 Quanxiao Li
Quanxiao Li Jun Lu
Jun Lu Jinyao Li
Jinyao Li Baohong Zhang
Baohong Zhang Yanling Wu
Yanling Wu Tianlei Ying
Tianlei Ying
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 April 2023
Sec. Cancer Molecular Targets and Therapeutics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1157345
Regulatory T cells (Tregs) are among the most abundant suppressive cells, which infiltrate and accumulate in the tumor microenvironment, leading to tumor escape by inducing anergy and immunosuppression. Their presence has been correlated with tumor progression, invasiveness and metastasis. Targeting tumor-associated Tregs is an effective addition to current immunotherapy approaches, but it may also trigger autoimmune diseases. The major limitation of current therapies targeting Tregs in the tumor microenvironment is the lack of selective targets. Tumor-infiltrating Tregs express high levels of cell surface molecules associated with T-cell activation, such as CTLA4, PD-1, LAG3, TIGIT, ICOS, and TNF receptor superfamily members including 4-1BB, OX40, and GITR. Targeting these molecules often attribute to concurrent depletion of antitumor effector T-cell populations. Therefore, novel approaches need to improve the specificity of targeting Tregs in the tumor microenvironment without affecting peripheral Tregs and effector T cells. In this review, we discuss the immunosuppressive mechanisms of tumor-infiltrating Tregs and the status of antibody-based immunotherapies targeting Tregs.
Regulatory T cells (Tregs) constitute 1–2% of the peripheral CD4+ lymphocyte population, which express high levels of the regulatory transcription factor forkhead box protein 3 (FOXP3), CD25 (IL-2Rα), cytotoxic T lymphocyte (CTL)-associated antigen-4 (CTLA4), glucocorticoid induced tumor necrosis factor receptor (GITR), and CD39 (1, 2). Tregs intrinsically function to maintain immune homeostasis, support self-tolerance, and downregulate excessive immune responses against auto-antigens via different mechanisms (3). They are produced by the thymus and then exported to the periphery. In healthy individuals, Tregs suppress the potential self-reactive T cells through active regulation.
Despite critical importance, the immunosuppressive activities of Tregs are not desired in the tumor microenvironment (TME). Tregs are considered to play key roles in tumor immune escape by hampering tumor-specific T-cell responses and promoting tumor growth (4). Changes of nutrient composition, oxygen availability, and cytokines and chemokines released in TME favor Treg infiltration and effector T cell (Teff) exhaustion (5). This correlation also exists in patients that do not respond to immune checkpoint inhibitor (ICI) treatment. Hence, Treg infiltration in TME has been appreciated as a biomarker and a predictive factor for tumor progression and therapy response (6, 7). Removal of Tregs can evoke effective antitumor immunity in tumor-bearing animals (8). In humans, cancer immunotherapy by depleting Tregs is under clinical trial. Here, we review Treg biology, immunosuppressive mechanisms, and interactions in the TME context. We also summarize the current antibody-based immunotherapies and their efficacy of targeting Tregs in the treatment of tumors.
Tregs are categorized in three distinct subsets according to their sites of development. The thymic Tregs (tTregs) are developed in the thymus from the precursors of CD4+ helper T (Th) cells, which show an enrichment of T-cell receptors (TCRs) with high affinity for self-antigens (9, 10). Peripheral Tregs (pTregs) differentiate from mature CD4+ Th cells in the periphery upon encounter with antigens. A third Treg type is known as induced Tregs (iTregs), which are developed from naïve T cells and require antigen stimulation in the presence of transforming growth factor-beta (TGF-β) and interleukin-2 (IL-2) (11).
By their functions, Tregs can be further specified in two main subsets, central Tregs (cTregs) and effector Tregs (eTregs), in peripheral lymphoid organs (12). cTregs are thymic emigrants that have not been activated, which exhibit a naïve phenotype (CD45RA+ FOXP3lo) with low suppressive activity. They are enriched in lymphoid tissues where they express lymphoid-tissue homing molecules involved in trafficking to secondary lymphoid organs (such as CD62L and CCR7) and are dependent on IL-2 to maintain their state of rest (13, 14). After antigen engagement by TCRs, cTregs differentiate into eTregs (CD45RA- FOXP3hi), which upregulate activation marker CD44, effector molecules CTLA4, GZMB and KLRG1, and immunosuppressive cytokines (15, 16). By upregulating these molecules, eTregs further direct the migration and localization to non-lymphoid peripheral sites in response to specific stimuli (17).
Some conventional T cells (Tconvs) also express FOXP3 at low levels in the blood without exhibiting suppressive activity (18). To distinguish between suppressive and non-suppressive subtypes, CD4+FOXP3+ Tregs could be classified in three categories in the blood of healthy individuals by their different expression levels of FOXP3 (and CD25) and CD45RA. Naïve/resting Tregs, defined by CD45RA+ FOXP3loCD25++ cells (Fraction (Fr.) 1, see Figure 1), derive from thymus with weak suppressive activity. After TCR stimulation in the draining lymph node, naïve Tregs proliferate and differentiate into highly suppressive and terminally differentiated eTregs (Fr. 2), defined by CD45RA−FOXP3hiCD25+++. In general, the frequency of eTregs of CD4+ T cells in humans is 1%-5% in the peripheral blood but approximately 10%-50% in most TME (19). Non-Tregs (Fr. 3) are defined by FOXP3loCD45RA−CD25++ cells (18).
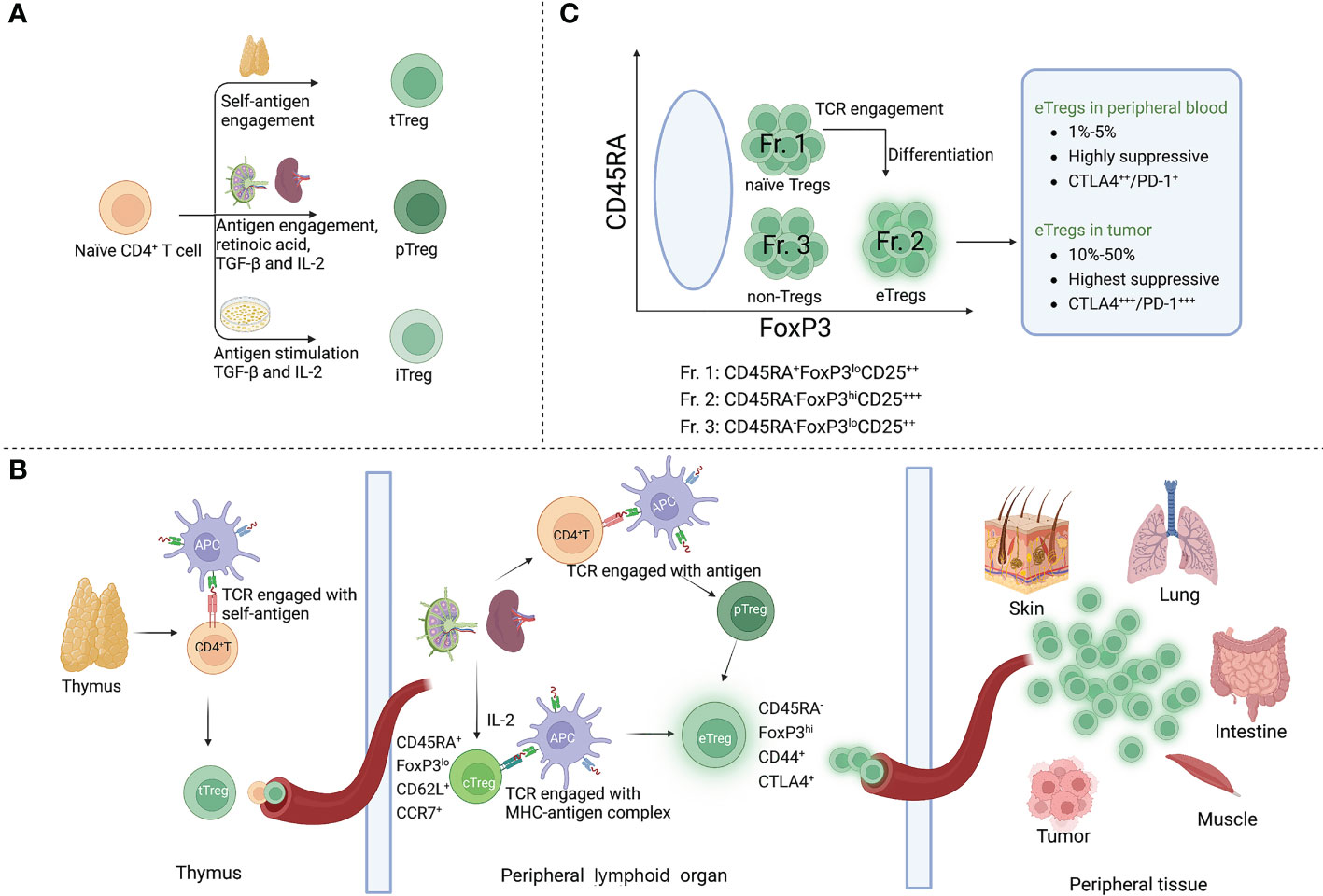
Figure 1 Phenotype and development of Tregs. (A) There are three Treg subtypes according to the sites of development: tTregs are developed in the thymus from the precursors of CD4+ T cells; pTregs are differentiated from mature CD4+ Th cells in the periphery upon encounter with antigens and other factors (IL-2, TGF-β, and retinoic acid); iTregs are developed from naïve T cells and require antigen stimulation in the presence of TGF-β and IL-2. (B) Development of cTregs and eTregs: tTregs migrate to peripheral lymphoid organs after thymus development and become cTregs, maintaining a naïve phenotype in the presence of IL-2 (CD45RA+, FOXP3lo, CD62L+, CCR7+). When cTregs and pTregs encounter TCR stimulation, they further differentiate into eTregs with expression of activation molecules CD44 and CTLA4. eTregs migrate and localize to non-lymphoid peripheral sites in response to specific stimuli. (C) Subfractions of human FOXP3+ Tregs: Fr. 1, CD45RA+ FOXP3lo CD25++ naïve Tregs; when naïve Tregs engage with antigens, they differentiate into Fr. 2 eTregs (CD45RA- FOXP3hi CD25+++); Fr. 3, non-Tregs, defined by CD45RA- FOXP3lo CD25++ cells.
Previous studies have demonstrated that Tregs have different immunosuppressive mechanisms, which can be summarized in three aspects. First, Tregs express immune checkpoint receptors, such as CTLA4, by which they interact with and suppress Teffs and antigen-presenting cells (APCs). Additionally, Tregs secrete immunomodulatory cytokines (IL-10, IL-35, and TGF-β) to repress the immune response and cytotoxic molecules (perforin, granzymes A and B), and directly induce apoptosis of effector immune cells. Moreover, Tregs interfere with the metabolism of effector cells thereby affecting their functions (see Figure 2) (20).
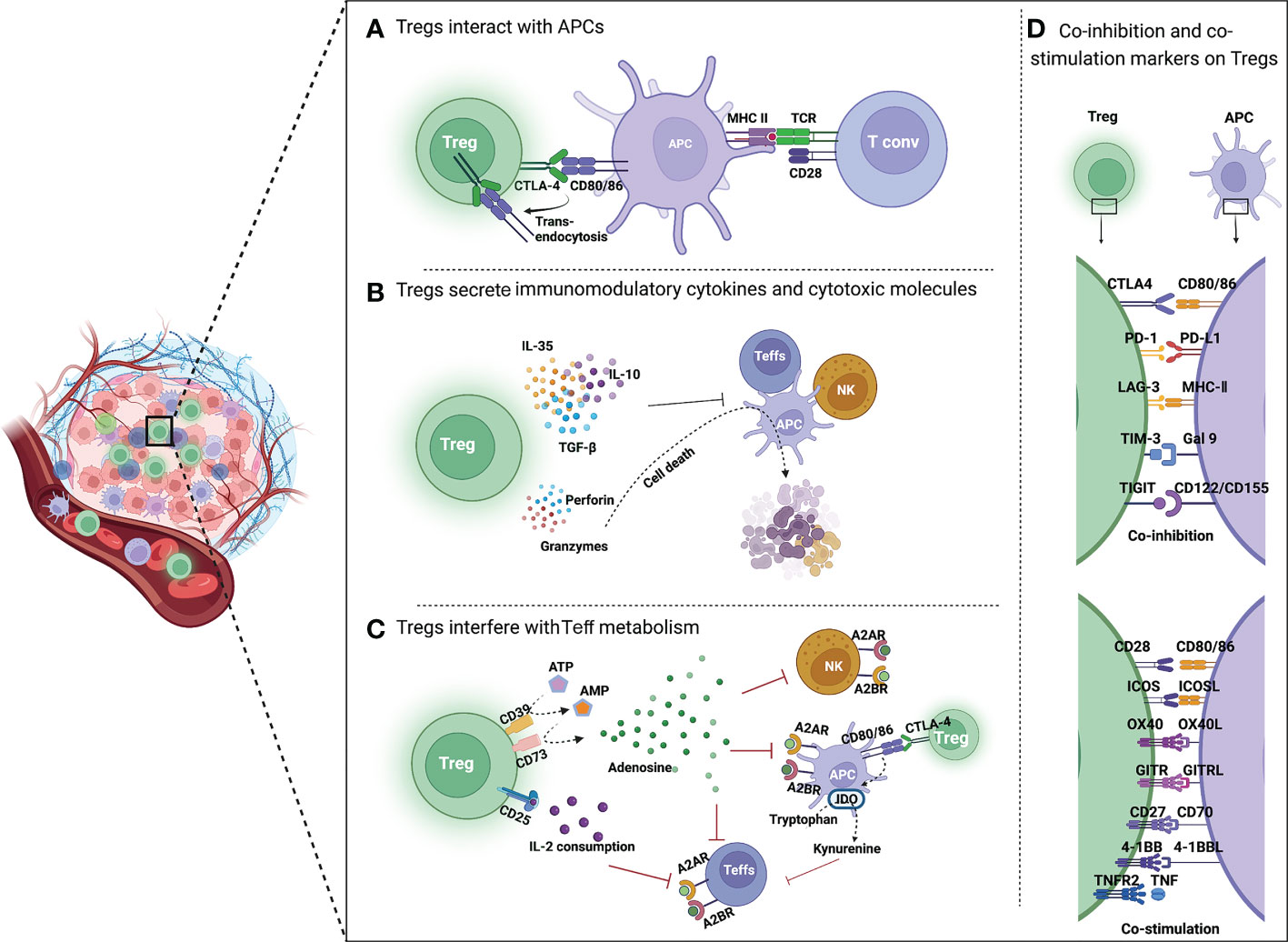
Figure 2 Mechanisms of Treg-mediated immunosuppression in TME. (A) Tregs inhibit the function of APCs and Teffs through immune checkpoints. CTLA4 is highly expressed on Tregs, which binds to CD80/86 on APCs, reduces CD80/86 expressed by APCs through trans-endocytosis, and inhibits the activation of T cells by APCs. (B) Tregs secrete immunomodulatory cytokines and cytotoxic molecules. Tregs directly inhibit the activation of Teffs, NK cells and APCs by secreting immunosuppressive cytokines such as IL-35, IL-10 and TGF-β. Moreover, Tregs can kill immune cells by directly secreting perforin and granzyme, leading to cell apoptosis. (C) Tregs interfere with Teff metabolism. CD39 converts extracellular ATP to generate AMP, which is then cleaved by CD73 to produce immunosuppressive molecular adenosine. Adenosine binds to receptors (A2A or A2B) on the surface of Teffs, APCs and NK cells, resulting in their immunosuppression. CTLA4 induces IDO production by APCs, which can oxidize tryptophan to kynurenine. Tryptophan is an essential amino acid for maintaining T cell activation. (D) Co-inhibition and co-stimulation molecules expressed by Tregs and their ligands. Tregs express a range of co-inhibition molecules of immune checkpoints (CTLA4, PD-1, LAG3, TIM3, and TIGIT) and co-stimulation molecules of Ig superfamily (CD28 and ICOS) as well as TNFR superfamily (OX40, GITR, CD27, 4-1BB, and TNFR2) on the surface.
CTLA4 expressed by Tregs shows higher affinity than CD28 when binding to CD80/86 on APCs, thus preventing Tconv activation through the interaction between CD80/CD86 and CD28. Removal of CD80/CD86 by trans-endocytosis results in impaired co-stimulation of T cells via CD28, leading to immune suppressive (21).
PD-1 is a transmembrane immunoinhibitory protein of the CD28 Ig superfamily and plays a major role in tumor immune escape. It is expressed at a low level by Tregs in the blood and upregulated in tumor-infiltrating Tregs (22). Different from CTLA4, Teffs in tumor express PD-1 at baseline or higher levels compared to tumor-infiltrating Tregs, potentially indicating exhaustion status. PD-1 engages with its ligands PD-L1/PD-L2 expressed on activated APCs to inhibit T-cell proliferation, survival, and effector functions (23) and induce apoptosis of tumor-specific T cells (24). The regulation and compartmentalization of PD-1 discriminate CD4+CD25+ resting Tregs from activated T cells. PD-1 signaling pathway is also important for the maintenance of the suppressive capacity of Tregs (25). PD-1 blockade enhanced PD-1+ Treg cell-mediated immunosuppression (26), and PD-1 expression in Tregs could be regulated by lactic acid in the highly glycolytic TME (27).
Tregs directly suppress the activity of T and B lymphocytes, dendritic cells (DCs), and macrophages by secreting anti-inflammatory soluble cytokines such as TGF-β, IL-10, and IL-35 (28, 29).
IL-10 secretion by Tregs impedes antigen-presenting effect of DCs. This effect was restored by anti-IL-10 treatment, and antigen-specific Tregs from IL-10−/− mice failed in decreasing antigen presentation of DCs (11). IL-10 can also downregulate MHC-II expression on the surface of mononuclear cells and reduce their antigen-presenting function, which impairs T-lymphocyte activity (11). Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion (28).
TGF-β can regulate the expression of FOXP3. iTregs rapidly lost the ability to express FOXP3 in the absence of TGF-β in vitro. TGF-β is important for Treg-induced inhibition of granule exocytosis and cytolytic function of CTLs (30). TGF-β1 is mainly secreted by Tregs. Tregs can express the docking receptor glycoprotein A repetitions predominant (GARP) and αv integrins that activate the cytokines, which triggers differentiation of other cell subsets, including T helper 17 (Th17) cells (29). IL-10 and TGF-β can also induce regulatory gamma-delta T cells, which is an innate-like T-cell population with a suppressive phenotype (31).
IL-35, a member of the IL-12 family, is preferentially secreted by mouse and human Tregs (28). IL-35 can induce Tconv conversion into suppressive CD4+ FOXP3− Treg population (32). In an IL-35-reporter mouse model, researchers found that the tumor was infiltrated with many IL-35+ Tregs, and tumor growth in both humans and mice was suppressed by neutralizing IL-35 with an antibody. Additionally, IL-35 limits antigen-specific antitumor T-cell responses by downregulating interferon-gamma (IFNγ) and tumor necrosis factor alpha (TNFα) secreted by CTLs. IL-35 also promotes the expression of exhaustion markers PD-1, TIM3, and LAG3 on T cells in TME (33).
In addition to secreting immunosuppressive cytokines, Tregs may directly kill APCs that express the target antigen by secreting perforin (34), while granzymes A and B induce apoptosis in target cells such as CD4+ and CD8+ T cells, monocytes, and DCs (35). Alternatively, Tregs can induce T cell apoptosis through TRAIL-DR5 pathway or via expression of galectin-1 (36) or FasL (37).
Tregs can suppress immune-cell activation through metabolic intermediates, including extracellular production of adenosine (38) and indoleamine 2,3-dioxygenase (IDO) (39).
CD39 is an ectonucleotidase overexpressed on the Tregs, which contributes to the conversion of adenosine triphosphate (ATP) to adenosine (40). Adenosine binds to the A2A receptor (A2AR) or A2B receptor (A2BR) on APCs, Teffs, and NK cells, resulting in immunosuppression. Apoptotic Tregs in TME will release large amounts of adenosine, which results in stronger suppression of antitumor immunity (41).
In addition, CTLA4 induces APCs to produce IDO, an essential enzyme in the kynurenine pathway of tryptophan metabolism, which converts tryptophan to kynurenine and leads to “death by starvation” of Teffs with cell cycle arrest (42, 43). IDO expression may also lead to the formation of a tolerant phenotype of IDO-expressing cells themselves (44). This is supported by in vitro studies, which showed that tryptophan consumption leads to inhibition of the immunoregulatory kinases mammalian target of rapamycin (mTOR) and protein kinase C, as well as induction of autophagy and Treg activation (45).
CD25 is the high affinity, alpha-chain of the heterotrimer IL-2 receptor (46). High expression of CD25 in Tregs may result in depletion of IL-2 from the surrounding microenvironment, which is detrimental to Teffs that rely on IL-2 for proliferation and activation (19, 47).
Tregs are also able to inhibit the cytotoxicity of NK cells through a cell-cell contact-dependent mechanism (48). In addition, Tregs directly reduce the activation of monocytes and macrophages through Fas/FasL interaction, resulting in decreased cytokine secretion and expression of stimulatory molecules, and induced monocyte apoptosis (49).
At present, antibodies are used as a rational approach to targeting tumor-infiltrating Tregs, and the involved mechanisms are to affect the immunosuppressive effect of Tregs in tumors by blocking or depleting Tregs. Table 1 summarizes some of the antibody-based immunotherapies that have been approved by Food and Drug Administration (FDA) or are currently under clinical investigation.
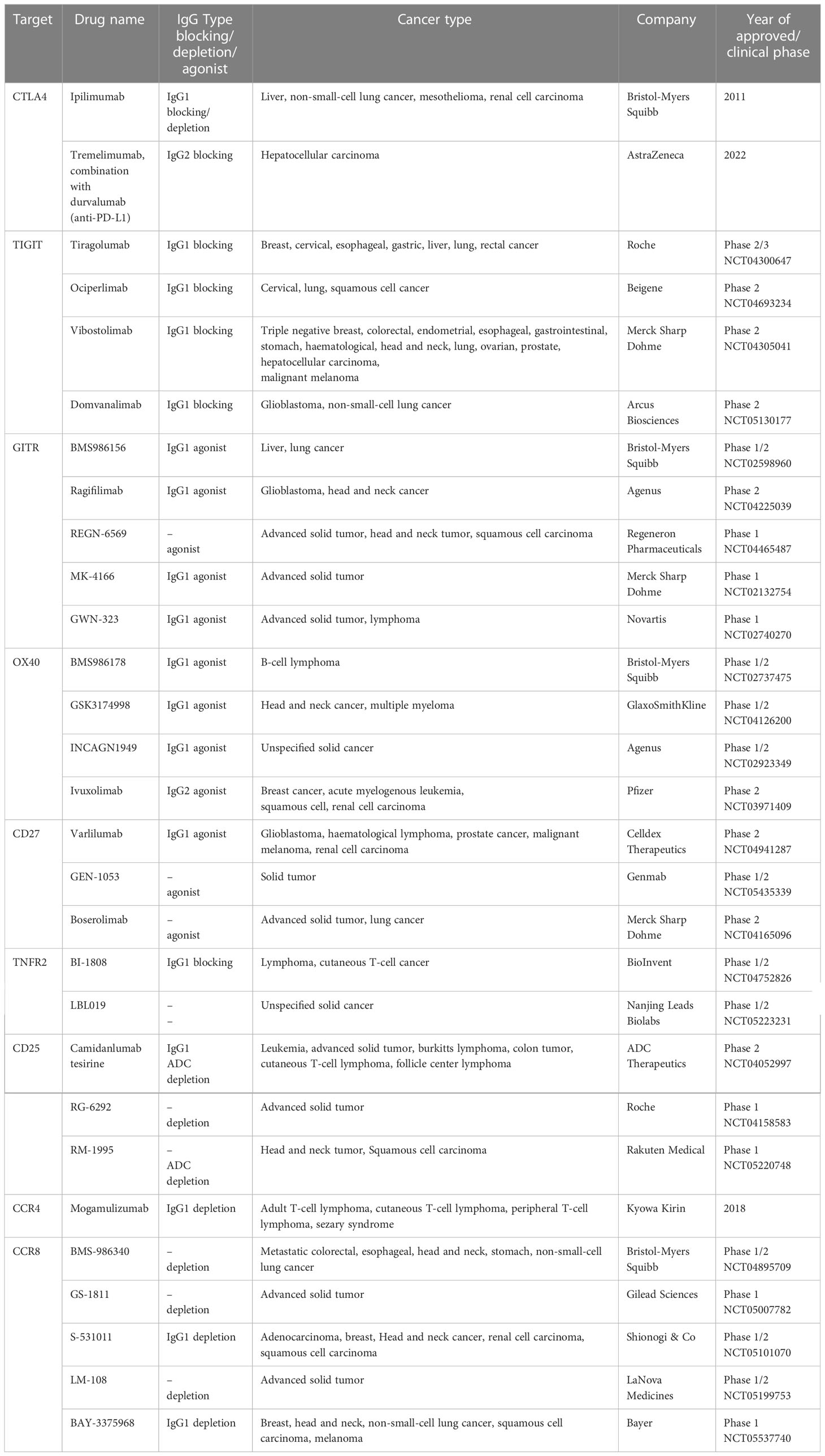
Table 1 Treg-targeting antibody drugs approved by FDA or in clinical investigation.
Since intratumoral Tregs upregulate ICIs, they are a key target for ICIs (50). The main targets of current immune checkpoint therapies include CTLA4, LAG3, TIM3, TIGIT, and IDO.
CTLA4
The anti-CTLA4 humanized IgG1 monoclonal antibody ipilimumab was approved by FDA in 2011 for the treatment of advanced melanoma, yet its therapeutic mechanism is still controversial. Previous studies have generally suggested that ipilimumab acts by inhibiting naïve T cell-intrinsic negative regulatory signaling from interacting with CD80/86-CTLA4, or blocking the immune checkpoint thereby activating effector cells. In human tumors, ipilimumab induces intratumoral CD4+ and CD8+ Teff infiltration without Treg depletion in TME (51). However, preclinical studies in mouse models revealed that the antitumor efficacy of ipilimumab was dependent on the depletion of CTLA4+ Tregs in tumors through antibody-dependent cellular cytotoxicity (ADCC) (52–54). CD16-V158F single-nucleotide polymorphisms was associated with higher response rates in inflamed or highly infiltrated tumors in patients with advanced melanoma. These results illustrated that at least in part, anti-CTLA-4 engage with FcγRs and depletion of Tregs (55).
Another anti-CTLA4 antibody tremelimumab, an IgG2 monoclonal antibody without ADCC activity, was approved in 2015 by FDA as an “orphan drug” for treating malignant mesothelioma. In 2022, tremelimumab was approved to treat unresectable hepatocellular carcinoma in combination with durvalumab, an anti-PD-L1 antibody (56).
TIGIT
Similar to other co-inhibitory molecules, TIGIT is highly expressed on Tregs (57). There are three ligands of TIGIT, CD155, CD112, and CD113, which are expressed on tumor cells and APCs. TIGIT competitively inhibits binding of CD226 to CD112 and CD155, leading to reduced T-cell activation and proliferation (58).
In vitro experiments identified FOXP3+ Tregs expressing TIGIT as a distinct Treg subset that specifically suppresses pro-inflammatory Th1 and Th17 cells compared to TIGIT− Tregs (59). Adoptive transfer experiments using TIGIT−/− Tregs exhibited tumor inhibition and enhanced pro-inflammatory cytokine production by CD8+ tumor-infiltrating lymphocytes (TILs) (60), which confirmed that TIGIT inhibits anti-tumor immunity predominantly by regulating Treg function.
In 2021, the anti-TIGIT antibody tiragolumab was approved by FDA as “breakthrough therapy designation” to treat patients with non-small-cell lung cancer in combination with anti-PD-L1 antibody atezolizumab. Currently, various TIGIT monoclonal antibodies are being evaluated in clinical trials as therapeutic agents for refractory solid tumors, either as single drugs or in combination with other agents.
In addition to co-inhibitory immune checkpoint molecules, tumor-infiltrating Tregs also express TNFRs on the surface, such as glucocorticoid-induced TNFR-related protein (GITR), OX40, 4-1BB, inducible co-stimulator (ICOS), TNFR superfamily member 1B (TNFR2), and CD27. Most of them are co-stimulatory receptors that play essential roles in many facets of immune response such as T-cell-antigen priming, expansion, survival, differentiation, and effector functions (59). Agonistic antibodies against these receptors have been demonstrated effective in inhibiting tumor progression in preclinical mouse model.
GITR
GITR is a co-stimulatory immune modulating receptor expressed on various immune cell subsets, with particularly high expression on Tregs (61, 62). In mature Tregs, FOXP3 promotes high-level GITR expression (63). GITR activation by its ligand GITRL in CD4+ Teffs and CTLs increases cell proliferation and effector function. GITR stimulation in Tregs leads to instability and decreased suppressive function and depletion of Tregs (64). Targeting GITR in Tregs using an agonistic antibody promotes Treg differentiation into Teffs, alleviates Treg-mediated suppression of antitumor immune response, and induces potent antitumor effector cells in glioblastoma (65). A pan-tumor study demonstrated GITR expression variability on tumor-infiltrating Tregs and lymphocytes across tumor types and suggested that patients with non-small-cell lung cancer, renal cell carcinoma, and melanoma may mostly benefit from anti-GITR therapies (66).
DTA-1, a rat IgG2a antibody targeting GITR, was validated for antitumor effect in multiple mouse tumor models including B16, CT26, and MC38. Analysis of TIL subsets after DTA-1 injection found that DTA-1 could reduce the proportion of Tregs in tumors and promote tumor regression. When the effect of ADCC was abolished by N297A mutation of Fc fragment, tumor regression was inhibited (67). This phenomenon indicated that the anti-GITR antibody plays an antitumor role by killing Teffs through Fc-mediated ADCC effect.
Currently, several anti-GITR antibodies are being investigated in early-phase clinical trials. One of them, BMS-986156, has shown to be well tolerated with no dose-limiting toxicities in Phase I/IIa alone or in combination with nivolumab to treat advanced malignant solid neoplasm (68). In addition, a humanized GITR monoclonal antibody MK-4166 was selected to bind to an epitope analogue of DTA-1, which reduced both Treg level and suppressive phenotype while enhancing effector responsiveness in preclinical research (69). However, recent studies on GITR agonists MK-4166, BMS-986156 and GWN323 showed that although they were well tolerated in patients with advanced solid tumors, objective responses were not observed with monotherapy but only in combination treatment with ICIs. The possible reason is that the proportion of Tregs in TME is low (<1% in patients with GWN323 treatment) (70), and only targeting Tregs cannot achieve a therapeutic effect.
OX40
OX40 is expressed on the TIL surface of various tumor tissues (71). Murine Tregs constitutively express OX40 and human Tregs upregulate OX40 upon activation (72, 73). OX40 signaling also impacts the generation of Tregs by strongly antagonizing TGF-β-driven FOXP3 mRNA and antigen mediated-conversion of naïve T-cells into FOXP3+ Tregs (72). OX40 signaling inhibits FOXP3 expression and Tregs induction through two distinct molecular pathways. OX40 can upregulate BATF3 to produce a closed chromatin configuration that is dependent on Sirt1/7. OX40 can also activate the AKT-mTOR pathway to mediate phosphorylation and nuclear exclusion of the transcription factor Foxo1 (74).
Currently, there are no anti-OX40 antibodies approved by FDA for clinical use, but several are under investigation, such as BMS986178, GSK3174998, INCAGN1949 and Ivuxolimab. BMS-986178 ± nivolumab and/or ipilimumab appeared to have a controllable safety feature, but no clear therapeutic effect was found (75). Moreover, INCAGN01949, a fully human IgG1κ anti-OX40 agonist monoclonal antibody, was designed to promote tumor immunity by Teff activation and Fcγ receptor-mediated Treg depletion. The preliminary efficacy showed that among 87 patients with colorectal, ovarian, and non-small-cell lung cancers, one patient with metastatic gallbladder cancer achieved a partial response, and 23 patients achieved stable disease. However, INCAGN01949 did not increase T-cell activation or decrease Tregs in the peripheral circulation, nor did it show any significant effect on T-cell subsets in the tumor biopsies (76). Therefore, the mechanism of this antibody targeting OX40 needs to be further investigated. Studies in mice have shown that complete Treg depletion was observed after treatment with an anti-OX40 containing a murine IgG2a Fc domain in the colon carcinoma tumor model CT26, whereas only partial depletion was observed with IgG1 Fc domain or Fc was point mutation that eliminates Fc binding activity. In this study, the ratio of CD8+ cells in the TME did not decrease. On the contrary, CD8+ T expanded significantly, possibly because the OX40 expression of effector T cells in the tumor microenvironment was much lower than Tregs (77).
CD27
CD27 is another member of the TNFR superfamily and a key receptor in T-cell costimulatory (78). By binding to CD70 on APC, CD27 can lead to Th1 and CTL activation and proliferation (79–81). However, the amount of CD27+ Tregs is higher than CD27+ conventional T cells in mice (82). CD27 expression on human Tregs from the peripheral blood and inflamed synovia has been shown with potent suppressive activity (83, 84). The signaling of CD27 stimulation is partially characterized with TNFR-associated factor (TRAF)-mediated and JNK- and NIK-dependent activation of the NFκB pathway (85). Depleting Tregs in a temporal through a mixed bone marrow chimeric mouse model showed that CD27+ Tregs prevent the breakdown of peripheral tolerance and limit antitumor immunity. Furthermore, inhibition of Treg-expressed CD27 acts synergistically with PD-1-checkpoint inhibition to improve CTL-mediated immunity against solid tumors (86), suggesting that CD27 is a useful marker in cancer immunotherapy.
Varlilumab is an IgG1-directed CD27 monoclonal antibody used against a variety of solid tumors. A Phase I study showed that the biologic activity of varlilumab is consistent with CD27-engaged chemokine secretion, T-cell activation, and Treg depletion at all dose levels (87). Two other monoclonal antibodies targeting CD27, GEN-1053 and boserolimab, are being evaluated in Phase I/II clinical trials.
TNFR2
Previous studies showed that TNF‐α/TNFR2 signaling pathway induced the activation and expansion of mouse and human Tregs (88, 89) and the subset of TNFR2+ Tregs were identified with maximal suppressive activity (90). TNFR2 plays a critical role in regulating FOXP3 expression and maintaining the phenotypic and functional stability of Tregs in an inflammatory environment (91). In lung cancer patients, 90% of Tregs in the peripheral blood were TNFR2-positive with highly expressed CTLA4, indicating a strong suppressive phenotype. Compared to TNFR2− Tregs, TNFR2+ Tregs significantly inhibited the proliferation of CTL and IFN‐γ secretion (92). Single‐cell RNA sequencing revealed high levels of TNFR2+ tumor-infiltrating Tregs in gastric cancer. In vitro studies revealed that TNFR2 increased the FOXP3 expression in Tregs and promoted TGF‐β production, which thus enhanced the immunosuppressive phenotype of Tregs (93). It was shown that human TNFR2-specific antagonistic antibodies can inhibit the proliferation of Tregs and promote the expansion of Teffs (94).
In a preclinical study, the anti-TNFR2 antibody BI-1808 demonstrated potent antitumor efficacy in CT26, MC38, and B16 cancer models as a single agent or in combination with an anti-PD-1 antibody. BI-1808 depleted early tumor-infiltrating Tregs and induced promotion of Teffs at the tumor site, which improved the ratio of CD8+ Teffs to Tregs and tumor regression (95). Currently, BI-1808 is being studied in Phase I/II clinical trials. Another antibody targeting TNFR2, LBL019, has also entered clinical studies for the treatment of an undisclosed solid tumor.
The cytokine receptors that are highly expressed on Tregs drive the migration ability of Tregs, which is a key factor to regulate tissue inflammation. One exception is CD25, which inhibits Teff activation by consumption of IL-2 in the environment. TCR-trafficking studies showed that a large proportion of tumor-infiltrating Tregs are derived from the peripheral circulation. Therefore, targeting cytokine receptors and impeding Treg migration into TME should be a primary consideration to induce a beneficial antitumor immune response in patients with solid cancer.
CD25
CD25, a high-affinity receptor alpha chain of IL-2Rα, was the first surface marker used to identify and isolate Tregs prior to the discovery of FOXP3 (46). It is also the most extensively studied target for depleting Tregs. Whereas CD25 is constitutively expressed at high levels on Tregs and absent on naïve Teffs, transient upregulation has been described upon activation of Teffs (96).
Using anti-CD25 antibodies with enhanced binding affinity to FcγRs led to effective depletion of tumor-infiltrating Tregs, which increased the ratio of Teffs to Tregs and enhanced the control of established tumors (97). RG6292, an anti-CD25 antibody, is optimized to deplete Tregs whilst preserving IL-2-STAT5 signaling on Teffs. In both non-human primates and humanized mouse models, efficient Treg depletion by RG6292 was observed with no overt immune-related toxicities (98). This antibody is in a Phase I clinical trial. These studies demonstrated CD25 as a therapeutic target and promising substrate in immune oncology.
As a proof of concept, camidanlumab tesirine, a DNA-damaging toxin that is based on pyrrolobenzodiazepine (PBD) dimer and targeting CD25, is the first antibody-drug conjugate (ADC) as immunotherapeutic agent to directly target immune cells rather than tumor cells. Camidanlumab tesirine is able to deplete Tregs and eradicate established tumors via antitumor immunity (99). In a Phase I clinical study, camidanlumab tesirine has been shown to be effective in treating Hodgkin’s lymphoma, with an overall response rate (ORR) of 86% and a complete response rate (CR) of 49% in patients with an acceptable safety (100). Another ADC, RM-1995, is composed of an antibody targeting CD25 and a photoactivating dye (IRDye® 700DX [IR700]) that could be activated by illumination with 690 nm nonthermal red light (101). The RM-1995 photoimmunotherapy was designed to specifically kill CD25- Tregs within solid tumors once activated with illumination.
CCR4
The CC chemokine receptor 4 (CCR4) is expressed on Tregs and other Th cells and binds to its two ligands CCL22 and CCL17. It is a key chemokine receptor in mediating Treg trafficking into TME (102–104) and has long been regarded as a potential therapeutic target for allergic diseases and tumor.
In addition, 80% and 73% of Tregs express cutaneous lymphocyte antigen (CLA) and CCR6, respectively, which is the phenotype of skin-homing T cells (105). Mice with CCR4-/- Tregs developed lymphocyte infiltration and severe inflammatory disease in the skin and lungs (106). These findings suggest that elimination of CCR4+ Tregs may severely disturb skin homeostasis.
Mogamulizumab is a fully humanized, defucosylated anti-CCR4 antibody that was approved by FDA in 2018 for the treatment of relapsed/refractory cutaneous T-cell lymphoma. Mogamulizumab has a high affinity for the N-terminus of CCR4. Once bound to CCR4, mogamulizumab exerts potent antitumor effects through ADCC, inducing cell-mediated lysis and depletion of CCR4-expressing malignant T cells and Tregs (107).The therapeutic efficacy of Mogamulizumab on adult T-cell leukemia and cutaneous T-cell lymphoma is also considered to be partly due to depletion of immunosuppressive Tregs (108, 109).
CCR8
CCR8 is a chemokine receptor that has recently been identified as a potential specific marker for tumor-infiltrating Tregs (110) and a core member of the interferon regulatory factor 4 (IRF4)-dependent “effector” Treg gene program (111). CCR8 expression marks highly active Tregs featuring the highest levels of inhibitory receptors. Researchers compared the RNA-seq data of Tregs in tumor tissues, normal tissues, and peripheral blood of breast cancer patients and found that Tregs at the tumor site had a high CCR8 expression (112).
CCR8-/- mice did not have altered numbers and proportions of Tregs in the tumor and spleen, nor did CCR8 deletion affect the growth of MC38-transplanted tumors. These results indicate that CCR8 does not influence the migration ability and function of Tregs in TME (113). Therefore, the mechanism of CCR8-targeting therapeutic antibodies for cancer may rely on Fc region-mediated ADCC and ADCP.
On another equally important note, 40% of peripheral blood CCR8+ Tregs are phenotypically similar to tumor-infiltrating CCR8+ Tregs and share many TCR clonotypes (114, 115), which suggests that tumor-specific Tregs recirculate between the tumor tissue and blood compartments, providing a rationale for targeting CCR8 on circulating Tregs.
In comparison to CCR4, T cells expressing CCR8 are much less common, being primarily restricted to skin T cells and tumor Tregs. Based on these considerations, it is possible that CCR8-directed antibodies may be better tolerated in patients and have acceptable safety profiles.
At present, there are 5 therapeutic antibodies targeting CCR8 in clinical studies. All of them eliminate CCR8+ Tregs through the action of ADCC or ADCP to exert antitumor effect.
Since Tregs lack specific targets, researchers are developing dual- or triple-targeted antibodies binding receptors that are highly expressed on tumor-infiltrating Tregs (116, 117). Compared to monoclonal antibodies, bispecific antibodies have two main advantages: Firstly, compared to monoclonal antibodies, bispecific antibodies have stronger specificity and targeting, which can reduce off-target toxicity and improve drug safety. Secondly, compared to combination therapy with monoclonal antibodies, dual- or triple-targeted antibodies can also effectively reduce the cost of drug development, clinical trials, and treatment. For example, PD-1 supports tumor immune evasion and increases FOXP3 expression in vitro (118, 119). However, since tumor-infiltrating CD8+ T and Tregs express PD-1 at equivalent high level, blocking this signaling pathway will not only activate CD8+ Teffs, but also enhance the immunosuppressive activity of Tregs (22). Therefore, anti-PD-1 antibody is usually used in combination with Treg-depletion antibody to increase the ratio of Teffs to Tregs and achieve better antitumor effect. Nowadays, there are several bispecific antibodies that simultaneously target PD-1 and a marker of Tregs, such as CTLA4 (Cadonilimab) (120), LAG3 (Tebotelimab and RG-6139), TIM3 (RG-7769), TIGIT (AZD2936), and CD25 (IBI-363) (Table 2).
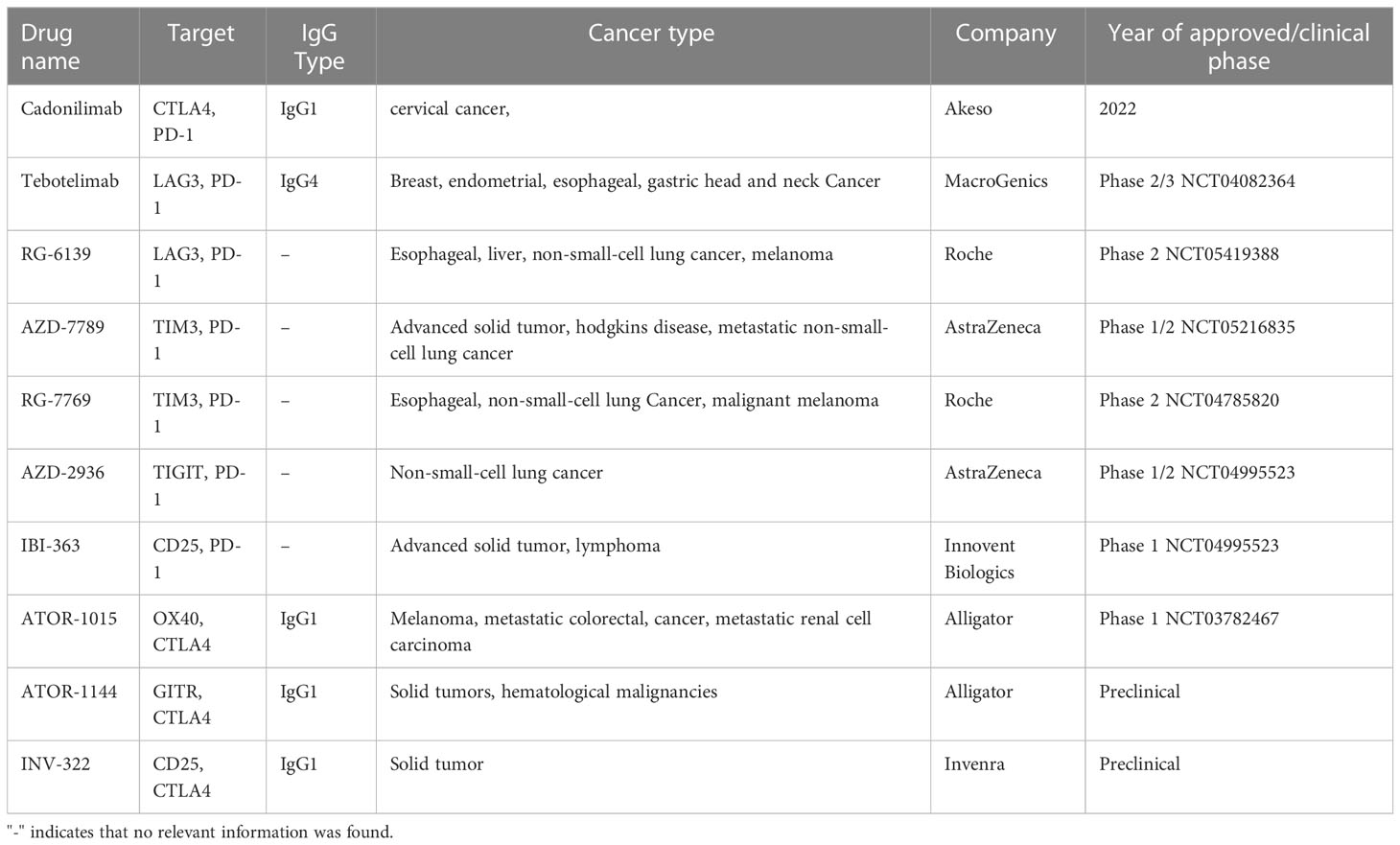
Table 2 Next-generation bispecific antibodies targeting Tregs in tumor immunotherapy.
In addition to PD-1, CTLA4 is also a popular target of bispecific antibodies. ATOR-1015 is a human CTLA4- and OX40-targeting IgG1 bispecific antibody generated by linking a natural CTLA4 ligand, the Ig-like V-type domain of human CD86, to an agonistic OX40 antibody. ATOR-1015 treatment reduced the frequency of Tregs and induced the activation of CD8+ T cells, which reduced tumor growth and improved survival in bladder, colon and pancreas cancer models (121). Furthermore, a novel GITR×CTLA4 bispecific antibody ATOR-1144 has shown to deplete Tregs in vitro through ADCC mechanisms (122). Moreover, the bispecific antibody INV322, engineered using Invenra’s fully human B-Body® platform, co-targets CTLA4 and CD25. In vitro studies have characterized avidity-mediated binding of INV322 to these two tumor Treg antigens, as well as ADCC-mediated mode of action. In vivo studies using a murine surrogate also showed potent antitumor responses after a single dose and selective Treg depletion in syngeneic mice (123). These preliminary data demonstrate the potential of bispecific antibodies against multiple targets expressed on Tregs.
Tregs correlate with poor prognosis in solid cancers, such as non-small-cell lung cancer, ovarian cancer, gastric cancer, and melanoma (110), which signifies the importance of targeting Tregs in cancer therapy. Tregs play several immunosuppressive roles in the tumor microenvironment, not only inhibiting the function and activation of T cells, APC and NK, but also secreting some immunosuppressive factors such as IL-35, IL-10 and TGF-β. Otherwise, some researchers support the longstanding hypothesis that Tregs play an important immunosuppressive role in the failure of immune checkpoint inhibitor-based immunotherapy in clinical trials, especially for many patients who have no response after anti-PD-1 treatment. Part of the reason is that anti-PD-1 can not only activate T cells, but also activate Tregs, making Tregs have stronger immunosuppressive activity (26).Therefore, regulating the proportion of Tregs or their signaling pathways in TME to reverse the immunosuppressive microenvironment may have a positive effect on the treatment by immune checkpoint inhibitors. Treg is targeted first and then combined with T cell engagers, better therapeutic effects may be achieved.
Nearly a century has passed since Tregs were discovered to play a role in self-tolerance and homeostasis distinct from conventional T cells. So far, the immunosuppressive effects of Tregs in cancer have been well investigated, with several studies affirming the difference between peripheral blood and tumor-infiltrating Tregs. But there are still many issues in targeting Tregs to treat cancer and autoimmunity. One main open question is how to control Tregs to augment tumor immunity without influencing autoimmunity, or to suppress the latter without inhibiting the former. A better understanding of the heterogeneity and function of Tregs in autoimmunity and cancer may help to find solutions. Many studies have concluded that there are barely specific targets on Tregs, and most of the targets of tumor-infiltrating Tregs are expressed on Teffs or NK cells in TME, especially for immune checkpoint molecules and co-stimulatory molecules. Targeting these molecules has the potential to equally damage effector cells. For example, mogamulizumab administered to patients in clinical studies may not differentiate between targeting effector Tregs and central memory CD8+ T cells, especially at high doses (124) However, currently, most antibodies used to eliminate Tregs increase rather than decrease the proportion of CD8+ T cells while eliminating Tregs, such as RG6292 (98). The reason may because that the expression level of these targets on effector T cells is much lower than tumor infiltrating Tregs. However, this cannot rule out that some Teffs are cleared by antibodies (98). Many researches have been evaluated to address this issue. Some novel immunotherapies target two or more markers on tumor-infiltrating Tregs, such as CTLA4/OX40 or CTLA4/PD-1, which improves specificity and reduces off-target effects, or adjusting the affinity of the antibodies so that they bind weakly to low-density targets and strongly to high-density targets, another method is to adjust the dosage to avoid excessive killing of Teffs. In addition, some ADC-based therapies directly kill Tregs through toxin to fix the insufficient ADCC effect caused by the exhaustion of Teffs in TME.
Another important question in tumor immunity is how to eliminate tumor-infiltrating Tregs without affecting Tregs in the peripheral blood. Compared to Tregs in the peripheral blood, intratumoral Tregs exhibit a more proliferative and immunosuppressive phenotype and are characterized by elevated expression of CTLA4, CD25, GITR, OX40, LAG3, TIM3, TIGIT, CCR4, CCR8, and PD-1. The affinity of antibodies to these targets should be carefully considered when screening antibodies. Antibodies with high affinity may cause disorder of peripheral Tregs, while antibodies with low affinity may not achieve the desired immunotherapy outcome and cause off-target effects. For instance, RG-6139 is a bispecific antibody that targets both LAG3 and PD-1. It has a higher affinity for PD-1 than for LAG3, which allows the bispecific antibody to target activated tumor-infiltrating T cells while effectively targeting Tregs.
The biological function of Tregs in TME is complex, and there are still many issues to be solved in the future to develop new treatment and immune-precision drugs for each cancer patient. Whether the next generation of drugs targeting Tregs can further improve patient outcomes while maintaining tolerance and particularly low risk of autoimmunity will be of paramount importance. It is hoped to make cancer immunotherapy more effective with fewer side effects in the future.
QL, YW and TY conceived and wrote the paper. JLu, J.Li and BZ revised and edited the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Key R&D Program of China (2019YFA0904400), National Natural Science Foundation of China (32270984), Shanghai Municipal Health Commission (GWV-10.2-XD01, GWV-10.2-YQ06), New Zealand Ministry of Education, New Zealand-China Tripartite Partnership Fund, and Royal Society of New Zealand Catalyst Seeding Fund (21-AUT-005-CSG).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Holm TL, Nielsen J, Claesson MH. Cd4+Cd25+ regulatory T cells: I. Phenotype Physiol APMIS (2004) 112(10):629–41. doi: 10.1111/j.1600-0463.2004.apm1121001.x
2. Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J (2012) 26(6):2253–76. doi: 10.1096/fj.11-193672
3. Iglesias-Escudero M, Arias-Gonzalez N, Martinez-Caceres E. Regulatory cells and the effect of cancer immunotherapy. Mol Cancer (2023) 22(1):26. doi: 10.1186/s12943-023-01714-0
4. Dikiy S, Rudensky AY. Principles of regulatory T cell function. Immunity (2023) 56(2):240–55. doi: 10.1016/j.immuni.2023.01.004
5. Nishikawa H, Koyama S. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer (2021) 9(7):e002591. doi: 10.1136/jitc-2021-002591
6. Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol (2022) 15(1):104. doi: 10.1186/s13045-022-01322-3
7. Shan F, Somasundaram A, Bruno TC, Workman CJ, Vignali DAA. Therapeutic targeting of regulatory T cells in cancer. Trends Cancer (2022) 8(11):944–61. doi: 10.1016/j.trecan.2022.06.008
8. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell (2018) 174(5):1293–308.e36. doi: 10.1016/j.cell.2018.05.060
9. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol (2017) 18(2):173–83. doi: 10.1038/ni.3646
10. Togashi Y, Nishikawa H. Regulatory T cells: molecular and cellular basis for immunoregulation. Curr Top Microbiol Immunol (2017) 410:3–27. doi: 10.1007/82_2017_58
11. Shevach EM, Thornton AM. Ttregs, ptregs, and itregs: similarities and differences. Immunol Rev (2014) 259(1):88–102. doi: 10.1111/imr.12160
12. Smigiel KS, Srivastava S, Stolley JM, Campbell DJ. Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol Rev (2014) 259(1):40–59. doi: 10.1111/imr.12170
13. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. Ccr7 provides localized access to il-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med (2014) 211(1):121–36. doi: 10.1084/jem.20131142
14. Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol (2014) 14(3):154–65. doi: 10.1038/nri3605
15. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity (2019) 50(2):493–504.e7. doi: 10.1016/j.immuni.2019.01.001
16. Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
17. Huehn J, Hamann A. Homing to suppress: address codes for treg migration. Trends Immunol (2005) 26(12):632–6. doi: 10.1016/j.it.2005.10.001
18. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human Cd4+ T cells expressing the Foxp3 transcription factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
19. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol (2019) 16(6):356–71. doi: 10.1038/s41571-019-0175-7
20. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
21. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of Cd80 and Cd86: a molecular basis for the cell-extrinsic function of ctla-4. Science (2011) 332(6029):600–3. doi: 10.1126/science.1202947
22. Cai J, Wang D, Zhang G, Guo X. The role of pd-1/Pd-L1 axis in treg development and function: implications for cancer immunotherapy. Onco Targets Ther (2019) 12:8437–45. doi: 10.2147/OTT.S221340
23. Francisco LM, Sage PT, Sharpe AH. The pd-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
24. Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, et al. Pd-1 and pd-L1 upregulation promotes Cd8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer (2011) 128(4):887–96. doi: 10.1002/ijc.25397
25. Raimondi G, Shufesky WJ, Tokita D, Morelli AE, Thomson AW. Regulated compartmentalization of programmed cell death-1 discriminates Cd4+Cd25+ resting regulatory T cells from activated T cells. J Immunol (2006) 176(5):2808–16. doi: 10.4049/jimmunol.176.5.2808
26. Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The pd-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of pd-1 blockade therapies. Nat Immunol (2020) 21(11):1346–58. doi: 10.1038/s41590-020-0769-3
27. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes pd-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell (2022) 40(2):201–18.e9. doi: 10.1016/j.ccell.2022.01.001
28. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of il-10(+) and il-35(+) treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol (2019) 20(6):724–35. doi: 10.1038/s41590-019-0346-9
29. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, Li Z. Transforming growth factor-Beta1 in regulatory T cell biology. Sci Immunol (2022) 7(69):eabi4613. doi: 10.1126/sciimmunol.abi4613
30. Wan YY, Flavell RA. Tgf-beta and regulatory T cell in immunity and autoimmunity. J Clin Immunol (2008) 28(6):647–59. doi: 10.1007/s10875-008-9251-y
31. Okeke EB, Uzonna JE. The pivotal role of regulatory T cells in the regulation of innate immune cells. Front Immunol (2019) 10:680. doi: 10.3389/fimmu.2019.00680
32. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. Il-35-Mediated induction of a potent regulatory T cell population. Nat Immunol (2010) 11(12):1093–101. doi: 10.1038/ni.1952
33. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 limits anti-tumor immunity. Immunity (2016) 44(2):316–29. doi: 10.1016/j.immuni.2016.01.013
34. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21(4):589–601. doi: 10.1016/j.immuni.2004.09.002
35. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol (2020) 20(3):158–72. doi: 10.1038/s41577-019-0232-6
36. Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by Cd4+Cd25+ T cells. Blood (2007) 109(5):2058–65. doi: 10.1182/blood-2006-04-016451
37. Janssens W, Carlier V, Wu B, VanderElst L, Jacquemin MG, Saint-Remy JM. Cd4+Cd25+ T cells lyse antigen-presenting b cells by fas-fas ligand interaction in an epitope-specific manner. J Immunol (2003) 171(9):4604–12. doi: 10.4049/jimmunol.171.9.4604
38. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by Cd39 and Cd73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
39. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi: 10.1038/nm934
40. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase Cd39 by Foxp3+ treg cells: hydrolysis of extracellular atp and immune suppression. Blood (2007) 110(4):1225–32. doi: 10.1182/blood-2006-12-064527
41. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and pd-L1-Blockade resistance in tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
42. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4(12):1206–12. doi: 10.1038/ni1003
43. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. Ctla-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol (2002) 3(11):1097–101. doi: 10.1038/ni846
44. Kurniawan H, Soriano-Baguet L, Brenner D. Regulatory T cell metabolism at the intersection between autoimmune diseases and cancer. Eur J Immunol (2020) 50(11):1626–42. doi: 10.1002/eji.201948470
45. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. Ido inhibits a tryptophan sufficiency signal that stimulates mtor: a novel ido effector pathway targeted by d-1-Methyl-Tryptophan. Oncoimmunology (2012) 1(9):1460–8. doi: 10.4161/onci.21716
46. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing il-2 receptor alpha-chains (Cd25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64. doi: 10.1177/1753425910361989
47. Tanaka A, Sakaguchi S. Targeting treg cells in cancer immunotherapy. Eur J Immunol (2019) 49(8):1140–6. doi: 10.1002/eji.201847659
48. Trzonkowski P, Szmit E, Mysliwska J, Dobyszuk A, Mysliwski A. Cd4+Cd25+ T regulatory cells inhibit cytotoxic activity of T Cd8+ and nk lymphocytes in the direct cell-to-Cell interaction. Clin Immunol (2004) 112(3):258–67. doi: 10.1016/j.clim.2004.04.003
49. Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Human Cd4+Cd25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol (2006) 177(9):6540–7. doi: 10.4049/jimmunol.177.9.6540
50. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19(1):116. doi: 10.1186/s12943-020-01234-1
51. Sharma A, Subudhi SK, Blando J, Vence L, Wargo J, Allison JP, et al. Anti-Ctla-4 immunotherapy does not deplete Foxp3(+) regulatory T cells (Tregs) in human cancers-response. Clin Cancer Res (2019) 25(11):3469–70. doi: 10.1158/1078-0432.CCR-19-0402
52. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells Co-defines the efficacy of anti-Ctla-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi: 10.1084/jem.20130579
53. Liu Y, Zheng P. How does an anti-Ctla-4 antibody promote cancer immunity? Trends Immunol (2018) 39(12):953–6. doi: 10.1016/j.it.2018.10.009
54. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med (2013) 210(9):1685–93. doi: 10.1084/jem.20130573
55. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-Ctla-4 antibodies. Cancer Cell (2018) 33(4):649–63.e4. doi: 10.1016/j.ccell.2018.02.010
56. Kudo M. Durvalumab plus tremelimumab in unresectable hepatocellular carcinoma. Hepatobil Surg Nutr (2022) 11(4):592–6. doi: 10.21037/hbsn-22-143
57. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein tigit suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
58. Harjunpaa H, Guillerey C. Tigit as an emerging immune checkpoint. Clin Exp Immunol (2020) 200(2):108–19. doi: 10.1111/cei.13407
59. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule tigit selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
60. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. Tigit predominantly regulates the immune response Via regulatory T cells. J Clin Invest (2015) 125(11):4053–62. doi: 10.1172/JCI81187
61. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of Cd25(+)Cd4(+) regulatory T cells through gitr breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi: 10.1038/ni759
62. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. Cd4(+)Cd25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced tnf receptor. Immunity (2002) 16(2):311–23. doi: 10.1016/s1074-7613(02)00280-7
63. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in Cd4+Cd25+ T regulatory cells. Nat Immunol (2003) 4(4):337–42. doi: 10.1038/ni909
64. Coe D, Begom S, Addey C, White M, Dyson J, Chai JG. Depletion of regulatory T cells by anti-gitr mab as a novel mechanism for cancer immunotherapy. Cancer Immunol Immunother (2010) 59(9):1367–77. doi: 10.1007/s00262-010-0866-5
65. Amoozgar Z, Kloepper J, Ren J, Tay RE, Kazer SW, Kiner E, et al. Targeting treg cells with gitr activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun (2021) 12(1):2582. doi: 10.1038/s41467-021-22885-8
66. Vence L, Bucktrout SL, Fernandez Curbelo I, Blando J, Smith BM, Mahne AE, et al. Characterization and comparison of gitr expression in solid tumors. Clin Cancer Res (2019) 25(21):6501–10. doi: 10.1158/1078-0432.CCR-19-0289
67. Knee DA, Hewes B, Brogdon JL. Rationale for anti-gitr cancer immunotherapy. Eur J Cancer (2016) 67:1–10. doi: 10.1016/j.ejca.2016.06.028
68. Heinhuis KM, Carlino M, Joerger M, Di Nicola M, Meniawy T, Rottey S, et al. Safety, tolerability, and potential clinical activity of a glucocorticoid-induced tnf receptor-related protein agonist alone or in combination with nivolumab for patients with advanced solid tumors: a phase 1/2a dose-escalation and cohort-expansion clinical trial. JAMA Oncol (2020) 6(1):100–7. doi: 10.1001/jamaoncol.2019.3848
69. Papadopoulos KP, Autio K, Golan T, Dobrenkov K, Chartash E, Chen Q, et al. Phase I study of mk-4166, an anti-human glucocorticoid-induced tnf receptor antibody, alone or with pembrolizumab in advanced solid tumors. Clin Cancer Res (2021) 27(7):1904–11. doi: 10.1158/1078-0432.CCR-20-2886
70. Piha-Paul SA, Geva R, Tan TJ, Lim DW, Hierro C, Doi T, et al. First-in-Human phase I/Ib open-label dose-escalation study of Gwn323 (Anti-gitr) as a single agent and in combination with spartalizumab (Anti-Pd-1) in patients with advanced solid tumors and lymphomas. J Immunother Cancer (2021) 9(8):e002863. doi: 10.1136/jitc-2021-002863
71. Willoughby J, Griffiths J, Tews I, Cragg MS. Ox40: structure and function - what questions remain? Mol Immunol (2017) 83:13–22. doi: 10.1016/j.molimm.2017.01.006
72. So T, Croft M. Cutting edge: Ox40 inhibits tgf-beta- and antigen-driven conversion of naive Cd4 T cells into Cd25+Foxp3+ T cells. J Immunol (2007) 179(3):1427–30. doi: 10.4049/jimmunol.179.3.1427
73. Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, et al. Ox40 costimulation turns off Foxp3+ tregs. Blood (2007) 110(7):2501–10. doi: 10.1182/blood-2007-01-070748
74. Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, et al. Ox40 costimulation inhibits Foxp3 expression and treg induction Via Batf3-dependent and independent mechanisms. Cell Rep (2018) 24(3):607–18. doi: 10.1016/j.celrep.2018.06.052
75. Gutierrez M, Moreno V, Heinhuis KM, Olszanski AJ, Spreafico A, Ong M, et al. Ox40 agonist bms-986178 alone or in combination with nivolumab and/or ipilimumab in patients with advanced solid tumors. Clin Cancer Res (2021) 27(2):460–72. doi: 10.1158/1078-0432.CCR-20-1830
76. Davis EJ, Martin-Liberal J, Kristeleit R, Cho DC, Blagden SP, Berthold D, et al. First-in-Human phase I/Ii, open-label study of the anti-Ox40 agonist Incagn01949 in patients with advanced solid tumors. J Immunother Cancer (2022) 10(10):e004235. doi: 10.1136/jitc-2021-004235
77. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. Ox40 engagement depletes intratumoral tregs Via activating fcgammars, leading to antitumor efficacy. Immunol Cell Biol (2014) 92(6):475–80. doi: 10.1038/icb.2014.26
78. Denoeud J, Moser M. Role of Cd27/Cd70 pathway of activation in immunity and tolerance. J Leukoc Biol (2011) 89(2):195–203. doi: 10.1189/jlb.0610351
79. Borst J, Hendriks J, Xiao Y. Cd27 and Cd70 in T cell and b cell activation. Curr Opin Immunol (2005) 17(3):275–81. doi: 10.1016/j.coi.2005.04.004
80. van Oosterwijk MF, Juwana H, Arens R, Tesselaar K, van Oers MH, Eldering E, et al. Cd27-Cd70 interactions sensitise naive Cd4+ T cells for il-12-Induced Th1 cell development. Int Immunol (2007) 19(6):713–8. doi: 10.1093/intimm/dxm033
81. Schildknecht A, Miescher I, Yagita H, van den Broek M. Priming of Cd8+ T cell responses by pathogens typically depends on Cd70-mediated interactions with dendritic cells. Eur J Immunol (2007) 37(3):716–28. doi: 10.1002/eji.200636824
82. Dhainaut M, Coquerelle C, Uzureau S, Denoeud J, Acolty V, Oldenhove G, et al. Thymus-derived regulatory T cells restrain pro-inflammatory Th1 responses by downregulating Cd70 on dendritic cells. EMBO J (2015) 34(10):1336–48. doi: 10.15252/embj.201490312
83. Duggleby RC, Shaw TN, Jarvis LB, Kaur G, Gaston JS. Cd27 expression discriminates between regulatory and non-regulatory cells after expansion of human peripheral blood Cd4+ Cd25+ cells. Immunology (2007) 121(1):129–39. doi: 10.1111/j.1365-2567.2006.02550.x
84. Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, et al. Coexpression of Cd25 and Cd27 identifies Foxp3+ regulatory T cells in inflamed synovia. J Exp Med (2005) 201(11):1793–803. doi: 10.1084/jem.20050085
85. Bullock TN. Stimulating Cd27 to quantitatively and qualitatively shape adaptive immunity to cancer. Curr Opin Immunol (2017) 45:82–8. doi: 10.1016/j.coi.2017.02.001
86. Muth S, Klaric A, Radsak M, Schild H, Probst HC. Cd27 expression on treg cells limits immune responses against tumors. J Mol Med (Berl) (2022) 100(3):439–49. doi: 10.1007/s00109-021-02116-9
87. Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and activity of varlilumab, a novel and first-in-Class agonist anti-Cd27 antibody, in patients with advanced solid tumors. J Clin Oncol (2017) 35(18):2028–36. doi: 10.1200/JCO.2016.70.1508
88. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of tnf with tnf receptor type 2 promotes expansion and function of mouse Cd4+Cd25+ T regulatory cells. J Immunol (2007) 179(1):154–61. doi: 10.4049/jimmunol.179.1.154
89. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous expansion of human T-regulatory cells Via tumor necrosis factor receptor 2. Sci Rep (2013) 3:3153. doi: 10.1038/srep03153
90. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of Tnfr2 defines a maximally suppressive subset of mouse Cd4+Cd25+Foxp3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol (2008) 180(10):6467–71. doi: 10.4049/jimmunol.180.10.6467
91. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. Tnfr2 is critical for the stabilization of the Cd4+Foxp3+ regulatory T. Cell Phenotype Inflammatory Environ J Immunol (2013) 190(3):1076–84. doi: 10.4049/jimmunol.1202659
92. Yan F, Du R, Wei F, Zhao H, Yu J, Wang C, et al. Expression of Tnfr2 by regulatory T cells in peripheral blood is correlated with clinical pathology of lung cancer patients. Cancer Immunol Immunother (2015) 64(11):1475–85. doi: 10.1007/s00262-015-1751-z
93. Qu Y, Wang X, Bai S, Niu L, Zhao G, Yao Y, et al. The effects of tnf-Alpha/Tnfr2 in regulatory T cells on the microenvironment and progression of gastric cancer. Int J Cancer (2022) 150(8):1373–91. doi: 10.1002/ijc.33873
94. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting Tnfr2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated tregs. Sci Signal (2017) 10(462):eaaf8608. doi: 10.1126/scisignal.aaf8608
95. Linda Mårtensson KC, Semmrich M, Kovacek M, Holmkvist P, Svensson C, Demiri M, et al. Targeting Tnfr2 for cancer immunotherapy: ligand blocking depletors versus receptor agonists. Proceedings of the AACR Annual Meeting 2020; April 27-28, 2020 and June 22-24, 2020; Philadelphia, PA (2020).
96. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12(3):180–90. doi: 10.1038/nri3156
97. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-Cd25 depletes tumor-infiltrating regulatory T cells and synergizes with pd-1 blockade to eradicate established tumors. Immunity (2017) 46(4):577–86. doi: 10.1016/j.immuni.2017.03.013
98. Solomon I, Amann M, Goubier A, Arce Vargas F, Zervas D, Qing C, et al. Cd25-Treg-Depleting antibodies preserving il-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat Cancer (2020) 1(12):1153–66. doi: 10.1038/s43018-020-00133-0
99. Zammarchi F, Havenith K, Bertelli F, Vijayakrishnan B, Chivers S, Van Berkel PH. Cd25-targeted antibody–drug conjugate depletes regulatory T cells and eliminates established syngeneic tumors Via antitumor immunity. J ImmunoTher Cancer (2020) 8(2):e000860. doi: 10.1136/jitc-2020-000860
100. Goldberg AD, Atallah E, Rizzieri D, Walter RB, Chung KY, Spira A, et al. Camidanlumab tesirine, an antibody-drug conjugate, in Relapsed/Refractory Cd25-positive acute myeloid leukemia or acute lymphoblastic leukemia: a phase I study. Leuk Res (2020) 95:106385. doi: 10.1016/j.leukres.2020.106385
101. Tahara M, Okano S, Enokida T, Ueda Y, Fujisawa T, Shinozaki T, et al. Single-center, open-label study of rm-1929 photoimmunotherapy in Japanese patients with recurrent head and neck squamous cell carcinoma. Int J Clin Oncol (2021) 26(10):1812–21. doi: 10.1007/s10147-021-01960-6
102. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi: 10.1038/nm1093
103. Imai T, Baba M, Nishimura M, Kakizaki M, Takagi S, Yoshie O. The T cell-directed cc chemokine tarc is a highly specific biological ligand for cc chemokine receptor 4. J Biol Chem (1997) 272(23):15036–42. doi: 10.1074/jbc.272.23.15036
104. Imai T, Chantry D, Raport CJ, Wood CL, Nishimura M, Godiska R, et al. Macrophage-derived chemokine is a functional ligand for the cc chemokine receptor 4. J Biol Chem (1998) 273(3):1764–8. doi: 10.1074/jbc.273.3.1764
105. Hirahara K, Liu L, Clark RA, Yamanaka K, Fuhlbrigge RC, Kupper TS. The majority of human peripheral blood Cd4+Cd25highfoxp3+ regulatory T cells bear functional skin-homing receptors. J Immunol (2006) 177(7):4488–94. doi: 10.4049/jimmunol.177.7.4488
106. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med (2007) 204(6):1335–47. doi: 10.1084/jem.20070081
107. Duvic M, Pinter-Brown LC, Foss FM, Sokol L, Jorgensen JL, Challagundla P, et al. Phase 1/2 study of mogamulizumab, a defucosylated anti-Ccr4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood (2015) 125(12):1883–9. doi: 10.1182/blood-2014-09-600924
108. Ishida T, Ito A, Sato F, Kusumoto S, Iida S, Inagaki H, et al. Stevens-Johnson syndrome associated with mogamulizumab treatment of adult T-cell leukemia / lymphoma. Cancer Sci (2013) 104(5):647–50. doi: 10.1111/cas.12116
109. Issa F, Hester J, Milward K, Wood KJ. Homing of regulatory T cells to human skin is important for the prevention of alloimmune-mediated pathology in an in vivo cellular therapy model. PloS One (2012) 7(12):e53331. doi: 10.1371/journal.pone.0053331
110. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity (2016) 45(5):1135–47. doi: 10.1016/j.immuni.2016.10.021
111. Alvisi G, Brummelman J, Puccio S, Mazza EM, Tomada EP, Losurdo A, et al. Irf4 instructs effector treg differentiation and immune suppression in human cancer. J Clin Invest (2020) 130(6):3137–50. doi: 10.1172/JCI130426
112. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity (2016) 45(5):1122–34. doi: 10.1016/j.immuni.2016.10.032
113. Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, et al. Ccr8 marks highly suppressive treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology (2021) 163(4):512–20. doi: 10.1111/imm.13337
114. Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanovic S, Robbins PF, et al. Tumor-infiltrating human Cd4(+) regulatory T cells display a distinct tcr repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol (2019) 4(31):eaao4310. doi: 10.1126/sciimmunol.aao4310
115. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell (2017) 169(7):1342–56.e16. doi: 10.1016/j.cell.2017.05.035
116. Deshaies RJ. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature (2020) 580(7803):329–38. doi: 10.1038/s41586-020-2168-1
117. Runcie K, Budman DR, John V, Seetharamu N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol Med (2018) 24(1):50. doi: 10.1186/s10020-018-0051-4
118. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. Pd-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
119. Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The Pdl1-Pd1 axis converts human Th1 cells into regulatory T cells. Sci Transl Med (2011) 3(111):111ra20. doi: 10.1126/scitranslmed.3003130
120. Keam SJ. Cadonilimab: first approval. Drugs (2022) 82(12):1333–9. doi: 10.1007/s40265-022-01761-9
121. Kvarnhammar AM, Veitonmaki N, Hagerbrand K, Dahlman A, Smith KE, Fritzell S, et al. The ctla-4 X Ox40 bispecific antibody ator-1015 induces anti-tumor effects through tumor-directed immune activation. J Immunother Cancer (2019) 7(1):103. doi: 10.1186/s40425-019-0570-8
122. Millan-Perez Pena L, Martin PS, Herrera-Camacho I, Bandala C, Anaya-Ruiz M. Colon carcinoma treatment using bispecific anti-Gitr/Ctla-4 antibodies: a patent evaluation of Wo2018091739. Expert Opin Ther Pat (2020) 30(5):307–11. doi: 10.1080/13543776.2020.1732352
123. Invenra to present preclinical data on Inv322, a next generation tumor treg depleter (2022). Available at: https://www.businesswire.com/news/home/20220920005953/en.
Keywords: antibody, cancer, immunotherapy, regulatory (Treg) cell, immune checkpoint inhibitors
Citation: Li Q, Lu J, Li J, Zhang B, Wu Y and Ying T (2023) Antibody-based cancer immunotherapy by targeting regulatory T cells. Front. Oncol. 13:1157345. doi: 10.3389/fonc.2023.1157345
Received: 02 February 2023; Accepted: 13 April 2023;
Published: 27 April 2023.
Edited by:
Massimo Fantini, Precision Biologics, Inc., United StatesReviewed by:
Shohei Koyama, Osaka University, JapanCopyright © 2023 Li, Lu, Li, Zhang, Wu and Ying. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tianlei Ying, dGx5aW5nQGZ1ZGFuLmVkdS5jbg==; Yanling Wu, eWFubGluZ3d1QGZ1ZGFuLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.