
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 14 June 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.949407
This article is part of the Research TopicGlobal Excellence in Inflammatory Diseases: Latin America 2021View all 6 articles
 Telma Saraiva-Santos1,2
Telma Saraiva-Santos1,2 Tiago H. Zaninelli1,2,3
Tiago H. Zaninelli1,2,3 Marília F. Manchope1
Marília F. Manchope1 Ketlem C. Andrade1
Ketlem C. Andrade1 Camila R. Ferraz1
Camila R. Ferraz1 Mariana M. Bertozzi1Nayara A. Artero1
Mariana M. Bertozzi1Nayara A. Artero1 Anelise Franciosi1
Anelise Franciosi1 Stephanie Badaro-Garcia1
Stephanie Badaro-Garcia1 Larissa Staurengo-Ferrari1
Larissa Staurengo-Ferrari1 Sergio M. Borghi1,4
Sergio M. Borghi1,4 Graziela S. Ceravolo5Avacir Casanova Andrello6Janaína Menezes Zanoveli7Michael S. Rogers3
Graziela S. Ceravolo5Avacir Casanova Andrello6Janaína Menezes Zanoveli7Michael S. Rogers3 Rubia Casagrande8
Rubia Casagrande8 Felipe A. Pinho-Ribeiro2*
Felipe A. Pinho-Ribeiro2* Waldiceu A. Verri Jr1*
Waldiceu A. Verri Jr1*Background: Lipoxin A4 (LXA4) has anti-inflammatory and pro-resolutive roles in inflammation. We evaluated the effects and mechanisms of action of LXA4 in titanium dioxide (TiO2) arthritis, a model of prosthesis-induced joint inflammation and pain.
Methods: Mice were stimulated with TiO2 (3mg) in the knee joint followed by LXA4 (0.1, 1, or 10ng/animal) or vehicle (ethanol 3.2% in saline) administration. Pain-like behavior, inflammation, and dosages were performed to assess the effects of LXA4 in vivo.
Results: LXA4 reduced mechanical and thermal hyperalgesia, histopathological damage, edema, and recruitment of leukocytes without liver, kidney, or stomach toxicity. LXA4 reduced leukocyte migration and modulated cytokine production. These effects were explained by reduced nuclear factor kappa B (NFκB) activation in recruited macrophages. LXA4 improved antioxidant parameters [reduced glutathione (GSH) and 2,2-azino-bis 3-ethylbenzothiazoline-6-sulfonate (ABTS) levels, nuclear factor erythroid 2-related factor 2 (Nrf2) mRNA and Nrf2 protein expression], reducing reactive oxygen species (ROS) fluorescent detection induced by TiO2 in synovial fluid leukocytes. We observed an increase of lipoxin receptor (ALX/FPR2) in transient receptor potential cation channel subfamily V member 1 (TRPV1)+ DRG nociceptive neurons upon TiO2 inflammation. LXA4 reduced TiO2‐induced TRPV1 mRNA expression and protein detection, as well TRPV1 co-staining with p-NFκB, indicating reduction of neuronal activation. LXA4 down-modulated neuronal activation and response to capsaicin (a TRPV1 agonist) and AITC [a transient receptor potential ankyrin 1 (TRPA1) agonist] of DRG neurons.
Conclusion: LXA4 might target recruited leukocytes and primary afferent nociceptive neurons to exert analgesic and anti-inflammatory activities in a model resembling what is observed in patients with prosthesis inflammation.
Total joint replacement recovers joint function, reduces pain, and improves quality of life (1–4). Total knee arthroplasty is a common procedure for joint replacement, which is expected to increase in the coming years (5, 6). In Europe, 2.5 million knee arthroplasties were recorded from 1975 to 2018 (7), and by the year 2030, 3.5 million procedures are expected in the United States (8). Despite the success of arthroplasty, deterioration of prosthetic components is the most associated complication. This event is characterized by the release of metallic nanoparticles that promote osteolysis, necessitating arthroplasty revision (8–10). Titanium is widely used in the production of orthopedic prostheses (11). However, TiO2 is the main trigger in prosthesis wear process-induced arthritis. Resident macrophages are activated and release tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) upon TiO2 phagocytosis (12). Intra-articular (i.a.) administration of TiO2 induces chronic arthritis and phenocopies the articular inflammation and pain caused by the release of prosthesis components upon wear (13). The available therapies for prosthesis-induced arthritis patients include non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, and opioids. These drugs promote tolerance, but are accompanied by adverse effects or addiction, affecting life quality and economic cost (14, 15). Therefore, investigating novel candidates for prosthesis-induced arthritis treatment is crucial. If a novel therapy presents different side effects, it might benefit patients whose need are not well-served by the current treatments due to side effects.
In fact, pain is a debilitating symptom of arthritis with consequences not only in the productivity, but also in the lifestyle and social interactions of patients. Pain is also one of the major reasons that patients seek medical care (16, 17). In peripheral inflammatory pain, the initiating factor of inflammation can, for instance, activate tissue-resident cells such as macrophages, which will produce inflammatory molecules, including cytokines and reactive oxygen species (ROS) (18). Cytokines such as TNF-α, IL-1β, and IL-6 as well as ROS such as superoxide anions have a role in recruiting leukocytes, thus changing the cellular profile in the inflammatory foci (19, 20). Not only do these mediators activate nuclear factor kappa B (NF-κB); NF-κB also induces their production (21). Cytokines and ROS can also sensitize the primary nociceptor sensory neurons causing hyperalgesia (18, 22). Nociceptor neuron sensitization can involve both enhancement of the function and increased production of ion channels that facilitate neuronal firing (23). Transient receptor potential (TRP) channels such as TRPV1 and TRPA1 are examples of ion channels expressed in the axons and cell bodies of primary afferent nociceptor neurons. TRP channels have been studied as targets for novel analgesics in cancer and neuropathic and chronic pain (24, 25).
Lipoxin A4 (LXA4) is a specialized pro-resolving lipid mediator (SPM) derived from arachidonic acid (26). This endogenous molecule plays anti-inflammatory and resolutive roles in inflammation (27, 28). LXA4 acts in the nanogram range, diminishing cell recruitment, chemotaxis, and polymorphonuclear cell adhesion, thus controlling inflammatory tissue damage (29). For example, in an acute liver failure model, LXA4 reduces pro-inflammatory cytokine levels and inhibits apoptosis (30). In addition, LXA4 reduces inflammatory pain by suppressing mechanical and thermal hyperalgesia (31, 32). LXA4 potently blocks ROS action via nuclear factor erythroid 2-related factor 2 (Nrf2)-dependent mechanisms in several animal models (33–37). LXA4 also reduces NF-κB activity, accounting for an essential anti-inflammatory mechanism (30, 38–40). LXA4 acts through G protein coupled receptors (GPCR) for LXA4 (ALXR), also known as FPRL1 and FPR2 (41–43). The activation of ALX/FPR2 receptor explains most of the anti-inflammatory, pro-resolving, and protective actions of LXA4 (29, 42, 44). The multiple sites of actions and cellular mechanisms demonstrate that LXA4 has relevant properties for therapeutic development (29). Some of the LXA4 mechanisms are relevant in the disease development in TiO2 articular inflammation such as oxidative stress and cytokine production (13). Therefore, we reason that LXA4 merits investigation of its anti-inflammatory and analgesic activities in the context of prosthesis wearing process-released components like TiO2, which we pursued in the present study.
For detailed materials and methods, please refer to the Supplementary Data 1. Briefly, Figure 1 shows the experimental protocols in which male Swiss (20–25 g) received the administration of intra-articular TiO2 (3 mg/10 µl/knee joint) as previously described (13) to induce aseptic arthritis. The first experiments were dedicated to determining the disease phenotype upon LXA4 treatment (Fig ure 1; protocol 1); for this, we performed the treatment with LXA4 (0.1, 1, or 10 ng) or vehicle (3.2% ethanol/saline) [100 µl per animal, intraperitoneal (i.p.)] 24 h after TiO2 stimulus. Mechanical hyperalgesia was assessed using an electronic esthesiometer (45) in different time points for 30 days to perform a dose–response curve. The most effective dose of LXA4 (10 ng/animal, every 48 h) was used for succeeding experiments. Edema was evaluated using measurements of the transverse diameters using a caliper and thermal hyperalgesia by Hargreaves apparatus. Knee joint lavages were collected on the 30th day to assess the total and differential leukocyte recruitment (46). Stomach was collected to assess myeloperoxidase (MPO) activity (gastric damage) (47) and blood samples were used to assess serum levels of aspartate transaminase (AST), alanine transaminase (ALT) (liver damage), urea, and creatinine (renal damage) (48). Hematoxylin–eosin (HE) staining was performed on knee joint samples for histopathology analysis (49).
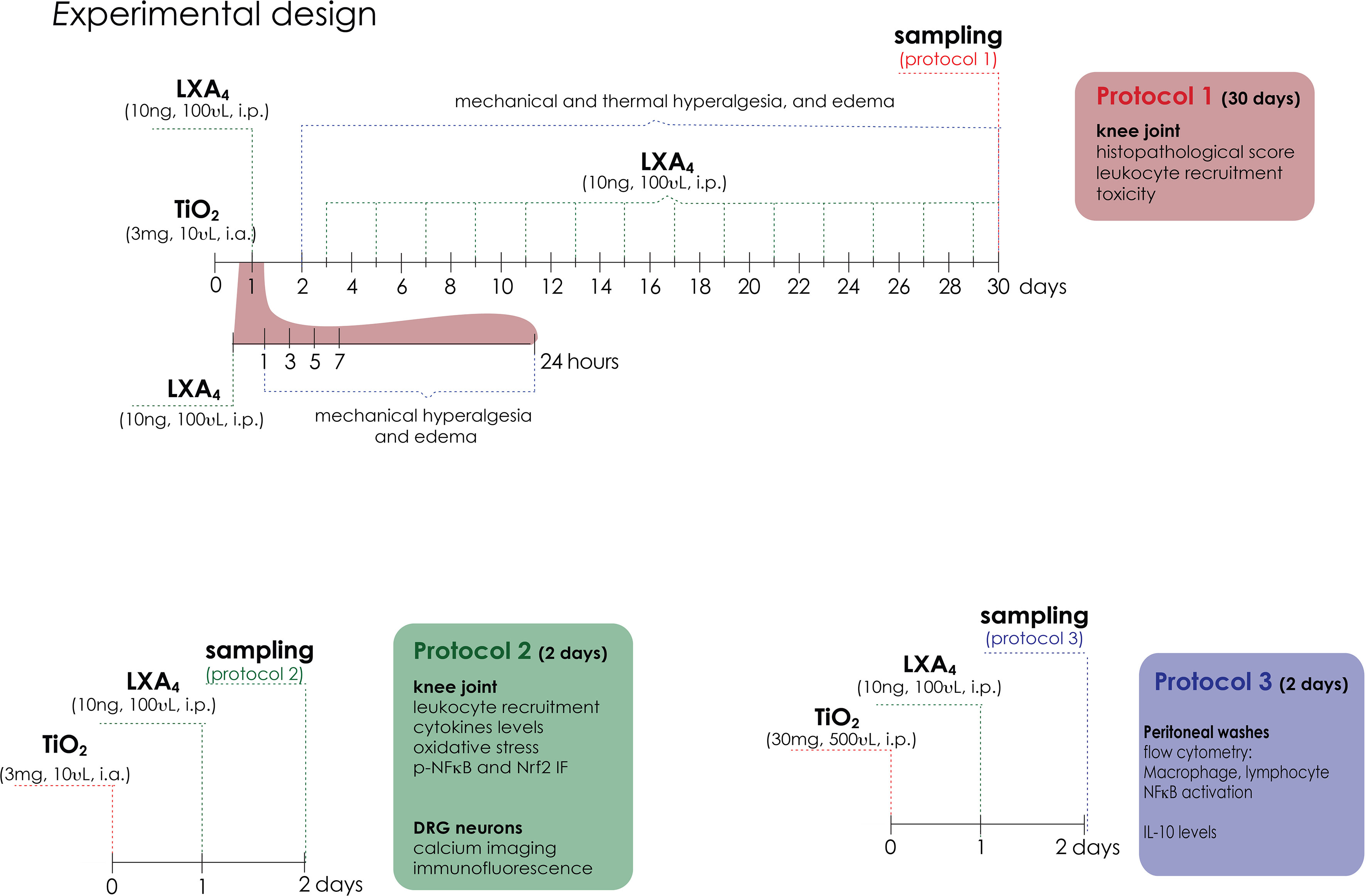
Figure 1 Experimental design. Protocol 1 is a 30-day experimental design. Mice were treated for 30 days with LXA4 (0.1, 1, and 10 ng/animal, i.p.) or vehicle (ethanol) starting 24 h after i.a. injection of TiO2 (3 mg/joint). Mechanical hyperalgesia and edema were evaluated 1, 3, 5, 7, 24 h (day 1), and subsequently every 2 days until the 30th day. Thermal hyperalgesia was evaluated on day 1 and every 3 days until day 30. On day 30, the knee joint was collected for histopathological analysis and toxicity assays, and the knee joint wash was collected for leukocyte recruitment. Protocol 2 is a 2-day experimental design. Mice were treated with a single treatment of LXA4 (10 ng/animal) starting 24 h after i.a. injection of TiO2 (3 mg/joint). On the 2nd day, knee joint wash was collected for leukocyte recruitment, knee joint cytokine levels, NF-κB phosphorylation, and oxidative stress (GSH, ABTS, ROS assay, and Nrf2 expression and activation). On the 2nd day of the model, DRG samples (L4–L6) were collected for calcium imaging (TRPV1 and TRPA1 agonists) and dissected for immunofluorescence (ALX/FPR2 receptor co-stained with TRPV1; TRPV1 and p-NF-κB co-staining with TRPV1 and TRPA1) and RT-qPCR. Protocol 3 is TiO2-induced peritonitis. Mice were given an i.p. injection of TiO2 (30 mg/500 µl), and after 24 h, the animals were treated with LXA4 (10 ng/animal) or vehicle (saline) (100 µl per animal, i.p.). Peritoneal washes were collected on the 2nd day to count total recruited leukocytes, for differential cell counts in stained slices, and for flow cytometry of lymphocytes (CD45+ CD4+), macrophages (CD45+ F4/80+), and p-NF-κB.
Inflammation and pain were present by the 2nd day of TiO2 arthritis and LXA4 activity could be observed. Chronic alterations were already studied with the experimental approach described in the previous paragraph. Considering these points, we reasoned that mechanistic studies could be performed on the 2nd day of TiO2 arthritis to reduce the duration of inflammation to which the animals were exposed. Therefore, potential mechanisms of LXA4 were studied in the early stages of TiO2-induced pain and inflammation (Figure 1; protocol 2). To this end, we collected the knee joint in the 2nd day after stimulus injection to determine leukocyte recruitment, and to assess the cytokine levels by enzyme-linked immunosorbent assay (ELISA) (TNF-α, IL-1β, IL-6, and IL-10 levels). Oxidative stress was measured by reduced glutathione (GSH), 2,2-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) measurement (50–52), and Nrf2 mRNA expression by reverse transcriptase-quantitative real-time polymerase chain reaction (RT-qPCR). Synovial fluid leukocytes were collected for p-NFκB and Nrf2 staining by immunofluorescence (53), and total ROS was measured using the probe 2’,7’-dichlorofluorescein diacetate (DCF-DA). Ipsilateral dorsal root ganglia (DRG) (corresponding to L4–L6 segments) were also dissected 2 days after TiO2 to perform calcium influx imaging using confocal microscopy (54), transient receptor potential cation channel subfamily V member 1 (TRPV1) mRNA expression by RT-qPCR, and TRPV1, TRPA1, ALX/FPR2, and p-NFκB staining by immunofluorescence.
The limited number of cells in the synovial fluid led us to use a TiO2-triggered peritonitis model to assess the cellular profile of recruited leukocytes and NF-κB activation in macrophages. We also determined if treatment with LXA4 could modulate the responses triggered by TiO2 via flow cytometry (Figure 1; protocol 3). For this approach, mice received an i.p. injection of TiO2 (30 mg/500 µl), and 24 h after TiO2 stimulus (post-treatment), mice were treated with LXA4 (10 ng) or vehicle (saline) (100 µl per animal, i.p.). After an additional 24 h, peritoneal washes were collected in FACS buffer (10 ml per animal), and total leukocyte recruitment was counted, and flow cytometry (54) for CD45, CD4, F4/80, and p-NFκB staining was performed. All experimental conditions were standardized by our laboratory as previously published (13, 49, 54, 55) and in preliminary experiments performed for this manuscript.
For in vivo experiments, we used 6, 8, or 10 mice in each group per experiment depending on the methodology (indicated in the figure legends). In vitro experiments with DRG samples were performed using an n of 4 pools (10 mice to form 1 pool) per group. Two-way ANOVA followed by Tukey’s post-test was used to compare all groups and doses when responses were measured at different times after the stimulus injection. The analyzed factors were treatments, time, and time versus treatment interaction. Parametric results were evaluated by one-way ANOVA followed by Tukey’s post-test for data from a single time point. Kruskal–Wallis followed by Dunn post-test or two-way were used for non-parametric results. p < 0.05 was considered significant.
A dose–response curve was performed to assess the potential analgesic and anti-inflammatory effects of LXA4 in TiO2-induced arthritis. Treatment started 24 h after i.a. TiO2 injection. We could still observe significant analgesia by the 24th hour after LXA4 treatment, which was reduced by the 48th hour (data not shown). Therefore, treatments with LXA4 were performed every 48 h. The injection of 3 mg/joint of TiO2 induced mechanical hyperalgesia, and treatment with LXA4 reduced the mechanical hyperalgesia in a dose-dependent (0.1, 1, or 10 ng/animal, 100 μl i.p.) manner. The most effective dose was 10 ng/animal, which was chosen for the subsequent experiments (Figure 2A). TiO2 also induced thermal hyperalgesia that was reduced by LXA4 10 ng/animal treatment. The reduction of thermal hyperalgesia was observed from the 4th day onwards, with complete inhibition from the 7th to the 30th day (Figure 2B).
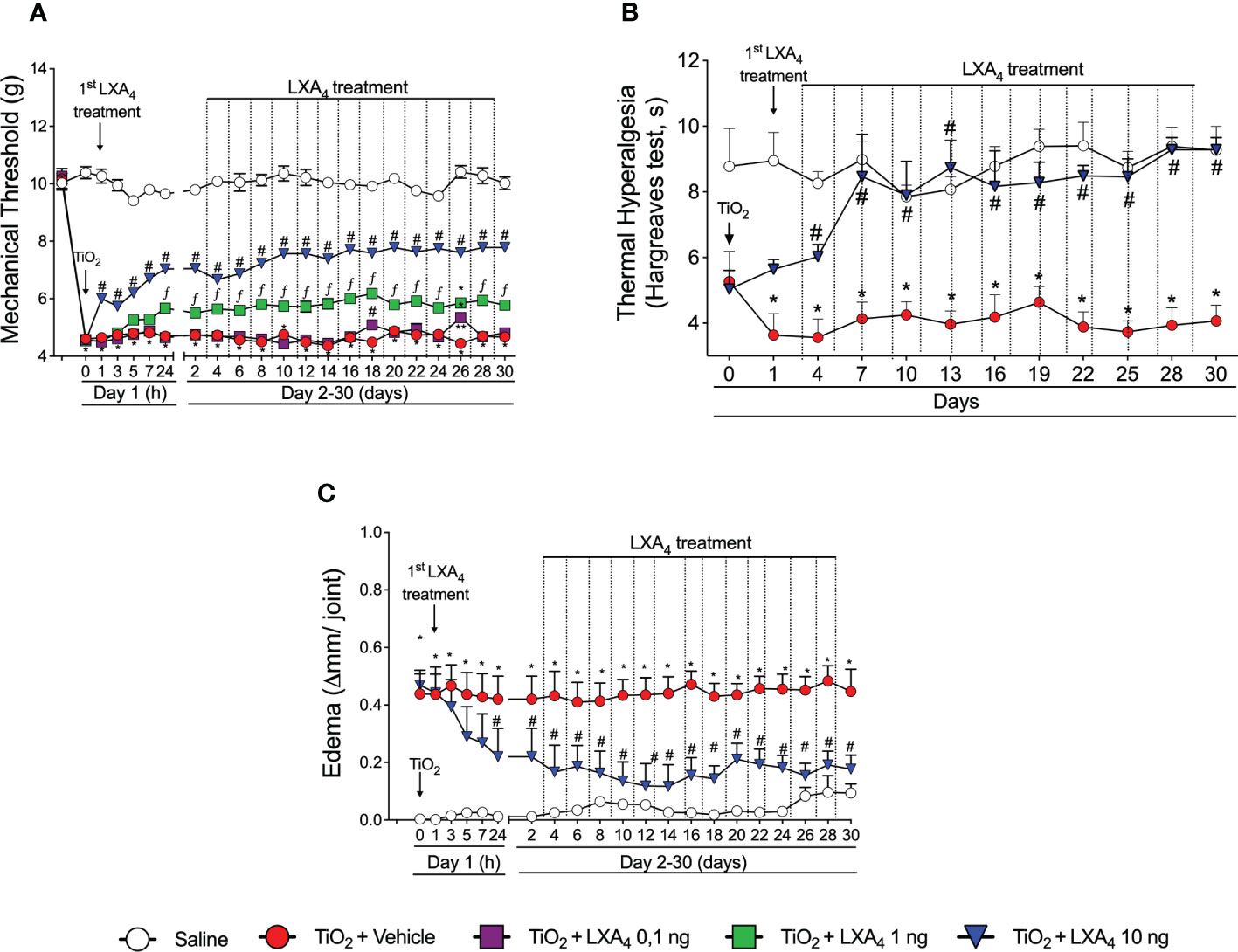
Figure 2 LXA4 inhibits TiO2-induced articular mechanical hyperalgesia, thermal hyperalgesia, and edema in the knee joint. Mice were treated for 30 days with LXA4 (0.1, 1, and 10 ng/animal, i.p.; 48-h intervals) or vehicle (ethanol) starting 24 h after i.a. injection of TiO2 (3 mg/joint). Mechanical hyperalgesia (A) was evaluated 1, 3, 5, 7, 24 h (day 1), and subsequently every 2 days until day 30. Thermal hyperalgesia (B) was evaluated on day 1 and every 3 days until day 30. Results are expressed as mean ± SEM, n = 6 mice per group per experiment and are representative of two separate experiments [*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group; **p < 0.05 vs. TiO2 and LXA4 (10 ng) groups; fp < 0.05 vs. TiO2 and LXA4 (10 and 1 ng) groups, repeated measures two-way ANOVA followed by Tukey’s post-test]. Edema (C) was evaluated 1, 3, 5, 7, 24 h (day 1), and subsequently every 2 days until day 30.
We also investigated if the treatment with LXA4 reduces knee joint edema. A dose of 10 ng/animal of LXA4 significantly reduced TiO2-induced articular edema 24 h after the first treatment, with persistent anti-inflammatory effect until the 30th day of arthritis (Figure 2C). The saline-injected group did not develop edema (Figure 2C).
Mice were treated with LXA4 (10 ng/animal, i.p., every 48h) or vehicle (3.2% ethanol in saline) 24 h after i.a. TiO2 (3 mg) injection. On the 30th day, the knee joint was collected for HE histopathology evaluation (Figures 3A–G). LXA4 reduced TiO2-induced synovial hyperplasia, inflammatory infiltrates, and vascular proliferation observed in the histopathological index analyses (Figure 3A). Treatment with vehicle showed no effect on TiO2-induced histopathological changes.
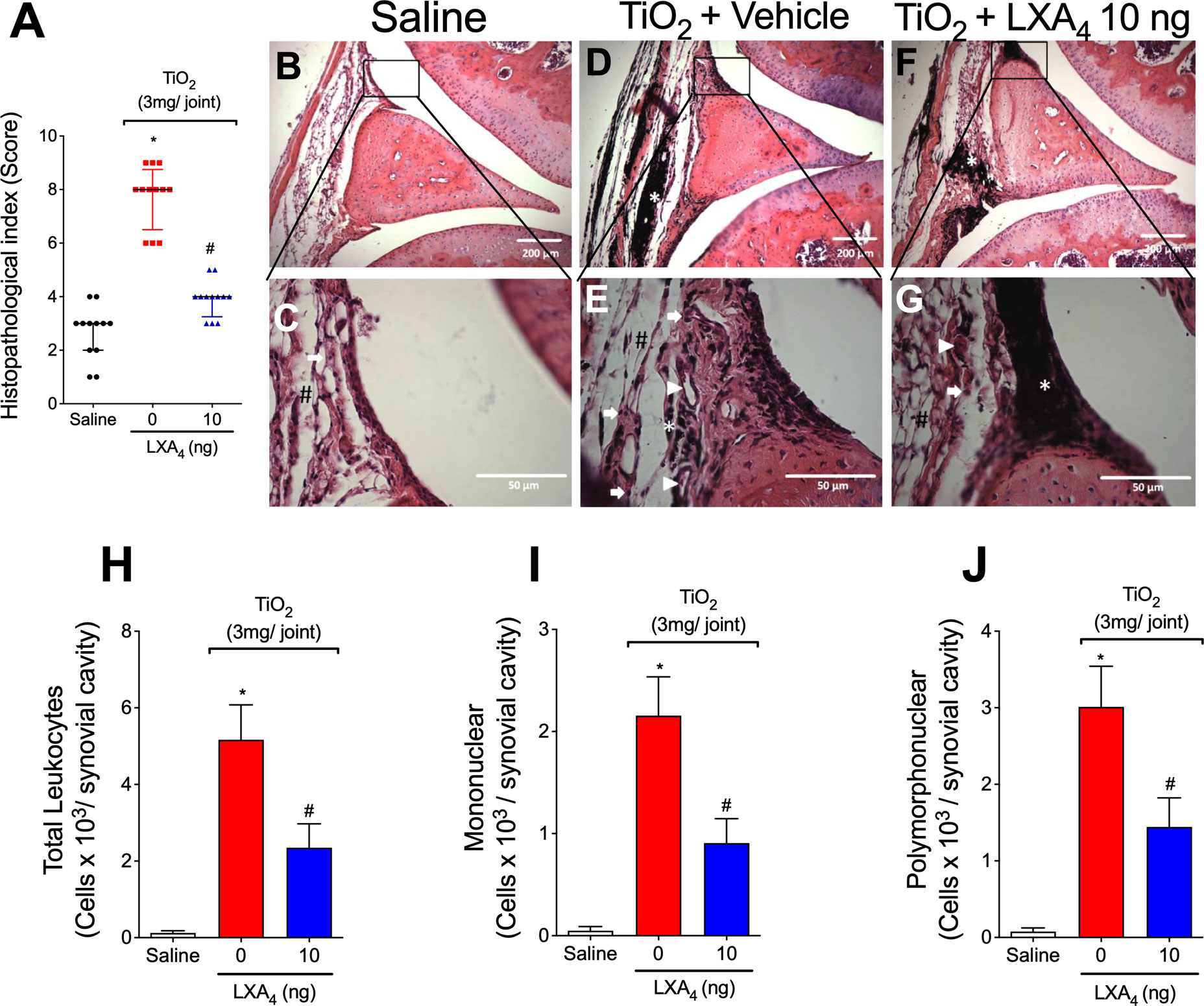
Figure 3 LXA4 reduces TiO2-induced histopathological damage and recruitment in the knee joint. Mice were treated with LXA4 (10 ng/animal, i.p.) or vehicle (ethanol) 24 h after i.a. TiO2 (3 mg) injection and on alternate days for 30 days. On the 30th day, the knee joints were collected and stained with HE. Histopathological index (A) and analysis (B–G). The panel shows: saline (B, C), TiO2-injected treated with vehicle (D, E), and TiO2-injected treated with LXA4 (F, G). The representative image demonstrated the invasive pannus (#), leukocyte infiltration (arrow), angiogenesis (arrowhead), and TiO2 nanoparticles (asterisk). Original magnification 10× (B, D, F) and 40× (C, E, G). Results are expressed as mean ± SEM, n = 12 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, Kruskal–Wallis followed by Dunn’s post-test). On the 30th day, knee joint washes were collected to count total leukocytes (H), mononuclear (I), and polymorphonuclear cells (J). Results are expressed as mean ± SEM, n = 6 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, one-way ANOVA followed by Tukey’s post-test).
Leukocyte recruitment to the knee joint is a hallmark of arthritis (56). To investigate the effect of LXA4 on leukocyte recruitment 30 days post-TiO2 stimulus, knee joint washes were collected to evaluate the total number of leukocytes and mononuclear and polymorphonuclear cells. The injection of TiO2 significantly increases the number of leukocytes recruited to the knee joint 30 days after the stimulus (Figures 3H–J). Our results show that the treatment with LXA4 at 10 ng/animal reduced TiO2-induced recruitment of total leukocyte (Figure 3H) and mononuclear (Figure 3I) and polymorphonuclear cells (Figure 3J).
Thirty days after TiO2 stimulus, serum samples and stomach were collected to evaluate whether the chronic treatment with LXA4 would induce gastric, hepatic, or renal damage, which are common side effects of non-steroidal anti-inflammatory drugs (56). Toxicity was assessed through the concentrations of AST, ALT, urea, and creatinine, and MPO activity (Figure 4). The treatment with 10 ng/animal qod of LXA4 did not modify the serum concentration of AST, ALT (Figures 4A, B), urea, creatinine (Figures 4C, D), or MPO activity in the stomach compared with positive controls (Figure 4E). Therefore, our data suggest that chronic treatment does not induce detectable gastric, hepatic, or renal lesion/damage.
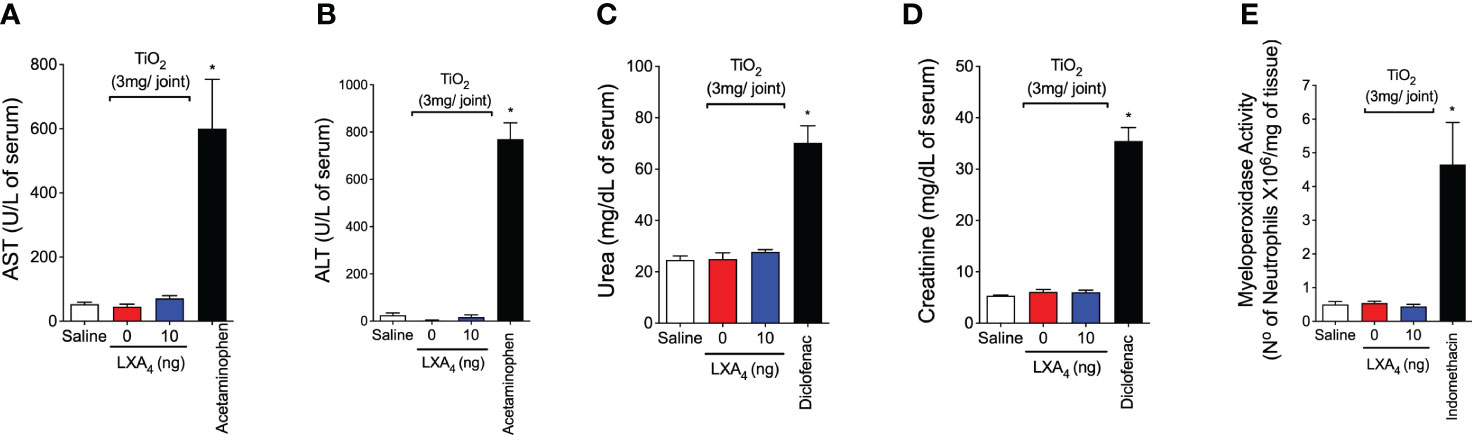
Figure 4 LXA4 chronic treatment does not induce toxicity. Mice were treated for 30 days with LXA4 (10 ng/animal, i.p. q.o.d.) starting 24 h after i.a. injection of TiO2 (3 mg/joint), and serum and stomach were collected. AST (A), ALT (B), urea (C), and creatinine (D) serum levels and MPO activity in the stomach (E) were determined to evaluate treatment toxicity. As positive drug control for gastric, hepatic, and renal toxicity, indomethacin (2.5 mg/kg, i.p., diluted in tris/HCl buffer, for 7 days), acetaminophen (650 mg/kg, i.p., diluted in saline), and diclofenac (200 mg/kg, p.o., diluted in saline) were used, respectively. Results are expressed as mean ± SEM, n = 6 mice per group per experiment, two independent experiments (*p < 0.05 vs. all groups, one-way ANOVA followed by Tukey’s post-test).
In the following experiments, we opted to reduce the treatment period to investigate the inflammatory and pain mechanisms of LXA4. We considered that Figures 2–4 established the beneficial effect of LXA4 treatment during a chronic period and that inflammation and pain achieved significant development by the second day of arthritis. This approach allowed us to reduce the suffering of animals and investigate the mechanisms involved in LXA4 post-treatment of ongoing TiO2 arthritis.
Given the role of recruited leukocytes in inflammatory pain and oxidative burst (57), we next assessed the efficacy of LXA4 in modulating TiO2-induced leukocyte recruitment after a single treatment. In this case, recruitment was evaluated on the 2nd day (Figures 5A–C) to further support that this time point is adequate and mimics all inflammatory features of TiO2 arthritis together with the pain and edema observed in Figure 2. The injection of TiO2 significantly increased the number of total leukocytes recruited on the 2nd day after the stimulus (Figures 5A–C). Our results show that the treatment with LXA4 at 10 ng/animal reduced TiO2-induced recruitment of total leukocyte (Figure 5A) and mononuclear (Figure 5B) and polymorphonuclear cells (Figure 5C). These data show that most leukocytes recruited to the joint were mononuclear cells (90%). Compared with the 30th day data (Figures 5A–C), 10.6-fold more leukocytes migrated in the knee joint on the 2nd day, indicating that this time point is suitable for investigating inflammatory mechanisms. Indeed, on the 2nd day, higher mononuclear cells than neutrophil counts were already established. The pathophysiological mechanisms underlying this unusual cellular profile deserves further investigation in future studies.
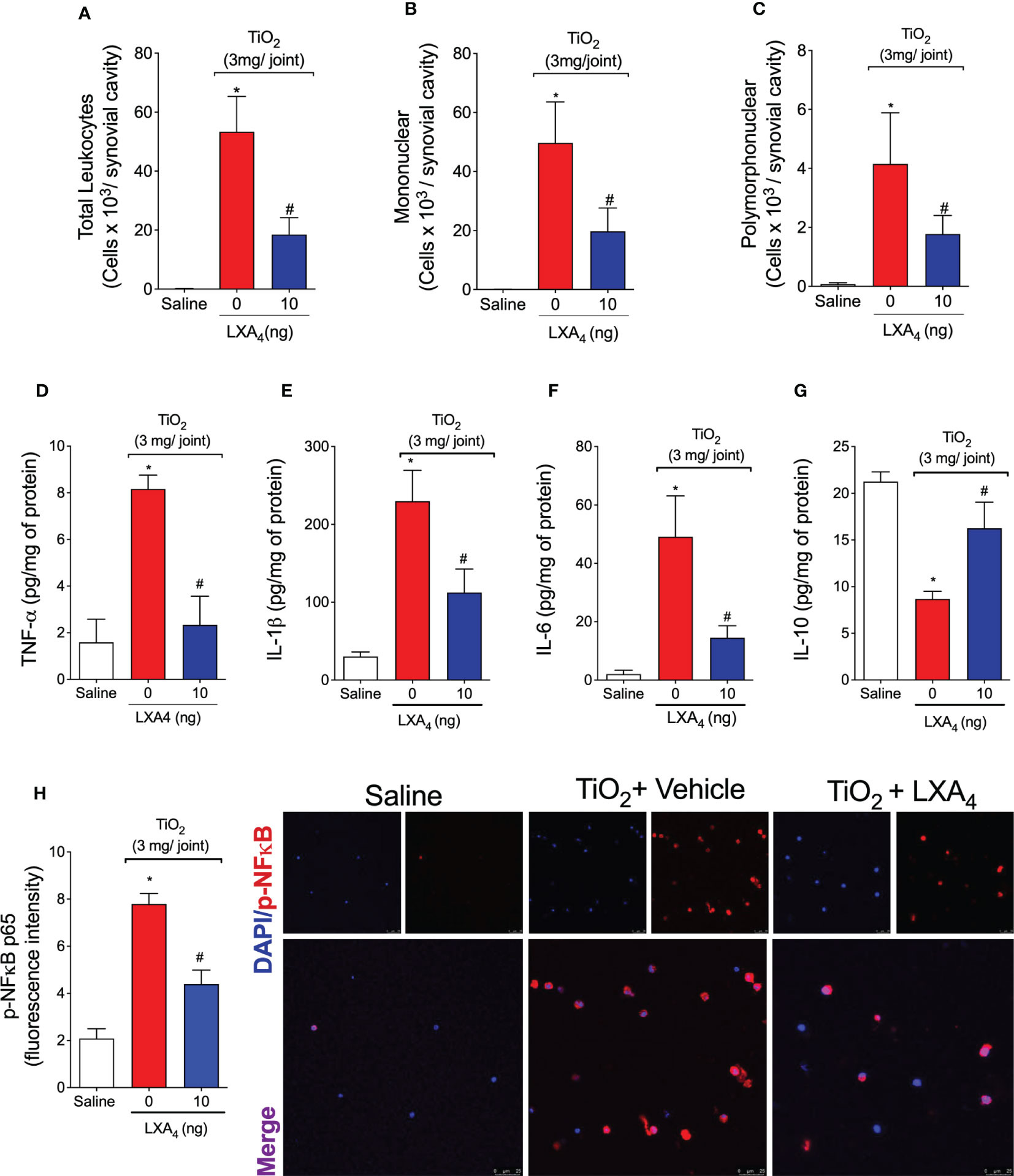
Figure 5 LXA4 modulates TiO2-induced leukocyte recruitment, cytokine production, and NF-κB activation. Mice received a single treatment of LXA4 (10 ng/animal) 24 h after i.a. injection of TiO2 (3 mg/joint), and on the 2nd day, knee joint washes were collected to count total leukocytes (A), mononuclear (B), and polymorphonuclear cells (C). The knee joint was collected, and TNF-α (D), IL-1β (E), IL-6 (F), and IL-10 (G) were measured by ELISA. Knee joint washes were used to perform an immunofluorescence assay. (H) shows the representative images of p-NF-κB p65 (red) with nuclear staining by DAPI and the quantitation. Fluorescence intensity (H) was analyzed by a confocal microscope at 63× magnification. Results are expressed as mean ± SEM, n = 6 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, one-way ANOVA followed by Tukey’s post-test).
The potential of LXA4 to modulate pro-inflammatory cytokine (TNF-α, IL-1β, and IL-6) and anti-inflammatory cytokine (IL-10) production in the joint tissue was evaluated on the 2nd day (Figures 5D–G). The i.a. injection of TiO2 induced a significant increase in TNF-α (Figure 5D), IL-1β (Figure 5E), and IL-6 (Figure 5F). A single treatment with LXA4 was enough to reduce the levels of these pro-inflammatory cytokines induced by TiO2 (Figures 5D–F). Thus, the effect of LXA4 in reducing the production of essential cytokines represents one of its mechanisms to reduce pain, edema, and recruitment of leukocytes (58). In addition, IL-10 production was increased by LXA4 (Figure 5G), evidencing this lipid mediator’s anti-inflammatory and immunoregulatory capacity with a single treatment.
Synovial fluid leukocytes were collected on the 2nd day, and the phosphorylated (p) form of NF-κB was determined by immunofluorescence assay (Figure 5H). Treatment with LXA4 reduced the fluorescence intensity of the p-NFκB p65 subunit induced by TiO2 (Figure 5H). Therefore, these data suggest that inhibiting NF-κB activation is, at least, one of the mechanisms by which LXA4 ameliorates TiO2-induced inflammation and pain. This underscores the importance of this transcription factor to cytokine production (Figures 5D–G) and leukocyte recruitment (Figures 5A–C).
The number of recovered cells in synovial washes was insufficient to perform a flow cytometry analysis in our hands. To enable further assessment of the cellular profile of leukocytes recruited upon TiO2 stimulation, NF-κB activation, and the effect of LXA4, we standardized a peritonitis model to mimic the TiO2-induced inflammation. The increased volume of the peritoneal cavity allows the recruitment of larger numbers of leukocytes than the knee joint. We performed a TiO2 dose–response (data not shown) and found that 30 mg per animal induced significant leukocyte recruitment. We performed a single post-treatment with 10 ng of LXA4 (similar to what was performed for the arthritis), which was sufficient to reduce the leukocyte recruitment (Figures 6A–C). TiO2 recruited mostly mononuclear cells, so we evaluated the ratio of recruited macrophages and lymphocytes, and the modulation by LXA4. Although we observed a similar percentage of positive cells in all analyzed groups, when we corrected the percentages by the total number of cells in the peritoneal washes, the results revealed significant differences between the groups (Figure 6D). We show that TiO2 increased the number of CD45+ F4/80+ macrophages (Figure 6E) and CD45+ CD4+ lymphocytes (Figure 6F), and treatment with LXA4 reduced the number of recruited CD45+ F4/80+ macrophages (Figure 6E), but not of CD45+ CD4+ lymphocytes (Figure 6F). The proportion of macrophages represents 70% of the total recruited leukocytes, and lymphocytes represent 10% of the total recruited CD45+ leukocyte population (Figure 6G). Thus, CD45+ F4/80+ macrophages represent the majority of leukocytes recruited by TiO2.

Figure 6 TiO2 increases CD45+ F4/80+ macrophages and CD45+ CD4 + lymphocyte counts that, in part, are down-modulated by LXA4. TiO2 peritonitis was induced with an i.p. injection of TiO2 (30 mg/500 µl), and after 24 h, the animals received treatment with LXA4 (10 ng) or vehicle (saline) (100 µl per animal, i.p.). Peritoneal washes were collected on the 2nd day to count total recruited leukocytes (A), mononuclear cells (B), and polymorphonuclear cells (C). Flow cytometry for total leukocyte cells, CD45+ cells (D), macrophages [CD45+ F4/80+ cells (E)], and lymphocytes [CD45+ CD4 + cells (F)] corrected by the total recruited leukocytes. (G) shows the representative gates. Results are expressed as mean ± SEM, n = 10 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, one-way ANOVA followed by Tukey’s post-test).
Since LXA4 reduced the recruitment of CD45+ F4/80+ macrophages and these are main cell population in the peritoneal cavity, we reasoned that these cells could be a cellular target of the LXA4-mediated reduction in activated NF-κB. To check this possibility, we performed flow cytometry and show that TiO2 increased NF-κB activation/phosphorylation in CD45+ F4/80+ macrophages (pNF-κB+ F4/80+ cells) (Figure 7B). Quite interestingly, CD45+ F4/80+ pNF-κB+ macrophages represent 85% of the total NF-κB+ CD45+ cells (Figures 7A, C), and LXA4 treatment reduced this activation. Although we have not exhaustively investigated the role of each cell type in TiO2 inflammation and LXA4 activity, these data show that CD45+ F4/80+ macrophages are the main mononuclear cell population in TiO2 inflammation and a target of LXA4 activity with respect to both cellular recruitment and NF-κB activation.
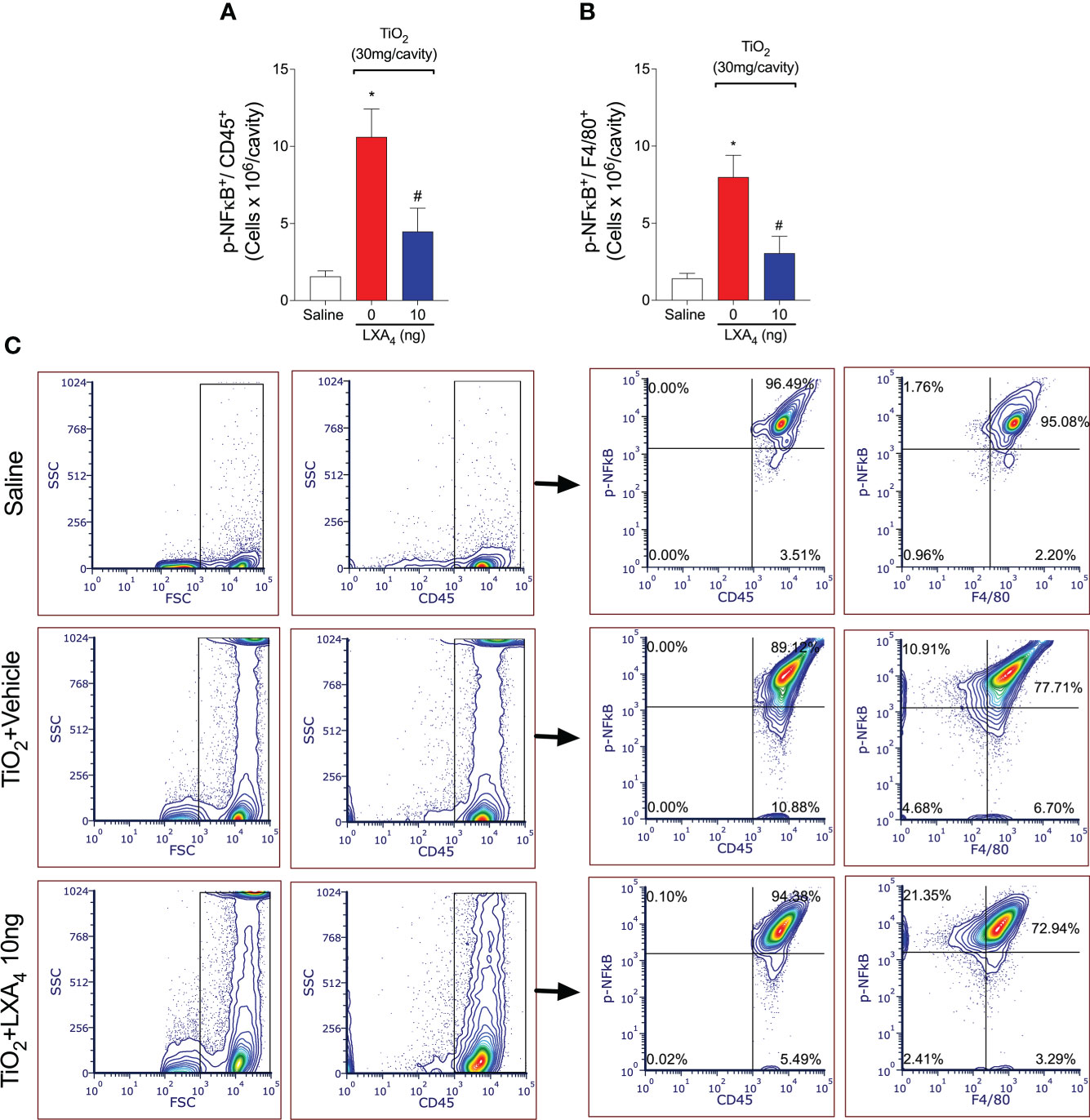
Figure 7 Treatment with LXA4 reduces TiO2-triggered NF-κB activation in CD45+ F4/80+ macrophages. TiO2 peritonitis was induced with an i.p. injection of TiO2 (30 mg/500 µl), and after 24 h, the animals were treated with 10 ng LXA4 or vehicle (saline) (100 µl per animal, i.p.). Flow cytometry for p-NFκB p65 in total leukocytes [CD45+ p-NFκB+ cells (A)] and macrophages [CD45+ F4/80+ p-NFκB+ cells (B)] corrected by the total number of recruited leukocytes. (C) shows the representative gates. Results are expressed as mean ± SEM, n = 10 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, one-way ANOVA followed by Tukey’s post-test).
Knee joint samples were collected on the 2nd day of TiO2 arthritis, and antioxidant capacity was measured with GSH and ABTS assays (Figures 8A, B). In other models, TiO2 induces the production of ROS and, consequently, oxidative stress in various organs (59–61). Herein, we show that TiO2 stimulus reduced the levels of endogenous antioxidants in the knee joint tissues as observed in free radical scavenging ability and GSH levels (Figures 8A, B). On the 2nd day, a single treatment with LXA4 significantly restored the levels of ABTS and GSH (Figures 8A, B), demonstrating that treatment with LXA4 reestablished the antioxidant ability to scavenge free radicals such as ABTS cationic radical and positively upregulates the endogenous antioxidant GSH. GSH is upregulated by the transcription factor Nrf2 (62), and we observed that LXA4 increases the Nrf2 mRNA expression (Figure 8C). Then, because these phenomena were observed in the knee joint tissue and recruited leukocytes have a major role in those alterations, we analyzed the recruited leukocytes.
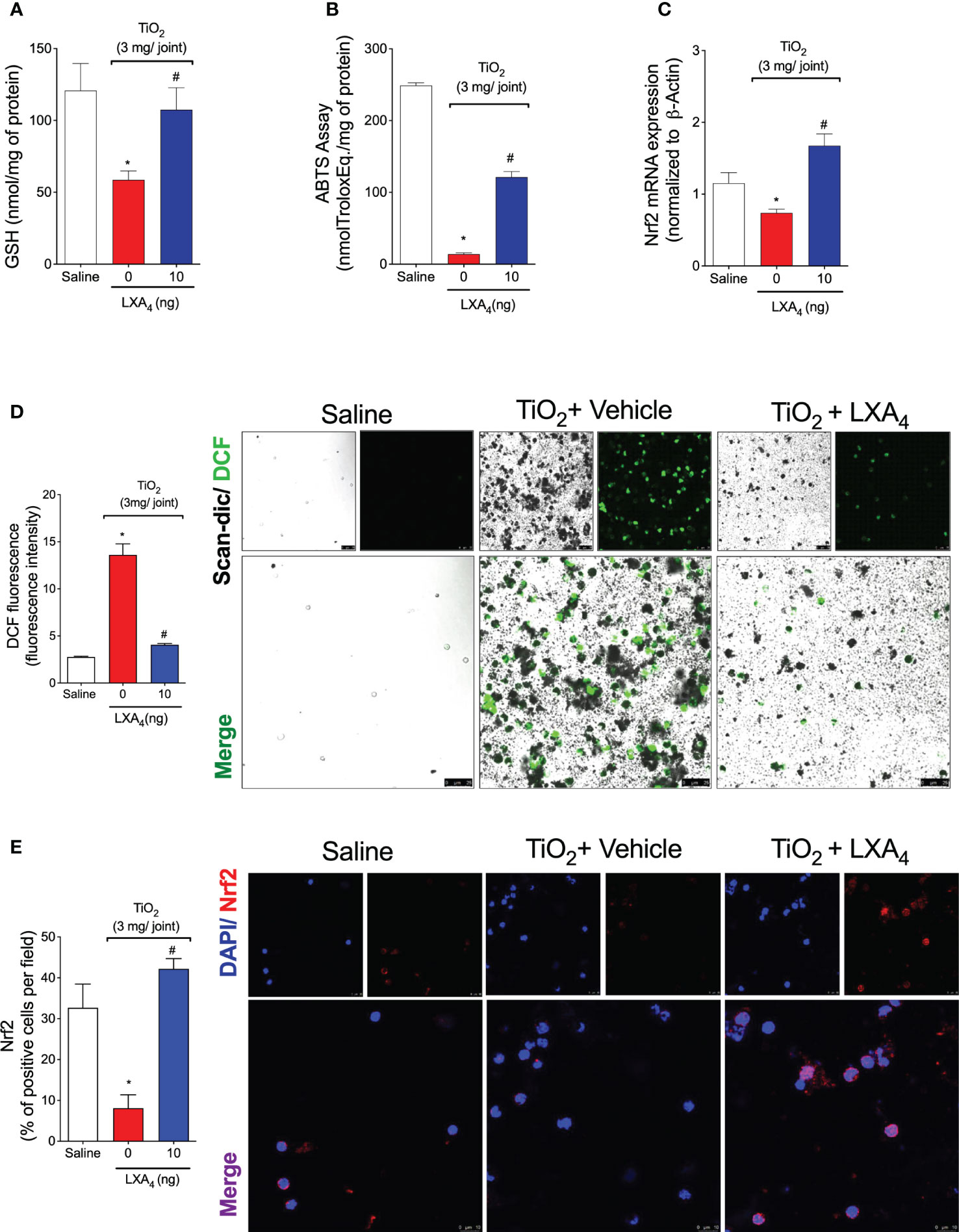
Figure 8 LXA4 inhibits TiO2-induced oxidative stress, improving antioxidant capacity. Mice received a single treatment of LXA4 (10 ng/animal) 24 h after i.a. injection of TiO2 (3 mg/joint), and on the 2nd day, the knee joint was collected, and the antioxidant effect of LXA4 was measured using GSH levels (A) and ABTS (B). Nrf2 mRNA expression was quantitated by RT-qPCR (C). Dihydrofluorescein diacetate (DCF-DA) was added to knee joint wash cells for 30 min, and intracellular ROS levels from intact cells were analyzed using the scan-dic and green channel in a confocal microscope at 63× magnification. DCF fluorescence intensity (D) indicates ROS production, which was quantitated. Representative images show DCF fluorescence for the negative control, TiO2, and LXA4 groups (D). Knee joint washes were used to perform an immunofluorescence assay. (E) shows the representative images of Nrf2 (red) with nuclear staining by DAPI, and the quantitation by % of positive cells per field. The data (E) were analyzed by a confocal microscope at 63× magnification with 1.5× zoom. TiO2 nanoparticles are the black pigments. Results are expressed as mean ± SEM, n = 6 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, one-way ANOVA followed by Tukey’s post-test).
ROS production was measured in the synovial fluid leukocytes using a DCF-DA probe, which, when oxidized, generates a fluorescence product (DCF) proportional to overall intracellular ROS levels. We observed that treatment with LXA4 reduced DCF fluorescence intensity (Figure 8D), demonstrating that treatment with LXA4 inhibits TiO2-induced production of ROS (Figure 8D). Articular fluids were collected on the 2nd day, and Nrf2 was determined by immunofluorescence assay (Figure 8E). Supporting the qPCR data, we observed that treatment with LXA4 increased the percentage of Nrf2-positive cells per field (Figure 8E).
LXA4 acts through the receptor ALX/FPR2 in peripheral tissues and regulates cellular responses of interest in inflammation and resolution (29). ALX/FPR2 is expressed in tissues and cell types such as immune cells, fibroblasts, epithelial cells, and astrocytes (29, 32). The effect of LXA4 in reducing mechanical and thermal hyperalgesia indicates that it could, eventually, act on nociceptor neurons. To suggest a neuronal effect of LXA4, it was necessary to determine (1) if the nociceptor sensory neurons express ALX/FPR2 and (2) whether LXA4 shapes the neuronal profile and activity. These were our next steps. We investigated the expression of ALX/FPR2 receptor in the DRG by immunofluorescence staining for ALX/FPR2 receptor and TRPV1, which is a TRP channel expressed by nociceptive C-fibers (Figure 9). Our data show that TiO2 increased the expression of ALX/FPR2 receptor in the DRG of mice (Figures 9A, C). We also found that TiO2 increases the percent of double positive ALXR/TRPV1 cells, indicating nociceptor sensory neurons express ALX/FPR2 receptor, which is enhanced in this neuronal population in TiO2 inflammation (Figures 9B, C). Altogether, these results suggest that nociceptive TRPV1+ neurons are targets of the action of LXA4 during TiO2-induced arthritis.
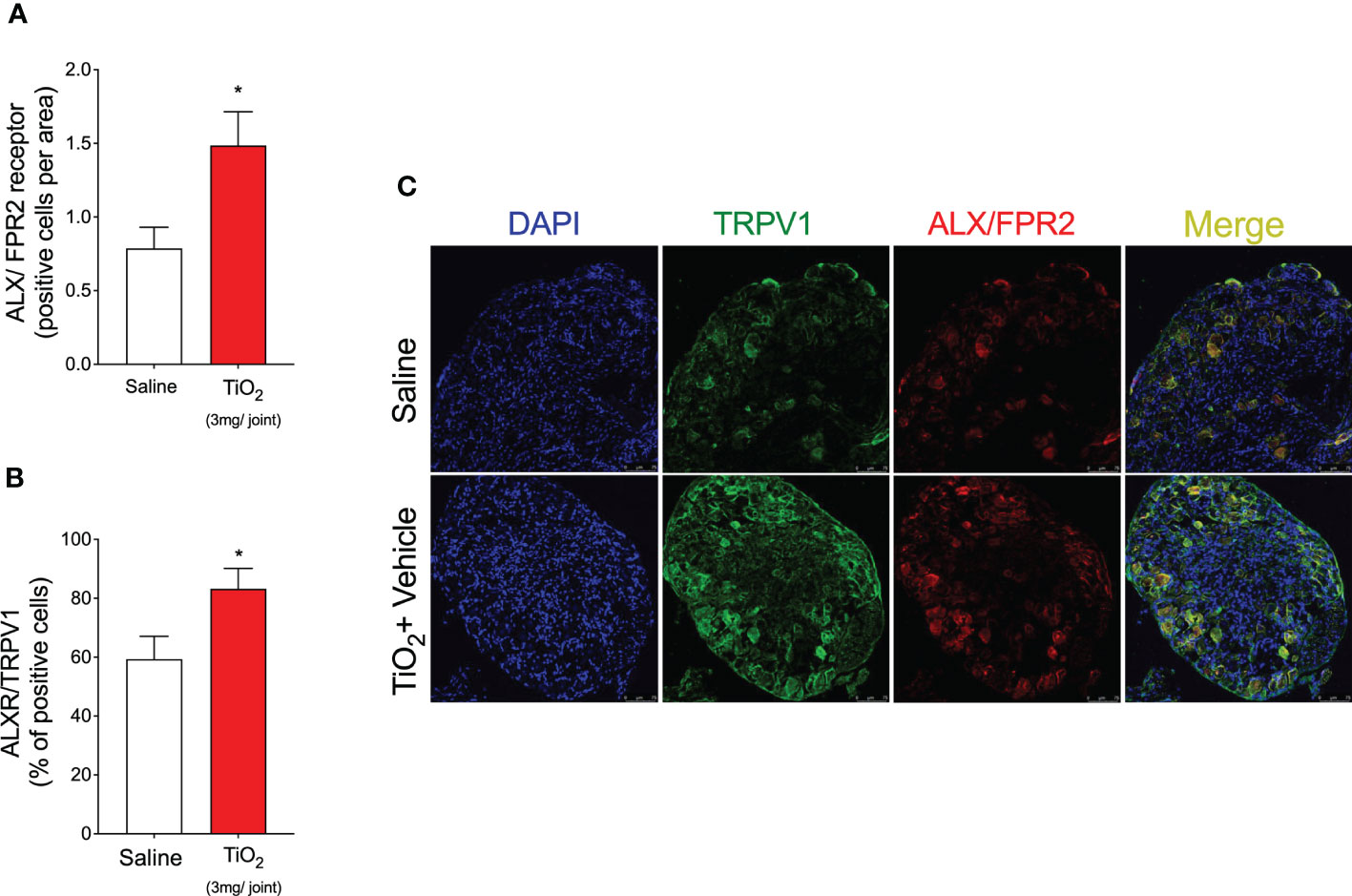
Figure 9 TiO2 increases ALX/FPR2 expression on nociceptive neurons. On the 2nd day of the model, DRGs samples (L4–L6) were dissected and immunofluorescence staining for TRPV1 and ALX/FPR2 was performed. Panels (A, B) show the quantitative analyses of the number of ALX/FPR2 receptor-positive cells per area (A) and co-staining with TRPV1 (as a percent of positive cells) (B). (C) shows the representative images of TRPV1+ cells (green), ALX/FPR2 receptor-positive cells (red), and the merge of double labeling of TRPV1 and ALX/FPR2 on DRG (20× magnification with 1.0 zoom in). Results are expressed as mean ± SEM, n = 8 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group, Student’s t-test).
Considering the results of Figure 9, our next step was to assess neuronal activation. This was quantified using calcium influx as measured by a fluorescent probe Fluo-4 AM in DRG neurons (63). We investigated whether DRG neurons from TiO2‐stimulated mice would present an increase in the baseline calcium levels and response to capsaicin (TRPV1 agonist) stimulation compared to saline-injected controls mice, and the ability of LXA4 to modulate this response (Figure 10). DRG neurons from vehicle-treated mice presented a higher baseline level of calcium influx than saline mice or LXA4-treated DRGs (Figures 10A–C). These data suggest that LXA4 reduces the activation of DRG neurons in TiO2-induced inflammation because the increase in calcium influx is indicative of DRG neuron activity (Figures 10A–C). Notably, in addition to the diminished basal level of calcium, LXA4 treatment also reduced the responsiveness of DRG neurons to capsaicin, which is a TRPV1 agonist (Figures 10A–C). The treatment with LXA4 reduced by 49% the capsaicin-responsive neurons compared to the TiO2 + vehicle group as per the Venn diagram (Figure 10D). Corroborating with the reduction of neuronal activation and diminished response to capsaicin, we demonstrated that treatment with LXA4 inhibited the increase of TRPV1 (Figure 11B) mRNA expression induced by TiO2, as well as TRPV1 staining in the DRG (Figures 11A, C). Therefore, LXA4 inhibits TiO2-induced DRG protein detection, mRNA expression, and activity of a critical ion channel (TRPV1) to nociceptor sensory neuron sensitization (64), which resulted in a functional outcome of reduced neuronal responsiveness and pain upon LXA4 treatment.
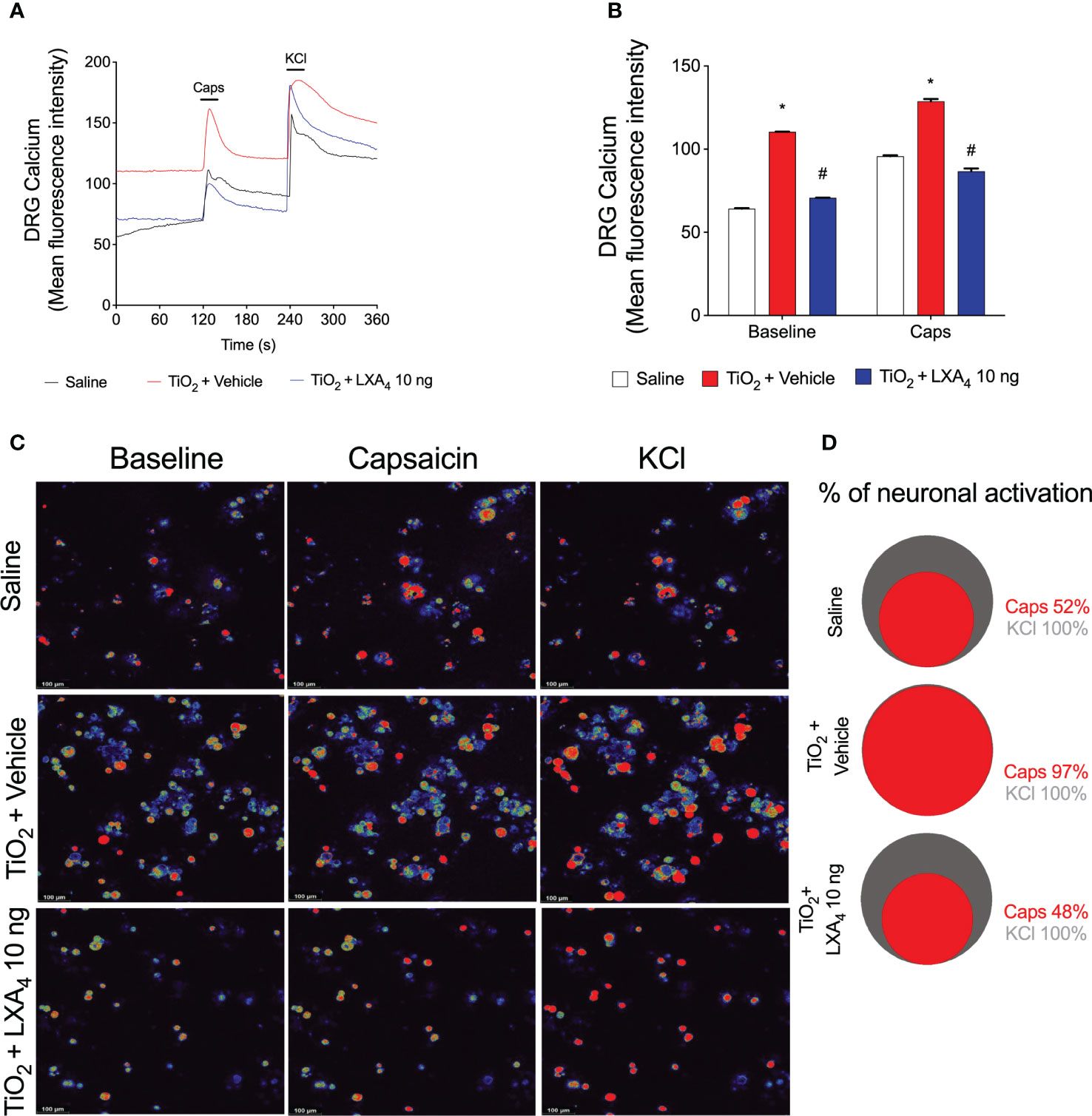
Figure 10 LXA4 reduces TiO2-induced TRPV1 activation on DRG neurons. Mice received a single treatment of LXA4 (10 ng/animal) 24 h after i.a. injection of TiO2 (3 mg/joint). On the 2nd day, the DRGs (L4–L6) were collected for calcium imaging using Fluo-4 AM probe. The fluorescence intensity traces of calcium-fluo-4 in representative DRG fields during the 6 min of recording is shown in panel (A). (B) displays the mean fluorescence intensity of calcium-fluo-4 at baseline (0‐s mark) and that following the stimulus with capsaicin (a TRPV1 agonist, 120‐s mark). Response to KCl (activates all neurons) begins at the 240‐s mark. (C) shows representative fields of DRG neurons (baseline fluorescence, the fluorescence after capsaicin, and after KCl). (D) shows a Venn diagram comparing the percent of the neuronal population that is capsaicin-responsive (red) within those neurons that responded to KCl control (gray). Results are expressed as mean ± SEM, n = 4 DRG seeded plates (each plate is a neuronal culture pooled from 10 mice) per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, two-way ANOVA followed by Tukey’s post-test).
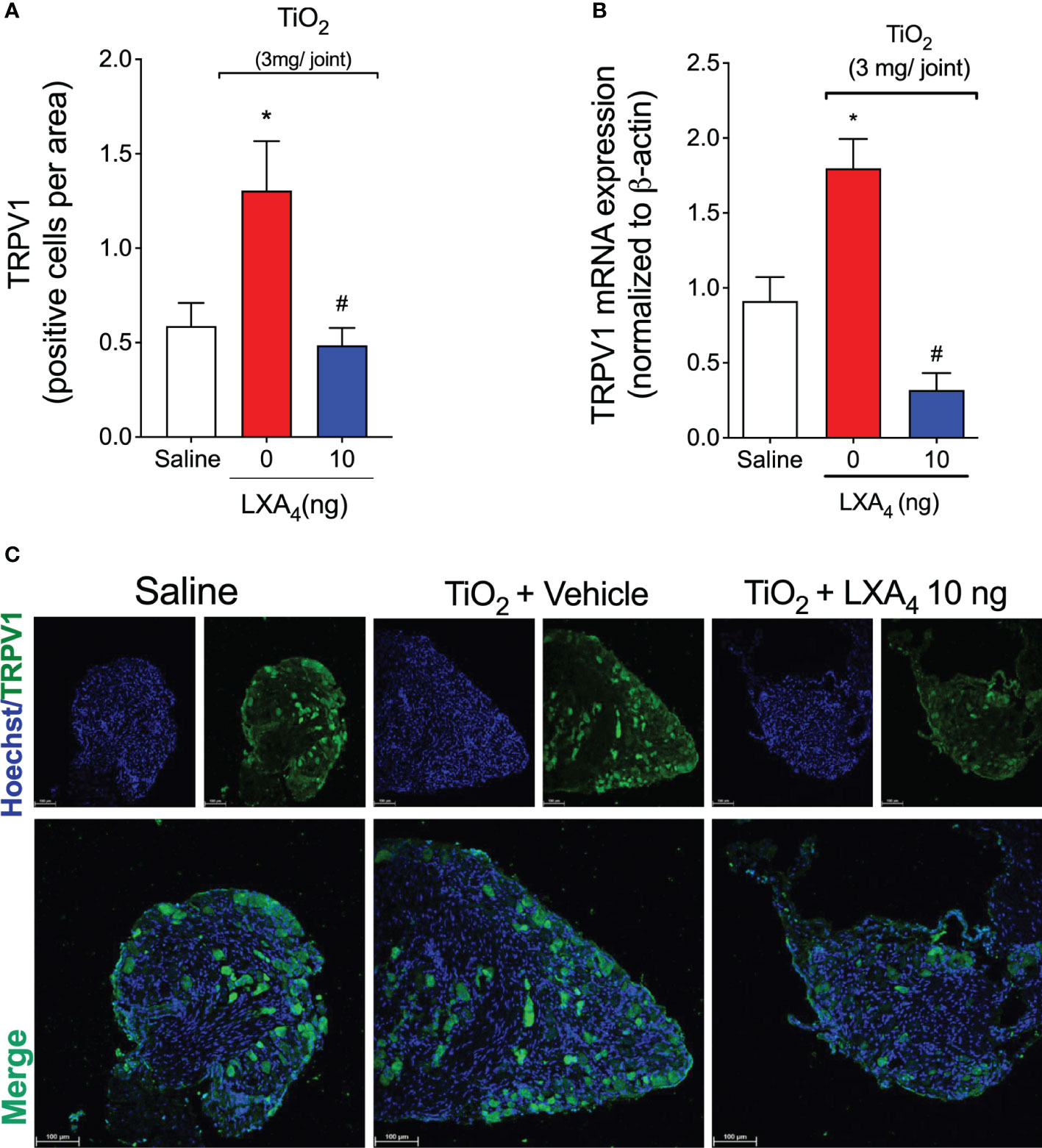
Figure 11 LXA4 inhibits TiO2-induced TRPV1 expression in DRG neurons. On the 2nd day of the model, DRG samples (L4–L6) were dissected for TRPV1 staining by immunofluorescence and for mRNA expression by RT-qPCR. (A) (quantitation) and (C) (representative images) show the number of positive cells per area TRPV1 (green) with nuclear staining by Hoechst 33342 in DRGs (20× magnification with 1.0 zoom in). (B) shows the DRG RT‐qPCR data, demonstrating that LXA4 reduced TiO2‐induced TRPV1 mRNA expression. Results are expressed as mean ± SEM, n = 8 mice per group per experiment, and RT‐qPCR used n = 6 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, one-way ANOVA followed by Tukey’s post-test).
We also investigated whether TRPV1+ neurons co-expressed p-NFκB in the TiO2-induced DRG as a surrogate marker of neuronal activation. Immunofluorescence shows that the intra-articular injection of TiO2 increased the percentage of TRPV1+ neurons co-stained with p-NFκB, and that treatment with LXA4 can reduce it (Figures 12A, B). These data further demonstrate that TRPV1+ neurons are activated in TiO2 inflammation and that LXA4 treatment reduces their activation.
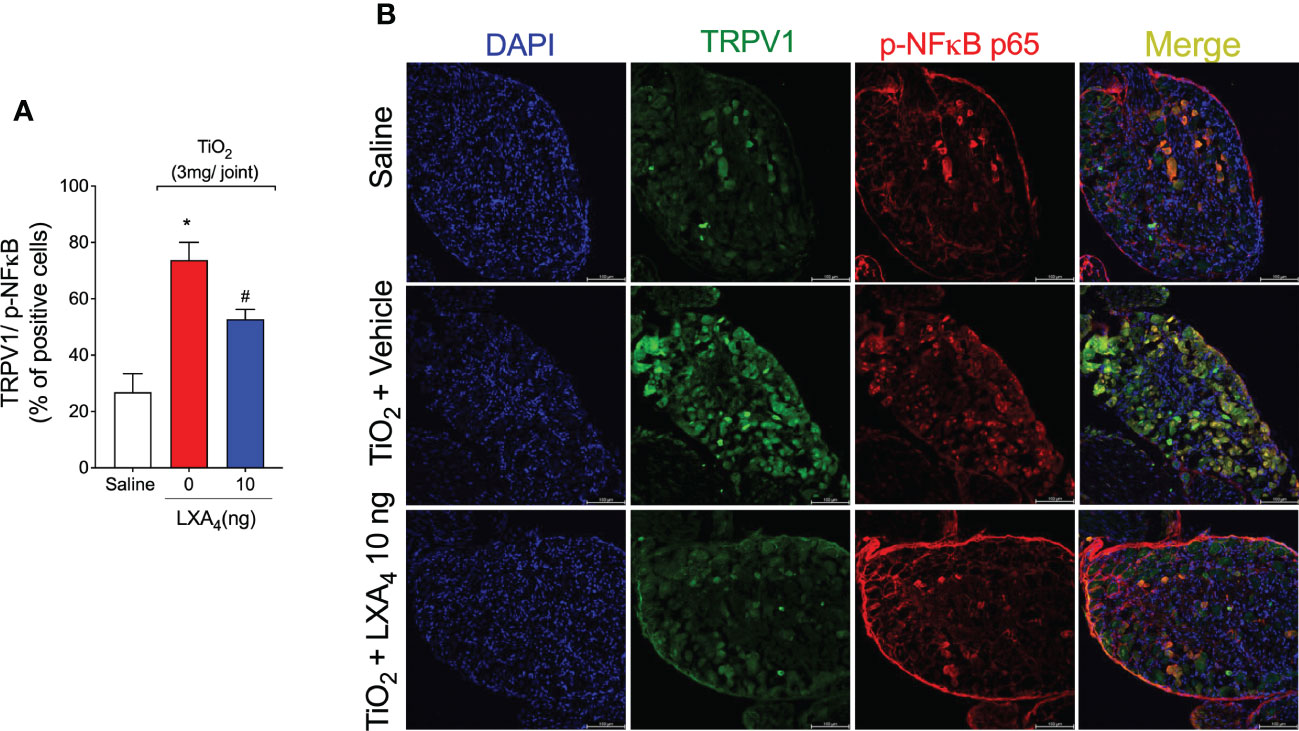
Figure 12 LXA4 reduces TiO2-induced NF-κB activation in TRPV1 positive neurons. On the 2nd day of the model, DRG samples (L4–L6) were dissected for immunofluorescent TRPV1 and p65 p-NF-κB staining. (A) shows the percent of positive cells co-stained with p65 p-NF-κB. (B) shows representative images of TRPV1+ cells (green), p65 p-NF-κB positive cells (red), and the merge of double labeling (TRPV1 and NF‐κB) in DRG samples (20× magnification with 1.0× zoom). DAPI was used for nuclear staining. Results are expressed as mean ± SEM, n = 8 mice per group per experiment, two independent experiments (*p < 0.05 vs. saline group; #p < 0.05 vs. TiO2 group, one-way ANOVA followed by Tukey’s post-test).
TRPA1+ neurons in the dorsal root ganglion are involved in inflammation-induced hyperalgesia in peripheral tissues (65–68). Therefore, to further explore the neuronal mechanism involved in the model and the role of LXA4, we investigated whether LXA4 modulates TRPA1 channels in TiO2-induced arthritis. To achieve this aim, we investigated whether DRG neurons from stimulated mice would present increase of calcium levels in response to AITC (a TRPA1 agonist) stimulation and the modulation by LXA4 (Figure S1). We observed that LXA4 treatment reduced the increased responsiveness of DRG neurons to AITC induced by TiO2 (Figures S1A–C). The treatment reduced by 37% the number of responsive neurons to AITC compared to the TiO2 + vehicle group as per the Venn diagram (Figure S1D). Moreover, TRPA1 staining was enhanced in DRG neurons in the TiO2 group, and one treatment with LXA4 decreased the density of TRPA1 stained neurons (Figures S2A, C). Co-staining of TRPA1 with p-NFκB showed that TiO2 did not induce the activation of NF-κB in TRPA1+ nociceptive neurons (Figures S2B, C). These data show (Figures 9–12; Figures S1, S2) the importance of the function of TRPV1 and TRPA1 ion channels in this model of inflammatory pain and that LXA4 reduced the activity of both TRP ion channels. These results also show that the transcription factor regulation occurring in nociceptive neurons can be different depending on the neuronal population profile, an observation that merits further investigation.
LXA4 administration reduced chronic ongoing TiO2-induced joint edema, mechanical and thermal hyperalgesia, leukocyte recruitment, and histopathological changes. LXA4 activity was explained by a reduction in pro-inflammatory cytokines (TNF‐α, IL‐1β, and IL-6) and an increase in the anti-inflammatory cytokine IL-10. Corroborating these data, LXA4 reduced NFκB activation in synovial fluid leukocytes. In the TiO2 inflammation context, we demonstrated that CD45+ F4/80+ macrophages are the main recruited leukocyte type induced by TiO2, and that 85% of NFκB activation occurs in these cells. LXA4 reduced both cell recruitment and activation of NFκB in CD45+ F4/80+ macrophages. Furthermore, a single treatment with LXA4 significantly restored free-radical scavenging ability (ABTS) and GSH levels. It reduced the production of ROS, accompanied by increased Nrf2 mRNA expression in the knee joint tissue and protein staining in synovial fluid leukocytes, supporting an antioxidant effect. Altogether, this demonstrates that LXA4 has anti-inflammatory and antioxidant effects in TiO2-induced arthritis. Moreover, we show that TiO2 injection increased the production of the LXA4 receptor protein, ALX/FPR2, by TRPV1+ neurons. In DRGs, LXA4 decreased TiO2‐induced mRNA expression and protein staining of the pain‐related ion channel TRPV1. In terms of neuronal function, LXA4 reduced the activation of DRG neurons, as determined by lower baseline levels of calcium influx in DRG, and reduced responsiveness to TRPV1 activation by capsaicin stimulation, and to TRPA1 activation by AITC stimulation. Furthermore, treatment with LXA4 did not induce gastric, hepatic, or renal damage, indicating its safety compared to common side effects of non-steroidal anti-inflammatory drugs.
Intra-articular administration of TiO2 induces a response that resembles prosthesis joint inflammation and pain (13). Pain is a cardinal symptom of joint inflammation and is a direct cause of the decision to seek medical care, limitation of limb function, and decreased quality of life (69). Therefore, the development of novel therapeutics that are effective for optimal pain management is critical in prosthesis wear process-induced arthritis. TiO2 arthritis is, in principle, an aseptic inflammation and the opposite of septic arthritis such as that induced by intraarticular injection of Staphylococcus aureus. Evidence demonstrates that limiting the endogenous production and action of LXA4 by genetic deletion of 5-lipoxygenase and antagonizing the ALX/FPR2 receptor with BOC-2, respectively, improve the immune response against S. aureus by avoiding the downregulation of dendritic cells’ recruitment by LXA4 (70). However, that context is different from that of the present study. Here, we demonstrate a beneficial effect of exogenous LXA4 treatment in aseptic prosthesis arthritis, while that previous study showed that endogenous LXA4 has a detrimental role in septic arthritis. Exemplifying aseptic inflammatory conditions, LXA4 levels were decreased in synovial fluid of patients with rheumatoid arthritis and osteoarthritis, suggesting that downmodulation of LXA4 is a permissive factor to aseptic chronic joint diseases with high and low inflammation profiles (71). These observations agree with the present findings.
Prosthesis wear process-released particles, such as TiO2, activate macrophages to produce various pro-inflammatory mediators, growth factors, and pro-inflammatory lipids (57, 72), and these molecules orchestrate the inflammatory response (73). LXA4 and other agonists of ALX/FPR2 can downregulate those inflammatory mechanisms. LXA4 inhibited synoviocyte proliferation and also decreased the levels of IL-6, IL-1β, and TNF-α in rheumatoid arthritis (74). Of interest, LXA4 downregulates TNF-α-directed neutrophil trafficking (75). The ALX/FPR2 agonist (AT-01-KG) reduced neutrophilic inflammation, CXCL1, and IL-1β production and enhanced neutrophil apoptosis in a model of gout arthritis (76). LXA4 diminishes pain in the non-compressive lumbar disc herniation model by inhibiting production of pro-inflammatory cytokines (TNF-α, and IL-1β) and upregulating IL-10 and transforming growth factor-beta (TGF-β) (38). Treatment with LXA4 also increases anti-inflammatory cytokine (TGF-β and IL-10) levels after exposition to ultraviolet light (77). IL-10 restricts the polarization of M1 macrophages, blocks the IL-33/ST2 axis during arthritis (78), and inhibits neutrophil recruitment, matrix metalloproteinases activity, edema (79), and pain (80). We show that LXA4 reduced TNF-α, IL-1β, and IL-6 levels and increased IL-10 levels in TiO2-induced arthritis. Thus, reducing pro-inflammatory cytokines and increasing anti-inflammatory cytokines, which orchestrate the inflammatory and nociceptive responses, might contribute to LXA4 alleviation of leukocyte recruitment, edema, and mechanical and thermal hyperalgesia. A limitation of the present study is that we did not identify the cells in which cytokine production was downregulated by LXA4. Macrophages and lymphocytes are important cells in the production of both pro-inflammatory (81) and anti-inflammatory (82, 83) cytokines. For instance, macrophages produce pro-inflammatory cytokines in response to TiO2 stimulus (12, 84). Also, M2 macrophages (85, 86) and regulatory T (Treg) cells (87, 88) produce IL-10 to limit inflammation. LXA4 has differential actions in M1 and M2 macrophages. LXA4 reduces the gene expression of pro-inflammatory cytokines in M1 macrophages and increases the IL-10 mRNA expression in M2 macrophages derived from THP-1 cells (89). LXA4 also induces M2 polarization in a model of osteoarthritis (90). Thus, LXA4 can both induce macrophage polarization towards the M2 profile and stimulate these cells to produce IL-10. The overexpression of 15-lipoxygenase in mesenchymal stem cells (MSC) can enhance LXA4 production, and consequently, MSC overexpressing 15-lipoxygeanse can shape the balance between Th17/Treg by increasing Treg and IL-10 production (91). Thus, macrophages and lymphocytes are potential sources of cytokines and targets of LXA4 activity.
Administration of TiO2 particles bypasses the wait for prosthesis wear and reduces the number of animals needed to investigate mechanisms and novel treatments for this condition. Chronic inflammation is responsible for peri-prosthetic osteolysis and aseptic loosening of the prosthesis (92, 93). Macrophage-like synoviocytes are resident cells in the synovium lining. They are responsible for the phagocytosis of prosthetic wear particles, production of pro-inflammatory cytokines such as IL-1β and TNF-α, triggering inflammation, recruitment of immune cells, and activation of fibroblast-like synoviocytes (94, 95). During aseptic loosening, a significant number of macrophages infiltrate into peri-implant tissues (96). In our model, total leukocytes in the synovial cavity were higher in the early stage (2nd day) than in the late stage (30th day). We also observed higher counts of mononuclear cells than neutrophils on the 2nd day post-TiO2 administration, which is unexpected considering the leukocyte recruitment kinetics of most inflammatory responses. We show that the CD45+ F4/80+ macrophage population is the main (up to 70%) recruited leukocyte type in the TiO2-inflammation and seems to be an important target of LXA4 anti-inflammatory effects. Further studies are necessary to investigate the underlying mechanisms for these specific leukocyte kinetics in TiO2 arthritis. LXA4 reduced inflammatory cytokine production induced by TiO2, which lined up well with the reduced p-NFκB staining in synovial fluid leukocytes. NF-κB exerts its transcription factor activity and regulates the expression of various genes encoding pro-inflammatory cytokines, which have been shown to play essential roles in inflammation. Diminished NF-κB activation reduces the production of pro-inflammatory cytokines and downmodulates inflammatory reactions (21). Our findings corroborate prior evidence that LXA4 inhibits NF-κB in other disease models (97–99). Furthermore, LXA4 suppresses the LPS-induced proliferation of RAW264.7 macrophages by targeting the NF-κB pathway (100). The treatment with LXA4 reduced LPS-evoked TNF-α production and inhibited NF-κB activation in a coculture system using RAW264.7 cells and human colon carcinoma cell line (Caco-2) (101). We observed that TiO2 induced NF-κB activation mostly in CD45+ F4/80+ macrophages (85% of the total CD45+ pNF-κB+ cells). LXA4 treatment reduced the NF-κB activation in CD45+ F4/80+ macrophages triggered by TiO2.
Oxidative stress has an essential role in inflammatory pain (102). Reactive oxygen and nitrogen species (ROS and RNS, respectively) produced during inflammation contribute directly to nociceptor neuron activation (103). TiO2 induces lipid peroxidation, DNA damage, and protein breakdown, corroborating the presence of oxidative stress (104). LXA4 increases antioxidant capacity via Nrf2 in various models (33, 37, 77, 105). Herein, we demonstrated the in vivo antioxidant effect of LXA4 and induction of Nrf2, explaining the mechanism of protection against oxidative stress by increasing endogenous antioxidants as per GSH and ABTS assays. GSH is a downstream target of Nrf2 activity (106), and our data on GSH together with the literature (33, 77) guided the choice of investigating Nrf2. In arthritis, synovial fluid cells are crucial in the production of ROS, which can increase the level of NF-κB-dependent pro-inflammatory cytokines and promote the formation of an amplification loop that feeds back to further elevation of additional ROS (107). Prosthesis wear particles can induce oxidative stress in macrophage culture (108). On the other hand, LXA4 treatment increases nuclear translocation of Nrf2 in cardiomyocytes (109). In cultured cortical astrocytes exposed to oxygen-glucose deprivation/recovery insults, LXA4 reduced oxidative stress by enhancing the Nrf2 pathway (37). We show that LXA4 inhibits TiO2-triggered ROS generation and enhances Nrf2 in synovial fluid leukocytes. Altogether, these data indicate that LXA4 enhances Nrf2 expression, and reduces cytokine, ROS production, and, importantly, TiO2-triggered NF-κB activation. The modulation of p-NFκB and Nrf2 by LXA4 may also involve their competition to bind to CREB (cAMP-responsive element-binding protein) (62).
LXA4 has an analgesic effect in various conditions, ranging from acute inflammation (31, 32) to neuropathic pain (110). Our data show that LXA4 has an analgesic effect on ongoing prothesis-wearing-like chronic arthritis (30 days) at 10 ng/animal. LXA4 reduced mechanical and thermal hyperalgesia and provided 2 days of analgesia per treatment. Other SPMs such as maresin (MaR) MaR1 and MaR2 and resolvin D1 and D2 reduce inflammatory pain by inhibiting the expression and/or activity of DRG neurons’ TRPV1 and TRPA1 (54, 111–113). Thus, some SPMs can modulate ion channels to induce analgesia, suggesting that this mechanism should also be investigated for LXA4 in TiO2-induced arthritis. However, to that end, we first needed to ascertain if nociceptive neurons express the ALX/FPR2 receptor in TiO2 arthritis. We observed that ALX/FPR2 receptor staining was increased in TiO2-induced arthritis, and more specifically, TRPV1+ nociceptive neurons express ALX/FPR2 receptor and that TiO2 inflammation enhances the percentage of ALXR+/TRPV1+ neurons. Thus, DRG TRPV1+ neurons are likely more susceptible to LXA4 action during TiO2 arthritis than when uninflamed, supporting the analgesic effect of LXA4. A single post-treatment with LXA4 reduced ongoing DRG neuronal activation (baseline calcium levels) and prevented capsaicin-induced TRPV1 activation of DRG neurons. Explaining the diminished neuronal activation by LXA4, this SPM reduced TiO2‐induced TRPV1 mRNA expression and protein staining (and co-stained with p-NFκB p65) in DRG neurons. To our knowledge, this is the first work to demonstrate that LXA4 reduces TRPV1 channel mRNA expression and protein staining in DRG neurons. Notably, this resulted in diminished TRPV1 activity, causing analgesia. TRPV1 is expressed by approximately 54% of DRG neurons, and TRPA1 is expressed by approximately 22% of DRG neurons. Most TRPA1 channels are co-expressed with TRPV1 in DRG neurons (114) and there is evidence that they can also dimerize as a mechanism of nociceptor sensitization (115). Those TRPA1 channels that are not co-expressed with TRPV1 represent a sub-population of neurons involved in neuropathic pain and not in inflammatory pain (115). Corroborating the literature about the role of TRPA1 in pain and its interaction with TRPV1 (67, 114, 115), as well as the present results on TRPV1, we also observed that TiO2 enhances the neuronal activation by a TRPA1 agonist and TRPA1 staining, both of which were inhibited by LXA4. However, in contrast to what was observed in TRPV1+ neurons, TiO2 did not induce an increase of p-NFκB in TRPA1+ neurons, indicating that the role of the transcription factor NF-κB is likely not the same in all DRG nociceptor neuron populations. Thus, the mechanism of action of LXA4 depends, at least in part, on down-modulating the activity of TRP channels essential to nociceptor neuronal sensitization and chronic pain (116). Our study also contributes to building the concept that targeting ion channels is part of the mechanisms of action of SPM.
We demonstrated that LXA4 has therapeutic effects against ongoing chronic TiO2 arthritis, favorably altering knee joint pathology. Figure 13 is a schematic representation of the mechanism of action of LXA4 in TiO2-induced arthritis. TiO2 triggered the production of cytokines and ROS to induce inflammation and pain. The activation of NF-κB and down-modulation of Nrf2 are mechanisms occurring, at least, in synovial fluid leukocytes that amplify inflammatory cytokines and oxidative stress pathways in response to TiO2. LXA4 targets these pathways. LXA4 could reduce recruitment and NF-κB activation mainly in CD45+ F4/80+ macrophages. We further observed that LXA4 attenuated the staining of the nociceptor-neuron-sensitization-related ion channels TRPV1 and TRPA1, unveiling a hitherto unknown nociceptor neuron mechanism of LXA4. To sum up, this study demonstrated that LXA4 is a promising approach to treating complications related to prosthesis-induced inflammation and pain by inhibiting the activation of synovial fluid leukocytes and primary afferent nociceptor sensory neurons.
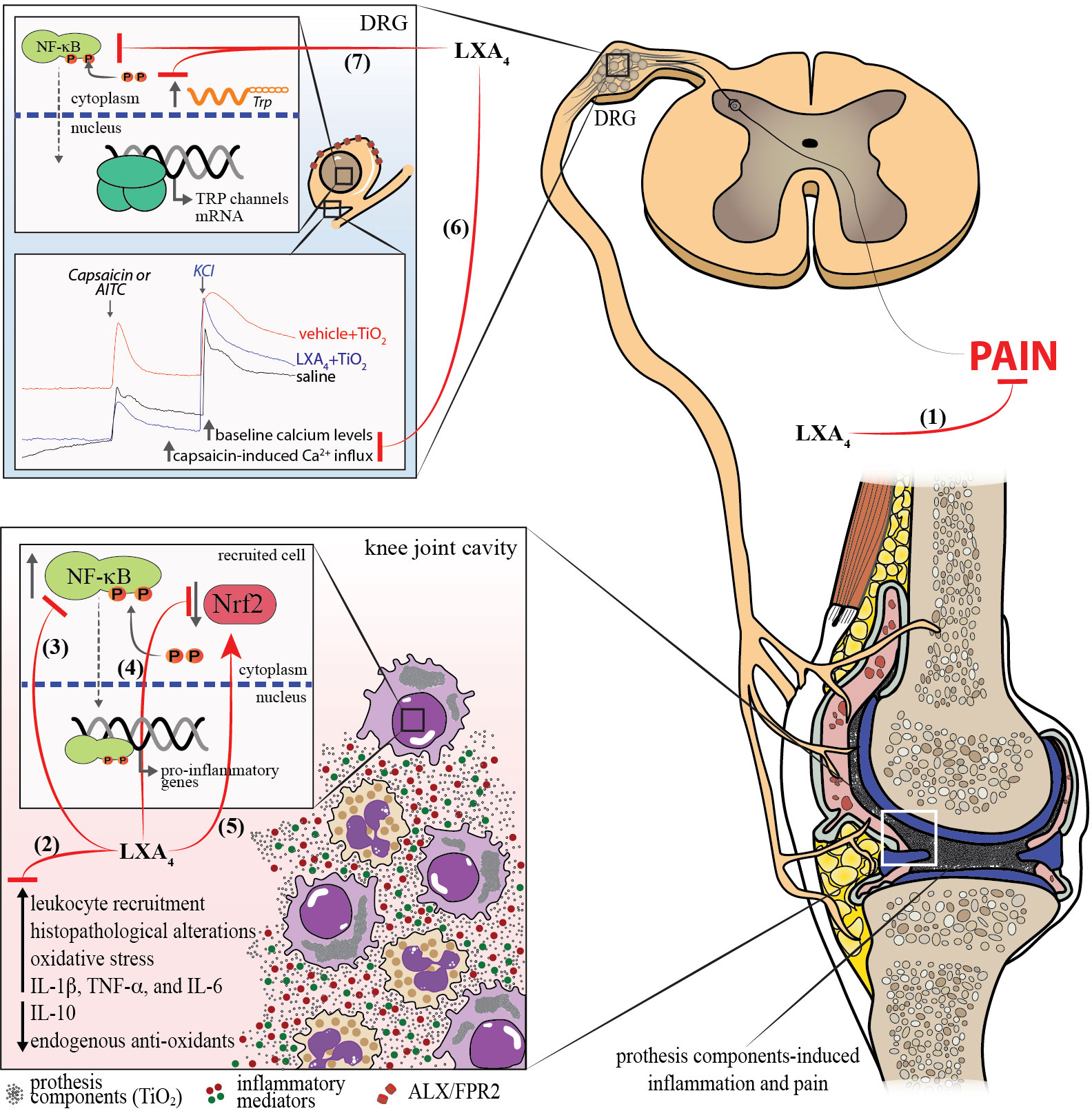
Figure 13 Mechanism of action of LXA4 in TiO2-induced arthritis. (1) LXA4 treatment reduces chronic articular pain induced by TiO2. (2) LXA4 reduces leukocyte recruitment to the knee joint at early and late TiO2-induced arthritis stages, histopathological alterations, oxidative stress, and IL-1β, TNF-α, and IL-6 production, and increases production of endogenous antioxidants and IL-10. These anti-inflammatory findings were supported by the (3) decreased NF-κB activation in macrophages. (4, 5) LXA4 increases the Nrf2 mRNA expression and activation, which were reduced by TiO2. (6) We also demonstrated that LXA4 reduces the activation of DRG neurons in TiO2 inflammation by decreasing the baseline neuronal activation and capsaicin/AITC-induced calcium influx (7) and increasing TRPV1 mRNA expression and protein staining (and co-stained with p-NFκB+), and TRPA1 staining induced by TiO2. TRPV1+ nociceptive neurons express ALX/FPR2 receptor, and TiO2 inflammation enhances the ALXR+/TRPV1+ neurons. Finally, all these mechanisms explain the analgesic (1) and anti-inflammatory (2) effects of LXA4 in this animal model of prosthesis-wearing-process-released components (e.g., TiO2)-induced arthritis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Londrina State University Ethics Committee on Animal Research and Welfare (process number 11147.2016.40).
Performed experiments: TS-S, THZ, MFM, KCA, CRF, MMB, NAA, AF, SB-G, LS-F, SMB. Methodological support: GSC, ACA, JMZ, MSR, RC, FAP-R, WAV. Data analysis: TS-S, THZ, MFM, KCA, CRF, MMB, NAA, AF, SB-G, LS-F, SMB, GSC, ACA, JMZ, MSR, RC, FAP-R, WAV. Supervision: RC, FAP-R, WAV. Funding: GSC, ACA, JMZ, MSR, RC, FAP-R, WAV. Conceived the study: RC, FAP-R, WAV. Wrote the first draft: TS-S, WAV. Editing and approval of final version: All authors.
This study was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; #309633/2021-4; #307852/2019-9; #307689/2022-0; #405027/2021-4; #427946/2018-2); Financiadora de Estudos e Projetos-Apoio à Infraestrutura (FINEP CT-INFRA); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; finance code 001); Programa de Pesquisa para o Sistema Único de Saúde (PPSUS) grant supported by Ministério da Ciência, Tecnologia e Inovação (MCTI), Secretaria da Saúde do Estado do Paraná (SESA-PR), and Parana State Government (Brazil) (agreements #041/2017); and Programa de Apoio a Grupos de Excelência (PRONEX) grant supported by SETI/Fundação Araucária and MCTI/CNPq, and Governo do Estado do Paraná (agreement #014/2017).
The authors thank the core facility CMLP-UEL for access to equipment free of charge and Prof. Mário Sérgio Mantovani for expert opinion on flow cytometry.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.949407/full#supplementary-material
1. Bruyère O, Ethgen O, Neuprez A, Zègels B, Gillet P, Huskin JP, et al. Health-related quality of life after total knee or hip replacement for osteoarthritis: a 7-year prospective study. Arch Orthop Trauma Surg (2012) 132:1583–7. doi: 10.1007/S00402-012-1583-7
2. Fortin PR, Clarke AE, Joseph L, Liang MH, Tanzer M, Ferland D, et al. OUTCOMES OF TOTAL HIP AND KNEE REPLACEMENT preoperative functional status predicts outcomes at six months after surgery. Arthritis Rheum (1999) 42:1722–8. doi: 10.1002/1529-0131
3. Dailiana ZH, Papakostidou I, Varitimidis S, Liaropoulos L, Zintzaras E, Karachalios T, et al. Patient-reported quality of life after primary major joint arthroplasty: a prospective comparison of hip and knee arthroplasty. BMC Musculoskelet Disord (2015) 16:1–8. doi: 10.1186/S12891-015-0814-9/TABLES/6
4. Jones CA, Voaklander DC, Johnston WC, Suarez-Almazor ME. The effect of age on pain, function, and quality of life after total hip and knee arthroplasty. Arch Intern Med (2001) 161:454–60. doi: 10.1001/ARCHINTE.161.3.454
5. Canovas F, Dagneaux L. Quality of life after total knee arthroplasty. Orthop Traumatol Surg Res (2018) 104:S41–6. doi: 10.1016/J.OTSR.2017.04.017
6. van Onna M, Öztürk B, Starmans M, Peeters R, Boonen A. Disease and management beliefs of elderly patients with rheumatoid arthritis and comorbidity: a qualitative study. Clin Rheumatol (2018) 37:2367–72. doi: 10.1007/S10067-018-4167-2/TABLES/1
7. Lübbeke A, Silman AJ, Barea C, Prieto-Alhambra D, Carr AJ. Mapping existing hip and knee replacement registries in Europe. Health Policy (2018) 122:548–57. doi: 10.1016/J.HEALTHPOL.2018.03.010
8. Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the united states from 2005 to 2030. J Bone Joint Surg Am (2007) 89:780–5. doi: 10.2106/JBJS.F.00222
9. Hellman EJ, Capello WN, Feinberg JR. Omnifit cementless total hip arthroplasty. a 10-year average followup. Clin Orthop Relat Res (1999) 364:164–74. doi: 10.1097/00003086-199907000-00022
10. Sansone V, Pagani D, Melato M. The effects on bone cells of metal ions released from orthopaedic implants. a review. Clin Cases Miner Bone Metab (2013) 10:34–40. doi: 10.11138/ccmbm/2013.10.1.034
11. Grande F, Tucci P. Titanium dioxide nanoparticles: a risk for human health? Mini Rev Med Chem (2016) 16:762–9. doi: 10.2174/1389557516666160321114341
12. Dörner T, Haas J, Loddenkemper C, von Baehr V, Salama A. Implant-related inflammatory arthritis. Nat Clin Pract Rheumatol (2006) 2:53–6. doi: 10.1038/NCPRHEUM0087
13. Borghi SM, Mizokami SS, Pinho-Ribeiro FA, Fattori V, Crespigio J, Clemente-Napimoga JT, et al. The flavonoid quercetin inhibits titanium dioxide (TiO2)-induced chronic arthritis in mice. J Nutr Biochem (2018) 53:81–95. doi: 10.1016/J.JNUTBIO.2017.10.010
14. Hawkey CJ. Nonsteroidal anti-inflammatory drug gastropathy. Gastroenterology (2000) 119:521–35. doi: 10.1053/gast.2000.9561
15. Zobdeh F, Eremenko II, Akan MA, Tarasov VV, Chubarev VN, Schiöth HB, et al. Pharmacogenetics and pain treatment with a focus on non-steroidal anti-inflammatory drugs (NSAIDs) and antidepressants: a systematic review. Pharmaceutics (2022) 14. doi: 10.3390/PHARMACEUTICS14061190
16. Bas DB, Su J, Wigerblad G, Svensson CI. Pain in rheumatoid arthritis: models and mechanisms. Pain Manag (2016) 6:265–84. doi: 10.2217/PMT.16.4
17. Dowell D, Ragan KR, Jones CM, Baldwin GT, Chou R. CDC Clinical practice guideline for prescribing opioids for pain - united states, 2022. MMWR Recomm Rep (2022) 71:1–95. doi: 10.15585/mmwr.rr7103a1
18. Pinho-Ribeiro FA, Verri WA, Chiu IM. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol (2017) 38:5–19. doi: 10.1016/J.IT.2016.10.001
19. Kany S, Vollrath JT, Relja B. Cytokines in inflammatory disease. Int J Mol Sci (2019) 20. doi: 10.3390/IJMS20236008
20. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal (2014) 20:1126. doi: 10.1089/ARS.2012.5149
21. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther (2017) 2:17023. doi: 10.1038/SIGTRANS.2017.23
22. Verri WA, Cunha TM, Parada CA, Poole S, Cunha FQ, Ferreira SH. Hypernociceptive role of cytokines and chemokines: targets for analgesic drug development? Pharmacol Ther (2006) 112:116–38. doi: 10.1016/J.PHARMTHERA.2006.04.001
23. Wood JN, Boorman JP, Okuse K, Baker MD. Voltage-gated sodium channels and pain pathways. J Neurobiol (2004) 61:55–71. doi: 10.1002/NEU.20094
24. Schumacher MA. Transient receptor potential channels in pain and inflammation: therapeutic opportunities. Pain Pract (2010) 10:185–200. doi: 10.1111/J.1533-2500.2010.00358.X
25. Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta (BBA) - Mol Basis Dis (2007) 1772:989–1003. doi: 10.1016/J.BBADIS.2007.01.008
26. Brennan EP, Nolan KA, Borgeson E, Gough OS, McEvoy CM, Docherty NG, et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFbetaR1. J Am Soc Nephrol (2013) 24:627–37. doi: 10.1681/ASN.2012060550
27. Chandrasekharan JA, Sharma-Walia N. Lipoxins: nature’s way to resolve inflammation. J Inflamm Res (2015) 8:181–92. doi: 10.2147/JIR.S90380
28. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510:92–101. doi: 10.1038/nature13479
29. Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids (2005) 73:141–62. doi: 10.1016/j.plefa.2005.05.002
30. Jiang X, Li Z, Jiang S, Tong X, Zou X, Wang W, et al. Lipoxin A4 exerts protective effects against experimental acute liver failure by inhibiting the NF-kappaB pathway. Int J Mol Med (2016) 37:773–80. doi: 10.3892/ijmm.2016.2483
31. Abdelmoaty S, Wigerblad G, Bas DB, Codeluppi S, Fernandez-Zafra T, El-Awady el S, et al. Spinal actions of lipoxin A4 and 17(R)-resolvin D1 attenuate inflammation-induced mechanical hypersensitivity and spinal TNF release. PloS One (2013) 8:e75543. doi: 10.1371/journal.pone.0075543
32. Svensson CI, Zattoni M, Serhan CN. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J Exp Med (2007) 204:245–52. doi: 10.1084/jem.20061826
33. Han X, Yao W, Liu Z, Li H, Zhang ZJ, Hei Z, et al. Lipoxin A4 preconditioning attenuates intestinal ischemia reperfusion injury through Keap1/Nrf2 pathway in a lipoxin A4 receptor independent manner. Oxid Med Cell Longev (2016) 2016:9303606. doi: 10.1155/2016/9303606
34. Jin W, Jia Y, Huang L, Wang T, Wang H, Dong Y, et al. Lipoxin A4 methyl ester ameliorates cognitive deficits induced by chronic cerebral hypoperfusion through activating ERK/Nrf2 signaling pathway in rats. Pharmacol Biochem Behav (2014) 124:145–52. doi: 10.1016/j.pbb.2014.05.023
35. Wu SH, Wang MJ, Lu J, Chen XQ. Signal transduction involved in lipoxin A4induced protection of tubular epithelial cells against hypoxia/reoxygenation injury. Mol Med Rep (2017) 15:1682–92. doi: 10.3892/mmr.2017.6195
36. Ye W, Zheng C, Yu D, Zhang F, Pan R, Ni X, et al. Lipoxin A4 ameliorates acute pancreatitis-associated acute lung injury through the antioxidative and anti-inflammatory effects of the Nrf2 pathway. Oxid Med Cell Longev (2019) 2019:2197017. doi: 10.1155/2019/2197017
37. Wu L, Li HH, Wu Q, Miao S, Liu ZJ, Wu P, et al. Lipoxin A4 activates Nrf2 pathway and ameliorates cell damage in cultured cortical astrocytes exposed to oxygen-glucose Deprivation/Reperfusion insults. J Mol Neurosci (2015) 56:848–57. doi: 10.1007/s12031-015-0525-6
38. Miao GS, Liu ZH, Wei SX, Luo JG, Fu ZJ, Sun T. Lipoxin A4 attenuates radicular pain possibly by inhibiting spinal ERK, JNK and NF-kappaB/p65 and cytokine signals, but not p38, in a rat model of non-compressive lumbar disc herniation. Neuroscience (2015) 300:10–8. doi: 10.1016/j.neuroscience.2015.04.060
39. Walker J, Dichter E, Lacorte G, Kerner D, Spur B, Rodriguez A, et al. Lipoxin a4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock (2011) 36:410–6. doi: 10.1097/SHK.0b013e31822798c1
40. Wu SH, Liao PY, Dong L, Chen ZQ. Signal pathway involved in inhibition by lipoxin A(4) of production of interleukins induced in endothelial cells by lipopolysaccharide. Inflamm Res (2008) 57:430–7. doi: 10.1007/s00011-008-7147-1
41. McMahon B, Mitchell S, Brady HR, Godson C. Lipoxins: revelations on resolution. Trends Pharmacol Sci (2001) 22:391–5. doi: 10.1016/S0165-6147(00)01771-5
42. Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med (1994) 180:253. doi: 10.1084/JEM.180.1.253
43. Chiang N, Fierro IM, Gronert K, Serhan CN. Activation of lipoxin a4 receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J Exp Med (2000) 191:1197. doi: 10.1084/JEM.191.7.1197
44. Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med (1997) 185:1693–704. doi: 10.1084/JEM.185.9.1693
45. Guerrero ATG, Verri WA, Cunha TM, Silva TA, Rocha FAC, Ferreira SH, et al. Hypernociception elicited by tibio-tarsal joint flexion in mice: a novel experimental arthritis model for pharmacological screening. Pharmacol Biochem Behav (2006) 84:244–51. doi: 10.1016/J.PBB.2006.05.008
46. Cobelli N, Scharf B, Crisi GM, Hardin J, Santambrogio L. Mediators of the inflammatory response to joint replacement devices. Nat Rev Rheumatol (2011) 7:600–8. doi: 10.1038/NRRHEUM.2011.128
47. Wallace JL, McKnight GW, Bell CJ. Adaptation of rat gastric mucosa to aspirin requires mucosal contact. Am J Physiol (1995) 268:G134–8. doi: 10.1152/AJPGI.1995.268.1.G134
48. Fattori V, Borghi SM, Guazelli CFS, Giroldo AC, Crespigio J, Bussmann AJC, et al. Vinpocetine reduces diclofenac-induced acute kidney injury through inhibition of oxidative stress, apoptosis, cytokine production, and NF-κB activation in mice. Pharmacol Res (2017) 120:10–22. doi: 10.1016/J.PHRS.2016.12.039
49. Manchope MF, Artero NA, Fattori V, Mizokami SS, Pitol DL, Issa JPM, et al. Naringenin mitigates titanium dioxide (TiO2)-induced chronic arthritis in mice: role of oxidative stress, cytokines, and NFkappaB. Inflamm Res (2018) 67:997–1012. doi: 10.1007/s00011-018-1195-y
50. Borghi SM, Carvalho TT, Staurengo-Ferrari L, Hohmann MS, Pinge-Filho P, Casagrande R, et al. Vitexin inhibits inflammatory pain in mice by targeting TRPV1, oxidative stress, and cytokines. J Nat Prod (2013) 76:1141–9. doi: 10.1021/np400222v
51. Casagrande R, Georgetti SR, Verri WA Jr., Jabor JR, Santos AC, Fonseca MJ. Evaluation of functional stability of quercetin as a raw material and in different topical formulations by its antilipoperoxidative activity. AAPS PharmSciTech (2006) 7:E10. doi: 10.1208/pt070110
52. Hohmann MS, Cardoso RD, Pinho-Ribeiro FA, Crespigio J, Cunha TM, Alves-Filho JC, et al. 5-lipoxygenase deficiency reduces acetaminophen-induced hepatotoxicity and lethality. BioMed Res Int (2013) 2013:627046. doi: 10.1155/2013/627046
53. Bussmann AJC, Borghi SM, Zaninelli TH, dos Santos TS, Guazelli CFS, Fattori V, et al. The citrus flavanone naringenin attenuates zymosan-induced mouse joint inflammation: induction of Nrf2 expression in recruited CD45+ hematopoietic cells. Inflammopharmacol (2019) 27:1229–42. doi: 10.1007/S10787-018-00561-6
54. Fattori V, Pinho-Ribeiro FA, Staurengo-Ferrari L, Borghi SM, Rossaneis AC, Casagrande R, et al. The specialised pro-resolving lipid mediator maresin 1 reduces inflammatory pain with a long-lasting analgesic effect. Br J Pharmacol (2019) 176:1728–44. doi: 10.1111/bph.14647
55. Ferraz CR, Carvalho TT, Fattori V, Saraiva-Santos T, Pinho-Ribeiro FA, Borghi SM, et al. Jararhagin, a snake venom metalloproteinase, induces mechanical hyperalgesia in mice with the neuroinflammatory contribution of spinal cord microglia and astrocytes. Int J Biol Macromol (2021) 179:610–9. doi: 10.1016/j.ijbiomac.2021.02.178
56. Bindu S, Mazumder S, Bandyopadhyay U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem Pharmacol (2020) 180. doi: 10.1016/J.BCP.2020.114147
57. Chen O, Donnelly CR, Ji RR. Regulation of pain by neuro-immune interactions between macrophages and nociceptor sensory neurons. Curr Opin Neurobiol (2020) 62:17–25. doi: 10.1016/j.conb.2019.11.006
58. Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin (2007) 45:27–37. doi: 10.1097/AIA.0b013e318034194e
59. Hwang YJ, Jeung YS, Seo MH, Yoon JY, Kim DY, Park JW, et al. Asian Dust and titanium dioxide particles–induced inflammation and oxidative DNA damage in C57BL/6 mice. Inhal Toxicol (2010) 22:1127–33. doi: 10.3109/08958378.2010.528805
60. Sang X, Zheng L, Sun Q, Li N, Cui Y, Hu R, et al. The chronic spleen injury of mice following long-term exposure to titanium dioxide nanoparticles. J BioMed Mater Res A (2012) 100A:894–902. doi: 10.1002/JBM.A.34024
61. Huang KT, Wu CT, Huang KH, Lin WC, Chen CM, Guan SS, et al. Titanium nanoparticle inhalation induces renal fibrosis in mice via an oxidative stress upregulated transforming growth factor-β pathway. Chem Res Toxicol (2015) 28:354–64. doi: 10.1021/TX500287F/ASSET/IMAGES/LARGE/TX-2014-00287F_0010.JPEG
62. Staurengo-Ferrari L, Badaro-Garcia S, Hohmann MSN, Manchope MF, Zaninelli TH, Casagrande R, et al. Contribution of Nrf2 modulation to the mechanism of action of analgesic and anti-inflammatory drugs in pre-clinical and clinical stages. Front Pharmacol (2018) 9:1536. doi: 10.3389/fphar.2018.01536
63. Chiu IM, Heesters BA, Ghasemlou N, von Hehn CA, Zhao F, Tran J, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature (2013) 501:52–7. doi: 10.1038/nature12479
64. Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature (1999) 398:436–41. doi: 10.1038/18906
65. McMahon SB, Wood JN. Increasingly irritable and close to tears: TRPA1 in inflammatory pain. Cell (2006) 124:1123–5. doi: 10.1016/J.CELL.2006.03.006
66. Silverman HA, Chen A, Kravatz NL, Chavan SS, Chang EH. Involvement of neural transient receptor potential channels in peripheral inflammation. Front Immunol (2020) 11:590261/BIBTEX. doi: 10.3389/FIMMU.2020.590261/BIBTEX
67. Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell (2006) 124:1269–82. doi: 10.1016/J.CELL.2006.02.023
68. Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron (2004) 41:849–57. doi: 10.1016/S0896-6273(04)00150-3
69. Hadler NM. Knee pain is the malady - not osteoarthritis. Ann Intern Med (1992) 116:598–9. doi: 10.7326/0003-4819-116-7-598
70. Boff D, Oliveira VLS, Queiroz Junior CM, Galvao I, Batista NV, Gouwy M, et al. Lipoxin A4 impairs effective bacterial control and potentiates joint inflammation and damage caused by staphylococcus aureus infection. FASEB J (2020) 34:11498–510. doi: 10.1096/fj.201802830RR
71. Hashimoto A, Hayashi I, Murakami Y, Sato Y, Kitasato H, Matsushita R, et al. Antiinflammatory mediator lipoxin A4 and its receptor in synovitis of patients with rheumatoid arthritis. J Rheumatol (2007) 34:2144–53.
72. Nich C, Takakubo Y, Pajarinen J, Ainola M, Salem A, Sillat T, et al. Macrophages-key cells in the response to wear debris from joint replacements. J BioMed Mater Res A (2013) 101:3033–45. doi: 10.1002/jbm.a.34599
73. Montecucco F, Mach F. Common inflammatory mediators orchestrate pathophysiological processes in rheumatoid arthritis and atherosclerosis. Rheumatol (Oxford) (2009) 48:11–22. doi: 10.1093/rheumatology/ken395
74. Li J, Sun Q, Zheng C, Bai C, Liu C, Zhao X, et al. Lipoxin A4-mediated p38 MAPK signaling pathway protects mice against collagen-induced arthritis. Biochem Genet (2021) 59:346–65. doi: 10.1007/s10528-020-10016-9
75. Hachicha M, Pouliot M, Petasis NA, Serhan CN. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: regulators of a cytokine-chemokine axis. J Exp Med (1999) 189:1923–30. doi: 10.1084/jem.189.12.1923
76. Galvao I, Melo EM, de Oliveira VLS, Vago JP, Queiroz-Junior C, de Gaetano M, et al. Therapeutic potential of the FPR2/ALX agonist AT-01-KG in the resolution of articular inflammation. Pharmacol Res (2021) 165:105445. doi: 10.1016/j.phrs.2021.105445
77. Martinez RM, Fattori V, Saito P, Melo CBP, Borghi SM, Pinto IC, et al. Lipoxin A4 inhibits UV radiation-induced skin inflammation and oxidative stress in mice. J Dermatol Sci (2018) 91:164–74. doi: 10.1016/j.jdermsci.2018.04.014
78. Chen S, Chen B, Wen Z, Huang Z, Ye L. IL-33/ST2-mediated inflammation in macrophages is directly abrogated by IL-10 during rheumatoid arthritis. Oncotarget (2017) 8:32407–18. doi: 10.18632/oncotarget.16299
79. Sodin-Semrl S, Taddeo B, Tseng D, Varga J, Fiore S. Lipoxin A4 inhibits IL-1 beta-induced IL-6, IL-8, and matrix metalloproteinase-3 production in human synovial fibroblasts and enhances synthesis of tissue inhibitors of metalloproteinases. J Immunol (2000) 164:2660–6. doi: 10.4049/jimmunol.164.5.2660
80. Poole S, Cunha FQ, Selkirk S, Lorenzetti BB, Ferreira SH. Cytokine-mediated inflammatory hyperalgesia limited by interleukin-10. Br J Pharmacol (1995) 115:684–8. doi: 10.1111/j.1476-5381.1995.tb14987.x
81. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta (BBA) - Mol Cell Res (2014) 1843:2563–82. doi: 10.1016/J.BBAMCR.2014.05.014
82. Duque GA, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol (2014) 5:491/BIBTEX. doi: 10.3389/FIMMU.2014.00491/BIBTEX
83. Shaw DM, Merien F, Braakhuis A, Dulson D. T-Cells and their cytokine production: the anti-inflammatory and immunosuppressive effects of strenuous exercise. Cytokine (2018) 104:136–42. doi: 10.1016/J.CYTO.2017.10.001
84. Schoenenberger AD, Schipanski A, Malheiro V, Kucki M, Snedeker JG, Wick P, et al. Macrophage polarization by titanium dioxide (TiO2) particles: size matters. ACS Biomater Sci Eng (2016) 2:908–19. doi: 10.1021/ACSBIOMATERIALS.6B00006/ASSET/IMAGES/LARGE/AB-2016-00006S_0006.JPEG
85. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(Lps+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol (2019) 10:1084/BIBTEX. doi: 10.3389/FIMMU.2019.01084/BIBTEX
86. da Silva MD, Bobinski F, Sato KL, Kolker SJ, Sluka KA, Santos ARS. IL-10 cytokine released from M2 macrophages is crucial for analgesic and anti-inflammatory effects of acupuncture in a model of inflammatory muscle pain. Mol Neurobiol (2015) 51:19–31. doi: 10.1007/S12035-014-8790-X
87. Ng THS, Britton GJ, Hill EV, Verhagen J, Burton BR, Wraith DC. Regulation of adaptive immunity; the role of interleukin-10. Front Immunol (2013) 4:129/BIBTEX. doi: 10.3389/FIMMU.2013.00129/BIBTEX
88. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity (2011) 34:566. doi: 10.1016/J.IMMUNI.2011.03.018
89. Aubeux D, Tessier S, Pérez F, Geoffroy V, Gaudin A. In vitro phenotypic effects of lipoxin A4 on M1 and M2 polarized macrophages derived from THP-1. Mol Biol Rep (2023) 50:1–10. doi: 10.1007/S11033-022-08041-5
90. Shen P, Jia S, Wang Y, Zhou X, Zhang D, Jin Z, et al. Mechanical stress protects against chondrocyte pyroptosis through lipoxin A4 via synovial macrophage M2 subtype polarization in an osteoarthritis model. BioMed Pharmacother (2022) 153. doi: 10.1016/J.BIOPHA.2022.113361
91. Özdemir AT, Nalbantsoy A, Özgül Özdemir RB, Berdeli A. Effects of 15-lipoxygenase overexpressing adipose tissue mesenchymal stem cells on the Th17 / treg plasticity. Prostaglandins Other Lipid Mediat (2022) 159. doi: 10.1016/J.PROSTAGLANDINS.2021.106610
92. Gallo J, Goodman SB, Konttinen YT, Raska M. Particle disease: biologic mechanisms of periprosthetic osteolysis in total hip arthroplasty. Innate Immun (2013) 19:213–24. doi: 10.1177/1753425912451779
93. Ulrich SD, Seyler TM, Bennett D, Delanois RE, Saleh KJ, Thongtrangan I, et al. Total hip arthroplasties: what are the reasons for revision? Int Orthop (2008) 32:597–604. doi: 10.1007/s00264-007-0364-3
94. Tu J, Hong W, Zhang P, Wang X, Körner H, Wei W. Ontology and function of fibroblast-like and macrophage-like synoviocytes: how do they talk to each other and can they be targeted for rheumatoid arthritis therapy? Front Immunol (2018) 9:1467/BIBTEX. doi: 10.3389/FIMMU.2018.01467/BIBTEX
95. Landgraeber S, Jäger M, Jacobs JJ, Hallab NJ. The pathology of orthopedic implant failure is mediated by innate immune system cytokines. Mediators Inflamm (2014) 2014. doi: 10.1155/2014/185150
96. Jämsen E, Pajarinen J, Lin Th, Lo CW, Nabeshima A, Lu L, et al. Effect of aging on the macrophage response to titanium particles. J Orthopaedic Research® (2020) 38:405–16. doi: 10.1002/JOR.24461
97. Song Y, Yang Y, Cui Y, Gao J, Wang K, Cui J. Lipoxin A4 methyl ester reduces early brain injury by inhibition of the nuclear factor kappa b (NF-κB)-Dependent matrix metallopeptidase 9 (MMP-9) pathway in a rat model of intracerebral hemorrhage. Med Sci Monit (2019) 25:1838–47. doi: 10.12659/MSM.915119
98. Miao G, Liu Z, Wei S, Luo J, Fu Z, Sun T. Lipoxin A4 attenuates radicular pain possibly by inhibiting spinal ERK, JNK and NF-κB/p65 and cytokine signals, but not p38, in a rat model of non-compressive lumbar disc herniation. Neuroscience (2015) 300:10–8. doi: 10.1016/J.NEUROSCIENCE.2015.04.060
99. Jiang X, Li Z, Jiang S, Tong X, Zou X, Wang W, et al. Lipoxin A4 exerts protective effects against experimental acute liver failure by inhibiting the NF-b pathway. Int J Mol Med (2016) 37:773–80. doi: 10.3892/IJMM.2016.2483/HTML
100. Huang YH, Wang HM, Cai ZY, Xu FY, Zhou XY. Lipoxin A4 inhibits NF-kappaB activation and cell cycle progression in RAW264.7 cells. Inflammation (2014) 37:1084–90. doi: 10.1007/s10753-014-9832-2
101. Kure I, Nishiumi S, Nishitani Y, Tanoue T, Ishida T, Mizuno M, et al. Lipoxin A(4) reduces lipopolysaccharide-induced inflammation in macrophages and intestinal epithelial cells through inhibition of nuclear factor-kappaB activation. J Pharmacol Exp Ther (2010) 332:541–8. doi: 10.1124/jpet.109.159046
102. Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med (2011) 51:951–66. doi: 10.1016/J.FREERADBIOMED.2011.01.026
103. Maioli NA, Zarpelon AC, Mizokami SS, Calixto-Campos C, Guazelli CF, Hohmann MS, et al. The superoxide anion donor, potassium superoxide, induces pain and inflammation in mice through production of reactive oxygen species and cyclooxygenase-2. Braz J Med Biol Res (2015) 48:321–31. doi: 10.1590/1414-431X20144187
104. Dubey A, Goswami M, Yadav K, Chaudhary D. Oxidative stress and nano-toxicity induced by TiO2 and ZnO on WAG cell line. PloS One (2015) 10:e0127493. doi: 10.1371/journal.pone.0127493
105. Zong H, Li X, Lin H, Hou C, Ma F. Lipoxin A4 pretreatment mitigates skeletal muscle ischemia-reperfusion injury in rats. Am J Transl Res (2017) 9:1139–50.
106. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal (2018) 29:1727–45. doi: 10.1089/ars.2017.7342
107. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res (2011) 21:103–15. doi: 10.1038/CR.2010.178
108. Luo G, Li Z, Wang Y, Wang H, Zhang Z, Chen W, et al. Resveratrol protects against titanium particle-induced aseptic loosening through reduction of oxidative stress and inactivation of NF-κB. Inflammation (2016) 39:775–85. doi: 10.1007/S10753-016-0306-6/FIGURES/6
109. Chen XQ, Wu SH, Zhou Y, Tang YR. Lipoxin A4-induced heme oxygenase-1 protects cardiomyocytes against Hypoxia/Reoxygenation injury via p38 MAPK activation and Nrf2/ARE complex. PloS One (2013) 8:e67120. doi: 10.1371/JOURNAL.PONE.0067120
110. Martini AC, Berta T, Forner S, Chen G, Bento AF, Ji RR, et al. Lipoxin A4 inhibits microglial activation and reduces neuroinflammation and neuropathic pain after spinal cord hemisection. J Neuroinflamm (2016) 13. doi: 10.1186/S12974-016-0540-8
111. Zaninelli TH, Fattori V, Saraiva-Santos T, Badaro-Garcia S, Staurengo-Ferrari L, Andrade KC, et al. RvD1 disrupts nociceptor neuron and macrophage activation and neuroimmune communication, reducing pain and inflammation in gouty arthritis in mice. Br J Pharmacol (2022) 179:4500–15. doi: 10.1111/BPH.15897
112. Fattori V, Zaninelli TH, Ferraz CR, Brasil-Silva L, Borghi SM, Cunha JM, et al. Maresin 2 is an analgesic specialized pro-resolution lipid mediator in mice by inhibiting neutrophil and monocyte recruitment, nociceptor neuron TRPV1 and TRPA1 activation, and CGRP release. Neuropharmacology (2022) 216. doi: 10.1016/J.NEUROPHARM.2022.109189
113. Payrits M, Horváth Á, Biró-Sütő T, Erostyák J, Makkai G, Sághy É, et al. Resolvin D1 and D2 inhibit transient receptor potential vanilloid 1 and ankyrin 1 ion channel activation on sensory neurons via lipid raft modification. Int J Mol Sci (2020) 21:5019. doi: 10.3390/IJMS21145019
114. Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Högestätt ED, et al. Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci U.S.A. (2005) 102:12248–52. doi: 10.1073/PNAS.0505356102
115. Patil MJ, Salas M, Bialuhin S, Boyd JT, Jeske NA, Akopian AN. Sensitization of small-diameter sensory neurons is controlled by TRPV1 and TRPA1 association. FASEB J (2020) 34:287–302. doi: 10.1096/FJ.201902026R
Keywords: lipoxin A4, TiO2, ALX/FPR2, inflammation, TRPV1, ROS
Citation: Saraiva-Santos T, Zaninelli TH, Manchope MF, Andrade KC, Ferraz CR, Bertozzi MM, Artero NA, Franciosi A, Badaro-Garcia S, Staurengo-Ferrari L, Borghi SM, Ceravolo GS, Andrello AC, Zanoveli JM, Rogers MS, Casagrande R, Pinho-Ribeiro FA and Verri WA Jr (2023) Therapeutic activity of lipoxin A4 in TiO2-induced arthritis in mice: NF-κB and Nrf2 in synovial fluid leukocytes and neuronal TRPV1 mechanisms. Front. Immunol. 14:949407. doi: 10.3389/fimmu.2023.949407
Received: 20 May 2022; Accepted: 25 May 2023;
Published: 14 June 2023.
Edited by:
Roberto César Pereira Lima-Júnior, Federal University of Ceara, BrazilReviewed by:
Takayoshi Masuoka, Kanazawa Medical University, JapanCopyright © 2023 Saraiva-Santos, Zaninelli, Manchope, Andrade, Ferraz, Bertozzi, Artero, Franciosi, Badaro-Garcia, Staurengo-Ferrari, Borghi, Ceravolo, Andrello, Zanoveli, Rogers, Casagrande, Pinho-Ribeiro and Verri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Waldiceu A. Verri Jr, d2F2ZXJyaUB1ZWwuYnI=; Felipe A. Pinho-Ribeiro, ZGZlbGlwZUB3dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.