 Diyuan Qin
Diyuan Qin- 1Cancer Center, Clinical Trial Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China
- 2Cancer Center, National Medical Products Administration Key Laboratory for Clinical Research and Evaluation of Innovative Drugs, West China Hospital, Sichuan University, Chengdu, Sichuan, China
- 3Division of Thoracic Tumor Multimodality Treatment, Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China
Immunotherapies have revolutionized the landscape of cancer treatment. Regulatory T cells (Tregs), as crucial components of the tumor immune environment, has great therapeutic potential. However, nonspecific inhibition of Tregs in therapies may not lead to enhanced antitumor responses, but could also trigger autoimmune reactions in patients, resulting in intolerable treatment side effects. Hence, the precision targeting and inhibition of tumor-infiltrating Tregs is of paramount importance. In this overview, we summarize the characteristics and subpopulations of Tregs within tumor microenvironment and their inhibitory mechanisms in antitumor responses. Furthermore, we discuss the current major strategies targeting regulatory T cells, weighing their advantages and limitations, and summarize representative clinical trials targeting Tregs in cancer treatment. We believe that developing therapies that specifically target and suppress tumor-infiltrating Tregs holds great promise for advancing immune-based therapies.
1 Introduction
In recent years, novel therapies such as immunological checkpoint inhibitors (PD-1, CTLA-4), tumor dendritic cell (DC) vaccines, chimeric antigen receptor T (CAR-T) cells, TCR-T cells, and tumor-infiltrating lymphocytes (TILs) have achieved notable therapeutic success by amplifying the antitumor activities of effector cells (1–6). The tumor microenvironment (TME) is multifaceted, containing both antitumor immune cells and immune-inhibitory components like regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages, and tumor-associated fibroblasts (7–9). Therapies targeting immune-suppressive cells also hold significant potential in cancer treatment (10–13). This review will primarily delve into immunotherapies targeting intratumoral Tregs (Ti-Tregs) and their future prospects.
Treg cells are widely distributed throughout the body, not just in lymphoid organs, but also in the lungs, intestines, mucosal skin, and notably, within the tumor microenvironment (14). Tregs possess a bifunctional role in immunity. On one hand, they can maintain immunological homeostasis inside the body and avoid excessive immune activation. Deficiency or malfunction of Tregs may lead to autoimmune diseases (15). Treg cells, on the other hand, have immunosuppressive functions in cancer. Multiple evidence demonstrate that Tregs not only reduce the antitumor activity of effector T cells (Teff) in the TME, but also suppress DCs and macrophages (16–18). As a result, eliminating or inhibiting Ti-Treg cells in cancer patients may improve the effectiveness of immunotherapy against malignancies (10). Determining how to efficiently and selectively target or suppress tumor-infiltrating Tregs is an important research topic. This review focuses on the phenotypic and functional distinctions among Treg subgroups, as well as the mechanism by which Tregs suppress immunological responses. We also review and analysis the features of various Tregs- targeting immunotherapy strategies, as well as the possibilities for future treatments.
2 Characteristics and subpopulations of Tregs
Gershon and Kondo revealed half a century ago that T cells in mice not only operate as effectors, but also dampen the immune response (19). Mice approximately three days old undergoing thymectomy, leading to partial dysfunction of T cells, suffer a variety of autoimmune disorders (20). Building upon this, Sakaguchi and colleagues identified the CD25 molecule (IL2 receptor alpha chain) within this suppressive T cell population, thereby better characterizing such unique subset. Notably, the autoimmune disorders in thymectomized mice were reduced when they were reinfused with CD25-expressing CD4 T cells (21).
Following CD25 discovery, the identification and research of the transcription factor Foxp3, a crucial regulator in the formation and function of Tregs, marked another significant milestone in Treg research (22–25). Mutations in the Foxp3 gene, even just a two-base insertion, result in Scurfy mice. These mice, with impaired Treg production and function, display severe autoimmune reactions, including significant skin inflammation, enhanced T cell activity, and a marked expansion of T cells in both spleen and lymph nodes (22, 26). Similarly, Foxp3 gene mutation in humans could also lead to Tregs impairment and thus severe systemic autoimmune diseases such as IPEX syndrome (Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome), whose clinical manifestations were first reported in 1982 (27).
Hence, CD25 and Foxp3 double positivity served as identification markers for the Treg population. However, the functionality and surface markers among CD4+ regulatory T cells, identified by CD25+Foxp3+, exhibit highly heterogeneous. For instance, some conventional effector T cells also transiently express CD25 and Foxp3 after activation by TCR signaling, which can still secrete IL-2 and IFN-γ without immunosuppressive effects (28, 29).
The expression of CD127 is negatively correlated with the suppressive function of Treg and the expression of FOXP3, some research teams suggested using CD127 (the alpha subunit of the IL-7 receptor) in combination with CD25 and CD4 as markers for human Tregs (30, 31). However, naïve T cells can also transiently express low levels of foxp3 and CD127 after activation by TCR signals (32), Consequently, this approach might face challenges in effectively distinguishing Tregs from certain activated conventional T cells (Tconvs).
To further distinguish Tregs subpopulations, Sakaguchi et al. subdivided Treg cells into natural Treg cells (nTreg) and peripheral Treg cells (pTreg) based on their origin and suppressive activity (16). Among them, nTregs, which are also known as thymic Tregs (tTregs), primarily derive from the thymus, and the main function of nTreg is to maintain immunological homeostasis and autoimmune tolerance. As previously indicated, after thymectomy in mice, nTregs are reduced, and autoimmune disorders occur spontaneously. pTregs evolve from peripheral Tconvs under the influence of TCR signaling and TGF-β stimulation (33, 34). Due to their distinct origins, nTregs and pTregs exhibit significant differences in their TCR repertoires. nTregs, developing in the thymus under the influence of self-antigens, have a TCR repertoire skewed towards self-antigens (35, 36). In contrast, pTregs, derived from peripheral Tconv cells, exhibit a TCR repertoire more similar to Tconv cells, offering greater diversity. TCR repertoire analysis shows that both nTreg and pTreg contribute to the TCR repertoire of intratumoural Treg (37).
Furthermore, researchers have cocultured mouse CD4+ T cells together with retinoic acid or TGF-β in vitro and obtained Foxp3-positive Treg cells, namely, induced Tregs (iTregs), which is also called pTreg in some other studies (34, 38, 39).
To more accurately characterize the immunosuppressive status of regulatory T cells, researchers thus utilized CD45RA, CD25, and Foxp3 with different expression intensities to characterize the functional subpopulations of Tregs, namely, naïve or resting Tregs, effector Tregs (eTregs), and non-Tregs (10, 40). Naïve Tregs originate from the thymus, have not been activated by TCR, have a phenotype of CD45RA+CD25LOWFOXP3LOW, and exhibit minimal immunosuppressive ability. However, once these naïve Tregs migrate to the peripheral tissues and contact antigens in draining lymph nodes, they proliferate and differentiate into effector Tregs (eTregs). These eTregs are prevalent in most solid tumors (41), exerting a powerful immunosuppressive effect and characterized by a CD45RA-CD25highFOXP3high. It is worth noting that for most solid tumor types, higher intratumoral FOXP3 expression correlates negatively with patient prognosis (42–44). However, in certain unique tumor types, such as colorectal cancer as well as few head and neck tumors (45–47), heightened intratumoral FOXP3 expression indicates a more favorable prognosis. Subsequent studies into Tregs in these unique cases indicated that the predominant phenotype of FOXP3+ CD4+ T cells among such colorectal cancers was CD45RA-CD25LOWFOXP3LOW, labeled as non-Tregs. These Tregs lack immunosuppressive effects but are capable of secreting inflammatory cytokines such as IFN-γ and IL-17 (46, 48), whose presence is associated with better outcomes for tumor patients.
It is worth noting that the relationship between Tregs and the prognosis of hematological malignancies is intricate as well. Researchers have extensively investigated Treg cells across various hematological malignancies. Due to the Treg heterogeneity, limitations in Treg detection methods and variations in patient tissue specimens, the impact of Tregs on disease prognosis varies (49). While in classical Hodgkin lymphoma tissues, the expression of FOXP3 alone may not serve as an independent prognostic factor, a multivariate analysis model including FOXP3, PD-1, and granzyme B demonstrates certain predictive value for the prognosis of classical Hodgkin lymphoma patients, with higher levels of FOXP3 expression often correlating with longer overall survival (50). Additionally, in follicular lymphoma (FL) and germinal center (GC) diffuse large B-cell lymphomas (DLBCL), a higher Treg cell count is linked to a favorable prognosis. In pediatric acute lymphoblastic leukemia patients, there is a weak negative correlation between Treg cells in the bone marrow and the percentage of primitive cells in the peripheral blood, but in chronic lymphocytic leukemia, an increased number of Tregs is associated with poorer outcomes (49, 51).
3 Immunosuppression mechanism of Tregs
3.1 Molecules associated with Treg immunosuppression
Tumor-infiltrating eTregs are activated and possess robust immunosuppressive properties (52), with high expression of molecules like CD25, CTLA4, PD-1, ICOS, LAG-3, TIGIT, and members of the TNF receptor superfamily, like GITR, 4-1BB, and OX-40 (53). Moreover, they express chemokine receptors, such as CCR4, CCR5 and CCR10, which facilitate chemotactic migration of Tregs into the tumor microenvironment (54). CTLA-4 is an important inhibitory function-related molecule, which is constitutively and highly expressed in Tregs and belongs to the same family as CD28. Ti-Tregs utilize CTLA-4 molecules with a higher affinity than CD28 to engage with the B7-1/B7-2 (CD80/CD86) ligand molecules on antigen-presenting cells (APCs), thereby competitively reducing the CD28 stimulation signal on Tconvs (55). Moreover, by virtue of CTLA4 molecules, Tregs hinder the maturation process of APCs, reducing CD80/86 expression and elevating inhibitory signaling on APCs, thereby dampening their ability to activate conventional T cells (56, 57), leading them to an anergic state with hyporesponsive to antigen stimulation.
In tumor, PD-1 molecules inhibit TCR and CD28 signaling, which causes Tconv dysfunction or exhaustion (58). PD-1 is also highly expressed in Tregs and exhibit inhibit ability in Treg activity. Antagonistic therapy against PD-1 promotes the activation and expansion of Treg cells (59, 60). For instance, mice autoimmune pancreatitis was alleviated by specifically knocking out PD-1 in Treg cells, which increased Treg suppression capacity (61). It is reported that in patients receiving targeted immune checkpoint therapies, particularly anti-PD-1 treatments, a subset of patients’ tumors exhibit unexpected rapid progression, known as hyperprogressive disease (HPD) (62), whose mechanism has not been fully elucidated. Several studies have found that HPD during PD-1 monoclonal antibody treatment is correlated with marked activation and proliferation of Tregs within tumor, whereas patients without HPD exhibit diminished eTreg proliferation (60, 63, 64). The precise role of PD-1 in Tregs requires further investigation to be fully utilized in Treg-targeting cancer treatment.
CD25, also known as the alpha subunit of the IL-2 receptor, is highly expressed on Tregs. This subunit, in conjunction with the beta (CD122) and gamma (CD132) subunits, forms the high-affinity IL-2 receptor predominantly found on Tregs. The IL-2 receptor on Tconvs, on the other hand, lacks the alpha subunit and is made up of solely the beta and gamma subunits. The trimeric configuration of IL-2 receptor allows Tregs to bind the IL-2 cytokine with significantly higher affinity, particularly in the TME, compared to that on Tconvs. The distinction in IL-2 receptor composition and affinity plays a crucial role in the functional differentiation and immune response modulation between Tregs and Tconvs. Tregs compete with conventional T cells and result in a relative shortage of IL-2 for Tconvs, which is crucial for their activation and proliferation processes (65, 66).
Other molecules such as lymphocyte-activation gene 3 (LAG-3), T cell immunoglobulin and mucin domain-containing 3 (TIM-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), inducible costimulator (ICOS), glucocorticoid-induced tumor necrosis factor-related receptor (GITR) and CD27 are unregulated on Ti-Tregs and contributed to their immunosuppressive activity in tumor treatment (67). By binding to MHC-II molecules with high affinity, LAG-3 plays a role in regulating the activation and proliferation of T cells (68). High expression of TIM-3 on Treg cells biases them towards an effector Treg (eTreg) phenotype, endowing them with more potent immunosuppressive capabilities to inhibit the proliferation and survival of CD8 T cells (69). Elevated TIGIT expression in Treg results in the transcription factor CEBPα overexpression, which in turn boosts the secretion of the soluble effector molecule fibrinogen-like protein 2 (Fgl2), facilitating the Treg-mediated selective suppression of pro-inflammatory Th1 and Th17 cells (70). Tregs expressing high levels of ICOS demonstrate significantly increased proliferative activity (71). In ICOS knock-out mice, the proportion of Tregs is substantially reduced compared to their wild-type counterparts. Furthermore, ICOS signaling plays a crucial role in enhancing Foxp3 transcription, thereby supporting the survival and augmenting the suppressive functionality of Tregs (72). GITR also promotes the survival and proliferation of Treg cells (73). The presence of CD27 on Treg cells plays a vital role in preserving the immune balance of peripheral CD8 T cells. Blocking CD27 signaling in combination with anti-PD-1 therapy has been shown to increase the number of CD8+ T cells within tumors and improve the CD8+ T cell/Treg ratio, thereby enhancing the anti-tumor efficacy (74).
V-domain immunoglobulin suppressor of T cell activation (VISTA) is also an immune checkpoint expressed on multiple cells such as T cells, myeloid cells and tumor cells. Specifically, VISTA is highly expressed in naïve CD4+ T cells and Treg cells, and it can increase FOXP3 expression. Treg proliferation can be inhibited by VISTA blockage (75).
All of the above-mentioned markers can serve as potential targets in strategies aimed at harnessing Tregs for anti-tumor therapy.
3.2 Immunosuppressive cytokines
Treg cells can also suppress immune reactions in TME by releasing anti-inflammatory cytokines such as IL-10, TGF-β, and IL-35. These cytokines suppress the activity of Teffs and APCs (76, 77).
TGF-β not only diminishes the cytotoxicity of Teffs and NK cells but also promotes the differentiation of peripheral naïve T cells into induced Treg cells (iTregs) (78–80). Furthermore, TGF-β augments the immunosuppressive function of Treg cells by activating Foxp3 (81). Consequently, it is feasible to directly target the TGF-β cytokine using blocking antibodies, or to inhibit the TGF-β receptor through antibodies or small molecule drugs, thereby disrupting the TGF-β signaling cascade and its facilitative function in Treg-mediated immunosuppression (82, 83).
In the tumor microenvironment, IL-10 and IL-35, primarily secreted by different Tregs subsets, are instrumental in modulating immune responses. IL-10, known for its ability to suppress the activity of effector T cells and antigen-presenting cells, plays a significant role in diminishing anti-tumor immunity. IL-35, another key cytokine, works in conjunction with IL-10 to induce an exhausted state in tumor-infiltrating lymphocytes and impairs the formation of T cell memory. However, IL-35-producing Tregs are vital for maintaining tolerance in auto-reactive T cells. Therefore, targeting IL-35 to inhibit Treg activity in cancer therapy necessitates careful consideration of potential autoimmune reactions (84).
Beyond cytokine secretions, Tregs can directly eliminate other effector immune cells by releasing cytotoxic components such as perforin and granzyme B (85).
3.3 Chemokines and chemokine receptors
Chemokines are also pivotal in the immunosuppressive process of Tregs. Treg cells exhibit high levels of chemokine receptors. Through chemotaxis like CCL17/CCL22-CCR4 (86, 87), CCL28-CCR10, CCL5-CCR5 et al. (88, 89), Tregs are recruited to the tumor microenvironment. For instance, CCR4 binds with ligands like CCL17 and CCL22 secreted by tumor cells, facilitating Treg infiltration (90). In a Pan02 pancreatic cancer mouse model, characterized by the secretion of the chemokines CCL17 and CCL22, the use of a CCR4 antagonist (CCR4-351) has shown significant effects in disrupting the CCR4-CCL17/CCL22 chemotactic axis, which effectively blocks the recruitment of Tregs to the TME (91).
In addition to chemotactic function, the CCR8-CCL1/CCL18 signaling axis, although not directly involved in the chemotactic aggregation of Tregs (which can chemotactically attract Th2 cells) (92, 93), is crucial for sustaining Treg homeostasis and immunosuppressive functions. Research suggests that the interaction between CCR8 and CCL1 enable the upregulation of FOXP3, IL-10, CD39 and granzyme B, resulting in proliferation and activation of Treg cells (94). Notably, CCR8 is highly expressed in Ti-Tregs but is rarely expressed by Teffs or naïve Tregs. Around 30-80% of Ti-Tregs possess CCR8 (95). Targeting intratumoral CCR8-positive Treg cells with an anti-CCR8 mAb could potentially induce tumor regression and promote lasting antitumor memory (95, 96).
3.4 Angiogenesis
Angiogenesis also influences the immunosuppressive activity of Ti-Tregs. Hypoxia can amplify the expression of CCL28, attracting CCR10-positive Treg cells into the tumor (88), suppressing the Teffs function, and also elevating the levels of VEGFA in the TME (97). When binding to its receptor VEGFR2, VEGFA suppresses the functions of APCs and Teffs (98–100), while also promoting the expansion and infiltration of Tregs within the TME (101, 102). VEGFA can further induce the expression of neuropilin 1 (NRP1) in Tregs, which promotes the aggregation of Tregs around tumor blood vessels, thereby promoting Treg aggregation around tumor blood vessels (103).
3.5 Metabolism adaptation
Tregs, utilizing their elevated expression of CD39 and CD73, which are also named ectonucleoside triphosphate diphosphohydrolase 1 (NTPDase1) and 5’-nucleotidase separately, to metabolize ATP and ADP within the tumor microenvironment into AMP. Subsequently, the CD73 molecule further converts AMP to adenosine (104, 105). Upon binding to the corresponding A2A receptors on Teffs, DC cells or NK cells, adenosine inhibits their activity and proliferation (106, 107). Furthermore, adenosine facilitates the proliferation of MDSCs and promotes the formation of M2 macrophages (108, 109), establishing an immunosuppressive microenvironment that attenuates the antitumor response (110).
Tregs also regulate metabolism within the tumor microenvironment through upregulating indoleamine 2,3-dioxygenase (IDO). IDO plays a crucial role in the metabolism of intermediate product tryptophan among the kynurenine pathway (111). IDO degrades tryptophan in the TME, resulting in Tonvs dysfunction (112). The engagement of CTLA-4 with CD80/CD86 lead to an further elevated expression of IDO in DCs (113). The consequential tryptophan reduction and kynurenine accumulation within the TME further induces exhaustion and compromised functionality of effector T cells.
It is worth mentioning that Tregs exhibit different metabolism preference and a superior metabolic adaptability compared to conventional T cells to sustain functional activity in tumor microenvironments characterized by hypoxia, glucose deficiency, lactic acid accumulation and the presence of various immunosuppressive metabolic products (114). Within the tumor microenvironment, tumor cells possess enhanced glycolytic capacity, leading to greater glucose consumption and consequent glucose restriction for TILs (115). Reduced glucose availability hampers mTOR activity, glycolytic function, and IFN-γ production in TILs, rendering TILs functionally ineffective (116). Through intrinsic regulation, Tregs demonstrate heightened Glut1 expression, glucose uptake, glycolysis rates, and fatty acid synthesis compared to Tconvs, preferentially utilizing glucose and fatty acids as substrates for energy production and biosynthesis (117, 118). Moreover, Foxp3 in Tregs was able to inhibit the expression of the Myc gene, enhancing oxidative phosphorylation and elevating the NAD : NADH ratio, leading to increased energy production (119).
Regarding lactate metabolism, Foxp3 was able to limit LDH’s reductive reaction from pyruvate to lactate, decreasing the production of NADH and promoting the oxidation reaction of L-lactate to pyruvate (119). Under high lactate conditions, Treg cells upregulate the expression of genes related to lactate metabolism, such as lactate dehydrogenase (LDH) and monocarboxylic acid transporter protein 1 (MCT1, lactate transporter protein) (120, 121), and are able to metabolize lactate as an alternative source of energy for the generation of intermediates needed for the tricarboxylic acid cycle (TCA) and phosphoenolpyruvate (PEP). Treg cells also highly express the CD36 molecule, a transporter protein for free fatty acids, which transports fatty acids and lipoprotein into the mitochondria. This protein shuttles fatty acids and lipoproteins to the mitochondria, facilitating mitochondrial energy metabolism and biological synthesis. This process alone with the peroxisome proliferator-activated receptor-β (PPAR-β) signaling pathway, support the survival and function of Ti-Treg cells (122). Such metabolic adaptations allow Tregs to better survive in environments with low glucose and high lactic acid or fatty acid levels, and they can resist the inhibitory effects of lactate on Treg function and proliferation.
However, it’s important to note that the immunosuppressive activity of Tregs also plays a critical role in maintaining immune balance and tolerance in the body. There is growing research interest in harnessing the suppressive functions of Tregs to reduce immune rejection in organ transplantation and to treat allergic or autoimmune diseases that are often caused by excessive immune activation (123–125). Treg can also be adopted to minimize immune-related adverse events (irAEs) in cancer immunotherapy. Scientists have developed pH-sensitive CTLA-4 antibodies, which bind to CTLA-4 on Treg cell surfaces and undergo internalization. In the acidic lysosomal environment, their affinity to CTLA-4 decreases, leading to dissociation. This process allows CTLA-4 to be recycled back to the Treg cell membrane, preserving the immunosuppressive role of Tregs in peripheral. Simultaneously, this antibody reactive exhausted Tconv cells, enabling their effector function. This approach, by preserving the peripheral immunosuppressive function of Tregs, mitigates irAEs while maintaining anti-tumor efficacy (126, 127).
In summary, Tregs possess a variety of immunosuppressive mechanisms that can diminish the efficacy of anti-tumor therapies (Figure 1). Targeting these mechanisms opens up possibilities for developing strategies that could significantly improve anti-tumor treatments.
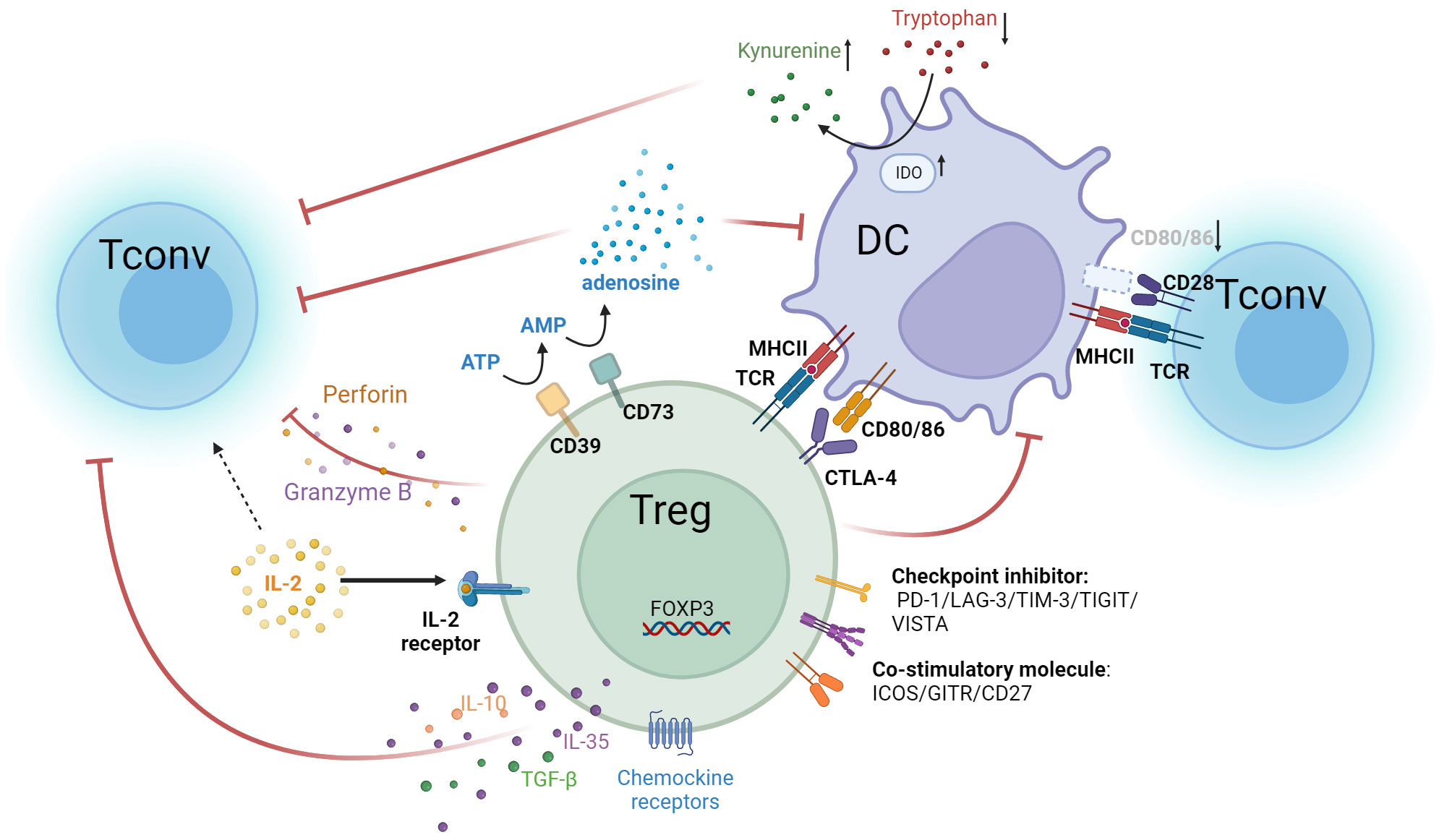
Figure 1 Multiple immunosuppressive mechanisms of Tregs. Treg cells express high-affinity IL-2 receptors, with CD25 as the α subunit of this receptor. In contrast, the IL-2 receptors on Tconv comprise only β and γ subunits and are of lower affinity. Tregs competitively bind IL-2 in the tumor microenvironment, causing a relative deficiency of IL-2 in Tconv and suppressing T cell effector functions. Tregs highly express CTLA-4 and bind to CD80/86 ligands on APCs. This not only inhibits APC function but also reduces the binding of CD80/86 expressed by APCs to CD28 molecules on T cells. As a result, the activation of Teff is attenuated. Additionally, suppression of DC cells by Tregs leads to overexpression of IDO, which decreases tryptophan and leads to kynurenine accumulation in the TME. This results in effector T cell exhaustion and dampens anti-tumor immunity. Tregs secrete immunosuppressive cytokines, such as IL-10, IL-35, and TGF-β, which inhibit the activity of T cells and APC cells. They also secrete perforin and granzyme B to directly lyse effector immune cells. Additionally, Tregs overexpress CD39 and CD73, converting ATP in the TME into adenosine, which inhibits the functions of various immune cells, such as DC and Tconv. Tregs also upregulate chemokines like CCL4, CCR5, CCR8, and CCR10, facilitating their accumulation in the tumor. In addition, Tregs overexpress several immune checkpoint molecules, including PD-1, LAG-3, TIM-3, TIGIT, and VISTA, which suppress anti-tumor effector cells. Treg also upregulate co-stimulatory molecules such as ICOS, GITR, and CD27, which enhance Treg activity and proliferation.
4 Targeting Ti-Tregs
4.1 Specific depletion or inhibition of Tregs
In peripheral blood, Treg cells account for 2-5% of all CD4+ T cells. While within tumor tissues, the Tregs proportion can reach up to 50%, and most of them are eTregs, which are crucial in establishing the immunosuppressive microenvironment of tumors (41). Specifically, eliminating or inhibition intratumoral eTreg cells can relieve the immunosuppression of the tumor microenvironment.
Tregs highly express molecules such as CTLA-4, CD25, PD-1, ICOS, LAG-3, TIGIT, GITR, 4-1BB, and OX40, which are ideal targets in immunotherapy (53). For example, the monoclonal antibody targeting the immune checkpoint CTLA-4, which has been used in the clinic, was primarily designed to block the immune checkpoint inhibitory signaling of effector T cells, thereby amplifying the Teff antitumor responses (128). Additionally, when antibody’s Fab region binds to the epitope on the surface of target cells, the Fc region recruits effector cells (such as NK cells and macrophages) and interacts with their Fc receptors (FcγR). This interaction mediates the direct killing of target cells by the effector cells, a process known as antibody-dependent cellular cytotoxicity (ADCC). Subsequent studies revealed that several CTLA-4 monoclonal antibodies can also eradicate Ti-Tregs via the ADCC effect, resulting in enhanced therapeutic outcomes (129–131). However, the efficacy of targeting CTLA-4 to deplete Tregs is still controversial. Some clinical studies have indicated that the number of Tregs within tumors does not decrease after CTLA-4 antibodies administration (132). It is also reported that CTLA-4 blocking antibody promoted the CD28-dependent expansion of Ti-Tregs (133). Additionally, as other immune cells within the microenvironment, such as activated Teffs and DCs (134), also express CTLA-4, nonselectively targeting CTLA-4 might inadvertently diminish antitumor immune cells. Thus, more detailed and comprehensive studies are still required to achieve favorable results with strategies targeting CTLA-4 to deplete Tregs.
Numerous studies have explored anti-tumor strategies targeting CD25 or combining IL-2 with cytotoxic molecules or CD25-ADC (antibody-drug conjugate) to deplete Treg cells. However, these approaches show variable effectiveness across different contexts (135–139). For instance, clinical trials using a fusion protein of IL-2 with diphtheria toxin (Denileukin Diftitox) for treating metastatic melanoma indicate that it cannot effectively eliminate intratumoral Treg cells, resulting in inadequate antitumor responses (136). Furthermore, the motifs in Denileukin Diftitox’s molecular structure may facilitate its binding to endothelial cells, resulting in endothelial damage and subsequent vascular leak syndrome, characterized by the leakage of plasma or proteins into surrounding tissues, leading to tissue edema, hypotension, and in severe cases, organ failure. This dose-limiting toxicity also restricts the anti-tumor application of this therapy (140). Since regular effector T cells also express the CD25 molecule, targeting CD25 could lead to the clearance of these conventional T cells. Some strategies involve optimizing the Fc segment of anti-CD25 monoclonal antibodies to favor binding with intratumoral FcγRIIb, specifically boosting the ADCC effect against intratumoral Tregs and promoting their elimination (141). Besides, employing a non-IL-2 blocking anti-CD25 antibody, Tregs can be eliminated while retaining the responsiveness of effector T cells to IL-2 signaling, thus amplifying the proliferation and function of anti-tumor T cells (142).
Similarly, other targets highly expressed on Treg cells, such as immune checkpoints LAG3, TIM3, VISTA and TIGIT, members of the tumor necrosis factor receptor superfamily (OX40, 4-1BB), immunoglobulins (ICOS, CD28) and G-protein coupled receptors, can all serve as potential targets for Treg cell elimination or inhibition. The combination of multi-target therapies might further enhance the efficacy of Treg-targeting treatment (143–145).
Anti-PD-1 therapy enhances the effector activity of Tconv cells and has shown promising outcomes in cancer treatment. However, it may also result in Treg activation and contribute to HPD (60, 63, 146). Studies have shown that in mouse models with a high PD-1+ Treg to PD-1+ CD8+ T cell ratio, PD-1 blockade could lead to tumor enlargement (147). Therefore, a combined approach targeting both PD-1 and other Treg markers to augment Teff activity while inhibiting Treg functions might be an effective strategy to enhance anti-tumor efficacy.
CTLA-4 and PD-1 are key immune checkpoints with distinct mechanisms in regulating T cell activity. CTLA-4 primarily functions during the immune priming phase, where its blockade enhances the activation and proliferation of Teffs as well as reducing Treg-mediated immune suppression. Conversely, PD-1 primarily functions during the effector phase, reversing exhausted T cell to an effector state (148). In melanoma patients with immune checkpoint blockade therapy, the peripheral blood CD8+ T cell TCR repertoire analysis provides insightful observations. Specifically, in the majority of patients (10 out of 12) who clinically benefited from PD-1 blockade therapy, there were no significant alterations in the frequency or diversity of circulating melanoma-reactive CD8+ T cell responses. However, the blockade of CTLA-4 resulted in a noticeable increase in the number of detectable melanoma-specific CD8+ T cell (149, 150). Theoretically, combining CTLA-4 and PD-1 blockade therapies is hypothesized to synergistically enhance anti-tumor effects. This has been confirmed in clinical settings, where combined therapy using ipilimumab (CTLA-4 antibody) and nivolumab (PD-1 antibody) has demonstrated greater efficacy than monotherapy in treating advanced melanoma (151). Numerous clinical trials are currently exploring this combination approach. However, it’s crucial to recognize that although the combination of CTLA-4 and PD-1 therapies can enhance effector T cell function and diminish Treg activity, it also disrupts immune homeostasis and leads to pronounced immune-related adverse events (irAEs), highlighting the necessity for diligent management and monitoring of patients receiving these combined treatments (152).
PD-1 blockade therapy has been shown to increase the expression of other immune checkpoints, such as TIM-3, Hammerman and colleagues discovered that prolonged exposure to PD-1 blockade antibodies escalates TIM-3 expression in non-small cell lung cancer patients (153, 154). Similarly, Xu et al. also found a significant increase in TIM3 expression within melanoma tumors of patients resistant to PD-1 treatment (155, 156). Preclinical models have confirmed that combining PD-1 and TIM-3 targeting in anti-tumor therapy is more effective than using either approach individually (154, 156).
Studies have constructed fusion proteins by combining PD-1 antibody and the GITR ligand (157). PD-1-GITR-L bispecific molecules were able to enhance the activation, proliferation, and memory formation of Tconvs in vivo and also boost the cytotoxicity of NK cells while reducing Tregs and exhausted T cells within the TME.
Studies have shown that increased TGF-β expression in tumors leads to an increase in PD-L1 positive cells within the TME, which, in turn, diminishes the effectiveness of PD-1/PD-L1 therapies (158). Further research found that the resistance to anti-PD-1 therapy in tumors correlates with increased TGF-β secretion by tumor fibroblasts and a decrease in CD8+ T cell infiltration (159). It has been established that inhibiting TGF-β signaling can significantly improve the efficacy of PD-1 antibody treatments (160). In addition, bispecific antibodies (TM101 and BiTP) targeting both TGF-β and PD-L1 have been shown to be more effective than anti-PD-L1 or anti-TGF-β monotherapy in various murine tumor models. This dual-targeting strategy can reduce collagen accumulation in tumors and promote T cell tumor infiltration, with great potential for application (161, 162).
In a preclinical model of head and neck squamous carcinoma, a VISTA blocking antibody monotherapy enabled the reversal of CD8+ T cell exhaustion into an effector state. However, it did not inhibit the recruitment of Tregs into the tumor and thus cannot effectively inhibit tumor growth. As CTLA-4 blockade significantly reduces Treg infiltration, the he combined administration of VISTA and CTLA-4 antibodies increases the CD8+T/Treg ratio and the CD4+Tconv/Treg ratio, significantly inhibiting tumor growth. Additionally, in prostate cancer patients treated with ipilimumab, a high expression of VISTA has been observed, indicating that the combined blockade of CTLA-4 and VISTA presents a promising strategy for enhancing anti-tumor efficacy (163).
4.2 Reduced recruitment of Tregs
Treg cells highly express certain chemokine receptors (CCR4, CCR5, CCR8, CCR10), which facilitates their preferential migration to the tumor microenvironment (164, 165). By inhibiting the chemokine signaling pathway, the accumulation of Tregs within tumors can be mitigated. For example, targeting CCR10 or CCR5 with receptor-blocking monoclonal antibodies significantly delayed tumor growth in mouse models (88, 166, 167). Moreover, certain monoclonal antibodies can also specifically eradicate Tregs through ADCC. Clinical trials with monoclonal antibodies against CCR4 have further validated their potential in depleting Treg cells (168, 169). Interestingly, tumors with naturally low expression of chemokines like CCL17/CCL22 demonstrate elevated chemokine expression following immune checkpoint blockade treatment (91). In such scenario, dual-targeting therapy involving chemokine receptors and immune checkpoints might aid in enhancing the efficacy of anti-tumor immunotherapy. In a clinical trial that combined mogamulizumab with nivolumab, 96 participants received dual-targeted therapy against CCR4 and PD-1, showing good safety profile and effectiveness in reducing eTregs and enhancing CD8+ T cell infiltration in tumors. Additionally, a study using an oral CCR4 antagonist combined with ICB (anti-CTLA-4 or anti-PD-1) therapy in a mouse tumor model demonstrated that the combination therapy is more effective against tumors than monotherapy (170, 171).
4.3 Attenuates the immunosuppressive function of Tregs
Treg cells, by highly expressing CD39 and CD73, adapt to the microenvironment and eventually convert intratumoral ATP to adenosine (104). Type 2 (P2) purinergic receptors, which recognize and bind to extracellular ATP, are categorized into two subtypes: P2X and P2Y. ATP, when acting on P2XR of Tregs, can induce Treg apoptosis and suppress Treg function (172, 173). CD39 reduces ATP levels in the TME, indirectly protecting Treg cells from ATP-triggered apoptosis. Meanwhile, CD73 elevates the adenosine concentration in the microenvironment. Adenosine binds to A2aR and A2bR on effector T cell membranes, suppressing their activity (106). Therefore, targeting CD39 and CD73 or using P2XR activators or antibodies blocking A2aR and A2bR can all alleviate the immunosuppressive effects of Tregs (107, 110).
Moreover, IDO acts as a tryptophan-consuming enzyme, leading to diminished levels of tryptophan within the TME. This amino acid is vital for the activation of T cells and their antitumor potentials (111). Notably, overexpression IDO in TME after immune checkpoint inhibitor administration, resulting in Tonvs dysfunction and heightened activation and proliferation of Tregs and MDSCs, establishing an immunosuppressive milieu (111, 174). Clinical trials have already been initiated for several IDO-targeting inhibitors (175).
Targeting molecules associated with Treg’s metabolic functions to reduce their activity offers a promising therapeutic strategy. For instance, Treg cells utilize the fatty acid transporter (CD36) and the lactic acid transporter MCT1 to transport free fatty acids and lactic acid from the glucose-deprived TME into cells, facilitating energy metabolism essential for their functionality (120–122). Studies targeting CD36 and MCT1 have demonstrated that focusing on these molecules can amplify the therapeutic effects of ICB treatments (120, 122).
In addition to targeting markers or key molecules highly expressed in Tregs, some studies have identified specific signaling pathways vital for Treg survival and functionality. Tregs with a specific deficiency or inhibition of the PI3Kδ molecule have restricted immunosuppressive capacities (176). Targeting PI3Kδ with small molecule inhibitors can diminish the Treg population within the TME, simultaneously boosting the activity of Teffs, reducing tumor progression, and suppressing tumor metastasis. Furthermore, Treg survival depends on sustained TCR activation signals, and there are differences in this signaling pathway between Tregs and Tconv cells (177, 178). Owing to the influence of the Foxp3 molecule, key components in this signaling pathway, such as LCK, ZAP70, and SLP76, are expressed at lower levels than in Tconvs, thus preventing apoptosis of T cells triggered by enduring TCR signal stimulation (179). Clinically, there are reports indicating that certain TKI inhibitors, like imatinib and dasatinib, due to their inhibitory effect on LCK, leading to a significant reduction in Treg cells (180, 181).
Targeting the VEGF-VEGFR pathway can also inhibit Treg cell functions and reduce their proportion in the TME. The monoclonal antibody against VEGFR2, ramucirumab, by blocking the VEGF-VEGFR2 signaling pathway, can decrease immunosuppressive cells such as Tregs, MDSCs, and M2 macrophages in the TME, while enhancing the presence of mature DCs (102, 182). Studies analyzing gastro-intestinal tumor samples pre- and postadministration of VEGFR2 monoclonal antibodies observed a notable reduction in eTreg cells within the TME (102). Additionally, after inhibiting the angiogenesis signaling pathway, there is a decline in the expression of effector T cell exhaustion markers (183). Clinically, there is a promising trend to co-administer antiangiogenic treatments with immune checkpoint inhibitors for enhanced antitumor therapy (184). Notably, the combination strategy achieves excellent results in treating hepatocellular carcinoma, renal cell carcinoma, non-small-cell lung cancer and colorectal cancer (185–188).
4.4 Treatment strategies modified based on kinetics and spatial distinction
While there are currently many treatment strategies for Tregs, few have yielded definitive results. One reason is that the characteristic molecules and metabolic pathways of Treg cells are not unique to themselves. Other antitumor effector cells within the immune system may also employ the same mechanisms for their function. Thus, a systemic drug delivery approach might result in the elimination or suppression of non-tumor Tregs outside the tumor as well as other effector cells. It may not only suppress pTreg functions, disrupting immunological homeostasis and triggering autoimmune inflammation, but also attenuate anti-tumor efficiency (189). Current research indicates that although certain cellular molecules are expressed in both Tregs and effector immune cells, there is a difference in their expression levels and kinetics. For instance, molecules like CD25 and CTLA-4 are consistently highly expressed in Tregs within tumors, while Tconv cells primarily express them at minimal levels in resting state but elevated levels upon activation (190).
Given the distinct expression patterns of CD25 between Tconvs and Tregs, the optimal timing for drug administration is chosen to specifically target Tregs while minimizing effects on Tconv. This concept was validated in clinical trials using CD25 monoclonal antibodies in conjunction with tumor antigen peptide vaccines, in which patients were administered a single dose of CD25 monoclonal antibody one week prior to vaccine treatment (135). That is, the drug clears CD25+ Tregs before antitumor-specific T cells are activated. This treatment effectively cleared Tregs and extended the progression-free survival of patients suffering from metastatic breast cancer. Clinical studies of anti-CTLA4 monoclonal antibodies along with tumor peptide vaccines also have proven that using CTLA-4 monotherapy before vaccination strengthens the antitumor reaction, enhancing therapeutic results (190).
Local drug delivery techniques can also be applied to improve treatment precision. for instance, utilizing interventional techniques to inject antibodies or drugs targeting Tregs locally or employing photodynamic drugs, coupling the photoactive dye IR700 with antibodies that target Tregs (191–193), and then irradiating the tumor site with near-infrared light, ensuring that the clearing antibodies locally exert their effects, leading to the suppression of both syngeneic and allogeneic tumors, even those untreated with near-infrared light.
5 Conclusion
This review encompasses a range of therapeutic strategies targeting Tregs for cancer treatment (Table 1). Given the varying roles and proportions of Tregs across different tumors, it is evident that no single Treg-targeting strategy can guarantee efficacy for every tumor type. Understanding the tumor immune microenvironment and selecting specialized therapeutic methods for Tregs is crucial. Moreover, the introduction of innovative drugs, together with comprehensive research on Treg subtypes and functions, promises significant advancements in Treg-targeted cancer therapies.
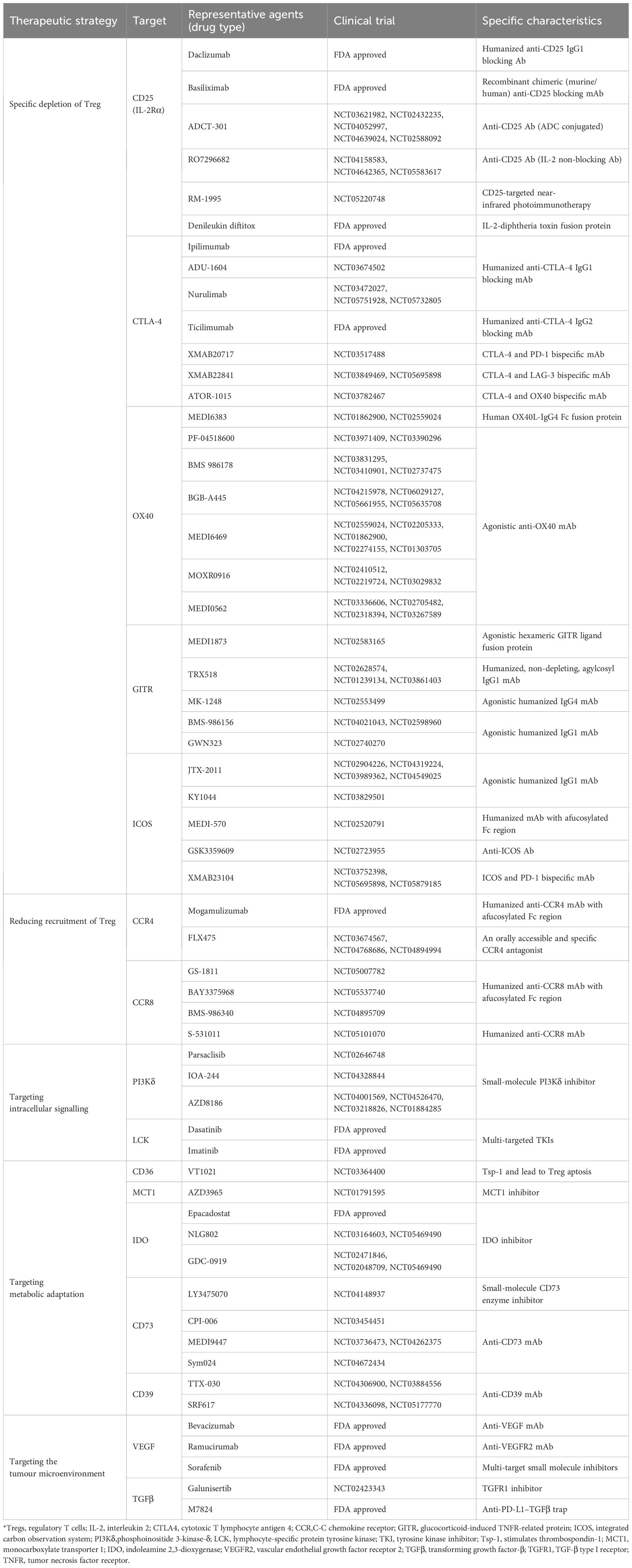
Table 1 Therapies targeting Tregs that are FDA approved or in clinical trials.
Author contributions
DQ: Writing – original draft. YZ: Writing – original draft. PS: Writing – original draft. YL: Writing – review & editing. XL: Writing – review & editing. YW: Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the China Postdoctoral Science Foundation (No. 2021M702346), the Post-Doctor Research Project, West China Hospital, Sichuan University (No. 2021HXBH092) and the Fundamental Research Funds for the Central Universities (No. 2023SCU12051).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Filin IY, Kitaeva KV, Rutland CS, Rizvanov AA, Solovyeva VV. Recent advances in experimental dendritic cell vaccines for cancer. Front In Oncol. (2021) 11:730824. doi: 10.3389/fonc.2021.730824
2. Coukos G. TIL therapy entering the mainstream. New Engl J Med. (2022) 387(23):2185–6. doi: 10.1056/NEJMe2214655
3. Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered t cell therapy in solid tumors: State of the art and perspectives. Sci Adv. (2023) 9(7):eadf3700. doi: 10.1126/sciadv.adf3700
4. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti- CD19 chimeric antigen receptor t-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematology. (2020) 7(11):e816–e826. doi: 10.1016/S2352-3026(20)30277-5
5. Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discovery. (2022) 21(7):509–28. doi: 10.1038/s41573-021-00345-8
6. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory hodgkin's lymphoma. New Engl J Med. (2015) 372(4):311–9. doi: 10.1056/NEJMoa1411087
7. Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol Off J Eur Soc For Med Oncol. (2016) 27(8):1482–92. doi: 10.1093/annonc/mdw168
8. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. (2020) 52(1):17–35. doi: 10.1016/j.immuni.2019.12.011
9. Lv B, Wang Y, Ma D, Cheng W, Liu J, Yong T, et al. Immunotherapy: Reshape the tumor immune microenvironment. Front Immunol. (2022) 13:844142. doi: 10.3389/fimmu.2022.844142
10. Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory t cells as targets of cancer immunotherapy. Cancer Cell. (2023) 41(3):450–65. doi: 10.1016/j.ccell.2023.02.014
11. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduction Targeted Ther. (2021) 6(1):362. doi: 10.1038/s41392-021-00670-9
12. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery. (2019) 18(2):99–115. doi: 10.1038/s41573-018-0004-1
13. Pittet MJ, Michielin O, Migliorini D. Clinical relevance of tumour- associated macrophages. Nat Rev Clin Oncol. (2022) 19(6):402–21. doi: 10.1038/s41571-022-00620-6
14. Savage PA, Klawon DEJ, Miller CH. Regulatory t cell development. Annu Rev Immunol. (2020) 38:421–53. doi: 10.1146/annurev-immunol-100219-020937
15. Dominguez-Villar M, Hafler DA. Regulatory t cells in autoimmune disease. Nat Immunol. (2018) 19(7):665–73. doi: 10.1038/s41590-018-0120-4
16. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory t cells and immune tolerance. Cell. (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
17. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory t cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol (Baltimore Md. 1950). (2008) 180(9):5916–26.
18. Okeke EB, Uzonna JE. The pivotal role of regulatory t cells in the regulation of innate immune cells. Front Immunol. (2019) 10:680. doi: 10.3389/fimmu.2019.00680
19. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. (1970) 18(5):723–37.
20. Nishizuka Y, Sakakura T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Sci (New York N.Y.). (1969) 166(3906):753–5.
21. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self- tolerance maintained by activated t cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (Baltimore Md. 1950). (1995) 155(3):1151–64.
22. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27(1):68–73. doi: 10.1038/83784
23. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ t regulatory cells. Nat Immunol. (2003) 4(4):337–42. doi: 10.1038/ni909
24. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory t cells. Nat Immunol. (2003) 4(4):330–6. doi: 10.1038/ni904
25. Hori S, Nomura T, Sakaguchi S. Control of regulatory t cell development by the transcription factor Foxp3. Science. (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
26. Godfrey VL, Wilkinson JE, Rinchik EM, Russell LB. Fatal lymphoreticular disease in the scurfy (sf) mouse requires t cells that mature in a sf thymic environment: potential model for thymic education. Proc Natl Acad Sci USA. (1991) 88(13):5528–32. doi: 10.1073/pnas.88.13.5528
27. Powell BR, Buist NR, Stenzel P. An x-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. (1982) 100(5):731–7.
28. Mayer E, Bannert C, Gruber S, Klunker S, Spittler A, Akdis CA, et al. Cord blood derived CD4+ CD25(high) t cells become functional regulatory t cells upon antigen encounter. PloS One. (2012) 7(1):e29355. doi: 10.1371/journal.pone.0029355
29. Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory t cells are not mirror images of each other. Trends Immunol. (2008) 29(9):429–35. doi: 10.1016/j.it.2008.06.005
30. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated t cells. J Exp Med. (2006) 203(7):1693–700.
31. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ t reg cells. J Exp Med. (2006) 203(7):1701–11. doi: 10.1084/jem.20060772
32. Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. (2007) 7(2):144–54.
33. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 t cells by t-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. (2007) 110(8):2983–90. doi: 10.1182/blood-2007-06-094656
34. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive t cells to CD4+CD25+ regulatory t cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
35. Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, et al. Analysis of the t-cell receptor repertoires of tumor-infiltrating conventional and regulatory t cells reveals no evidence for conversion in carcinogen-induced tumors. Cancer Res. (2011) 71(3):736–46. doi: 10.1158/0008-5472.CAN-10-1797
36. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ t cell receptors. Immunity. (2004) 21(2):267–77. doi: 10.1016/j.immuni.2004.07.009
37. Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, et al. Tumor-infiltrating human CD4+ regulatory t cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. (2019) 4(31). doi: 10.1126/sciimmunol.aao4310
38. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory t cell differentiation mediated by retinoic acid. Science. (2007) 317(5835):256–60. doi: 10.1126/science.1145697
39. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. (2014) 259(1):88–102. doi: 10.1111/imr.12160
40. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ t cells expressing the FoxP3 transcription factor. Immunity. (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
41. Togashi Y, Shitara K, Nishikawa H. Regulatory t cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16(6):356–71. doi: 10.1038/s41571-019-0175-7
42. Cunha LL, Morari EC, Nonogaki S, Soares FA, Vassallo J, Ward LS. Foxp3 expression is associated with aggressiveness in differentiated thyroid carcinomas. Clinics (Sao Paulo). (2012) 67(5):483–8. doi: 10.6061/clinics/2012(05)13
43. Wolf D, Wolf AM, Rumpold H, Fiegl H, Zeimet AG, Muller-Holzner E, et al. The expression of the regulatory t cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res. (2005) 11(23):8326–31. doi: 10.1158/1078-0432.Ccr-05-1244
44. Saleh R, Elkord E. FoxP3(+) t regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. (2020) 490:174–85. doi: 10.1016/j.canlet.2020.07.022
45. Ma GF, Miao Q, Liu YM, Gao H, Lian JJ, Wang YN, et al. High FoxP3 expression in tumour cells predicts better survival in gastric cancer and its role in tumour microenvironment. Br J Cancer. (2014) 110(6):1552–60. doi: 10.1038/bjc.2014.47
46. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) t cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. (2016) 22(6):679–84. doi: 10.1038/nm.4086
47. Seminerio I, Descamps G, Dupont S, de Marrez L, Laigle JA, Lechien JR, et al. Infiltration of FoxP3+ regulatory t cells is a strong and independent prognostic factor in head and neck squamous cell carcinoma. Cancers (Basel). (2019) 11(2). doi: 10.3390/cancers11020227
48. Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, et al. A distinct subpopulation of CD25(-) t-follicular regulatory cells localizes in the germinal centers. Proc Natl Acad Sci USA. (2017) 114(31):E6400–e6409. doi: 10.1073/pnas.1705551114
49. D'Arena G, Vitale C, Coscia M, Festa A, Di Minno NMD, De Feo V, et al. Regulatory t cells and their prognostic relevance in hematologic malignancies. J Immunol Res. (2017) 2017:1832968. doi: 10.1155/2017/1832968
50. Muenst S, Hoeller S, Dirnhofer S, Tzankov A. Increased programmed death- 1+ tumor-infiltrating lymphocytes in classical hodgkin lymphoma substantiate reduced overall survival. Hum Pathol. (2009) 40(12):1715–22. doi: 10.1016/j.humpath.2009.03.025
51. Niedzwiecki M, Budzilo O, Zielinski M, Adamkiewicz-Drozynska E, Maciejka-Kemblowska L, Szczepanski T, et al. CD4(+)CD25(high)CD127(low/-)FoxP(3)(+) regulatory t cell subpopulations in the bone marrow and peripheral blood of children with ALL: Brief report. J Immunol Res. (2018) 2018:1292404. doi: 10.1155/2018/1292404
52. Ohue Y, Nishikawa H, Regulatory T. (Treg) cells in cancer: Can treg cells be a new therapeutic target? Cancer Sci. (2019) 110(7):2080–9. doi: 10.1111/cas.14069
53. Lucca LE, Dominguez-Villar M. Modulation of regulatory t cell function and stability by co-inhibitory receptors. Nat Rev Immunol. (2020) 20(11):680–93. doi: 10.1038/s41577-020-0296-3
54. Wei S, Kryczek I, Zou W. Regulatory t-cell compartmentalization and trafficking. Blood. (2006) 108(2):426–31. doi: 10.1182/blood-2006-01-0177
55. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. (1994) 1(9):793–801. doi: 10.1016/s1074-7613(94)80021-9
56. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of t cell responses. Nat Rev Immunol. (2011) 11(12):852–63. doi: 10.1038/nri3108
57. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332(6029):600–3. doi: 10.1126/science.1202947
58. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. (2016) 375(18):1767–78. doi: 10.1056/NEJMra1514296
59. Vick SC, Kolupaev OV, Perou CM, Serody JS. Anti-PD-1 checkpoint therapy can promote the function and survival of regulatory t cells. J Immunol (Baltimore Md. 1950). (2021) 207(10):2598–607. doi: 10.4049/jimmunol.2001334
60. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1(+) regulatory t cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci USA. (2019) 116(20):9999–10008. doi: 10.1073/pnas.1822001116
61. Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci USA. (2016) 113(30):8490–5. doi: 10.1073/pnas.1608873113
62. Park HJ, Kim KW, Won SE, Yoon S, Chae YK, Tirumani SH, et al. Definition, incidence, and challenges for assessment of hyperprogressive disease during cancer treatment with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Netw Open. (2021) 4(3):e211136. doi: 10.1001/jamanetworkopen.2021.1136
63. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria JC, et al. Hyperprogressive disease: recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol. (2018) 15(12):748–62. doi: 10.1038/s41571-018-0111-2
64. Togashi Y, Kamada T, Sasaki A, Nakamura Y, Fukuoka S, Tada Y, et al. Clinicopathological, genomic and immunological features of hyperprogressive disease during PD-1 blockade in gastric cancer patients. J Clin Oncol. (2018) 36(15_suppl):4106–6. doi: 10.1200/JCO.2018.36.15_suppl.4106
65. Höfer T, Krichevsky O, Altan-Bonnet G. Competition for IL-2 between regulatory and effector t cells to chisel immune responses. Front Immunol. (2012) 3:268. doi: 10.3389/fimmu.2012.00268
66. Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. (2018) 18(10):648–59. doi: 10.1038/s41577-018-0046-y
67. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating t regulatory cells. Immunity. (2016) 45(5):1135–47. doi: 10.1016/j.immuni.2016.10.021
68. Huang C-T, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory t cells. Immunity. (2004) 21(4):503–13.
69. Banerjee H, Nieves-Rosado H, Kulkarni A, Murter B, McGrath KV, Chandran UR, et al. Expression of tim-3 drives phenotypic and functional changes in treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep. (2021) 36(11):109699. doi: 10.1016/j.celrep.2021.109699
70. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
71. Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam K-P, Coyle AJ, et al. ICOS controls the pool size of effector-memory and regulatory t cells. J Immunol (Baltimore Md. 1950). (2008) 180(2):774–82.
72. Chen Q, Mo L, Cai X, Wei L, Xie Z, Li H, et al. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory t cells. Int J Med Sci. (2018) 15(7):666–73. doi: 10.7150/ijms.23940
73. Ronchetti S, Ricci E, Petrillo MG, Cari L, Migliorati G, Nocentini G, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein: a key marker of functional regulatory t cells. J Immunol Res. (2015) 2015:171520. doi: 10.1155/2015/171520
74. Muth S, Klaric A, Radsak M, Schild H, Probst HC. CD27 expression on treg cells limits immune responses against tumors. J Mol Med (Berl). (2022) 100(3):439–49. doi: 10.1007/s00109-021-02116-9
75. Mortezaee K, Majidpoor J, Najafi S. VISTA immune regulatory effects in bypassing cancer immunotherapy: Updated. Life Sci. (2022) 310:121083. doi: 10.1016/j.lfs.2022.121083
76. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory t-cell function. Nature. (2007) 450(7169):566–9. doi: 10.1038/nature06306
77. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10(+) and IL-35(+) t(reg) cells cooperatively promotes tumor t cell exhaustion. Nat Immunol. (2019) 20(6):724–35. doi: 10.1038/s41590-019-0346-9
78. Cuende J, Lienart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J, et al. Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory t cells in vivo. Sci Transl Med. (2015) 7(284):284ra56. doi: 10.1126/scitranslmed.aaa1983
79. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory t cells. J Exp Med. (2005) 201(7):1061–7. doi: 10.1084/jem.20042276
80. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. (2010) 10(8):554–67. doi: 10.1038/nri2808
81. Wang J, Zhao X, Wan YY. Intricacies of TGF-beta signaling in treg and Th17 cell biology. Cell Mol Immunol. (2023) 20(9):1002–22. doi: 10.1038/s41423-023-01036-7
82. Polanczyk MJ, Walker E, Haley D, Guerrouahen BS, Akporiaye ET. Blockade of TGF-beta signaling to enhance the antitumor response is accompanied by dysregulation of the functional activity of CD4(+)CD25(+)Foxp3(+) and CD4(+)CD25(-)Foxp3(+) t cells. J Transl Med. (2019) 17(1):219. doi: 10.1186/s12967-019-1967-3
83. Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF-beta pathway. J Hematol Oncol. (2021) 14(1):55. doi: 10.1186/s13045-021-01053-x
84. Wei X, Zhang J, Gu Q, Huang M, Zhang W, Guo J, et al. Reciprocal expression of IL-35 and IL-10 defines two distinct effector treg subsets that are required for maintenance of immune tolerance. Cell Rep. (2017) 21(7):1853–69. doi: 10.1016/j.celrep.2017.10.090
85. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. And perforin are important for regulatory t cell-mediated suppression of tumor clearance. Immunity. (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
86. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory t cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. (2009) 69(5):2000–9. doi: 10.1158/0008-5472.Can-08-2360
87. Faget J, Biota C, Bachelot T, Gobert M, Treilleux I, Goutagny N, et al. Early detection of tumor cells by innate immune cells leads to t(reg) recruitment through CCL22 production by tumor cells. Cancer Res. (2011) 71(19):6143–52. doi: 10.1158/0008-5472.Can-11-0573
88. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and t(reg) cells. Nature. (2011) 475(7355):226–30. doi: 10.1038/nature10169
89. Halvorsen EC, Hamilton MJ, Young A, Wadsworth BJ, LePard NE, Lee HN, et al. Maraviroc decreases CCL8-mediated migration of CCR5(+) regulatory t cells and reduces metastatic tumor growth in the lungs. Oncoimmunology. (2016) 5(6):e1150398. doi: 10.1080/2162402x.2016.1150398
90. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory t cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10(9):942–9. doi: 10.1038/nm1093
91. Marshall LA, Marubayashi S, Jorapur A, Jacobson S, Zibinsky M, Robles O, et al. Tumors establish resistance to immunotherapy by regulating t(reg) recruitment via CCR4. J Immunother Cancer. (2020) 8(2). doi: 10.1136/jitc-2020-000764
92. Islam SA, Ling MF, Leung J, Shreffler WG, Luster AD. Identification of human CCR8 as a CCL18 receptor. J Exp Med. (2013) 210(10):1889–98. doi: 10.1084/jem.20130240
93. Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, et al. CCR8 marks highly suppressive treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology. (2021) 163(4):512–20. doi: 10.1111/imm.13337
94. Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J, et al. CCR8(+)FOXp3(+) t(reg) cells as master drivers of immune regulation. Proc Natl Acad Sci USA. (2017) 114(23):6086–91. doi: 10.1073/pnas.1621280114
95. Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, et al. CCR8-targeted specific depletion of clonally expanded treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci USA. (2022) 119(7). doi: 10.1073/pnas.2114282119
96. Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, et al. Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory t cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer. (2021) 9(2). doi: 10.1136/jitc-2020-001749
97. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. (1992) 359(6398):843–5. doi: 10.1038/359843a0
98. Han Z, Dong Y, Lu J, Yang F, Zheng Y, Yang H. Role of hypoxia in inhibiting dendritic cells by VEGF signaling in tumor microenvironments: mechanism and application. Am J Cancer Res. (2021) 11(8):3777–93.
99. Bourhis M, Palle J, Galy-Fauroux I, Terme M. Direct and indirect modulation of t cells by VEGF-a counteracted by anti-angiogenic treatment. Front Immunol. (2021) 12:616837. doi: 10.3389/fimmu.2021.616837
100. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-a drives TOX-dependent t cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol. (2019) 4(41). doi: 10.1126/sciimmunol.aay0555
101. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory t-cell proliferation in colorectal cancer. Cancer Res. (2013) 73(2):539–49. doi: 10.1158/0008-5472.CAN-12-2325
102. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 with ramucirumab strongly impacts effector/ activated regulatory t cells and CD8(+) t cells in the tumor microenvironment. J Immunother Cancer. (2018) 6(1):106. doi: 10.1186/s40425-018-0403-1
103. Djordjevic S, Driscoll PC. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discovery Today. (2013) 18(9-10):447–55. doi: 10.1016/j.drudis.2012.11.013
104. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. (2017) 276(1):121–44. doi: 10.1111/imr.12528
105. Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D, et al. Human CD4+ CD39+ regulatory t cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol. (2014) 177(2):531–43. doi: 10.1111/cei.12354
106. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory t cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol. (2012) 3:190. doi: 10.3389/fimmu.2012.00190
107. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6(1):57. doi: 10.1186/s40425-018-0360-8
108. Devi VJ, Radhika A, Biju PG. Adenosine receptor activation promotes macrophage class switching from LPS-induced acute inflammatory M1 to anti- inflammatory M2 phenotype. Immunobiology. (2023) 228(3):152362. doi: 10.1016/j.imbio.2023.152362
109. Sarkar OS, Donninger H, Al Rayyan N, Chew LC, Stamp B, Zhang X, et al. Monocytic MDSCs exhibit superior immune suppression via adenosine and depletion of adenosine improves efficacy of immunotherapy. Sci Adv. (2023) 9(26):eadg3736. doi: 10.1126/sciadv.adg3736
110. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discovery. (2014) 4(8):879–88. doi: 10.1158/2159-8290.Cd-14-0341
111. Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3- dioxygenase inhibition for cancer therapy. Eur J Cancer. (2017) 76:167–82. doi: 10.1016/j.ejca.2017.01.011
112. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9(10):1269–74. doi: 10.1038/nm934
113. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4(10):762–74. doi: 10.1038/nri1457
114. Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory t cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol. (2022) 15(1):104. doi: 10.1186/s13045-022-01322-3
115. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
116. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. (2008) 84(4):949–57. doi: 10.1189/jlb.0108024
117. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance t(reg) cell anabolic metabolism for suppression. Nat Immunol. (2016) 17(12):1459–66. doi: 10.1038/ni.3577
118. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory t cell expansion during tumor growth. Proc Natl Acad Sci USA. (2018) 115(28):E6546–E6555. doi: 10.1073/pnas.1720113115
119. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms t cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25(6):1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
120. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory t cells in highly glycolytic tumor microenvironments. Cancer Cell. (2022) 40(2):201–218.e9. doi: 10.1016/j.ccell.2022.01.001
121. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory t cells by lactic acid. Nature. (2021) 591(7851):645–51. doi: 10.1038/s41586-020-03045-2
122. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory t cell survival and function in tumors. Nat Immunol. (2020) 21(3):298–308. doi: 10.1038/s41590-019-0589-5
123. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory t-cell-based cell therapy in living donor liver transplantation. Hepatol (Baltimore Md.). (2016) 64(2):632–43. doi: 10.1002/hep.28459
124. Baeten P, Van Zeebroeck L, Kleinewietfeld M, Hellings N, Broux B. Improving the efficacy of regulatory t cell therapy. Clin Rev Allergy Immunol. (2022) 62(2):363–81. doi: 10.1007/s12016-021-08866-1
125. Nahm DH. Regulatory t cell-targeted immunomodulatory therapy for long-term clinical improvement of atopic dermatitis: Hypotheses and perspectives. Life (Basel). (2023) 13(8). doi: 10.3390/life13081674
126. Altman A, Kong KF. pH-sensitive anti-CTLA4 antibodies: yes to efficacy, no to toxicity. Cell Res. (2019) 29(8):601–2. doi: 10.1038/s41422-019-0198-8
127. Zhang Y, Du X, Liu M, Tang F, Zhang P, Ai C, et al. Hijacking antibody- induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res. (2019) 29(8):609–27. doi: 10.1038/s41422-019-0184-1
128. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
129. Quezada SA, Peggs KS. Lost in translation: Deciphering the mechanism of action of anti-human CTLA-4. Clin Cancer Res. (2019) 25(4):1130–1132. doi: 10.1158/1078-0432.Ccr-18-2509
130. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. (2018) 33(4):649–663.e4. doi: 10.1016/j.ccell.2018.02.010
131. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory t cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210(9):1695–710. doi: 10.1084/jem.20130579
132. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti- CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory t cells (Tregs) in human cancers. Clin Cancer Res. (2019) 25(4):1233–8. doi: 10.1158/1078-0432.Ccr-18-0762
133. Marangoni F, Zhakyp A, Corsini M, Geels SN, Carrizosa E, Thelen M, et al. Expansion of tumor-associated treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell. (2021) 184(15):3998–4015 e19. doi: 10.1016/j.cell.2021.05.027
134. Wang XB, Fan ZZ, Anton D, Vollenhoven AV, Ni ZH, Chen XF, et al. CTLA4 is expressed on mature dendritic cells derived from human monocytes and influences their maturation and antigen presentation. BMC Immunol. (2011) 12:21. doi: 10.1186/1471-2172-12-21
135. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr., et al. CD25 blockade depletes and selectively reprograms regulatory t cells in concert with immunotherapy in cancer patients. Sci Transl Med. (2012) 4(134):134ra62. doi: 10.1126/scitranslmed.3003330
136. Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin diftitox, DAB389IL-2, ONTAK) to eliminate regulatory t lymphocytes in patients with melanoma. J Immunother. (2005) 28(6):582–92. doi: 10.1097/01.cji.0000175468.19742.10
137. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ t cells: a common basis between tumor immunity and autoimmunity. J Immunol. (1999) 163(10):5211–8.
138. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. (1999) 59(13):3128–33.
139. Zammarchi F, Havenith K, Bertelli F, Vijayakrishnan B, Chivers S, van Berkel PH. CD25-targeted antibody-drug conjugate depletes regulatory t cells and eliminates established syngeneic tumors via antitumor immunity. J Immunother Cancer. (2020) 8(2). doi: 10.1136/jitc-2020-000860
140. Baluna R, Rizo J, Gordon BE, Ghetie V, Vitetta ES. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc Natl Acad Sci USA. (1999) 96(7):3957–62. doi: 10.1073/pnas.96.7.3957
141. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory t cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. (2017) 46(4):577–86. doi: 10.1016/j.immuni.2017.03.013
142. Solomon I, Amann M, Goubier A, Arce Vargas F, Zervas D, Qing C, et al. CD25-t(reg)-depleting antibodies preserving IL-2 signaling on effector t cells enhance effector activation and antitumor immunity. Nat Cancer. (2020) 1(12):1153–66. doi: 10.1038/s43018-020-00133-0
143. Ritter A, Marshal N, Pulukkunat D, Pereira N, Rakhmilevich A, Gawdzik J, et al. 500 INV322, a TME selective CD25 x CTLA4 bispecific antibody approach for depletion of tumor restricted tregs. Regular Young Investigator Award Abstracts. (2022) 10(suppl 2):A521–1. doi: 10.1136/jitc-2022-SITC2022.0500
144. Berezhnoy A, Sumrow BJ, Stahl K, Shah K, Liu D, Li J, et al. Development and preliminary clinical activity of PD-1-Guided CTLA-4 blocking bispecific DART molecule. Cell Rep Med. (2020) 1(9):100163. doi: 10.1016/j.xcrm.2020.100163
145. Zhang T, Lin Y, Gao Q. Bispecific antibodies targeting immunomodulatory checkpoints for cancer therapy. Cancer Biol Med. (2023) 20(3):181–95. doi: 10.20892/j.issn.2095-3941.2023.0002
146. Wang X, Wang F, Zhong M, Yarden Y, Fu L. The biomarkers of hyperprogressive disease in PD-1/PD-L1 blockage therapy. Mol Cancer. (2020) 19(1):81. doi: 10.1186/s12943-020-01200-x
147. Wakiyama H, Kato T, Furusawa A, Okada R, Inagaki F, Furumoto H, et al. Treg-dominant tumor microenvironment is responsible for hyperprogressive disease after PD-1 blockade therapy. Cancer Immunol Res. (2022) 10(11):1386–1397. doi: 10.1158/2326-6066.CIR-22-0041
148. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
149. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ t cell response. Sci Trans Med. (2014) 6(254):254ra128. doi: 10.1126/scitranslmed.3008918
150. Gangaev A, Rozeman EA, Rohaan MW, Isaeva OI, Philips D, Patiwael S, et al. Differential effects of PD-1 and CTLA-4 blockade on the melanoma-reactive CD8 t cell response. Proc Natl Acad Sci USA. (2021) 118(43). doi: 10.1073/pnas.2102849118
151. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl J Med. (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
152. Willsmore ZN, Coumbe BGT, Crescioli S, Reci S, Gupta A, Harris RJ, et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur J Immunol. (2021) 51(3):544–56. doi: 10.1002/eji.202048747
153. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
154. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting tim-3 and PD-1 pathways to reverse t cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
155. Huang L, Xu Y, Fang J, Liu W, Chen J, Liu Z, et al. Targeting STAT3 abrogates tim-3 upregulation of adaptive resistance to PD-1 blockade on regulatory t cells of melanoma. Front Immunol. (2021) 12:654749. doi: 10.3389/fimmu.2021.654749
156. Liu J, Luan Y, Deng H, Wang F, Wang C, Zhang Z. A bivalent tim- 3/PD-1 bispecific antibody for the treatment of PD-1 antibody resistant or refractory NSCLC. J Clin Oncol. (2022) 40(16_suppl):e14597–7. doi: 10.1200/JCO.2022.40.16_suppl.e14597
157. Chan S, Belmar N, Ho S, Rogers B, Stickler M, Graham M, et al. An anti- PD-1-GITR-L bispecific agonist induces GITR clustering-mediated t cell activation for cancer immunotherapy. Nat Cancer. (2022) 3(3):337–54. doi: 10.1038/s43018-022-00334-9
158. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia- Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554(7693):538–43. doi: 10.1038/nature25492
159. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of t cells. Nature. (2018) 554(7693):544–8. doi: 10.1038/nature25501
160. Terabe M, Robertson FC, Clark K, De Ravin E, Bloom A, Venzon DJ, et al. Blockade of only TGF-beta 1 and 2 is sufficient to enhance the efficacy of vaccine and PD-1 checkpoint blockade immunotherapy. Oncoimmunology. (2017) 6(5):e1308616. doi: 10.1080/2162402X.2017.1308616
161. Yi M, Wu Y, Niu M, Zhu S, Zhang J, Yan Y, et al. Anti-TGF-beta/PD-L1 bispecific antibody promotes t cell infiltration and exhibits enhanced antitumor activity in triple-negative breast cancer. J Immunother Cancer. (2022) 10(12). doi: 10.1136/jitc-2022-005543
162. Yi M, Zhang J, Li A, Niu M, Yan Y, Jiao Y, et al. The construction, expression, and enhanced anti-tumor activity of YM101: a bispecific antibody simultaneously targeting TGF-beta and PD-L1. J Hematol Oncol. (2021) 14(1):27. doi: 10.1186/s13045-021-01045-x
163. Kondo Y, Ohno T, Nishii N, Harada K, Yagita H, Azuma M. Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol. (2016) 57:54–60. doi: 10.1016/j.oraloncology.2016.04.005
164. Korbecki J, Grochans S, Gutowska I, Barczak K, Baranowska- Bosiacka I. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 ligands. Int J Mol Sci. (2020) 21(20). doi: 10.3390/ijms21207619
165. Korbecki J, Kojder K, Simińska D, Bohatyrewicz R, Gutowska I, Chlubek D, et al. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int J Mol Sci. (2020) 21(21). doi: 10.3390/ijms21218412
166. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory t cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. (2009) 182(3):1746–55. doi: 10.4049/jimmunol.182.3.1746
167. de Oliveira CE, Gasparoto TH, Pinheiro CR, Amor NG, Nogueira MRS, Kaneno R, et al. CCR5-dependent homing of t regulatory cells to the tumor microenvironment contributes to skin squamous cell carcinoma development. Mol Cancer Ther. (2017) 16(12):2871–80. doi: 10.1158/1535-7163.MCT-17-0341
168. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase ia study of FoxP3+ CD4 treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. (2015) 21(19):4327–36. doi: 10.1158/1078-0432.Ccr-15-0357
169. Maeda S, Motegi T, Iio A, Kaji K, Goto-Koshino Y, Eto S, et al. Anti- CCR4 treatment depletes regulatory t cells and leads to clinical activity in a canine model of advanced prostate cancer. J Immunother Cancer. (2022) 10(2). doi: 10.1136/jitc-2021-003731
170. Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, et al. A phase i study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res. (2019) 25(22):6614–22. doi: 10.1158/1078-0432.Ccr-19-1090
171. Robles O, Jackson JJ, Marshall L, Talay O, Chian D, Cutler G, et al. Novel piperidinyl-azetidines as potent and selective CCR4 antagonists elicit antitumor response as a single agent and in combination with checkpoint inhibitors. J Med Chem. (2020) 63(15):8584–607. doi: 10.1021/acs.jmedchem.0c00988
172. Schenk U, Frascoli M, Proietti M, Geffers R, Traggiai E, Buer J, et al. ATP inhibits the generation and function of regulatory t cells through the activation of purinergic P2X receptors. Sci Signal. (2011) 4(162):ra12. doi: 10.1126/scisignal.2001270
173. Aswad F, Kawamura H, Dennert G. High sensitivity of CD4+CD25+ regulatory t cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: a role for P2X7 receptors. J Immunol. (2005) 175(5):3075–83. doi: 10.4049/jimmunol.175.5.3075
174. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep. (2015) 13(2):412–24. doi: 10.1016/j.celrep.2015.08.077
175. Komiya T, Huang CH. Updates in the clinical development of epacadostat and other indoleamine 2,3-dioxygenase 1 inhibitors (IDO1) for human cancers. Front Oncol. (2018) 8:423. doi: 10.3389/fonc.2018.00423
176. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110δ breaks regulatory t-cell-mediated immune tolerance to cancer. Nature. (2014) 510(7505):407–11. doi: 10.1038/nature13444
177. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory t cell function. Nat Immunol. (2014) 15(11):1070–8. doi: 10.1038/ni.3004
178. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous t cell receptor signals maintain a functional regulatory t cell pool. Immunity. (2014) 41(5):722–36. doi: 10.1016/j.immuni.2014.10.012
179. Tanaka A, Maeda S, Nomura T, Llamas-Covarrubias MA, Tanaka S, Jin L, et al. Construction of a t cell receptor signaling range for spontaneous development of autoimmune disease. J Exp Med. (2023) 220(2). doi: 10.1084/jem.20220386
180. Tanaka A, Nishikawa H, Noguchi S, Sugiyama D, Morikawa H, Takeuchi Y, et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory t cells. J Exp Med. (2020) 217(2). doi: 10.1084/jem.20191009
181. Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. (2015) 2(12):e528–35. doi: 10.1016/s2352-3026(15)00196-9
182. Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA. Cytokine levels correlate with immune cell infiltration after anti- VEGF therapy in preclinical mouse models of breast cancer. PloS One. (2009) 4(11):e7669. doi: 10.1371/journal.pone.0007669
183. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-a modulates expression of inhibitory checkpoints on CD8+ t cells in tumors. J Exp Med. (2015) 212(2):139–48. doi: 10.1084/jem.20140559
184. Song Y, Fu Y, Xie Q, Zhu B, Wang J, Zhang B. Anti-angiogenic agents in combination with immune checkpoint inhibitors: A promising strategy for cancer treatment. Front Immunol. (2020) 11:1956. doi: 10.3389/fimmu.2020.01956
185. Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: An open-label, dose-escalation, and dose-expansion phase ib trial (REGONIVO, EPOC1603). J Clin Oncol. (2020) 38(18):2053–61. doi: 10.1200/jco.19.03296
186. Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. (2019) 393(10189):2404–15. doi: 10.1016/s0140-6736(19)30723-8
187. Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. (2019) 7(5):387–401. doi: 10.1016/s2213-2600(19)30084-0
188. Lee M, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Randomised efficacy and safety results for atezolizumab (Atezo) + bevacizumab (Bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Ann Oncol. (2019) 30. doi: 10.1093/annonc/mdz394.030
189. Tanaka A, Sakaguchi S. Targeting treg cells in cancer immunotherapy. Eur J Immunol. (2019) 49(8):1140–6. doi: 10.1002/eji.201847659
190. Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human treg and effector t cells in tumor immunity by fc- engineered anti-CTLA-4 antibody. Proc Natl Acad Sci USA. (2019) 116(2):609–18. doi: 10.1073/pnas.1812186116
191. van Hooren L, Sandin LC, Moskalev I, Ellmark P, Dimberg A, Black P, et al. Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer. Eur J Immunol. (2017) 47(2):385–93. doi: 10.1002/eji.201646583
192. Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJM. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ t-cell- dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res an Off J Am Assoc For Cancer Res. (2013) 19(19):5381–9. doi: 10.1158/1078-0432.CCR-12-0781
Keywords: regulatory T cells, immunotherapy, cancer, microenvironment, metabolism, checkpoint inhibitor, cytokine, chemokine
Citation: Qin D, Zhang Y, Shu P, Lei Y, Li X and Wang Y (2024) Targeting tumor-infiltrating tregs for improved antitumor responses. Front. Immunol. 15:1325946. doi: 10.3389/fimmu.2024.1325946
Received: 22 October 2023; Accepted: 16 February 2024;
Published: 04 March 2024.
Edited by:
Luigi Cari, University of Perugia, ItalyReviewed by:
Ming Yi, Zhejiang University, ChinaMaciej Zieliński, Department of Medical Immunology Medical University of Gdańsk, Poland
Copyright © 2024 Qin, Zhang, Shu, Lei, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongsheng Wang, wangys@scu.edu.cn
†These authors have contributed equally to this work and share first authorship