 Dan Long
Dan Long Chenhan Mao
Chenhan Mao Yin Xu
Yin Xu Ying Zhu1*
Ying Zhu1*- 1Department of Gastroenterology, The First Hospital of Hunan University of Chinese Medicine, Changsha, Hunan, China
- 2Affiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, China
Ulcerative colitis (UC) is characterized by chronic non-recessive inflammation of the intestinal mucosa involving both innate and adaptive immune responses. Currently, new targeted therapies are urgently needed for UC, and neutrophil extracellular traps (NETs) are new therapeutic options. NETs are DNA-based networks released from neutrophils into the extracellular space after stimulation, in which a variety of granule proteins, proteolytic enzymes, antibacterial peptides, histones, and other network structures are embedded. With the deepening of the studies on NETs, their regulatory role in the development of autoimmune and autoinflammatory diseases has received extensive attention in recent years. Increasing evidence indicates that excess NETs exacerbate the inflammatory response in UC, disrupting the structure and function of the intestinal mucosal barrier and increasing the risk of thrombosis. Although NETs are usually assigned a deleterious role in promoting the pathological process of UC, they also appear to have a protective role in some models. Despite such progress, comprehensive reviews describing the therapeutic promise of NETs in UC remain limited. In this review, we discuss the latest evidence for the formation and degradation of NETs, focusing on their double-edged role in UC. Finally, the potential implications of NETs as therapeutic targets for UC will be discussed. This review aims to provide novel insights into the pathogenesis and therapeutic options for UC.
1 Introduction
Ulcerative colitis (UC) is a complex chronic inflammatory bowel disease (IBD) affecting the mucosa of the rectum and colon. Its main clinical symptoms are recurrent diarrhea, mucopurulent bloody stool, and may be accompanied by extra-intestinal manifestations of varying degrees of severity (1). UC is characterized by recurrent episodes and remissions, which may result in prolonged and burdensome complications that seriously affect patients’ quality of life. A substantial body of evidence suggests that multiple factors are involved in the pathogenesis of UC, including autoimmune disorders, genetic susceptibility, defects in the intestinal epithelial mucosal barrier, and imbalances in the gut microbiota (2–4). The main pathological change in UC is an abnormal mucosal immune and inflammatory response, the main features of which include pro-inflammatory cytokines, oxygen free radicals, nitrogen production, and activation of inflammatory cells. Among them, neutrophils play a key role in intestinal homeostasis (5).
Neutrophils, as one of the most important immune cells in innate immunity, are also the body’s first line of defense against external microorganisms. It mediates antimicrobial activity through phagocytosis and degranulation, resulting in immune defense and killing of pathogens. In addition, activated neutrophils release neutrophil extracellular traps (NETs), which are complex networks comprised of DNA, histones, and granule proteins (6). NETs effectively capture pathogens and secrete antimicrobial proteins to kill them (7, 8). On the other hand, over-activated NETs may further amplify the inflammatory response, leading to tissue damage (9). Apart from infectious diseases, NETs have successively been reported to play a significant role in numerous non-infectious diseases, such as autoimmune diseases (10, 11), cancers (12–15), cardiovascular diseases (16–20), and so on. UC, an autoinflammatory disease, has not been fully elucidated in terms of its specific pathogenesis. The potential role of NETs in the pathogenesis of UC is a relatively new area of research. Here, we focus on the latest evidence for the formation and degradation of NETs, the mechanisms associated with their involvement in UC pathophysiology, and their potential role in UC therapeutic targets.
2 The role of neutrophils in UC
Neutrophils are the most abundant immune cells in the body, accounting for approximately 70% of peripheral blood leukocytes. Neutrophils are known for their rapid recruitment to sites of infection or tissue damage to accommodate pathogens. They can activate pathways that ultimately facilitate sustained inflammation reduction and mucosal healing (21–23). However, persistent activation and over-recruitment of neutrophils is a common feature of numerous inflammatory diseases. Neutrophils produce inflammatory factors and large amounts of reactive oxygen species (ROS), disrupt the intestinal epithelial mucosal barrier, recruit and activate other immune cells, and activate redox-sensitive inflammatory pathways (24–27). Neutrophil infiltration is associated with disease activity in UC (21, 28). Neutrophils are a widely used and reliable component of the disease scoring system for UC (29).
Neutrophils exhibit a dual role in UC, either favoring the abatement of inflammation or being deleterious when over-activated, leading to collateral tissue damage. In other words, both functional defects and hyperreactivity of neutrophils contribute to intestinal inflammation, and functional neutrophils are essential to maintain intestinal homeostasis.
3 Neutrophil extracellular traps
In 1996, Takei et al. (30) first discovered that neutrophils exhibit special morphological changes such as the dissociation of lobulated nuclei and rupture of nuclear and cell membranes under the stimulation of phorbol myristate acetate (PMA). In 2004, Brinkmann et al. (6) defined this new type of specific cell death, different from apoptosis and necrosis, as NETs by further validation. With subsequent studies, NETs are thought to have a DNA backbone in which a variety of active proteins are embedded, including histones, cathepsin G (CG), neutrophil elastase (NE), matrix metalloproteinase 9 (MMP-9), myeloperoxidase (MPO), calprotectin and other granule proteins, protein hydrolases, antimicrobial peptides, histones, etc. Although the NET proteome composition is fairly stable, it may be enriched with different protein components depending on the stimulus received (31).
3.1 NET formation
The generation of NETs by neutrophils is regulated by neutrophil intrinsic and extrinsic factors and pathways (32). The release of NETs occurs through a cell death process named NETosis (33). It is triggered by a variety of stimulants such as bacteria, viruses (34–36), fungi (37), activated platelets (38–40), immune complexes, cytokines, and chemokines (41). Studies have shown that the mechanism of NETs formation may vary depending on the initial stimulus that activates the neutrophils (42). There are two main modes of NET formation: lytic or suicidal NET formation and nonlytic or vital NET formation (43, 44).
3.1.1 Lytic NETs formation
Lytic NET formation involves morphological changes in neutrophils, where activated neutrophils become flattened, lose their nucleoli, have their chromatin decondensed, and die after NETs are formed (summarized in Figure 1). In contrast to other cell death processes, such as apoptosis, necrosis, or pyroptosis, chromatin decondensation is the main defining feature of NETosis. NETs are generated by neutrophils stimulated by bacteria, fungi, cytokines, lipopolysaccharide (LPS) and PMA, with PMA being the most inducible (45). After stimulation with PMA or IL-8, calcium is released from the endoplasmic reticulum (ER), followed by the entrance of extracellular calcium through channels in the cellular membrane (46–49). As a result, the intracellular calcium concentrations increase in activated neutrophils (50). Interestingly, the activation of peptidyl arginine deiminase-4 (PAD4) depends on high levels of intracellular calcium concentration (51). As is well known, PAD4 is a key driver of NETosis, which causes histone citrullination and thus induces chromatin decondensation process (52, 53). Citrullinated histone H3 (Cit-H3) is generally considered as a specific marker of NET (54–57).
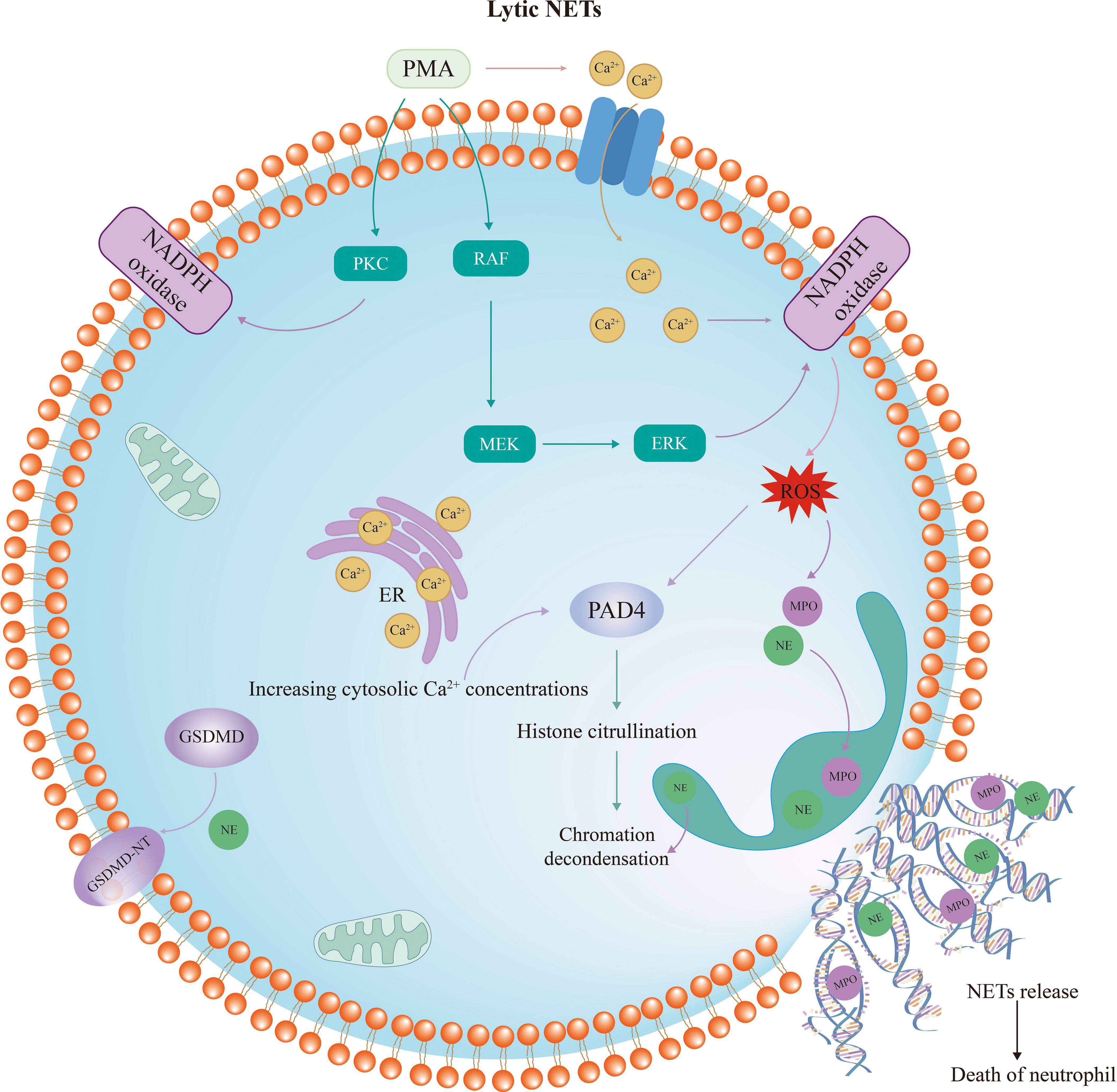
Figure 1 Diagram of lytic neutrophil extracellular trap formation. The RAF/MEK/ERK pathway and protein kinase C are activated by PMA, resulting in the phosphorylation of NADPH oxidase and ROS formation. This process depends on high calcium concentrations. Subsequently, PAD4 is activated and NE and MPO are translocated from the azurophilic granules to the nucleus. NE, MPO and PAD4 lead to histone citrullination and chromatin decondensation. Upon rupture of the nuclear membrane, the decondensed chromatin enters the cytoplasm and mixes with granular proteins. Finally, the cytoplasma membrane ruptures and the modified chromatin is released from neutrophils, marking the completion of NETosis. PMA, phorbol myristate acetate; PKC, protein kinase C; NADPH, nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species; NE, neutrophil elastase; MPO, myeloperoxidase; PAD4, peptidyl arginine deiminase-4; GSDMD, Gasdermin D.
Apart from high intracellular calcium concentration, the mechanism of NETosis is closely related to nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. PMA stimulation leads to the activation of the RAF/MEK/ERK pathway and protein kinase C (PKC), which in turn contributes to the phosphorylation of NADPH oxidase and the production of ROS (45). Then, NADPH oxidase and ROS complexes induce the translocation of NE and MPO from neutrophil granules into the nucleus (58), along with the activation of PAD4 (33). Although MPO does not directly decondense chromatin in isolated nuclei or degrade histones in vitro, it was demonstrated to augment chromatin decondensation mediated by NE (59). It is reported that MPO binds to chromatin and activates NE in small azurophilic structures visible in vitro and in vivo. NE disrupts actin filaments in the cytoplasm while translocating to the nucleus along with MPO (58). In the nucleus, serine proteases and NE degrade histones, further promoting chromatin decondensation. Upon rupture of the nuclear membrane, the decondensed chromatin enters the cytoplasm and mixes with granular proteins to form NET (33). Finally, the cell membrane ruptures, releasing NET and causing neutrophil death. This process is dependent on NADPH oxidase to promote ROS production and occurs slowly, typically taking three to four hours.
Interestingly, emerging evidence suggests that Gasdermin D (GSDMD), which is often considered an execution factor in pyroptosis, also plays a crucial role in cell lysis and NET release (60, 61). Inhibition of GSDMD with disulfiram or genic deletion has been shown to eliminate NET formation (62). In neutrophils, GSDMD is usually cleaved and activated by two pathways. When initiated by classical stimulus such as PMA or extracellular pathogens, NE converts GSDMD to the active form GSDMD-NT, which mediates the formation of pores in the nuclear, granular, and plasma membranes and enhances the release of NE and other granular components (60, 63). Additionally, it is well known that GSDMD could be cleaved by caspases (64). When stimulated by LPS or cytosolic Gram-negative bacteria, GSDMD is activated by caspases. Among them, Caspase-11 is required for GSDMD-dependent NET formation (61). It has been reported that the specific deletion of Caspase-11 in neutrophils significantly inhibited NET formation (65). Caspase-11 and GSDMD are not only required for plasma membrane rupture in neutrophils during the final stages of NET release, but are also essential for the early features of NETosis, including nuclear foliation and DNA amplification, which are mediated by nuclear membrane permeabilization and histone degradation induced by Caspase-11 and GSDMD (61).
3.1.2 Nonlytic NETs release
A nonlytic and NADPH-oxidase-independent mechanism of NET formation was subsequently discovered, which appears to maintain neutrophil integrity and viability by actively releasing DNA-containing vesicles (33, 43). Unlike lytic NET formation, this process releases NETs less than an hour after stimulation. Nonlytic NET release plays a more important role in regulating pathogen infection. This form of NET formation is triggered by a sudden elevation of intracellular calcium that results in the expulsion of nuclear chromatin and granule proteins, causing anucleate cytoplasm to remain capable of migration and phagocytosis (summarized in Figure 2). Staphylococcus aureus and Candida albicans activate Toll-like receptor 2 (TLR2) and complement receptors, respectively, while platelets activated by LPS and Escherichia coli activate TLR4 (66, 67). TLRs and complement receptors activate PAD4 and activated PAD4 triggers histone citrullination, resulting in chromatin decondensation. The decondensed chromatin enters into the cytoplasm and binds granzymes such as MPO, NE, and other proteins, eventually releasing into the extracellular space. However, instead of plasma membrane disruption through vesicle release, protein-modified chromatin is secreted through vesicles. The nuclear and cellular membranes remain intact during this process, and neutrophil viability and functions such as phagocytosis and chemotaxis are unaffected (44). Although this pathway is not dependent on NADPH oxidase-derived ROS, a recent study has demonstrated that calcium ion carriers induce the production of mitochondrial ROS (68). After being stimulated by granulocyte-macrophage colony-stimulating factor or LPS, neutrophils release mitochondrial DNA (mtDNA) mixed with granule proteins (69). Additionally, it has been proven that deletion of mtDNA results in a significant reduction in NADPH-oxidase-independent NET production, suggesting that mitochondria are also important in NET formation (70). The specific mechanism of mitochondria in this process requires further study.
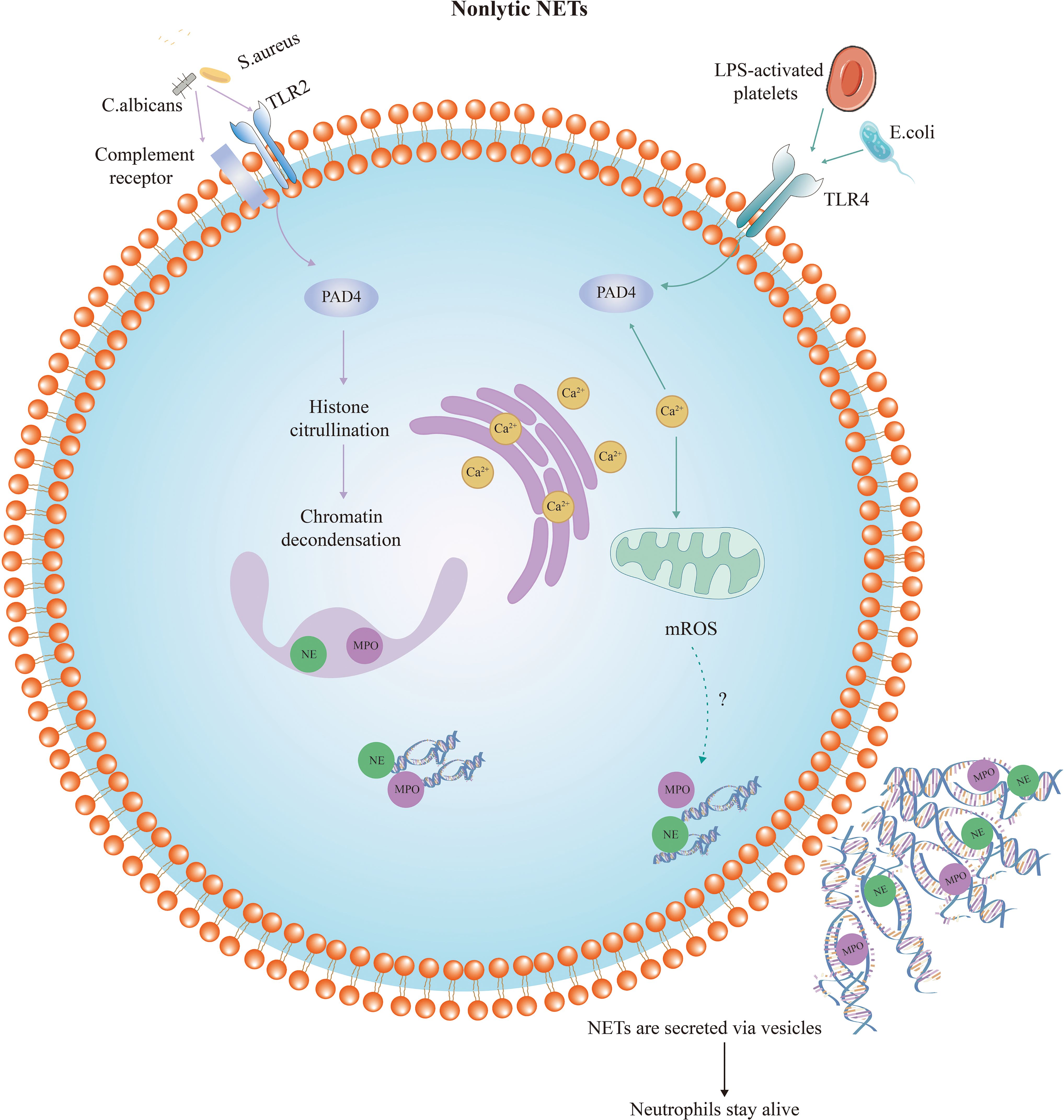
Figure 2 Diagram of nonlytic neutrophil extracellular trap formation. Compared with suicidal NETosis, nonlytic NET formation is completed in a shorter period of time and may occur in the absence of NADPH oxidase and ROS. This form of NET formation is also triggered by a sudden elevation of intracellular calcium levels. Nonlytic NET formation is initiated by stimuli such as Staphylococcus aureus activating TLR2, or Escherichia coli activating TLR4. PAD4 is activated and NE and MPO translocate to the nucleus to stimulate chromatin decondensation. Decondensed chromatin decorated with granulin and histones is packaged in vesicles budding from the nucleus. Then, these vesicles are expelled from intact neutrophils and form NETs near the neutrophils. Thus, the neutrophils remain intact and maintain their function.TLR, Toll-like receptor; LPS, lipopolysaccharide; NE, neutrophil elastase; MPO, myeloperoxidase; PAD4, peptidyl arginine deiminase-4.
3.2 NET degradation
Although numerous researchers have been focusing on the molecular mechanisms of NET formation, little is known regarding NET degradation. Actually, the balance between the formation and degradation of NETs is critical for maintaining NET homeostasis. Since DNA is the main component of NETs, DNA degradation enzymes play an instrumental role in NET degradation. In autoimmune diseases, reduced Deoxyribonuclease (DNase) activity is strongly correlated with NET accumulation. DNase is regarded as an essential enzyme for degrading NET in vivo by cleaving circulating free DNA (71). DNase consists of two families, DNase I and DNase II. The DNase I family is composed of four members including DNase I, DNase1L1, DNase1L2, and DNase1L3, whereas the DNase II family consists of DNase II α and DNase II β (72). The kinetics of hydrolysis are influenced by the structure and sequence of the DNA substrate. Double-stranded DNA (dsDNA) is cleaved by DNase I 100-500 times faster than single-stranded DNA (ssDNA) (73, 74). It was reported that DNase1 inhibitors or anti-NET antibodies preventing DNase1 entry into NETs both resulted in impaired NET clearance (71). Li et al. revealed that the downregulation of DNase1L3 expression is a critical cause of impaired DNA clearance in NETs (75). In addition, the accumulation of NETs and their components is associated with the formation of anti-dsDNA, anti-histones, and anti-nucleosome antibodies, which are recognized as pathogenic factors in systemic lupus erythematosus (SLE) (76).
Apart from the DNase family, other enzymes have been revealed to disrupt the structure of NETs, such as 3′-exonucleases (TREX1 and TREX2) (77, 78). Interestingly, macrophages and dendritic cells were found to exert an important role in the intracellular and extracellular degradation of NETs. NET degradation mediated by extracellular DNase1L3 was observed in dendritic cells, while intracellular degradation of NETs by macrophages relied on the function of TREX1 (77). A growing number of researches emphasized the prominent role of macrophages in NET degradation (79, 80). Farrera et al. confirmed that macrophages effectively removed NETs by taking up extracellular DNA (79). Macrophage-secreted DNase I facilitates this process by extracellularly digesting large fragments of NETs, whereas complement factor 1q (C1q) promotes NETs opsonization. NETs phagocytosed by macrophages are degraded in the lysosomal compartment. Interestingly, a recent study indicated that the polarization of macrophages affected their ability to degrade NETs (81). In particular, proinflammatory polarization promotes NET degradation through enhanced macropinocytosis.
3.3 Functions of NETs
NETs were originally described as host defense mechanisms for trapping or killing neutrophilic pathogenic microorganisms. NETs encapsulate proteins such as histone, NE, MPO, and CG, which trap pathogens and prevent them from spreading to secondary sites of infection, exerting anti-bacterial, fungal, and viral effects (6, 31). In neutrophils, NADPH is produced by glucose 6-phosphate in the oxidation branch of the pentose phosphate pathway. A recent study by Ulrich and colleagues demonstrated that low levels of NADPH and ROS in patients with defects in glucose-6-phosphate dehydrogenase caused impaired formation of NETs and greatly increased the risk of bacterial infection (82). Indeed, impairing NET formation might exacerbate inflammation and worsen conditions (7, 83). NET also modulates other immune cells. For instance, by inhibiting dendritic cell activation, it promotes the Th2 response and facilitates inflammation to subside (84). However, NET is a double-edged sword. There is growing evidence that when there is an imbalance between the formation and removal of NETs, the deposition of excess NETs in tissues and organs promotes an inflammatory response (85, 86) and drives disease progression (9, 87, 88). NETs recruit macrophages and other pro-inflammatory cells or proteins, promote the release of inflammatory factors such as IL-1β and IL-6, and activate NLRP3 inflammatory vesicles to further amplify the inflammatory response (89–91). Overall, the anti-inflammatory effects of NETs are counteracted by pro-inflammatory effects in disease. A growing body of research suggests that NETs are associated with the progression of several diseases and may serve as potential biomarkers for disease diagnosis and prognosis (92–95).
4 Increased NETs formation in UC
A growing number of studies have shown that NET-related proteins have enhanced expression in the inflamed colon of UC patients (96–99). Proteomic studies revealed that the abundance of NET-associated proteins in colonic mucosal samples from patients with UC was, on average, 42.2 times higher than in normal colons (100). Western blotting analysis and quantitative immunoblotting indicated that PAD4 expression was dramatically enhanced in inflamed mucosa of UC patients compared to Crohn’s disease (CD) patients or healthy individuals (98). Western blotting analysis has also shown that the expression of NE, MPO, and Cit-H3 was significantly up-regulated in colon samples from UC patients. Moreover, the double-immunofluorescence assay indicated that these three proteins were co-localized in UC mucosa. Similarly, confocal microscopy analysis also exhibited positive staining for citH3 and NE, overlapping with diffuse DNA scaffolding, confirming NET deposition in the colon of patients with active UC (101). Additionally, recent studies suggest that plasma levels of NET are significantly elevated in UC patients (101, 102). Other evidence points out that neutrophils isolated from UC patients are more likely to form NETs when stimulated in vitro (98, 101).
Apart from patients with UC, it has been demonstrated that NETs and related proteins are significantly increased in a mouse model of experimental colitis (98, 103). A recent study by Li and colleagues showed elevated serum levels of cell-free DNA (cfDNA) and increased NET formation in mice with dextran sulfate sodium (DSS)-induced colitis (101). Mechanistically, LINC00668, which is highly enriched in intestinal epithelial cell (IEC)-derived exosomes, mediates the translocation of NE from cytoplasmic granules to the nucleus, thus stimulating histone cleavage and chromatin decondensation and triggering NETs release (104). Other evidence suggests that high levels of antineutrophil cytoplasm autoantibodies (ANCA) in serum induce neutrophil aggregation and foster NET generation (105).
Damage-associated molecular patterns (DAMPs) are molecules released in response to cellular stress or tissue damage and have been demonstrated to function as direct pro-inflammatory mediators (106, 107). Several researches have identified DAMPs capable of inducing NET formation in UC (108). For example, cfDNA is a DAMP capable of enhancing NET generation (109). Interestingly, cfDNA is significantly increased in the serum of UC patients as well as in murine UC models, and its plasma concentration is positively correlated with clinical severity (110, 111). cfDNA has a strong potential to trigger NETosis, resulting in endothelial cell injury (112). High-mobility group box 1 (HMGB1) is another DAMP known for its ability to promote NETs (113, 114). Recent evidence suggests that TLR4 and C-X-C motif chemokine receptor 4 (CXCR4) are specific receptors for HMGB1 (115). Notably, HMGB1 not only induces NETosis but is also involved in the extrusion of NETs. Chen et al. detected that HMGB1 was significantly higher in the inflamed colon of UC patients (116). They also demonstrated that anti-HMGB1 neutralizing-antibody improved intestinal barrier function and inflammation in DSS-induced colitis mice.
Moreover, in UC, cytokines are important mediators in inducing NETosis. As is well known, IL-1β, IL-6, tumour necrosis factor α (TNF-α), and other cytokines are expressed at relatively higher levels in the intestinal tissues of UC patients (117). These cytokines trigger macrophages and neutrophils to phagocytose and to release effector mediators. It has been demonstrated that neutrophils from UC patients produced significantly more NETs under the stimulation of TNF-α (98). In addition, NET formation was significantly reduced in colons of UC patients treated with infliximab, a TNF-α inhibitor, indicating a strong stimulatory effect of TNF-α in NETosis (98). UC patients show significantly enhanced expression of IL-8 (118, 119), a cytokine that not only stimulates neutrophil recruitment but also triggers NETosis (120, 121). Similarly, IL-6, a cytokine widely increased in UC patients and colitis mice (122, 123), serves as another potent inducer of NETosis (124, 125). However, there are still substantial gaps in the upstream mechanisms of NETosis induction in UC, which need to be further explored.
5 Impaired clearance of NETs in UC
As mentioned above, several autoimmune diseases have been linked to dysregulated NET clearance (126). The balance between generation and degradation of NETs is critical in UC and other autoimmune diseases. Some findings suggest that in UC, the increase in NETs may be attributed to decreased clearance rather than merely increased production (101). Li et al. (101)detected impaired degradation of NETs in the plasma of patients with active UC. DNase I activity has been reported to be significantly lower in IBD patients than in healthy individuals (127). DNase I activity is strongly negatively correlated with the serum concentration of anti-nucleosome antibodies. Impaired DNaseI-mediated NET clearance is probably related to the pathogenesis of IBD. Thus, the ability of UC patients to clear NETs and specific mechanisms deserve further research.
6 The double role of NETs in UC
6.1 The role of NETs in the pathogenesis of UC
6.1.1 NETs exacerbate inflammatory response in UC
UC mainly manifests as a chronic, recurrent, non-specific inflammatory response in the intestinal mucosa. Neutrophil infiltration correlates with endoscopic severity and systemic inflammatory indices, whereas serum levels of C-reactive protein and fecal lactoferrin and calprotectin may serve as sensitive markers of intestinal inflammation (128–130). Notably, both lactoferrin and calprotectin are key components of NETs (128). NET is associated with diverse immune and inflammatory diseases, including UC, and is thought to maintain mucosal inflammation in this disease (98).
It has also been shown in vitro that NETs enhance the secretion of TNF-α and IL-1β in UC lamina propria mononuclear cells by promoting the phosphorylation of ERK (98). As shown in Figure 3, NETs induce the release of pro-inflammatory cytokines from macrophages, thus stimulating local and systemic inflammatory responses (131). A recent study showed that NETs significantly boosted the response of monocyte-derived macrophages to low-dose LPS and increased the release of TNF-α, IL-6, and monocyte chemotactic protein-1 (MCP-1) (101). PAD4 is considered a biomarker of NETosis as its suppression or gene deficiency in neutrophils inhibits this process (132). Numerous researchers have been focusing on the critical role of PAD4 in NETs of UC. Considerable studies have revealed that PAD4 expression is significantly elevated in colonic samples from UC patients (96, 133). Immunohistochemistry analysis of paired colon sections from UC patients showed more pronounced PAD4 expression in inflamed areas compared to uninvolved mucosa from the same patients (98). Blocking the formation of NETs by PAD4 knockout alleviates clinical colitis indices, intestinal inflammation, and barrier dysfunction (134).
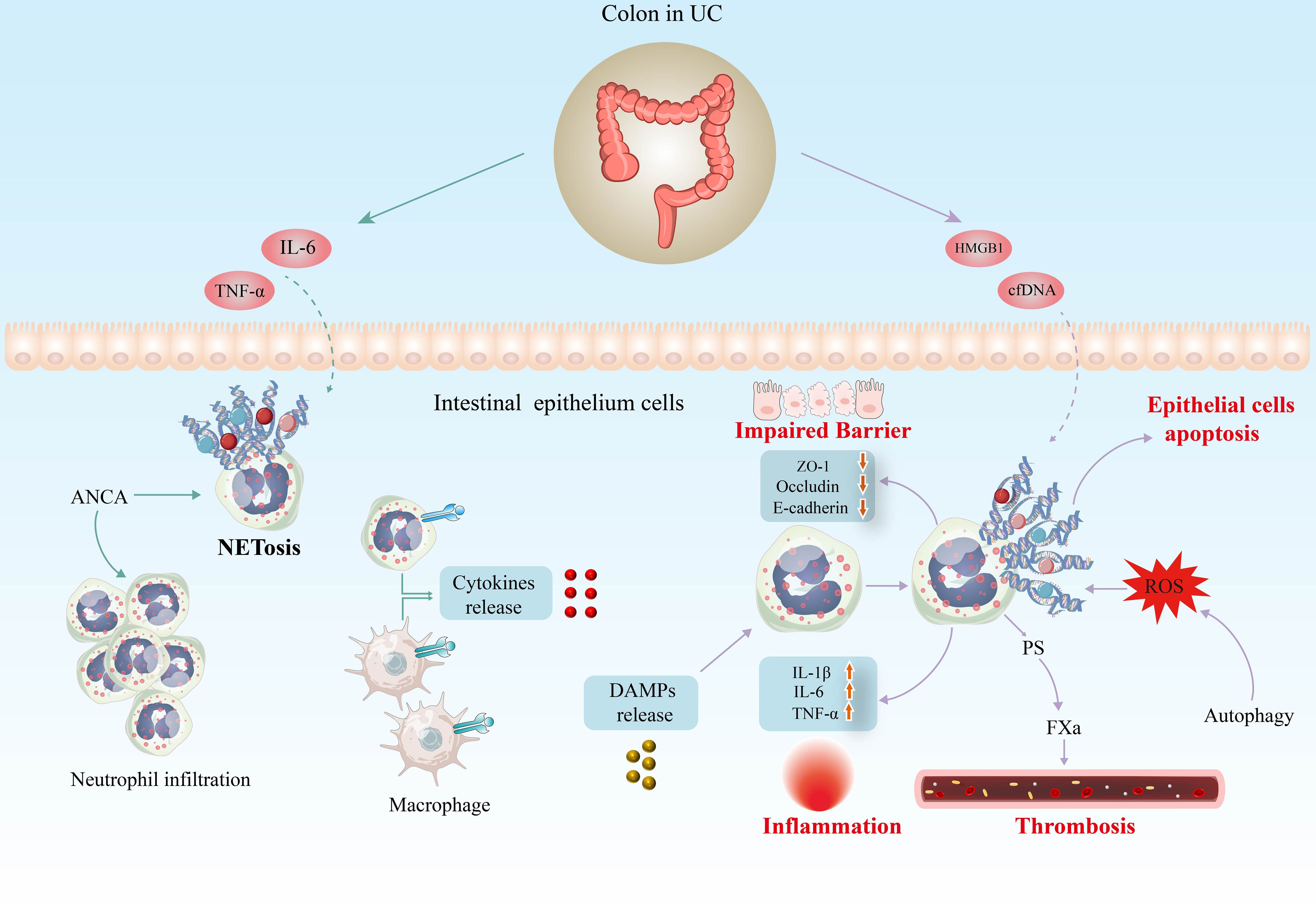
Figure 3 Potential implications of NETs in the pathogenesis of UC. Inflammatory factors such as IL-6 and TNF-α are potent inducers of NETosis in UC models. In addition, high levels of antineutrophil cytoplasm autoantibodies (ANCA) in serum induce neutrophil infiltration and foster NET generation. Several researches have identified DAMPs capable of inducing NET formation in UC, including cfDNA and HMGB1. Increasing evidence indicates that excess NETs exacerbate the inflammatory response in UC, disrupting the structure and function of the intestinal mucosal barrier and increasing the risk of thrombosis. IL-6, interleukin 6; TNF-α, tumour necrosis factor α; cfDNA, cell-free DNA; HMGB1, high-mobility group box 1; DAMP, damage-associated molecular pattern; ROS, reactive oxygen species; PS, phosphatidylserine.
Increasing studies have shown that cellular autophagy may play an essential role in the formation of NETs (135–137). Inhibition of autophagy leads to reduced generation of ROS and NETs (138). Another researcher revealed that inhibition of ATG7, a critical autophagy-associated protein, suppressed autophagy and significantly reduced NET formation (139). Moreover, increased NET formation in UC patients has been demonstrated to be autophagy-dependent and is associated with increased expression of regulated in development and DNA damage response 1 (REDD1) in neutrophils. The REDD1/autophagy/NETs axis is involved in the IL-1β-driven inflammatory response in UC patients (97).
6.1.2 NETs impair epithelial barrier function in UC
Impaired intestinal epithelial barrier function due to IEC injury is one of the key features of the pathophysiology of UC. Also, damage to the vascular endothelium cell results in delayed healing of the localized ulcerated mucosa, exacerbating intestinal damage. It was found that in mouse with DSS-induced colitis, the presence of NET structures was not limited to the lamina propria and epithelium, but also in the intestinal lumen (103). NETs increase bacterial translocation by increasing intestinal barrier permeability and disrupting colonic epithelial barrier function. Further studies revealed that the NET-induced deterioration of barrier function was attributed to the promotion of apoptosis in IECs (103). Compared with control mice, expression of the tight junction proteins occludin and ZO-1 as well as the adhesion junction component E-cadherin was reduced in Caco-2 monolayers treated with NETs or histones (103). NETs-associated histones alter the integrity of tight junctions and adhere to junctional proteins and induce IEC death, leading to increased intestinal epithelial permeability (140). Whether NETs are cytotoxic to endothelial cells, leading to intestinal tissue damage in UC, remains to be investigated.
6.1.3 NETs and thrombotic tendency in UC
Patients with IBD face three times the risk of thromboembolic events than the general population (141). In recent years, the association between NETs and arteriovenous thrombosis has attracted widespread attention. The network structure of NETs provides a scaffold for the deposition of platelets, erythrocytes, fibrinogen, platelet adhesion factor, and other substances that favor thrombosis (142). At the same time, a number of components of NETs actively trigger platelet activation and blood coagulation. There is already considerable evidence of a link between NETs and thrombosis in IBD (101, 143, 144). Research has shown that incubation of normal platelets with NETs from patients with active UC and CD resulted in a significant 32% increase in their procoagulant activity and a 42% increase in their ability to support fibrin formation. Another study (101) revealed that NET-induced platelet activation is mediated by signaling through TLR2 and TLR4. NETs from patients with active UC trigger increased exposure to phosphatidylserine in endothelial cells in a time-dependent manner, which promotes thrombin generation as well as the production of intrinsic and exogenous FXa complexes. Researchers found a significant increase in NETs in inferior vena cava thrombosis in DSS-induced IBD mice (104). Further studies discovered that this was related to exosomes released by IECs under inflammatory conditions. The specific mechanism of NETs on the thrombosis tendency in UC deserves further in-depth study.
6.2 The beneficial role of NETs in UC
Although NETs are often assigned a deleterious role in promoting inflammatory responses, there is growing evidence that they exert beneficial anti-inflammatory effects in a variety of conditions. For example, during severe sepsis, NETs have proved indispensable in trapping circulating bacteria to prevent systemic infection (67). NETs have been demonstrated to promote the regression of gouty inflammation through the degradation of cytokines and chemokines by serine proteases (7). When NET formation is impaired, monosodium urate crystals induce uncontrolled production of inflammatory mediators. Hahn and other researchers have also found that NETs work through a similar mechanism to reduce inflammation in periodontitis (145). In rheumatoid arthritis, NETs inhibit IL-6 secretion from LPS-activated macrophages and induce secretion of the anti-inflammatory factor IL-10 (146).
The formation of NETs during necrotizing enterocolitis has been reported to reduce intestinal bacterial translocation and attenuate systemic inflammation (83). Conversely, the reduction of NETs may lead to increased systemic inflammation in mice, ultimately resulting in the severe consequences of bacteremia and significantly reduced survival. Another study confirmed that activation of triggering receptor expressed on myeloid cell-1 (TREM-1) clears pathogens and protects the intestinal barrier by increasing the production of NETs and IL-22 by CD177 neutrophils, which is likely to be an effective therapeutic strategy for IBD (147). In addition, a recent study showed that PAD4-deficient mice had an exacerbated course of colitis and a significant increase in rectal hemorrhage compared to controls (133). The PAD4-dependent NET formation is closely associated with the remodeling of the blood clot into a secondary immune thrombus, thereby preventing rectal hemorrhage in UC. Therefore, caution must be exercised when targeting NET as a treatment for UC.
7 NETs as potential therapeutic targets in UC
The higher expression of NETs in UC plays an influential part in its pathogenesis and development, so targeting NETs to treat UC has gradually become a research hotspot. The regulation of NET generation or DNA degradation appears to be two possible effective strategies in UC therapy, and a balance of these two approaches may be better for positive outcomes. Recent studies have identified a number of drugs, including phytomedicine, that suppress UC through targeting NETs, as outlined in Table 1.
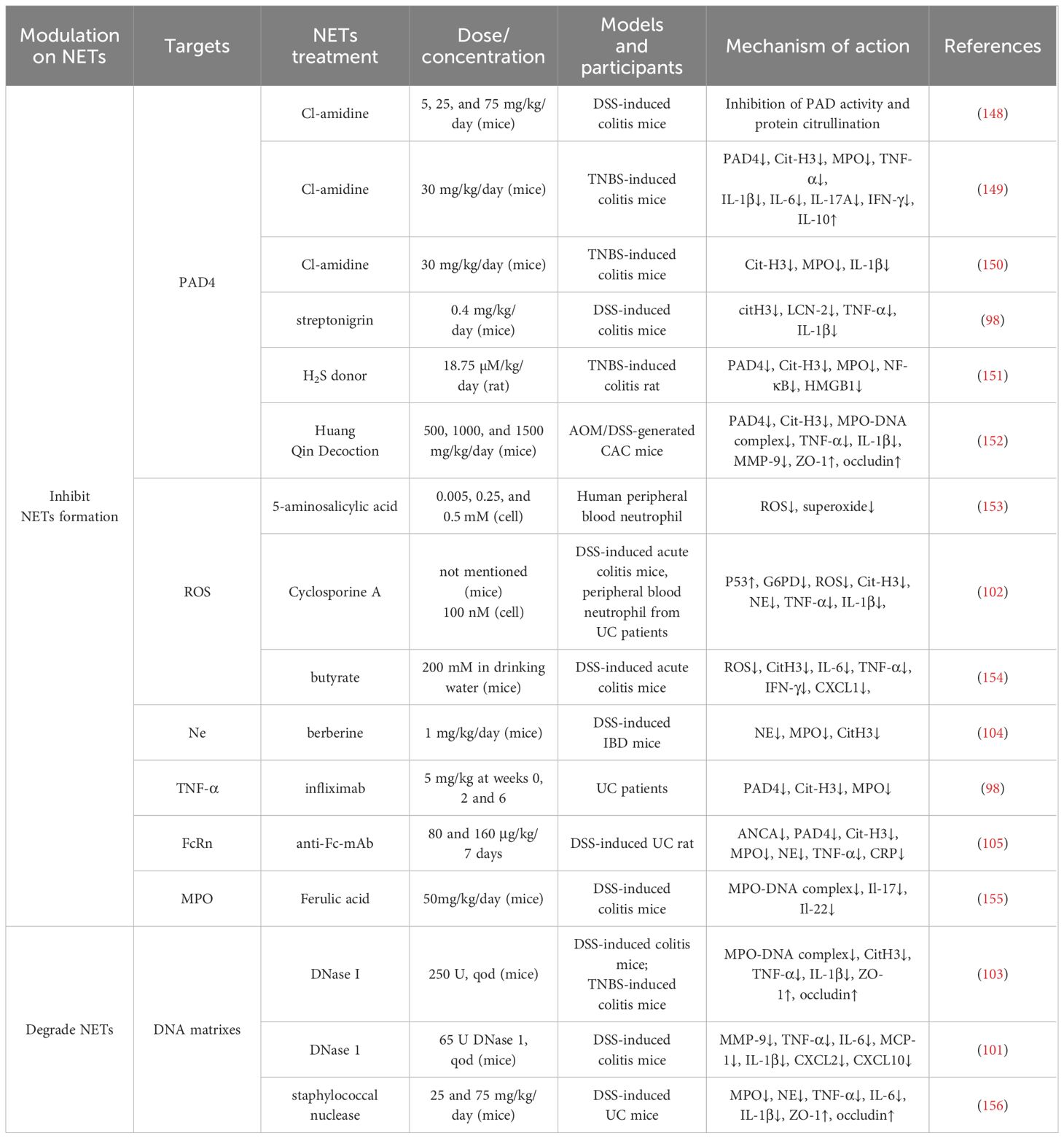
Table 1 Summary of potential NETs-targeted therapies in UC.
7.1 PAD4 inhibitors
Theoretically, any important link in the formation of NETs may become a potential target for the treatment of UC (summarized in Table 1). As mentioned above, PAD4 is a key enzyme in the formation of NETs. Therefore, it is also an important target for blocking the pathological effects of NETs. At present, the most representative PAD inhibitor in the UC field is the irreversible inhibitor Cl-amidine. According to reports, Cl-amidine blocked the formation of NET structure, reduced the expression of PAD4 and cit-H3 in the colon tissue, and effectively alleviated clinical colitis index in TNBS-induced colitis mice (149). The inhibition of PAD4 mediated by CL-amidine reduces the expression of pro-inflammatory cytokines such as TNF- α, IL-1 β, and IL-6, while the expression of anti-inflammatory factor IL-10 is also upregulated (149). CL-amidine also has antioxidant consequences in a colitis mouse model, with the ability to suppress leukocyte activation and prevent colon epithelial DNA damage (157).
In addition, streptonigrin, a selective PAD4 inhibitor, has also been shown to reduce the expression of pro-inflammatory cytokines such as TNF-α and IL-1 β, as well as NETs-associated proteins, alleviating colonic inflammation (98). Interestingly, a recent study showed that H2S donor significantly inhibited PAD4, Cit-H3 and MPO expression in TNBS-induced colitis rats (151). It appears to exert an anti-inflammatory effect by inhibiting NET formation and downregulating NF-κB and HMGB1 expression.
7.2 NADPH/ROS inhibitors
Similarly, NET formation is suppressed by inhibiting the expression of proteins such as Ne (158) and ROS (153). Cyclosporine A (CsA), a well-known immunosuppressive drug, is routinely prescribed for the treatment of patients with steroid-refractory acute severe ulcerative colitis (ASUC) (159, 160). It not only suppresses IL-2 secretion by T cells, but also interferes with dendritic cell (DC) migration (161, 162). Strikingly, CsA suppresses a variety of neutrophil processes, including ROS production and NET formation (70, 163). It has been demonstrated to decrease ROS generation in isolated human neutrophils (159, 164). A recent study revealed that CsA inhibits apoptosis and migration as well as the release of ROS, MPO, and antimicrobial peptides from neutrophils in ASUC patients (165). Mechanistically, CsA inhibits Sirtuin 6 (SIRT6) expression and subsequently promotes hypoxia‐inducible factor‐1α (HIF-1α) expression in neutrophils as well as glycolysis and the tricarboxylic acid cycle to limit neutrophil overactivation, thus alleviating mucosal inflammation in ASUC patients. Recently, Xu et al. (102) identified that CsA directly reduces the activity of pentose phosphate pathway (PPP) rate-limiting enzyme G6PD via activating P53 protein and represses PPP metabolism to produce ROS, thereby reducing ROS-dependent NETs release and attenuating colitis.
7.3 DNase I
In addition, several studies have demonstrated that DNase I is a promising treatment for UC (101, 103). DNase I is an enzyme that dissolves reticulated DNA filaments of NET. In mice with DSS-induced colitis, DNase I improved fecal consistency and reduced fecal occult blood and rectal bleeding (103). In addition to improved intestinal inflammation, DSS mice treated with DNase I showed significantly increased expression levels of tight junction protein occludin and ZO-1. Meanwhile, the disruption of NET structure by DNase I was demonstrated to also restore the structure and function of the intestinal mucosal barrier in mice with TNBS-induced colitis, reduce the levels of pro-inflammatory cytokines, and alleviate intestinal inflammation (103).
7.4 Bioextracts
The treatment of autoimmune and inflammatory diseases by modulating NETs in traditional Chinese medicine (TCM) is an area of great interest. For instance, crocetin inhibits PMA-induced NETs formation, as evidenced by reduced expression of NE, PAD4, and CitH3, and alleviates symptoms in adjuvant-induced arthritis mice (166). Apart from inhibiting the activation of the nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, the mechanism involves inhibiting the formation of NETs by suppressing autophagy. Moreover, celastrol is a triterpenoid compound. Celastrol has been shown to completely inhibit neutrophil oxidative burst and NET formation induced by TNF-α. Celastrol downregulates the activation of spleen tyrosine kinase (SYK) and the concomitant phosphorylation of MAPKK/MEK and ERK, as well as the citrullination of histones (167). It suggests that celastrol probably has the potential to modulate inflammation involving neutrophils and NETs and to have therapeutic implications for autoimmune diseases. HMGB1 protein secreted by NETs promotes the release of cytokines and chemokines, causing inflammatory responses. While celastrol effectively suppresses the expression of HMGB1, NF-κB and IL-1β to reduce inflammatory pain (168). Triptolide alleviates chronic arthritis by reducing neutrophil recruitment and suppressing the expression of IL-6 and TNF-α (169). It also inhibits the expression of pro-inflammatory cytokines and NETosis in neutrophils. Furthermore, glycyrrhizin has been demonstrated to inhibit TLR9/MyD88 activation by decreasing HMGB1 expression, thereby reducing NET formation and alleviating sepsis-induced acute respiratory distress syndrome (170). Moreover, forsythiaside B improves coagulopathy in septic rats by inhibiting the formation of PAD4-dependent NETs (171). Luteolin significantly inhibits superoxide anion production, ROS generation, NE release, and NET formation in human neutrophils (172). Ginsenoside Rg5 reduces the inflammatory response by inhibiting the activation of the ERK/NF-κB signaling pathway while lowering the cellular Ca2+ concentration, thereby suppressing the activity and expression of PAD4 to inhibit NETosis (173).
The potential of these active ingredients to mitigate inflammation by modulating NETs is evident. Modern pharmacological studies suggest that numerous natural products have significant advantages in the treatment of UC (174–178). For example, baicalin was shown to exert its anti-inflammatory effects in UC rats by modulating the IKK/IKB/NF-kB signaling pathway and apoptosis-related proteins (179). Puerarin is one of the major isoflavonoid components of the root of Pueraria lobata. In DSS-induced colitis mice, it was demonstrated to exert an anti-inflammatory effect via inhibition of MPO activity and down-regulation of NF-κB and pro-inflammatory mediator secretion. It also possesses antioxidant potential as well as improves intestinal epithelial barrier function (180). Berberine, an isoquinoline alkaloid extracted from Coptidis Rhizoma, exerts a therapeutic role in UC that may involve several aspects such as anti-inflammatory, anti-oxidative stress, maintenance of the structure and function of the intestinal mucosal barrier, modulation of intestinal mucosal immune homeostasis, and regulation of intestinal flora (181–184). Interestingly, berberine has been found to suppress the nuclear translocation of NE and subsequent formation of NETs by inhibiting the interaction of LINC00668 with NE, thus exerting its antithrombotic effect in IBD (104).
7.5 Compound prescriptions in TCM
Huang Qin Decoction, a traditional Chinese prescription for UC, inhibits colonic neutrophil infiltration, restores the levels of the intestinal tight junction proteins Occludin and ZO-1, and alleviates intestinal inflammation induced by TNF-α, and IL-1β (152). At the same time, it down-regulates the expression of PAD4 and citH3 to inhibit the production of NETs, which in turn inhibits colitis-associated carcinogenesis (152). In addition, Sijunzi Decoction is also likely to treat UC by decreasing the level of intestinal NETs (185). It has also been found to inhibit the expression of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) while promoting the expression of anti-inflammatory cytokines (IL-10, IL-37, TGF-β). In light of this evidence, we propose compound prescriptions and active ingredients as a promising therapeutic approach for targeting NETs.
In conclusion, various biological agents and drugs seem to inhibit the formation of NET, but the mechanism needs further investigation. The changes in other physical activities during NET inhibition and the consequences of NET inhibition should not be ignored. Indeed, further studies on the balance between NET induction, inhibition, and degradation are necessary to pharmacologically target NET and its compounds without damaging the immune defenses of patients.
8 Conclusions and future perspectives
Excessive NETs cause abnormal activation of the body’s immune system, leading to tissue damage. However, increasing evidence suggests that NETs exert their anti-inflammatory effects by capturing, killing, and clearing pathogenic microorganisms. NETs play an important role in the pathophysiology of many diseases, including UC. NETs are closely related to the inflammatory response, disruption of intestinal epithelial barrier function, and thrombotic trend in UC.
Although NETs have long been shown to be involved in UC, their potential role in UC pathogenesis remains elusive. Obviously, it is necessary to explore the multifaceted biology of neutrophils, especially the regulatory mechanisms controlling the formation and degradation of NETs in the context of UC, to expand our understanding of the pathways leading to increased NETs. In particular, little is known about NET clearance in UC.
In summary, the balance between NET generation and clearance is essential for health. And NETs seem to play a dual role in UC. Targeting NETs opens the door to new therapeutic options for UC. Meanwhile, the beneficial effects of moderate amounts of NETs in UC and the serious consequences of excessive inhibition of its formation cannot be ignored. Therefore, the treatment methods for UC need to be carefully balanced rather than eliminating neutrophil responses.
Author contributions
DL: Conceptualization, Formal analysis, Investigation, Methodology, Software, Writing – original draft. CM: Conceptualization, Formal analysis, Investigation, Methodology, Software, Writing – original draft. YX: Project administration, Supervision, Writing – review & editing. YZ: Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from the National Natural Science Foundation of China (81874466, 81904176 and 82374426), Natural Science Foundation of Hunan Province (2021JJ30531, 2023JJ60044), Hunan Province Administration of Traditional Chinese Medicine (B2023079), Clinical Medical Technology Innovation Guide Project of Hunan Province (2021SK51413), and domestic first-class construction discipline of Chinese Medicine in Hunan University of Chinese Medicine.
Acknowledgments
The authors thank Jun Xu for her assistance with language.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ASUC, severe ulcerative colitis; CD, Crohn’s disease; cfDNA, cell-free DNA; CG, cathepsin G; Cit-H3, citrullinated histone H3; CsA, Cyclosporine A; DAMP, damage-associated molecular pattern; DNase, Deoxyribonuclease; dsDNA, double-stranded DNA; GSDMD, gasdermin D; HIF-1α, hypoxia‐inducible factor‐1α; HMGB1, high-mobility group box 1; IBD, inflammatory bowel disease; IEC, intestinal epithelial cell; IL, interleukin; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MMP-9, matrix metalloproteinase 9; MPO, myeloperoxidase; NADPH, nicotinamide adenine dinucleotide phosphate; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NF-κB, nuclear factor-κB; PAD4, peptidyl arginine deiminase-4; PKC, protein kinase C; PMA, phorbol myristate acetate; ROS, reactive oxygen species; TCM, traditional Chinese medicine; TLR, Toll-like receptor; TLR4, Toll-like receptor 4; TNF-α, tumour necrosis factor α; UC, ulcerative colitis.
References
1. Le Berre C, Honap S, Peyrin-Biroulet L. Ulcerative colitis. Lancet. (2023) 402:571–84. doi: 10.1016/S0140-6736(23)00966-2
2. Kobayashi T, Siegmund B, Le Berre C, Wei SC, Ferrante M, Shen B, et al. Ulcerative colitis. Nat Rev Dis Primers. (2020) 6:74. doi: 10.1038/s41572-020-0205-x
3. Li MX, Li MY, Lei JX, Wu YZ, Li ZH, Chen LM, et al. Huangqin decoction ameliorates DSS-induced ulcerative colitis: Role of gut microbiota and amino acid metabolism, mTOR pathway and intestinal epithelial barrier. Phytomedicine. (2022) 100:154052. doi: 10.1016/j.phymed.2022.154052
4. Cui L, Guan X, Ding W, Luo Y, Wang W, Bu W, et al. Scutellaria baicalensis Georgi polysaccharide ameliorates DSS-induced ulcerative colitis by improving intestinal barrier function and modulating gut microbiota. Int J Biol Macromol. (2021) 166:1035–45. doi: 10.1016/j.ijbiomac.2020.10.259
5. Loh JT, Lee KG, Lee AP, Teo J, Lim HL, Kim SS, et al. DOK3 maintains intestinal homeostasis by suppressing JAK2/STAT3 signaling and S100a8/9 production in neutrophils. Cell Death Dis. (2021) 12:1054. doi: 10.1038/s41419-021-04357-5
6. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
7. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. (2014) 20:511–7. doi: 10.1038/nm.3547
8. Podolska MJ, Mahajan A, Hahn J, Knopf J, Maueröder C, Petru L, et al. Treatment with DNases rescues hidden neutrophil elastase from aggregated NETs. J Leukoc Biol. (2019) 106:1359–66. doi: 10.1002/JLB.3AB0918-370R
9. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol. (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
10. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis. (2016) 75:1866–75. doi: 10.1136/annrheumdis-2015-208356
11. Frangou E, Chrysanthopoulou A, Mitsios A, Kambas K, Arelaki S, Angelidou I, et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann Rheum Dis. (2019) 78:238–48. doi: 10.1136/annrheumdis-2018-213181
12. Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H, et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. (2019) 79:5626–39. doi: 10.1158/0008-5472.CAN-19-0800
13. Stehr AM, Wang G, Demmler R, Stemmler MP, Krug J, Tripal P, et al. Neutrophil extracellular traps drive epithelial-mesenchymal transition of human colon cancer. J Pathol. (2022) 256:455–67. doi: 10.1002/path.5860
14. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
15. Xia X, Zhang Z, Zhu C, Ni B, Wang S, Yang S, et al. Neutrophil extracellular traps promote metastasis in gastric cancer patients with postoperative abdominal infectious complications. Nat Commun. (2022) 13:1017. doi: 10.1038/s41467-022-28492-5
16. He L, Liu R, Yue H, Zhang X, Pan X, Sun Y, et al. Interaction between neutrophil extracellular traps and cardiomyocytes contributes to atrial fibrillation progression. Signal Transduct Target Ther. (2023) 8:279. doi: 10.1038/s41392-023-01497-2
17. Tang X, Wang P, Zhang R, Watanabe I, Chang E, Vinayachandran V, et al. KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression. J Clin Invest. (2022) 132:e147191. doi: 10.1172/JCI147191
18. Zhang Z, Ding S, Wang Z, Zhu X, Zhou Z, Zhang W, et al. Prmt1 upregulated by Hdc deficiency aggravates acute myocardial infarction via NETosis. Acta Pharm Sin B. (2022) 12:1840–55. doi: 10.1016/j.apsb.2021.10.016
19. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. (2015) 36:1405–14. doi: 10.1093/eurheartj/ehv007
20. Langseth MS, Opstad TB, Bratseth V, Solheim S, Arnesen H, Pettersen AÅ, et al. Markers of neutrophil extracellular traps are associated with adverse clinical outcome in stable coronary artery disease. Eur J Prev Cardiol. (2018) 25:762–9. doi: 10.1177/2047487318760618
21. Zhou G, Yu L, Fang L, Yang W, Yu T, Miao Y, et al. CD177(+) neutrophils as functionally activated neutrophils negatively regulate IBD. Gut. (2018) 67:1052–63. doi: 10.1136/gutjnl-2016-313535
22. Kühl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. (2007) 133:1882–92. doi: 10.1053/j.gastro.2007.08.073
23. McCracken JM, Allen LA. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death. (2014) 7:15–23. doi: 10.4137/JCD.S11038
24. Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur J Clin Invest. (2018) 48 Suppl 2:e12951. doi: 10.1111/eci.12951
25. Ren J, Yan D, Wang Y, Zhang J, Li M, Xiong W, et al. Inhibitor of differentiation-2 protein ameliorates DSS-induced ulcerative colitis by inhibiting NF-κB activation in neutrophils. Front Immunol. (2021) 12:760999. doi: 10.3389/fimmu.2021.760999
26. Zhou G, Zhu F, Zhang H, Wang Y, Yang Y, Jin G, et al. PTK2B regulates immune responses of neutrophils and protects mucosal inflammation in ulcerative colitis. FASEB J. (2023) 37:e22967. doi: 10.1096/fj.202201995RR
27. Raab Y, Gerdin B, Ahlstedt S, Hällgren R. Neutrophil mucosal involvement is accompanied by enhanced local production of interleukin-8 in ulcerative colitis. Gut. (1993) 34:1203–6. doi: 10.1136/gut.34.9.1203
28. Akpinar MY, Ozin YO, Kaplan M, Ates I, Kalkan IH, Kilic Z, et al. Platelet-to-lymphocyte ratio and neutrophil-to-lymphocyte ratio predict mucosal disease severity in ulcerative colitis. J Med Biochem. (2018) 37:155–62. doi: 10.1515/jomb-2017-0050
29. Fukunaga S, Kuwaki K, Mitsuyama K, Takedatsu H, Yoshioka S, Yamasaki H, et al. Detection of calprotectin in inflammatory bowel disease: Fecal and serum levels and immunohistochemical localization. Int J Mol Med. (2018) 41:107–18. doi: 10.3892/ijmm.2017.3244
30. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. (1996) 59:229–40. doi: 10.1002/jlb.59.2.229
31. Dwyer M, Shan Q, D’Ortona S, Maurer R, Mitchell R, Olesen H, et al. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J Innate Immun. (2014) 6:765–79. doi: 10.1159/000363242
32. Poli V, Zanoni I. Neutrophil intrinsic and extrinsic regulation of NETosis in health and disease. Trends Microbiol. (2023) 31:280–93. doi: 10.1016/j.tim.2022.10.002
33. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
34. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. (2012) 12:109–16. doi: 10.1016/j.chom.2012.05.015
35. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217:e20201129. doi: 10.1084/jem.20201129
36. Aymonnier K, Ng J, Fredenburgh LE, Zambrano-Vera K, Münzer P, Gutch S, et al. Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Adv. (2022) 6:2001–13. doi: 10.1182/bloodadvances.2021005949
37. Smolarz M, Zawrotniak M, Satala D, Rapala-Kozik M. Extracellular nucleic acids present in the Candida albicans biofilm trigger the release of neutrophil extracellular traps. Front Cell Infect Microbiol. (2021) 11:681030. doi: 10.3389/fcimb.2021.681030
38. Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res. (2022) 1:732–47. doi: 10.1038/s44161-022-00108-7
39. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. (2014) 12:2074–88. doi: 10.1111/jth.12710
40. Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Trans Med. (2018) 10:eaao3089. doi: 10.1126/scitranslmed.aao3089
41. Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem. (2004) 279:44123–32. doi: 10.1074/jbc.M405883200
42. Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. (2017) 6:e24437. doi: 10.7554/eLife.24437
43. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (Baltimore Md: 1950). (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
44. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
45. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. (2011) 7:75–7. doi: 10.1038/nchembio.496
46. Schappe MS, Szteyn K, Stremska ME, Mendu SK, Downs TK, Seegren PV, et al. Chanzyme TRPM7 mediates the Ca(2+) influx essential for lipopolysaccharide-induced toll-like receptor 4 endocytosis and macrophage activation. Immunity. (2018) 48:59–74.e5. doi: 10.1016/j.immuni.2017.11.026
47. Immler R, Simon SI, Sperandio M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur J Clin Invest. (2018) 48 Suppl 2:e12964. doi: 10.1111/eci.12964
48. Kandasamy K, Bezavada L, Escue RB, Parthasarathi K. Lipopolysaccharide induces endoplasmic store Ca2+-dependent inflammatory responses in lung microvessels. PloS One. (2013) 8:e63465. doi: 10.1371/journal.pone.0063465
49. Schorr W, Swandulla D, Zeilhofer HU. Mechanisms of IL-8-induced Ca2+ signaling in human neutrophil granulocytes. Eur J Immunol. (1999) 29:897–904. doi: 10.1002/(SICI)1521-4141(199903)29:03<897::AID-IMMU897<3.0.CO;2-5
50. Krause KH, Campbell KP, Welsh MJ, Lew DP. The calcium signal and neutrophil activation. Clin Biochem. (1990) 23:159–66. doi: 10.1016/0009-9120(90)80030-M
51. Kearney PL, Bhatia M, Jones NG, Yuan L, Glascock MC, Catchings KL, et al. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry. (2005) 44:10570–82. doi: 10.1021/bi050292m
52. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. (2010) 207:1853–62. doi: 10.1084/jem.20100239
53. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. (2009) 184:205–13. doi: 10.1083/jcb.200806072
54. Kim SW, Lee H, Lee HK, Kim ID, Lee JK. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol Commun. (2019) 7:94. doi: 10.1186/s40478-019-0747-x
55. Cao D, Qian K, Zhao Y, Hong J, Chen H, Wang X, et al. Association of neutrophil extracellular traps with fistula healing in patients with complex perianal fistulizing Crohn’s disease. J Crohn’s Colitis. (2023) 17:580–92. doi: 10.1093/ecco-jcc/jjac171
56. Schroder AL, Chami B, Liu Y, Doyle CM, El Kazzi M, Ahlenstiel G, et al. Neutrophil extracellular trap density increases with increasing histopathological severity of Crohn’s disease. Inflamm Bowel Dis. (2022) 28:586–98. doi: 10.1093/ibd/izab239
57. Poli V, Pui-Yan Ma V, Di Gioia M, Broggi A, Benamar M, Chen Q, et al. Zinc-dependent histone deacetylases drive neutrophil extracellular trap formation and potentiate local and systemic inflammation. iScience. (2021) 24:103256. doi: 10.1016/j.isci.2021.103256
58. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. (2014) 8:883–96. doi: 10.1016/j.celrep.2014.06.044
59. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. (2010) 191:677–91. doi: 10.1083/jcb.201006052
60. Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6689. doi: 10.1126/sciimmunol.aar6689
61. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6676. doi: 10.1126/sciimmunol.aar6676
62. Silva C, Wanderley C, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. (2021) 138:2702–13. doi: 10.1182/blood.2021011525
63. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. (2018) 22:2924–36. doi: 10.1016/j.celrep.2018.02.067
64. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. (2020) 180:941–955.e20. doi: 10.1016/j.cell.2020.02.002
65. Wang Y, Li Y, Chen Z, Yuan Y, Su Q, Ye K, et al. GSDMD-dependent neutrophil extracellular traps promote macrophage-to-myofibroblast transition and renal fibrosis in obstructive nephropathy. Cell Death Dis. (2022) 13:693. doi: 10.1038/s41419-022-05138-4
66. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
67. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. (2012) 12:324–33. doi: 10.1016/j.chom.2012.06.011
68. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A. (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
69. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
70. Vorobjeva N, Galkin I, Pletjushkina O, Golyshev S, Zinovkin R, Prikhodko A, et al. Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165664. doi: 10.1016/j.bbadis.2020.165664
71. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
72. Lauková L, Konečná B, Janovičová Ľ, Vlková B, Celec P. Deoxyribonucleases and their applications in biomedicine. Biomolecules. (2020) 10:1036. doi: 10.3390/biom10071036
73. Yasuda T, Awazu S, Sato W, Iida R, Tanaka Y, Kishi K. Human genetically polymorphic deoxyribonuclease: purification, characterization, and multiplicity of urine deoxyribonuclease I. J Biochem. (1990) 108:393–8. doi: 10.1093/oxfordjournals.jbchem.a123212
75. Li N, Zheng X, Chen M, Huang L, Chen L, Huo R, et al. Deficient DNASE1L3 facilitates neutrophil extracellular traps-induced invasion via cyclic GMP-AMP synthase and the non-canonical NF-κB pathway in diabetic hepatocellular carcinoma. Clin Transl Immunol. (2022) 11:e1386. doi: 10.1002/cti2.1386
76. Ciesielski O, Biesiekierska M, Panthu B, Soszyński M, Pirola L, Balcerczyk A. Citrullination in the pathology of inflammatory and autoimmune disorders: recent advances and future perspectives. Cell Mol Life Sci. (2022) 79:94. doi: 10.1007/s00018-022-04126-3
77. Lazzaretto B, Fadeel B. Intra- and extracellular degradation of neutrophil extracellular traps by macrophages and dendritic cells. J Immunol (Baltimore Md: 1950). (2019) 203:2276–90. doi: 10.4049/jimmunol.1800159
78. Jani D, Lutz S, Hurt E, Laskey RA, Stewart M, Wickramasinghe VO. Functional and structural characterization of the mammalian TREX-2 complex that links transcription with nuclear messenger RNA export. Nucleic Acids Res. (2012) 40:4562–73. doi: 10.1093/nar/gks059
79. Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol (Baltimore Md: 1950). (2013) 191:2647–56. doi: 10.4049/jimmunol.1300436
80. Grégoire M, Uhel F, LesouHaitier M, Gacouin A, Guirriec M, Mourcin F, et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J. (2018) 52:1702590. doi: 10.1183/13993003.02590-2017
81. Haider P, Kral-Pointner JB, Mayer J, Richter M, Kaun C, Brostjan C, et al. Neutrophil extracellular trap degradation by differently polarized macrophage subsets. Arterioscler Thromb Vasc Biol. (2020) 40:2265–78. doi: 10.1161/ATVBAHA.120.314883
82. Siler U, Romao S, Tejera E, Pastukhov O, Kuzmenko E, Valencia RG, et al. Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J Allergy Clin Immunol. (2017) 139:212–219.e3. doi: 10.1016/j.jaci.2016.04.041
83. Chaaban H, Burge K, Eckert J, Keshari RS, Silasi R, Lupu C, et al. Neutrophil extracellular trap inhibition increases inflammation, bacteraemia and mortality in murine necrotizing enterocolitis. J Cell Mol Med. (2021) 25:10814–24. doi: 10.1111/jcmm.15338
84. Barrientos L, Bignon A, Gueguen C, de Chaisemartin L, Gorges R, Sandré C, et al. Neutrophil extracellular traps downregulate lipopolysaccharide-induced activation of monocyte-derived dendritic cells. J Immunol. (2014) 193:5689–98. doi: 10.4049/jimmunol.1400586
85. Kim TS, Silva LM, Theofilou VI, Greenwell-Wild T, Li L, Williams DW, et al. Neutrophil extracellular traps and extracellular histones potentiate IL-17 inflammation in periodontitis. J Exp Med. (2023) 220:e20221751. doi: 10.1084/jem.20221751
86. Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, et al. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight. (2020) 5:e134796. doi: 10.1172/jci.insight.134796
87. Cho Y, Bukong TN, Tornai D, Babuta M, Vlachos IS, Kanata E, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. (2023) 78:28–44. doi: 10.1016/j.jhep.2022.08.029
88. Chu C, Wang X, Yang C, Chen F, Shi L, Xu W, et al. Neutrophil extracellular traps drive intestinal microvascular endothelial ferroptosis by impairing Fundc1-dependent mitophagy. Redox Biol. (2023) 67:102906. doi: 10.1016/j.redox.2023.102906
89. Hu Q, Shi H, Zeng T, Liu H, Su Y, Cheng X, et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res Ther. (2019) 21:9. doi: 10.1186/s13075-018-1800-z
90. Cui Y, Yang Y, Tao W, Peng W, Luo D, Zhao N, et al. Neutrophil extracellular traps induce alveolar macrophage pyroptosis by regulating NLRP3 deubiquitination, aggravating the development of septic lung injury. J Inflammation Res. (2023) 16:861–77. doi: 10.2147/JIR.S366436
91. Liu D, Yang P, Gao M, Yu T, Shi Y, Zhang M, et al. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin Sci (Lond). (2019) 133:565–82. doi: 10.1042/CS20180600
92. Zhang Y, Guo L, Dai Q, Shang B, Xiao T, Di X, et al. A signature for pan-cancer prognosis based on neutrophil extracellular traps. J Immunother Cancer. (2022) 10:e004210. doi: 10.1136/jitc-2021-004210
93. de Bont CM, Stokman M, Faas P, Thurlings RM, Boelens WC, Wright HL, et al. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J Autoimmun. (2020) 113:102484. doi: 10.1016/j.jaut.2020.102484
94. Nomura K, Miyashita T, Yamamoto Y, Munesue S, Harashima A, Takayama H, et al. Citrullinated histone H3: early biomarker of neutrophil extracellular traps in septic liver damage. J Surg Res. (2019) 234:132–8. doi: 10.1016/j.jss.2018.08.014
95. Janssen P, Tosi I, Hego A, Maréchal P, Marichal T, Radermecker C. Neutrophil extracellular traps are found in bronchoalveolar lavage fluids of horses with severe asthma and correlate with asthma severity. Front Immunol. (2022) 13:921077. doi: 10.3389/fimmu.2022.921077
96. Abd El Hafez A, Mohamed AS, Shehta A, Sheta H. Neutrophil extracellular traps-associated protein peptidyl arginine deaminase 4 immunohistochemical expression in ulcerative colitis and its association with the prognostic predictors. Pathol Res Pract. (2020) 216:153102. doi: 10.1016/j.prp.2020.153102
97. Angelidou I, Chrysanthopoulou A, Mitsios A, Arelaki S, Arampatzioglou A, Kambas K, et al. REDD1/autophagy pathway is associated with neutrophil-driven IL-1β Inflammatory response in active ulcerative colitis. J Immunol. (2018) 200:3950–61. doi: 10.4049/jimmunol.1701643
98. Dinallo V, Marafini I, Di Fusco D, Laudisi F, Franzè E, Di Grazia A, et al. Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis. J Crohn’s Colitis. (2019) 13:772–84. doi: 10.1093/ecco-jcc/jjy215
99. Neuenfeldt F, Schumacher JC, Grieshaber-Bouyer R, Habicht J, Schröder-Braunstein J, Gauss A, et al. Inflammation induces pro-NETotic neutrophils via TNFR2 signaling. Cell Rep. (2022) 39:110710. doi: 10.1016/j.celrep.2022.110710
100. Bennike TB, Carlsen TG, Ellingsen T, Bonderup OK, Glerup H, Bøgsted M, et al. Neutrophil extracellular traps in ulcerative colitis: A proteome analysis of intestinal biopsies. Inflammation Bowel Dis. (2015) 21:2052–67. doi: 10.1097/MIB.0000000000000460
101. Li T, Wang C, Liu Y, Li B, Zhang W, Wang L, et al. Neutrophil extracellular traps induce intestinal damage and thrombotic tendency in inflammatory bowel disease. J Crohn’s Colitis. (2020) 14:240–53. doi: 10.1093/ecco-jcc/jjz132
102. Xu C, Ye Z, Jiang W, Wang S, Zhang H. Cyclosporine A alleviates colitis by inhibiting the formation of neutrophil extracellular traps via the regulating pentose phosphate pathway. Mol Med (Cambridge Mass). (2023) 29:169. doi: 10.1186/s10020-023-00758-8
103. Lin EY, Lai HJ, Cheng YK, Leong KQ, Cheng LC, Chou YC, et al. Neutrophil extracellular traps impair intestinal barrier function during experimental colitis. Biomedicines. (2020) 8:275. doi: 10.3390/biomedicines8080275
104. Zhang L, Zheng B, Bai Y, Zhou J, Zhang XH, Yang YQ, et al. Exosomes-transferred LINC00668 contributes to thrombosis by promoting NETs formation in inflammatory bowel disease. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2023) 10:e2300560. doi: 10.1002/advs.202300560
105. Wen C, Hu H, Yang W, Zhao Y, Zheng L, Jiang X, et al. Targeted inhibition of FcRn reduces NET formation to ameliorate experimental ulcerative colitis by accelerating ANCA clearance. Int Immunopharmacol. (2022) 113:109474. doi: 10.1016/j.intimp.2022.109474
106. Bale S, Verma P, Varga J, Bhattacharyya S. Extracellular matrix-derived damage-associated molecular patterns (DAMP): implications in systemic sclerosis and fibrosis. J Invest Dermatol. (2023) 143:1877–85. doi: 10.1016/j.jid.2023.04.030
107. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. (2015) 62:600–14. doi: 10.1002/hep.27841
108. Nanini HF, Bernardazzi C, Castro F, de Souza H. Damage-associated molecular patterns in inflammatory bowel disease: From biomarkers to therapeutic targets. World J Gastroenterol. (2018) 24:4622–34. doi: 10.3748/wjg.v24.i41.4622
109. Ngo AT, Skidmore A, Oberg J, Yarovoi I, Sarkar A, Levine N, et al. Platelet factor 4 limits neutrophil extracellular trap- and cell-free DNA-induced thrombogenicity and endothelial injury. JCI Insight. (2023) 8:e171054. doi: 10.1172/jci.insight.171054
110. Koike Y, Uchida K, Tanaka K, Ide S, Otake K, Okita Y, et al. Dynamic pathology for circulating free DNA in a dextran sodium sulfate colitis mouse model. Pediatr Surg Int. (2014) 30:1199–206. doi: 10.1007/s00383-014-3607-6
111. Rauh P, Rickes S, Fleischhacker M. Microsatellite alterations in free-circulating serum DNA in patients with ulcerative colitis. Digest Dis (Basel Switzerland). (2003) 21:363–6. doi: 10.1159/000075361
112. Paunel-Görgülü A, Wacker M, El Aita M, Hassan S, Schlachtenberger G, Deppe A, et al. cfDNA correlates with endothelial damage after cardiac surgery with prolonged cardiopulmonary bypass and amplifies NETosis in an intracellular TLR9-independent manner. Sci Rep. (2017) 7:17421. doi: 10.1038/s41598-017-17561-1
113. Denorme F, Portier I, Rustad JL, Cody MJ, de Araujo CV, Hoki C, et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J Clin Invest. (2022) 132:e154225. doi: 10.1172/JCI154225
114. Zhan Y, Ling Y, Deng Q, Qiu Y, Shen J, Lai H, et al. HMGB1-mediated neutrophil extracellular trap formation exacerbates intestinal ischemia/reperfusion-induced acute lung injury. J Immunol (Baltimore Md: 1950). (2022) 208:968–78. doi: 10.4049/jimmunol.2100593
115. Publisher Correction to: Acta Neuropathologica Communications, volume 7. Acta Neuropathol Commun. (2019) 7:131. doi: 10.1186/s40478-019-0784-5
116. Chen L, Li J, Ye Z, Sun B, Wang L, Chen Y, et al. Anti-high mobility group box 1 neutralizing-antibody ameliorates dextran sodium sulfate colitis in mice. Front Immunol. (2020) 11:585094. doi: 10.3389/fimmu.2020.585094
117. Nakase H, Sato N, Mizuno N, Ikawa Y. The influence of cytokines on the complex pathology of ulcerative colitis. Autoimmun Rev. (2022) 21:103017. doi: 10.1016/j.autrev.2021.103017
118. Chapuy L, Bsat M, Rubio M, Sarkizova S, Therrien A, Bouin M, et al. IL-12 and Mucosal CD14+ Monocyte-Like Cells Induce IL-8 in Colonic Memory CD4+ T Cells of Patients With Ulcerative Colitis but not Crohn’s Disease. J Crohn’s Colitis. (2020) 14:79–95. doi: 10.1093/ecco-jcc/jjz115
119. Walana W, Ye Y, Li M, Wang J, Wang B, Cheng JW, et al. IL-8 antagonist, CXCL8(3-72)K11R/G31P coupled with probiotic exhibit variably enhanced therapeutic potential in ameliorating ulcerative colitis. BioMed Pharmacother. (2018) 103:253–61. doi: 10.1016/j.biopha.2018.04.008
120. Shu Q, Zhang N, Liu Y, Wang X, Chen J, Xie H, et al. IL-8 triggers neutrophil extracellular trap formation through an nicotinamide adenine dinucleotide phosphate oxidase- and mitogen-activated protein kinase pathway-dependent mechanism in uveitis. Invest Ophthalmol Vis Sci. (2023) 64:19. doi: 10.1167/iovs.64.13.19
121. de Andrea CE, Ochoa MC, Villalba-Esparza M, Teijeira Á, Schalper KA, Abengozar-Muela M, et al. Heterogenous presence of neutrophil extracellular traps in human solid tumours is partially dependent on IL-8. J Pathol. (2021) 255:190–201. doi: 10.1002/path.5753
122. Fan Q, Li M, Zhao W, Zhang K, Li M, Li W. Hyper α2,6-sialylation promotes CD4(+) T-cell activation and induces the occurrence of ulcerative colitis. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2023) 10:e2302607. doi: 10.1002/advs.202302607
123. Chen X, Xu T, Lv X, Zhang J, Liu S. Ginsenoside Rh2 alleviates ulcerative colitis by regulating the STAT3/miR-214 signaling pathway. J Ethnopharmacol. (2021) 274:113997. doi: 10.1016/j.jep.2021.113997
124. Ohyama A, Osada A, Kawaguchi H, Kurata I, Nishiyama T, Iwai T, et al. Specific increase in joint neutrophil extracellular traps and its relation to interleukin 6 in autoimmune arthritis. Int J Mol Sci. (2021) 22:7633. doi: 10.3390/ijms22147633
125. Keir HR, Chalmers JD. IL-6 trans-signalling: how Haemophilus surfs the NET to amplify inflammation in COPD. Eur Respir J. (2021) 58:2102143. doi: 10.1183/13993003.02143-2021
126. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. (2000) 25:177–81. doi: 10.1038/76032
127. Malíčková K, Duricová D, Bortlík M, Hrušková Z, Svobodová B, Machková N, et al. Impaired deoxyribonuclease I activity in patients with inflammatory bowel diseases. Autoimmune Dis. (2011) 2011:945861. doi: 10.4061/2011/945861
128. Fiorino G, Danese S, Peyrin-Biroulet L, Sans M, Bonelli F, Calleri M, et al. LIAISON(®) Calprotectin for the prediction of relapse in quiescent ulcerative colitis: The EuReCa study. United Eur Gastroenterol J. (2022) 10:836–43. doi: 10.1002/ueg2.12268
129. Hart L, Chavannes M, Kherad O, Maedler C, Mourad N, Marcus V, et al. Faecal calprotectin predicts endoscopic and histological activity in clinically quiescent ulcerative colitis. J Crohns Colitis. (2020) 14:46–52. doi: 10.1093/ecco-jcc/jjz107
130. Mortensen JH, Sinkeviciute D, Manon-Jensen T, Domislović V, McCall K, Thudium CS, et al. A specific calprotectin neo-epitope [CPa9-HNE] in serum from inflammatory bowel disease patients is associated with neutrophil activity and endoscopic severity. J Crohns Colitis. (2022) 16:1447–60. doi: 10.1093/ecco-jcc/jjac047
131. Hu Z, Murakami T, Tamura H, Reich J, Kuwahara-Arai K, Iba T, et al. Neutrophil extracellular traps induce IL-1β production by macrophages in combination with lipopolysaccharide. Int J Mol Med. (2017) 39:549–58. doi: 10.3892/ijmm.2017.2870
132. Mondal S, Thompson PR. Protein arginine deiminases (PADs): biochemistry and chemical biology of protein citrullination. Acc Chem Res. (2019) 52:818–32. doi: 10.1021/acs.accounts.9b00024
133. Leppkes M, Lindemann A, Gößwein S, Paulus S, Roth D, Hartung A, et al. Neutrophils prevent rectal bleeding in ulcerative colitis by peptidyl-arginine deiminase-4-dependent immunothrombosis. Gut. (2022) 71:2414–29. doi: 10.1136/gutjnl-2021-324725
134. Wang P, Liu D, Zhou Z, Liu F, Shen Y, You Q, et al. The role of protein arginine deiminase 4-dependent neutrophil extracellular traps formation in ulcerative colitis. Front Immunol. (2023) 14:1144976. doi: 10.3389/fimmu.2023.1144976
135. Sharma A, Simonson TJ, Jondle CN, Mishra BB, Sharma J. Mincle-mediated neutrophil extracellular trap formation by regulation of autophagy. J Infect Dis. (2017) 215:1040–8. doi: 10.1093/infdis/jix072
136. Xu F, Zhang C, Zou Z, Fan E, Chen L, Li Y, et al. Aging-related Atg5 defect impairs neutrophil extracellular traps formation. Immunology. (2017) 151:417–32. doi: 10.1111/imm.12740
137. Itakura A, McCarty OJ. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol. (2013) 305:C348–54. doi: 10.1152/ajpcell.00108.2013
138. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. (2011) 21:290–304. doi: 10.1038/cr.2010.150
139. Ma R, Li T, Cao M, Si Y, Wu X, Zhao L, et al. Extracellular DNA traps released by acute promyelocytic leukemia cells through autophagy. Cell Death Dis. (2016) 7:e2283. doi: 10.1038/cddis.2016.186
140. Lai HJ, Doan HT, Lin EY, Chiu YL, Cheng YK, Lin YH, et al. Histones of neutrophil extracellular traps directly disrupt the permeability and integrity of the intestinal epithelial barrier. Inflammation Bowel Dis. (2023) 29:783–97. doi: 10.1093/ibd/izac256
141. Giannotta M, Tapete G, Emmi G, Silvestri E, Milla M. Thrombosis in inflammatory bowel diseases: what’s the link. Thromb J. (2015) 13:14. doi: 10.1186/s12959-015-0044-2
142. Wang Y, Luo L, Braun OÖ, Westman J, Madhi R, Herwald H, et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. (2018) 8:4020. doi: 10.1038/s41598-018-22156-5
143. He Z, Si Y, Jiang T, Ma R, Zhang Y, Cao M, et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb Haemost. (2016) 115:738–51. doi: 10.1160/TH15-09-0710
144. Cao M, Yu M, Zhang Y, Tong D, Shi J. Neutrophil extracellular traps exacerbate inflammatory responses and thrombotic tendency in both a murine colitis model and patients with inflammatory bowel disease. Blood. (2017) 130:994–4. doi: 10.1182/blood.V130.Suppl_1.994.994
145. Hahn J, Schauer C, Czegley C, Kling L, Petru L, Schmid B, et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. (2019) 33:1401–14. doi: 10.1096/fj.201800752R
146. Ribon M, Seninet S, Mussard J, Sebbag M, Clavel C, Serre G, et al. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J Autoimmun. (2019) 98:122–31. doi: 10.1016/j.jaut.2019.01.003
147. Seo DH, Che X, Kim S, Kim DH, Ma HW, Kim JH, et al. Triggering receptor expressed on myeloid cells-1 agonist regulates intestinal inflammation via Cd177(+) neutrophils. Front Immunol. (2021) 12:650864. doi: 10.3389/fimmu.2021.650864
148. Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, et al. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G929–938. doi: 10.1152/ajpgi.00435.2010
149. Zhang T, Mei Y, Dong W, Wang J, Huang F, Wu J. Evaluation of protein arginine deiminase-4 inhibitor in TNBS- induced colitis in mice. Int Immunopharmacol. (2020) 84:106583. doi: 10.1016/j.intimp.2020.106583
150. Yasuda H, Uno A, Tanaka Y, Koda S, Saito M, Sato EF, et al. Neutrophil extracellular trap induction through peptidylarginine deiminase 4 activity is involved in 2,4,6-trinitrobenzenesulfonic acid-induced colitis. Naunyn-Schmiedeberg’s Arch Pharmacol. (2023) 397(5):3127–40. doi: 10.1007/s00210-023-02800-2
151. Török S, Almási N, Valkusz Z, Pósa A, Varga C, Kupai K. Investigation of H(2)S donor treatment on neutrophil extracellular traps in experimental colitis. Int J Mol Sci. (2021) 22:12729. doi: 10.3390/ijms222312729
152. Pan Z, Xie X, Chen Y, Pan S, Wu Z, Yang C, et al. Huang Qin Decoction inhibits the initiation of experimental colitis associated carcinogenesis by controlling the PAD4 dependent NETs. Phytomedicine. (2022) 107:154454. doi: 10.1016/j.phymed.2022.154454
153. Kirchner T, Hermann E, Möller S, Klinger M, Solbach W, Laskay T, et al. Flavonoids and 5-aminosalicylic acid inhibit the formation of neutrophil extracellular traps. Mediators Inflamm. (2013) 2013:710239. doi: 10.1155/2013/710239
154. Li G, Lin J, Zhang C, Gao H, Lu H, Gao X, et al. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes. (2021) 13:1968257. doi: 10.1080/19490976.2021.1968257
155. Han D, Wu Y, Lu D, Pang J, Hu J, Zhang X, et al. Polyphenol-rich diet mediates interplay between macrophage-neutrophil and gut microbiota to alleviate intestinal inflammation. Cell Death Dis. (2023) 14:656. doi: 10.1038/s41419-023-06190-4
156. Dong W, Liu D, Zhang T, You Q, Huang F, Wu J. Oral delivery of staphylococcal nuclease ameliorates DSS induced ulcerative colitis in mice via degrading intestinal neutrophil extracellular traps. Ecotoxicol Environ Saf. (2021) 215:112161. doi: 10.1016/j.ecoenv.2021.112161
157. Witalison EE, Cui X, Hofseth AB, Subramanian V, Causey CP, Thompson PR, et al. Inhibiting protein arginine deiminases has antioxidant consequences. J Pharmacol Exp Ther. (2015) 353:64–70. doi: 10.1124/jpet.115.222745
158. Okeke EB, Louttit C, Fry C, Najafabadi AH, Han K, Nemzek J, et al. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials. (2020) 238:119836. doi: 10.1016/j.biomaterials.2020.119836
159. Yang Y, Hu N, Gao XJ, Li T, Yan ZX, Wang PP, et al. Dextran sulfate sodium-induced colitis and ginseng intervention altered oral pharmacokinetics of cyclosporine A in rats. J Ethnopharmacol. (2021) 265:113251. doi: 10.1016/j.jep.2020.113251
160. Song EM, Oh EH, Hwang SW, Park SH, Yang DH, Byeon JS, et al. Comparison of outcomes of cyclosporine A and infliximab for steroid-refractory acute severe ulcerative colitis. J Gastroenterol Hepatol. (2021) 36:2463–70. doi: 10.1111/jgh.15508
161. Liu J, Farmer JD Jr., Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. (1991) 66:807–15. doi: 10.1016/0092-8674(91)90124-H
162. Chen T, Guo J, Yang M, Han C, Zhang M, Chen W, et al. Cyclosporin A impairs dendritic cell migration by regulating chemokine receptor expression and inhibiting cyclooxygenase-2 expression. Blood. (2004) 103:413–21. doi: 10.1182/blood-2003-07-2412
163. Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PloS One. (2014) 9:e97088. doi: 10.1371/journal.pone.0097088
164. Greenblatt MB, Aliprantis A, Hu B, Glimcher LH. Calcineurin regulates innate antifungal immunity in neutrophils. J Exp Med. (2010) 207:923–31. doi: 10.1084/jem.20092531
165. Lu H, Lin J, Xu C, Sun M, Zuo K, Zhang X, et al. Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1α-glycolysis axis to alleviate severe ulcerative colitis. Clin Transl Med. (2021) 11:e334. doi: 10.1002/ctm2.334
166. Jiang H, Lu Q, Xu J, Huo G, Cai Y, Geng S, et al. Sinomenine ameliorates adjuvant-induced arthritis by inhibiting the autophagy/NETosis/inflammation axis. Sci Rep. (2023) 13:3933. doi: 10.1038/s41598-023-30922-3
167. Yu Y, Koehn CD, Yue Y, Li S, Thiele GM, Hearth-Holmes MP, et al. Celastrol inhibits inflammatory stimuli-induced neutrophil extracellular trap formation. Curr Mol Med. (2015) 15:401–10. doi: 10.2174/1566524015666150505160743
168. Zhang X, Zhao W, Liu X, Huang Z, Shan R, Huang C. Celastrol ameliorates inflammatory pain and modulates HMGB1/NF-κB signaling pathway in dorsal root ganglion. Neurosci Lett. (2019) 692:83–9. doi: 10.1016/j.neulet.2018.11.002
169. Huang G, Yuan K, Zhu Q, Zhang S, Lu Q, Zhu M, et al. Triptolide inhibits the inflammatory activities of neutrophils to ameliorate chronic arthritis. Mol Immunol. (2018) 101:210–20. doi: 10.1016/j.molimm.2018.06.012
170. Gu J, Ran X, Deng J, Zhang A, Peng G, Du J, et al. Glycyrrhizin alleviates sepsis-induced acute respiratory distress syndrome via suppressing of HMGB1/TLR9 pathways and neutrophils extracellular traps formation. Int Immunopharmacol. (2022) 108:108730. doi: 10.1016/j.intimp.2022.108730
171. He W, Xi Q, Cui H, Zhang P, Huang R, Wang T, et al. Forsythiaside B ameliorates coagulopathies in a rat model of sepsis through inhibition of the formation of PAD4-dependent neutrophil extracellular traps. Front Pharmacol. (2022) 13:1022985. doi: 10.3389/fphar.2022.1022985
172. Yang SC, Chen PJ, Chang SH, Weng YT, Chang FR, Chang KY, et al. Luteolin attenuates neutrophilic oxidative stress and inflammatory arthritis by inhibiting Raf1 activity. Biochem Pharmacol. (2018) 154:384–96. doi: 10.1016/j.bcp.2018.06.003
173. Chen Z, Wang G, Xie X, Liu H, Liao J, Shi H, et al. Ginsenoside Rg5 allosterically interacts with P2RY(12) and ameliorates deep venous thrombosis by counteracting neutrophil NETosis and inflammatory response. Front Immunol. (2022) 13:918476. doi: 10.3389/fimmu.2022.918476
174. Guo R, Meng Q, Wang B, Li F. Anti-inflammatory effects of Platycodin D on dextran sulfate sodium (DSS) induced colitis and E. coli Lipopolysaccharide (LPS) induced inflammation. Int Immunopharmacol. (2021) 94:107474. doi: 10.1016/j.intimp.2021.107474
175. El-Akabawy G, El-Sherif NM. Zeaxanthin exerts protective effects on acetic acid-induced colitis in rats via modulation of pro-inflammatory cytokines and oxidative stress. BioMed Pharmacother. (2019) 111:841–51. doi: 10.1016/j.biopha.2019.01.001
176. Fan H, Gao Z, Ji K, Li X, Wu J, Liu Y, et al. The in vitro and in vivo anti-inflammatory effect of osthole, the major natural coumarin from Cnidium monnieri (L.) Cuss, via the blocking of the activation of the NF-κB and MAPK/p38 pathways. Phytomedicine. (2019) 58:152864. doi: 10.1016/j.phymed.2019.152864
177. Gao Z, Yu C, Liang H, Wang X, Liu Y, Li X, et al. Andrographolide derivative CX-10 ameliorates dextran sulphate sodium-induced ulcerative colitis in mice: Involvement of NF-κB and MAPK signalling pathways. Int Immunopharmacol. (2018) 57:82–90. doi: 10.1016/j.intimp.2018.02.012
178. Zhang CL, Zhang S, He WX, Lu JL, Xu YJ, Yang JY, et al. Baicalin may alleviate inflammatory infiltration in dextran sodium sulfate-induced chronic ulcerative colitis via inhibiting IL-33 expression. Life Sci. (2017) 186:125–32. doi: 10.1016/j.lfs.2017.08.010
179. Shen J, Cheng J, Zhu S, Zhao J, Ye Q, Xu Y, et al. Regulating effect of baicalin on IKK/IKB/NF-kB signaling pathway and apoptosis-related proteins in rats with ulcerative colitis. Int Immunopharmacol. (2019) 73:193–200. doi: 10.1016/j.intimp.2019.04.052
180. Jeon YD, Lee JH, Lee YM, Kim DK. Puerarin inhibits inflammation and oxidative stress in dextran sulfate sodium-induced colitis mice model. BioMed Pharmacother. (2020) 124:109847. doi: 10.1016/j.biopha.2020.109847
181. Li H, Feng C, Fan C, Yang Y, Yang X, Lu H, et al. Intervention of oncostatin M-driven mucosal inflammation by berberine exerts therapeutic property in chronic ulcerative colitis. Cell Death Dis. (2020) 11:271. doi: 10.1038/s41419-020-2470-8
182. Dong Y, Fan H, Zhang Z, Jiang F, Li M, Zhou H, et al. Berberine ameliorates DSS-induced intestinal mucosal barrier dysfunction through microbiota-dependence and Wnt/β-catenin pathway. Int J Biol Sci. (2022) 18:1381–97. doi: 10.7150/ijbs.65476
183. Sun X, Zhang Y, Cheng G, Zhu T, Zhang Z, Xiong L, et al. Berberine improves DSS-induced colitis in mice by modulating the fecal-bacteria-related bile acid metabolism. BioMed Pharmacother. (2023) 167:115430. doi: 10.1016/j.biopha.2023.115430
184. Li H, Fan C, Lu H, Feng C, He P, Yang X, et al. Protective role of berberine on ulcerative colitis through modulating enteric glial cells-intestinal epithelial cells-immune cells interactions. Acta Pharm Sin B. (2020) 10:447–61. doi: 10.1016/j.apsb.2019.08.006
Keywords: ulcerative colitis, neutrophils, neutrophil extracellular traps, PAD4, inflammation, immunology
Citation: Long D, Mao C, Xu Y and Zhu Y (2024) The emerging role of neutrophil extracellular traps in ulcerative colitis. Front. Immunol. 15:1425251. doi: 10.3389/fimmu.2024.1425251
Received: 29 April 2024; Accepted: 22 July 2024;
Published: 07 August 2024.
Edited by:
Wenyi Gu, The University of Queensland, AustraliaReviewed by:
Dhirendra Kumar Singh, University of North Carolina at Chapel Hill, United StatesVeronica Azcutia, Complutense University of Madrid, Spain
Copyright © 2024 Long, Mao, Xu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yin Xu, MzExMTE4QGhudWNtLmVkdS5jbg==; Ying Zhu, emh1eWluZzA4OUAxMjYuY29t