 Cui Zhao
Cui Zhao Chen Liu
Chen Liu- Department of Neonatology, Children’s Hospital Affiliated to Shandong University, Jinan, China
Background: Neonatal-onset multisystem inflammatory disease (NOMID) is a rare and severe autoinflammatory disease caused by mutations of the NLRP3 gene and is characterized by a skin rash, fever, arthropathy, and neurologic manifestations. We herein report a neonatal case with recurrent rash, fever, and meningitis from 12 h after birth, and NOMID was diagnosed during the neonatal period. We also reviewed the clinical characteristics and genetic mutations of previously reported Chinese neonates with NOMID.
Case presentation and literature review: NOMID is rare in China, and there have been over 100 cases uncovered thus far, including ours. The patient we reported here was the youngest among the confirmed Chinese cases and had the de novo mutation c.1210G>C (p.V404L) in exon 4 of the NLRP3 gene, which has not been reported previously. All 25 patients manifested recurrent urticaria-like rash, and 24 were febrile. Of the 23 patients with genetic data available, all had NLRP3 mutations. The primary treatment of these patients entailed glucocorticoids and immunosuppressants; however, the IL-1 inhibitor was rarely used due to its current unavailability in China. One patient was cured by umbilical cord blood stem cell transplantation (UCBT), which provided an alternative treatment.
Conclusion: We recommend that NOMID be considered for neonates with recurrent rash, fever, and aseptic meningitis. However, further research on underlying mechanisms and therapeutic regimens in China is necessary to provide improved management.
1 Introduction
Neonatal-onset multisystem inflammatory disease (NOMID), also called chronic infantile neurological cutaneous articular (CINCA) syndrome, is a rare autoinflammatory disease that may be grouped into working categories according to its major pathogenesis. Two other conditions referred to as familial cold autoinflammatory syndrome (FCAS) and Muckle-Wells syndrome (MWS)—together with NOMID—are known collectively as cryopyrin-associated periodic syndromes (CAPS), the subserving mechanism of which is associated with disorders of inflammasomes and related interleukin (IL)-1 family cytokines due to mutations in the NLRP3 gene. The clinical features overlap and include recurrent episodes of unexplained fever, urticaria rash, and joint involvement, with NOMID the severest and rarest form of CAPS. Although characteristics usually appear within the first hours/days of life, they are rarely diagnosed at the neonatal stage, particularly in China, and the youngest patient we found was three months of age. Herein we describe a neonatal case of NOMID with a novel NLRP3 mutation and present a review of all cases of NOMID reported in China.
2 Case presentation
A newborn girl, 15 days of age, was transferred to the neonatal intensive care unit (NICU) of our hospital because of recurrent fever, rash, and abnormal cerebrospinal fluid (CSF) parameters.
The patient was born to a 20-year-old mother by spontaneous vaginal delivery after an uncomplicated gestation of 39 + 3/7 weeks. Membranes ruptured 19h prior to delivery. Because of a rash covering her entire body starting 12 hours after birth, the girl was admitted to the NICU of the local hospital. Initial laboratory results showed a white blood cell (WBC) count of 23.07×109/L (60.9% neutrophils) and a C-reactive protein (CRP) concentration of 23.7 mg/L. Due to the possibility of infection, piperacillin-tazobactam was started empirically, and on day three the baby developed fever; sepsis was then diagnosed, and a lumbar puncture was subsequently performed. CSF showed a WBC count of 292×106/L, a glucose concentration of 2.62 mmol/L (reference range, 2.8 to 4.4), and protein of 1.29 g/L (reference range, 0.15 to 0.45). Since the CSF findings suggested bacterial meningitis, meropenem replaced piperacillin-tazobactam, and intravenous immunoglobulin (IVIG) was administered. However, at day 15, her temperature and rash were not completely controlled, and CRP remained positive, although the CSF WBC count decreased to 112×106/L. The patient was then transferred to our hospital for further evaluation and treatment.
On arrival at the NICU of our hospital, we noted that the patient was afebrile, showed stable cardio-respiratory function, and presented a normal neurologic status. Scattered rashes were still present on her axillary skin. Based on her symptoms and laboratory findings, although the baby was diagnosed with neonatal bacterial meningitis and received meropenem plus vancomycin, treatment was unsuccessful; the neonate still suffered an obvious urticarial-like rash (Figure 1) and chronic recurrent fever. In addition, the CSF WBC rose to 1719×106/L, and CRP, erythrocyte sedimentation rate (ESR), and serum amyloid A (SAA) were also elevated. All microbiologic tests were negative, including blood culture, CSF culture, and metagenomic next-generation sequencing (mNGS) of CSF. IgM antibodies to toxoplasmosis, cytomegalovirus (CMV), rubella, and herpes simplex virus (HSV) were negative. Hepatitis B serology was negative. Serum levels of procalcitonin (PCT), (1,3)-beta-D-glucan, alanine aminotransferase, aspartate aminotransferase, direct bilirubin, and coagulation tests were normal—as were the results of antinuclear antibodies. The baby’s electrocardiogram, echocardiography, electroencephalogram, and ophthalmologic evaluations were also normal—as were auditory evoked potentials (AEPs) and visual evoked potentials (VEPs). Cerebral MRI was normal except for enlarged peri-cerebral spaces. Due to the unsatisfactory effect of anti-infection and absence of evidence of infection (including bacteria, virus, fungus, tuberculosis, etc.), autoinflammatory diseases quickly rose to the top of differential diagnosis, particularly when we discovered that dexamethasone could partially relieve the symptoms. Genetic testing ultimately confirmed our suspicion, showing the heterozygote mutation c.1210G>C (p.V404L) in exon 4 of the NLRP3 gene (Figure 2) and the electropherograms of the V404L mutation in samples from the patient, father, and mother are shown in Figure 3. The REVEL and PolyPhen-2 software tools were used for prediction of functional effects of this mutation. Although the algorithms showed that the mutation was benign, the variant was de novo, it was absent from a large general population by searching publicly available population databases (including the gnomAD v2.1 data set and the 1000 Genomes data set). Therefore, this mutation was determined to be likely pathogenic according to the ACMG Standards and Guidelines. The gene-mutation analysis findings, together with the clinical features, led to the diagnosis of NOMID.
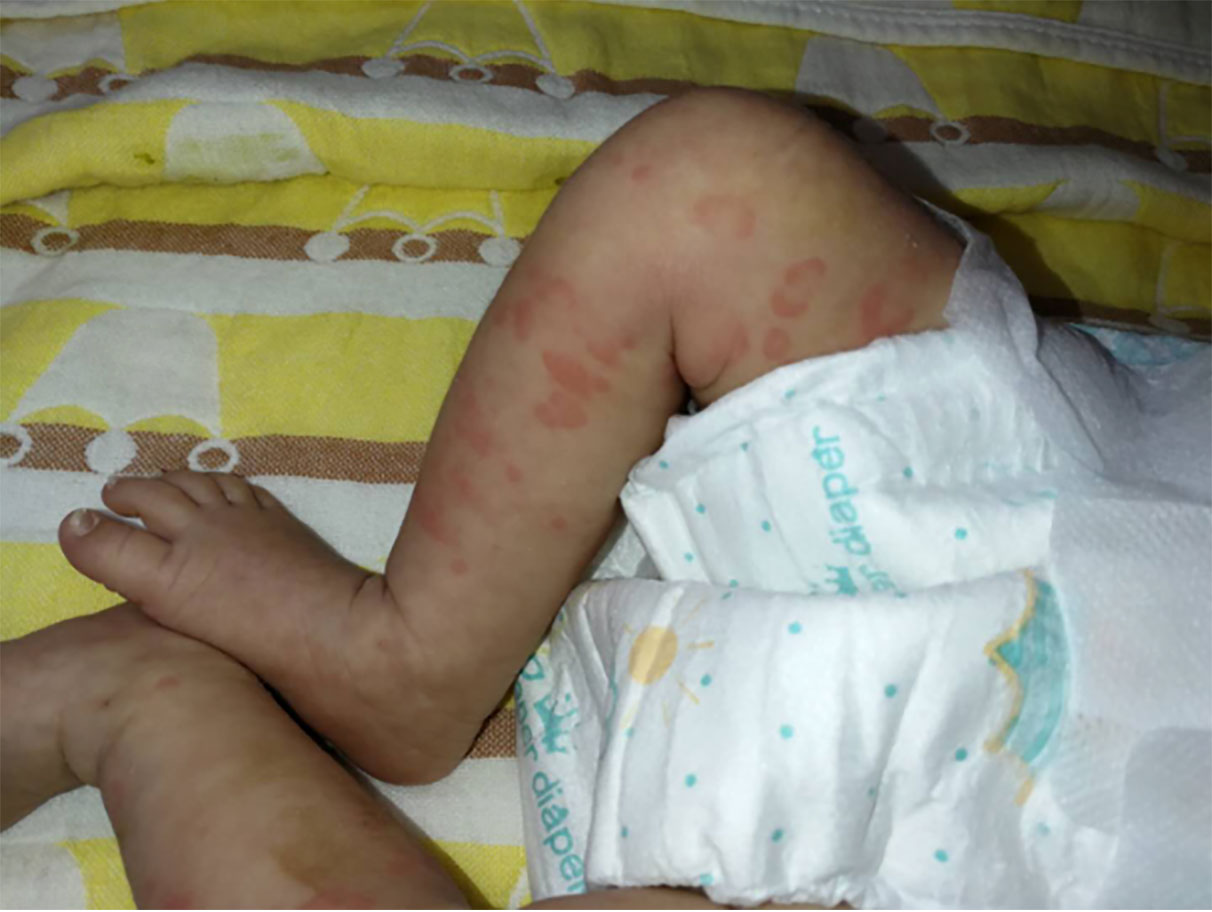
Figure 1 Cutaneous rash in neonatal NOMID patient at 18 days of life.
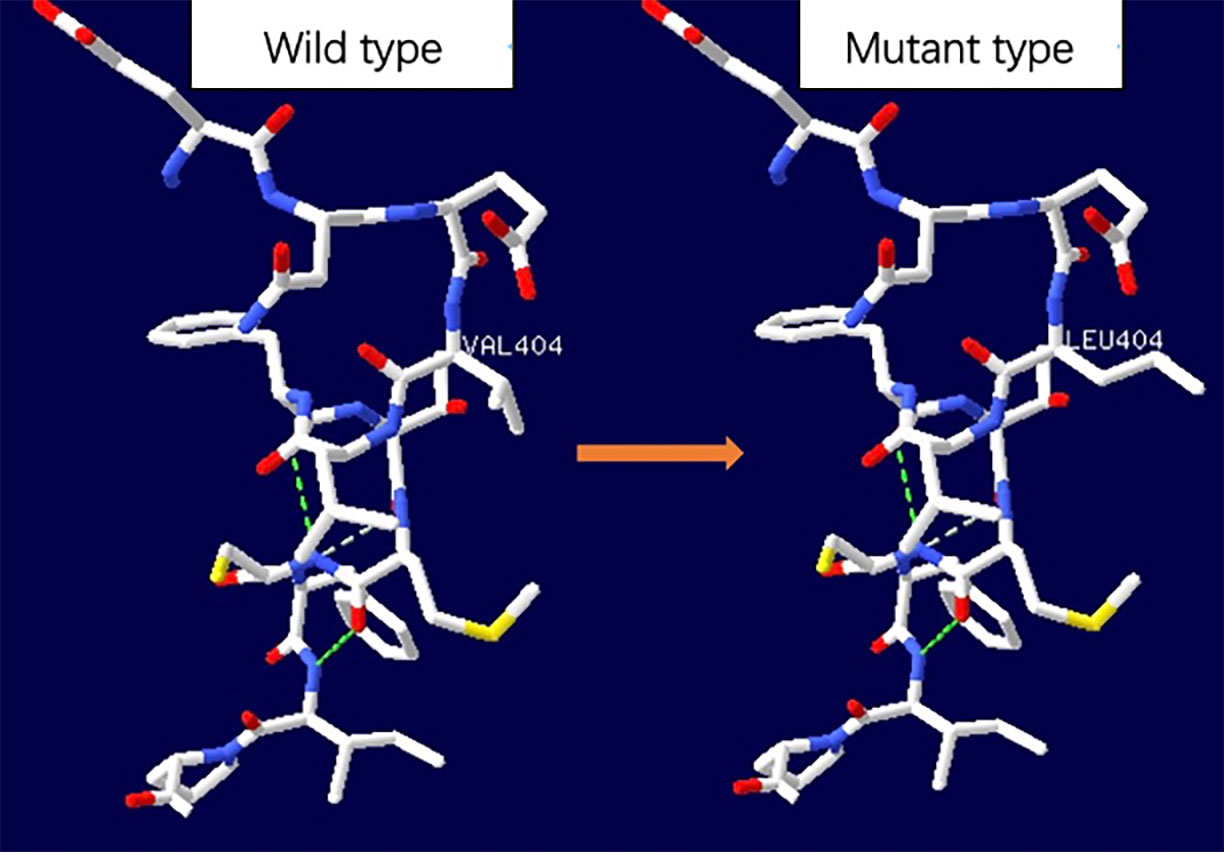
Figure 2 Space distribution of missense variant in NLRP3 protein. The dotted line represented hydrogen bond. No significant hydrogen bond loss was found as predicted by SWISS-MODEL, but the side chain changed.
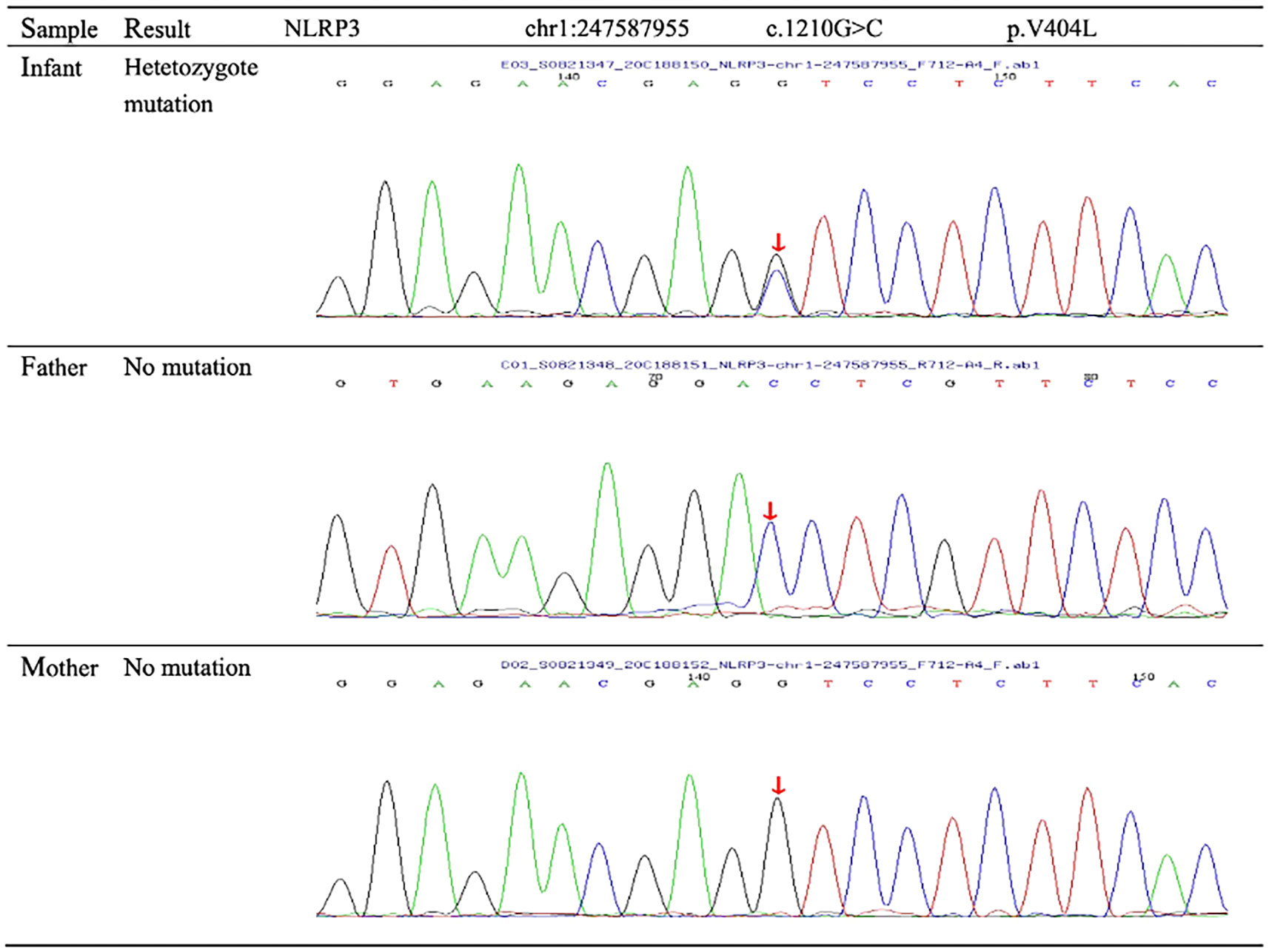
Figure 3 Sequence diagrams for the patient and parents. Genetic testing revealed the patient’s novel missense mutation of c.1210G>C (p.V404L) in the NLRP3 gene. No mutations of the NLRP3 gene were found in the parents.
On day 58 (i.e., after 43 days of treatment), the baby was discharged home with a normal temperature, reduced urticarial rash, and normal CRP, SAA, and ESR—although CSF WBC count was still abnormal at 45×106/L. The baby also showed recurrent subtle rashes and mild anemia but a normal temperature and normal acute-phase reactants when we followed up after two weeks of leaving the hospital. Due to the unavailability of anakinra and canakinumab in China and the difficulty to purchase them from abroad due to COVID-19, the baby went to another hospital to receive anti-TNF-a agent (etanercept), which had help partial patients improve symptoms.
3 Literature review
We collected previous Chinese reports of NOMID by searching PubMed, Wanfang, and China National Knowledge Infrastructure online literature databases and presented a summary of key baseline indices, clinical manifestations, laboratory features, and genetic analyses (Table 1). We have thus far counted 25 NOMID patients from Chinese reports, including our current case and the onset time ranged from 3 hours after birth to 6 years of age, with the age at diagnosis from one month to 16 years. All patients had recurrent urticaria-like rash, and 24 had fever—with other manifestations that included meningitis or CNS manifestations in 22 patients; musculoskeletal involvement in 17 patients; eye involvement in 15 patients; hearing loss in 15 patients; impaired growth in 14 patients; abnormal facies with frontal bossing, protruding eyes, and saddle-shaped nose in 13 patients; cognitive disability in 8 patients; and lymphadenopathy or hepatosplenomegaly in 8 patients. The onset time of the other 24 NOMID patients reported in China ranged from 3 hours after birth to 6 years. Most of them presented clinical features within half one month. Two patients had later onset time: one was 2 years and 5 months old, the other one was 6 years old. They were diagnosed with NOMID not MWS based on the severe clinical features (the frontal bossing, impaired growth, CNS manifestations) and NLRP3 mutation. Despite the early onset time, the average time at diagnosis was delayed to 6.1 years. Of the 23 cases with genotype available, all exhibited NLRP3 mutations, including the novel variant c.1210G>C (p.V404L) in our case, which had not been previously reported. One or more drugs were administered to the 25 patients, including corticosteroids, methotrexate, cyclosporine, and thalidomide. Only 2 patients received treatment with canakinumab (one of the anti-IL-treatment medications) to control the symptoms. Two patients were provided etanercept (an anti-TNF-a therapeutic), successfully alleviating clinical symptoms and systemic inflammation. One patient was cured by umbilical cord blood stem cell transplantation (UCBT).
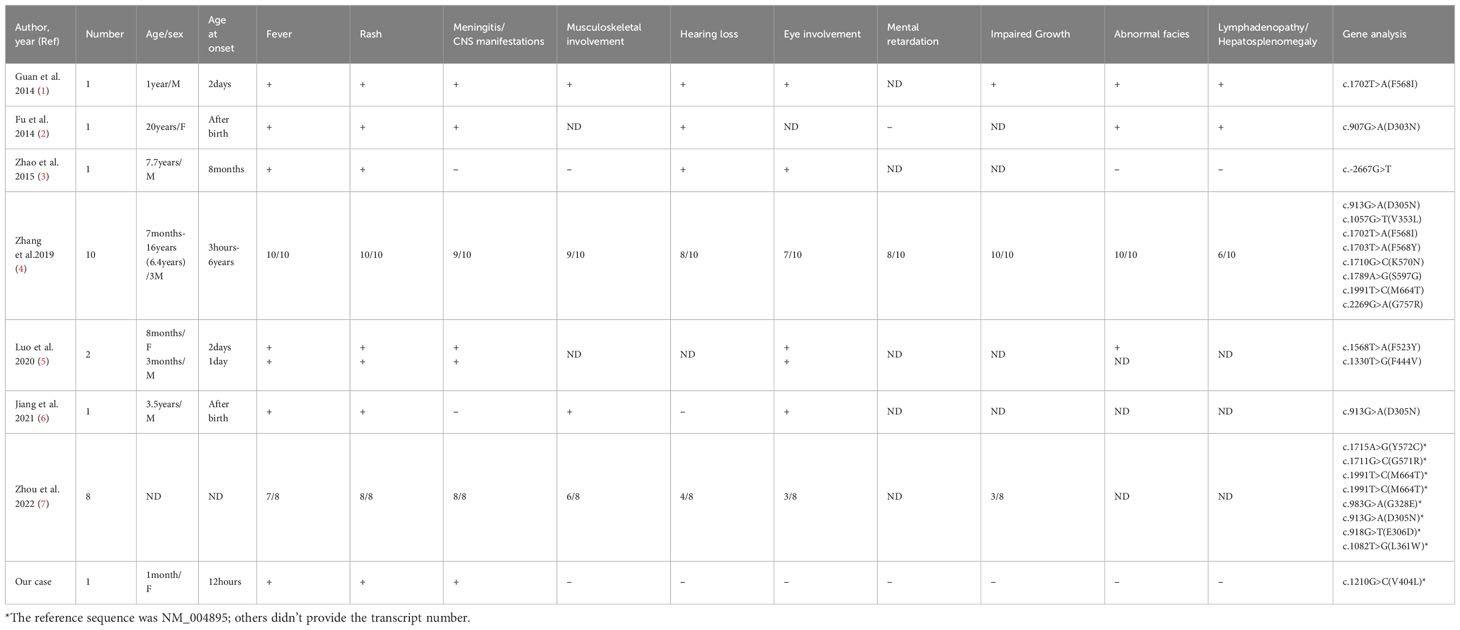
Table 1 Clinical symptoms and laboratory findings in 25 Chinese patients with NOMID.
4 Discussion
NOMID, as the rarest but most severe CAPS phenotype, has been reported in over 100 cases since 1973 when NOMID was first described (4, 8–11), and the first Chinese patient with NOMID was reported in 2014 (1). However, an insufficient understanding and lack of effectively targeted treatment in China often led to permanent organ damage and a poor prognosis (4). We recommend that more doctors (and not only clinical immunologists) pay close attention to this orphan disease because these patients may initially consult various medical providers, including rheumatologists, neonatologists, dermatologists, ophthalmologists, and otolaryngologists.
NOMID patients are generally present at or near the time of birth with recurrent fever, urticarial-like rash, and sustained elevations of acute phase reactants that are easily misdiagnosed as neonatal infections. Intrauterine-onset necrotizing funisitis was recently reported as the first symptom of a newborn with NOMID (12). Although the patients can be identified by typical “facies”, central nervous system (CNS) symptoms, progressive hearing and vision loss, impaired growth, and amyloidosis (8, 13), we expect to be able to make a diagnosis before organ damage occurs. When there is no response to antibiotic therapy, NOMID should be evoked as a differential diagnosis by astute clinicians. In our case, the baby was misdiagnosed with bacterial meningitis initially because of her abnormal CSF, but the diagnosis was questioned since the trend observed for the CSF was irrelevant to anti-infective treatment. Fortunately, we correctly diagnosed the disease with the assistance of clinical immunologists and rheumatologists before the appearance of irreversible organ damage and disability. The onset time of the other 24 NOMID patients reported in China ranged from 3 hours after birth to 6 years; however, the average time at diagnosis was delayed to 6.1 years. Therefore, clinical features such as short stature, intellectual disability, chronic aseptic meningitis, sensorineural hearing loss, eye involvement, lymphadenopathy, hepatosplenomegaly, musculoskeletal symptoms, and skeletal abnormalities are often observed in these patients, but amyloidosis has not been noted thus far. The clinical features noted above have been reported frequently, but there may now be novel unreported complications that relate to the newly described genotype variant of NOMID—including thyroid carcinoma (14). According to a study by Goldbach-Mansky et al. (15), NOMID patients were defined as who present with at least two of the following clinical manifestations: urticarial rash, CNS involvement (e.g., papilledema, pleocytosis in the cerebrospinal fluid, and sensorineural hearing loss), or epiphyseal or patellar overgrowth on radiography. The diagnosis of NOMID can be confirmed by genetic testing for NLRP3 mutations, but this has not yet replaced clinical evaluation.
NLRP3 is located on chromosome 1q44 and encodes the NLRP3 protein, also called cryopyrin (16), which serves as a scaffold for the assembly of the NLRP3 inflammasome (17). The NLRP3 inflammasome can make pro-IL-1β and pro-IL-18 into their bioactive forms, then release into the environment (18). Once released, IL-1β elicits neutrophilic inflammation through a cascade of downstream signals. NOMID patients possess the NLRP3 gene (with point mutations) that encodes aberrant cryopyrin and promotes the formation of the hyperactive inflammasome and inappropriate production of active IL-1β, leading to amplification of inflammation—and even loss of control—following a series of manifestations of organ inflammation. Infevers database (https://infevers.umai-montpellier.fr/) listed 264 NLRP3 variants in August 2023, and a majority are found in either NOMID or other NLRP3-associated autoinflammatory diseases. The de novo missense mutation in our patient was located in exon 4 of the NLRP3 gene and was not reported previously. NLRP3 mutations were detected in all the patients with genotype available reported in China. However, approximately 40-65% of NOMID patients lack detectable mutations in the NLRP3 as determined by Sanger sequencing (8, 19). This is because some of these individuals are carriers of somatic mosaicism, and the proportion is up to 69.2% in patients presenting with symptoms of NOMID and who tested negative by Sanger sequencing (20, 21). Deep sequencing may be needed to detect these somatic mutations to make an accurate genetic diagnosis (22). Low-penetrance NLRP3 variants may be found in asymptomatic healthy individuals (20, 23), whose management and prognosis may be complicated (22).
Early and effective treatment after making a definite diagnosis is crucial in delaying and preventing organ damage. Since IL-1 plays a central role in NOMID pathogenesis, anti-IL-1 treatment is recommended for NOMID (22). Current anti-IL-1 treatment includes IL-1 receptor antagonists (e.g., anakinra), anti-IL-1β monoclonal antibody (canakinumab), and IL-1 traps (e.g., rilonacept). Although Anakinra has been approved for NOMID by the US Food and Drug Administration (FDA) and for CAPS by the European Medicines Agency (EMA) in 2013 (24, 25), it is not yet available in China. The drug has improved partial signs and symptoms related to inflammation in some (13, 15, 26–28), but not all cases (29), and in severe cases where irreversible lesions developed, its efficacy was greatly diminished (13). The biggest hindrance to long-term compliance with treatment is its short half-life, leading to the regimen of daily injections. Canakinumab, which exhibits a long half-life, improves the acceptability of anti-IL-1 treatment (30). However, it is difficult to achieve full remission for some NOMID patients with severe CNS inflammation, and it is speculated that the penetration of canakinumab into CSF may take longer to reach a steady-state level because of its long half-life (31, 32). The differences in the inhibition of CSF biomarkers between anakinra and canakinumab suggest that anakinra is more effective in the intrathecal compartment (33). Canakinumab has also only been approved by the EMA for patients with NOMID (25), and it is not yet available in China. Another IL-1 inhibitor, rilonacept, is only available in the United States for FCAS and MWS, but there is no evidence supporting its efficacy in NOMID (8, 30, 34). Due to the current unavailability of the aforementioned IL-1-targeted biologics in China, most patients accept conventional anti-inflammatories, immunosuppressants, and antihistamines, but all of these medications have elicited disappointing results (4, 35). High-dose corticosteroids and thalidomide have partially improved symptoms (36), but their administration is always interrupted due to their adverse effects. Some centers have attempted to use anti-TNF-a therapeutic, but only a few animal researches (37) and a few case reports (5) support it. Suffering from occasional fever and subtle rashes, our patient received etanercept in another hospital. The acute-phase reactants were normal before and after the therapy. However, the long-term effects of this treatment on this patient are still needed to be observed. UCBT may be an alternative treatment for children with NOMID in China. Jiang et al. (6) observed that a 4 year old boy with NOMID with a heterozygous mutation of c.913G>A (p. D305N) achieved full remission in fever and urticarial-like rash during a 28-month follow-up after UCBT, and he had no sensorineural hearing loss and eye involvement. Further investigation is required to determine its long-term efficacy. There are now novel treatment approaches under investigation that focus on inhibitors of the NLRP3 inflammasome pathway due to its crucial role in NOMID pathogenesis; these include MCC950, β-hydroxybutyrate, tranilast, autophagy, and microRNAs (38–40).
To increase patient quality of life, Romano M et al. (22) recommend regular monitoring of disease activity to adjust the management strategy, fostering of self-management skills and medical decision-making, and initiating a transition programme to adult specialist care in adolescent patients. Owing to the unavailability of anti-IL-1 therapy in China, the long-term monitoring goals are difficult to achieve. Monitoring for infection is recommended, because patients with NOMID may have an increased susceptibility to pneumococcal pneumoniae. However, pneumococcal vaccines, unlike other vaccines, may lead to exacerbations of inflammation or flare-ups of the disease (41). Hence, clinicians must balance the potential benefits of vaccination against the risks in this population.
In conclusion, the appropriate prognosis of NOMID depends upon early diagnosis and the initiation of aggressive treatment for NOMID patients. Therefore, the features of cutaneous rash, arthritis, and meningitis-like symptoms but ineffectiveness of antibiotic treatment should also be noted by neonatologists—not solely by immunologists and rheumatologists—to initiate treatment of neonates. Specific therapy—especially anti-IL-1 therapies—has proven successful, and recommendations for management have been optimized continuously to improve quality of life. Patients cannot get IL-1blockers from the hospital or chemist’s shop in China. They must purchase them from abroad, and that is difficult for most patients. The high price of IL-1blockers is also another hindrance to long-term compliance with treatment. There are increasing reports of incomplete clinical responses to anakinra and canakinumab emphasizing the need for further understanding of the disease mechanisms and novel drug development. We posit that NOMID patients worldwide should achieve optimal management, including targeted therapy and multidisciplinary cooperation.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by The Research Ethics Boards of Children’s Hospital Affiliated to Shandong University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
CZ: Formal analysis, Writing – original draft, Writing – review & editing, Data curation, Methodology, Project administration. CL: Writing – review & editing, Formal analysis, Investigation, Project administration, Resources. XL: Writing – review & editing, Conceptualization, Methodology, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by Natural Science Foundation of Shandong Province (ZR2022MH235).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Guan N, Li B, Wu Y. Report of a child with neonatal-onset multisystem inflammatory disease and review of the literature. Chin J Pediatr (2014) 52(12):932–6. doi: 10.3760/cma.j.issn.0578-1310.2014.12.012
2. Fu Q, Chen J, Tacu C, Chen S. A platform setup for genetic diagnosis of Cryoyrin associated recurrent syndrome and a case report. Chin J Rheumatol (2014) 18(1):29–33. doi: 10.3760/cma.j.issn.1007-7480.2014.01.007
3. Zhao PW, Ding Y, Yin W, Yue X, He XL. Cryopyrin-associated periodic syndrome: one case report. J Clin Pediatr (2015) 6):579–82. doi: 10.3969/j.issn.1000-3606.2015.06.020
4. Zhang JM, Li CF, Tan XH, Piao YR, Han TX, Kuang WY, et al. Analyses of gene mutation, clinical phenotype, treatment and follow up ot 10 cases with chronic infantile neurologic, cutaneous, articular syndrome. Chin J Rheumatol (2019) 23(08):536–9. doi: 10.3760/cma.j.issn.1007-7480.2019.08.007
5. Luo XY, Chen AW, Cai JH, Fang J, Wang H. Tumor necrosis factor-alpha blockade ameliorates inflammatory response in two children with chronic infantile neurological, cutaneous and articular syndrome. J Dermatol (2020) 47(8):903–6. doi: 10.1111/1346-8138.15414
6. Jiang WJ, Man J, Qian XW, Wang HS, Wang P, Zhai XW. Umbilical cord blood stem cell transplantation for CINCA syndrome and literature review. Fudan Univ J Med Sci (2021) 48(3):349–55. doi: 10.3969/j.issn.1672-8467.2021.03.011
7. Zhou Y, Wang W, Zhong L, Wang L, Ma M, Tang X, et al. et al: Clinical and genetic spectrum of 14 cases of NLRP3-associated autoinflammatory disease (NLRP3-AID) in China and a review of the literature. Orphanet J Rare Dis (2022) 17(1):214. doi: 10.1186/s13023-022-02364-z
8. Finetti M, Omenetti A, Federici S, Caorsi R, Gattorno M. Chronic Infantile Neurological Cutaneous and Articular (CINCA) syndrome: a review. Orphanet J Rare Dis (2016) 11(1):167. doi: 10.1186/s13023-016-0542-8
9. Paccaud Y, Berthet G, Von Scheven-Gete A, Vaudaux B, Mivelaz Y, Hofer M, et al. Neonatal treatment of CINCA syndrome. Pediatr Rheumatol Online J (2014) 12:52. doi: 10.1186/1546-0096-12-52
10. Turel O, Goknar N, Uzuner S, Kasapcopur O, Cuisset L. CINCA syndrome in an infant presenting with hydrocephalus. Int J Rheum Dis (2014) 17(3):346–8. doi: 10.1111/1756-185X.12308
11. Aoyama K, Amano H, Takaoka Y, Nishikomori R, Ishikawa O. Cryopyrin-associated periodic syndrome: a case report and review of the Japanese literature. Acta Derm Venereol (2012) 92(4):395–8. doi: 10.2340/00015555-1322
12. Yokoi K, Minamiguchi S, Honda Y, Kobayashi M, Kobayashi S, Nishikomori R. Necrotizing Funisitis as an Intrauterine manifestation of Cryopyrin-Associated Periodic Syndrome: a case report and review of the literature. Pediatr Rheumatol Online J (2021) 19(1):77. doi: 10.1186/s12969-021-00578-2
13. Neven B, Marvillet I, Terrada C, Ferster A, Boddaert N, Couloignier V, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum (2010) 62(1):258–67. doi: 10.1002/art.25057
14. Salehzadeh F, Barak M, Hosseiniasl S, Shahbazfar E. CINCA syndrome with new NLRP3 mutation and unreported complication of thyroid carcinoma. Clin Med Insights Case Rep (2019) 12:1179547619854705. doi: 10.1177/1179547619854705
15. Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med (2006) 355(6):581–92. doi: 10.1056/NEJMoa055137
16. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol (2017) 18(8):832–42. doi: 10.1038/ni.3777
17. Kuemmerle-Deschner JB. CAPS–pathogenesis, presentation and treatment of an autoinflammatory disease. Semin Immunopathol (2015) 37(4):377–85. doi: 10.1007/s00281-015-0491-7
18. Mathur A, Hayward JA, Man SM. Molecular mechanisms of inflammasome signaling. J Leukoc Biol (2018) 103(2):233–57. doi: 10.1189/jlb.3MR0617-250R
19. Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol (2019) 39(3):277–86. doi: 10.1007/s10875-019-00638-z
20. Tanaka N, Izawa K, Saito MK, Sakuma M, Oshima K, Ohara O, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum (2011) 63(11):3625–32. doi: 10.1002/art.30512
21. Rowczenio DM, Gomes SM, Arostegui JI, Mensa-Vilaro A, Omoyinmi E, Trojer H, et al. Late-Onset cryopyrin-Associated periodic syndromes caused by somatic NLRP3 mosaicism-UK single center experience. Front Immunol (2017) 8:1410. doi: 10.3389/fimmu.2017.01410
22. Romano M, Arici ZS, Piskin D, Alehashemi S, Aletaha D, Barron KS, et al. The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis (2022) 81(7):907–21. doi: 10.1136/annrheumdis-2021-221801
23. Kuemmerle-Deschner JB, Verma D, Endres T, Broderick L, de Jesus AA, Hofer F, et al. Clinical and molecular phenotypes of low-Penetrance variants of NLRP3: diagnostic and therapeutic challenges. Arthritis Rheumatol (2017) 69(11):2233–40. doi: 10.1002/art.40208
24. Hansmann S, Lainka E, Horneff G, Holzinger D, Rieber N, Jansson AF, et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: a German PRO-KIND initiative. Pediatr Rheumatol Online J (2020) 18(1):17. doi: 10.1186/s12969-020-0409-3
25. ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis (2015) 74(9):1636–44. doi: 10.1136/annrheumdis-2015-207546
26. Wiken M, Hallen B, Kullenberg T, Koskinen LO. Development and effect of antibodies to anakinra during treatment of severe CAPS: sub-analysis of a long-term safety and efficacy study. Clin Rheumatol (2018) 37(12):3381–6. doi: 10.1007/s10067-018-4196-x
27. Sibley CH, Plass N, Snow J, Wiggs EA, Brewer CC, King KA, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine three- and five-year outcomes. Arthritis Rheum (2012) 64(7):2375–86. doi: 10.1002/art.34409
28. Lepore L, Paloni G, Caorsi R, Alessio M, Rigante D, Ruperto N, et al. Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with Anakinra. J Pediatr (2010) 157(2):310–315.e311. doi: 10.1016/j.jpeds.2010.02.040
29. Matsubara T, Hasegawa M, Shiraishi M, Hoffman HM, Ichiyama T, Tanaka T, et al. A severe case of chronic infantile neurologic, cutaneous, articular syndrome treated with biologic agents. Arthritis Rheum (2006) 54(7):2314–20. doi: 10.1002/art.21965
30. Malcova H, Strizova Z, Milota T, Striz I, Sediva A, Cebecauerova D, et al. IL-1 inhibitors in the treatment of monogenic periodic fever syndromes: from the past to the future perspectives. Front Immunol (2020) 11:619257. doi: 10.3389/fimmu.2020.619257
31. Sibley CH, Chioato A, Felix S, Colin L, Chakraborty A, Plass N, et al. A 24-month open-label study of canakinumab in neonatal-onset multisystem inflammatory disease. Ann Rheum Dis (2015) 74(9):1714–9. doi: 10.1136/annrheumdis-2013-204877
32. Yokota S, Imagawa T, Nishikomori R, Takada H, Abrams K, Lheritier K, et al. Long-term safety and efficacy of canakinumab in cryopyrin-associated periodic syndrome: results from an open-label, phase III pivotal study in Japanese patients. Clin Exp Rheumatol (2017) 35 Suppl 108(6):19–26.
33. Rodriguez-Smith J, Lin YC, Tsai WL, Kim H, Montealegre-Sanchez G, Chapelle D, et al. Cerebrospinal fluid cytokines correlate with aseptic meningitis and blood-Brain barrier function in neonatal-Onset multisystem inflammatory disease: central nervous system biomarkers in neonatal-Onset multisystem inflammatory disease correlate with central nervous system inflammation. Arthritis Rheumatol (2017) 69(6):1325–36. doi: 10.1002/art.40055
34. Arnold DD, Yalamanoglu A, Boyman O. Systematic review of safety and efficacy of IL-1-targeted biologics in treating immune-mediated disorders. Front Immunol (2022) 13:888392. doi: 10.3389/fimmu.2022.888392
35. Yu JR, Leslie KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep (2011) 11(1):12–20. doi: 10.1007/s11882-010-0160-9
36. Kallinich T, Hoffman HM, Roth J, Keitzer R. The clinical course of a child with CINCA/NOMID syndrome improved during and after treatment with thalidomide. Scand J Rheumatol (2005) 34(3):246–9. doi: 10.1080/03009740410010236
37. McGeough MD, Wree A, Inzaugarat ME, Haimovich A, Johnson CD, Pena CA, et al. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J Clin Invest (2017) 127(12):4488–97. doi: 10.1172/JCI90699
38. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi: 10.3389/fphar.2015.00262
39. Vande Walle L, Stowe IB, Sacha P, Lee BL, Demon D, Fossoul A, et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PloS Biol (2019) 17(9):e3000354. doi: 10.1371/journal.pbio.3000529
40. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
Keywords: NOMID/CINCA, NLRP3 gene, novel mutation, neonate, aseptic meningitis
Citation: Zhao C, Liu C and Li X (2024) Clinical characteristics of Chinese neonates with neonatal-onset multisystem inflammatory disease: a case report and literature review. Front. Immunol. 14:1291345. doi: 10.3389/fimmu.2023.1291345
Received: 09 September 2023; Accepted: 12 December 2023;
Published: 05 January 2024.
Edited by:
Daniela Bosisio, University of Brescia, ItalyReviewed by:
Marco Cattalini, University of Brescia, ItalyRenshuai Zhang, The Affiliated Hospital of Qingdao University, China
Adriana Almeida de Jesus, National Institute of Allergy and Infectious Diseases (NIH), United States
Copyright © 2024 Zhao, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoying Li, bHh5X2puQGVtYWlsLnNkdS5lZHUuY24=