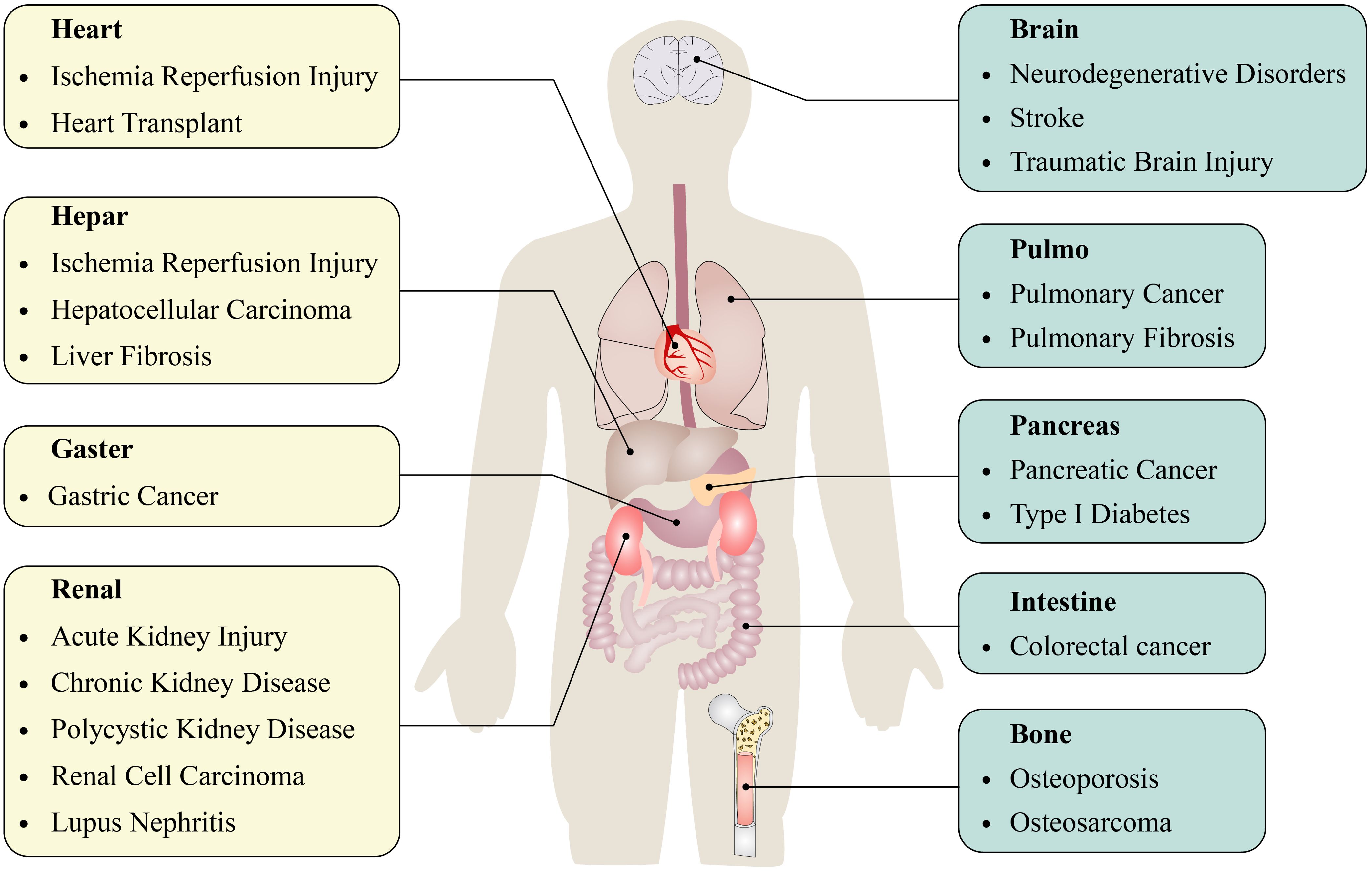
Figure 1. Autophagy is involved in the development and progression of multiple diseases.
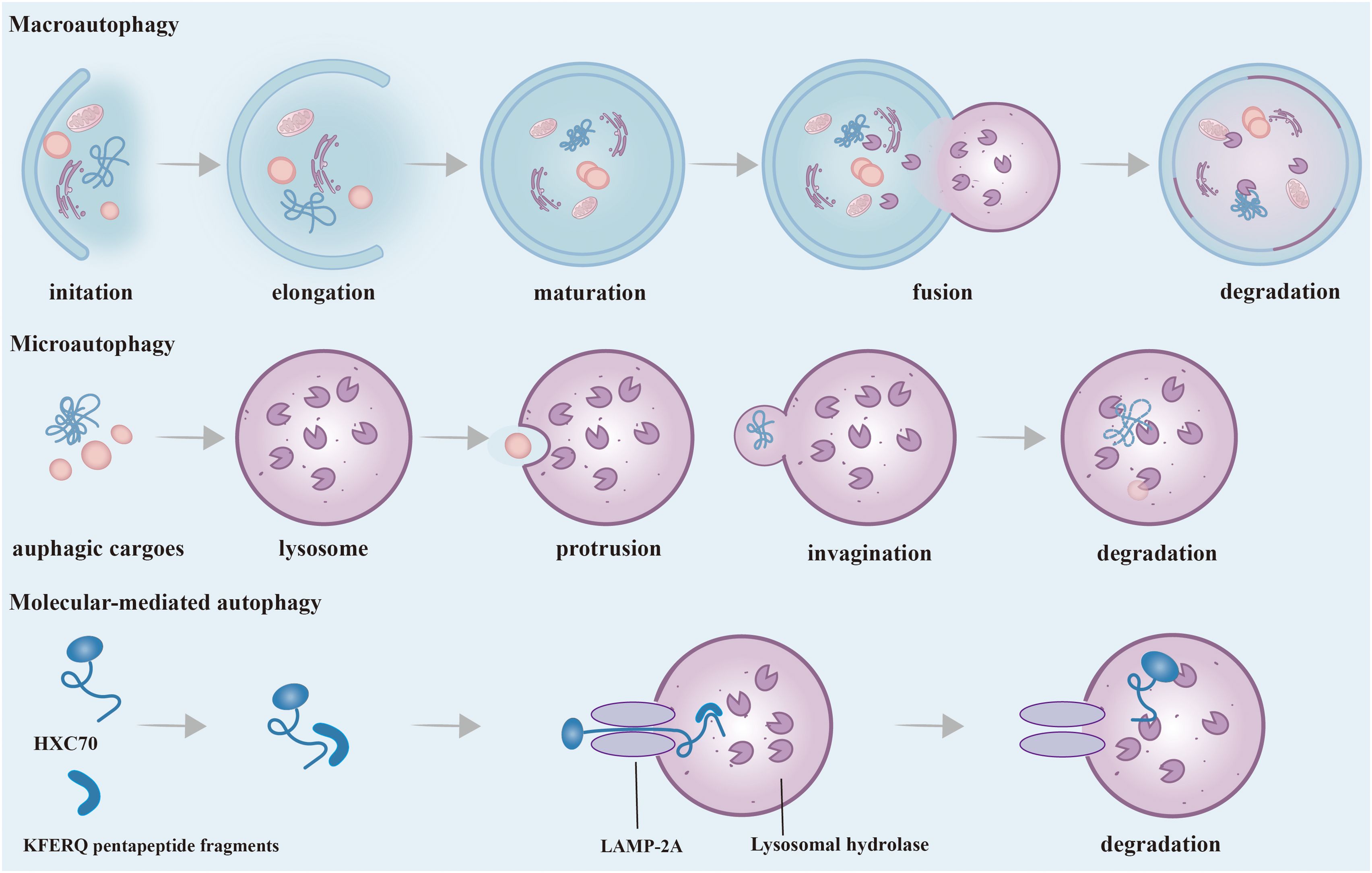
Figure 2. Phases and Classification of Autophagy. According to the different ways of transporting substrates to lysosomes, autophagy can be divided into three main ways: macroautophagy, microautophagy and CMA. Macroautophagy: It starts as autophagy-related substances accumulate around misfolded and aggregated proteins, pathogens, non-essential amino acids, etc. to form a barrier membrane. Dysfunctional organelles as well as proteins are surrounded by an isolation membrane and gradually form a bilayer membrane structure, called autophagosomes. The outer membrane of autophagosomes then fuses with lysosomes, and internal material is degraded in autolysosomes. Microautophagy: The process by which membranes of lysosomes encapsulate cargo by direct protrusion or invagination and are degraded in lysosomes. CMA: Substrate proteins containing the KFERQ-like pentapeptide sequence are first recognized by HSC70, then bind to LAMP-2A on the lysosomal membrane and enter the lysosome and eventually are degraded. CMA, Chaperone-mediated autophagy; HSC70, Heat shock cognate protein 70; LAMP-2A, Lysosomal membrane-associated protein 2A.
This review highlights the most recent developments in the molecular control and the role of autophagy in lung diseases. Additionally, we explore how autophagy-related proteins and regulatory processes may contribute to either the protection against or the advancement of human lung diseases, offering new insights for targeted treatment options.
Phases and classification of autophagy
Autophagy is essential for the process of protein degradation with relatively short half-lives. Morphologically, a significant quantity of dissociative membranous structures appears in the cytoplasm of cells that are about to undergo autophagy, which are called proautophagosomes. The proautophagosome gradually develops into a vacuole with a double membrane structure, which is surrounded by degraded organelles and some cytoplasm (2, 15). This double membrane structure is referred to as the autophagosome (2). Next, after autophagosomes fuse with lysosomes, the inner membranes and their encapsulated substances enter the lysosome and undergo hydrolysis by lysosome enzymes. The lysosomes found in this phagocyte are called autolysosomes. This process leads to the retrieval of soluble cytoplasmic proteins, mitochondria, peroxides, Golgi complexes, and portions of the endoplasmic reticulum, while some digested fragments are released into the cytoplasm for biosynthesis (3, 5, 16).
According to the different ways of transporting substrates to lysosomes, autophagy can be divided into three main ways: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (17). Macroautophagy is the most common autophagy in eukaryotic cells by forming a double-layer membrane around misfolded and aggregated protein pathogens, and non-essential amino acids, and fusing with lysosomes for degradation. Many stresses, such as nutritional deficiency, infection, oxidative stress, and toxic stimulation, can stimulate the occurrence of macroautophagy, which is generally referred to as autophagy. Different from macroautophagy, there is no formation process of autophagy membrane in microautophagy. A characteristic aspect is that the lysosome membrane is straight taken in by lysosomes and late endosomes via membrane protrusion and invagination, and it is then broken down within the endolysosomal lumen. During the dependent multivesicular body (MVB) formation, a significant quantity of cytoplasmic proteins is selectively integrated into the lumens of endosomes in substantial amounts (18). CMA represents a highly selective mechanism of autophagy with two core members: the heat shock cognate protein 70 (HSC70) and the lysosomal membrane-associated protein 2A(LAMP-2A). HSC70 is a molecular chaperone protein. The process of CMA degrades proteins that contain KFERQ pentapeptide fragments in the peptide chain. First, the heat shock protein HSC70 specifically recognizes and binds to proteins containing KFERQ five-peptide fragments, and transports the target protein into the lysosome for degradation through interaction with LAMP2A (17). Macroautophagy is considered to be the predominant form of autophagy compared to microautophagy and molecular-mediated autophagy, and this has also been the subject of extensive research. Therefore, what we usually call “autophagy” is macroautophage.
In addition, autophagy can be classified into selective autophagy, aggregative autophagy, and xenophagy, etc. Recent research has demonstrated that several denatured proteins, organelles, and certain bacteria can be selectively destroyed by autophagy. This process is called “selective autophagy”, the most representative of which is mitophagy (17, 19, 20). Mitophagy is a specific degradation targeting depolarized mitochondria (21). Xenophagy involves the digestion of extracellular components containing pathogens or bacteria that invade the body (22).
Molecular involvement in autophagy and molecular regulation
The process of autophagy is modulated and governed by various relative proteins. In mammalian cells, starvation-induced autophagy is regulated by approximately 20 core Atg genes (23). These gene products are persistently incorporated into vacuoles and assembled to construct pre-autophagosomes. In addition, the modification of microtubule-associated protein-1 light chain 3 (LC3) is an important step in forming autophagic vacuole. In autophagosomes, LC3 and its homologues act on autophagic substrates or proteins to facilitate the selection of autophagic cargoes (24).
The elongation stage of autophagosome formation relies on two ubiquitin-like conjugation systems (Figure 3), (2, 3). Besides the proteins mentioned in Figure 3, the maturation and fusion of autophagosomes also depend on various other proteins, such as small GTPases (like Rab7), class C Vps proteins, ultraviolet radioresistance-associated gene protein (UVRAG), and lysosome-associated membrane proteins (for instance, LAMP-2A) (25, 26). In recent years, additional proteins associated with autophagy have been progressively identified alongside the aforementioned proteins. In a complicated regulatory network, these proteins regulate the initiation and execution of autophagy (2, 27). We will describe this in detail in the following paragraphs (Figure 3).
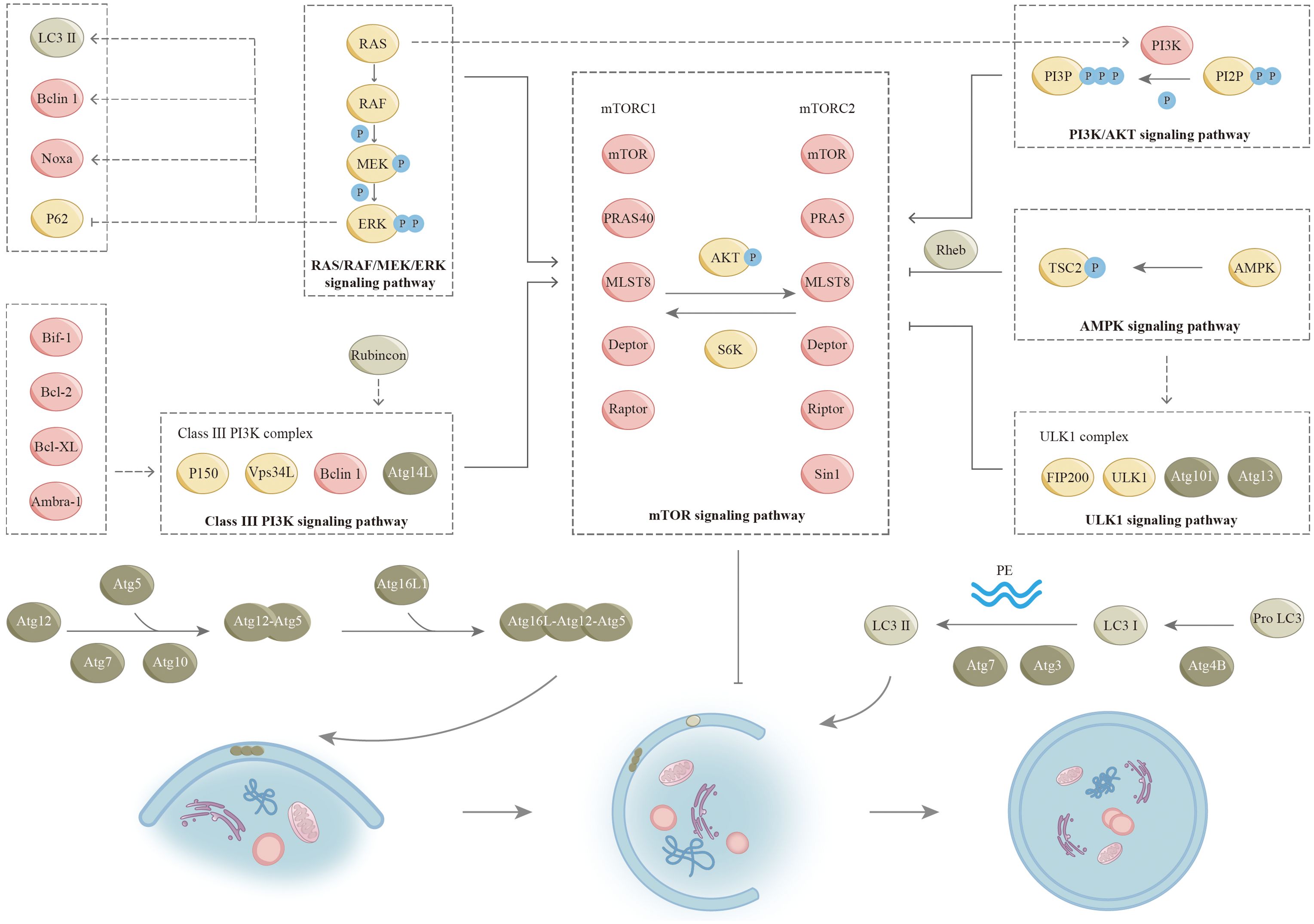
Figure 3. Signaling pathways for autophagy. The process of autophagy is regulated by many signaling pathways (as shown), and there is also complex crosstalk between various pathways. Two ubiquitin-like conjugation systems involved in the formation of autophagosome: In the first system, the ubiquitin-like protein Atg12 is enzymatically coupled to Atg5 by Atg7 (E1 ubiquitin-activating enzyme-like) and Atg10 (E2 ubiquitin-conjugating enzyme-like) to produce the Atg5-Atg12 complex. The Atg5-Atg12 complex interacts with Atg16L1 to form a complex that plays a role in the formation of autophagic membranes. As part of the maturation process, these factors are separated from autophagosomes. The second coupling system requires the ubiquitin-like protein LC3. LC3 and its homologues, including the isozymes of LC3 and associated proteins (e.g., GABARAP), are modified by cellular lipid PE. An important regulatory step in the formation of autophagosomes is the transformation of LC3-I (free form) to LC3-II (PE conjugated form). The precursor form of LC3 is cleaved by the protease ATG4B to yield LC3- I (not shown). ATG7 and ATG3 participate in conjugating PE with LC3-I to LC3-II. LC3-II cytoplasmic redistribution, characterized by punctate LC3 staining, is indicative of autophagosome formation. GABARAP, (GABA type A receptor-associated protein); LC3, (Microtubule-associated protein 1 light chain 3; PE, (Phosphatidylethanolamine).
Mammalian target of rapamycin signaling pathway
Many studies have demonstrated that mTOR negatively regulates autophagy in nutrient-rich environments (28). mTOR is an atypical serine/threonine protein kinase that is evolutionarily relatively conserved. Cell cycle regulation, proliferation, differentiation, motility, and invasion are among its physiological functions. Two unique complexes, mTORC1 and mTORC2, exist within the cell and are distinguished by distinct components. mTORC1 and mTORC2 are two signaling complexes that play a major role in the mTOR pathway. Ribosomal protein S6 kinase (S6K) and kinase B (AKT) are key enzymes in the interaction between mTORC1 and mTORC2. S6K is activated by mTORC1, which subsequently activates mTORC2. Conversely, mTORC2 facilitates the phosphorylation of AKT, leading to the activation of mTORC1. mTORC1 is responsive to energy levels and stress, and it is significantly inhibited by rapamycin. A substantial body of research has indicated that mTORC1 exerts an inhibitory influence on the process of autophagy (29, 30). Unlike mTORC1, mTORC2 is not susceptible to both rapamycin and nutrients because of the presence of Rictor (31). However, long-term rapamycin treatment ultimately inhibits mTORC2 activity (32). mTOR is a key molecule during autophagy induction. Many signaling pathways have the capacity to either promote or inhibit the process of autophagy through their interactions with mTOR (Figure 3). Nonetheless, the enhancement or suppression of autophagy by these pathways is not definitive. In some specific cases, the opposite effect may also be exerted. We will describe it further in the following sections.
mTOR is integral to numerous physiological functions, such as cell proliferation, survival, and autophagy, which is intricately linked to various lung diseases through its regulatory effects on cell growth, inflammation, and fibrosis. We will describe it in the following sections. The mTOR signaling pathway plays a critical role in the development and maintenance of lung function. It regulates the growth and differentiation of lung epithelial cells and fibroblasts to maintain normal lung function. Moreover, mTOR signaling is involved in the immune response to pulmonary pathogens, which regulates the activation of immune cells and the inflammatory response. Given its central role in lung diseases, mTOR signaling has become a target for therapeutic intervention.
The phosphoinositide-3-kinase protein/kinase B signaling pathway
PI3K/AKT was discovered in the 1980s and plays an important role in major physiological activities of cells (33). PI3K-AKT signaling mainly involves two metabolites, phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-bisphosphate (PIP3), and two coding genes, lipid phosphatase (PTEN) and 3-phosphoinositide-dependent protein kinase-1 (PDK1). PIP2 is converted to PIP3 by phosphorylation in response to PI3K. Next, PIP3 on autophagosome membranes recruits ATG18 and binds to bilayer membranes, allowing autophagosomes to extend and complete (34). PDK1 is a key regulatory molecule of the PI3K-AKT signal transduction pathway and plays an important part in the activation of AKT (35–37). In addition, mTORC2 can directly activate AKT by phosphorylating Ser-473 (36). PTEN is an important negative regulator of PIP2 conversion to PIP3. PTEN acts to promote dephosphorylation of PIP3 thereby inhibiting its accumulation in cells (38). Once activated, AKT acts on various cytoplasmic proteins to mediate cell growth and survival. The main downstream effector is mTOR. Furthermore, AKT influences the interaction between phosphorylated tuberous sclerosis complex 1 (TSC1) and phosphorylated tuberous sclerosis complex 2 (TSC2), consequently facilitating the activation of mTORC1 via the H-Ras-like GTPase (Rheb) (39). Subsequently, active mTORC1 inhibits autophagy by blocking the uncoordinated 51-like protein kinase (ULK1) (40).
The PI3K/AKT signaling has a tight relationship in regulating cell growth, survival, and metabolism. This pathway is involved in numerous cellular processes and has significant implications for various lung diseases. In certain pathological conditions, the PI3K/AKT signaling pathway is frequently activated in reaction to inflammatory stimuli and oxidative stress, which results in airway remodeling and contributes to the pathophysiology of the disease, including mucus hypersecretion and smooth muscle cell proliferation, therefore enhancing the survival of inflammatory cells in the lungs. The PI3K/AKT pathway presents multiple potential therapeutic targets for treating lung diseases. Inhibitors of PI3K, AKT, or associated pathways are currently undergoing investigation to reduce inflammation, fibrosis, and tumor growth in lung diseases.
RAS/RAF/MEK/ERK signaling pathway
As a significant signaling pathway of mitogen-activated protein kinase (MAPK), RAS/RAF/MEK/ERK is involved in regulating cell proliferation, differentiation, apoptosis, and numerous signaling pathways (41, 42). RAS is a small GTPase that is activated by several factors, including receptor tyrosine kinases, growth factors, heterotrimeric G proteins, integrins, serpentine receptors, and cytokine receptors. Furthermore, oxidative stress activates the RAS/RAF/MEK/ERK signaling pathway. Notably, certain growth receptors are not required for ROS-induced RAS activation (43). In addition, ROS can uncouple MAPK pathway activity from RAS expression (44). Activated RAS further recruits RAF (MAPKKK) to the plasma membrane for activation. Following this, RAF activates and phosphorylates MEK (MAPKK), followed by ERK (MAPK). As ERK is activated, it translocates to the nucleus and triggers transcription and expression of target genes (45, 46). The expression products of these genes regulate various physiological functions of cells, including the regulation of autophagy (45, 46). PI3K and TSC2 are regulated by the RAS/RAF/MEK/ERK pathway, thereby activating mTORC1 activity. In addition, the activated RAS/RAF/MEK/ERK signaling pathway up-regulates LC3, Beclin1, and Noxa, and directly down-regulates p62 to induce autophagy (47, 48). Following induction by lindane, the formation of autophagosomes within cells is closely linked to the prolonged activation of ERK (49). Notably, this phenomenon occurs independently of both mTOR and p38 (49). These seemingly contradictory findings indicate that specific environmental conditions may directly influence the regulation of autophagy via the RAS/RAF/MEK/ERK signaling pathway.
Dysregulation of this pathway has been implicated in various diseases, including lung cancer. Mutations in genes encoding components of this pathway, such as KRAS, BRAF, and MEK, are commonly found in lung cancer patients. Mutations in BRAF and MEK are also observed in a subset of lung cancer patients. Furthermore, aberrant activation of the RAS/RAF/MEK/ERK pathway has been linked to other lung diseases, such as pulmonary fibrosis and COPD. In these conditions, dysregulated signaling through this pathway can lead to inflammation, tissue remodeling, and fibrosis in the lungs.
Adenosine 5’monophosphate-activated protein kinase signaling pathway
AMPK is recognized as one of the primary substrates of LKB1 (liver kinase B1), which functions as an intrinsic energy sensor and regulator of cellular homeostasis (50, 51). AMPK is a heterotrimeric serine/threonine kinase that consists of a catalytic αsubunit and two regulatory subunits, which are β and γ. The activation of AMPK occurs in reaction to elevated levels of intracellular AMP and reduced levels of ATP, particularly during conditions of nutrients. LKB1 implements the involvement of this process by phosphorylating the α-activating loop (52). The activation of AMPK affects multiple processes, including mTOR pathway regulation and p53 phosphorylation (53). Further, AMPK is capable of directly phosphorylating Raptor or TSC2. Next, TSC2 signals to inhibit mTOC1 activity (44, 45, 54). In this pathway, AMPK negatively regulates mTORC1 by adenosine 5’ -monophosphate levels, thereby positively regulating autophagy upon energy depletion (55). Research indicates that AMPK exerts direct regulation over ULK1 in a manner that is sensitive to nutrient availability, thereby contributing to the intricate nature of regulatory mechanisms, as elaborated upon in the subsequent sections (56–58).
AMPK is a dominant y regulator of cellular energy metabolism and plays a crucial role in maintaining cellular homeostasis. Within the realm of pulmonary disorders, AMPK signaling has been demonstrated to exhibit both preventive and pathogenic effects. In several lung diseases, including COPD, asthma, and pulmonary fibrosis, dysregulation of AMPK signaling has been implicated. However, the relationship between AMPK signaling and lung disease is complex and disease-specific. Additional investigation is necessary to elucidate the specific mechanisms by which AMPK influences lung function, as well as to assess the feasibility of marking this pathway for therapeutic strategies.
Uncoordinated-51-like protein kinase signaling pathway
ULK1 is a master regulator of autophagy initiation among mTORC1 downstream regulatory targets (59). Among the components of autophagy, ATG1, ATG13, and ATG17 are critical regulators of autophagy initiation (44, 60–62). In mammals, ULK1 and ULK2 are homologues of ATG1, and mATG13 and 200 kDa adhesion kinase family interacting protein (FIP200) are homologues of ATG13 and ATG17, respectively (59, 63–66). The importance of ULK1 in the autophagy pathway is reflected in its involvement in forming mTOR substrate complexes (60, 66, 67). mTORC1 has been reported to inhibit its pre-autophagic effect by phosphorylating ULK1 under normal and nutrient-rich conditions (68). mTORC1 is also able to directly phosphorylate and inhibit ATG13, one of the activators of ULK1. ULK1 can activate autophagy by phosphorylating Beclin-1 indirectly involved in the formation of VPS34-Beclin-1-ATG14 (29, 69, 70). In addition, AMPK can directly interact with ULK1 to regulate ULK1 in a nutrient-sensitive manner. Activating ULK1 by phosphorylating Ser 317/Ser 777, AMPK acts by triggering autophagy in response to glucose and amino acid starvation (58). Interestingly, mTORC1 blocks the cellular collection between ULK1 and AMPK by phosphorylating Ser 757. Consequently, it can be inferred that ULK1 equips cells with the capacity to effectively respond to intricate environmental alterations in conjunction with mTORC1 and AMPK.
Recent studies denote that ULK1 signaling may be implicated in the development of several pulmonary disorders. In conditions like IPF, COPD, and lung cancer, dysregulation of autophagy, including ULK1 signaling, has been implicated in disease progression. Investigating the function of ULK1 signaling in lung diseases may facilitate the creation of targeted therapies designed to regulate autophagy and improve outcomes for patients with these conditions.
Type III phosphatidylinositol triphosphate kinase signaling pathway
Autophagosome formation is closely dependent on class III PI3K complexes. Activated class III PI3K complexes lead to increased PI3P formation, and PI3P-recruiting protein factors initiate autophagosome formation, including WD repeat protein interacting with inosine phosphate (WIPI-1/2), Atg18, and protein 1 containing double FYVE (DFCP1) (71, 72). Class III PI3K complexes exist in two distinct types in mammalian cells, where complexes consisting of VPS34L, p150, Beclin1, and ATG14L are closely associated with autophagy. We refer here to this complex collectively as the class III PI3K complex. Beclin 1 serves as a significant regulator of autophagy. It is also defined as a tumor suppressor protein, exhibiting the capacity to engage with a wide variety of proteins, including ATG14L, ultraviolet resistance-associated gene protein (UVRAG), Rubicon, and Bcl-2 (73–76). Three domains play important roles in Beclin1 function, including the Bcl-2 homology 3 (BH3) domain and the central coiled-coil domain (CCD) that mediates interactions with ATG14L and UVRAG (77–80). In addition, the active ULK1 results in the recruitment of class III PI3K complexes to autophagosomes, forming alternating Beclin 1-Vps34L complexes with UVRAG and promoting autophagy (75, 81–83). Evolutionarily conserved domain (ECD) mediates communication between Beclin 1 and VPS34, which in turn activates VPS34 kinase to regulate autophagosome formation. Furthermore, class III PI3K complexes can negatively regulate autophagy in response to the newly identified factor Rubicon (84, 85). Ambra-1, Bif-1, Bcl-2, and Bcl-XL can also act on class III PI3K complexes to modulate their activity (76, 86, 87).
In the context of lung disease, dysregulation of Class III PI3K signaling has been implicated in various respiratory conditions. In diseases like IPF, COPD, and lung cancer, altered Class III PI3K signaling has been associated with disease pathogenesis and progression. Understanding the role of Class III PI3K signaling in lung diseases is important for identifying potential therapeutic targets and developing targeted interventions to modulate this pathway for the treatment of respiratory disorders.
Wild-type p53 signaling pathway
p53 functions as a tumor suppressor protein and serves as a transcription factor that regulates gene networks in response to various cellular stresses, thereby maintaining genome stability and integrity. However, p53 not only prevents tumorigenesis but also plays a critical regulatory part in primary signaling and metabolic adaptation (88, 89). The effect of wild-type p53 on autophagy is complex, highly dependent on the environment, and determined by the cellular microenvironment and stressful conditions. The progression of the cell cycle and the subcellular localization of p53 serve dual functions in the regulation of autophagy. The dual effect of wild-type p53 on autophagy is reflected in its transcriptional activity against a range of downstream target genes with autophagic regulatory effects. The dual role of p53 in autophagy is presented in Table 1.
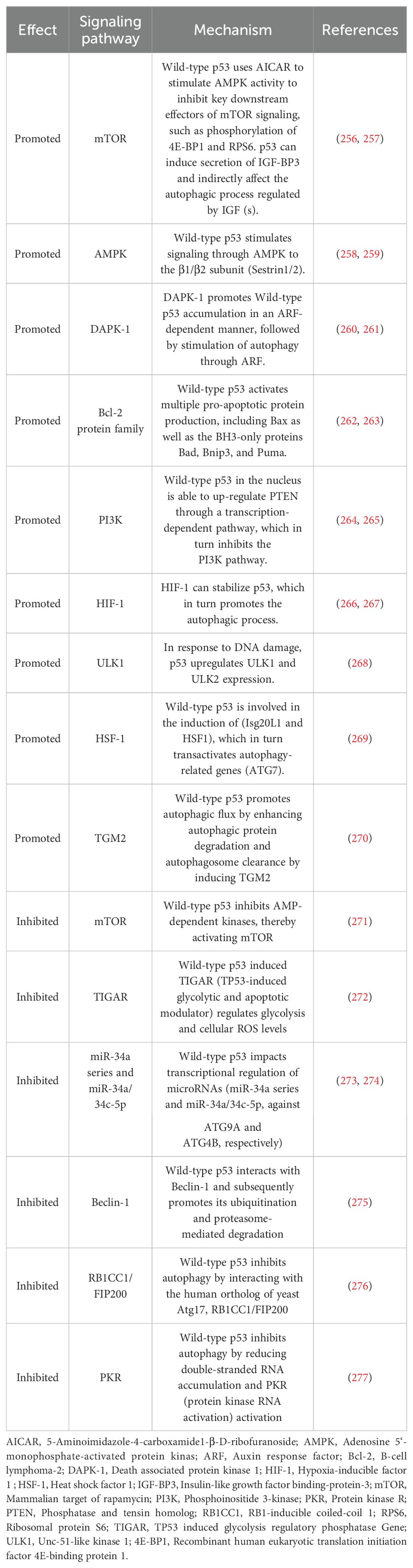
Table 1. The dual effects of wild-type p53 on autophagy.
p53 signaling plays a significant role in the development and progression of various lung diseases. Mutations in the p53 gene can disrupt its tumor-suppressor function, leading to uncontrolled cell growth, evasion of cell death, and genomic instability, all of which are hallmarks of cancer. Dysregulated p53 signaling has been linked to a poorer prognosis in lung cancer patients and resistance to certain anticancer therapies. Dysregulation of p53 contributes to abnormal repair processes in the lung tissue, leading to excessive collagen deposition, fibrosis, and impaired lung function. Moreover, activation of p53 can promote cell cycle arrest, DNA repair, or apoptosis, depending on the extent of damage while disruption of p53 function may impair the lung’s ability to repair and regenerate, exacerbating lung injury and contributing to disease progression.
Function of autophagy
Autophagy serves as a mechanism for maintaining a stable pool of organelles by regenerating metabolic precursors and eliminating subcellular debris in response to diverse environmental stressors. In the presence of such stress, autophagy initiates cellular defense mechanisms by facilitating the removal of damaged organelles and ubiquitinated protein aggregates (90, 91). Under specific conditions of glucose or amino acid starvation, autophagy is compensatory to participate in the basic metabolic cycle of cells by acting on intracellular proteins, lipids, and other organic macromolecules (5). Specifically, autophagy plays a very important role in apoptosis, inflammation, and immunity. We will describe this in detail below.
Autophagy in apoptosis
Cells can undergo apoptosis in response to intracellular signaling, extracellular signaling, and endoplasmic reticulum (ER) stress (Figure 4). Cysteine protease (caspase) induction and activation play a critical role in apoptosis. Endogenous apoptosis is also known as the mitochondrial pathway due to a mechanism closely related to the permeability of the mitochondrial membrane. This process is also strongly associated with the Bcl-2 protein family. Bcl-2, Bcl-XL, and Mcl-1 are negative regulators of apoptosis and protect cells from apoptosis when multiple types of cells are stimulated. Bax and Bak can undergo apoptosis by penetrating the mitochondrial membrane, releasing cytochrome c, and subsequently activating caspases. However, the exact mechanism by which these proteins promote apoptosis is unknown. Exogenous apoptosis requires the formation of a critical complex, the death-inducing signaling complex (DISC). Death receptors, including Fas, TNFR1, and TRAIL, are located on the cell surface and mediate apoptosis when activated. The production of DISC is initiated by the binding of death receptors to their corresponding ligands. When Fas binds to its ligand, activated Fas forms a DISC by binding connexin to the death domain (FADD). DISC then binds recruited pro-caspase 8 by interacting with another motif called the death effector domain (DED). Next, Pro-caspase 8 dimerizes and gains catalytic activity after degrading downstream substrates, producing and releasing heterotetrameric active caspase 8. Eventually, cells undergo extrinsic apoptosis. The ER stress pathway involves the buildup of incorrectly folded or unfolded proteins within the ER, which can arise from various factors such as infection, hypoxia, starvation, chemical influences, and deviations from homeostatic regulation of ER secretory functions. This accumulation leads to ER stress-induced apoptosis and triggers the unfolded protein response (UPR) pathway in response to the misfolding of proteins within the ER.
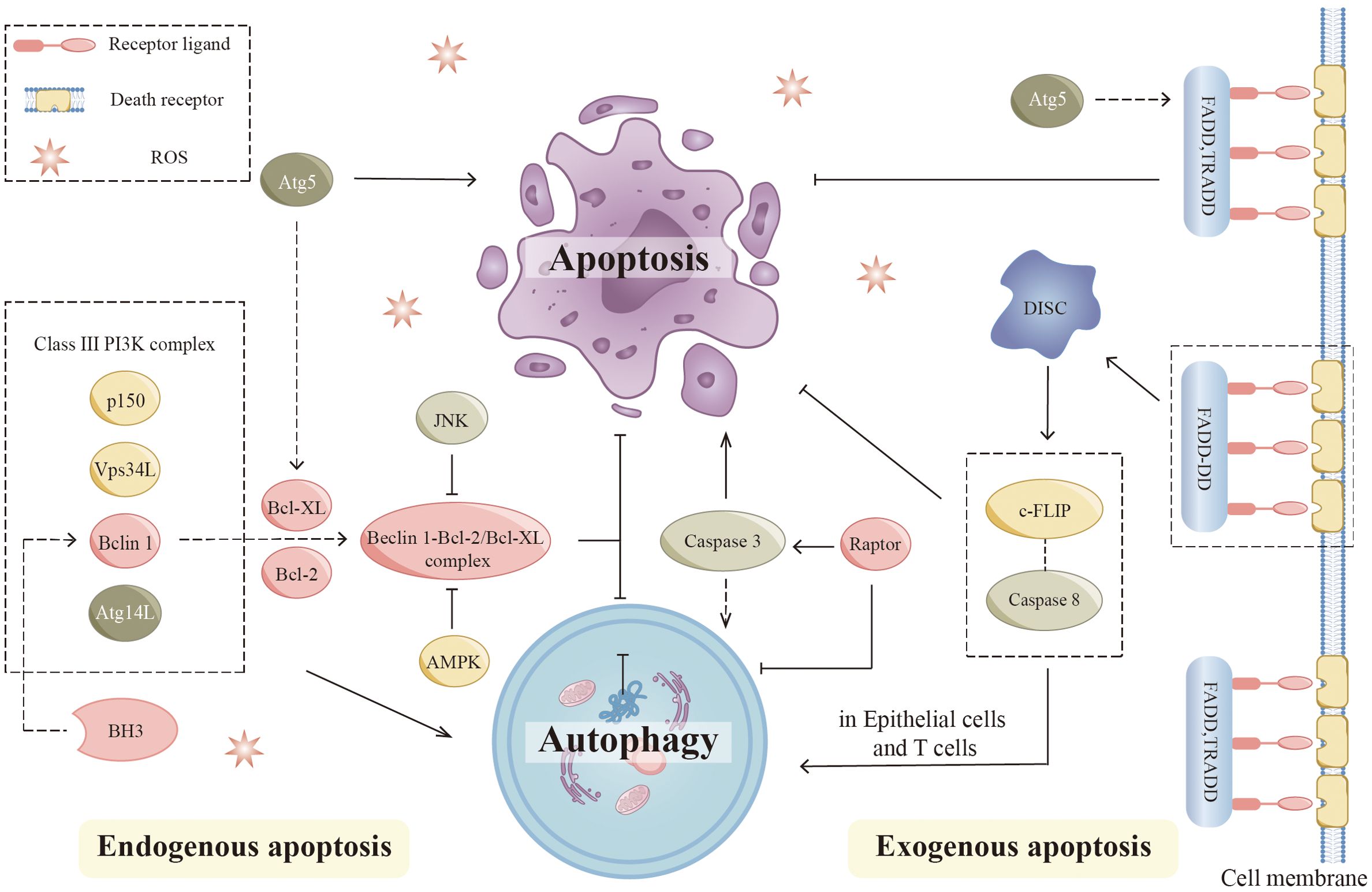
Figure 4. Autophagy and apoptosis. In endogenous apoptosis, the interaction of autophagic proteins with apoptotic proteins regulates this process. Bcl-2 family members, including Bcl-2 and Bcl-XL, can directly interact with Beclin 1 by binding to the BH3 domain. The JNK pathway promotes autophagy by preventing the association between Beclin 1 and Bcl-2 family proteins. AMPK also dissociates the Bcl-2-Beclin1 complex and promotes Beclin1-PI3K complex formation. Apoptosis signaling pathways may be affected by various autophagic proteins such as Atg5. Proteolytic fragments of Atg5 are able to promote apoptosis by inhibiting Bcl-XL. In extrinsic apoptosis, key components of DISC regulate autophagy during this process. Apoptosis and autophagy are affected by mutations in FADD, which create DD. The mutant (FADD-DD) was recruited to DISC in the absence of DED. By interacting with caspase 8 precursor and c-FLIP, this domain prevents the development of death receptor-induced apoptosis, while it can lead to excessive autophagy in epithelial cells and T cells. Atg5 can form a complex with FADD to affect the apoptosis process. AMPK, (Adenosine 5’-monophosphate-activated protein kinase); Bcl-2, (B-cell lymphoma-2); BH3, (Bcl-2 homolog3r); Caspase, (Cysteine protease); c-FLIP, (Cellular FADD-like IL-1β-converting enzyme-inhibitory protein); DD, (Death domain); DED, (Death effector domain); DISC, (Death-inducing signaling complex); JNK, (c-Jun-NH2-terminal kinase).
Recent research has indicated a strong connection between autophagy and apoptosis (92, 93). According to different experimental models, autophagy is associated with anti-apoptotic and pro-apoptotic effects (94). Several signaling mechanisms interact between apoptosis and autophagy. Autophagy proteins are involved in the regulation of apoptosis, while apoptotic proteins also influence the process of autophagy (95). Bcl-2 family members Bcl-2 and Bcl-XL, can directly interact with Beclin 1 by binding to the BH3 domain in the intrinsic apoptotic pathway (96, 97). Further studies showed that the anti-autophagic function of Bcl-2 mainly occurs in the ER and stabilizes Beclin-1 interaction with Bcl-2 through its 2Fe-2S cluster binding to Bcl-2. The c-Jun-NH2-terminal kinase (JNK) pathway is closely linked to apoptosis signaling. The JNK pathway can regulate the function of autophagy by affecting several key proteins (98). The JNK pathway promotes autophagy by preventing the association between Beclin 1 and Bcl-2 family proteins (99). In addition, AMPK can dissociate the Bcl-2-Beclin1 complex and promote the formation of the Beclin1-PI3K complex (100). Notably, mTOR is key in linking apoptosis and autophagy. It has been shown that loss of Raptor activates caspase 3, leading to mitochondrial abnormalities, which positively regulate apoptosis and autophagy (101).
Additionally, there is a complex link between the extrinsic apoptotic pathway and autophagy. Critical components of DISC regulate autophagy in this process. Excess autophagy occurs in fibroblasts, macrophages, and T cells when caspase 8 is inhibited or deficient (102, 103). DED is a protein interaction domain that can be found in pro-caspases and proteins in the apoptotic cascade that regulate caspase activation. Apoptosis and autophagy are also affected by FADD mutations, which produce abnormal death domains (DD). Mutants (FADD-DD) were recruited to DISC without DED. By interacting with pro-caspase 8 and cellular FADD-like IL-1β converting enzyme inhibitor protein (c-FLIP), this domain prevented the development of death receptor-induced apoptosis. Besides, it can cause excessive autophagy in epithelial cells and T cells (102, 104). Exogenous apoptotic signaling pathways can be affected by several autophagic proteins such as Atg5 (105). The knockdown of Atg5 exerts different effects on cell survival under different study conditions (106). Typically, proteolytic fragments of Atg5 can promote apoptosis by inhibiting Bcl-XL (107).
Autophagy in inflammation and immunity
The autophagy process influences immune and inflammatory responses in many diseases (Figure 5) (7). There is a complex interrelationship among autophagy, immunity, and inflammation. Autophagic proteins play a role in inducing and suppressing immune and inflammatory responses. Similarly, immune and inflammatory signals play a role in inducing and inhibiting autophagy. Autophagy provides new insights into the prevention and treatment of infectious, autoimmune, and inflammatory diseases by balancing the benefits and drawbacks of immune and inflammatory responses.
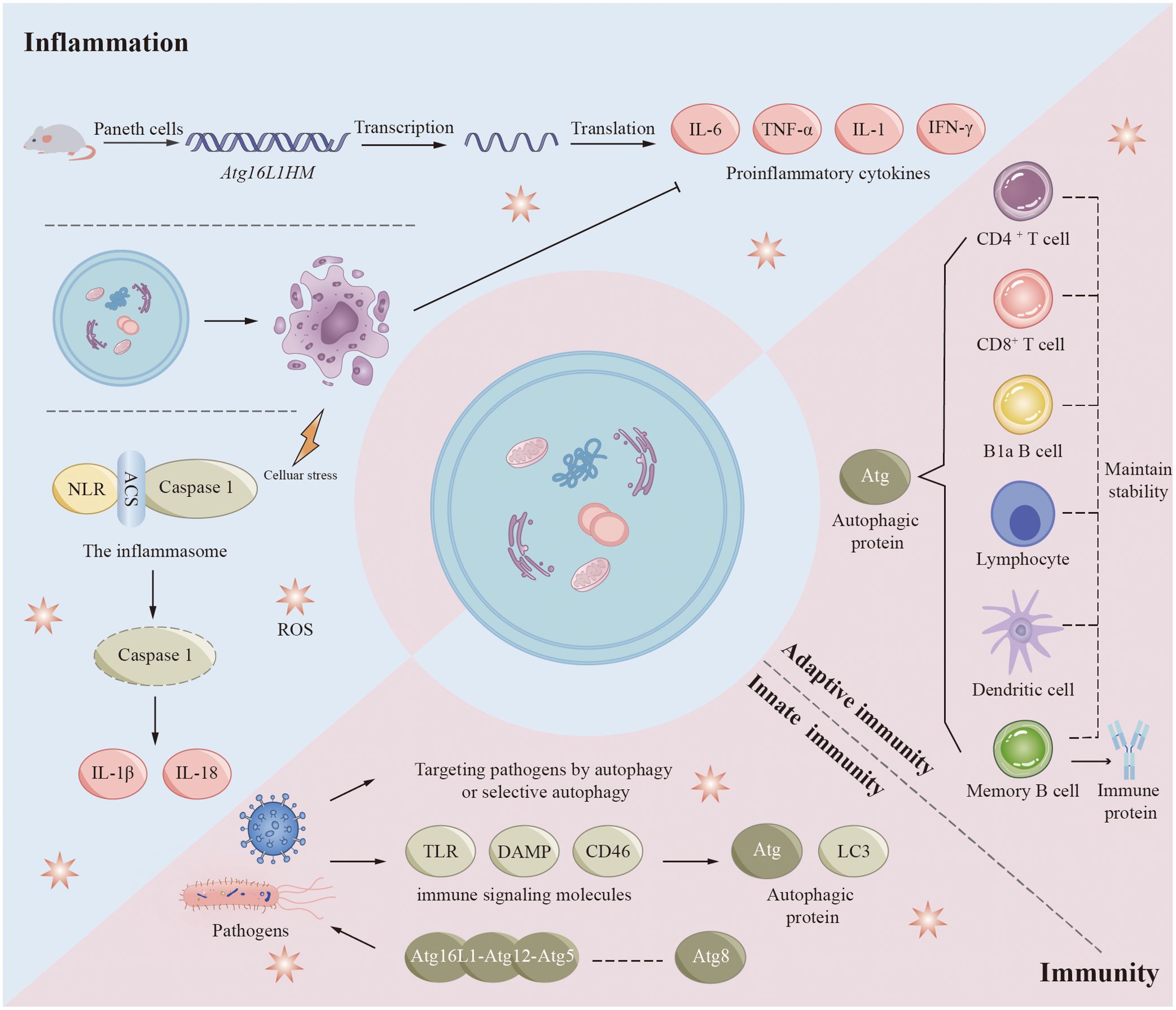
Figure 5. Autophagy in inflammation and immunity. Autophagy proteins play a role in inducing and suppressing immune and inflammatory responses, and immune and inflammatory signals play a role in inducing and inhibiting autophagy. Autophagic proteins play an important role in adaptive immunity, mainly including maintaining the normal number and function of immune-related cells such as B1a B cells, CD4+ T cells, and CD8+ T cells. Autophagy also plays a role in innate immunity when pathogens such as bacteria and viruses invade the human body. However, some pathogens are able to achieve their own survival by inhibiting, evading or even utilizing the autophagic process. Autophagy pathways and associated proteins also play crucial roles in regulating inflammatory responses. Increased transcription of pro-inflammatory cytokines and adipokines has been observed in mouse Paneth cells (Atg16L1HM), which contribute to the development of inflammation. Inflammasomes are important substances in the development of inflammation, and inflammasomes activated by various factors mediate the degradation and activation of caspase-1 and ultimately promote the synthesis and secretion of inflammatory factors (IL-1β and IL-18). Autophagy also removes cell debris generated by apoptosis, which in turn inhibits tissue inflammation.
Autophagy regulates various immune responses during infection. In many experiments, we have found that mutations in autophagy genes increase susceptibility to certain diseases (108–112). Studies performed on human genetics have revealed important clues regarding xenophagy, autophagic proteins that affect pathogen replication or survival, and the general immune system. Numerous studies have demonstrated the significance of autophagy in the human cellular defense mechanisms against mycobacterial infections (113). Autophagy genes play a significant role in regulating host genes for Mycobacterium tuberculosis (Mtb) replication (114). Autophagy may be a crucial component of TB drug resistance. At the same time, to survive in vivo, some viruses and bacteria have evolved different methods of adaptation to autophagy. They can prevent the occurrence of autophagy by inhibiting the foremost steps of autophagy or/and the production of autophagosomes, avoid protein modification or interfere with the recognition of autophagy by autophagy signaling, and even promote self-replication and survival using autophagy-related proteins. For example, human immunodeficiency virus (HIV), Kaposi’s sarcoma-associated herpes virus inhibits antiviral capacity and immune properties in vivo by affecting key pathways of autophagy. HIV envelope proteins activate mTOR signaling and prevent HIV transfer to CD4 + T cells. Kaposi’s sarcoma-associated herpesvirus prevents LC3-II production by interacting with Atg3 (115). Bacteria also have multiple strategies to avoid degradation. By disguising themselves, several bacteria can evade autophagic recognition in the cytoplasm. For example, VirG is a protein present on the bacterial surface and is necessary for Shigella to be targeted by autophagosomes. Atg5 can prevent its interaction with VirG by competitively binding to IcsB, an effector of Shigella (116). Several cytoskeletal proteins of cells are ActA-dependent (117). This feature allows bacteria to masquerade as their host organelle (117). Listeria protein ActA interacts with the intracytoplasmic actin polymerization machinery, thereby blocking binding to ubiquitin, recruitment of p62, and autophagy targeting (117). Several pathogens are also able to benefit themselves using components of autophagy in membrane trafficking, including poliovirus, rotavirus, coronavirus, dengue virus, and hepatitis B and C viruses (113, 118).
Autophagy is also regulated by immune signaling molecules, including innate and adaptive immunity. Although the regulatory mechanism of autophagy by most immune-related signaling molecules is currently unknown, some findings provide clues. NOD1 and NOD2, two typical NLRs (NOD-like receptor cryopyrin protein), can be activated by specific components of bacterial peptidoglycan. In response to bacterial infection, activated NOD1 and NOD2 interact with ATG16L1 to induce autophagy (119). NOD2 mutations associated with Crohn’s disease have been found to influence ATG16L1 recruitment and bacterial co-localization with LC3 (119). Presumably, in innate immunity, the ATG5-ATG12-ATG16L1 complex interacts with members of the ATG8 family and may stimulate pathogen-induced autophagy or enhance the ability of selective autophagy to target pathogens (120). Various cytokines, including but not limited to CLCF1, LIF, IGF1, FGF2, and the chemokine SDF1 (also called CXCL12) may have a broader role in controlling autophagy (121). Autophagy also plays a crucial role in adaptive immunity. Multiple regulatory pathways of autophagy possess the capacity to affect both the functionality and stability of the immune system, in addition to influencing antigen presentation. B1a B cells, CD4 + T cells, CD8 + T cells, and fetal hematopoietic stem cells rely on autophagic proteins to maintain their numbers (122–124). Thymic clearance of autoreactive T cells is an important function of autophagy in immune system development and homeostasis (123). Epithelial cells of the thymus are highly autophagic. Mutations in Atg5 in thymic epithelial cells result in altered autoimmunity and specific immunity of certain MHC class II-restricted T cells (125). In addition, autophagy may play an important role in the differentiation of lymphocytes by indirectly affecting the expression of cytokines. During antigen presentation, autophagy proteins present endogenous antigen MHC class II to CD4 + T cells, enhance cross-presentation of antigen-providing cells with CD8 + T cells, and facilitate cross-presentation of phagocytosed antigens by dendritic cells to CD4 + T cells (126–128). Autophagy also contributes to memory B cell maintenance and regulates immunoglobulin secretion (129–131).
Recent findings have shown that autophagy is closely associated with the development of certain chronic inflammatory diseases, such as Crohn’s disease, systemic lupus erythematosus (SLE), and other autoimmune diseases (132, 133). In animal models and human diseases, autophagic failure is usually characterized by dysregulation of inflammation (134). Its main role is to regulate inflammatory transcriptional responses. Increased transcription of proinflammatory cytokines and adipokines has been observed in Paneth cells (Atg16L1HM) of Atg16L1 subtype mice (119, 135–137). Inflammasomes are another important target of autophagic proteins in inflammatory signaling. Inflammasomes are multiprotein complexes containing NLR, adaptor protein ASC, and caspase 1. Inflammasomes can be stimulated by infection or other stress-related pathways. Activated inflammasomes mediate the degradation and activation of caspase-1 and ultimately promote synthesis and secretion of IL-1β and IL-18 (138, 139). In addition, activation of the NALP3 inflammasome is increased in Beclin 1 and LC3B gene-deficient monocytes (140, 141). This enhancement ultimately facilitates the activation of IL-1β and IL-18. The autophagic process can also suppress tissue inflammation by removing apoptotic corpses. During developmentally programmed cell death, autophagy induces xenophagic clearance in dying apoptotic cells by generating ATP-dependent phagocytic signals (142). Increasing evidence suggests that autophagic proteins are required for TLRs mediated phagolysosomal pathways (142). To clear inflammatory sources such as exogenous inflammatory sources (e.g., bacterial viruses) and endogenous pro-inflammatory sources (e.g., damaged organelles, aggregates), autophagic cargoes are usually regulated by ubiquitin and are regulated by a type called SLR (sequestrate-like receptor: p62 [SQSTM1], NBR1, OPTN, NDP52, TAX1BP1, etc.) (143).
Methods for measuring autophagic activity
Currently, the most effective methods for analyzing autophagy in vitro and in vivo remain significantly controversial, due to the complexity of the autophagic process. The measurement of autophagic activity can be divided into two categories: counting autophagosomes and measuring autophagic flux.
Currently, three primary methodologies are employed to assess the number of autophagosomes: electron microscopy (144), Western blot (WB) analysis (145), and fluorescent protein labeling techniques (146). Electron microscopy observation of autophagic structures is the most traditional method. Morphological alterations occurring at various stages of autophagy can be directly visualized using a transmission electron microscope, allowing for an initial assessment of the autophagic phase. Electron microscopy showed damaged organelles in cells undergoing autophagy. In the case of mitochondria, vacuolated bilayer membrane-like structures, or vacuolated structures of bilayer membranes, i.e., autophagosomes, can be observed around them (146, 147). LC3 runs through the whole autophagic process and is currently recognized as an autophagic marker. Changes in the LC3-II/I ratio can be detected using WB to assess the intensity of autophagy. In addition, autophagy can be detected using the property of green fluorescent protein (GFP) quenching in acidic environments (146). Based on GFP-LC3, the researchers developed the GFP-RFP-LC3 tool, a method that allows observation of autophagy in individual cells. Keima is a unique fluorescent protein that is independent of LC3 and suitable for monitoring nonselective autophagy and microautophagy (148). Keima can additionally serve as a tool for the detection of organelle autophagy when conjugated with organelle-specific markers. Scholarly investigations have indicated that an increased presence of autophagosomes or LC3B-II within the system correlates with enhanced proteolytic activity. However, there is no clear correlation between autophagy activity and the abundance of autophagosomes or LC3B proteins (24, 146, 149). For this reason, dynamic measurements of autophagic flux are required (146).
A prominent contemporary approach for assessing autophagic flux involves the observation of LC3 turnover. This approach relies on LC3B-II pooling at autophagosome membranes. When cells were treated with lysosomal reagents (e.g., ammonium chloride) or lysosomal protease inhibitors (e.g., chloroquine), the degradation of LC3-II was blocked, resulting in the accumulation of LC3-II. Thus, the difference in LC3-II amounts between samples represents the amount of LC3 that is delivered to lysosomes for degradation (150). Second, the amount of total cellular LC3 can be quantified by immunoblot analysis or flow cytometry or qualitatively observed by fluorescence microscopy, which is inversely proportional to autophagic flux. In addition to LC3, several groups have developed some specific macrophage substrates to monitor autophagic flux, such as p62/SQSTM1 (151, 152), BRCA1 gene 1 protein (NBR1) (151), betaine-homocysteine s-methyltransferase (153), and polyglutamine protein aggregates (154).
Autophagy in lung diseases
COPD
COPD is a chronic inflammatory pulmonary disease connected with smoking, which is the third most common death factor around the world and consists of 3 primary disease states: chronic bronchitis or proximal airway mucus hypersecretion; emphysema or peripheral lung destruction and loss of alveolar attachments; and small airway disease characterized by inflammation and airway remodeling (Figure 6) (155, 156).
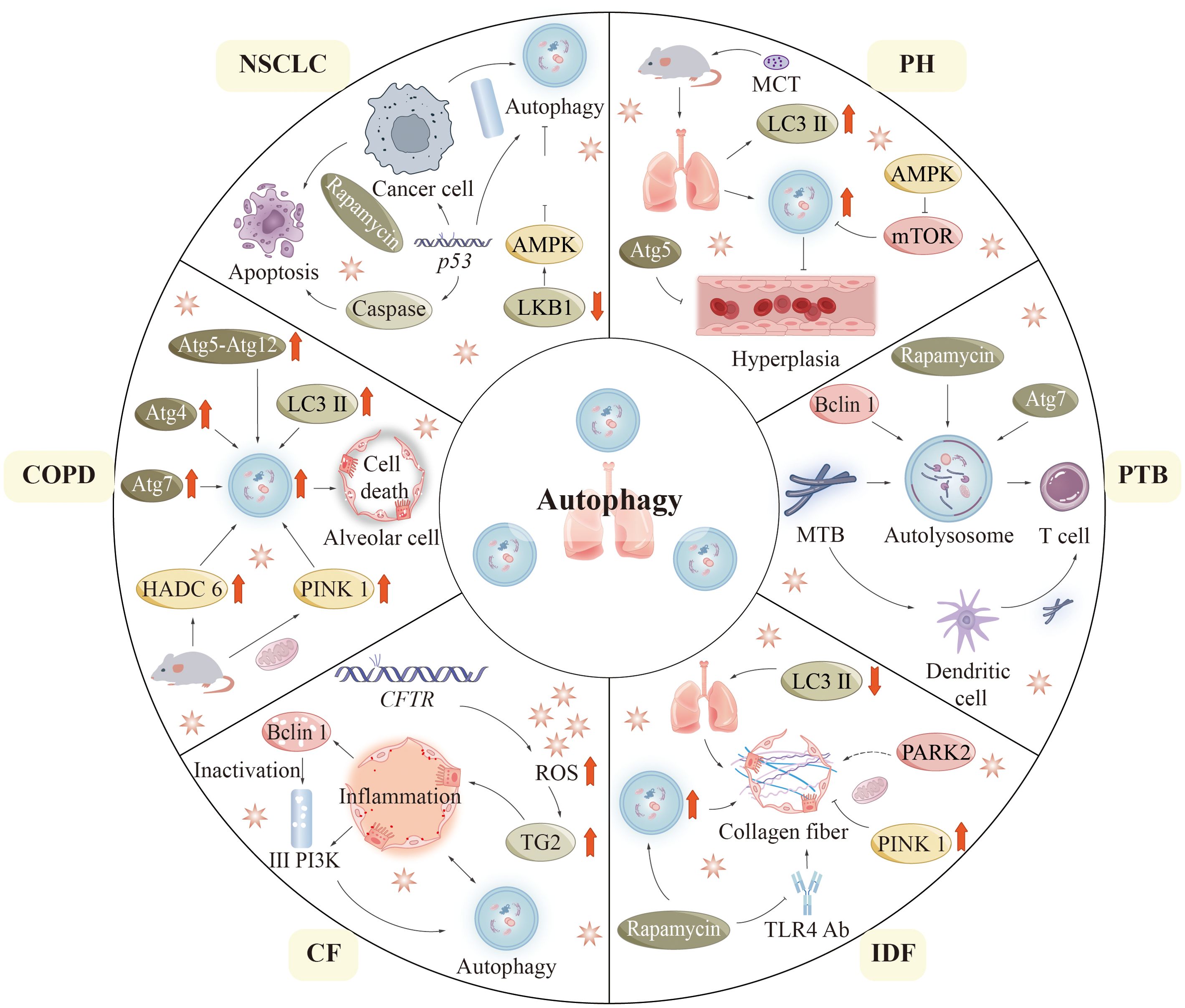
Figure 6. Autophagy in lung diseases. In this figure, we summarize the pathogenesis related to the process of autophagy in six pulmonary diseases: COPD, CF, IPF, PTB, PH, and NSCLC. CF, (Cystic fibrosis); COPD, (Chronic obstructive pulmonary disease); IPF, (Idiopathic pulmonary fibrosis); NSCLC, (Non-small cell lung cancer); PH, (Pulmonary hypertension); PTB, (Pulmonary tuberculosis).
In 2000, autophagic vacuoles were detected in liver specimens lacking alpha-1 antitrypsin, indicating the potential role of autophagy in lung disease (157). COPD pathogenesis is not fully understood but may be associated with aberrant cellular responses of bronchial cells and lung cells to CS (cigarette smoke) (158, 159). In the setting of COPD, autophagy-promoting epithelial cell death was shown to be a potential mechanism (160, 161). As compared to healthy individuals, COPD patients have increased levels of LC3B-II and autophagy-related proteins including ATG4, ATG5-ATG12, and ATG7 (160). In addition, it was observed under electron microscopy that the formation of autophagosomes was also markedly increased in lung tissues from COPD patients compared with control lung tissues (160). Mice exposed to CS are usually used as an experimental model of COPD. In lung tissue, mice subjected to cigarette smoke exposure exhibit elevated levels of autophagic proteins and an increased presence of autophagosomes. Interestingly, mice deficient in LC3B and autophagy proteins are resistant to CS-induced pathological changes (161, 162). These findings indicate that the autophagy pathway may contribute to the progress toward COPD in some specific circumstances (160, 161).
Histone deacetylase 6 (HDAC6) is a critical regulator of primary ciliary uptake. Studies have shown that HDAC6 is involved in the degradation of autophagy in cells (162). Shortened cilia and increased HDAC6 are observed in respiratory epithelial cells treated with CS exposure. Cilia shortening induced by CS is inhibited in mice that lack autophagic protein and HDAC6 (162). This result reflects the importance of pathological changes of HDAC6 in respiratory epithelial cells. Consequently, autophagy plays a role in the HDAC6-mediated degradation of cilia within airway epithelial cells in experimental models of COPD (163). Some studies have reported the involvement of mitochondrial in (164, 165). A key mitophagy protein, phosphatase and angiotensin homolog (PTEN) -induced putative kinase 1 (PINK1), has been found to be increased in the lungs of COPD patients (166). Genetic defects in PINK1 and inhibition of mitophagy with drugs showed resistance to COPD pathology in CS-exposed mice (166).
Cystic fibrosis
Cystic fibrosis (CF) is an autosomal recessive disorder due to mutations in the CF gene located in the 7th pair of chromosomes, which can cause serious damage to the lungs, digestive system, and other organs of the body (167–169). Mutations in related CF genes affect the expression of cystic fibrosis transmembrane conductance regulators (CFTR). It is most typical to have a 508-phenylalanine deletion in the CFTR gene (CFTRF508del) (168). The primary characteristic of cystic fibrosis (CF) within the respiratory system is the overproduction and subsequent accumulation of mucus in the airways. This pathological change can secondarily cause recurrent bronchial infections and airway obstruction. In epithelial cells, mutations in CFTR lead to increased ROS formation. Accumulated ROS promotes tissue transglutaminase 2 (TG2) production. Excessive TG2 is an important cause of inflammatory reactions in CF (170, 171). These complex responses lead to the loss of Beclin 1 and class III PI3K complex function, further affecting autophagic function. Notably, the enhancement of autophagy through the overexpression of Beclin 1 has been shown to enhance inflammatory responses (170), indicating that the autophagic system is essential for the clearance of protein aggregates. In a similar vein, mice with the F508del-CFTR mutation demonstrate reduced levels of Beclin 1 expression (172). Significant risk of morbidity and mortality exists in CF patients due to pseudomonas aeruginosa infection. Experimentally, defective autophagic function resulting from reduced levels or loss of function of BECN1 renders mice lungs more vulnerable to pseudomonas aeruginosa infection (173).
Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is the most common form of idiopathic interstitial pneumonia in clinical practice and is a chronic pulmonary disease of unrecognized etiology (174, 175). Although IPF has epithelial origins, it displays abnormal adaptive immune responses, such as T-cell and B-cell dysregulation (176). The levels of LC3-II expression in lung tissue from patients diagnosed with IPF are significantly reduced compared to those observed in healthy individuals (160, 177). Autophagy seems to play a protective role in the development of IPF. In experimental models of IPF, rapamycin inhibits lung fibroblasts’ expression of fibronectin and alpha-smooth muscle actin through its up-regulation of autophagy (178). In addition, the pro-autophagic effect of rapamycin is shown to promote collagen formation in lung epithelial cells (177). Rapamycin also inhibits pulmonary fibrosis induced by Toll-like receptor 4 (TLR4) antibodies or bleomycin in mice (177–179). Furthermore, in the absence of autophagy genes or when autophagy is suppressed pharmacologically, transforming growth factor (TGF) activates lung fibroblasts (177, 178).
In IPF, researchers found clusters of malformed mitochondria in lung epithelial cells, particularly in alveolar type II cells (AECIIs) (180). In addition, microarray analysis showed decreased PINK1 in lung tissues from IPF patients (180). The knock-down PINK1 mice displayed increased mitochondrial depolarization and expression of pro-fibrotic factors (180, 181). The mechanism of the antifibrotic effect of PINK1 in lung epithelial cells is reflected in the prevention of cell death by preserving the morphology and function of mitochondria (180, 181). It has been suggested that PARK2, an important mitophagy-related molecule may be linked to the pathogenesis of IPF (182). Mitophagy is activated in alveolar macrophages from IPF patients and mice treated with bleomycin. Whereas increased apoptosis of macrophages is found in mitophagy-deficient mice, which prevents them from pulmonary fibrosis.
Pulmonary hypertension
Pulmonary hypertension (PH) is a disease of abnormally high blood pressure in the pulmonary arteries. PH is predominantly defined by the remodeling of pulmonary vasculature, a multifaceted and progressive phenomenon that ultimately results in right heart failure and mortality. Studies have investigated how autophagy acts in PAH pathogenesis, but conclusions remain disputed. Experimental mice with chronic hypoxia showed increased levels of LC3B and autophagosomes in their lungs (183). Furthermore, a higher prevalence of autophagic vacuoles was noted in lung tissue subjected to hypoxia. Mice deficient in MAP1LC3B (MAP1LC3B −/−) that were exposed to chronic hypoxic conditions demonstrated more pronounced PH values in comparison to their wild-type counterparts. PH values included right ventricular systolic pressure and vessel wall thickness. An elevation in angiogenesis within pulmonary artery endothelial cells has been noted in cases of persistent PH in Beclin 1-null mice (184). ATG5-targeting siRNA has been found to directly disrupt autophagy to inhibit the proliferation process of rat pulmonary artery smooth muscle cells. The AMPK signaling pathway, recognized as a crucial component of autophagy, significantly results in the process of autophagy in cardiomyocytes. Research has shown that pharmacological inhibition of the AMPK pathway increases cardiomyocyte mortality, suggesting a protective effect on AMPK-associated cardiomyocyte autophagy. Monocrotaline (MCT) is a commonly used drug in animal experimental models of induced PH. Recent research indicates that in rats treated with monocrotaline (MCT), the expression of phospho-mTOR in the right ventricle is down-regulated, while the expression of phospho-AMPK is elevated at the 2 and 4-week marks. Conversely, at the 6-week interval, there is an up-regulation of phospho-mTOR expression and a decrease in phospho-AMPK expression in the right ventricle of MCT-treated rats (185). This suggests that the AMPK-mTOR autophagy signaling pathway is involved in regulating autophagy in pulmonary hypertension rats. It has already been demonstrated that rapamycin treatment can prevent right ventricular hypertrophy and dysfunction through activation of the autophagy pathway in animal models. The findings indicate that autophagy could potentially be a contributing factor to human vascular disease (186). However, these findings are derived from static measurements. Additional experimental investigations are necessary to elucidate the relationship between human vascular disease and autophagy (183).
Pulmonary tuberculosis
PTB is a chronic and long-term pulmonary disease caused by Mtb infecting the human lung, and it is the predominant manifestation of tuberculosis. Mtb is classified as an intracellular pathogen that releases a diverse array of effector proteins within host cells. These proteins subsequently disrupt cellular signaling pathways, thereby influencing normal cellular functions. It ultimately promotes its survival in host cells and leads to host cell pathology. During the initial stages of infection, innate immune responses are stimulated, and inflammatory cells are recruited to the lungs. Mtb evades and eliminates innate immune cells, spreads to the draining lymph nodes, and triggers a specific T-cell response that promotes the formation of granulomas at the sites of pulmonary infection (187). Inflammatory granulomas are believed to lead to lung tissue damage, and pulmonary fibrosis, and progressively develop chronic and persistent clinical manifestations of PTB (188).
Mtb inhibits phagosome-lysosome fusion, allowing it to persist in the phagosome during maturation. Autophagy is important for the elimination of Mtb. In vitro, rapamycin or starvation-induced autophagy promotes the conversion of Mtb phagosomes into autolysosomes, which contain more antimicrobial chambers (e.g., antimicrobial peptides) than conventional phagosomes (129, 189). Macrophages are also enhanced in their ability to present mycobacterial antigens by autophagy (197). Moreover, phagolysosomal fusion is found to be inhibited when cells are infected with Mtb in macrophages lacking Beclin 1 and ATG7 (189). This result could preliminarily prove that autophagy is advantageous for killing tubercle bacilli. However, the specific mechanism of the defense effect of autophagy proteins on Mtb in humans is unknown. In addition, recent in vitro studies have demonstrated increased Mtb replication in HIV-infected macrophages co-infected with Mtb when autophagy is activated by the suppression of the mTOR pathway (190). A recent study found that certain autophagic mechanisms acting on phagocytes are critical mechanisms to target Mtb, known as xenophagy. The embryonic exogenic homeobox 1 (ESX-1) secretion system is a virulence factor of Mtb (191). ESX-1 causes Mtb DNA exposure to the host cytoplasm through phagosome permeation (192). DNA exposed to the cytoplasm is detected and ubiquitinated by cytoplasmic DNA sensor molecules (e.g., STING) (192). Ubiquitinated DNA attaches to LC3 via several proteins like p62 and nucleoporin 52. Consequently, it is encapsulated in autophagosomes to fuse with lysosomes (192).
Non-small cell lung cancer
The mechanism of action of autophagy in cancer has repeatedly been described as a double-edged sword. The role that autophagy-related cellular pathways play in the pathological progression of NSCLC is being extensively investigated. Mutations in genes involved in the mTOR signaling pathway may be associated with malignant proliferation of cells. Mutations in genes in the mTOR pathway, such as KRAS, epidermal growth factor receptor (EGFR), LKB1, PTEN, PIK3CA, AKT1, EGFR, PIK3CA, and PTEN, have some relationship with the development of NSCLC (29). Research indicates that the anticancer efficacy of LKB1 is diminished in NSCLC. The researchers propose that this reduction may facilitate tumor proliferation via the LKB1-AMPK-mTOR signaling pathway (193). Rapamycin causes endogenous apoptosis of cancerous cells, which in turn inhibits tumor growth in mouse models of NSCLC (194, 195). In vitro lung cancer models, rapamycin enhances apoptosis and autophagy (196). The PI3K signaling pathway serves as a primary regulator of autophagy, with its activation leading to the inhibition of autophagic processes in cancerous cells. Furthermore, the activation of this pathway enhances the production of tumor-promoting antigens, thereby facilitating the process of carcinogenesis. Mutations in p53 are also important correlates of tumorigenesis. p53 is one of the most frequently mutated genes and is present in 45% – 70% of adenocarcinomas and 60% – 80% of squamous cell carcinomas (196). The pathogenesis of NSCLC depends heavily on the absence of the p53 gene (197). p53 can be present in the cytoplasm and nucleus. Under cellular stress, p53 can translocate into the nucleus (197). The p53 protein, located in the nucleus, experiences conformational alterations that enable it to function as a transcription factor. This activity facilitates the upregulation of numerous pro-apoptotic proteins, thereby making sense in the regulation of both endogenous and exogenous apoptotic pathways (197). p53 can also translocate to the mitochondrial surface and directly bind to Bcl-2 family proteins to promote endogenous apoptosis (197, 198). In addition to this, p53 promotes the expression of Apaf-1 and caspase-6 and promotes extrinsic apoptosis (198). p53 also plays a role in regulating the autophagic process. An autophagy-induced response was observed in mice whose p53 expression was blocked using cypermethrin-α. And p53-null cells also showed enhanced autophagy compared with wild-type cells. In addition, cytoplasmic p53 can interact with FIP200, which in turn competitively inactivates autophagy.
Other lung diseases
In addition to the above lung diseases, autophagy is also greatly related to the occurrence and development of many other lung diseases, such as asthma, COVID-19, and atypical pneumonia.
Autophagy plays a complex role in the pathophysiology of asthma and may be deleterious or beneficial. Autophagy can affect inflammatory response, airway remodeling, immune regulation, and other aspects, and is an important field of asthma treatment research. Polymorphisms in the autophagy-related gene Atg5 are strongly associated with asthma (199). In respiratory epithelial cells of patients with severe asthma, the expression level of Atg5 protein is increased, and this phenomenon is closely related to the deepening of the degree of fibrosis in the lower cell layer as well as the increase of collagen-1 expression (200). IL-13 plays a crucial role in the development of T2 asthma (201). In vitro, IL-13 stimulated goblet cell production and secretion of MUC5AC protein from human respiratory epithelial cells (202). This process is associated with activation of the autophagic process, which is blocked when expression of Atg5 is inhibited. In addition, inhibition of autophagy also affects IL-13 production in response to ROS (203). In addition, in asthmatic patients, airway epithelial cells initiate autophagy by inhibiting mTORC1 signaling in response to IL-13 or IL-33 (204). Bronchial fibroblasts showed enhanced mitophagy accompanied by increased expression levels of PINK1 and Parkin protein in severe asthma, which may be an adaptive response against mitochondrial dysfunction in asthmatic cells (205).
COVID-19 is caused by a coronavirus called SARS-CoV-2, and prior research has indicated that autophagy may play a dual role in the context of coronavirus infection (206). Autophagy can degrade coronavirus, enhance inflammatory responses, and modulate inflammation in neutrophils (207). At the same time, it also promotes antigen presentation and provokes immunity against coronavirus (208). However, double-membrane vesicles of autophagosomes facilitate the sequestration of the virus from external immune responses, thereby serving as sites for viral replication and transcription (207). In addition, nonstructural protein 6 (NSP6) of novel coronavirus assists the virus in escaping host innate immunity by activating autophagy. Recent studies have shown that overexpression of SARS-CoV-2 papain-like protease cleaves ULK1 and disrupts the formation of ULK1-ATG13 complex to block intact host autophagy (209). Corona virus also inhibits BECN1 and activates autophagy inhibitors (SKP2 and AKT1) to prevent autophagosome fusion with lysosomes to limit autophagy signal transduction (210, 211). Interestingly, compared with classical SARS-CoV, ORF3a of SARS-CoV-2 can separate homotypic fusion and protein classification components, thereby inhibiting fusion of autophagosomes and lysosomes, which is a unique feature of SARS-CoV-2 inhibition of autophagy (212, 213).
Autophagy is also important in atypical pneumonia, for example, infections caused by Chlamydia pneumonia(CP), Mycoplasma pneumonia, and Legionella. In CP, it has been shown to limit intracellular CP growth in vitro by inhibiting autophagy, but in vivo, research has demonstrated that the impairment of autophagy in myeloid cells is associated with increased mortality, potentially resulting from intricate antagonistic interactions between inflammasomes and autophagy (214). Post-infection with Mycoplasma pneumonia, the activation of autophagy may correlate with the severity of the disease, and both excessive activation and suppression of autophagy could influence the progression of the illness. Membrane lipids of Mycoplasma pneumonia can activate autophagy through TLR4 and promote the production of inflammatory factors such as TNF-α and IL-1β, exacerbating the inflammatory response (215). Legionella can inject effector proteins into host cells via its type IV secretion system (Dot/Icm) to avoid autophagy and survive. Nevertheless, the autophagy gene Atg7 can also exert its effect by assisting macrophages to clear bacteria (216).
Autophagy as a potential therapeutic target for the treatment of lung diseases
In the human body, autophagy is essential to maintain the normal functioning of tissues and organs as well as the development of diseases. Thus, targeting autophagy may be useful in the treatment of disease, but may also exacerbate disease deterioration. Because the autophagic process can help clear harmful protein aggregates and damaged organelles. However, excessive autophagy or dysregulation of autophagy may be harmful to cells. In lung diseases, the role of autophagy is particularly complex. On the one hand, it can clear pathogens and damaged cells in the lungs and help resist infection and inflammation. On the other hand, if the autophagic process is dysregulated, it may lead to damage and dysfunction of lung tissue. Therefore, using autophagy as a target for the treatment of lung diseases requires great caution. If the treatment strategy is not appropriate, it may exacerbate the condition rather than improve it. How to balance the activation and inhibition of autophagy to achieve the best therapeutic effect is currently a major difficulty in autophagy-targeted therapy (Table 2).
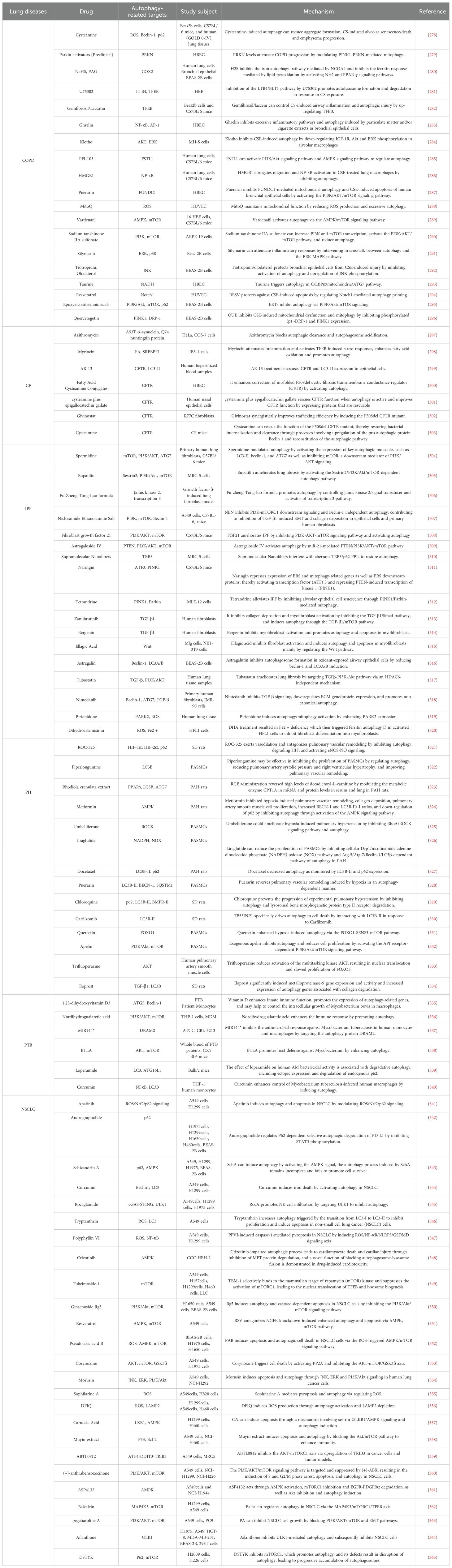
Table 2. The mechanism of autophagy-related targets in lung diseases.
COPD
Based on the available findings, it may be possible to hypothesize that selective targeting of autophagy-related proteins at the genetic or pharmacological level may serve as a basis for the formulation of novel therapies for COPD. In mouse models, many studies have attempted to mitigate the occurrence of abnormal autophagy during smoke exposure by different approaches. However, these investigations have primarily focused on preventive interventions related to the duration of smoking. These studies included the chemical chaperone 4-phenylbutyrate (162); antioxidant drug, cysteine (217); arachidonic acid-derived epoxyeicosatrienoic acid (218); HDAC6 inhibitor tubastatin (219); mitophagy inhibitor Mdivi134; and sodium channel inhibitor carbamazepine. In addition, studies using the mTOR inhibitor rapamycin suggested that increasing autophagy during CS exposure could reduce lung tissue inflammation, which may be of assistance. However, rapamycin increased the number of apoptotic and inflammatory cells compared with controls at baseline. To clarify the pathophysiological function of autophagy in disease, it is essential to carefully time the activation of autophagy and the targeting of lung cells. Further investigation is required to assess the impact of these agents on dysregulated autophagy in COPD.
CF
Treatments for CF have been extensively investigated. The restoration of autophagic functionality may provide additional therapeutic options for treating CF. The antioxidant n-acetyl-l-cysteine has been shown to improve airway phenotypes in CFTR mutant mice. In addition, oral cysteamine was found to restore Beclin 1 expression and prolonged the survival of CFTR F508del mutant mice (220). Hence, it may be worthwhile to investigate cysteamine drugs’ mechanism through restoring autophagy (221). In addition, regular and continuous use of azithromycin has been demonstrated to enhance the health condition of CF patients (222, 223). However, It has been reported that mycobacterial infection increases synchronously with the onset of CF in some studies (224, 225). The seemingly contradictory results observed in cystic fibrosis patients treated with azithromycin may be attributed to the drug’s capacity to inhibit lysosomal acidification. This inhibition subsequently disrupts autophagy and the degradation processes within phagosomes. More therapeutic options targeting autophagy-related pathways need to be investigated in greater depth.
IPF
Promoting autophagy may be beneficial in the treatment of IPF. Currently, drugs that may be active include IL17A neutralizing antibodies (177), MIR449A(microRNA 449a) (226), or PDGFRB (platelet-derived growth factor receptor beta) inhibitors. Bleomycin-mediated increases in mortality and decreases in fibrotic resistance in mice have been observed in experimental models (178, 227). However, rapamycin appears to potentiate silica-induced effects, exacerbating inflammation and fibrosis (228). In individuals diagnosed with IPF, the rapamycin analog everolimus has been observed to contribute to the progression of the disease. Therefore, further studies are required to assess whether targeted autophagy agents are beneficial in IPF.
Pulmonary hypertension
Numerous studies utilizing animal models have investigated the impact of rapamycin, mTOR inhibitors, and autophagy activators on the prevention of PAH development (229). In clinical settings, everolimus, a derivative of rapamycin, has been shown to enhance outcomes in patients with strict PH resulting from chronic thromboembolic disease, while also decreasing pulmonary vascular resistance (230). The suppression of the autophagy pathway may offer potential targeted therapeutic strategies for the disease.
Pulmonary tuberculosis
Therapeutic regimens targeting pulmonary tuberculosis by autophagy-related pathways are being widely investigated. Vitamin D stimulates autophagy activation in Mycobacterium tuberculosis by inducing antibiotics (231). Vitamin D deficiency has been linked to a heightened threat of active TB (232). The conversion time of sputum cultures is not affected by vitamin D supplementation according to recent research (233, 234). However, it has also been reported that vitamin D supplementation given to patients with vitamin D receptor polymorphisms shortens sputum culture conversion time (234). Isoniazid and pyrazinamide are recognized as primary agents in the treatment of tuberculosis. These compounds facilitate the activation of autophagy and the maturation of autophagosomes within host cells infected by Mtb (235). This may constitute a component of the fundamental mechanism associated with the treatment involving these agents. Although the activation of autophagy has been previously proposed as a viable therapeutic approach for patients infected with Mtb, recent findings cast uncertainty on this hypothesis and impede the progression of further research.
Non-small cell lung cancer
The development of novel compounds aimed at targeting mutant p53 and reinstating its wild-type functionality represents a promising therapeutic approach for cancer treatment, particularly in the context of NSCLC, which is characterized by a significant mutation frequency (236–238). This therapeutic potential has already been demonstrated in many compounds. Nutlins are cis-imidazoline analogs that inhibit the interaction between MDM2 and wild-type p53 in vivo, which in turn enhances the anti-tumor ability of p53 (239). We speculate that the development of targeted agents against aberrant p53 or promoting anti-tumor activity of wild-type p53 may be helpful in the treatment of cancer. RETRA was found to inhibit the malignant proliferation of cancer cells carrying aberrant p53 via a p73-dependent salvage pathway (240). The reactivating small molecule PRIMA-1 of mutant p53 can be combined to convert it into a wild-type construct, thereby achieving inhibition of tumor growth (241, 242). In addition, restoring and stabilizing the DNA binding domain (DBD) of p53 is also a promising tumor suppressor strategy.
Rapamycin has great potential in cancer therapy, which activates mitochondria-mediated apoptosis independent of p53 in NSCLC cells, stressing its effectiveness in disease (29). It has been shown that mouse models of NSCLC have reduced tumor growth and apoptosis following rapamycin treatment (232). Furthermore, certain anticancer agents have demonstrated markedly enhanced efficacy when administered in conjunction with rapamycin. This includes Bcl-2 inhibitors such as ABT-737, pemetrexed, and lipophilic bisphosphonates (196). Additionally, EGFR tyrosine kinase inhibitors (TKIs) have received approval for the treatment of patients with NSCLC who possess particular EGFR mutations (243). However, resistance to this drug is a major problem in clinical treatment. Notably, erlotinib combined with rapamycin enhanced autophagy and restored sensitivity to EGFR-TKIs (244). Erlotinib in combination with rapamycin has also been shown to help overcome resistance due to p53 deficiency in vitro.
In addition to rapamycin, drugs targeting proteins related to other signaling pathways of autophagy, such as AZD8055 (PI3K inhibitor), NVP-BEZ235 (PI3K and mTORC1 inhibitors), perifosine (AKT inhibitor), and GSK-690693 (AKT inhibitor), have been investigated in NSCLC. In certain instances, these targeted therapies modulate autophagy-related pathways as a component of the treatment regimen for NSCLC (245–248). The effects of some traditional Chinese medicine compounds in NSCLC have also been largely investigated. Curcumin is a phenolic compound derived from the plant Curcuma longa (249, 250). Curcumin treatment showed a promoting effect of autophagy as well as a pro-apoptotic effect in lung adenocarcinoma A549 cells, allowing us to speculate its therapeutic potential in NSCLC (251). Cytoprotective autophagy in NSCLC cells is also activated by cucurbitacin E and glycerinic acid (252). Licochalcone A, a flavonoid derived from the traditional Chinese medicinal plant Glycyrrhiza uralensis Fisch, has been shown to promote apoptosis and autophagy through the induction of ER stress (253). Thick acid from Poria cocos halted lung cancer cell growth by boosting ROS and activating JNK (254). Platycodin-D can induce autophagy in H460 and A549 NSCLC cells, as shown by stimulating the formation of ATG3, ATG7, Beclin-1, and LC3-II (255).
Conclusion
Autophagy plays a dual role in lung diseases, exhibiting both potentially harmful effects in certain pathophysiological conditions and serving as a protective mechanism that promotes cell survival. Recent advancements in research have significantly enhanced our understanding of the role of autophagy in the pathophysiological mechanisms underlying various diseases, thereby offering novel insights for the development of targeted therapies for pulmonary diseases. However, assessing the actual clinical effects of targeted agents for autophagy-related pathways on lung diseases is challenging. The challenge arises from the observation that autophagic responses occurring in various compartments of the lung may yield markedly distinct effects. Accurate measurements of autophagy also need to be updated. It is critical to revise the precise assessments of autophagy. Understanding the impact of highly specific autophagy modulators on disease models characterized by particular autophagy deficiencies is vital for formulating clinically relevant approaches to either stimulate or suppress autophagy.
Author contributions
LL: Writing – original draft. YL: Writing – original draft. ZH: Writing – review & editing, Writing – original draft. KW: Writing – original draft. SZ: Writing – original draft. ZW: Writing – original draft. SW: Writing – review & editing. HC: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
2. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. (2010) 90:1383–435. doi: 10.1152/physrev.00030.2009
3. Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. (2009) 1793:664–73. doi: 10.1016/j.bbamcr.2008.07.014
4. Pattison CJ, Korolchuk VI. Autophagy: ‘Self-eating’ Your way to longevity. Subcell Biochem. (2018) 90:25–47. doi: 10.1007/978-981-13-2835-0_2
5. Rabinowitz JD, White E. Autophagy and metabolism. Science. (2010) 330:1344–8. doi: 10.1126/science.1193497
6. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
7. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. (2011) 469:323–35. doi: 10.1038/nature09782
8. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. (2007) 39:596–604. doi: 10.1038/ng2032
9. Alula KM, Theiss AL. Autophagy in crohn’s disease: converging on dysfunctional innate immunity. Cells. (2023) 12:1779. doi: 10.3390/cells12131779
10. Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. (2009) 104:304–17. doi: 10.1161/CIRCRESAHA.108.188318
11. Tang Y, Xu W, Liu Y, Zhou J, Cui K, Chen Y. Autophagy protects mitochondrial health in heart failure. Heart Fail Rev. (2024) 29:113–23. doi: 10.1007/s10741-023-10354-x
12. Diab R, Pilotto F, Saxena S. Autophagy and neurodegeneration: Unraveling the role of C9ORF72 in the regulation of autophagy and its relationship to ALS-FTD pathology. Front Cell Neurosci. (2023) 17:1086895. doi: 10.3389/fncel.2023.1086895
13. Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. (2011) 32:955–63. doi: 10.1093/carcin/bgr031
14. Liang C. Negative regulation of autophagy. Cell Death Differ. (2010) 17:1807–15. doi: 10.1038/cdd.2010.115
15. Lin Z, Long F, Kang R, Klionsky DJ, Yang M, Tang D. The lipid basis of cell death and autophagy. Autophagy. (2024) 20:469–88. doi: 10.1080/15548627.2023.2259732
16. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. (2007) 8:931–7. doi: 10.1038/nrm2245
17. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
18. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. (2011) 20:131–9. doi: 10.1016/j.devcel.2010.12.003
19. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. (2013) 368:1845–6. doi: 10.1056/NEJMra1205406
20. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. (2014) 16:495–501. doi: 10.1038/ncb2979
21. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. (2023) 24:167–85. doi: 10.1038/s41580-022-00542-2
22. Rubio-Tomás T, Sotiriou A, Tavernarakis N. The interplay between selective types of (macro)autophagy: Mitophagy and xenophagy. Int Rev Cell Mol Biol. (2023) 374:129–57. doi: 10.1016/bs.ircmb.2022.10.003
23. Tanida I. Autophagy basics. Microbiol Immunol. (2011) 55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x
24. Li W, Zhou C, Yu L, Hou Z, Liu H, Kong L, et al. Tumor-derived lactate promotes resistance to bevacizumab treatment by facilitating autophagy enhancer protein RUBCNL expression through histone H3 lysine 18 lactylation (H3K18la) in colorectal cancer. Autophagy. (2024) 20:114–30. doi: 10.1080/15548627.2023.2249762
25. Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. (2008) 10:776–87. doi: 10.1038/ncb1740
26. Grochowska KM, Sperveslage M, Raman R, Failla AV, Głów D, Schulze C, et al. Chaperone-mediated autophagy in neuronal dendrites utilizes activity-dependent lysosomal exocytosis for protein disposal. Cell Rep. (2023) 42:112998. doi: 10.1016/j.celrep.2023.112998
27. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. (2010) 22:124–31. doi: 10.1016/j.ceb.2009.11.014
28. Jung CH, Ro SH, Cao J, Otto NM, Kim DH, Mtor Regulation of Autophagy. mTOR regulation of autophagy. FEBS Lett. (2010) 584:1287–95. doi: 10.1016/j.febslet.2010.01.017
29. Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. (2014) 90:197–207. doi: 10.1016/j.bcp.2014.05.011
30. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. (2014) 15:155–62. doi: 10.1038/nrm3757
31. Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. (2002) 10:457–68. doi: 10.1016/S1097-2765(02)00636-6
32. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. (2011) 36:320–8. doi: 10.1016/j.tibs.2011.03.006
33. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. (2002) 2:489–501. doi: 10.1038/nrc839
34. Proikas-Cezanne T, Haas ML, Pastor-Maldonado CJ, Schüssele DS. Human WIPI β-propeller function in autophagy and neurodegeneration. FEBS Lett. (2024) 598:127–39. doi: 10.1002/1873-3468.14782
35. Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. (1995) 81:727–36. doi: 10.1016/0092-8674(95)90534-0
36. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. (2005) 307:1098–101. doi: 10.1126/science.1106148
37. Fei C, Zhen X, Shiqiang Z, Jun P. Frontier knowledge and future directions of programmed cell death in clear cell renal cell carcinoma. Cell Death Discovery. (2024) 10:113. doi: 10.1038/s41420-024-01880-0
38. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discovery. (2009) 8:627–44. doi: 10.1038/nrd2926
39. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. (2002) 4:648–57. doi: 10.1038/ncb839
40. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
41. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, Malignant transformation and drug resistance. Biochim Biophys Acta. (2007) 1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001
42. Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur J Med Chem. (2016) 109:314–41. doi: 10.1016/j.ejmech.2016.01.012
43. Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL. Redox regulation of cell signalling. Nature. (1996) 381:380–1. doi: 10.1038/381380a0
44. Livingston MJ, Zhang M, Kwon SH, Chen JK, Li H, Manicassamy S, et al. Autophagy activates EGR1 via MAPK/ERK to induce FGF2 in renal tubular cells for fibroblast activation and fibrosis during maladaptive kidney repair. Autophagy. (2024) 20:1032–53. doi: 10.1080/15548627.2023.2281156
46. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. (1998) 94:491–501. doi: 10.1016/S0092-8674(00)81590-1
47. Goulielmaki M, Koustas E, Moysidou E, Vlassi M, Sasazuki T, Shirasawa S, et al. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget. (2016) 7:9188–221. doi: 10.18632/oncotarget.6942
48. Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. (2011) 42:23–35. doi: 10.1016/j.molcel.2011.02.009
49. Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, et al. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. (2006) 66:6861–70. doi: 10.1158/0008-5472.CAN-05-3557
50. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. (2004) 6:91–9. doi: 10.1016/j.ccr.2004.06.007
51. Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. (2003) 13:2004–8. doi: 10.1016/j.cub.2003.10.031
52. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. (2009) 9:563–75. doi: 10.1038/nrc2676
53. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. (2005) 18:283–93. doi: 10.1016/j.molcel.2005.03.027
54. Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. (2004) 18:1533–8. doi: 10.1101/gad.1199104
55. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
56. Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PloS One. (2010) 5:e15394. doi: 10.1371/journal.pone.0015394
57. Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. (2011) 108:4788–93. doi: 10.1073/pnas.1100844108
58. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
59. Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. (2007) 282:25464–74. doi: 10.1074/jbc.M703663200
60. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. (2010) 22:132–9. doi: 10.1016/j.ceb.2009.12.004
61. Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy. (2009) 5:758–65. doi: 10.4161/auto.8709
62. Yao W, Chen Y, Chen Y, Zhao P, Liu J, Zhang Y, et al. TOR-mediated Ypt1 phosphorylation regulates autophagy initiation complex assembly. EMBO J. (2023) 42:e112814. doi: 10.15252/embj.2022112814
63. Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. (2008) 181:497–510. doi: 10.1083/jcb.200712064
64. Young AR, Chan EY, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. (2006) 119:3888–900. doi: 10.1242/jcs.03172
65. Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. (2009) 284:12297–305. doi: 10.1074/jbc.M900573200
66. Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. (2009) 5:973–9. doi: 10.4161/auto.5.7.9296
67. Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. (2009) 29:157–71. doi: 10.1128/MCB.01082-08
68. Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. (2009) 20:2004–14. doi: 10.1091/mbc.e08-12-1250
69. Randall-Demllo S, Chieppa M, Eri R. Intestinal epithelium and autophagy: partners in gut homeostasis. Front Immunol. (2013) 4:301. doi: 10.3389/fimmu.2013.00301
70. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. (2006) 22:159–68. doi: 10.1016/j.molcel.2006.03.029
71. Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. (2004) 23:9314–25. doi: 10.1038/sj.onc.1208331
72. Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. (2010) 6:506–22. doi: 10.4161/auto.6.4.11863
73. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. (1999) 402:672–6. doi: 10.1038/45257
74. Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. (2005) 1:46–52. doi: 10.4161/auto.1.1.1542
75. He C, Levine B. The beclin 1 interactome. Curr Opin Cell Biol. (2010) 22:140–9. doi: 10.1016/j.ceb.2010.01.001
76. Prerna K, Dubey VK. Beclin1-mediated interplay between autophagy and apoptosis: New understanding. Int J Biol Macromol. (2022) 204:258–73. doi: 10.1016/j.ijbiomac.2022.02.005
77. Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. (1998) 72:8586–96. doi: 10.1128/JVI.72.11.8586-8596.1998
78. Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol. (2007) 372:223–35. doi: 10.1016/j.jmb.2007.06.069
79. Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, et al. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun. (2012) 3:662. doi: 10.1038/ncomms1648
80. González-King H, Rodrigues PG, Albery T, Tangruksa B, Gurrapu R, Silva AM, et al. VHL suppresses autophagy and tumor growth through PHD1-dependent Beclin1 hydroxylation. EMBO J. (2024) 43:931–55. doi: 10.1038/s44318-024-00051-2
81. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. (2006) 8:688–99. doi: 10.1038/ncb1426
82. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. (2008) 19:5360–72. doi: 10.1091/mbc.e08-01-0080
83. Itakura E, Mizushima N. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy. (2009) 5:534–6. doi: 10.4161/auto.5.4.8062
84. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. (2009) 11:468–76. doi: 10.1038/ncb1854
85. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. (2009) 11:385–96. doi: 10.1038/ncb1846
86. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. (2007) 26:2527–39. doi: 10.1038/sj.emboj.7601689
87. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. (2005) 122:927–39. doi: 10.1016/j.cell.2005.07.002
88. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. (2007) 8:275–83. doi: 10.1038/nrm2147
89. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. (2012) 26:1268–86. doi: 10.1101/gad.190678.112
90. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. (2010) 40:280–93. doi: 10.1016/j.molcel.2010.09.023
91. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. (2011) 7:279–96. doi: 10.4161/auto.7.3.14487
92. Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. (2009) 16:12–20. doi: 10.1038/cdd.2008.101
93. Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med. (2008) 8:78–91. doi: 10.2174/156652408783769616
94. Chen R, Zou J, Zhong X, Li J, Kang R, Tang D. HMGB1 in the interplay between autophagy and apoptosis in cancer. Cancer Lett. (2024) 581:216494. doi: 10.1016/j.canlet.2023.216494
95. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. (2007) 8:741–52. doi: 10.1038/nrm2239
96. Shvets E, Elazar Z. Flow cytometric analysis of autophagy in living mammalian cells. Methods Enzymol. (2009) 452:131–41. doi: 10.1016/S0076-6879(08)03609-4
97. Farkas T, Høyer-Hansen M, Jäättelä M. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy. (2009) 5:1018–25. doi: 10.4161/auto.5.7.9443
98. Shimizu S, Konishi A, Nishida Y, Mizuta T, Nishina H, Yamamoto A, et al. Involvement of JNK in the regulation of autophagic cell death. Oncogene. (2010) 29:2070–82. doi: 10.1038/onc.2009.487
99. Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. (2008) 4:949–51. doi: 10.4161/auto.6788
100. He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. (2013) 62:1270–81. doi: 10.2337/db12-0533
101. Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. (2011) 123:1073–82. doi: 10.1161/CIRCULATIONAHA.110.977066
102. Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A. (2008) 105:16677–82. doi: 10.1073/pnas.0808597105
103. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. (2004) 304:1500–2. doi: 10.1126/science.1096645
104. Thorburn J, Moore F, Rao A, Barclay WW, Thomas LR, Grant KW, et al. Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol Biol Cell. (2005) 16:1189–99. doi: 10.1091/mbc.e04-10-0906
105. Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. (2005) 280:20722–9. doi: 10.1074/jbc.M413934200
106. Wang F, Trosdal ES, Paddar MA, Duque TLA, Allers L, Mudd M, et al. The role of ATG5 beyond Atg8ylation and autophagy. Autophagy. (2024) 20:448–50. doi: 10.1080/15548627.2023.2273703
107. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. (2006) 8:1124–32. doi: 10.1038/ncb1482
108. Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. (2008) 4:458–69. doi: 10.1016/j.chom.2008.10.003
109. Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. (2009) 30:588–98. doi: 10.1016/j.immuni.2009.02.009
110. Orvedahl A, MacPherson S, Sumpter R Jr., Tallóczy Z, Zou Z, Levine B, et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. (2010) 7:115–27. doi: 10.1016/j.chom.2010.01.007
111. Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. (2008) 9:908–16. doi: 10.1038/ni.1634
112. Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, et al. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci U S A. (2009) 106:14564–9. doi: 10.1073/pnas.0813319106
113. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. (2009) 5:527–49. doi: 10.1016/j.chom.2009.05.016
114. Dwivedi R, Baindara P. Differential regulation of TFEB-induced autophagy during mtb infection and starvation. Microorganisms. (2023) 11:2944. doi: 10.3390/microorganisms11122944
115. Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. (2009) 11:1355–62. doi: 10.1038/ncb1980
116. Sumpter R Jr., Levine B. Autophagy and innate immunity: triggering, targeting and tuning. Semin Cell Dev Biol. (2010) 21:699–711. doi: 10.1016/j.semcdb.2010.04.003
117. Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. (2009) 11:1233–40. doi: 10.1038/ncb1967
118. Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle. (2010) 9:1295–307. doi: 10.4161/cc.9.7.11109
119. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. (2010) 11:55–62. doi: 10.1038/ni.1823
120. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. (2010) 466:68–76. doi: 10.1038/nature09204
121. Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. (2010) 18:1041–52. doi: 10.1016/j.devcel.2010.05.005
122. Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. (2009) 10:461–70. doi: 10.1038/ni.1726
123. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. (2010) 12:823–30. doi: 10.1038/ncb0910-823
124. Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. (2010) 116:4806–14. doi: 10.1182/blood-2010-06-288589
125. Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. (2008) 455:396–400. doi: 10.1038/nature07208
126. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. (2010) 32:227–39. doi: 10.1016/j.immuni.2009.12.006
127. Münz C. Antigen processing via autophagy–not only for MHC class II presentation anymore? Curr Opin Immunol. (2010) 22:89–93. doi: 10.1016/j.coi.2010.01.016
128. Lo TH, Weng IC, Chen HL, Liu FT. The role of galectins in the regulation of autophagy and inflammasome in host immunity. Semin Immunopathol. (2024) 46:6. doi: 10.1007/s00281-024-01018-5
129. Deretic V, Kimura T, Timmins G, Moseley P, Chauhan S, Mandell M. Immunologic manifestations of autophagy. J Clin Invest. (2015) 125:75–84. doi: 10.1172/JCI73945
130. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. (2013) 14:298–305. doi: 10.1038/ni.2524
131. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. (2014) 20:503–10. doi: 10.1038/nm.3521
132. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
133. Keller CW, Adamopoulos IE, Lünemann JD. Autophagy pathways in autoimmune diseases. J Autoimmun. (2023) 136:103030. doi: 10.1016/j.jaut.2023.103030
134. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. (2018) 14:243–51. doi: 10.1080/15548627.2017.1402992
135. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. (2008) 456:264–8. doi: 10.1038/nature07383
136. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. (2008) 456:259–63. doi: 10.1038/nature07416
137. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. (2005) 307:593–6. doi: 10.1126/science.1104904
138. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
139. Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. (2010) 189:925–35. doi: 10.1083/jcb.201002021
140. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
141. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
142. Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. (2007) 128:931–46. doi: 10.1016/j.cell.2006.12.044
143. Birgisdottir Å B, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. (2013) 126:3237–47. doi: 10.1242/jcs.126128
144. Swanlund JM, Kregel KC, Oberley TD. Investigating autophagy: quantitative morphometric analysis using electron microscopy. Autophagy. (2010) 6:270–7. doi: 10.4161/auto.6.2.10439
145. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. (2007) 3:542–5. doi: 10.4161/auto.4600
146. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. (2010) 140:313–26. doi: 10.1016/j.cell.2010.01.028
147. Martinet W, De Meyer GR, Andries L, Herman AG, Kockx MM. In situ detection of starvation-induced autophagy. J Histochem Cytochem. (2006) 54:85–96. doi: 10.1369/jhc.5A6743.2005
148. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. (2017) 18:1865. doi: 10.3390/ijms18091865
149. Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer. Autophagy. (2009) 5:585–9. doi: 10.4161/auto.5.5.8823
150. Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, et al. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy. (2011) 7:629–42. doi: 10.4161/auto.7.6.15100
151. Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. (2010) 6:784–93. doi: 10.4161/auto.6.6.12510
152. Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. (2009) 452:181–97. doi: 10.1016/S0076-6879(08)03612-4
153. Dennis PB, Mercer CA. The GST-BHMT assay and related assays for autophagy. Methods Enzymol. (2009) 452:97–118. doi: 10.1016/S0076-6879(08)03607-0
154. Ju JS, Miller SE, Jackson E, Cadwell K, Piwnica-Worms D, Weihl CC. Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy. (2009) 5:511–9. doi: 10.4161/auto.5.4.7761
155. Burney PG, Patel J, Newson R, Minelli C, Naghavi M. Global and regional trends in COPD mortality, 1990-2010. Eur Respir J. (2015) 45:1239–47. doi: 10.1183/09031936.00142414
156. Lareau SC, Fahy B, Meek P, Wang A. Chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med. (2019) 199:P1–p2. doi: 10.1164/rccm.1991P1
157. Teckman JH, Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. (2000) 279:G961–74. doi: 10.1152/ajpgi.2000.279.5.G961
158. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. (2009) 4:435–59. doi: 10.1146/annurev.pathol.4.110807.092145
159. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. (2008) 8:183–92. doi: 10.1038/nri2254
160. Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PloS One. (2008) 3:e3316. doi: 10.1371/journal.pone.0003316
161. Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. (2010) 107:18880–5. doi: 10.1073/pnas.1005574107
162. Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. (2013) 123:5212–30. doi: 10.1172/JCI69636
163. Cloonan SM, Lam HC, Ryter SW, Choi AM. Ciliophagy”: The consumption of cilia components by autophagy. Autophagy. (2014) 10:532–4. doi: 10.4161/auto.27641
164. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2015) 136:769–80. doi: 10.1016/j.jaci.2015.01.046
165. Sureshbabu A, Bhandari V. Targeting mitochondrial dysfunction in lung diseases: emphasis on mitophagy. Front Physiol. (2013) 4:384. doi: 10.3389/fphys.2013.00384
166. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. (2014) 124:3987–4003. doi: 10.1172/JCI74985
167. Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. (2015) 372:351–62. doi: 10.1056/NEJMra1300109
168. Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. (2015) 16:45–56. doi: 10.1038/nrg3849
169. Fiedorczuk K, Chen J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science. (2022) 378:284–90. doi: 10.1126/science.ade2216
170. Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. (2010) 12:863–75. doi: 10.1038/ncb2090
171. Feng Y, et al. Interplay of energy metabolism and autophagy. Autophagy. (2024) 20:4–14. doi: 10.1080/15548627.2023.2247300
172. De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy. (2014) 10:2053–74. doi: 10.4161/15548627.2014.973737
173. Junkins RD, Shen A, Rosen K, McCormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS One. (2013) 8:e72263. doi: 10.1371/journal.pone.0072263
174. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. (2014) 9:157–79. doi: 10.1146/annurev-pathol-012513-104706
175. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
176. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. (2001) 345:517–25. doi: 10.1056/NEJMra003200
177. Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. (2011) 187:3003–14. doi: 10.4049/jimmunol.1004081
178. Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PloS One. (2012) 7:e41394. doi: 10.1371/journal.pone.0041394
179. Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. (2012) 180:275–92. doi: 10.1016/j.ajpath.2011.09.019
180. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. (2015) 125:521–38. doi: 10.1172/JCI74942
181. Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PloS One. (2015) 10:e0121246. doi: 10.1371/journal.pone.0121246
182. Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J Immunol. (2016) 197:504–16. doi: 10.4049/jimmunol.1600265
183. Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. (2011) 183:649–58. doi: 10.1164/rccm.201005-0746OC
184. Teng RJ, Du J, Welak S, Guan T, Eis A, Shi Y, et al. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L651–63. doi: 10.1152/ajplung.00177.2011
185. Deng Y, Wu W, Guo S, Chen Y, Liu C, Gao X, et al. Altered mTOR and Beclin-1 mediated autophagic activation during right ventricular remodeling in monocrotaline-induced pulmonary hypertension. Respir Res. (2017) 18:53. doi: 10.1186/s12931-017-0536-7
186. Lee SJ, Kim HP, Jin Y, Choi AM, Ryter SW. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy. (2011) 7:829–39. doi: 10.4161/auto.7.8.15598
187. Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb). (2011) 91:497–509. doi: 10.1016/j.tube.2011.03.007
188. Sasindran SJ, Torrelles JB. Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front Microbiol. (2011) 2:2. doi: 10.3389/fmicb.2011.00002
189. Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. (2011) 3:104ra102. doi: 10.1126/scitranslmed.3003045
190. Andersson AM, Andersson B, Lorell C, Raffetseder J, Larsson M, Blomgran R. Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Sci Rep. (2016) 6:28171. doi: 10.1038/srep28171
191. Tzfadia O, Gijsbers A, Vujkovic A, Snobre J, Vargas R, Dewaele K, et al. Single nucleotide variation catalog from clinical isolates mapped on tertiary and quaternary structures of ESX-1-related proteins reveals critical regions as putative Mtb therapeutic targets. Microbiol Spectr. (2024) 12:e0381623. doi: 10.1128/spectrum.03816-23
192. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
193. Dong LX, Sun LL, Zhang X, Pan L, Lian LJ, Chen Z, et al. Negative regulation of mTOR activity by LKB1-AMPK signaling in non-small cell lung cancer cells. Acta Pharmacol Sin. (2013) 34:314–8. doi: 10.1038/aps.2012.143
194. Wang L, Wang R. Effect of rapamycin (RAPA) on the growth of lung cancer and its mechanism in mice with A549. Int J Clin Exp Pathol. (2015) 8:9208–13.
195. Abadi AH, Mahdavi M, Khaledi A, Esmaeili SA, Esmaeili D, Sahebkar A. Study of serum bactericidal and splenic activity of Total-OMP- CagA combination from Brucella abortus and Helicobacter pylori in BALB/c mouse model. Microb Pathog. (2018) 121:100–5. doi: 10.1016/j.micpath.2018.04.050
196. Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Combined Bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin Cancer Res. (2009) 15:6096–105. doi: 10.1158/1078-0432.CCR-09-0589
197. Kruse JP, Gu W. Modes of p53 regulation. Cell. (2009) 137:609–22. doi: 10.1016/j.cell.2009.04.050
198. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. (2012) 149:1536–48. doi: 10.1016/j.cell.2012.05.014
199. Zhao H, Dong F, Li Y, Ren X, Xia Z, Wang Y, et al. Inhibiting ATG5 mediated autophagy to regulate endoplasmic reticulum stress and CD4(+) T lymphocyte differentiation: Mechanisms of acupuncture’s effects on asthma. BioMed Pharmacother. (2021) 142:112045. doi: 10.1016/j.biopha.2021.112045
200. Poon AH, Choy DF, Chouiali F, Ramakrishnan RK, Mahboub B, Audusseau S, et al. Increased autophagy-related 5 gene expression is associated with collagen expression in the airways of refractory asthmatics. Front Immunol. (2017) 8:355. doi: 10.3389/fimmu.2017.00355
201. Chung KF, Dixey P, Abubakar-Waziri H, Bhavsar P, Patel PH, Guo S, et al. Characteristics, phenotypes, mechanisms and management of severe asthma. Chin Med J (Engl). (2022) 135:1141–55. doi: 10.1097/CM9.0000000000001990
202. Dickinson JD, Alevy Y, Malvin NP, Patel KK, Gunsten SP, Holtzman MJ, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. (2016) 12:397–409. doi: 10.1080/15548627.2015.1056967
203. Dickinson JD, Sweeter JM, Warren KJ, Ahmad IM, De Deken X, Zimmerman MC, et al. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. (2018) 14:272–84. doi: 10.1016/j.redox.2017.09.013
204. Li W, Wu Y, Zhao Y, Li Z, Chen H, Dong L, et al. MTOR suppresses autophagy-mediated production of IL25 in allergic airway inflammation. Thorax. (2020) 75:1047–57. doi: 10.1136/thoraxjnl-2019-213771
205. Eltokhy AK, Toema O, El-Deeb OS. The correlation between PINK-1/parkin mediated mitophagy, endoplasmic reticulum stress and total polyamines in pediatric bronchial asthma: an integrated network of pathways. Mol Biol Rep. (2022) 49:227–35. doi: 10.1007/s11033-021-06861-5
206. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. (2018) 16:341–54. doi: 10.1038/s41579-018-0003-6
207. García-Pérez BE, González-Rojas JA, Salazar MI, Torres-Torres C, Castrejón-Jiménez NS. Taming the autophagy as a strategy for treating COVID-19. Cells. (2020) 9:2679. doi: 10.3390/cells9122679
208. Ahmad L, Mostowy S, Sancho-Shimizu V. Autophagy-virus interplay: from cell biology to human disease. Front Cell Dev Biol. (2018) 6:155. doi: 10.3389/fcell.2018.00155
209. Mohamud Y, Xue YC, Liu H, Ng CS, Bahreyni A, Jan E, et al. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem Biophys Res Commun. (2021) 540:75–82. doi: 10.1016/j.bbrc.2020.12.091
210. Gassen NC, Niemeyer D, Muth D, Corman VM, Martinelli S, Gassen A, et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat Commun. (2019) 10:5770. doi: 10.1038/s41467-019-13659-4
211. Gassen NC, Papies J, Bajaj T, Emanuel J, Dethloff F, Chua RL, et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat Commun. (2021) 12:3818. doi: 10.1038/s41467-021-24007-w
212. Miao G, Zhao H, Li Y, Ji M, Chen Y, Shi Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. (2021) 56:427–442.e5. doi: 10.1016/j.devcel.2020.12.010
213. Zhao Z, Lu K, Mao B, Liu S, Trilling M, Huang A, et al. The interplay between emerging human coronavirus infections and autophagy. Emerg Microbes Infect. (2021) 10:196–205. doi: 10.1080/22221751.2021.1872353
214. Crother TR, Porritt RA, Dagvadorj J, Tumurkhuu G, Slepenkin AV, Peterson EM, et al. Autophagy limits inflammasome during chlamydia pneumoniae infection. Front Immunol. (2019) 10:754. doi: 10.3389/fimmu.2019.00754
215. Shimizu T. Inflammation-inducing factors of mycoplasma pneumoniae. Front Microbiol. (2016) 7:414. doi: 10.3389/fmicb.2016.00414
216. Thomas DR, Newton P, Lau N, Newton HJ. Interfering with Autophagy: The Opposing Strategies Deployed by Legionella pneumophila and Coxiella burnetii Effector Proteins. Front Cell Infect Microbiol. (2020) 10:599762. doi: 10.3389/fcimb.2020.599762
217. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. (2018) 314:C73–c87. doi: 10.1152/ajpcell.00110.2016
218. Li Y, Yu G, Yuan S, Tan C, Lian P, Fu L, et al. Cigarette smoke-induced pulmonary inflammation and autophagy are attenuated in ephx2-deficient mice. Inflammation. (2017) 40:497–510. doi: 10.1007/s10753-016-0495-z
219. Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF, Hsueh HW, et al. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann Surg. (2013) 257:352–63. doi: 10.1097/SLA.0b013e318269d0e2
220. Chen X, Tsvetkov AS, Shen HM, Isidoro C, Ktistakis NT, Linkermann A, et al. International consensus guidelines for the definition, detection, and interpretation of autophagy-dependent ferroptosis. Autophagy. (2024) 20:1213–46. doi: 10.1080/15548627.2024.2319901
221. Maule G, Arosio D, Cereseto A. Gene therapy for cystic fibrosis: progress and challenges of genome editing. Int J Mol Sci. (2020) 21:3903. doi: 10.3390/ijms21113903
222. Martinez FJ, Curtis JL, Albert R. Role of macrolide therapy in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. (2008) 3:331–50. doi: 10.2147/COPD.S681
223. McArdle JR, Talwalkar JS. Macrolides in cystic fibrosis. Clin Chest Med. (2007) 28:347–60. doi: 10.1016/j.ccm.2007.02.005
224. Esther CR Jr., Esserman DA, Gilligan P, Kerr A, Noone PG. Chronic Mycobacterium abscessus infection and lung function decline in cystic fibrosis. J Cyst Fibros. (2010) 9:117–23. doi: 10.1016/j.jcf.2009.12.001
225. Roux AL, Catherinot E, Ripoll F, Soismier N, Macheras E, Ravilly S, et al. Multicenter study of prevalence of nontuberculous mycobacteria in patients with cystic fibrosis in France. J Clin Microbiol. (2009) 47:4124–8. doi: 10.1128/JCM.01257-09
226. Han R, Ji X, Rong R, Li Y, Yao W, Yuan J, et al. MiR-449a regulates autophagy to inhibit silica-induced pulmonary fibrosis through targeting Bcl2. J Mol Med (Berl). (2016) 94:1267–79. doi: 10.1007/s00109-016-1441-0
227. Gui YS, Wang L, Tian X, Li X, Ma A, Zhou W, et al. mTOR overactivation and compromised autophagy in the pathogenesis of pulmonary fibrosis. PloS One. (2015) 10:e0138625. doi: 10.1371/journal.pone.0138625
228. Liu H, Cheng Y, Yang J, Wang W, Fang S, Zhang W, et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis. (2017) 8:e2657. doi: 10.1038/cddis.2017.78
229. Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. (2013) 123:3600–13. doi: 10.1172/JCI65592
230. Seyfarth HJ, Hammerschmidt S, Halank M, Neuhaus P, Wirtz HR. Everolimus in patients with severe pulmonary hypertension: a safety and efficacy pilot trial. Pulm Circ. (2013) 3:632–8. doi: 10.1086/674311
231. Samimi R, Hosseinpanahi A, Zaboli R, Peymani A, Rouhi S, Ahmadi Gooraji S, et al. Prevalence of vitamin D receptor genes polymorphisms in people with pulmonary tuberculosis: A systematic review and meta-analysis. Med J Islam Repub Iran. (2024) 38:32. doi: 10.47176/mjiri.38.32
232. Nnoaham KE, Clarke A. Low serum vitamin D levels and tuberculosis: a systematic review and meta-analysis. Int J Epidemiol. (2008) 37:113–9. doi: 10.1093/ije/dym247
233. Daley P, Jagannathan V, John KR, Sarojini J, Latha A, Vieth R, et al. Adjunctive vitamin D for treatment of active tuberculosis in India: a randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. (2015) 15:528–34. doi: 10.1016/S1473-3099(15)70053-8
234. Martineau AR, Timms PM, Bothamley GH, Hanifa Y, Islam K, Claxton AP, et al. High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet. (2011) 377:242–50. doi: 10.1016/S0140-6736(10)61889-2
235. Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. (2012) 11:457–68. doi: 10.1016/j.chom.2012.03.008
236. Pflaum J, Schlosser S, Müller M. p53 family and cellular stress responses in cancer. Front Oncol. (2014) 4:285. doi: 10.3389/fonc.2014.00285
237. Parrales A, Iwakuma T. Targeting oncogenic mutant p53 for cancer therapy. Front Oncol. (2015) 5:288. doi: 10.3389/fonc.2015.00288
238. Gibbons DL, Byers LA, Kurie JM. Smoking, p53 mutation, and lung cancer. Mol Cancer Res. (2014) 12:3–13. doi: 10.1158/1541-7786.MCR-13-0539
239. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. (2004) 303:844–8. doi: 10.1126/science.1092472
240. Kravchenko JE, Ilyinskaya GV, Komarov PG, Agapova LS, Kochetkov DV, Strom E, et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci U S A. (2008) 105:6302–7. doi: 10.1073/pnas.0802091105
241. Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. (2009) 15:376–88. doi: 10.1016/j.ccr.2009.03.003
242. Meher RK, Mir SA, Anisetti SS. In silico and in vitro investigation of dual targeting Prima-1(MET) as precision therapeutic against lungs cancer. J Biomol Struct Dyn. (2024) 42:4169–84. doi: 10.1080/07391102.2023.2219323
243. Nagano T, Tachihara M, Nishimura Y. Dacomitinib, a second-generation irreversible epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) to treat non-small cell lung cancer. Drugs Today (Barc). (2019) 55:231–6. doi: 10.1358/dot.2019.55.4.2965337
244. Fung C, Chen X, Grandis JR, Duvvuri U. EGFR tyrosine kinase inhibition induces autophagy in cancer cells. Cancer Biol Ther. (2012) 13:1417–24. doi: 10.4161/cbt.22002
245. Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. (2010) 70:288–98. doi: 10.1158/0008-5472.CAN-09-1751
246. Sini P, James D, Chresta C, Guichard S. Simultaneous inhibition of mTORC1 and mTORC2 by mTOR kinase inhibitor AZD8055 induces autophagy and cell death in cancer cells. Autophagy. (2010) 6:553–4. doi: 10.4161/auto.6.4.11671
247. Jin HO, Lee YH, Park JA, Kim JH, Hong SE, Kim HA, et al. Blockage of Stat3 enhances the sensitivity of NSCLC cells to PI3K/mTOR inhibition. Biochem Biophys Res Commun. (2014) 444:502–8. doi: 10.1016/j.bbrc.2014.01.086
248. Xu CX, Zhao L, Yue P, Fang G, Tao H, Owonikoko TK, et al. Augmentation of NVP-BEZ235’s anticancer activity against human lung cancer cells by blockage of autophagy. Cancer Biol Ther. (2011) 12:549–55. doi: 10.4161/cbt.12.6.16397
249. Wu SH, Hang LW, Yang JS, Chen HY, Lin HY, Chiang JH, et al. Curcumin induces apoptosis in human non-small cell lung cancer NCI-H460 cells through ER stress and caspase cascade- and mitochondria-dependent pathways. Anticancer Res. (2010) 30:2125–33.
250. Lin SS, Huang HP, Yang JS, Wu JY, Hsia TC, Lin CC, et al. DNA damage and endoplasmic reticulum stress mediated curcumin-induced cell cycle arrest and apoptosis in human lung carcinoma A-549 cells through the activation caspases cascade- and mitochondrial-dependent pathway. Cancer Lett. (2008) 272:77–90. doi: 10.1016/j.canlet.2008.06.031
251. Su Q, Pan J, Wang C, Zhang M, Cui H, Zhao X. Curcumin and baicalin co-loaded nanoliposomes for synergistic treatment of non-small cell lung cancer. Pharmaceutics. (2024) 16:973. doi: 10.3390/pharmaceutics16080973
252. Lin Y, Wang Y, Liu X, Yan J, Su L, Liu X. A novel derivative of tetrandrine (H1) induces endoplasmic reticulum stress-mediated apoptosis and prosurvival autophagy in human non-small cell lung cancer cells. Tumour Biol. (2016) 37:10403–13. doi: 10.1007/s13277-016-4950-0
253. Tang ZH, Chen X, Wang ZY, Chai K, Wang YF, Xu XH, et al. Induction of C/EBP homologous protein-mediated apoptosis and autophagy by licochalcone A in non-small cell lung cancer cells. Sci Rep. (2016) 6:26241. doi: 10.1038/srep26241
254. Ma J, Liu J, Lu C, Cai D. Pachymic acid induces apoptosis via activating ROS-dependent JNK and ER stress pathways in lung cancer cells. Cancer Cell Int. (2015) 15:78. doi: 10.1186/s12935-015-0230-0
255. Zhao R, Chen M, Jiang Z, Zhao F, Xi B, Zhang X, et al. Platycodin-D induced autophagy in non-small cell lung cancer cells via PI3K/akt/mTOR and MAPK signaling pathways. J Cancer. (2015) 6:623–31. doi: 10.7150/jca.11291
256. Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, et al. Induction of the Growth Inhibitor Igf-Binding Protein 3 by P53. Nature. (1995) 377:646–9. doi: 10.1038/377646a0
257. Drakos E, Atsaves V, Li J, Leventaki V, Andreeff M, Medeiros LJ, et al. Stabilization and Activation of P53 Downregulates Mtor Signaling through Ampk in Mantle Cell Lymphoma. Leukemia. (2009) 23:784–90. doi: 10.1038/leu.2008.348
258. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The Regulation of Ampk Beta1, Tsc2, and Pten Expression by P53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the Igf-1-Akt-Mtor Pathways. Cancer Res. (2007) 67:3043–53. doi: 10.1158/0008-5472.Can-06-4149
259. Budanov AV, Karin M. P53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and Mtor Signaling. Cell. (2008) 134:451–60. doi: 10.1016/j.cell.2008.06.028
260. Martoriati A, Doumont G, Alcalay M, Bellefroid E, Pelicci PG, Marine JC. Dapk1, Encoding an Activator of a P19arf-P53-Mediated Apoptotic Checkpoint, Is a Transcription Target of P53. Oncogene. (2005) 24:1461–6. doi: 10.1038/sj.onc.1208256
261. Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, et al. A Short Mitochondrial Form of P19arf Induces Autophagy and Caspase-Independent Cell Death. Mol Cell. (2006) 22:463–75. doi: 10.1016/j.molcel.2006.04.014
262. Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. Puma- and Bax-Induced Autophagy Contributes to Apoptosis. Cell Death Differ. (2009) 16:1135–45. doi: 10.1038/cdd.2009.28
263. Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, et al. Bh3-Only Proteins and Bh3 Mimetics Induce Autophagy by Competitively Disrupting the Interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy. (2007) 3:374–6. doi: 10.4161/auto.4237
264. Abraham AG, O'Neill E. Pi3k/Akt-Mediated Regulation of P53 in Cancer. Biochem Soc Trans. (2014) 42:798–803. doi: 10.1042/bst20140070
265. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of Pten Transcription by P53. Mol Cell. (2001) 8:317–25. doi: 10.1016/s1097-2765(01)00323-9
266. An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of Wild-Type P53 by Hypoxia-Inducible Factor 1alpha. Nature. (1998) 392:405–8. doi: 10.1038/32925
267. Chen D, Li M, Luo J, Gu W. Direct Interactions between Hif-1 Alpha and Mdm2 Modulate P53 Function. J Biol Chem. (2003) 278:13595–8. doi: 10.1074/jbc.C200694200
268. Gao W, Shen Z, Shang L, Wang X. Upregulation of Human Autophagy-Initiation Kinase Ulk1 by Tumor Suppressor P53 Contributes to DNA-Damage-Induced Cell Death. Cell Death Differ. (2011) 18:1598–607. doi: 10.1038/cdd.2011.33
269. Desai S, Liu Z, Yao J, Patel N, Chen J, Wu Y, et al. Heat Shock Factor 1 (Hsf1) Controls Chemoresistance and Autophagy through Transcriptional Regulation of Autophagy-Related Protein 7 (Atg7). J Biol Chem. (2013) 288:9165–76. doi: 10.1074/jbc.M112.422071
270. Yeo SY, Itahana Y, Guo AK, Han R, Iwamoto K, Nguyen HT, et al. Transglutaminase 2 Contributes to a Tp53-Induced Autophagy Program to Prevent Oncogenic Transformation. Elife. (2016) 5:e07101. doi: 10.7554/eLife.07101
271. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, et al. Regulation of Autophagy by Cytoplasmic P53. Nat Cell Biol. (2008) 10:676–87. doi: 10.1038/ncb1730
272. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. Tigar, a P53-Inducible Regulator of Glycolysis and Apoptosis. Cell. (2006) 126:107–20. doi: 10.1016/j.cell.2006.05.036
273. Xu J, Wang Y, Tan X, Jing H. Micrornas in Autophagy and Their Emerging Roles in Crosstalk with Apoptosis. Autophagy. (2012) 8:873–82. doi: 10.4161/auto.19629
274. Liao JM, Cao B, Zhou X, Lu H. New Insights into P53 Functions through Its Target Micrornas. J Mol Cell Biol. (2014) 6:206–13. doi: 10.1093/jmcb/mju018
275. Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 Controls the Levels of P53 by Regulating the Deubiquitination Activity of Usp10 and Usp13. Cell. (2011) 147:223–34. doi: 10.1016/j.cell.2011.08.037
276. Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Mariño G, Galluzzi L, et al. P53 Inhibits Autophagy by Interacting with the Human Ortholog of Yeast Atg17, Rb1cc1/Fip200. Cell Cycle. (2011) 10:2763–9. doi: 10.4161/cc.10.16.16868
277. Galluzzi L, Kepp O. Kroemer G. A New Role Cytoplasmic P53: Binding Destroying Double-Stranded Rna. Cell Cycle. (2010) 9:2491–2. doi: 10.4161/cc.9.13.12191
278. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette Smoke-Induced Autophagy Impairment Accelerates Lung Aging, Copd-Emphysema Exacerbations and Pathogenesis. Am J Physiol Cell Physiol. (2018) 314:C73–c87. doi: 10.1152/ajpcell.00110.2016
279. Araya J, Tsubouchi K, Sato N, Ito S, Minagawa S, Hara H, et al. Prkn-Regulated Mitophagy and Cellular Senescence During Copd Pathogenesis. Autophagy. (2019) 15:510–26. doi: 10.1080/15548627.2018.1532259
280. Wang Y, Liao S, Pan Z, Jiang S, Fan J, Yu S, et al. Hydrogen Sulfide Alleviates Particulate Matter-Induced Emphysema and Airway Inflammation by Suppressing Ferroptosis. Free Radic Biol Med. (2022) 186:1–16. doi: 10.1016/j.freeradbiomed.2022.04.014
281. Zhang L, Huang J, Dong R, Feng Y, Zhou M. Therapeutic Potential of Blt1 Antagonist for Copd: Involvement of Inducing Autophagy and Ameliorating Inflammation. Drug Des Devel Ther. (2019) 13:3105–16. doi: 10.2147/dddt.S215433
282. Bodas M, Patel N, Silverberg D, Walworth K, Vij N. Master Autophagy Regulator Transcription Factor Eb Regulates Cigarette Smoke-Induced Autophagy Impairment and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis. Antioxid Redox Signal. (2017) 27:150–67. doi: 10.1089/ars.2016.6842
283. Song B, Yan X, Li R, Zhang H. Ghrelin Ameliorates Chronic Obstructive Pulmonary Disease-Associated Infllammation and Autophagy. Biotechnol Appl Biochem. (2021) 68:356–65. doi: 10.1002/bab.1933
284. Li L, Zhang M, Zhang L, Cheng Y, Tu X, Lu Z. Klotho Regulates Cigarette Smoke-Induced Autophagy: Implication in Pathogenesis of Copd. Lung. (2017) 195:295–301. doi: 10.1007/s00408-017-9997-1
285. Liu Y, Xu J, Liu T, Wu J, Zhao J, Wang J, et al. Fstl1 Aggravates Cigarette Smoke-Induced Airway Inflammation and Airway Remodeling by Regulating Autophagy. BMC Pulm Med. (2021) 21:45. doi: 10.1186/s12890-021-01409-6
286. Le Y, Wang Y, Zhou L, Xiong J, Tian J, Yang X, et al. Cigarette Smoke-Induced Hmgb1 Translocation and Release Contribute to Migration and Nf-κb Activation through Inducing Autophagy in Lung Macrophages. J Cell Mol Med. (2020) 24:1319–31. doi: 10.1111/jcmm.14789
287. Wang L, Jiang W, Wang J, Xie Y, Wang W. Puerarin Inhibits Fundc1-Mediated Mitochondrial Autophagy and Cse-Induced Apoptosis of Human Bronchial Epithelial Cells by Activating the Pi3k/Akt/Mtor Signaling Pathway. Aging (Albany NY). (2022) 14:1253–64. doi: 10.18632/aging.203317
288. Chen S, Wang Y, Zhang H, Chen R, Lv F, Li Z, et al. The Antioxidant Mitoq Protects against Cse-Induced Endothelial Barrier Injury and Inflammation by Inhibiting Ros and Autophagy in Human Umbilical Vein Endothelial Cells. Int J Biol Sci. (2019) 15:1440–51. doi: 10.7150/ijbs.30193
289. Li W, Yan J, Xu J, Zhu L, Zhai C, Wang Y, et al. Vardenafil Alleviates Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease by Activating Autophagy Via the Ampk/Mtor Signalling Pathway: An in Vitro and in Vivo Study. In Vitro Cell Dev Biol Anim. (2023) 59:717–28. doi: 10.1007/s11626-023-00820-z
290. Han D, Wu X, Liu L, Shu W, Huang Z. Sodium Tanshinone Iia Sulfonate Protects Arpe-19 Cells against Oxidative Stress by Inhibiting Autophagy and Apoptosis. Sci Rep. (2018) 8:15137. doi: 10.1038/s41598-018-33552-2
291. Li D, Hu J, Wang T, Zhang X, Liu L, Wang H, et al. Silymarin Attenuates Cigarette Smoke Extract-Induced Inflammation Via Simultaneous Inhibition of Autophagy and Erk/P38 Mapk Pathway in Human Bronchial Epithelial Cells. Sci Rep. (2016) 6:37751. doi: 10.1038/srep37751
292. Chen CH, Li YR, Lin SH, Chang HH, Chai WH, Chan PC, et al. Tiotropium/Olodaterol Treatment Reduces Cigarette Smoke Extract-Induced Cell Death in Beas-2b Bronchial Epithelial Cells. BMC Pharmacol Toxicol. (2020) 21:74. doi: 10.1186/s40360-020-00451-0
293. Li X, Yang H, Sun H, Lu R, Zhang C, Gao N, et al. Taurine Ameliorates Particulate Matter-Induced Emphysema by Switching on Mitochondrial Nadh Dehydrogenase Genes. Proc Natl Acad Sci U.S.A. (2017) 114:E9655–e64. doi: 10.1073/pnas.1712465114
294. Zong DD, Liu XM, Li JH, Ouyang RY, Long YJ, Chen P, et al. Resveratrol Attenuates Cigarette Smoke Induced Endothelial Apoptosis by Activating Notch1 Signaling Mediated Autophagy. Respir Res. (2021) 22:22. doi: 10.1186/s12931-021-01620-3
295. Li Y, Yu G, Yuan S, Tan C, Xie J, Ding Y, et al. 14,15-Epoxyeicosatrienoic Acid Suppresses Cigarette Smoke Condensate-Induced Inflammation in Lung Epithelial Cells by Inhibiting Autophagy. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L970–l80. doi: 10.1152/ajplung.00161.2016
296. Son ES, Kim SH, Ryter SW, Yeo EJ, Kyung SY, Kim YJ, et al. Quercetogetin Protects against Cigarette Smoke Extract-Induced Apoptosis in Epithelial Cells by Inhibiting Mitophagy. Toxicol In Vitro. (2018) 48:170–8. doi: 10.1016/j.tiv.2018.01.011
297. Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K, et al. Azithromycin Blocks Autophagy and May Predispose Cystic Fibrosis Patients to Mycobacterial Infection. J Clin Invest. (2011) 121:3554–63. doi: 10.1172/jci46095
298. Signorelli P, Pivari F, Barcella M, Merelli I, Zulueta A, Dei Cas M, et al. Myriocin Modulates the Altered Lipid Metabolism and Storage in Cystic Fibrosis. Cell Signal. (2021) 81:109928. doi: 10.1016/j.cellsig.2021.109928
299. Assani K, Shrestha CL, Rinehardt H, Zhang S, Robledo-Avila F, Wellmerling J, et al. Ar-13 Reduces Antibiotic-Resistant Bacterial Burden in Cystic Fibrosis Phagocytes and Improves Cystic Fibrosis Transmembrane Conductance Regulator Function. J Cyst Fibros. (2019) 18:622–9. doi: 10.1016/j.jcf.2018.10.010
300. Vu CB, Bridges RJ, Pena-Rasgado C, Lacerda AE, Bordwell C, Sewell A, et al. Fatty Acid Cysteamine Conjugates as Novel and Potent Autophagy Activators That Enhance the Correction of Misfolded F508del-Cystic Fibrosis Transmembrane Conductance Regulator (Cftr). J Med Chem. (2017) 60:458–73. doi: 10.1021/acs.jmedchem.6b01539
301. Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, et al. A Novel Treatment of Cystic Fibrosis Acting on-Target: Cysteamine Plus Epigallocatechin Gallate for the Autophagy-Dependent Rescue of Class Ii-Mutated Cftr. Cell Death Differ. (2016) 23:1380–93. doi: 10.1038/cdd.2016.22
302. Hoch L, Bourg N, Degrugillier F, Bruge C, Benabides M, Pellier E, et al. Dual Blockade of Misfolded Alpha-Sarcoglycan Degradation by Bortezomib and Givinostat Combination. Front Pharmacol. (2022) 13:856804. doi: 10.3389/fphar.2022.856804
303. Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, et al. Cysteamine Re-Establishes the Clearance of Pseudomonas Aeruginosa by Macrophages Bearing the Cystic Fibrosis-Relevant F508del-Cftr Mutation. Cell Death Dis. (2017) 8:e2544. doi: 10.1038/cddis.2016.476
304. Baek AR, Hong J, Song KS, Jang AS, Kim DJ, Chin SS, et al. Spermidine Attenuates Bleomycin-Induced Lung Fibrosis by Inducing Autophagy and Inhibiting Endoplasmic Reticulum Stress (Ers)-Induced Cell Death in Mice. Exp Mol Med. (2020) 52:2034–45. doi: 10.1038/s12276-020-00545-z
305. Gong H, Lyu X, Liu Y, Peng N, Tan S, Dong L, et al. Eupatilin Inhibits Pulmonary Fibrosis by Activating Sestrin2/Pi3k/Akt/Mtor Dependent Autophagy Pathway. Life Sci. (2023) 334:122218. doi: 10.1016/j.lfs.2023.122218
306. Zhang X, Su J, Lin J, Liu L, Wu J, Yuan W, et al. Fu-Zheng-Tong-Luo Formula Promotes Autophagy and Alleviates Idiopathic Pulmonary Fibrosis by Controlling the Janus Kinase 2/Signal Transducer and Activator of Transcription 3 Pathway. J Ethnopharmacol. (2023) 314:116633. doi: 10.1016/j.jep.2023.116633
307. Pei X, Zheng F, Li Y, Lin Z, Han X, Feng Y, et al. Niclosamide Ethanolamine Salt Alleviates Idiopathic Pulmonary Fibrosis by Modulating the Pi3k-Mtorc1 Pathway. Cells. (2022) 11:346. doi: 10.3390/cells11030346
308. Qi J, Wu Y, Guo Z, Zhu S, Xiong J, Hu F, et al. Fibroblast Growth Factor 21 Alleviates Idiopathic Pulmonary Fibrosis by Inhibiting Pi3k-Akt-Mtor Signaling and Stimulating Autophagy. Int J Biol Macromol. (2024) 273:132896. doi: 10.1016/j.ijbiomac.2024.132896
309. Li T, Gao X, Jia R, Sun Y, Ding Y, Wang F, et al. Astragaloside Iv Inhibits Idiopathic Pulmonary Fibrosis through Activation of Autophagy by Mir-21-Mediated Pten/Pi3k/Akt/Mtor Pathway. Cell Mol Biol (Noisy-le-grand). (2024) 70:128–36. doi: 10.14715/cmb/2024.70.2.18
310. Zheng D, Guo J, Liang Z, Jin Y, Ding Y, Liu J, et al. Supramolecular Nanofibers Ameliorate Bleomycin-Induced Pulmonary Fibrosis by Restoring Autophagy. Adv Sci (Weinh). (2024) 11:e2401327. doi: 10.1002/advs.202401327
311. Wei Y, Sun L, Liu C, Li L. Naringin Regulates Endoplasmic Reticulum Stress and Mitophagy through the Atf3/Pink1 Signaling Axis to Alleviate Pulmonary Fibrosis. Naunyn Schmiedebergs Arch Pharmacol. (2023) 396:1155–69. doi: 10.1007/s00210-023-02390-z
312. Chu L, Zhuo J, Huang H, Chen W, Zhong W, Zhang J, et al. Tetrandrine Alleviates Pulmonary Fibrosis by Inhibiting Alveolar Epithelial Cell Senescence through Pink1/Parkin-Mediated Mitophagy. Eur J Pharmacol. (2024) 969:176459. doi: 10.1016/j.ejphar.2024.176459
313. Chen S, Wei Y, Li S, Miao Y, Gu J, Cui Y, et al. Zanubrutinib Attenuates Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the Tgf-β1 Signaling Pathway. Int Immunopharmacol. (2022) 109316. doi: 10.1016/j.intimp.2022.109316
314. Li X, Wang Y, Liang J, Bi Z, Ruan H, Cui Y, et al. Bergenin Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice Via Inhibiting Tgf-β1 Signaling Pathway. Phytother Res. (2021) 35:5808–22. doi: 10.1002/ptr.7239
315. Zhao X, Gao M, Liang J, Chen Y, Wang Y, Wang Y, et al. Slc7a11 Reduces Laser-Induced Choroidal Neovascularization by Inhibiting Rpe Ferroptosis and Vegf Production. Front Cell Dev Biol. (2021) 9:639851. doi: 10.3389/fcell.2021.639851
316. Cho IH, Choi YJ, Gong JH, Shin D, Kang MK, Kang YH. Astragalin Inhibits Autophagy-Associated Airway Epithelial Fibrosis. Respir Res. (2015) 16:51. doi: 10.1186/s12931-015-0211-9
317. Saito S, Zhuang Y, Shan B, Danchuk S, Luo F, Korfei M, et al. Tubastatin Ameliorates Pulmonary Fibrosis by Targeting the Tgfβ-Pi3k-Akt Pathway. PloS One. (2017) 12:e0186615. doi: 10.1371/journal.pone.0186615
318. Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, et al. Novel Mechanisms for the Antifibrotic Action of Nintedanib. Am J Respir Cell Mol Biol. (2016) 54:51–9. doi: 10.1165/rcmb.2014-0445OC
319. Kurita Y, Araya J, Minagawa S, Hara H, Ichikawa A, Saito N, et al. Pirfenidone Inhibits Myofibroblast Differentiation and Lung Fibrosis Development During Insufficient Mitophagy. Respir Res. (2017) 18:114. doi: 10.1186/s12931-017-0600-3
320. Yu N, Wang N, Zhang W, Xue J, Zhou Q, Hu F, et al. Dihydroartemisinin (Dha) Inhibits Myofibroblast Differentiation through Inducing Ferroptosis Mediated by Ferritinophagy. Heliyon. (2024) 10:e27276. doi: 10.1016/j.heliyon.2024.e27276
321. Bao C, Liang S, Han Y, Yang Z, Liu S, Sun Y, et al. The Novel Lysosomal Autophagy Inhibitor (Roc-325) Ameliorates Experimental Pulmonary Hypertension. Hypertension. (2023) 80:70–83. doi: 10.1161/hypertensionaha.122.19397
322. Ye W, Tang T, Li Z, Li X, Huang Q. Piperlongumine Attenuates Vascular Remodeling in Hypoxic Pulmonary Hypertension by Regulating Autophagy. J Cardiol. (2022) 79:134–43. doi: 10.1016/j.jjcc.2021.08.023
323. Ren HH, Niu Z, Guo R, Fu M, Li HR, Zhang XY, et al. Rhodiola Crenulata Extract Decreases Fatty Acid Oxidation and Autophagy to Ameliorate Pulmonary Arterial Hypertension by Targeting Inhibiton of Acylcarnitine in Rats. Chin J Nat Med. (2021) 19:120–33. doi: 10.1016/s1875-5364(21)60013-4
324. Liu Y, Xu Y, Zhu J, Li H, Zhang J, Yang G, et al. Metformin Prevents Progression of Experimental Pulmonary Hypertension Via Inhibition of Autophagy and Activation of Adenosine Monophosphate-Activated Protein Kinase. J Vasc Res. (2019) 56:117–28. doi: 10.1159/000498894
325. Shang P, Sun SB, Liu BH. Umbelliferone Improves Chronic Hypoxia-Induced Pulmonary Hypertension by Inhibiting the Rhoa/Rock Signaling Pathway and Autophagy. Sheng Li Xue Bao. (2022) 74:555–62.
326. Wu YC, Wang WT, Lee SS, Kuo YR, Wang YC, Yen SJ, et al. Glucagon-Like Peptide-1 Receptor Agonist Attenuates Autophagy to Ameliorate Pulmonary Arterial Hypertension through Drp1/Nox- and Atg-5/Atg-7/Beclin-1/Lc3β Pathways. Int J Mol Sci. (2019) 20:3435. doi: 10.3390/ijms20143435
327. Ibrahim YF, Shults NV, Rybka V, Suzuki YJ. Docetaxel Reverses Pulmonary Vascular Remodeling by Decreasing Autophagy and Resolves Right Ventricular Fibrosis. J Pharmacol Exp Ther. (2017) 363:20–34. doi: 10.1124/jpet.117.239921
328. Zhang X, Liu Q, Zhang C, Sheng J, Li S, Li W, et al. Puerarin Prevents Progression of Experimental Hypoxia-Induced Pulmonary Hypertension Via Inhibition of Autophagy. J Pharmacol Sci. (2019) 141:97–105. doi: 10.1016/j.jphs.2019.09.010
329. Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, et al. Chloroquine Prevents Progression of Experimental Pulmonary Hypertension Via Inhibition of Autophagy and Lysosomal Bone Morphogenetic Protein Type Ii Receptor Degradation. Circ Res. (2013) 112:1159–70. doi: 10.1161/circresaha.111.300483
330. Wang X, Ibrahim YF, Das D, Zungu-Edmondson M, Shults NV, Suzuki YJ. Carfilzomib Reverses Pulmonary Arterial Hypertension. Cardiovasc Res. (2016) 110:188–99. doi: 10.1093/cvr/cvw047
331. He Y, Cao X, Guo P, Li X, Shang H, Liu J, et al. Quercetin Induces Autophagy Via Foxo1-Dependent Pathways and Autophagy Suppression Enhances Quercetin-Induced Apoptosis in Pasmcs in Hypoxia. Free Radic Biol Med. (2017) 103:165–76. doi: 10.1016/j.freeradbiomed.2016.12.016
332. Zhang H, Gong Y, Wang Z, Jiang L, Chen R, Fan X, et al. Apelin Inhibits the Proliferation and Migration of Rat Pasmcs Via the Activation of Pi3k/Akt/Mtor Signal and the Inhibition of Autophagy under Hypoxia. J Cell Mol Med. (2014) 18:542–53. doi: 10.1111/jcmm.12208
333. Grobs Y, Awada C, Lemay SE, Romanet C, Bourgeois A, Toro V, et al. Preclinical Investigation of Trifluoperazine as a Novel Therapeutic Agent for the Treatment of Pulmonary Arterial Hypertension. Int J Mol Sci. (2021) 22:2919. doi: 10.3390/ijms22062919
334. Gomez-Arroyo J, Sakagami M, Syed AA, Farkas L, Van Tassell B, Kraskauskas D, et al. Iloprost Reverses Established Fibrosis in Experimental Right Ventricular Failure. Eur Respir J. (2015) 45:449–62. doi: 10.1183/09031936.00188013
335. Afsal K, Selvaraj P. Effect of 1,25-Dihydroxyvitamin D3 on the xpression of Mannose Receptor, Dc-Sign and Autophagy Genes in Pulmonary Tuberculosis. Tuberculosis (Edinb). (2016) 99:1–10. doi: 10.1016/j.tube.2016.03.010
336. Guzmán-Beltrán S, Rubio-Badillo M, Juárez E, Hernández-Sánchez F, Torres M. Nordihydroguaiaretic Acid (Ndga) and α-Mangostin Inhibit the Growth of Mycobacterium Tuberculosis by Inducing Autophagy. Int Immunopharmacol. (2016) 31:149–57. doi: 10.1016/j.intimp.2015.12.027
337. Kim JK, Lee HM, Park KS, Shin DM, Kim TS, Kim YS, et al. Mir144* Inhibits Antimicrobial Responses against Mycobacterium Tuberculosis in Human Monocytes and Macrophages by Targeting the Autophagy Protein Dram2. Autophagy. (2017) 13:423–41. doi: 10.1080/15548627.2016.1241922
338. Liu J, Ming S, Song W, Meng X, Xiao Q, Wu M, et al. B and T Lymphocyte Attenuator Regulates Autophagy in Mycobacterial Infection Via the Akt/Mtor Signal Pathway. Int Immunopharmacol. (2021) 91:107215. doi: 10.1016/j.intimp.2020.107215
339. Juárez E, Carranza C, Sánchez G, González M, Chávez J, Sarabia C, et al. Loperamide Restricts Intracellular Growth of Mycobacterium Tuberculosis in Lung Macrophages. Am J Respir Cell Mol Biol. (2016) 55:837–47. doi: 10.1165/rcmb.2015-0383OC
340. Bai X, Oberley-Deegan RE, Bai A, Ovrutsky AR, Kinney WH, Weaver M, et al. Curcumin Enhances Human Macrophage Control of Mycobacterium Tuberculosis Infection. Respirology. (2016) 21:951–7. doi: 10.1111/resp.12762
341. Xie C, Zhou X, Liang C, Li X, Ge M, Chen Y, et al. Apatinib Triggers Autophagic and Apoptotic Cell Death Via Vegfr2/Stat3/Pd-L1 and Ros/Nrf2/P62 Signaling in Lung Cancer. J Exp Clin Cancer Res. (2021) 40:266. doi: 10.1186/s13046-021-02069-4
342. Wang XR, Jiang ZB, Xu C, Meng WY, Liu P, Zhang YZ, et al. Andrographolide Suppresses Non-Small-Cell Lung Cancer Progression through Induction of Autophagy and Antitumor Immune Response. Pharmacol Res. (2022) 179:106198. doi: 10.1016/j.phrs.2022.106198
343. Zhu L, Wang Y, Lv W, Wu X, Sheng H, He C, et al. Schizandrin a Can Inhibit Non−Small Cell Lung Cancer Cell Proliferation by Inducing Cell Cycle Arrest, Apoptosis and Autophagy. Int J Mol Med. (2021) 48:214. doi: 10.3892/ijmm.2021.5047
344. Tang X, Ding H, Liang M, Chen X, Yan Y, Wan N, et al. Curcumin Induces Ferroptosis in Non-Small-Cell Lung Cancer Via Activating Autophagy. Thorac Cancer. (2021) 12:1219–30. doi: 10.1111/1759-7714.13904
345. Yan X, Yao C, Fang C, Han M, Gong C, Hu D, et al. Rocaglamide Promotes the Infiltration and Antitumor Immunity of Nk Cells by Activating Cgas-Sting Signaling in Non-Small Cell Lung Cancer. Int J Biol Sci. (2022) 18:585–98. doi: 10.7150/ijbs.65019
346. Zou Y, Zhang G, Li C, Long H, Chen D, Li Z, et al. Discovery of Tryptanthrin and Its Derivatives and Its Activities against Nsclc in Vitro Via Both Apoptosis and Autophagy Pathways. Int J Mol Sci. (2023) 24:1450. doi: 10.3390/ijms24021450
347. Teng JF, Mei QB, Zhou XG, Tang Y, Xiong R, Qiu WQ, et al. Polyphyllin Vi Induces Caspase-1-Mediated Pyroptosis Via the Induction of Ros/Nf-κb/Nlrp3/Gsdmd Signal Axis in Non-Small Cell Lung Cancer. Cancers (Basel). (2020) 12:193. doi: 10.3390/cancers12010193
348. Xu Z, Pan Z, Jin Y, Gao Z, Jiang F, Fu H, et al. Inhibition of Prkaa/Ampk (Ser485/491) Phosphorylation by Crizotinib Induces Cardiotoxicity Via Perturbing Autophagosome-Lysosome Fusion. Autophagy. (2024) 20:416–36. doi: 10.1080/15548627.2023.2259216
349. Liu X, Yin M, Dong J, Mao G, Min W, Kuang Z, et al. Tubeimoside-1 Induces Tfeb-Dependent Lysosomal Degradation of Pd-L1 and Promotes Antitumor Immunity by Targeting Mtor. Acta Pharm Sin B. (2021) 11:3134–49. doi: 10.1016/j.apsb.2021.03.039
350. Zhang C, Jin Y. Ginsenoside Rg5 Induces Nsclc Cell Apoptosis and Autophagy through Pi3k/Akt/Mtor Signaling Pathway. Hum Exp Toxicol. (2024) 43:9603271241229140. doi: 10.1177/09603271241229140
351. Li J, Fan Y, Zhang Y, Liu Y, Yu Y, Ma M. Resveratrol Induces Autophagy and Apoptosis in Non-Small-Cell Lung Cancer Cells by Activating the Ngfr-Ampk-Mtor Pathway. Nutrients. (2022) 14:2413. doi: 10.3390/nu14122413
352. Luo D, He F, Liu J, Dong X, Fang M, Liang Y, et al. Pseudolaric Acid B Suppresses Nsclc Progression through the Ros/Ampk/Mtor/Autophagy Signalling Pathway. BioMed Pharmacother. (2024) 175:116614. doi: 10.1016/j.biopha.2024.116614
353. Hou G, Hu W, Sang Y, Gan X, Xu H, Hu Q, et al. Corynoxine Triggers Cell Death Via Activating Pp2a and Regulating Akt-Mtor/Gsk3β Axes in Nsclc. Biochem Pharmacol. (2024) 222:116110. doi: 10.1016/j.bcp.2024.116110
354. Wang J, Liu X, Zheng H, Liu Q, Zhang H, Wang X, et al. Morusin Induces Apoptosis and Autophagy Via Jnk, Erk and Pi3k/Akt Signaling in Human Lung Carcinoma Cells. Chem Biol Interact. (2020) 331:109279. doi: 10.1016/j.cbi.2020.109279
355. Luo D, Dai X, Tian H, Fan C, Xie H, Chen N, et al. Sophflarine a, a Novel Matrine-Derived Alkaloid from Sophora Flavescens with Therapeutic Potential for Non-Small Cell Lung Cancer through Ros-Mediated Pyroptosis and Autophagy. Phytomedicine. (2023) 116:154909. doi: 10.1016/j.phymed.2023.154909
356. Huang HW, Bow YD, Wang CY, Chen YC, Fu PR, Chang KF, et al. Dfiq, a Novel Quinoline Derivative, Shows Anticancer Potential by Inducing Apoptosis and Autophagy in Nsclc Cell and in Vivo Zebrafish Xenograft Models. Cancers (Basel). (2020) 12:1348. doi: 10.3390/cancers12051348
357. O'Neill EJ, Sze NSK, MacPherson REK, Tsiani E. Carnosic Acid against Lung Cancer: Induction of Autophagy and Activation of Sestrin-2/Lkb1/Ampk Signalling. Int J Mol Sci. (2024) 25:1950. doi: 10.3390/ijms25041950
358. Kan Y, Song M, Cui X, Yang Q, Zang Y, Li Q, et al. Muyin Extract Inhibits Non-Small-Cell Lung Cancer Growth by Inducing Autophagy and Apoptosis in Vitro and in Vivo. Phytomedicine. (2022) 96:153834. doi: 10.1016/j.phymed.2021.153834
359. Muñoz-Guardiola P, Casas J, Megías-Roda E, Solé S, Perez-Montoyo H, Yeste-Velasco M, et al. The Anti-Cancer Drug Abtl0812 Induces Er Stress-Mediated Cytotoxic Autophagy by Increasing Dihydroceramide Levels in Cancer Cells. Autophagy. (2021) 17:1349–66. doi: 10.1080/15548627.2020.1761651
360. Li XQ, Cheng XJ, Wu J, Wu KF, Liu T. Targeted Inhibition of the Pi3k/Akt/Mtor Pathway by (+)-Anthrabenzoxocinone Induces Cell Cycle Arrest, Apoptosis, and Autophagy in Non-Small Cell Lung Cancer. Cell Mol Biol Lett. (2024) 29:58. doi: 10.1186/s11658-024-00578-6
361. Xia YC, Zha JH, Sang YH, Yin H, Xu GQ, Zhen J, et al. Ampk Activation by Asp4132 Inhibits Non-Small Cell Lung Cancer Cell Growth. Cell Death Dis. (2021) 12:365. doi: 10.1038/s41419-021-03655-2
362. Li J, Yan L, Luo J, Tong L, Gao Y, Feng W, et al. Baicalein Suppresses Growth of Non-Small Cell Lung Carcinoma by Targeting Map4k3. BioMed Pharmacother. (2021) 133:110965. doi: 10.1016/j.biopha.2020.110965
363. Wu Z, Li W, Tang Q, Huang L, Zhan Z, Li Y, et al. A Novel Aniline Derivative from Peganum Harmala L. Promoted Apoptosis Via Activating Pi3k/Akt/Mtor-Mediated Autophagy in Non-Small Cell Lung Cancer Cells. Int J Mol Sci. (2023) 24:12626. doi: 10.3390/ijms241612626
364. Fang C, Wu W, Ni Z, Liu Y, Luo J, Zhou Y, et al. Ailanthone Inhibits Non-Small Cell Lung Cancer Growth and Metastasis through Targeting Upf1/Gas5/Ulk1 Signaling Pathway. Phytomedicine. (2024) 128:155333. doi: 10.1016/j.phymed.2023.155333
Keywords: autophagy, pulmonary diseases, apoptosis, autophagosome, COPD
Citation: Lin L, Lin Y, Han Z, Wang K, Zhou S, Wang Z, Wang S and Chen H (2024) Understanding the molecular regulatory mechanisms of autophagy in lung disease pathogenesis. Front. Immunol. 15:1460023. doi: 10.3389/fimmu.2024.1460023
Received: 05 July 2024; Accepted: 07 October 2024;
Published: 31 October 2024.
Edited by:
Suraj P. Parihar, University of Cape Town, South AfricaReviewed by:
Claudia Carranza, National Institute of Respiratory Diseases-Mexico (INER), MexicoGagandeep Kaur, University of Rochester Medical Center, United States
Copyright © 2024 Lin, Lin, Han, Wang, Zhou, Wang, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongyu Han, aHp5Y3p5MTk5N0AxNjMuY29t; Siyu Wang, SG9yaXpvbndzeTAyMjJAMTI2LmNvbQ==; Haoran Chen, Y2hlbmhycnJAMTYzLmNvbQ==
†These authors have contributed equally to this work