
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 April 2024
Sec. Cytokines and Soluble Mediators in Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1331217
 James G. Krueger1Kilian Eyerich2,3Vijay K. Kuchroo4
James G. Krueger1Kilian Eyerich2,3Vijay K. Kuchroo4 Christopher T. Ritchlin5Maria T. Abreu6M. Merle Elloso7
Christopher T. Ritchlin5Maria T. Abreu6M. Merle Elloso7 Anne Fourie8Steven Fakharzadeh9Jonathan P. Sherlock10,11Ya-Wen Yang9
Anne Fourie8Steven Fakharzadeh9Jonathan P. Sherlock10,11Ya-Wen Yang9 Daniel J. Cua10*
Daniel J. Cua10* Iain B. McInnes12*
Iain B. McInnes12*Interleukin (IL)-23, an IL-12 cytokine family member, is a hierarchically dominant regulatory cytokine in a cluster of immune-mediated inflammatory diseases (IMIDs), including psoriasis, psoriatic arthritis, and inflammatory bowel disease. We review IL-23 biology, IL-23 signaling in IMIDs, and the effect of IL-23 inhibition in treating these diseases. We propose studies to advance IL-23 biology and unravel differences in response to anti–IL-23 therapy. Experimental evidence generated from these investigations could establish a novel molecular ontology centered around IL-23–driven diseases, improve upon current approaches to treating IMIDs with IL-23 inhibition, and ultimately facilitate optimal identification of patients and, thereby, outcomes.
Interleukin (IL)-23 is a member of the IL-12 cytokine family and is implicated in a cluster of immune-mediated inflammatory diseases (IMIDs) that includes psoriasis (PsO), psoriatic arthritis (PsA), and inflammatory bowel disease (IBD; namely Crohn’s disease [CD] and ulcerative colitis [UC]) (1–8). Because of its ability to reprogram immune cells in inducing and maintaining a highly proinflammatory state, IL-23 can be considered a dominant regulatory cytokine that acts as a rheostat, or a master switch, for initiating development and maintenance of these IMIDs.
In response to tissue injury or pathogenic insults, IL-23 is produced by tissue-resident myeloid cells and promotes expansion and survival of a subset of T lymphocytes, known as T helper (Th)17 cells, that produce and secrete IL-17A and other inflammatory mediators (Figure 1) (1, 13). Besides promoting proliferation and maintenance of Th17 cells, the cell type that has been studied most extensively with regard to IL-23 function, IL-23 can stimulate “natural” Th17 cells and IL-17+CD8+ T (Tc17) cells, as well as innate immune cells, such as γδ T cells, natural killer T cells (NKTs), mucosal-associated invariant T cells (MAITs), and IL-17–secreting innate lymphoid cells (ILC3s), all of which may be collectively termed type 3 immune cells or type-17 cells (2, 14, 15). This set of innate immune cells express the IL-23 receptor and retinoic acid receptor–related orphan receptor-γt (RORγt). The IL-23 receptor is also expressed on a small subset of interferon-γ (IFN-γ)–producing Th1 cells, where it also plays a critical role in inducing a colitogenic phenotype with induction of genes that have been implicated with human IBD by genome-wide association study (GWAS) (16). IL-23 stimulation of Th17 and type-17 cells induces local tissue inflammation via release of proinflammatory cytokines, such as IL-17A, IL-17F, IL-22, human granulocyte macrophage colony-stimulating factor (GM-CSF), IFN-γ, and tumor necrosis factor (TNF)-α. IL-23 can also suppress regulatory T (Treg) cell differentiation and promote expansion of tissue-resident memory T (TRM) cells, which have been linked to the chronicity of autoimmune and inflammatory diseases (9). IL-23 promotes programming of pathogenic Th17 cells that drive immune-mediated disease through the induction of genes encoding IL-23–dependent transcription factors, including the RORγt gene and the IL-23 receptor gene (IL23R), thus amplifying IL-23 signaling in a positive feedback loop and stabilizing production of proinflammatory effector molecules (17–19).
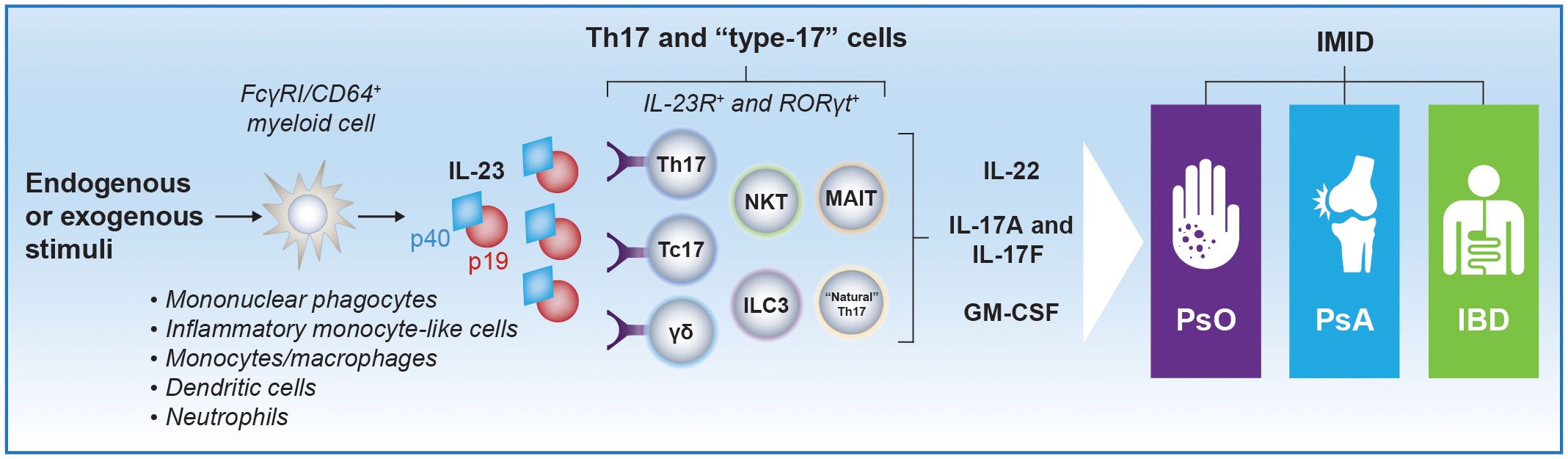
Figure 1 Schematic representation of IL-23–producing cells and IL-23 target cells implicated in PsO, PsA, and IBD pathogenesis. Myeloid cells are considered major producers of IL-23 in response to endogenous (eg, IL-6, TGF-β) or exogenous (eg, inflammation, infection) stimuli. Studies assessing the precise phenotypic and transcriptomic signature of IL-23–producing myeloid subsets in different IMIDs are ongoing. However, monocytes/macrophages, mononuclear phagocytes, and inflammatory monocyte–like cells that express FcγRI/CD64 have been identified as IL-23–producing cells in patients with psoriatic disease and IBD (9, 10). Semimature dendritic cells and neutrophils, which may also express FcγRI/CD64, have been identified as producers of IL-23 in PsO and IBD, respectively, as well (11, 12). High local concentrations of IL-23 produced by these cells can promote proliferation and survival of Th17 cells and stimulate other immune cells that express the IL-23 receptor and RORγt, including γδ T cells, NKTs, “natural” Th17 cells, Tc17 cells, and ILC3s, which collectively are termed type-17 cells (2). Th17 and type-17 cells induce local tissue inflammation via release of IL-17A, IL-17F, IL-22, and GM-CSF. Notably, induction of IL-17A and IL-17F production from Th17 cells does not require IL-23R signaling, but activation of the IL-23 receptor is required for pathogenic effector Th17 cell responses. FcγRI, Fc-γ receptor I; GM-CSF, granulocyte macrophage colony-stimulating factor; IBD, inflammatory bowel disease; IL, interleukin; IL-23R, IL-23 receptor; ILC, innate lymphoid cell; IMID, immune-mediated inflammatory disease; MAIT, mucosal-associated invariant T cell; NKT, natural killer T cell; PsA, psoriatic arthritis; PsO, psoriasis; RORγt, retinoic acid receptor–related orphan receptor-γt; Tc17, IL-17+ CD8+ T; Th, T helper.
Th17 effector cytokines drive inflammation in psoriatic disease (ie, PsO and PsA) and contribute to the development of spondyloarthritis (20). In IBD, IL-23 is one of the primary drivers of inflammation, although patients with CD and UC may have different cytokine expression profiles (21). Targeting IL-23 for the treatment of psoriatic disease and IBD may restore immune homeostasis and could have significant long-term anti-inflammatory effects. Inhibition of IL-23 reduces levels of IL-17A, IL-17F, and IL-22, and markedly improves clinical outcomes for patients with these IMIDs (1, 3–7, 22, 23). Moreover, robust joint and skin responses have been sustained long-term in patients with psoriatic disease receiving anti–IL-23 therapy (24–28). Although most patients respond to treatment, some show only a partial response. Furthermore, the therapeutic benefit of IL-23 inhibition beyond this immediate cluster of IMIDs is unclear. An attenuated effect with IL-23 inhibition in some cases may be due, in part, to the timing of therapeutic intervention over the course of disease and/or limitations of currently available anti–IL-23 therapies. Also, IL-23 may be a dominant regulatory cytokine in only type 3 IMIDs driven primarily by type 3 immune cells, pathogenic type-17 cells, and/or dysregulation of the IL-23/IL-17 axis. Additional studies are needed to establish a novel molecular ontology centered around IL-23–driven disease, advance new approaches to treating inflammatory diseases with IL-23 inhibition, and ultimately identify IMID patients who represent optimal candidates to receive treatment with anti–IL-23 therapy.
In 2000, Oppmann and colleagues identified IL-23, a novel member of the IL-12 family of cytokines comprising p19 and p40 subunits (Figure 2) (30). Although the p40 subunit is common to both, IL-23 (p19/p40) and IL-12 (p35/p40) are functionally distinct, with divergent roles in driving differentiation and maintenance of Th17 and Th1 cells, respectively (Figure 3) (1, 39, 41). While early studies using p40-/- mice and anti-p40 antibodies could not discriminate between IL-12 and IL-23, later studies in p19-/- (IL-23 deficient), p35-/- (IL-12 deficient), and p40-/- (IL-12 and IL-23 deficient) mice revealed that IL-23 was a critical cytokine driving inflammation in several models of IMIDs and exhibited functions that were pathogenetically discrete from IL-12 (1, 31, 39, 41, 45). In contrast to the role of IL-23 in driving inflammation in the skin, IL-12 receptor–mediated signaling appears protective in PsO (46). In some models of gastrointestinal inflammation, IL-12 has been shown to have a limited role, whereas other models suggest IL-12 is necessary to trigger inflammation, with IL-23 driving disease chronicity (1, 47). The roles of p19 heterodimeric cytokines paired with other subunits are not fully understood in IMIDs (1, 41).
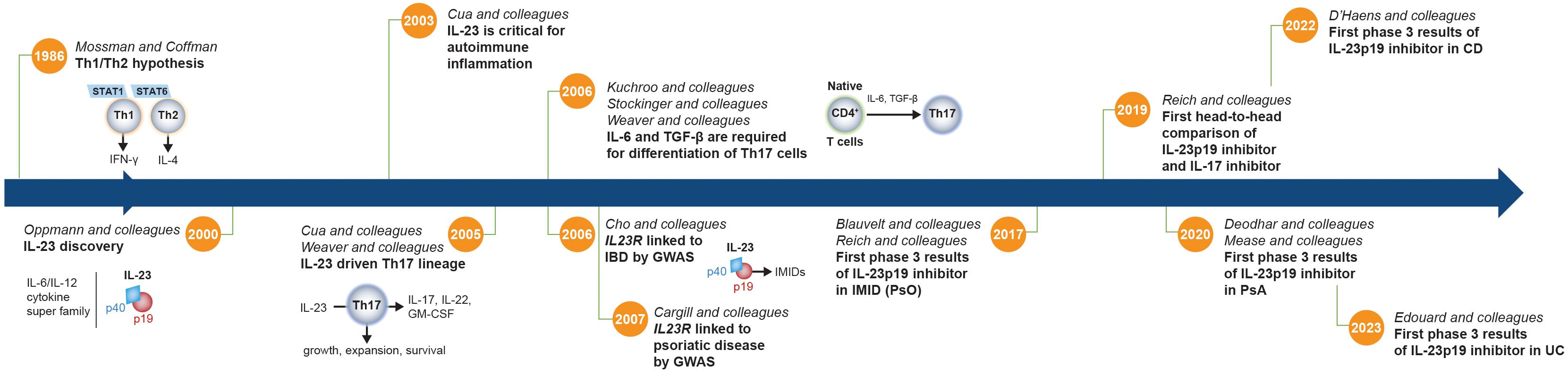
Figure 2 Timeline of critical discoveries related to the IL-23 pathway. Prior to the discovery of IL-23, the accepted paradigm proposed to divide CD4+ Th cells into 2 subsets (IFN-γ–producing Th1 cells and IL-4–producing Th2 cells) to explain the tendency of T cells to diverge into those that drive either a cellular or humoral immune response (29). However, this binary hypothesis did not explain the full spectrum of immune responses. IFN-γ–producing Th1 cells and IL-4–producing Th2 cells are dependent on STAT1 and STAT6, respectively, which did not adequately explain the regulation of STAT3-dependent CD4+ T cells in autoimmunity and infection. In addition, neutrophil recruitment and activation are major hallmarks of inflammation, and Th1 and Th2 cells do not promote significant neutrophil-mediated tissue injury. Following its discovery (30), IL-23 was shown to be essential for development of experimental autoimmune encephalomyelitis and was implicated in the growth, expansion, and survival of a subset of pathogenic T lymphocytes characterized by the production of IL-17A, IL-17F, IL-6, and TNF that is distinct from Th1 and Th2 cells, now known as Th17 cells (31, 32). TGF-β and IL-6 were later shown to be required to facilitate differentiation of naive T cells into Th17 cells (33–35), and the IL-23 signaling pathway was then linked to IMIDs by GWAS that identified variants of the IL23R gene that are associated with IBD and psoriatic disease susceptibility (36, 37). A little more than a decade later, the first phase 3 clinical trial results showing efficacy of an IL-23p19 subunit inhibitor (guselkumab) in an IMID (PsO) were published, followed over the next several years by results in PsA, CD, and UC (3–7, 38). CD, Crohn’s disease; GM-CSF, granulocyte macrophage colony-stimulating factor; GWAS, genome-wide association study; IBD, inflammatory bowel disease; IFN-γ, interferon-γ; IL, interleukin; IL23R, IL-23 receptor gene; IMID, immune-mediated inflammatory disease; PsA, psoriatic arthritis; PsO, psoriasis; STAT, signal transducer and activator of transcription; TGF-β, transforming growth factor-β; Th, T helper; UC, ulcerative colitis.
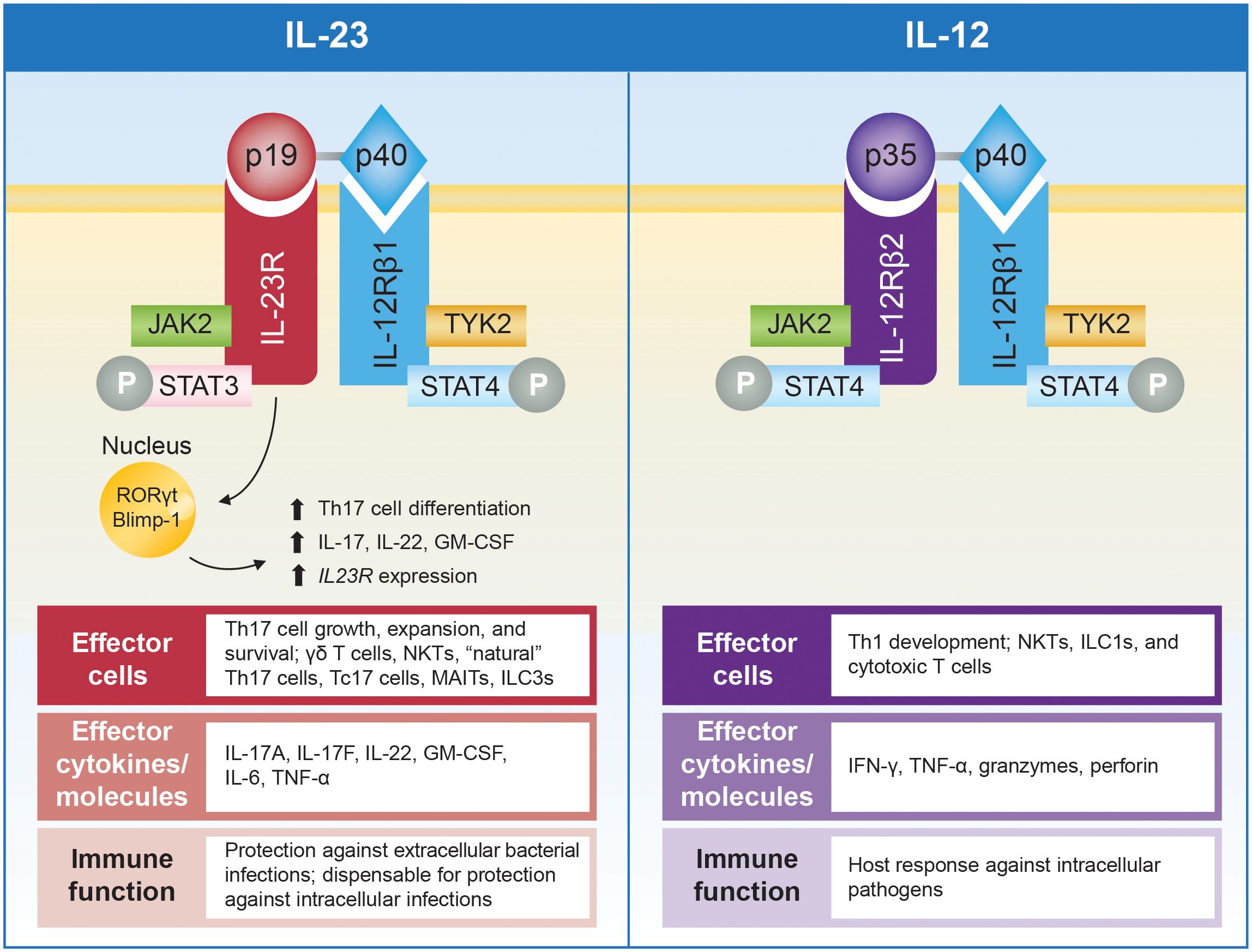
Figure 3 Schematic representation of IL-23 and IL-12 and their receptors, with summary of associated effector cells, cytokines, and immune function. IL-23 comprises the p19 and p40 subunits that bind to the IL-23R and IL-12Rβ1 subunits of the IL-23 receptor, respectively. IL-12 also contains the p40 subunit, which pairs with the p35 subunit that binds to IL-12Rβ2 (39). Engagement of the IL-23 receptor results in formation of a cytokine-receptor ternary complex that includes IL-23, IL-23R, and IL-12Rβ1 (40). Formation of this ternary complex activates a range of JAKs, including JAK2 and TYK2, which associate with intracellular domains of the receptor subunits and initiate specific signaling cascades (40–42). IL-23 activates pathogenic functions of CD4+ Th17 cells and stimulates other effector cells, including γδ T cells, NKTs, “natural” Th17 cells, Tc17 cells, MAITs, and ILC3s, that collectively produce IL-17A, IL-17F, IL-22, GM-CSF, IL-6, and TNF-α (32, 41). In contrast, IL-12 promotes differentiation of Th1 cells and activates NK cells, ILC1s, and cytotoxic T cells leading to production of IFN-γ, TNF-α, granzymes, and perforin (41). In addition, IL-23 activation of transcription factors, including STAT3, RORγt, and Blimp-1, enhances IL23R expression and, in a positive-feedback loop, can amplify IL-23 signaling (39, 43). RORγt represents a particularly notable IL-23–induced transcription factor essential for differentiation of naive T cells into Th17 cells. Simultaneous priming with TGF-β, IL-6, and IL-1 activates RORγt and induces expression of the IL-23 receptor, thus amplifying IL-23 signaling, which is critical for maturation and stabilization of the proinflammatory Th17 phenotype (18, 34, 35). The IL-23/Th17 pathway is important for protection against extracellular bacterial and fungal infections, while it appears to be dispensable for protection against most intracellular infections. In contrast, the IL-12/Th1 pathway is required for host responses to intracellular pathogens (44). Blimp-1, B lymphocyte–induced maturation protein 1; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; IL-12Rβ1, IL-12 receptor, β1 subunit; IL-12Rβ2, IL-12 receptor, β2 subunit; IL-23R, IL-23 receptor subunit; ILC, innate lymphoid cell; JAK, Janus kinase; MAIT, mucosal-activated invariant T cell; NKT, natural killer T cell; P, phosphoryl group; RORγt, retinoic acid receptor–related orphan receptor-γt; STAT, signal transducer and activator of transcription; Tc17, IL-17+ CD8+ T; Th, T helper; TNF-α, tumor necrosis factor-α; TYK, tyrosine kinase.
The IL-23/IL-17 axis is also involved in host defense against certain bacterial and fungal pathogens (2) and also plays a key role in gut tissue homeostasis by supporting barrier function, tissue repair, and maintenance (1, 48). IL-17A signaling contributes to maintenance of gut epithelial homeostasis and promotes neutrophil effector function to protect against certain mucosal infections. By corollary, IL-17A inhibition has been shown to have paradoxical effects in the gut and may induce or worsen bowel inflammation (48).
The role of IL-23 in maintaining barrier homeostasis in the skin and joints is less well understood. Keratinocytes from normal skin express low levels of IL-23 (49), but it is only in the context of inflammatory skin disease that IL-23 can stimulate IL-20 family members, resulting in TNF-regulated epidermal thickening (17, 50). In the gut, epithelial-like C-X3-C motif chemokine receptor 1(CX3CR1)+ macrophages are an important source of IL-23 and protect from effacing bacteria, likely via modification of gastrointestinal barrier function (51). In joints, CX3CR1+ macrophages play a role in maintaining tight junction-mediated barriers at the synovium lining, but how IL-23 may contribute to maintaining barrier function has not yet been elucidated (52). Only a small number of IL-23–responsive cells appear to be present in entheseal tissue from mice and humans (20, 53). In response to endogenous (eg, IL-6, TGF-β) or exogenous (eg, microbial-component lipopolysaccharides, bacterial infection) stimuli, IL-23 expression increases and activates IL-23 receptor–expressing cells to produce downstream effector cytokines, contributing to local tissue inflammation that plays a role in barrier homeostasis (1, 13).
The IL-23 receptor (Figure 3) is constitutively expressed on the surface of NKTs, ILCs, γδ T cells and MAITs, all of which recognize antigens via invariant T-cell receptors or other motifs (42). Engagement of the IL-23 receptor results in formation of a cytokine-receptor ternary complex that includes IL-23, IL-23R, and IL-12Rβ1 (40), and activates a range of Janus kinases (JAKs; eg, JAK2, TYK2) and signal transducers and activators of transcription (STATs; eg, STAT3, STAT4). In particular, TYK2 and STAT3 activation represent key proximal signaling events for upregulating production of cytokines (eg, TNF-α, IL-17, IL-6, IL-23) implicated in IMIDs (40–42). Therefore, blockade of JAKs, including TYK2, has emerged as a therapeutic target in IMIDs (2). However, JAK inhibitors broadly inhibit the signaling of multiple cytokines and are not specific to IL-23 activation.
Induction of IL-17A and IL-17F production by Th17 cells does not require IL-23R signaling, but activation of the IL-23 receptor is required for pathogenic effector Th17 cell responses, which include production of IL-17A, IL-17F, IL-22, GM-CSF, and IFN-γ (54, 55). Type-17 cells have been shown to be important in the pathogenesis of IMIDs, with Th17 cells being major producers of IL-17A in host defense (2). There is growing evidence that two different types of Th17 cells exist in vivo in murine models: non-pathogenic (homeostatic) and pathogenic Th17 cells (54). Nonpathogenic Th17 cells regulate barrier functions and control microbial invasion at mucosal surfaces. Pathogenic Th17 cells induce tissue inflammation and autoimmunity. IL-23 receptor expression differentiates pathogenic from nonpathogenic type 17 cells, with pathogenic cells demonstrating high levels of IL-23 receptor expression (16). IL-21 and IL-12 were shown to stimulate IL-23 receptor expression and, therefore, promote differentiation and activation of type 17 cells in vitro. In addition, the localization pattern of IL-23 receptor–positive cells within tissues, particularly in barrier tissues that have resident type-17 cells, may determine disease-specific patterns of clinical inflammation when the IL-23 pathway is dysregulated (42).
The roles of IL-17, IL-22, and other effector cytokines downstream of IL-23 vary by disease state (56, 57). Cytokine expression is dynamic, and expression patterns change over the course of disease; in particular, timing and location are important. The IL-17A receptor subunit (IL-17RA), which binds IL-17A, IL-17F, IL-17A/F, and IL-25 (IL-17E), is ubiquitously expressed by a variety of cell types, including endothelial cells, keratinocytes, and gut enterocytes. Activation of these cells by IL-17A and IL-17F, usually working in synergy with other activator cytokines, promotes the expression of IL-1, IL-6, IL-8, and TNF, which may induce and sustain chronic inflammatory processes. IL-23 also regulates production of IL-22 by Th17 cells, Th22 cells, and ILC3s; furthermore, CD8+ T cells, γδ T cells, and NKTs can produce IL-22 in the presence of IL-23 (56). In addition, a small subset of Th1 cells was shown to express IL-23R and activation of IL-23 resulted in the induction of multiple genes that are not typically expressed in Th17 cells, but which have been linked to IBD by GWAS analysis (16). IL-17 upregulates expression of genes encoding immune chemoattractants whereas IL-22 downregulates expression of genes needed for keratinocyte differentiation; both IL-17 and IL-22 can induce expression of antimicrobial protein-encoding genes (58). IL-22 has been implicated in promoting inflammation and the pathogenesis of several diseases (eg, PsO and select cancers) but plays a protective role in other conditions (eg, asthma and pancreatitis) (56). The potentially conflicting roles for IL-22, coupled with a lack of efficacy for IL-22 inhibition in studies in some disease states, raise questions about the therapeutic utility of targeting this cytokine.
In Tables 1, 2, we summarize the extensive experimental and clinical data characterizing the role of IL-23 and the therapeutic effect of targeting the IL-23p19 subunit in PsO and PsA. IL-23 plays an important role in the pathogenesis of psoriatic disease, in part, by contributing to recurrence of skin lesions and inducing enthesitis in PsO and PsA, respectively (20, 63, 90). Clinical data suggest that selective IL-23p19 subunit inhibitors are effective in psoriatic disease. In PsO, targeting the IL-23p19 subunit can restore favorable ratios of T-cell populations in lesional skin compared with IL-17A inhibition (9). In PsA, targeting the IL-23p19 subunit can modulate expression of disease-associated genes, with directionality toward normalization of whole blood transcriptomes towards those found in healthy volunteers (87). Selective blockade targeting of the IL-23p19 subunit has not elicited specific safety signals in either PsO or PsA (5, 6, 68, 71, 84, 85). The clinical efficacy of selective IL-23 inhibition has been established, with anti–IL-23 therapy approved for use in both PsO and PsA. In addition, head‐to‐head comparative data from clinical trials of patients with PsO suggest that IL-23p19 subunit inhibitors (ie, guselkumab, risankizumab) have superior efficacy over the TNF inhibitor adalimumab (72), the IL-17A inhibitor secukinumab (68, 71), and the IL-12/IL-23 inhibitor ustekinumab (91). Nevertheless, onset of efficacy with IL-23p19 subunit inhibitors has been shown to be less rapid than with IL-17A inhibitors, as observed in the head-to-head comparison of guselkumab and ixekizumab for PsO (71, 75). In PsA, there are no head-to-head comparisons of IL-23p19 subunit inhibitors versus other targeted PsA therapies. A systematic literature review and network meta-analysis of clinical trials in patients with PsA showed that IL-23p19 subunit inhibition with guselkumab provided better skin efficacy than most TNF and JAK inhibitors, while demonstrating joint efficacy comparable to IL-17A, JAK, and TNF inhibitors administered by subcutaneous injection (92).
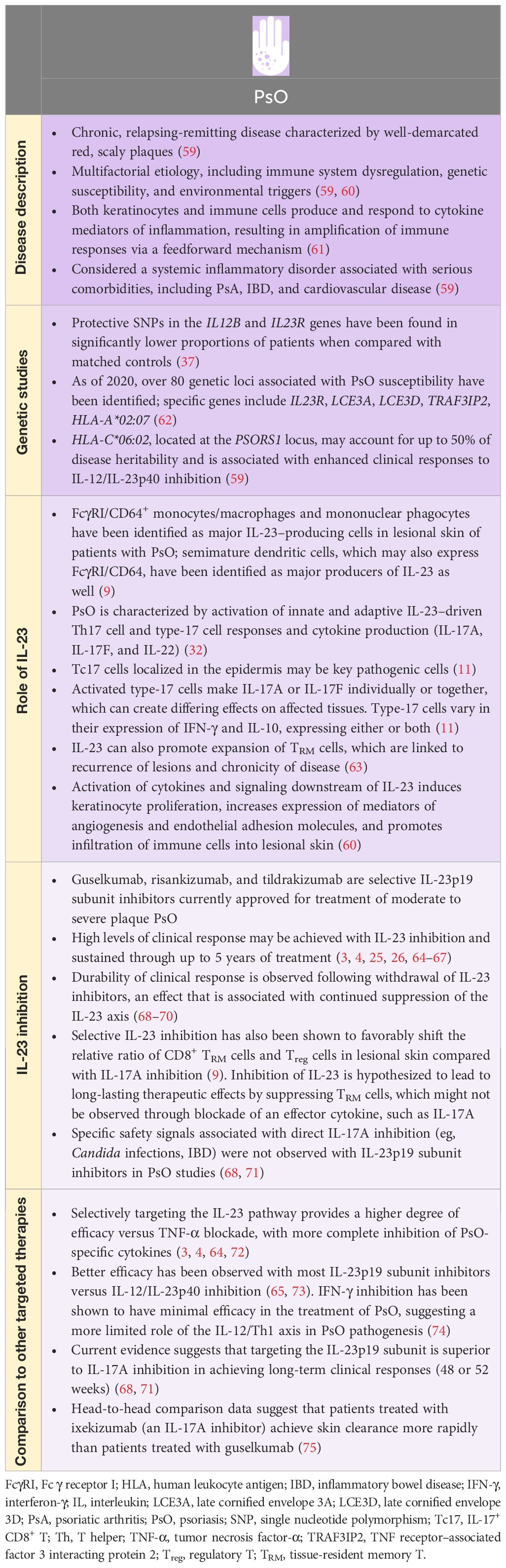
Table 1 PsO and IL-23.
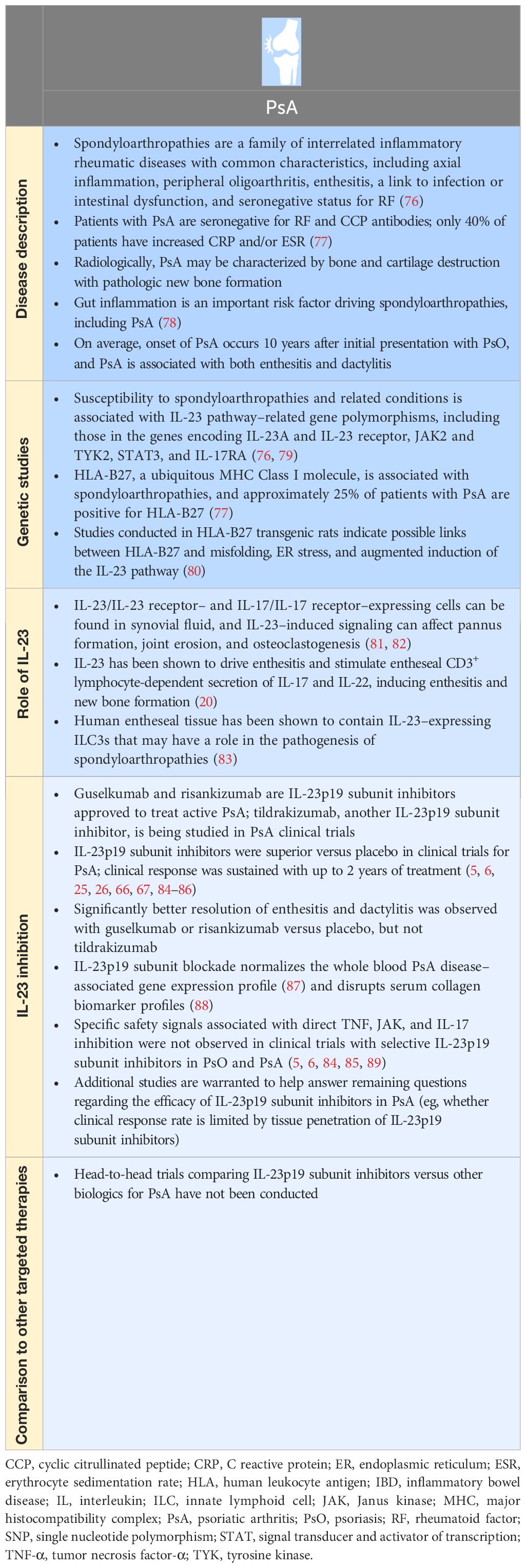
Table 2 PsA and IL-23.
Despite experimental evidence from animal models that suggests a pathogenic role for IL-23 in axial spondyloarthritis, clinical trials of IL-23p19 subunit and IL-12/IL-23p40 subunit inhibitors in patients with axial spondyloarthritis failed (93, 94). The lack of efficacy of IL-23 blockade in axial (axial spondyloarthritis) but not peripheral (PsA) spondyloarthritis may be due to differences in IL-23 responsive cell types involved in disease pathogenesis and the timing of therapeutic intervention in the course of these diseases (95). Limited penetration of large molecule therapeutic antibodies into joint tissues, such as synovium, may also limit efficacy. In contrast to trials in axial spondyloarthritis, post hoc analyses of selective IL-23p19 subunit inhibition with guselkumab in patients with axial PsA showed significant improvement in symptoms of axial involvement versus placebo (96). A prospective, controlled trial of guselkumab in patients with PsA with magnetic resonance imaging–confirmed axial involvement is ongoing to further expand upon this finding (ClinicalTrials.gov Identifier: NCT04929210).
In Table 3, the genetic, experimental, and clinical evidence supporting a role for IL-23 inhibition in IBD is summarized. In inflamed tissue, overexpression of IL-23 converts nonpathogenic Th17 cells into pathogenic effectors, resulting in impaired barrier function, and drives continued inflammation via downstream pathways, leading to survival and expansion of Th17 cells (1). Moreover, IL-23 signaling can suppress Treg cell differentiation, a cell population that is important in regulation and suppression of chronic gut inflammation (1). Risankizumab is an IL-23p19 subunit inhibitor approved for use in many countries for CD, while mirikizumab is an IL-23p19 inhibitor approved for treatment of UC in Japan and by the European Medicines Agency. IL-23p19 subunit inhibitors have generally demonstrated efficacy in clinical and/or endoscopic outcomes, with evidence to support maintenance of long-term clinical responses and a favorable safety profile (7, 8, 101, 102, 104, 105). One systematic review and network meta-analysis of clinical trials for biologic therapies for CD suggested that IL-23p19 subunit inhibition with risankizumab may yield better outcomes compared to other biologics and be preferred as second-line therapy for induction of clinical remission (106). However, head-to-head comparative studies of IL-23p19 subunit inhibitors versus other biologics for IBD are warranted to evaluate the relative efficacy of IL-23p19 subunit inhibitors versus other biologics.

Table 3 IBD and IL-23.
Other IMIDs in which IL-23 has been implicated through genetic association, tissue expression, animal models, and plausible biological profile include palmoplantar pustulosis, pityriasis rubra pilaris, rheumatoid arthritis, multiple sclerosis, giant cell arteritis, and type 1 diabetes (31, 45, 107–118). Guselkumab is an IL-23p19 subunit inhibitor approved for treatment of palmoplantar pustulosis in Japan and other Asian countries. Selective targeting of the IL-23p19 subunit improved clinical outcomes for patients with palmoplantar pustulosis and a small number of patients with pityriasis rubra pilaris (107, 118).
In contrast, IL-12/IL-23p40 subunit inhibition failed to show clinical benefit in multiple sclerosis, and IL-12/IL-23p40 or IL-23p19 subunit-targeted therapy failed to improve clinical outcomes in studies of patients with active rheumatoid arthritis, despite strong scientific rationale for this approach derived from animal models (31, 45, 108, 110). The IL-23/IL-17 axis has been implicated in driving intrinsic inflammatory activity and triggering the clinical onset of rheumatoid arthritis, primarily in animal models, suggesting that early intervention in the pre–rheumatoid arthritis phase could potentially lead to clinical improvement (111, 119). Observations in animal models of multiple sclerosis suggest that IL−23 is necessary for the induction of disease but not disease maintenance, and that lack of IL−23 is not protective once encephalitogenic T cells have developed (112). Patients with multiple sclerosis may therefore potentially benefit from targeting the IL-23/IL-17 axis at an earlier stage of disease. Treatment of multiple sclerosis is further complicated by the need for therapeutic agents that cross the blood-brain barrier (120). Nevertheless, IL12/23p40–targeted therapy in patients with established multiple sclerosis was ineffective (121), whereas the impact of IL-23p19 subunit inhibition in this patient population has yet to be determined. However, the anti–IL-17A therapeutic antibody secukinumab was shown to decrease the development of new lesions in a small, double-blind clinical trial of patients with multiple sclerosis (122).
Conflicting results regarding the efficacy of IL-12/IL-23p40–targeted therapy in patients with giant cell arteritis have been observed across trials, and further studies are needed for more definitive evidence (117, 123). A proof-of-concept study to evaluate targeting the IL-23p19 subunit with guselkumab in patients with new-onset or relapsing giant cell arteritis is ongoing (NCT04633447). IL-12/IL-23p40 inhibition is currently being assessed for treatment of new-onset type 1 diabetes in clinical studies (113, 114). IL-23 gene expression was shown to be increased in the pancreatic islet cells of a patient with type 1 diabetes, and targeting both the IL-23p19 and IL-12/IL-23p40 subunits has been shown to suppress the incidence of diabetes in nonobese diabetic mice (115, 116).
The pleiotropic nature of the biology of IL-23 also predicts its potentially important role in other diseases beyond IMIDs, such as obesity (124), atherosclerosis (125), stroke (126), cancer (1, 39), mood disorders (124), and neurodegenerative diseases (127). Although IL-23 could be involved in the pathogenesis of multiple disease states, it may act on different cell types or at different stages of disease in each, yielding nuanced effects, a wide range of clinical manifestations of disease, and varied implications of IL-23p19 subunit-targeted therapy.
Further investigation is needed to better understand the molecular biology of IL-23 to properly adapt anti–IL-23 therapy across the breadth of IMIDs. Herein, we propose a multiomics approach to advance the science of IL-23, including comprehensive studies to identify the spectrum of IL-23 receptor–expressing cells, and to provide insights into the role of IL-23 in driving IMIDs. Moreover, we identify gaps in the understanding of IL-23 signaling and cellular activity and discuss durability of treatment response and future prospects for anti–IL-23 therapeutic intervention, including potential approaches for combination therapy (Box 1).
The emergence of high-throughput technologies over the past 30 years has revolutionized medical research. The “omic” technologies provide a comprehensive assessment of genomics, epigenomics, transcriptomics (including single cell RNA-seq and spatial transcriptomics), proteomics, metabolomics, and microbiomics. Each of these “omic” technologies provides a global pattern of molecular signatures (genes, epigenetic factors, RNA transcripts, proteins, or metabolites) that are altered in disease, providing insight into biomarkers, pathways, or cell types involved in disease pathogenesis. Combining these “omic” technologies can provide a more complete picture of the molecular factors that drive functional consequences and interactions of IL-23 signaling underlying the pathogenesis and phenotypic manifestations of IMIDs (128–130). For instance, a recent genome-wide study identified allelic variants of the HLA-C upstream region (which includes PSORS1C3, MICA, and TNFA) that were associated with clinical response to anti-IL-12/IL-23 therapy (131). In addition, an interaction between gut microbiota composition and responsiveness to IL-17 and IL-23 inhibitors has been observed in patients with PsO (132). Another example derives from a multiomics study that used matched colonic biopsy tissue and serum samples at multiple time points from IBD patients treated with guselkumab or placebo. This application of a multiomics approach allowed deeper understanding of local (gastrointestinal tissues) versus systemic (serum protein) immune responses driven by IL-23 in IBD patients and revealed that guselkumab mainly suppressed local gastrointestinal tissue inflammation by normalization of gut epithelial cell-injury responses (manuscript in preparation).
A definitive atlas of IL-23 receptor–expressing cells across different tissues and at different phases of disease activity or progression in patients would be a valuable resource to further elucidate understanding of normal physiological and pathophysiological function of IL-23. For example, the role of IL-23 receptor signaling in T cells has been most extensively studied in experimental animal models, and the effect of IL-23 on diverse T-cell targets in human patients is not well understood (133). Generally, the IL-23 receptor is expressed at very low levels and is difficult to detect at the protein level; therefore, a combination of spatial transcriptomic and RNA sequencing data may elucidate IL-23 receptor–expressing cell populations of interest with high resolution. Data from single cells may then be interpreted in the context of broader sequencing data to better understand the IL-23 pathway gene expression profile. In turn, a recently developed and validated panel of anti–human IL-23 receptor antibodies is being utilized to identify and characterize unique IL-23 responding populations across inflamed skin, joint, and gut tissues (Lin et al, manuscript in preparation). These studies will complement IL-23 receptor transcriptomic analyses and contribute to further elucidating the role of IL-23 during disease pathogenesis.
Studies on IL-23 signaling and cellular activity are needed to further characterize the normal physiological function of IL-23 and to identify new targets and strategies for drug development independent of direct IL-23 engagement in order to more fully tap into the therapeutic potential of inhibiting the IL-23 pathway. Gaps currently exist with respect to the impact of therapeutic intervention at the level of downstream effectors, potential modulators of IL-23 signaling, and the molecular basis for inadequate response to IL-23 inhibition in some cases.
Inhibiting IL-23 directly may have broader utility than targeting downstream effectors of the IL-23 pathway. For example, targeting members of the IL-17 family of cytokines may not sufficiently or uniformly suppress inflammatory pathways owing to the functional redundancy of IL-17 family cytokines in promoting inflammation (134). However, not all inflammatory diseases in which IL-17A or IL-17F are considered pathogenic drivers are necessarily IL-23–dependent (eg, hidradenitis suppurativa). As aforementioned, the effect of IL-23 on diverse T-cell targets in patients is not yet well understood. Comparing and contrasting effector and regulatory pathway mediators across diseases in patients will better describe the impact of IL-23 and, in turn, IL-23 inhibition on broader T-cell populations. Although IL-23 signaling is generally highly conserved across species, experiments using human cells or tissues are essential to ensure relevance to human disease. Approaches to characterize IL-23–producing and IL-23–responsive cells from clinical tissue samples, such as single-cell transcriptomics, will better refine understanding of IL-23 signaling pathways and provide temporal and spatial maps of IL-23 pathway dysregulation in tissues (11).
Even within the cluster of IMIDs that is sensitive to selective IL-23p19 subunit targeting, not all patients respond adequately. Additional research on signaling pathways downstream of IL-23 may determine why some patients experience inadequate response and identify potential complementary therapeutic targets. In turn, it will be important to understand synergistic and antagonistic mediators of IL-23 receptor signaling, and how they are dependent on the local tissue microenvironment. For example, in a recent study, quantitative phospho-proteomic analysis of Th17 cells was used to identify potential targets downstream of IL-23 signaling (128). Similar approaches may be used to address questions such as whether there are nuances to IL-23 signaling in different cell types or tissues that may impact responses to therapeutics targeting the IL-23p19 subunit. Furthermore, signaling crosstalk between IL-23 and other cytokines, such as IL-1β, can augment and sustain IL-23 signaling (135); study of such interactions is now feasible given the availability of high-fidelity resolution techniques. In this context, other pathogenetic components, such as the skin and gut microbiomes, may influence IL-23 receptor signaling, given its critical roles in protecting against invading pathogens, educating the immune system, and metabolizing natural products in these tissues (136).
TRM cells are a subset of noncirculating memory T cells that persist over the long-term in peripheral tissues. TRM cells provide rapid, local immune responses on re-exposure to pathogens under healthy conditions, but also develop after sensitization to self-antigens and are involved in the pathogenesis of autoimmune disorders. TRM cells are a key reason for why some IMIDs, such as PsO, are chronic, life-long diseases, and why skin lesions in PsO tend to recur at the same sites upon relapse of disease. Treg cells help to maintain immune homeostasis and self-tolerance by suppressing inflammation and effector T cells. IL-23 has been shown to suppress the differentiation of Treg cells (9), and promote differentiation, survival, and expansion of pathogenic Th17 cells (13).
The durable, long-lasting therapeutic effect observed with selective targeting of IL-23p19 subunit is hypothesized to derive from suppression of TRM cells. IL-23 inhibition has been shown to favorably shift the relative ratio of CD8+ TRM cells and Treg cells in PsO lesions compared with IL-17A inhibition (9). Moreover, clinical studies have shown superior long-term maintenance of response with IL-23 inhibition over IL-17A blockade; IL-17A inhibitors, in essence targeting an effector cytokine, have less impact on the relative numbers of proinflammatory and anti-inflammatory T cells compared to IL-23 inhibitors (71). Nonetheless, further studies with larger numbers of samples over long-term duration of follow-up are needed to confirm these findings. Future studies are needed to determine whether selective IL-23 inhibition, if initiated in the optimal context, can result in the reduction or even elimination of pathogenic TRM cells and ultimately drive PsO and other IMIDs into long-term remission (9). The intriguing possibility of prolonged drug-free remission is particularly enticing in this context. The ongoing GUIDE study is evaluating the implications of early intervention with guselkumab in patients with PsO and short disease duration to achieve a super-response, extend dosing interval, and maintain disease control after treatment withdrawal, and is further exploring the impact of guselkumab treatment on the balance of TRM/Treg cell populations in patients with PsO over time (137).
Specific attributes of currently available and future IL-23 inhibitors may enhance blockade of IL-23 signaling and further improve clinical outcomes. The in vitro binding affinity for IL-23 and potency for blocking IL-23 signaling of available antibodies targeting the IL-23p19 subunit may be different, potentially driving differences in clinical performance of these drugs (138). However, the relevance of bioavailability and serum levels of IL-23 inhibitors to clinical efficacy is unclear given that IL-23 is produced and drives inflammation within the local tissue microenvironment. It is important to note, however, that IL-23–producing cells are also capable of trafficking to other tissue sites to extend or propagate disease pathogenesis (139). The mechanism by which an anti–IL-23 antibody localizes to inflamed tissue(s) to sequester and neutralize IL-23 at the site of its production and activity may represent an important attribute for consideration. For example, myeloid cells that express FcγRI/CD64 have been identified as major IL-23–producing cells in preclinical models as well as in lesional tissue from human patients with psoriatic disease and IBD (9, 10). Thus, anti–IL-23 monoclonal antibodies that can also bind to FcγRI/CD64 via their Fc region may be optimally localized within the inflamed tissue microenvironment to neutralize IL-23 at its cellular source and potentially provide a therapeutic advantage.
A potential alternative to directly targeting the IL-23p19 subunit is to target the IL-23 receptor itself. Orally bioavailable therapeutic peptides that bind the IL-23 receptor with high specificity and affinity offer promise (140–142). Small molecules and peptides may have an advantage over therapeutic antibodies by virtue of better tissue penetration, which could potentially improve outcomes for and expand the scope of IMIDs that are treatable through inhibition of IL-23 receptor signaling. An oral IL-23 receptor antagonist peptide (142) is currently being studied in PsO and UC in clinical trials, and future studies in other IMIDs may be warranted.
Although monotherapy with selective anti–IL-23 antibodies has been shown to be therapeutically effective across a variety of IMIDs, some patients fail to achieve adequate or durable responses. One approach to addressing remaining unmet needs across IMIDs is to also target complementary pathogenic pathways through combination therapy. IL-23 inhibitors may be useful in the context of combination therapy because of their potent ability to target a dominant, regulatory cytokine that serves as a rheostat to control the stream of inflammatory signals that can drive IMID pathogenesis. Moreover, the safety profile of IL-23 inhibitors has been thus far reassuring, which further supports pairing IL-23 inhibition with a second mechanism in a combination therapy approach. In turn, IL-23 blockade may synergize with inhibition of a pathway with a complementary mechanism of action, such as the TNF pathway, to achieve even higher levels of efficacy than those achievable using either approach alone, and potentially with minimal risk of compounding safety concerns, a problem that has beset prior combination therapeutic approaches for cytokine blockade.
Recently, the combination of IL-23 and TNF inhibition was shown to have a synergistic effect on preventing loss of body weight and reducing local colonic tissue inflammation in an anti-CD40 antibody–induced colitis mouse model (143). In that preclinical study, improvements in disease features were accompanied by normalization of gene expression patterns in shared molecular pathways for either treatment when used as monotherapy and when used in combination; however, normalization of expression of gene networks that were uniquely impacted in response to combination therapy was also observed. In the phase 2 VEGA study for UC, combination induction treatment with both an IL-23 inhibitor and a TNF inhibitor more effectively induced clinical response, remission, and endoscopic improvement than monotherapy with either agent alone (144). Further still, combination therapy was also shown to reverse the disease transcriptomic profile to a greater extent than either monotherapy alone (145). An additional ongoing clinical trial is studying the impact of IL-23 inhibition in combination with TNF inhibition in PsA (NCT05071664).
Remarkable progress over the last 20 years has advanced our understanding of the science of IL-23, leading to the development of transformational therapies that selectively block IL-23. IL-23 may represent a dominant regulatory cytokine in the context of type 3 IMIDs, with broad effects mediated by regulation of Th17 cells, other IL-23 receptor+ type-17 cells, and/or the IL-23/IL-17 axis. Extensive experimental and clinical evidence supports a role for IL-23 as a critical regulatory cytokine in a class of IMIDs that includes PsO, PsA, and IBD. The use of IL-23 inhibitors has been very successful in treating these IMIDs. However, not all patients respond adequately to currently available anti–IL-23 therapies and some patients may show only a partial response to treatment. IL-23 inhibition has been unsuccessful in clinical trials for some IMIDs, despite strong a priori scientific rationale for anti–IL-23 treatment. Studies to determine the therapeutic efficacy of IL-23 inhibition in yet other inflammatory diseases are needed. Factors contributing to these observations may include the timing of therapeutic intervention over the course of disease and limitations of currently available IL-23 inhibitor therapies.
We propose additional studies to address remaining challenges and unmet needs pertaining to anti–IL-23 therapy and to advance the science of IL-23. These include studies using a multiomics approach to: 1) further define the identities and roles of IL-23–expressing cells and IL-23 receptor–expressing cell targets in different inflammatory diseases; 2) characterize nuanced effects of IL-23 in different cell types or tissues; 3) describe changes in IL-23 receptor expression and signaling over the course of disease; and 4) characterize the mechanisms underlying tissue-specific responses and durability of response with IL-23 inhibitor therapy. Results from such studies hold strong potential to establish a novel molecular ontology centered around IL-23–driven disease, enhance and/or complement therapeutic approaches for IL-23 inhibition in treating IMIDs, further understand variability in achieving clinical responses, and ultimately better identify ideal patient candidates for anti–IL-23 therapies. The future appears promising, with the emergence of new strategies, including combination therapy, and ongoing progress in the understanding of the science of IL-23 to foster the next generation of therapeutics targeting the IL-23 pathway.
JK: Conceptualization, Writing – review & editing. KE: Conceptualization, Writing – review & editing. VK: Conceptualization, Writing – review & editing. CR: Conceptualization, Writing – review & editing. MA: Conceptualization, Writing – review & editing. ME: Conceptualization, Writing – review & editing. AF: Conceptualization, Writing – review & editing. SF: Conceptualization, Writing – review & editing. JS: Conceptualization, Writing – review & editing. Y-WY: Conceptualization, Writing – review & editing. DC: Conceptualization, Writing – review & editing. IM: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare that this study received funding from Janssen Global Services, LLC. The funder had the following involvement in the study: preparation of the article.
The authors wish to thank J. Matthew Kuczmarski, PhD, and Miranda Tradewell, PhD, of Lumanity Communications Inc. for their writing and editorial support in the preparation of this manuscript.
JK served as a consultant for and/or received honoraria from AbbVie, Allergan, Almirall, Amgen, Arena, Aristea, Asana, Aurigene, Biogen Idec, Boehringer Ingelheim, Bristol Myers Squibb, Escalier, Galapagos, Janssen, Eli Lilly, MoonLake Immunotherapeutics, Nimbus, Novartis, Pfizer, Sanofi, Sienna Biopharmaceuticals, Sun Pharmaceutical Industries, Target-Derm, UCB, Valeant, and Ventyx. KE received speaker fees from and/or served on an advisory board for AbbVie, Almirall, Boehringer Ingelheim, Bristol Myers Squibb, Galapagos, Eli Lilly, Janssen, Pfizer, Novartis, Sanofi, and UCB. VK served as a consultant for iTeos; has an ownership interest in and is a member of the scientific advisory board for Tizona Therapeutics; is a cofounder of and has an ownership interest in Celsius Therapeutics; and is a cofounder of Bicara Therapeutics. CR received grant/research support and consulting fees from UCB, AbbVie, and Amgen; and received consulting fees from Eli Lilly, Pfizer, Novartis, Gilead, and Janssen. MA is a consultant or served on advisory boards for AbbVie Inc, Arena Pharmaceuticals Inc now Pfizer, Bristol Myers Squibb, Celsius Therapeutics, Eli Lilly and Company, Gilead Sciences Inc, Janssen Pharmaceuticals, Janssen Global Services, Pfizer Pharmaceutical, Prometheus Biosciences, UCB Biopharma SRL. She has received fees for lecturing from Alimentiv, Janssen Pharmaceuticals, Prime CME and WebMD Global LLC. IM received consulting fees and grant/research support from AstraZeneca, Bristol Myers Squibb, Amgen, Eli Lilly, GSK, Janssen, Novartis, Roche, and UCB; received consulting fees from AbbVie, Cabaletta, Compugen, Gilead, Pfizer, and Sanofi; serves as a shareholder of Compugen and Causeway Therapeutics, and as a board member for National Health Service Greater Glasgow and Clyde; and is a trustee for Versus Arthritis. ME, AF, SF, JS, Y-WY, and DC are employees of Janssen and hold stock in Johnson & Johnson.
Box 1. Questions to consider for future investigation to advance the science of IL-23 and approaches to IL-23 inhibitor therapy.
A multiomics approach to advancing the science of IL-23
● Are there any trends in the literature that identify how the IL-23/IL-17 axis may function and drive nuanced effects in different cell types or tissues?
● Does the impact of this pathway change over time or with progression of disease, identifying an ideal timeframe or context in the course of disease to initiate IL-23 inhibitor treatment?
● Are there any differences in biomarker profiles between inadequate responders and responders to therapy that may be used to predict clinical outcomes?
● What is the epigenetic profile of IL-23 signaling in various immune and non-immune cell types and how may this impact disease pathogenesis, disease progression, or response to treatment?
● What post-translational modifications of downstream cytokines/effector molecules are associated with IL-23 signaling?
● What are the functional consequences of changes in multiomics profiles and associated changes in immune cell populations over the course of disease and in response to treatment?
“Broad sweep” of IL-23 receptor–expressing cells
● Which cells express the IL-23 receptor in healthy and inflamed tissues in humans?
● What is the role of IL-23 receptor signaling in diverse T-cell targets?
● How may a comprehensive assessment of IL-23 receptor expression across cell types and tissues identify new disease states that may be treatable through targeted intervention with IL-23 inhibition?
● How does IL-23 receptor expression change at various timepoints and tissue types across IMIDs and in response to treatment with IL-23 blockade?
● Is the IL-23 receptor coexpressed with other molecules of interest (ie, other cytokines or receptors) that may help to explain differences between currently available therapies and help design future treatments?
Gaps in understanding IL-23 signaling and cellular activity
● Are there different nuances to IL-23 signaling in different cell types or tissues that may have functional consequences impacting disease pathogenesis or response to treatment?
● Do these signaling nuances change over the course of disease progression, and normalize with treatment with an IL-23 inhibitor?
● What is the role of STAT3 and STAT4 signaling in the context of IL-23 receptor signaling in Treg cells?
● What is the relationship between tissue-specific microbiota and IL-23 receptor signaling?
Durability of response
● What is the effect of targeting IL-23p19 on TRM cells and the ratio of TRM/Treg cells across IMIDs?
● What are the mechanisms underlying the long-lasting effects of targeting IL-23p19?
● Can early intervention with IL-23 blockade after recent onset of disease modify the course of disease progression by suppressing TRM cells?
Future IL-23 inhibitor molecules
● What molecular attributes of IL-23 inhibitors may be relevant for optimizing inhibition of IL-23 across IMIDs?
● Are there therapeutic advantages in targeting the IL-23 receptor versus IL-23p19 subunit directly in treating IMIDs?
Combination therapy
● What potential therapeutic benefits may derive from combining IL-23 inhibition with blockade of another complementary inflammatory disease-driving pathway in treating IMIDs?
● What complementary pathway(s) would be optimal for blockade in combination with IL-23 inhibition for treatment of given IMIDs?
IL, interleukin; STAT, signal transducer and activator of transcription; Treg, regulatory T.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Neurath MF. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. (2019) 45:1–8. doi: 10.1016/j.cytogfr.2018.12.002
2. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
3. Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. (2017) 76:405–17. doi: 10.1016/j.jaad.2016.11.041
4. Reich K, Armstrong AW, Foley P, Song M, Wasfi Y, Randazzo B, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. (2017) 76:418–31. doi: 10.1016/j.jaad.2016.11.042
5. Deodhar A, Helliwell PS, Boehncke WH, Kollmeier AP, Hsia EC, Subramanian RA, et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. (2020) 395:1115–25. doi: 10.1016/s0140-6736(20)30265-8
6. Mease PJ, Rahman P, Gottlieb AB, Kollmeier AP, Hsia EC, Xu XL, et al. Guselkumab in biologic-naive patients with active psoriatic arthritis (DISCOVER-2): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. (2020) 395:1126–36. doi: 10.1016/s0140-6736(20)30263-4
7. D'Haens G, Panaccione R, Baert F, Bossuyt P, Colombel JF, Danese S, et al. Risankizumab as induction therapy for Crohn's disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet. (2022) 399:2015–30. doi: 10.1016/S0140-6736(22)00467-6
8. Ferrante M, Panaccione R, Baert F, Bossuyt P, Colombel JF, Danese S, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn's disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet. (2022) 399:2031–46. doi: 10.1016/S0140-6736(22)00466-4
9. Mehta H, Mashiko S, Angsana J, Rubio M, Hsieh YM, Maari C, et al. Differential changes in inflammatory mononuclear phagocyte and T-cell profiles within psoriatic skin during treatment with guselkumab vs. secukinumab. J Invest Dermatol. (2021) 141:1707–18.e9. doi: 10.1016/j.jid.2021.01.005
10. Chapuy L, Bsat M, Sarkizova S, Rubio M, Therrien A, Wassef E, et al. Two distinct colonic CD14+ subsets characterized by single-cell RNA profiling in Crohn's disease. Mucosal Immunol. (2019) 12:703–19. doi: 10.1038/s41385-018-0126-0
11. Kim J, Lee J, Kim HJ, Kameyama N, Nazarian R, Der E, et al. Single-cell transcriptomics applied to emigrating cells from psoriasis elucidate pathogenic versus regulatory immune cell subsets. J Allergy Clin Immunol. (2021) 148:1281–92. doi: 10.1016/j.jaci.2021.04.021
12. Kvedaraite E, Lourda M, Idestrom M, Chen P, Olsson-Akefeldt S, Forkel M, et al. Tissue-infiltrating neutrophils represent the main source of IL-23 in the colon of patients with IBD. Gut. (2016) 65:1632–41. doi: 10.1136/gutjnl-2014-309014
13. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. (2009) 10:314–24. doi: 10.1038/ni.1698
14. Constantinides MG, Link VM, Tamoutounour S, Wong AC, Perez-Chaparro PJ, Han SJ, et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science. (2019) 366:eaax6624. doi: 10.1126/science.aax6624
15. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. (2010) 10:479–89. doi: 10.1038/nri2800
16. Pawlak M, DeTomaso D, Schnell A, Meyer Zu Horste G, Lee Y, Nyman J, et al. Induction of a colitogenic phenotype in Th1-like cells depends on interleukin-23 receptor signaling. Immunity. (2022) 55:1663–79 e6. doi: 10.1016/j.immuni.2022.08.007
17. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. (2007) 445:648–51. doi: 10.1038/nature05505
18. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature. (2010) 467:967–71. doi: 10.1038/nature09447
19. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
20. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. (2012) 18:1069–76. doi: 10.1038/nm.2817
21. Geremia A, Arancibia-Cárcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. (2011) 208:1127–33. doi: 10.1084/jem.20101712
22. Visvanathan S, Baum P, Salas A, Vinisko R, Schmid R, Grebe KM, et al. Selective IL-23 inhibition by risankizumab modulates the molecular profile in the colon and ileum of patients with active Crohn's disease: results from a randomised phase II biopsy sub-study. J Crohn's colitis. (2018) 12:1170–9. doi: 10.1093/ecco-jcc/jjy099
23. Sofen H, Smith S, Matheson RT, Leonardi CL, Calderon C, Brodmerkel C, et al. Guselkumab (an IL-23-specific mAb) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J Allergy Clin Immunol. (2014) 133:1032–40. doi: 10.1016/j.jaci.2014.01.025
24. McInnes IB, Rahman P, Gottlieb AB, Hsia EC, Kollmeier AP, Xu XL, et al. Long-term efficacy and safety of guselkumab, a monoclonal antibody specific to the p19 subunit of interleukin-23, through two years: results from a phase III, randomized, double-blind, placebo-controlled study conducted in biologic-naive patients with active psoriatic arthritis. Arthritis Rheumatol. (2022) 74:475–85. doi: 10.1002/art.42010
25. Reich K, Gordon KB, Strober BE, Armstrong AW, Miller M, Shen YK, et al. Five-year maintenance of clinical response and health-related quality of life improvements in patients with moderate-to-severe psoriasis treated with guselkumab: results from VOYAGE 1 and VOYAGE 2. Br J Dermatol. (2021) 185:1146–59. doi: 10.1111/bjd.20568
26. Papp KA, Lebwohl MG, Puig L, Ohtsuki M, Beissert S, Zeng J, et al. Long-term efficacy and safety of risankizumab for the treatment of moderate-to-severe plaque psoriasis: interim analysis of the LIMMitless open-label extension trial beyond 3 years of follow-up. Br J Dermatol. (2021) 185:1135–45. doi: 10.1111/bjd.20595
27. Mease PJ, Kellner H, Morita A, Kivitz AJ, Aslanyan S, Padula SJ, et al. Long-term efficacy and safety of risankizumab in patients with active psoriatic arthritis: results from a 76-week phase 2 randomized trial. Rheumatol Ther. (2022) 9:1361–75. doi: 10.1007/s40744-022-00474-5
28. Ritchlin CT, Helliwell PS, Boehncke WH, Soriano ER, Hsia EC, Kollmeier AP, et al. Guselkumab, an inhibitor of the IL-23p19 subunit, provides sustained improvement in signs and symptoms of active psoriatic arthritis: 1 year results of a phase III randomised study of patients who were biologic-naïve or TNFα inhibitor-experienced. RMD Open. (2021) 7:e001457. doi: 10.1136/rmdopen-2020-001457
29. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. (1986) 136:2348–57. doi: 10.4049/jimmunol.136.7.2348
30. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. (2000) 13:715–25. doi: 10.1016/S1074-7613(00)00070-4
31. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. (2003) 421:744–8. doi: 10.1038/nature01355
32. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
33. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
34. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
35. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. (2006) 441:231–4. doi: 10.1038/nature04754
36. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. (2006) 314:1461–3. doi: 10.1126/science.1135245
37. Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. (2007) 80:273–90. doi: 10.1086/511051
38. Edouard L, Panaccione R, Parkes G, Peyrin-Biroulet L, Ferrante M, Takeuchi K, et al. Risankizumab induction therapy in patients with moderately to severely active ulcerative colitis: efficacy and safety in the randomized phase 3 INSPIRE study. Am J Gastroenterol. (2023) 118:S624–S5. doi: 10.14309/01.ajg.0000953020.05891.05
39. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. (2015) 21:719–29. doi: 10.1038/nm.3895
40. Bloch Y, Bouchareychas L, Merceron R, Składanowska K, Van den Bossche L, Detry S, et al. Structural activation of pro-inflammatory human cytokine IL-23 by cognate IL-23 receptor enables recruitment of the shared receptor IL-12Rβ1. Immunity. (2018) 48:45–58.e6. doi: 10.1016/j.immuni.2017.12.008
41. Jefremow A, Neurath MF. All are equal, some are more equal: targeting IL 12 and 23 in IBD - a clinical perspective. Immunotargets Ther. (2020) 9:289–97. doi: 10.2147/ITT.S282466
42. Sherlock JP, Cua DJ. Interleukin-23 in perspective. Rheumatol (Oxford). (2021) 60:iv1–3. doi: 10.1093/rheumatology/keab461
43. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, et al. Interleukin-23-induced transcription factor blimp-1 promotes pathogenicity of T helper 17 cells. Immunity. (2016) 44:131–42. doi: 10.1016/j.immuni.2015.11.009
44. Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity. (2009) 31:799–810. doi: 10.1016/j.immuni.2009.08.025
45. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. (2003) 198:1951–7. doi: 10.1084/jem.20030896
46. Kulig P, Musiol S, Freiberger SN, Schreiner B, Gyulveszi G, Russo G, et al. IL-12 protects from psoriasiform skin inflammation. Nat Commun. (2016) 7:13466. doi: 10.1038/ncomms13466
47. Eftychi C, Schwarzer R, Vlantis K, Wachsmuth L, Basic M, Wagle P, et al. Temporally distinct functions of the cytokines IL-12 and IL-23 drive chronic colon inflammation in response to intestinal barrier impairment. Immunity. (2019) 51:367–80.e4. doi: 10.1016/j.immuni.2019.06.008
48. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. (2015) 43:727–38. doi: 10.1016/j.immuni.2015.09.003
49. Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MBM. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. (2006) 176:1908–15. doi: 10.4049/jimmunol.176.3.1908
50. Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. (2006) 203:2577–87. doi: 10.1084/jem.20060244
51. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX₃CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. (2014) 211:1571–83. doi: 10.1084/jem.20140678
52. Culemann S, Grüneboom A, Nicolás-Ávila J, Weidner D, Lämmle KF, Rothe T, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature. (2019) 572:670–5. doi: 10.1038/s41586-019-1471-1
53. Bauche D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3+ regulatory T cells restrain interleukin-23-producing CX3CR1+ gut-resident macrophages during Group 3 innate lymphoid cell-driven colitis. Immunity. (2018) 49:342–52 e5. doi: 10.1016/j.immuni.2018.07.007
54. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. (2012) 13:991–9. doi: 10.1038/ni.2416
55. Schnell A, Huang L, Singer M, Singaraju A, Barilla RM, Regan BML, et al. Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell. (2021) 184:6281–98 e23. doi: 10.1016/j.cell.2021.11.018
56. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discovery. (2014) 13:21–38. doi: 10.1038/nrd4176
57. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. (2023) 23:38–54. doi: 10.1038/s41577-022-00746-9
58. Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suárez-Fariñas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. (2008) 159:1092–102. doi: 10.1111/j.1365-2133.2008.08769.x
60. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. (2020) 323:1945–60. doi: 10.1001/jama.2020.4006
61. Albanesi C, Madonna S, Gisondi P, Girolomoni G. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front Immunol. (2018) 9:1549. doi: 10.3389/fimmu.2018.01549
62. Ogawa K, Okada Y. The current landscape of psoriasis genetics in 2020. J Dermatol Sci. (2020) 99:2–8. doi: 10.1016/j.jdermsci.2020.05.008
63. Puig L, Costanzo A, Munoz-Elias EJ, Jazra M, Wegner S, Paul CF, et al. The biological basis of disease recurrence in psoriasis: a historical perspective and current models. Br J Dermatol. (2022) 186:773–81. doi: 10.1111/bjd.20963
64. Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaçi D, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. (2017) 390:276–88. doi: 10.1016/s0140-6736(17)31279-5
65. Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. (2018) 392:650–61. doi: 10.1016/s0140-6736(18)31713-6
66. Griffiths CEM, Papp KA, Song M, Miller M, You Y, Shen YK, et al. Continuous treatment with guselkumab maintains clinical responses through 4 years in patients with moderate-to-severe psoriasis: results from VOYAGE 1. J Dermatolog Treat. (2022) 33:848–856. doi: 10.1080/09546634.2020.1782817
67. Thaçi D, Piaserico S, Warren RB, Gupta AK, Cantrell W, Draelos Z, et al. Five-year efficacy and safety of tildrakizumab in patients with moderate-to-severe psoriasis who respond at week 28: pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2). Br J Dermatol. (2021) 185:323–34. doi: 10.1111/bjd.19866
68. Warren RB, Blauvelt A, Poulin Y, Beeck S, Kelly M, Wu T, et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): results from a phase III, randomized, open-label, efficacy-assessor-blinded clinical trial. Br J Dermatol. (2021) 184:50–9. doi: 10.1111/bjd.19341
69. Blauvelt A, Leonardi CL, Gooderham M, Papp KA, Philipp S, Wu JJ, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. (2020) 156:649–58. doi: 10.1001/jamadermatol.2020.0723
70. Gordon KB, Armstrong AW, Foley P, Song M, Shen YK, Li S, et al. Guselkumab efficacy after withdrawal is associated with suppression of serum IL-23-regulated IL-17 and IL-22 in psoriasis: VOYAGE 2 study. J Invest Dermatol. (2019) 139:2437–46.e1. doi: 10.1016/j.jid.2019.05.016
71. Reich K, Armstrong AW, Langley RG, Flavin S, Randazzo B, Li S, et al. Guselkumab versus secukinumab for the treatment of moderate-to-severe psoriasis (ECLIPSE): results from a phase 3, randomised controlled trial. Lancet. (2019) 394:831–9. doi: 10.1016/S0140-6736(19)31773-8
72. Reich K, Gooderham M, Thaçi D, Crowley JJ, Ryan C, Krueger JG, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. (2019) 394:576–86. doi: 10.1016/s0140-6736(19)30952-3
73. Langley RG, Papp K, Gooderham M, Zhang L, Mallinckrodt C, Agada N, et al. Efficacy and safety of continuous every 2-week dosing of ixekizumab over 52 weeks in patients with moderate-to-severe plaque psoriasis in a randomized phase 3 trial (IXORA-P). Br J Dermatol. (2018) 178:1315–23. doi: 10.1111/bjd.16426
74. Harden JL, Johnson-Huang LM, Chamian MF, Lee E, Pearce T, Leonardi CL, et al. Humanized anti-IFN-γ (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol. (2015) 135:553–6. doi: 10.1016/j.jaci.2014.05.046
75. Blauvelt A, Papp K, Gottlieb A, Jarell A, Reich K, Maari C, et al. A head-to-head comparison of ixekizumab vs. guselkumab in patients with moderate-to-severe plaque psoriasis: 12-week efficacy, safety and speed of response from a randomized, double-blinded trial. Br J Dermatol. (2020) 182:1348–58. doi: 10.1111/bjd.18851
76. McGonagle D, Watad A, Sharif K, Bridgewood C. Why inhibition of IL-23 lacked efficacy in ankylosing spondylitis. Front Immunol. (2021) 12:614255. doi: 10.3389/fimmu.2021.614255
77. Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. (2017) 376:957–70. doi: 10.1056/NEJMra1505557
78. Łukasik Z, Gracey E, Venken K, Ritchlin C, Elewaut D. Crossing the boundaries: IL-23 and its role in linking inflammation of the skin, gut and joints. Rheumatol (Oxford). (2021) 60:iv16–27. doi: 10.1093/rheumatology/keab385
79. McGonagle D, Aydin SZ, Gül A, Mahr A, Direskeneli H. 'MHC-I-opathy'-unified concept for spondyloarthritis and Behçet disease. Nat Rev Rheumatol. (2015) 11:731–40. doi: 10.1038/nrrheum.2015.147
80. DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. (2009) 60:2633–43. doi: 10.1002/art.24763
81. Raychaudhuri SP, Raychaudhuri SK. IL-23/IL-17 axis in spondyloarthritis-bench to bedside. Clin Rheumatol. (2016) 35:1437–41. doi: 10.1007/s10067-016-3263-4
82. Suzuki E, Mellins ED, Gershwin ME, Nestle FO, Adamopoulos IE. The IL-23/IL-17 axis in psoriatic arthritis. Autoimmun Rev. (2014) 13:496–502. doi: 10.1016/j.autrev.2014.01.050
83. Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, et al. Brief report: Group 3 innate lymphoid cells in human enthesis. Arthritis Rheumatol. (2017) 69:1816–22. doi: 10.1002/art.40150
84. Kristensen LE, Keiserman M, Papp K, McCasland L, White D, Lu W, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 1 trial. Ann Rheum Dis. (2022) 81:225–31. doi: 10.1136/annrheumdis-2021-221019
85. Östör A, Van den Bosch F, Papp K, Asnal C, Blanco R, Aelion J, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 2 trial. Ann Rheum Dis. (2022) 81:351–8. doi: 10.1136/annrheumdis-2021-221048
86. Coates LC, Gossec L, Theander E, Bergmans P, Neuhold M, Karyekar CS, et al. Efficacy and safety of guselkumab in patients with active psoriatic arthritis who are inadequate responders to tumour necrosis factor inhibitors: results through one year of a phase IIIb, randomised, controlled study (COSMOS). Ann Rheum Dis. (2022) 81:359–69. doi: 10.1136/annrheumdis-2021-220991
87. Siebert S, Sweet K, Ritchlin CT, Hsia EC, Kollmeier A, Xu XL, et al. Guselkumab treatment modulates core psoriatic arthritis gene expression in two phase 3 clinical trials. Ann Rheum Dis. (2021) 80:312. doi: 10.1136/annrheumdis-2021-eular.479
88. Schett G, Loza MJ, Palanichamy A, FitzGerald O, Ritchlin C, Barbaud F, et al. Collagen turnover markers are associated with active psoriatic arthritis and decrease with guselkumab treatment in a phase-3 clinical trial [abstract]. Arthritis Rheumatol. (2020) 72. doi: 10.1002/art.41538
89. Mease PJ, Chohan S, Fructuoso FJG, Luggen ME, Rahman P, Raychaudhuri SP, et al. Efficacy and safety of tildrakizumab in patients with active psoriatic arthritis: results of a randomised, double-blind, placebo-controlled, multiple-dose, 52-week phase IIb study. Ann Rheum Dis. (2021) 80:1147–57. doi: 10.1136/annrheumdis-2020-219014
90. Eyerich K, Eyerich S. Immune response patterns in non-communicable inflammatory skin diseases. J Eur Acad Dermatol Venereol. (2018) 32:692–703. doi: 10.1111/jdv.14673
91. Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. (2017) 376:1551–60. doi: 10.1056/NEJMoa1607017
92. Mease PJ, McInnes IB, Tam L-S, Eaton K, Peterson S, Schubert A, et al. Comparative effectiveness of guselkumab in psoriatic arthritis: results from systematic literature review and network meta-analysis. Rheumatology. (2021) 60:2109–21. doi: 10.1093/rheumatology/keab119
93. Baeten D, Østergaard M, Wei JC, Sieper J, Järvinen P, Tam LS, et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann Rheum Dis. (2018) 77:1295–302. doi: 10.1136/annrheumdis-2018-213328
94. Deodhar A, Gensler LS, Sieper J, Clark M, Calderon C, Wang Y, et al. Three multicenter, randomized, double-blind, placebo-controlled studies evaluating the efficacy and safety of ustekinumab in axial spondyloarthritis. Arthritis Rheumatol. (2019) 71:258–70. doi: 10.1002/art.40728
95. Baeten D, Adamopoulos IE. IL-23 inhibition in ankylosing spondylitis: where did it go wrong? Front Immunol. (2020) 11:623874. doi: 10.3389/fimmu.2020.623874
96. Mease PJ, Helliwell P, Gladman D, Poddubnyy D, Baraliakos X, Chakravarty SD, et al. Efficacy of guselkumab on axial involvement in patients with active psoriatic arthritis and sacroiliitis: a post-hoc analysis of the phase 3 DISCOVER-1 and DISCOVER-2 studies. Lancet Rheumatol. (2021) 3:E715–E23. doi: 10.1016/S2665-9913(21)00105-3
97. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med. (2020) 383:2652–64. doi: 10.1056/NEJMra2002697
98. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. (2019) 20:970–9. doi: 10.1038/s41590-019-0415-0
99. Sands BE. From symptom to diagnosis: clinical distinctions among various forms of intestinal inflammation. Gastroenterology. (2004) 126:1518–32. doi: 10.1053/j.gastro.2004.02.072
100. Peng L-L, Wang Y, Zhu F-L, Xu W-D, Ji X-L, Ni J. IL-23R mutation is associated with ulcerative colitis: a systemic review and meta-analysis. Oncotarget. (2017) 8:4849–63. doi: 10.18632/oncotarget.13607
101. Sandborn WJ, Ferrante M, Bhandari BR, Berliba E, Hibi T, D'Haens GR, et al. Efficacy and safety of continued treatment with mirikizumab in a phase 2 trial of patients with ulcerative colitis. Clin Gastroenterol Hepatol. (2022) 20:105–15.e14. doi: 10.1016/j.cgh.2020.09.028
102. Sandborn WJ, D'Haens GR, Reinisch W, Panes J, Chan D, Gonzalez S, et al. Guselkumab for the treatment of Crohn's disease: induction results from the phase 2 GALAXI-1 study. Gastroenterology. (2022) 162:1650–64 e8. doi: 10.1053/j.gastro.2022.01.047
103. Ferrante M, Feagan BG, Panés J, Baert F, Louis E, Dewit O, et al. Long-term safety and efficacy of risankizumab treatment in patients with Crohn's disease: results from the phase 2 open-label extension study. J Crohn's colitis. (2021) 15:2001–10. doi: 10.1093/ecco-jcc/jjab093
104. Danese S, Panaccione R, Feagan BG, Afzali A, Rubin DT, Sands BE, et al. Efficacy and safety of 48 weeks of guselkumab for patients with Crohn's disease: maintenance results from the phase 2, randomised, double-blind GALAXI-1 trial. Lancet Gastroenterol Hepatol. (2023) 9:133–46. doi: 10.1016/S2468-1253(23)00318-7
105. Peyrin-Biroulet L, Allegretti JR, Rubin DT, Bressler B, Germinaro M, Huang KG, et al. Guselkumab in patients with moderately to severely active ulcerative colitis: QUASAR phase 2b induction study. Gastroenterology. (2023) 165:1443–57. doi: 10.1053/j.gastro.2023.08.038
106. Singh S, Murad MH, Fumery M, Sedano R, Jairath V, Panaccione R, et al. Comparative efficacy and safety of biologic therapies for moderate-to-severe Crohn's disease: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. (2021) 6:1002–14. doi: 10.1016/S2468-1253(21)00312-5
107. Terui T, Kobayashi S, Okubo Y, Murakami M, Zheng R, Morishima H, et al. Efficacy and safety of guselkumab in Japanese patients with palmoplantar pustulosis: a phase 3 randomized clinical trial. JAMA Dermatol. (2019) 155:1153–61. doi: 10.1001/jamadermatol.2019.1394
108. Smolen JS, Agarwal SK, Ilivanova E, Xu XL, Miao Y, Zhuang Y, et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann Rheum Dis. (2017) 76:831–9. doi: 10.1136/annrheumdis-2016-209831
109. Najm A, McInnes IB. IL-23 orchestrating immune cell activation in arthritis. Rheumatology. (2021) 60:iv4–iv15. doi: 10.1093/rheumatology/keab266
110. Vollmer TL, Wynn DR, Alam MS, Valdes J. A phase 2, 24-week, randomized, placebo-controlled, double-blind study examining the efficacy and safety of an anti-interleukin-12 and -23 monoclonal antibody in patients with relapsing-remitting or secondary progressive multiple sclerosis. Mult Scler. (2011) 17:181–91. doi: 10.1177/1352458510384496
111. Pfeifle R, Rothe T, Ipseiz N, Scherer HU, Culemann S, Harre U, et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat Immunol. (2017) 18:104–13. doi: 10.1038/ni.3579
112. Longbrake EE, Racke MK. Why did IL-12/IL-23 antibody therapy fail in multiple sclerosis? Expert Rev Neurother. (2009) 9:319–21. doi: 10.1586/14737175.9.3.319
113. Marwaha AK, Chow S, Pesenacker AM, Cook L, Sun A, Long SA, et al. A phase 1b open-label dose-finding study of ustekinumab in young adults with type 1 diabetes. Immunother Adv. (2021) 2:ltab022. doi: 10.1093/immadv/ltab022
114. Gregory JW, Carter K, Cheung WY, Holland G, Bowen-Morris J, Luzio S, et al. Phase II multicentre, double-blind, randomised trial of ustekinumab in adolescents with new-onset type 1 diabetes (USTEK1D): trial protocol. BMJ Open. (2021) 11:e049595. doi: 10.1136/bmjopen-2021-049595
115. Mastracci TL, Turatsinze J-V, Book BK, Restrepo IA, Pugia MJ, Wiebke EA, et al. Distinct gene expression pathways in islets from individuals with short- and long-duration type 1 diabetes. Diabetes Obes Metab. (2018) 20:1859–67. doi: 10.1111/dom.13298
116. Fujihira K, Nagata M, Moriyama H, Yasuda H, Arisawa K, Nakayama M, et al. Suppression and acceleration of autoimmune diabetes by neutralization of endogenous interleukin-12 in NOD mice. Diabetes. (2000) 49:1998–2006. doi: 10.2337/diabetes.49.12.1998
117. Conway R, O'Neill L, Gallagher P, McCarthy GM, Murphy CC, Veale DJ, et al. Ustekinumab for refractory giant cell arteritis: a prospective 52-week trial. Semin Arthritis Rheum. (2018) 48:523–8. doi: 10.1016/j.semarthrit.2018.04.004
118. Cutler B, Strunck J, Greiling T. Pityriasis rubra pilaris treated with guselkumab: interim analysis of a single-arm trial. J Invest Dermatol. (2021) 141:S121. doi: 10.1016/j.jid.2021.02.725
119. Alivernini S, Firestein GS, McInnes IB. The pathogenesis of rheumatoid arthritis. Immunity. (2022) 55:2255–70. doi: 10.1016/j.immuni.2022.11.009
120. Pulgar VM. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front Neurosci. (2019) 12:1019. doi: 10.3389/fnins.2018.01019
121. Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. (2008) 7:796–804. doi: 10.1016/S1474-4422(08)70173-X
122. Havrdova E, Belova A, Goloborodko A, Tisserant A, Wright A, Wallstroem E, et al. Activity of secukinumab, an anti-IL-17A antibody, on brain lesions in RRMS: results from a randomized, proof-of-concept study. J Neurol. (2016) 263:1287–95. doi: 10.1007/s00415-016-8128-x
123. Matza MA, Fernandes AD, Stone JH, Unizony SH. Ustekinumab for the treatment of giant cell arteritis. Arthritis Care Res (Hoboken). (2021) 73:893–7. doi: 10.1002/acr.24200
124. Zafiriou E, Daponte AI, Siokas V, Tsigalou C, Dardiotis E, Bogdanos DP. Depression and obesity in patients with psoriasis and psoriatic arthritis: is IL-17-mediated immune dysregulation the connecting link? Front Immunol. (2021) 12:699848. doi: 10.3389/fimmu.2021.699848
125. Abbas A, Gregersen I, Holm S, Daissormont I, Bjerkeli V, Krohg-Sørensen K, et al. Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke. (2015) 46:793–9. doi: 10.1161/strokeaha.114.006516
126. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat Med. (2009) 15:946–50. doi: 10.1038/nm.1999
127. Nitsch L, Schneider L, Zimmermann J, Müller M. Microglia-derived interleukin 23: a crucial cytokine in Alzheimer's disease? Front Neurol. (2021) 12:639353. doi: 10.3389/fneur.2021.639353
128. Alvarez-Salamero C, Castillo-Gonzalez R, Pastor-Fernandez G, Mariblanca IR, Pino J, Cibrian D, et al. IL-23 signaling regulation of pro-inflammatory T-cell migration uncovered by phosphoproteomics. PloS Biol. (2020) 18::e3000646. doi: 10.1371/journal.pbio.3000646
129. Park YJ, Kim YH, Lee ES, Kim YC. Comparative analysis of single-cell transcriptome data reveals a novel role of keratinocyte-derived IL-23 in psoriasis. Front Immunol. (2022) 13:905239. doi: 10.3389/fimmu.2022.905239
130. Kim J, Lee J, Li X, Kunjravia N, Rambhia D, Cueto I, et al. Multi-omics segregate different transcriptomic impacts of anti-IL-17A blockade on type 17 T-cells and regulatory immune cells in psoriasis skin. Front Immunol. (2023) 14:1250504. doi: 10.3389/fimmu.2023.1250504
131. Morelli M, Galluzzo M, Scarponi C, Madonna S, Scaglione GL, Girolomoni G, et al. Allelic variants of HLA-C upstream region, PSORS1C3, MICA, TNFA and genes involved in epidermal homeostasis and barrier function influence the clinical response to anti-IL-12/IL-23 treatment of patients with psoriasis. Vaccines (Basel). (2022) 10:1977. doi: 10.3390/vaccines10111977
132. Huang YH, Chang LC, Chang YC, Chung WH, Yang SF, Su SC. Compositional alteration of gut microbiota in psoriasis treated with IL-23 and IL-17 inhibitors. Int J Mol Sci. (2023) 24:4568. doi: 10.3390/ijms24054568
133. Liang D, Zuo A, Shao H, Born WK, O'Brien RL, Kaplan HJ, et al. IL-23 receptor expression on γδ T cells correlates with their enhancing or suppressive effects on autoreactive T cells in experimental autoimmune uveitis. J Immunol. (2013) 191:1118–25. doi: 10.4049/jimmunol.1300626
134. Tollenaere MAX, Hebsgaard J, Ewald DA, Lovato P, Garcet S, Li X, et al. Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways. Br J Dermatol. (2021) 185:585–94. doi: 10.1111/bjd.20090
135. Pastor-Fernandez G, Mariblanca IR, Navarro MN. Decoding IL-23 signaling cascade for new therapeutic opportunities. Cells. (2020) 9:2044. doi: 10.3390/cells9092044
136. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. (2018) 16:143–55. doi: 10.1038/nrmicro.2017.157
137. Eyerich K, Weisenseel P, Pinter A, Schäkel K, Asadullah K, Wegner S, et al. IL-23 blockade with guselkumab potentially modifies psoriasis pathogenesis: rationale and study protocol of a phase 3b, randomised, double-blind, multicentre study in participants with moderate-to-severe plaque-type psoriasis (GUIDE). BMJ Open. (2021) 11:e049822. doi: 10.1136/bmjopen-2021-049822
138. Zhou L, Wang Y, Wan Q, Wu F, Barbon J, Dunstan R, et al. A non-clinical comparative study of IL-23 antibodies in psoriasis. MAbs. (2021) 13:1964420. doi: 10.1080/19420862.2021.1964420
139. Bridgewood C, Watad A, Russell T, Palmer TM, Marzo-Ortega H, Khan A, et al. Identification of myeloid cells in the human enthesis as the main source of local IL-23 production. Ann Rheum Dis. (2019) 78:929–33. doi: 10.1136/annrheumdis-2018-214944
140. Quiniou C, Domínguez-Punaro M, Cloutier F, Erfani A, Ennaciri J, Sivanesan D, et al. Specific targeting of the IL-23 receptor, using a novel small peptide noncompetitive antagonist, decreases the inflammatory response. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R1216–R30. doi: 10.1152/ajpregu.00540.2013
141. Cheng X, Lee T-Y, Ledet G, Zemade G, Tovera J, Campbell R, et al. Safety, tolerability, and pharmacokinetics of PTG-200, an oral GI-restricted peptide antagonist of IL-23 receptor, in normal healthy volunteers. Am J Gastroenterol. (2019) 114:S439–S40. doi: 10.14309/01.ajg.0000592540.43886.35
142. Fourie A, Cheng X, Chang L, Greving C, Patrick A, Knight B, et al. First-in-class oral peptide systemically targeting the IL-23 pathway. J Invest Dermatol. (2023) 143:S190. doi: 10.1016/j.jid.2023.03.1121
143. Perrigoue J, Muniz-Bongers L, Ong L, Chen Y, Chang L, Ngo K, et al. In silico evaluation and pre-clinical efficacy of anti-TNF and anti-IL-23 combination therapy in Inflammatory Bowel Disease. J Crohns Colitis. (2022) 16. doi: 10.1093/ecco-jcc/jjab232.455
144. Sands BE, Feagan BG, Sandborn WJ, Shipitofsky N, Marko M, Sheng S, et al. Efficacy and safety of combination induction therapy with guselkumab and golimumab in participants with moderately-to-severely active Ulcerative Colitis: Results through week 12 of a phase 2a randomized, double-blind, active-controlled, parallel-group, multicenter, proof-of-concept study. J Crohns Colitis (2022) 16:i042–i043. doi: 10.1093/ecco-jcc/jjab232.035
145. Desai P, Branigan P, Richards D, Mcgonagle D, Vetter M, Cua D, et al. Differential and combinatorial mechanism of action of Golimumab and Guselkumab in ulcerative colitis induction therapy: IL-23 blackade drives restoration of normal epithelium and mucosal healing. EULAR. (2023) 82:1147. doi: 10.1136/annrheumdis-2023-eular.2982
Keywords: IL-23, cytokine, immune-mediated inflammatory diseases, psoriasis, psoriatic arthritis, inflammatory bowel disease
Citation: Krueger JG, Eyerich K, Kuchroo VK, Ritchlin CT, Abreu MT, Elloso MM, Fourie A, Fakharzadeh S, Sherlock JP, Yang Y-W, Cua DJ and McInnes IB (2024) IL-23 past, present, and future: a roadmap to advancing IL-23 science and therapy. Front. Immunol. 15:1331217. doi: 10.3389/fimmu.2024.1331217
Received: 31 October 2023; Accepted: 21 March 2024;
Published: 15 April 2024.
Edited by:
Soren Skov, University of Copenhagen, DenmarkReviewed by:
Daniella Schwartz, University of Pittsburgh, United StatesCopyright © 2024 Krueger, Eyerich, Kuchroo, Ritchlin, Abreu, Elloso, Fourie, Fakharzadeh, Sherlock, Yang, Cua and McInnes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel J. Cua, REN1YUBJVFMuSk5KLmNvbQ==; Iain B. McInnes, aWFpbi5tY2lubmVzQGdsYXNnb3cuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.