 Jacob C. Bjorgen
Jacob C. Bjorgen Jenna K. Dick
Jenna K. Dick Ross Cromarty
Ross Cromarty Geoffrey T. Hart
Geoffrey T. Hart Joshua Rhein
Joshua Rhein
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 19 October 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1267774
In the setting of viral challenge, natural killer (NK) cells play an important role as an early immune responder against infection. During this response, significant changes in the NK cell population occur, particularly in terms of their frequency, location, and subtype prevalence. In this review, changes in the NK cell repertoire associated with several pathogenic viral infections are summarized, with a particular focus placed on changes that contribute to NK cell dysregulation in these settings. This dysregulation, in turn, can contribute to host pathology either by causing NK cells to be hyperresponsive or hyporesponsive. Hyperresponsive NK cells mediate significant host cell death and contribute to generating a hyperinflammatory environment. Hyporesponsive NK cell populations shift toward exhaustion and often fail to limit viral pathogenesis, possibly enabling viral persistence. Several emerging therapeutic approaches aimed at addressing NK cell dysregulation have arisen in the last three decades in the setting of cancer and may prove to hold promise in treating viral diseases. However, the application of such therapeutics to treat viral infections remains critically underexplored. This review briefly explores several therapeutic approaches, including the administration of TGF-β inhibitors, immune checkpoint inhibitors, adoptive NK cell therapies, CAR NK cells, and NK cell engagers among other therapeutics.
Natural killer (NK) cells are a type of lymphocyte notable for their cytotoxic activity and role as an innate immune cell that acts as an early responder to physiologically stressed cancer and virally infected cells (1). NK cells are particularly important in viral infections, given their ability to respond to them before the development of adaptive immune responses (2). Evidence for a role of NK cells in the protection against certain viral infections is appreciated in patients with selective NK cells deficiencies; the hallmark clinical feature in these patients are severe and recurrent viral infections (3, 4). For example, of a group of 19 NK cell deficient patients, 42% died prematurely while 53% had experienced severe herpesvirus infections including cytomegalovirus, Epstein-Barr virus, Varicella Zoster virus, and herpes simplex virus (5). The dramatically increased incidence rate of severe complications related to these infections in patients with NK cell deficiency is strongly illustrative of their important role in combating and containing viral infections.
Like other immune cells, NK cells originate from CD34+ hematopoietic stem cells, and most of their development occurs in discrete stages. Commitment to the NK cell lineage relies on loss of CD3 and gain of the natural cytotoxicity receptors NKp44 and NKp46 (6). Roughly 5-15% of all circulating lymphocytes is made up of NK cells, and NK cells are known to also reside in secondary lymphoid tissue, bone marrow, lungs, liver, spleen, and uterine tissues.
NK cells have been divided into different populations based on the relative expression of surface markers CD56 and CD16 (FcgrIIIa), with the two largest subsets being CD56bright and CD56dim (7). CD56bright NK cells constitute roughly 2-10% of all NK cells in the blood and are found mainly in the secondary lymphoid organs where they constitute 50% of the NK cells, whereas CD56dim NK cells comprise 90% of NK cells in the blood (8). CD56bright NK cells differentiate into CD56dim NK cells and then express CD16 (9, 10). Functionally, CD56bright NK cells are considered to be far less cytotoxic than their CD56dim counterparts, and play a role in the production of antiviral proinflammatory cytokines like IFN-γ and TNF-α (7, 8). In contrast, CD56dim NK cells are far more cytotoxic since they contain 10-fold more perforin and Granzyme B and due to their expression of CD16, which enables them to engage in antibody-dependent cellular cytotoxicity (ADCC) (11). CD16 is the only receptor known to activate NK cells on its own without activation through other receptors (12). Of note, CD56dim NK cells are also capable of producing proinflammatory cytokines like IFN-γ, typically producing them two to four hours after stimulation, whereas CD56bright NK cells predominantly produce cytokines 16 hours or more after stimulation (13). There is another subset of NK cells, termed adaptive (or memory-like) NK cells, that possess qualities of the adaptive immune system such as antigen-driven clonal expansion and long lived memory that have been recognized and will be examined in more detail later (14). Finally, there are also different subsets of NK cell populations that reside in peripheral tissues that carry distinct phenotypic markers (15).
NK cells are unique in their mechanism of recognition of dysregulated or infected cells. Instead of solely recognizing viral peptides presented by major histocompatibility (MHC) molecules like cytotoxic T lymphocytes, NK cell activation is dependent on the balance between different activating and inhibitory receptors to distinguish between virally infected and uninfected cells in a non-antigen-specific manner. Many NK cell receptors sense surface expression of MHC class I molecules as well as MHC class I chain related (MIC) molecules among others (16). NK cell activation will only occur if the activating signal overrides the inhibitory signal (Figure 1) (17). Importantly, NK cells can recognize virally infected cells or tumor targets without prior sensitization to antigens. Some viruses will down-regulate MHC-I to interfere with the presentation of viral antigens, thereby escaping cytotoxic T lymphocytes. This downregulation of MHC-I promotes the recognition and killing of virally infected cells by NK cells. This concept of recognizing cells that lack MHC-I is known as the “missing-self” hypothesis (18).
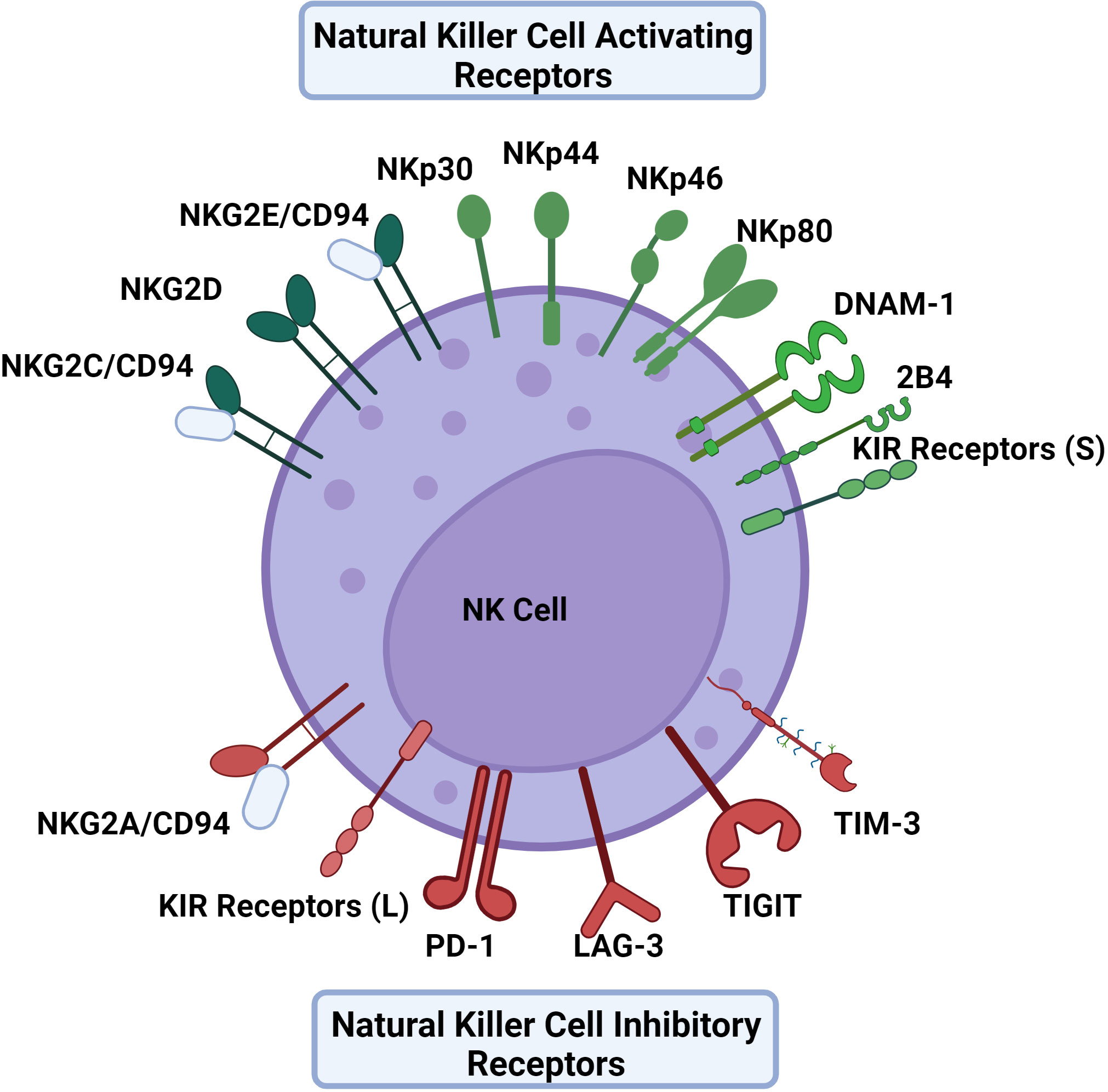
Figure 1 The activating and inhibitory receptors of NK cells. NK cell activation depends on a balance of activating and inhibitory signals using receptors that primarily bind HLA to distinguish between virally infected cells and uninfected cells in a non-antigen specific mechanism. Activating receptors include NKG2C, NKG2D, NKG2E, NKp30, NKp44, NKp46, NKp80, DNAM-1, and KIR receptors with short cytoplasmic tails. Inhibitory receptors include NKG2A, PD-1, LAG-3, TIGIT, TIM-3 and KIR receptors with long cytoplasmic tails excluding KIR2DL4 which is activating. The KIR receptor family contains both activating and inhibitory receptors, as well as receptors that can act as either. Similarly, 2B4 can also provide either an activating or inhibitory signal.
Activating receptors include natural cytotoxicity receptors (NCRs) such as NKp30, NKp44, NKp46, and NKp80, in addition to some receptors of the NKG2 family, including NKG2C, NKG2D, and NKG2E (7, 19). Resting NK cells can express NKp30, NKp46, and NKp80 whereas NKp44 is expressed by activated NK cells (20). The acquisition of natural cytotoxicity receptors during NK cell maturation is correlated with the development of cytotoxic activity against tumor cells depicting the receptor family’s importance in NK cell activation (21). NKG2C and NKG2E form heterodimers with CD94 which bind to HLA-E, an interaction that supports NK cell expansion in antiviral responses (17). Another activating receptor is DNAM-1 which binds nectin and nectin-like molecules (22).
Inhibitory receptors include the members of the killer cell immunoglobulin-like (KIR) receptor family that have long cytoplasmic tails (23). Of note, while many KIR receptors are inhibitory, a portion are activating and have short tails, and some can be both activating and inhibitory (24). Similarly 2B4 is another NK cell receptor that has been observed to be both activating and inhibitory depending on the presence of modulating co-receptors and signaling molecules (25). NKG2A/CD94 heterodimers also serve as another important inhibitory receptor that binds HLA-E as its ligand (26). In the blood, approximately 50% of NK cells express NKG2A/CD94 receptors (27). In terms of NK cell differentiation, it has been noted that the NK cell maturation process entails a stepwise decline in NKG2A with a simultaneous increase in killer immunoglobulin-like receptors (KIRs) (28). TIGIT, LAG-3, TIM-3, and PD-1 receptors are classically defined as inhibitory receptors on cytotoxic T lymphocytes, but can serve a role in modulating NK cell responses in an inhibitory or activating fashion depending on the context (29, 30).
Adding to the complexity of NK cell signaling, extensive genetic diversity exists within many NK cell receptors and ligands, particularly within the KIR receptor family and HLA genes (31–33). To elaborate, the KIR receptor family has been found to vary widely in terms of the presence of different KIR genes, copy number of KIR genes, and there exsists substantial polymorphism within these genes resulting in markedly different KIR repertoires between individuals (34, 35). Two primary haplotypes have been identified and termed group A and group B (36). The group A haplotype is relatively consistent in its makeup, consisting of the activating KIR receptor KIR2DS4 and the inhibitory KIR receptors KIR2DL1, KIR2DL3, KIR3DL1, KIR3DL2, and KIR2DL4 (31). In comparison, the B haplotype is less fixed and contains one or more activating KIR receptor along with genes encoding the inhibitory KIR receptors KIR2DL5A, KIR2DL5B, and KIR2DL2. Within haplotypes, group A varies in terms of allelic polymorphisms in its constituent genes, while group B varies more in terms of gene content and copy number, although allelic variation is also a consideration too (36). The HLA gene family, whose constituents serve as ligands for many NK cell receptors encompasses more than 35,000 different MHC alleles, making it the most polymorphic region in the human genome (37). The implications of this diversity on NK cell signaling are made very apparent by the observation that various combinations of KIR receptors and HLA ligand have displayed differential functional activation (32, 38–40), and have additionally been associated with infection outcome in human immunodeficiency virus (HIV) (41, 42), hepatitis C virus (HCV) (43), SARS-CoV-2 (44), and ebola virus (45).
A similar effect can be observed from the effect of genetic variation within the NKG2D/NKG2DL signaling axis. While NKG2D is evolutionarily conserved relative to the KIR receptor family, distinct haplotypes exist and have been implicated in predisposing different cancer risk (46), as well as being a prognostic factor for treatment free remission in the setting of chronic myeloid leukemia (47). In humans, the ligands for NKG2D are MIC and ULBP which are upregulated by virally infected cells, and both of these ligands display extensive polymorphism (48). Certain alleles in these ligands have been associated with diseases like Dengue shock syndrome (49, 50), colorectal cancer (51), oral squamous cell carcinoma (52, 53), breast cancer (54), and cervical cancer (55). Taking the examples of the KIR receptor family and NKG2D/NKG2DL axis together, it is very evident that the genetic content of an individual's NK cell repertoire plays a significant role in the regulation of NK cell function in disease.
Other notable NK cell markers include CD57, CD69, and CD107a. CD57 expressing NK cells are connected with terminal differentiation, senescence, and high cytotoxic potential (56). CD69 is a marker expressed by NK cells during acute activation (57, 58). Additionally, CD69 has been shown as a marker of tissue resident NK cells (59). CD107a is expressed on the inner membrane of cytotoxic granules containing molecules like granzymes and perforin, which are released upon NK cell activation. When the granule is released, its membrane fuses with the outer cell membrane making cells that released granules positive for CD107a. Therefore, CD107a as a marker is indicative of NK cell granule release (degranulation) (60).
NK cells primarily kill infected cells through the release of granules containing granzyme-B and perforin, a serine protease that activates apoptosis and a protein that forms pores in the membrane of targeted cells, respectively (61, 62). In this process, perforin enables granzymes to infiltrate past the membrane of target cells, where they cleave internal components including procaspases that then initiate apoptosis (63). ADCC is another important mechanism through which NK cells kill target cells. In ADCC, the target cell is bound by antibodies and the Fc region of these antibodies in turn binds CD16, ultimately triggering the release of cytotoxic effector molecules that kill the target cell as described above (64).
During the NK cell response to viral infection, the repertoire of NK cell activating and inhibitory receptors is altered, often with an impact on the functionality of these cells. The aim of this review is to summarize these changes, particularly those related to NK cell dysregulation and exhaustion in the context of HIV, HCV, influenza A viruses (IVA), cytomegalovirus (CMV), and SARS-CoV-2. These viruses were chosen due to their clinical relevance and the existence of a plethora of literature noting alterations in NK cell phenotype and repertoire during their pathogenesis. Each of these viruses represent different viral families, and furthermore, the selection offers perspective into the similarities and differences between acute and chronic viral infections’ impact on the NK cell population. Further, potential therapeutics that could help maintain, or enhance an effective NK cell response will be explored.
HIV belongs to the Retroviridae family and has a genome that consists of two single-stranded RNA molecules (65). HIV primarily targets CD4+ T cells and ultimately results in their depletion rendering those infected progressively immunocompromised and susceptible to opportunistic infections and rare forms of cancer (66). With contemporary antiretroviral therapy (ART), replication of the virus can be controlled to the extent that patients on ART are aviremic (66). However, ART is not curative due to the establishment of reservoirs of infected cells that persist in lymphoid tissue and are often latent (67). Significant attention has been placed on characterizing the role of NK cells in HIV infection within both the blood and lymphoid tissue given the nature of the infection.
In the context of HIV infection, and the general immune dysregulation incurred by depletion of CD4 T cells, NK cells undergo significant changes regarding their receptor expression and functional activity. HIV progression in terms of declining CD4 T cell counts is associated with a decline in the quantity of cytotoxic CD56dim NK cells (68, 69). The remaining CD56dim NK cell population has CD57 and KIR receptor expression characteristic of a more mature phenotype. However, these cells have decreased functional potential evidenced by their lower degranulation ability and perforin expression (70, 71). Regarding the CD56bright NK cell population, they express CCR7, a chemokine receptor associated with tissue trafficking; this has been noted to decrease. This decrease in CCR7 expression has been correlated with HIV viral load (72), supporting the idea that issues in NK cell trafficking contributes to a failure to contain HIV replication. Moreover, CD56bright NK cells have also been found to be more expressive of TGF-β during HIV infection, which is of note as TGF-β inhibits immune effector functions (73, 74). As HIV infection progresses, an expansion in the amount of CD56neg NK cells has also been observed (Figure 2) (71, 75, 76). In healthy subjects, the CD56neg NK cell subset constitutes roughly 5.7% of NK cells in the peripheral blood, which has been shown in a meta-analysis to increase by an average of 10.6% after HIV-1 infection (77). CD56neg NK cells have significantly impaired cytotoxic function due to expression of a relatively high quantity of inhibitory receptors (78). Furthermore, this cell population is known to produce a greater quantity of TGF-β, a cytokine associated with anti-inflammatory and homeostatic functions. TGF-β may also hinder autologous CD8 T cell functionality and potential to generate IFN-γ (79). The CD56neg NK cell population is also known to expand in other chronic viral infections including hepatitis C virus (HCV), Epstein-Barr virus (EBV), and chronic cytomegalovirus (CMV). This observation suggests that it is likely a common feature among chronic viral infections (80).
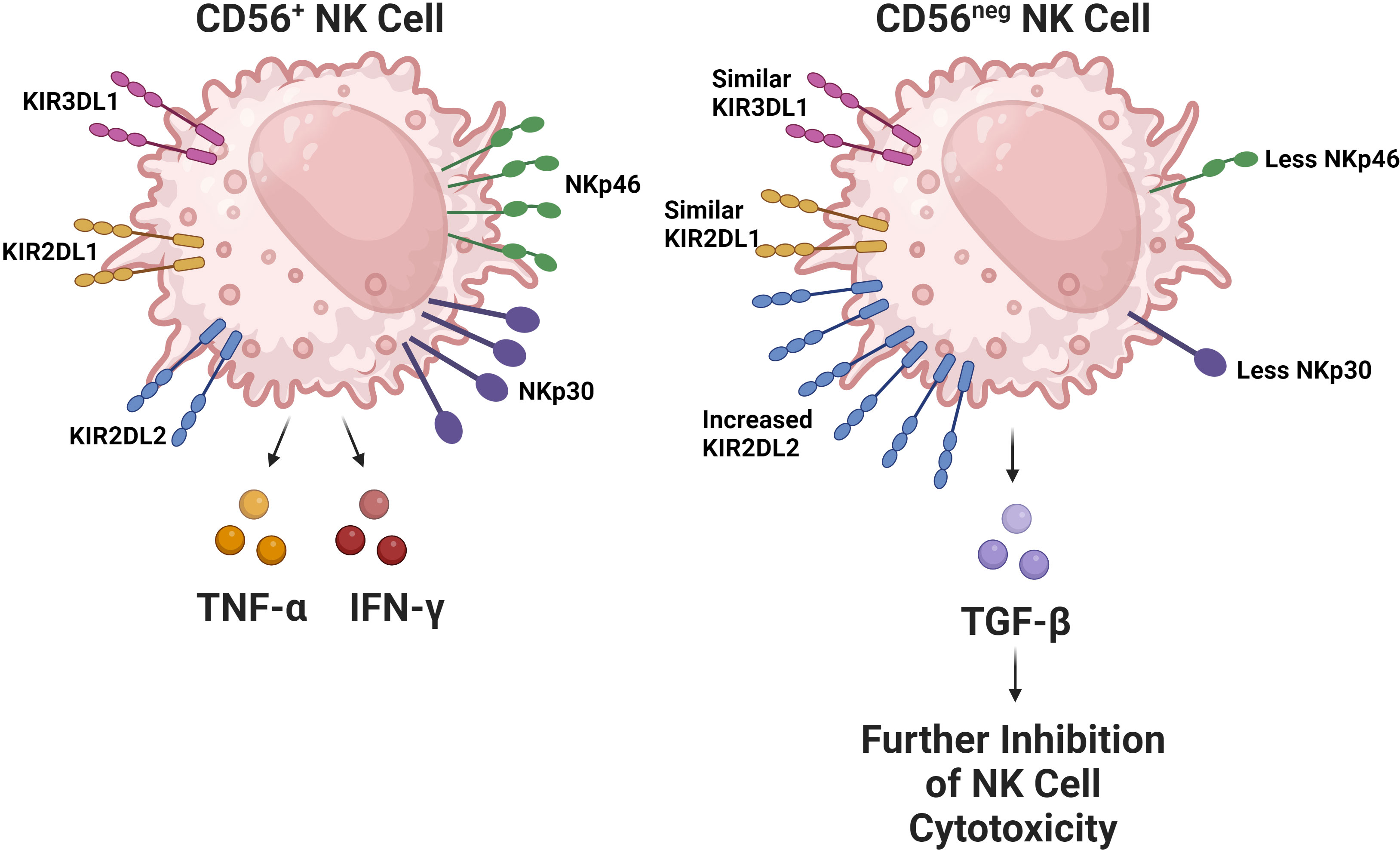
Figure 2 The CD56neg NK cell subset increases during chronic viral infections. This NK cell subset has been observed to expand in the setting of HIV, chronic HCV, EBV, and chronic CMV. The subset has substantially impaired cytolytic capabilities due to alterations in its receptor repertoire, particularly due to decreased expression of activating receptors like NKp30 and NKp46 in addition to the increased expression of the inhibitory KIR receptor KIR2DL2 (52). Interestingly, the expression of inhibitory KIR receptors KIR2DL1 and KIR3DL1 remain comparable between CD56+ and CD56neg NK cells. In HIV, this subset has been found to be more expressive of TGF-β, which contributes to the exhaustion of other NK cells.
During HIV infection, the expression of activating natural cytotoxicity receptors (NCRs) like NKp80 and NKp46 in NK cells are suppressed (81). Regarding inhibitory receptors, HIV viremia is associated with a significant increase in the expression of inhibitory KIR receptors, while NKG2A has been shown to decline, and taken together, the alterations in the expression of both activating and inhibitory receptors is associated with diminished cytolytic function (82). Additionally, the expression of CD69, CD18, and CD11b are also decreased, further suggesting functional impairment (83). A decrease in IFN-γ production by NK cells has also been observed, and coinfection with hepatitis C virus (HCV) is found to synergistically impair functional ability, with even further reduced IFN-γ and degranulation ability (84, 85).
To avoid targeting by cytotoxic T-lymphocytes, HIV selectively downregulates MHC-I molecules HLA-A and HLA-B, but largely retains HLA-E and HLA-C in some individuals in an attempt to avoid lysis by NK cells (86). This occurs in a mechanism dependent on HIV’s Nef protein and results in the aforementioned surface MHC-I molecules being rapidly internalized and degraded (87). Similarly, HLA-C has been shown to be downregulated by HIV-1’s viral protein U (Vpu) in some individuals, though not all (88, 89). HIV has been shown to induce the expression of ligands for NKG2D in infected cells by a mechanism dependent on its Vpr protein (90). The subsequent NK cell activation is speculated to contribute to CD4 T-cell depletion and NK cell dysfunction as sustained exposure to its ligand desensitizes the NKG2D receptor, resulting in its downregulation and diminished NK cell cytotoxic activity (91, 92). However, it is worth noting that NKG2D signaling alone is not enough to trigger NK cell degranulation as it requires coactivation with NK-T-B-antigen (NTB-A) which is downregulated through the action of Vpu enabling escape from the NK cell response (93). Also of note is that HLA-E is capable of displaying HIV antigen that blocks interactions with NKG2A limiting inhibitory NK cell signaling by this mechanism. This contrasts with HLA-C, which remains unblocked during HIV infection resulting in conserved inhibitory signaling via KIR receptors. This suggests that HIV+ cells are uniquely susceptible to NKG2A expressing NK cells that lack inhibitory KIR receptors (94), and therapeutics taking advantage of this possibility deserve further exploration. Similarly, the blocking of interactions between HLA-A and HLA-C with NK cell inhibitory receptors have enhanced cytotoxicity against HIV infected cells (95).
During early HIV infection, NK cell numbers and distribution remain relatively unaltered. KIR+ NK cells remain rare at this stage, and the relative naivety of the NK cell repertoire limits their ability to control the infection (96). As HIV infection progresses, there has been an observed increase in the number of CXCR5+ NK cells in lymphoid tissue (97). This population of NK cells highly express activating receptors, including NKG2D and NKp44, although they do demonstrate functional impairment, with reduced degranulation compared to CXCR5+ NK cells in non-infected individuals. Most notably, the frequency of these cells is negatively correlated with HIV-1 burden in the lymph tissue, illustrating their potential importance in controlling the HIV-1 reservoir.
Also found within lymph tissue is a subset of CD56bright NK cells which express a high quantity of NKG2A inhibitory receptors, as well as NKG2D, NKp46, and NKp30 activating receptors (98). This subset is also relatively immature in regards to its differentiation status and cytotoxic potential due to expressing only low quantities of CD57 and CD16. In humanized MISTRG-6-15 mice, after HIV-1 infection, NK cells in the lymphoid tissue were relatively less mature and similarly more dysfunctional when compared to NK cells in the nonlymphoid tissues, consistent with this previous result (99). However, this observation may be in part driven by mismatched trafficking receptors impairing infiltration of NK cells into lymphoid tissue in this mouse model. Within the gut mucosa, two distinct subsets of NK cells located in the intraepithelial spaces and lamina propria are significantly diminished in prevalence during chronic HIV infection which contrasts with spontaneous HIV controllers who have a stable intraepithelial subset (100).
In terms of KIR receptor expression, the expression of the activating KIR2DS4 has been shown to decrease while the expression of other inhibitory KIRs remains constant with exception of KIR3DL2 which increases (101). Interestingly failure to downregulate KIR2DS4 has been associated with increased viral replication possibly due to promoting excessive NK cell activation in chronic HIV (102, 103). Similarly, the possession of inhibitory KIR receptor KIR2DL3 has been associated with increased chronic immune activation, especially when an individual is also a carrier of HLA-C2 (104). This same study also noted that carriers of activating KIR3DS1 also display heightened immune activation during chronic HIV. Further study has shown that KIR3DS1+ NK cells in the presence of the receptor’s cognate ligand display enhanced ability to control HIV infection in vitro and result in lower viral set points in vivo (105, 106). Overall, these studies again highlight the importance of an individual’s KIR and HLA repertoire, as the presence of certain receptor-ligand genes are associated with different functionality in HIV.
Mounting evidence also suggests that HIV infection is associated with an increase and persistence of memory NK cells (107). For example, macaques infected with either SIV or SHIV have been found to develop a subset of antigen specific NK cells present in the spleen and liver that specifically lyse dendritic cells pulsed with viral antigens like GAG or ENV (108). This mechanistically occurs in an NKG2C dependent manner and markedly contrasts with NK cells isolated from healthy macaques that lysed fewer cells in a non-specific manner. In humans, the presence of rare HIV peptide specific NK cells has been observed and is also associated with lower viral load, higher CD4 counts, and stronger HIV specific T-cell responses (109, 110). Further study is required to elucidate the mechanism by which these cells arise and to explore their function in greater detail. An expansion in NKG2C+ memory NK cells have also been described in early HIV infection and patients with high NKG2C+ CD57+ NK cell frequencies have been found to show lower immune activation and HIV RNA one month after starting antiretroviral therapy (ART) (111). The patients in this study were seropositive for cytomegalovirus (CMV), so from this data it is indeterminable the degree to which this memory NK cell population was induced by HIV infection itself. Regardless, further characterization of the functional role of NKG2C+ CD57+ memory NK cells in HIV is needed.
ART does not normalize the phenotypic changes observed in the CD56dim, CD56negative, and CD56bright NK cell subsets caused by HIV infection (112). Deficiencies in the prevalence of CD56dim and CD56bright, as well as the expression of CD107a, Granzyme-B, NKG2D, and NKp46, remain following ART therapy suggesting prolonged NK cell functional impairment (113). In spite of functional impairment, NK cell activation persists following viral suppression by ART (114). However, the cytokine-induced expression of IFN-γ and activating receptor NKp44 are also reduced, and CD56bright cell counts are inversely correlated to the decreased T regulatory cell counts observed during ART, suggesting autoreactivity (115). In contrast, the relative expression of inhibitory receptors after ART does appear to return and is comparable to healthy donors (82). Despite ART, the dysregulation of NK cells in HIV is a clear component of the infection’s immunopathogenesis and is an avenue that many of the therapeutics discussed later are targeting for cure strategies.
Hepatitis C Virus (HCV) belongs to the Flaviviridae family and has a single-stranded RNA-genome (116). HCV primarily infects hepatocytes, but can also replicate in macrophages, B cells, and T cells, among other extrahepatic tissues (117). While HCV infection is often asymptomatic, chronic infection is frequently associated with the development of liver fibrosis and in severe cases cirrhosis (118).
In regards to the NK cell response, the relative prevalence of CD56bright NK cells increases, while the prevalence of CD56dim NK cells slightly decreases in the blood during acute HCV infection (119). Additionally, both subsets have been found to upregulate the expression of activating receptor NKG2D while demonstrating strong cytokine production and cytotoxic activity. Following acute infection, approximately 25% of individuals spontaneously clear the virus, though the virus persists in the majority of individuals leading to chronic infection (120). Self-clearing HCV infections have been associated with highly differentiated NK cell phenotypes with increased expression of CD57 and KIR receptors (KIR2D, KIR3DL1, and KIR3DL2) and decreased expression of NKG2A (Figure 3) (121). Furthermore, certain NK cell phenotypes, like the expression of NKp30, are associated with spontaneous clearance (122). Homozygosity in KIR2DL3 and HLA-C1 has also been strongly associated with spontaneous clearance (43). The expression of inhibitory receptors NKG2A and inhibitory KIR2DL3 has been found to increase during chronic HCV infection while the expression of NKp30 and NKp46 was decreased relative to healthy controls (123, 124). Additionally, possession of HLA-B*44, HLA-C*12, and KIR3DS1 have been associated with failure to spontaneously clear HCV (125). Overall, the correlation of self clearance with the presence of mature, functionally active NK cells, as well as the presence or absence of specific KIR receptors suggests NK cell involvement in determining the chronicity of HCV.
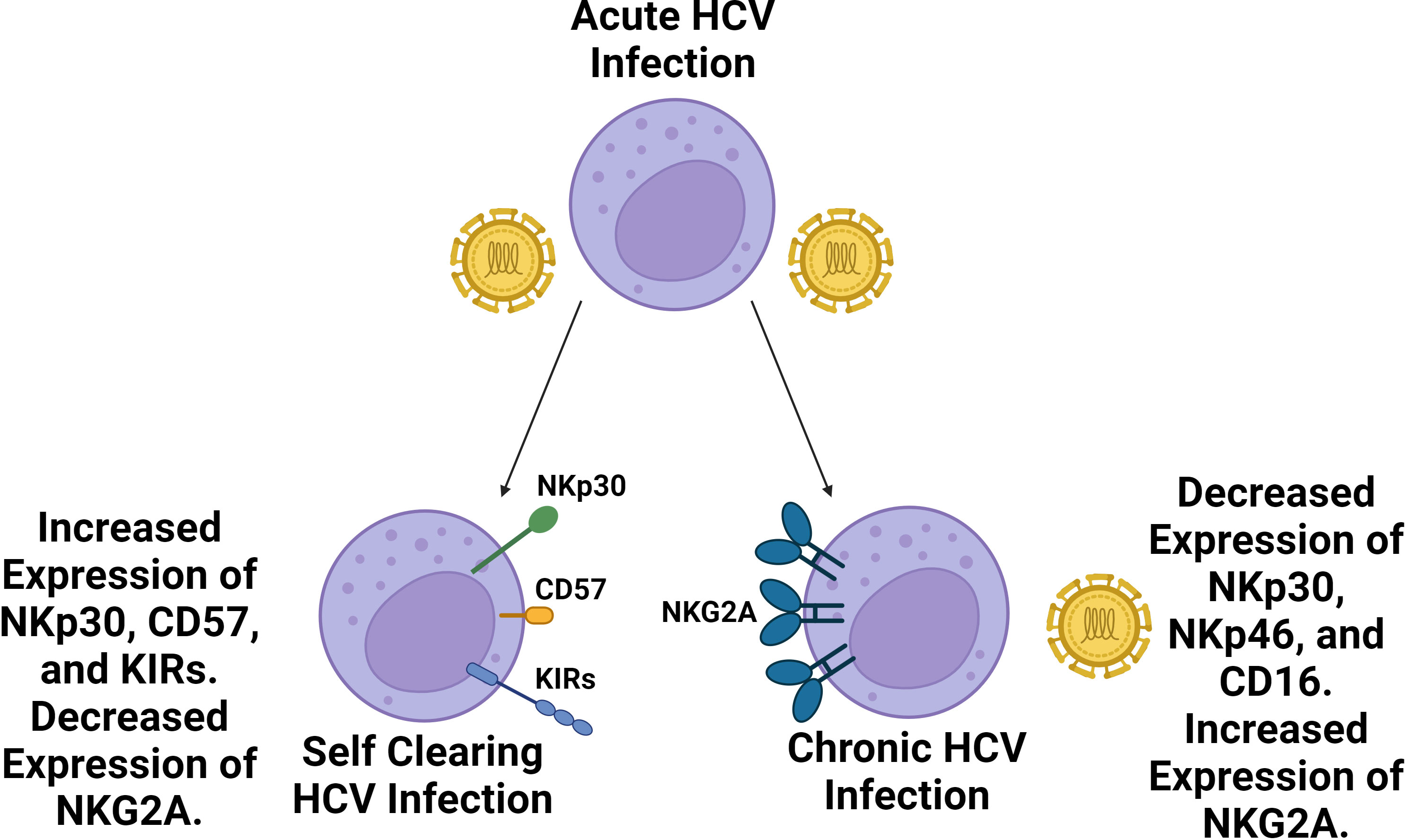
Figure 3 Self clearing HCV infections are associated with a more highly differentiated NK cell repertoire compared to chronic HCV infections. Following acute HCV infection, two outcomes may follow. In roughly a quarter of cases, HCV infection is spontaneously cleared whereas roughly three quarters of cases become chronic. Spontaneously cleared cases are associated with a more a more differentiated NK cell phenotype with increased prevalence of NKp30, CD57 and KIR expression as well as decreased expression of NKG2A. Chronic HCV cases are associated with diminished NK cell expression of NKp46, NKp30, and CD16 along with increased expression of NKG2A. NKG2A inhibitory signaling has been identified as a key source of NK cell exhaustion in HCV infection.
Chronic HCV infection has also been associated with a significant decline in the total number of NK cells, specifically the CD56dim subset in the peripheral blood (126, 127), similar to what occurs during chronic HIV infection. Additionally, the relative proportion of CD56bright NK cells in the blood appears to increase during chronic infection due to both expansion of the subset, and the decline of CD56dim NK cells in the blood (128, 129). For example, the CD56dim NK cell subset has been observed to shrink to 4.9% of total lymphocytes in the blood during chronic HCV infection from 9.0% in healthy controls (123). In an ex vivo study, extracellular HCV-core protein was found to be correlated with the frequency of CD56bright NK cells, suggesting that the protein attenuates NK cell differentiation and may help explain the increased prevalence of more naive CD56bright NK cells (130). This direct viral modulation, along with other explanations providing rationale for the changes in NK cell subset prevalence in the blood during chronic HCV infection like tissue homing, would benefit from further investigation.
NK cells in the liver are predominantly of the CD56bright subset, and liver fibrosis is associated with the diminished expression of activating receptors, including NKG2D and NKp46 (131). During chronic HCV infection, IL-26, has been found to be overexpressed (132). IL-26 is a cytokine that was recently identified as an inflammatory mediator, which binds extracellular DNA from damaged cells and induces activation of myeloid and lymphoid cells (133). The overexpression of IL-26 during HCV in turn upregulates TNF-related apoptosis-inducing ligand (TRAIL) expression on CD56bright NK cells, increasing their ability to kill HCV-infected hepatoma cells (132). Additionally, an increase in the number of intrahepatic NKp44+ NK cells has been observed during chronic HCV (134). These intrahepatic NKp44+ NK cells are potent producers of TNF-α, and their frequency has been positively correlated with both HCV-RNA and fibrosis stage, indicating they may play a role in liver damage. It has also been observed that the relative prevalence of resident liver NK cells relative to circulating NK cells increases in the setting of cirrhosis, further suggesting NK cell involvement in HCV related liver fibrosis (135). Further work exploring if the NK cell response in the liver is mediating liver fibrosis or attempting to inhibit it through IL-10 and other mechanisms is needed to better understand the pathogenesis.
Functionally speaking, NK cells from HCV infected patients have a reduced capability to degranulate and produce cytokines (136). As infection progresses, reduced expression of NKG2D, NKp30, NKp46, and an increase in the CD56neg NK cell subset and NKG2A expression is observed (137). Remaining CD56dim NK cells also express CD16 to a lesser degree, limiting their ADCC ability (128). Additionally, hepatocytes in humanized mice upregulate Qa-1 (a murine functional homolog to HLA-E), which significantly contributes to NK cell exhaustion through inhibitory NKG2A receptor signaling (138). Furthermore, anti-NKG2A antibodies have been shown to restore NK cell production of IFN-γ in mice, resulting in decreased viral replication, demonstrating the relative importance of this pathway in NK cell exhaustion. Such immune checkpoint inhibitors will be explored further later.
Direct acting antiviral (DAA) therapy has been shown to normalize many of the changes in the NK cell repertoire associated with chronic HCV infection. For example, the prevalence of CD56dim NK cells increases and the prevalence of the CD56bright NK cell subset decreases, reverting both to levels comparable to healthy controls (139, 140). DAA has also been associated with decreased NK cell activation in the periphery, as well as decreased IL-12, IL-18, NKp30, NKp46, and TRAIL expression (139, 141). This further supports the idea that DAA, and by extension the decline in viral titers it causes results in diminished NK cell activation, and also teases NK cell involvement in the mediation of liver damage, due to treatment resulting in declining NK cell activation.
Cytomegalovirus (CMV) is a member of the Herpesviridae family and has a large linear double-stranded genome (142). The virus infects a wide assortment of cells including epithelial cells, fibroblasts, monocytes, and macrophages and establishes a latent reservoir that is prone to later reactivation (143). The most notable feature of NK cells in CMV infection is the expansion of NKG2C+ CD57+ memory NK cells, also known as adaptive NK cells (144–147). These cells are phenotypically mature CD56dim NK cells that express NKG2C, and persist after acute infection (148). Further, this NKG2C NK cell population preferentially expresses the inhibitory KIR2DL receptors, which has been described as contributing to increased functionality (149). CMV seropositivity is also associated with a decline in the expression of NKp30 and an increase in NKp46 (Figure 4) (150).

Figure 4 Acute CMV Infection and Subsequent Reactivation is Associated with an Expansion in NKG2C+ memory-like NK cells. These cells are phenotypically mature, long lasting, and exhibit enhanced functional potential leading them to be coined adaptive or memory like NK cells. This population is phenotypically defined by the expression of NKG2C and is associated with a high level of expression of CD57, NKp46, and inhibitory KIR2DL receptors.
A similar expansion of NKG2C+ CD56dim NK cells has been observed during CMV coinfection with viral hepatitis (both hepatitis B and C), suggesting that CMV-related NKG2C expansion is prominent even in the setting of other viral infections (151). This NKG2C+ memory NK cell population has been found to be have a greater capacity for ADCC and a potent capability to produce TNF-α and IFN-γ (152). As described earlier, these NKG2C+ CD57+ NK cells have been coined adaptive NK cells due to their selective proliferation, persistence, and functional potency in the setting of CMV infection, comparable to that of the adaptive immune system (153). The emergence and persistence of NKG2C+ CD57+ NK cells secondary to CMV infection are thought to be driven by epigenetic changes (154, 155).
Hematopoietic stem cell transplant (HSCT) recipients who experienced CMV reactivation post-transplant show a similar increase in the proportion of adaptive NK cells (156). These adaptive NK cells persist in the body for at least a year in post-HSCT recipients (157). In contrast, CMV seronegative recipients retain a population of comparatively less mature NK cells, presumably offering these recipients less protection. This is especially important considering that NK cells are among the first lymphocytes to recover status post-HSCT (158). This expansion of NK cells associated with CMV reactivation is also linked to a significant increase in the number of all NK cell subsets that persists even without further viral reactivation (157). Furthermore, NKG2C+ NK cells transplanted from CMV seropositive donors display heightened function during CMV reactivation (159). This supports the idea that NKG2C+ memory NK cells are also potentially transplantable and have rationale as a therapeutic which deserves further exploration.
Influenza A Viruses are members of the Orthomyxoviridae family and have a segmented genome consisting of 8 linear RNA strands (160). They are further subtyped based on the hemagglutinin (HA) and neuraminidase (NA) they express, which are surface glycoproteins that help mediate viral entry and exit respectively (161). Influenza viruses primarily infect epithelial cells of the upper and lower respiratory tract, and severe cases can result in acute respiratory distress syndrome (162).
Influenza virus A (IVA) infection induces the infiltration of NK cells into lung tissue with a significant increase in the number of NK cells expressing CXCR3, CXCR6, and CCR5, all of which are markers of tissue homing (163, 164). In mice, this infiltration is also in part regulated by IL-15 as evidenced by the blocking of IL-15 delaying this infiltration (165). Severe infection is associated with a decline in the total number of NK cells in the peripheral blood, particularly among CD56bright NK cells, along with marked NK cell activation evidenced by an increase in the proportion of NK cells expressing CD69 (166, 167).
As a whole, there is a decrease in the number of NK cells expressing NKp46 but an increase in the number of cells expressing coreceptor 2B4 in the blood and these cells also show a greater degree of activation (168). Of the remaining NKp46 expressing NK cells, the proportion of NKp46+ NKp44+ NK cells expands while the proportion of NKp46+ NKp44- declines, contributing to the shift towards NK cell activation (169). An NKp46+ NKG2A+ NK cell subset has also been found to be induced by IVA infection. This subset of NK cells has demonstrated a memory phenotype in that upon reencountering previously exposed influenza antigen, they display decreased cytotoxicity and increased IFN-γ production (170). Influenza is also directly capable of infecting NK cells, triggering apoptosis, and infection has been shown to inhibit NK cell cytotoxic function (171–173).
In the lung, tissue resident NK cells comprise 10-25% of the NK cell population meaning that most NK cells in the lung are circulating NK cells (15). The majority of lung tissue resident NK cells are CD16- and have similarly been divided into CD56dim and CD56bright subsets in some studies (15). Functionally, the CD56bright lung tissue resident NK cells subset has been described as showing greater functionality, with increased degranulation and IFN-γ production in response to IVA infection compared to other NK cells ex vivo (174). The rationale behind this subsets enhanced function is poorly understood and whether or not it translates in vivo is unknown.
Differential activation of NK cells has been observed following coculture with replication deficient influenza A pseudotypes that express IVA HA and NA (175). Particularly, more robust upregulation of CD69, CD107a, and IFN-γ along with downregulation of NKp46 is observed with H5N1, and the 1918 H1N1 compared to 2009 H1N1 (Figure 5) (175). More broadly speaking, Influenza induces both degranulation and cytokine production, particularly in CD56bright NK cells, and while their activation contributes to host immunity, it may well contribute to tissue damage as well (176). For example, NK cell-depleted mice infected with a high dose of influenza A virus have decreased mortality and disease morbidity illustrating how the NK cell response can be deleterious (177). Conversely, a study looking at NK cell responses in chickens challenged by either a weakly pathogenic H9N2 avian influenza virus or a highly pathogenic H5N1 avian influenza virus found that the less pathogenic avian virus induced a more vigorously activated NK cell response (178). This shows that a notable NK cell response in influenza is not always associated with viral virulence and may even be protective. Additionally, CD94 expression has been negatively correlated with influenza symptom severity, suggesting that signaling through CD94 containing receptors plays a role in control of IVA infection (179). Overall, more work is required to characterize differential NK cell responses between influenza viruses, and what correlates are predictive of deleterious responses.
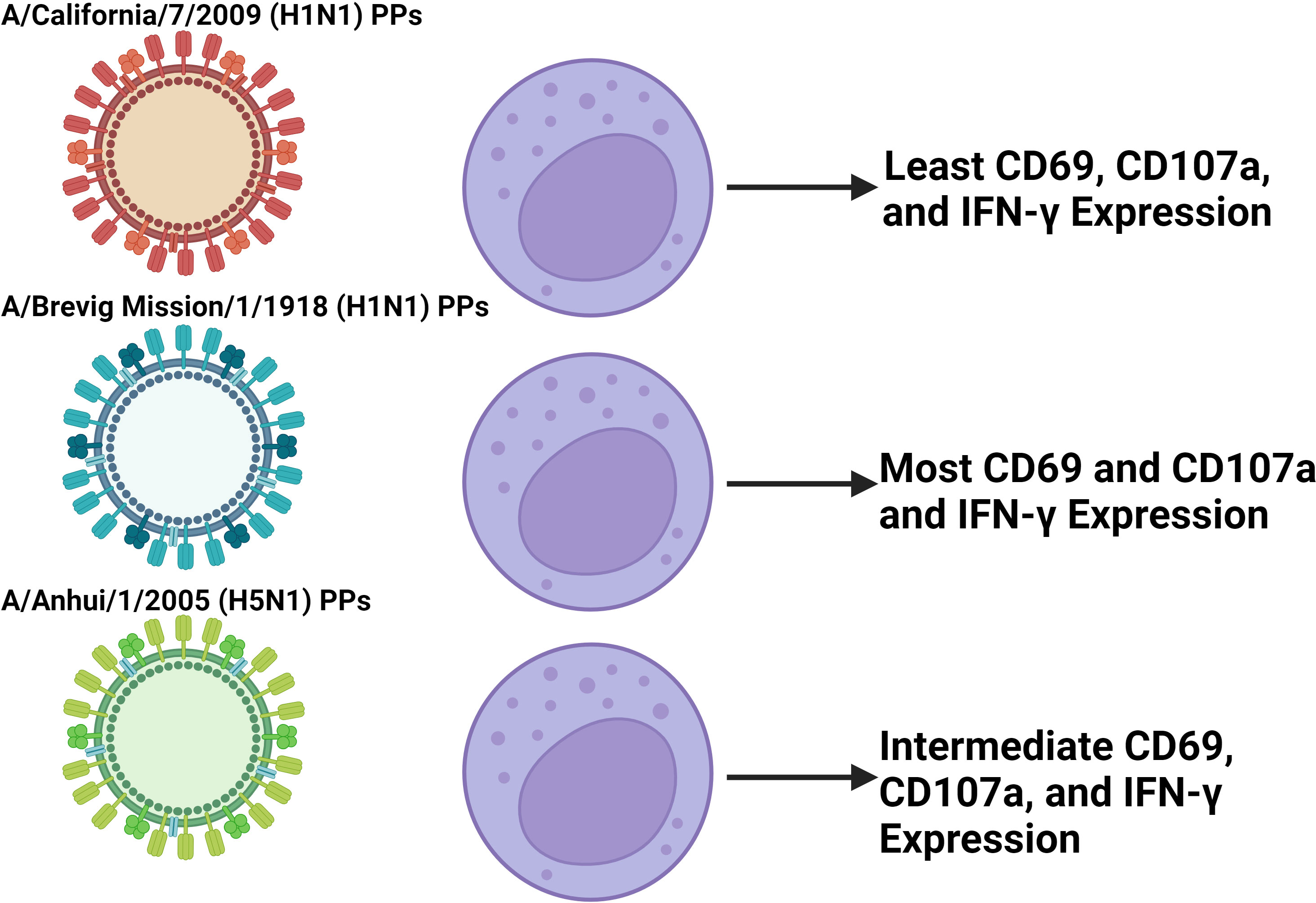
Figure 5 Coculture of NK cells with different IVA PPs induce differential NK cell activation. In vitro experiments coculturing NK cells isolated from PBMCs with pseudotype particles (PPs) of different IVA viruses found differential activation between PPs in terms of CD69, CD107a, and IFN-γ expression (139). The findings presented in the figure correspond to the 500 HAU/mL IVA coculture dose. The PPs modeling the more virulent 1918 H1N1 and H5N1 IVA viruses induced greater activation than the less virulent 2009 H1N1 PPs. This suggests that in IVA infection, disparities in early NK cell activation between influenza viruses may play a role in contributing to differential virulence between them. Further work is required to characterize this effect in vivo with whole virus that is not replication deficient due to IVA infecting and killing NK cells.
The IFN-γ response orchestrated by NK cells is particularly important in the context of infection. Notably, impairment of NK cell activation and IFN-γ secretion is associated with weakened cytotoxic T lymphocyte activity against influenza (180). This has been corroborated in mice, and the initial increase in production of IFN-γ after NK cell activation results in downstream cytotoxic function in CD8 and NK cells during influenza infection (181). This potent IFN-γ response orchestrated by NK cells has also been implicated in contributing to thymic atrophy during influenza A infection, showing it can also be deleterious (182).
Influenza vaccination has also been shown to prime NK cell function with patients displaying increased production of IFN-γ and enhanced activation after stimulation, while NK cell phenotype and subset is largely unaltered (183, 184). However, influenza vaccination does induce a proportion of CD56dim NK cells to become memory NK cells and express intracellular NKp46, offering a possible mechanism by which the IFN-γ NK cell response is enhanced after vaccination (185). These vaccine induced memory NK cells display a heightened IFN-γ response that persists 6 months after vaccination. It has also been found that NK cells in mice vaccinated with a multivalent influenza vaccine had greater expression of granzyme B and perforin and took on a more mature phenotype after being challenged by heterosubtypic influenza strains compared to unvaccinated mice (186). This again supports the notion that influenza vaccination primes NK cells to offer greater protection even against heterosubtypic strains where vaccine-derived humoral immunity may struggle. Changes in the NK cell population induced by other vaccines remain underexplored, and examining these NK cell phenotypic changes induced by vaccines as a correlate of immunity warrants more attention.
SARS-CoV-2 belongs to the Coronaviridae family and has a large single-stranded RNA genome (187). The virus primarily infects ACE2+ cells of the respiratory tract, particularly type II alveolar pneumocytes and in severe cases can result in acute respiratory distress syndrome (188). Severe COVID-19 disease has been linked to a significant decline in CD56+ CD16+ NK cells in the blood (189–191). This decrease in circulating NK cells is correlated with acute SARS-CoV-2 infection and is associated with disease severity (192, 193). In hospitalized patients, NK cell counts correlate directly with the speed of viral load decline. For example, those with normal NK cell numbers in the blood showed a faster decline of viral load compared to those with low NK cell numbers, suggesting that circulating NK cells may represent a prognostic clinical marker of COVID-19 severity and outcome in adult patients (194). When patients recover after COVID-19, their circulating NK cell counts return to normal. In patients affected by long COVID-19, NK cell counts have recovered, despite continued symptoms (195). In Multisystem Inflammatory Syndrome in Children (MIS-C), a severe post-infectious complication that occurs 4-6 weeks after COVID-19 infection, CD56dim NK cells are also decreased in number (196).
During COVID-19, NK cells are robustly activated, evidenced by an increase in the expression of CD69, and KI-67 a proliferation marker (192). Expression of the genes KLRD1 and KLRG1, which encode CD94 and killer cell lectin-like receptor G1 respectively, are also noted to decrease in many patients contributing towards NK cell activation (197).
SARS-CoV-2 infection has also been linked to an increase in the expression of NKG2C, creating a population similar to the earlier described memory NK cells (192, 198). Interestingly in patients with adaptive NK cells, there was no detectable circulating CMV DNA, suggesting that the expansion of adaptive NK cells in severe COVID-19 patients may be independent of CMV reactivation secondary to COVID-19. It is still to be determined if SARS-CoV-2 itself can drive the expansion of adaptive NK cells or if they are driven by the production of pro-inflammatory cytokines. In those affected by long COVID-19, there are increased levels of CD57+ NKG2C+ memory NK cells; these are one of the variables important for predicting a higher susceptibility to develop long COVID-19. However, despite greater populations of memory-like NK cells, their effector functions remained impaired (195).
The expression of cytotoxic effector molecules like perforin and Granzyme A are also significantly increased during most COVID-19 cases (189, 199, 200). While these cytotoxic effectors are upregulated, NK cells from COVID-19 patients have been shown to have reduced degranulation potential and cytotoxicity (201). Furthermore, in severe cases, the quantity of TNF-α and IFN-γ produced by NK cells is diminished (202–204). Inhibitory KIR and NKG2A receptors have been found to be upregulated during COVID-19 and likely contribute to NK cell exhaustion (205, 206) NKG2A is also highly upregulated in acute, severe COVID-19 patients, while mild and moderate patients show a recovery of NKG2A after resolution of the infection (198). Significant NK cell exhaustion is further supported by the increased frequency of programmed cell death protein 1 (PD-1), Lymphocyte-Activation Gene 3 (LAG-3), and TIGIT, as well as a decreased frequency of NKG2D. However, the level of PD-1 expression in NK cells isn’t correlated with disease severity (203, 204), while decreased expression of TIGIT and DNAM-1 have been associated with slower viral clearance (207). Overall, more work is needed to ascertain the degree to which exhaustion is responsible for NK cell’s diminished function in COVID-19, as opposed to issues in tissue homing.
Infection has also been linked to the expansion of the CD56dim CD16neg NK cell population, which has decreased cytotoxic potential compared to the standard CD56dim CD16+ NK cell subtype (Figure 6). It has also been shown that CD56dim CD16neg NK cells expand in the early phases of SARS-CoV-2 and then decrease in mild or moderate cases, but in severe COVID-19 cases, this subset continues to expand (208). The study also suggests that loss of CD16 is potentially being mediated by cleavage through Adam17 in these cells (208). In MIS-C, studies have shown that there is a decrease in the prevalence of CD16+ NK cells, suggesting that there is a decrease in NK cell ADCC function in MIS-C (200). However, further studies are needed to explore NK cell function in MIS-C directly beyond just immunophenotyping.
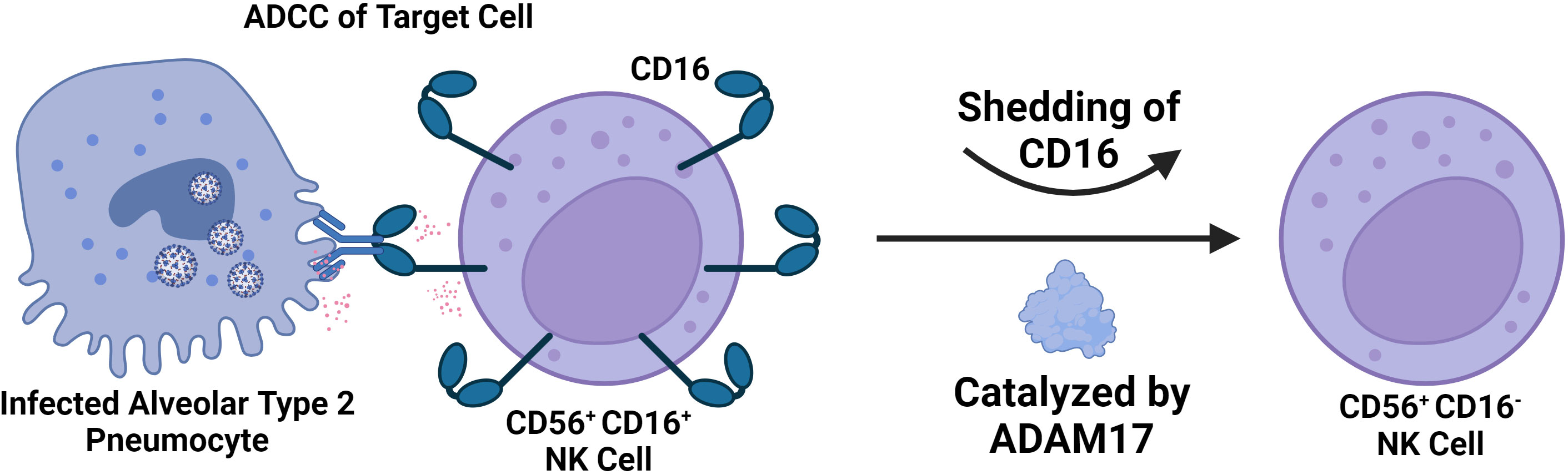
Figure 6 Severe SARS-CoV-2 is Associated with a Significant decrease in the number of CD56+ CD16+ NK Cells in the Blood. The decline in CD56+ CD16+ NK Cells is due to CD16 shedding catalyzed by ADAM17. This results in an accumulation of CD56+ CD16- NK cells of less cytotoxic potential and in severe cases likely contributes to impaired capability to engage in ADCC. CD16 mediates ADCC by binding to antibodies attached to target cells, ultimately resulting in degranulation.
In fatal COVID-19 cases, patients had impaired upregulation of perforin, granzyme-A, and KI-67, suggesting that defects in NK cell cytotoxic activity are associated with increased morbidity and mortality (209). Corroborating this is the observation that NK cell mediated ADCC appears to be impaired in severe COVID-19 (210). In the context of COVID-19 infection, TGF-β signaling has been associated with a stark reduction in NK cell effector functions, with patient serum having been found to inhibit NK cell cytotoxicity in a TGF-β dependent manner (194). Similarly, impairments in IFN-γ production appear to occur in a TGF-β dependent manner (211). This suggests that TGF-β signaling contributes significantly to NK cell dysregulation and exhaustion during COVID-19.
Given the extensive changes in NK cell phenotype and repertoire, which often lead to exhaustion and dysregulated NK responses in viral infections, several therapeutic approaches with the goal of promoting NK cell function are under investigation. The majority of NK cell-related therapies originate from the domain of cancer biology and have only recently been studied in the context of viral diseases.
The development of therapeutic TGF-β inhibitors largely stems from the implication of aberrant TGF-β signaling in the pathogenesis of cancer, cardiovascular disease, and fibrosis. As a therapeutic class, they span a wide variety of structures from small effector drugs to blocking antibodies targeting a diverse host of TGF-β pathway components (212, 213). TGF-β signaling in HIV infection has been documented to promote latency by upregulating the transcriptional repressor Blimp-1 (214, 215). Galunisertib, a TGF-β type I receptor inhibitor, was administered to SIV infected rhesus macaques, where it was shown to increase reactivation of the SIV latent reservoir, and enhance the subsequent immune response to SIV (216). TGF-β signaling during SARS-CoV-2 infection has also been associated with NK cell dysregulation that limits functional activity contributing to subsequent viral pathogenesis (194, 217), showing that TGF-β inhibitors may have rationale to be applied in acute viral infections too.
Broadly speaking, TGF-β signaling has been shown to impair NK cell cytotoxic function in part by restricting the IL-15 activation of the mTOR pathway (218). Additionally, deletion of the TGF-β receptor subunit TGF-βRII has been shown to promote the cytotoxic function of NK cells in response to IL-15, enhancing the ability of NK cells to limit metastasis in two different murine tumor models (218). It is worth noting that by inhibiting NK cell cytotoxic function and the secretion of proinflammatory cytokines like IFN-γ, TGF-β plays an important role in limiting excessive NK cell activation. The risk of NK cell hyperresponsiveness, and the subsequent inflammatory environment and tissue damage this induces, should be considered in the testing of TGF-β Inhibitors. However, considering the clear contribution of TGF-β signaling to NK cell dysregulation and evidence that restraining this signaling may actually promote NK cell cytotoxicity beneficially, there warrants further investigation into the use of TGF-β inhibitors as a potential therapeutic for treating viral infections.
A key component of NK cell dysregulation is exhaustion driven by inhibitory pathways. These inhibitory pathways have been termed immune checkpoints, and immune checkpoint inhibitors have been developed to interfere with these pathways in an effort to preserve immune function (219).
NKG2A is an important inhibitory receptor expressed on most NK cells and is considered an immune checkpoint for NK cells. NKG2A inhibitors like the monoclonal antibody monalizumab have been found to promote antitumor immunity in treating squamous cell carcinoma by enhancing the activity of NK and CD8 cells (220). Monalizumab has also demonstrated a similar response in the setting of murine colorectal cancer (221), and against HLA-E+ leukemia and lymphoma in mice (222). NKG2A blocking monoclonal antibodies have also shown promise in treating viral hepatitis, with cultured NK cells from patient’s demonstrating enhanced cytotoxicity (223). Similarly, NKG2A-blocking monoclonal antibodies have been shown to restore the production of IFN-γ by NK cells during HCV infection, and has been associated with diminished CD8 T cell exhaustion (138). This therapeutic also has rationale to be applied in treating COVID-19 due to NK and CD8 T cell exhaustion being driven in part by increased NKG2A expression (205, 224, 225).
Antibodies targeting inhibitory KIR receptors have also emerged as another NK cell related immune checkpoint therapy. One such KIR inhibitor named IPH2101 which targets inhibitory KIRs (KIR2DL-1, KIR2DL-2, and KIRT2Dl-3) recently underwent a phase 1 study where it was deemed safe and tolerable and in ex vivo assays demonstrated increased NK cell cytotoxicity against multiple myeloma (226). However, a phase 2 study of IPH2101 was terminated due to lack of clinical efficacy (227), and similarly, a trial for lirulumab, another anti-KIR antibody (which targets KIR2DL-1, KIR2DL-2, and KIRT2Dl-3) was terminated for failing to meet objective response criteria (228). IPH2101 has also be combined with the anti-tumor drug lenalidomide in further testing against multiple myeloma with early data suggesting a promising synergistic effect, although more study is required (229, 230). As this study highlighted, a key advantage of immune checkpoint inhibitors is that they can readily be combined with other therapeutics. IPH2101 has also been tested in the setting of acute myeloid leukemia, where a degree of early TNF-α and CD69 upregulation was detected (231). Another anti-KIR antibody named IPH4102, targeting KIR3DL2, has also been tested in relapsed and refractory cutaneous T-cell lymphoma, which showed its potential to elicit a promising clinical response in 36.4% of the study participants (232). Little testing of KIR inhibitors has been undertaken in the setting of viral infections, although they warrant study particularly in HIV due to HIV viremia being associated with an increase in the expression of inhibitory KIR receptors (82).
PD-1 signaling has been implicated as a hallmark of T-cell exhaustion and a contributing factor of immune escape in the setting of cancer, and is considered another important immune checkpoint (233, 234). PD-1 signaling has been shown to be of significance to NK cells in multiple myeloma (235), renal cell carcinoma (236), and in Kaposi sarcoma (237), where it contributes to an exhausted NK cell response (29). Several PD-1 checkpoint inhibitors have been approved by the FDA including pembrolizumab, nivolumab, and cemiplimab among many others, which together have been tested clinically in a diverse arrangement of cancers (238). Significant expression of PD-1 has been shown in chronic HIV (239), HCB (240), HCV (241), Influenza (242), and SARS-CoV-2 (243), with a similar exhaustive effect.
In a mice influenza challenge model, PD-1 inhibitors were shown to enhance CD8 T-cell function resulting in quicker viral clearance (244). In SIV infected rhesus macaques, the blockade of PD-1 was well tolerated and resulted in the expansion of functional SIV-specific CD8 T-cells in the blood and gut as well as the expansion of memory B-cells all of which was associated with lower viral load and prolonged survival (245). Similarly, in CD4+ humanized mice, PD-1 blockade was associated with lower HIV viral load and increased T-cell counts (246). In HBV, PD-1 blockade has also been shown to again promote CD8+ T-cell expansion and secretion of IFN-γ and IL-2 (247). Importantly, the upregulation of PD-1 in NK cells during acute febrile malaria has been associated with enhanced ADCC, but otherwise diminished degranulation (248). This finding hints that the impact of PD-1 blockade on NK cells, may be more nuanced in terms of impact on functional activity than CD8 T cell function. Overall, more work is needed to characterize the impact that PD-1 blockade has on the NK cell population in the setting of these viral infections; however it is clear based on the CD8 T-cell response alone that PD-1 blockade is a promising therapeutic that deserves further examination in the setting of infectious diseases (249).
Another therapeutic approach entails the administration of exogenous NK cells. The goal is to supply functionally intact cells to supplement NK cell functional activity during virally mediated NK cell dysfunction and exhaustion. The administration of allogenic NK cells has been safely and effectively used to treat leukemia (250). Delivery of exogenous NK cells have also demonstrated clinical responses in treating non-small cell lung cancer (251), platinum-resistant ovarian cancer (252), melanoma (253), and renal cell carcinoma (253). Very little work has been done studying adoptive NK cell therapies in infectious diseases. In SARS-CoV-2 and HIV-1, an additional consideration is the infusion of adaptive NK cells due to their enhanced cytotoxicity and persistence (198).
Recently, genetically engineered NK cell lines derived from induced pluripotent stem cells (iPSC) have emerged. Some of the gene edits in these cells include a non-cleavable CD16a receptor, a membrane-bound IL-15/IL-15R fusion protein, and knockout of coenzyme CD38 to promote the persistence and function of these cells after administration (254). In the setting of HIV, allogenic NK cells derived from human embryonic stem cells have been shown to decrease HIV replication during acute infection in humanized mice, and when combined with latency reversal, helped diminish the HIV reservoir (255, 256). Furthermore, phase 1 safety trials where haploidentical NK cells are infused into HIV infected ART suppressed patients in combination with either IL-2 or N-803 (an IL-15 superagonist) supportive therapy have been completed (NCT03346499, NCT03899480).
There are clinical trials using primary and iPSC-derived or “off-the-shelf” NK cells as therapeutics against SARS-CoV-2, suggesting that adoptive NK cells transfer may be a viable therapeutic in the setting of acute viral infection. An example of this is a phase 1 study in which a line of IPSC derived NK cells called FT516 administered to hypoxic COVID-19 patients to assess their safety and maximum tolerated dose. Results of this study (NCT04363346) are anticipated soon. Taken together, the demonstrated safety of utilizing exogenous NK cells, along with preliminary findings suggesting possible efficacy in treating viral infections, particularly in HIV, calls for further investigation.
Approaches to enhance NK cell anti-viral functions in vivo and prior to adoptive transfer of exogenous NK cells are of interest. In oncology, many cytokines have been used for the treatment of NK cell dysfunction, such as IL-12, IL-15, and IL-18 given their ability to promote NK cell expansion and maturation (257–259). In the context of SARS-CoV-2, a phase 2 clinical trial has been completed looking at the efficacy of low-dose IL-2 in acute respiratory distress syndrome (ARDS) related to COVID-19 (NCT04357444). NK cells treated with IL-15 and stimulated with an inactivated whole influenza virus were shown to mount a more vigorous NK cell response compared to when not treated with IL-15 (260). In HIV, NK cells stimulated with IL-15 demonstrated uniformly improved effector functions and the ability to kill HIV-infected cells that were treated with a latency reversal drug (261).
IL-12, IL-15 IL-18 are also of particular interest given their ability to induce changes in NK cells through transcriptional, epigenetic, and metabolic mechanisms that ultimately increase their cytotoxicity and result in memory like behavior (262). Cytokine induced memory like (CIML) NK cells have displayed increased IFN-γ responses in addition to enhanced recall responses even after multiple rounds of cell division (263, 264). They also display increased expression of NKG2C, NKG2A, CD69, and CD94, but not CD57 or KIR receptors (263). Due to the enhanced effector functions and relative ease at which CIML NK cells are generated, extensive study of the adoptive transfer of these cells, especially in the setting of myeloid malignancies, multiple myeloma, and head and neck squamous cell carcinoma has been undertaken (262). For example, infusion of 5 to 10 million cells/kg in patients with myeloid malignancies on lymphocyte depleting chemotherapy displayed a 10 to 50 fold expansion of NK cells that was determined to be independent of CMV reactivation and persisted for months (265). Other phase 1 studies of adoptively transferred CIML NK cells has also documented clinical responses. In the setting of acute myeloid leukemia, one study found clinical responses in 5 of 9 patients, of which included 4 complete remissions (259). Another study found complete remission in 4 of 8 patients (266), and when supported with 3 weeks of N-803 an 87% complete response rate was achieved by day 28 in a different study (267). The adoptive transfer of CIML NK cells has been understudied as a possible treatment for viral infections, and preclinical work, especially in the setting of HIV should be prioritized.
Due to the aforementioned benefit of IL-15 stimulation on NK cell effector functions, novel approaches to enhance IL-15 signaling in vivo have been investigated. One such therapeutic is the IL-15 superagonist ALT-803. In SIV-positive rhesus macaques, ALT-803 (now N-803), an IL-15 superagonist comprised of an N72D mutant IL-15 molecule attached to its alpha receptor and a human IgG1 fragment designed to increase IL-15 activity, was shown to increase the NK cell population in the peripheral blood and decreased viral loads (268). In SHIV-infected rhesus macaques, N-803 was shown to mediate migration of virus-specific CD8+ T and NK cells to B-cell follicles, however no latency reversal was observed (269). A recent phase-1 clinical trial of N-803, showed that this molecule is also safe and tolerable in people living with HIV that are ART-suppressed. Further, N-803 was associated with CD8+ T and NK cell activation, as well as induced HIV RNA expression, with concurrent reduction in HIV DNA in Lymphoid tissue over time (270). Future clinical trials are needed to test the effects of these molecules on HIV reservoirs due to the promising results of the aforementioned preclinical and phase 1 studies.
As mentioned above, viral infections, especially HIV, can deter the natural recognition of virally infected cells by NK cells due to the manipulation of activating and inhibitory receptors expressed by the infected cell, thereby evading the immune response (82, 86, 87, 90–95). Broadly neutralizing antibodies (bnAbs) have been identified to play a role in viral control (271), and are used as a treatment and prevention strategy (272, 273).
Furthermore, in the RV144 HIV vaccine trial, which showed a statistically significant decrease in HIV infection risk (274), one correlate of reduced risk was found to be increased IgG antibodies targeting the V1V2 loop of the HIV ENV protein (275, 276). In a follow-up analysis, it was shown that these IgG antibodies elicited Fc-mediated NK cell ADCC functions (277, 278). These data suggest that non-neutralizing antibodies play a role in HIV protection through mediating effective Fc-effector functions (279–281). Additionally, NK cell-mediated ADCC against HIV has been noted as an important mechanism for HIV control (282–284), despite NK cell dysfunction (75, 113). However, a major concern with HIV-specific bnAbs is that HIV mutates rapidly and can escape bnAb-mediated neutralization (285–288). Further, the majority of bnAbs, which are generally of the IgG1 isotype, have good neutralization capacity, but subpar Fc-effector functions compared to their IgG3 isotypes (289–291).
Engineered small molecules are a promising strategy to direct NK cell mediated killing. The cancer field has made great progress in this field with the development of NK cell engagers (NKCE), Bi- and Tri– specific killer engagers (BiKEs and TriKEs™) as well as multi-specific antibody-based constructs (ANKET and StitchMabs) (292–297), which bind to activating receptors on NK cells. Briefly, TriKEs™ consist of an anti-CD16 engaging molecule that directly binds the CD16 receptor, joined via a flexible linker to an IL-15 molecule, and in-turn joined via linkers to an antigen engaging molecule to target an antigen of interest (298). Similarly, BiKEs consist of a similar anti-CD16 engaging molecule that is joined via a linker to an analogous antigen engaging molecule (298). ANKET constructs consist of an Fab that targets an NK cell receptor, commonly NKp46, together with another Fab that targets an antigen of interest (292). As alluded to above, ANKET molecules can also be generated to display bivalent binding to one or two different antigens (292). Furthermore, T-cell based nano engagers have also been developed in the form of Bi-specific T cell engagers (BiTEs) (299), and dual affinity retargeting antibodies (DARTs) (300, 301), although this is outside the scope of this review and won’t be discussed further here.
If we consider these therapies for HIV infection, many of the NK cell engagers mentioned above use either NKp46 or CD16 as the NK cell engaging molecule. However, NK cells are dysregulated during HIV infection, with activating and inhibitory receptor expression modulated. Importantly, despite NK cell dysfunction during HIV infection, NK cells still retain expression of CD16, enabling them to mediate ADCC (82). Therefore, given NK dysfunction, the retention of CD16 and the importance of Fc-mediated effector functions to combat HIV, strategies that exploit these are promising. Engineered small molecules that can bind directly to NK cells via CD16 interaction have been recently developed (302). This has been achieved by utilizing the BiKE and TriKE™ constructs that had previously been designed to target cancers (303–308).
These molecules allow the targeting of a variety of antigens for multiple viral infections, while also mitigating potential issues that arise with Fc-mediated effector functions. Further the IL-15 molecule induces persistence, expansion, and activation of NK cells (257). Currently, a BiKE construct has been generated using the HIV binding region of the VRC01 bnAb. This VRC01 BiKE construct, was shown to bind the HIV infected cell lines HIV-IIIB and ACH-2, but not their uninfected counterparts H9 or CEMs. Further, this VRC01 BiKE mediated effective NK activation, assessed by degranulation via CD107a expression and IFN-γ and TNF-α cytokine production, and subsequent killing against these HIV infected cells (309). Further work generating and testing TriKE constructs making use of the VRC01 binding region as well as other known HIV bnAb binding regions is currently underway.
However, HIV can escape viral restriction via bnAbs, so breadth is an important consideration in the development of novel therapeutics. Importantly, all HIV variants must utilize the CD4 receptor for initial gp120 binding to the cell surface (310). Therefore, using the CD4 receptor as the HIV binding region in novel therapeutics could overcome HIV escape (285). However, as this is a naturally found receptor used by many cells, careful consideration must be taken in the design so not to induce self-targeting. Extensive work in this field has already been done (311–313), and a CD4 extracellular domain 1 (CD4ECD1) molecule, designated mD1.22, with potent HIV neutralization and little binding to MHC class 2 has been identified (314). When designed into a BiKE construct with CD16 binding capabilities, it induced NK cell degranulation and killed HIV target cells (315). A CD4ECD1 TRiKE is currently in development and testing. Further, these TriKEs can be used in combination with a checkpoint inhibitor therapy (308), or other cytokine based or adoptive NK transfer strategies, highlighting the vast potential of these NK cell engagers for combination therapies to combat viral infections.
Chimeric antigen receptor (CAR) cells are another promising treatment that have been popular in the cancer field for many years and are now starting to be studied as a potential therapeutic for viral diseases. Many researchers have begun to produce CAR NK cells as they are less expensive and less likely to cause cytokine storm than CAR T cells (316). One such example is a CAR-NK cell therapy that secretes IL-15 and expresses a CAR with an extracellular ACE2 domain to target the SARS-CoV-2 spike protein and has been promising against SARS-CoV-2 in vitro (317). There is also currently another clinical trial underway to test a CAR NK cell that expresses ACE2 on it surface to treat severe COVID-19 patients (NCT04324996).
CAR NK cells are also currently being used in a clinical trial to treat HIV (318). In the setting of HIV, CAR-NK cells have largely targeted a single epitope of the HIV envelope glycoprotein gp160. Recently, the development of universal CAR-NK cells that target a broader range of gp160 has shown greater activation and the ability to kill HIV infected human CD4+ T cells (319). A visual overview of the discussed therapeutics is available in Figure 7.
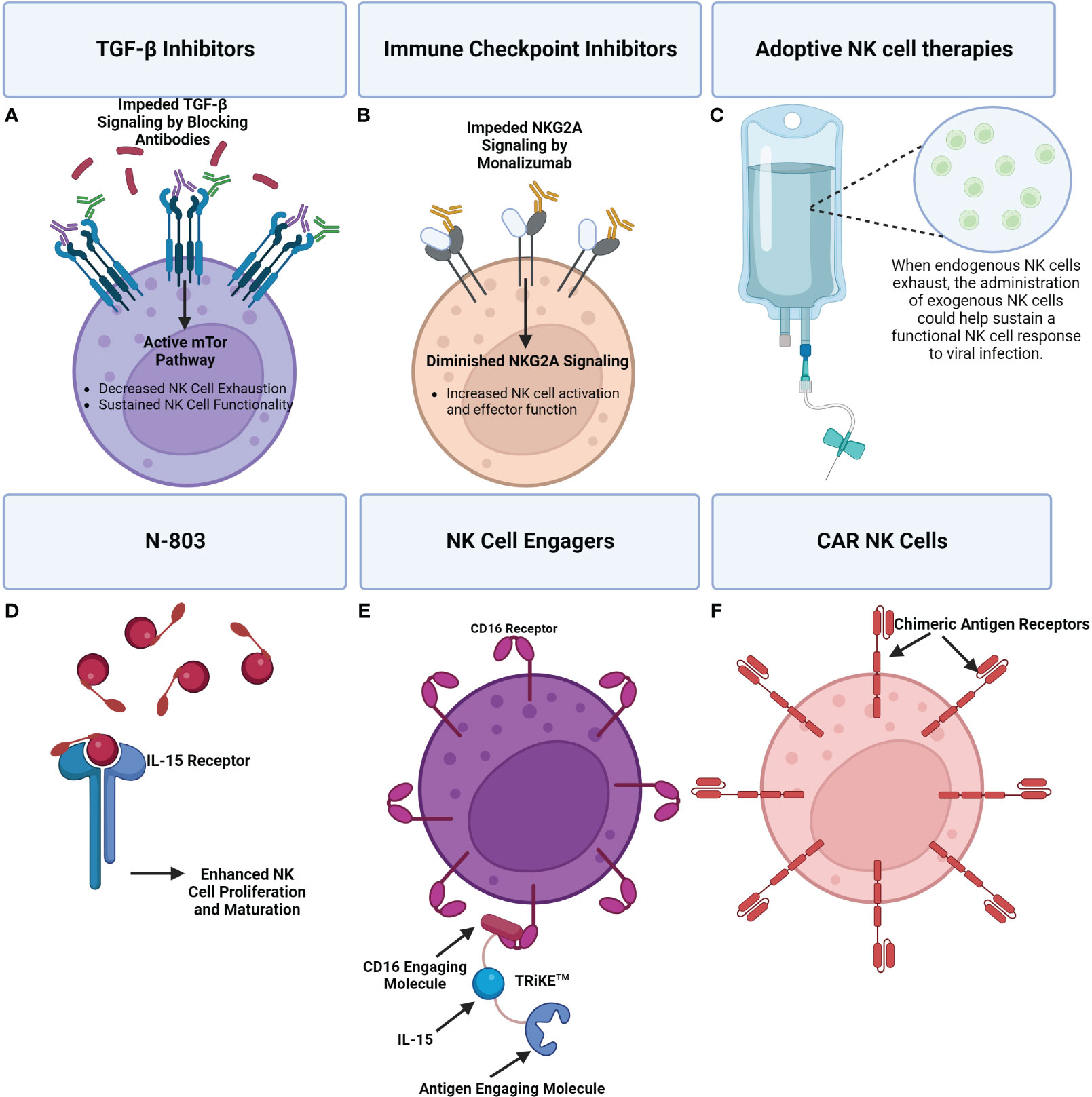
Figure 7 A visual overview of a few emerging therapeutics seeking to address NK cell dysregulation during viral infection. (A) TGF-β signaling inhibits the mTOR pathway which has been shown to result in impaired NK cell effector functions. The use of TGF-β inhibitors can promote mTOR signaling and in turn NK cell activity. (B) Immune checkpoint inhibitors can be applied to reduce inhibitory signaling that is causing NK cell exhaustion. For example, Monalizumab, an NKG2A inhibitor, can block inhibitory NKG2A contributing to a shift towards NK cell activation and effector functions. (C) Adoptive NK cell therapies entail the administration of exogenous NK cells to supplement the immune response. (D) N-803 is an IL-15 superagonist consisting of a mutant IL-15 molecule associated with an IL-15 receptor fusion protein. Administration of N-803 promotes NK cell proliferation and maturation. (E) An example of an NK cell engager are TRiKE™ constructs which consist of a CD16 engaging molecule linked to an IL-15 molecule that’s linked to an antigen linking molecule. Binding of the TRiKE™ to an NK cell CD16 molecule induces activation while IL-15 supports proliferation and maturation. The antigen linking molecule is designed to target an antigen of interest. (F) The extracellular antigen recognition domain of CAR receptors enables CAR NK cells to specifically target cells producing an antigen of interest. CAR NK cells can be administered to supplement an exhausted NK cell response, with less risk than CAR T-cells.
As described above, several lines of therapeutics including TGF- β inhibitors, adoptive cell therapies, cytokine therapies, NK cell engagers, and CAR NK cells have emerged in an effort to remedy NK cell dysregulation. While many of these therapeutics have been primarily tested in the setting cancer, they deserve further attention as possible remedies to preserve and restore NK cells function in the setting of viral infection. Furthermore, testing of these therapies in conjunction with each other as well as other therapeutics may prove particularly beneficial due to possible synergism.
Due to the extensive genetic differences present in NK cell receptors like the KIR family and ligands like HLA (31–33, 37), it is likely that there is also significant variation in the efficacy of these therapeutics depending on an individual’s personal repertoire of receptor and ligand genes. One of the clearest examples of this can be found in the case of KIR checkpoint inhibitors like IPH4102. Due to extensive variability in individuals’ KIR repertoire, responses to therapy between patients are also very variable, with only a subset achieving clinical responses (232). Similar variability in responses should be anticipated for the other therapeutics discussed here due to genetic variability in other NK cell receptors and ligands. Another consideration is that many of these therapeutics also run the risk of contributing to tissue damage and hyperinflammatory states by enhancing NK cell cytotoxic action and secretion of proinflammatory cytokines. Nonetheless, there may prove to be a balance between this risk and possible therapeutic benefit.
While NK cells play an important role in responding early to viral challenges, they can be dysregulated. This dysregulation, whether it be related to alterations in the prevalence of activating or inhibitory receptors, relative maturation in terms of subset prevalence, expression of functional effector proteins, or a product of an individual’s receptor and HLA genetics limits their ability to mount an effective antiviral response. For example, during chronic HIV and HCV, as well as acute SARS-CoV-2 there is an inherent depletion in the total prevalence of CD56+ CD16+ NK cells in the peripheral blood which is accompanied by markedly increased NK cell exhaustion. In contrast, aberrant NK cell activation in response to viral infection can also result in a direct contribution to viral disease pathogenesis by mediating excessive tissue damage, as was described as possibly being the case in some select influenza viruses.
While significant work has been done characterizing the NK cell repertoire correlated with disease state, less work has been done regarding elucidating the signaling environment that leads to changes in the NK cell population in vivo. As discussed, while cytokines like TGF-β are known to alter NK cell phenotype and function, the full landscape of signaling that leads to disease-specific NK cell repertoire requires more broad characterization. This rings especially true when considering the extensive genetic variability present in both HLA and important receptor families such as the KIR receptor family. The significant genetic variance in terms of NK cell repertoire and the subsequent corresponding differences in affinity between NK cell receptors and their ligands renders it extremely difficult to elucidate relationships between NK cell repertoire and disease. In essence, the state of an NK cell response, is determined not just by the relative expression of these receptors and their ligands, but by their genetic content and the relative receptor-ligand affinity it institutes. Despite these limitations, early preclinical and clinical work testing therapeutics seeking to address the changes in NK cell repertoire and activity associated with viral infection remains promising (Figure 8).

Figure 8 Schema of the Logic Underpinning the Use of NK Cell Therapeutics in Viral Infection. Many viral infections are associated with changes in the NK cell repertoire in terms of receptor expression and subset prevalence that result in altered NK cell functionality. Specific changes in NK cell repertoire can be targeted for therapeutic modulation to alter functionality in order to combat pathogenesis. For example, chronic HCV is associated with a significant increase in the expression of NKG2A, ultimately contributing to NK cell exhaustion, and a diminished antiviral response. The administration of NKG2A blockade interferes with inhibitory signaling in an effort to help restore effector functions to promote antiviral immunity.
JB: Conceptualization, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing. JD: Writing – original draft, Writing – review & editing. RC: Writing – original draft, Writing – review & editing. GH: Writing – review & editing. JR: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by NCATS UL1TR002494 (JD).
We thank BioRender which was used to create the figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
2. Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol (2017) 8:930. doi: 10.3389/fimmu.2017.00930
3. Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med (1989) 320(26):1731–5. doi: 10.1056/NEJM198906293202605
4. Orange JS. Human natural killer cell deficiencies. Curr Opin Allergy Clin Immunol (2006) 6(6):399–409. doi: 10.1097/ACI.0b013e3280106b65
5. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132(3):515–25. doi: 10.1016/j.jaci.2013.07.020
6. Yu J, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol (2013) 34(12):573–82. doi: 10.1016/j.it.2013.07.005
7. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/S1471-4906(01)02060-9
8. Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology (2009) 126(4):458–65. doi: 10.1111/j.1365-2567.2008.03027.x
9. Carson WE, Fehniger TA, Caligiuri MA. CD56bright natural killer cell subsets: characterization of distinct functional responses to interleukin-2 and the c-kit ligand. Eur J Immunol (1997) 27(2):354–60. doi: 10.1002/eji.1830270203
10. Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol (2007) 179(1):89–94. doi: 10.4049/jimmunol.179.1.89
11. Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol (2001) 31(10):3121–7. doi: 10.1002/1521-4141(2001010)31:10<3121::AID-IMMU3121>3.0.CO;2-4
12. Capuano C, Pighi C, Battella S, De Federicis D, Galandrini R, Palmieri G. Harnessing CD16-mediated NK cell functions to enhance therapeutic efficacy of tumor-targeting mAbs. Cancers (Basel) (2021) 13(10):2500. doi: 10.3390/cancers13102500
13. De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci USA (2011) 108(2):728–32. doi: 10.1073/pnas.1012356108
14. Mujal AM, Delconte RB, Sun JC. Natural killer cells: from innate to adaptive features. Annu Rev Immunol (2021) 39:417–47. doi: 10.1146/annurev-immunol-101819-074948
15. Franklin M, Connolly E, Hussell T. Recruited and tissue-resident natural killer cells in the lung during infection and cancer. Front Immunol (2022) 13:887503. doi: 10.3389/fimmu.2022.887503
16. Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology (2009) 128(1):7–15. doi: 10.1111/j.1365-2567.2009.03123.x
17. Ferez M, Knudson CJ, Lev A, Wong EB, Alves-Peixoto P, Tang L, et al. Viral infection modulates Qa-1b in infected and bystander cells to properly direct NK cell killing. J Exp Med (2021) 218(5):e20201782. doi: 10.1084/jem.20201782
18. Kaürre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319(6055):675–8. doi: 10.1038/319675a0
19. Chester C, Fritsch K, Kohrt HE. Natural killer cell immunomodulation: targeting activating, inhibitory, and co-stimulatory receptor signaling for cancer immunotherapy. Front Immunol (2015) 6:601. doi: 10.3389/fimmu.2015.00601
20. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol (2009) 9(8):568–80. doi: 10.1038/nri2604
21. Sivori S, Cantoni C, Parolini S, Marcenaro E, Conte R, Moretta L, et al. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur J Immunol (2003) 33(12):3439–47. doi: 10.1002/eji.200324533
22. Cifaldi L, Doria M, Cotugno N, Zicari S, Cancrini C, Palma P, et al. DNAM-1 activating receptor and its ligands: how do viruses affect the NK cell-mediated immune surveillance during the various phases of infection? Int J Mol Sci (2019) 20(15):3715. doi: 10.3390/ijms20153715
23. Middleton D, Curran M, Maxwell L. Natural killer cells and their receptors. Transpl Immunol (2002) 10(2-3):147–64. doi: 10.1016/S0966-3274(02)00062-X
24. Blunt MD, Khakoo SI. Activating killer cell immunoglobulin-like receptors: Detection, function and therapeutic use. Int J Immunogenet (2020) 47(1):1–12. doi: 10.1111/iji.12461
25. Waggoner SN, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front Immunol (2012) 3:377. doi: 10.3389/fimmu.2012.00377
26. Wang X, Xiong H, Ning Z. Implications of NKG2A in immunity and immune-mediated diseases. Front Immunol (2022) 13:960852. doi: 10.3389/fimmu.2022.960852
27. Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol (2020) 11:16. doi: 10.3389/fimmu.2020.00167
28. Béziat V, Descours B, Parizot C, Debré P, Vieillard V. NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PloS One (2010) 5(8):e11966. doi: 10.1371/journal.pone.0011966
29. Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The immune checkpoint PD-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers (Basel) (2020) 12(11):3285. doi: 10.3390/cancers12113285
30. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
31. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-like receptors (KIRs): their role in NK cell modulation and developments leading to their clinical exploitation. Front Immunol (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
32. Dubreuil L, Chevallier P, Retière C, Gagne K. Relevance of polymorphic KIR and HLA class I genes in NK-cell-based immunotherapies for adult leukemic patients. Cancers (Basel) (2021) 13(15):3767. doi: 10.3390/cancers13153767
33. Yawata M, Yawata N, Abi-Rached L, Parham P. Variation within the human killer cell immunoglobulin-like receptor (KIR) gene family. Crit Rev Immunol (2002) 22(5-6):463–82. doi: 10.1615/CritRevImmunol.v22.i5-6.70
34. Norman PJ, Stephens HA, Verity DH, Chandanayingyong D, Vaughan RW. Distribution of natural killer cell immunoglobulin-like receptor sequences in three ethnic groups. Immunogenetics (2001) 52(3-4):195–205. doi: 10.1007/s002510000281
35. Shilling HG, Guethlein LA, Cheng NW, Gardiner CM, Rodriguez R, Tyan D, et al. Allelic polymorphism synergizes with variable gene content to individualize human KIR genotype. J Immunol (2002) 168(5):2307–15. doi: 10.4049/jimmunol.168.5.2307
36. Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, et al. Human diversity in killer cell inhibitory receptor genes. Immunity (1997) 7(6):753–63. doi: 10.1016/S1074-7613(00)80394-5
37. Barker DJ, Maccari G, Georgiou X, Cooper MA, Flicek P, Robinson J, et al. The IPD-IMGT/HLA database. Nucleic Acids Res (2023) 51(D1):D1053–D60. doi: 10.1093/nar/gkac1011
38. Yawata M, Yawata N, Draghi M, Little AM, Partheniou F, Parham P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med (2006) 203(3):633–45. doi: 10.1084/jem.20051884
39. Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J Immunol (1998) 161(2):571–7. doi: 10.4049/jimmunol.161.2.571
40. Dubreuil L, Maniangou B, Chevallier P, Quéméner A, Legrand N, Béné MC, et al. Centromeric KIR AA individuals harbor particular KIR alleles conferring beneficial NK cell features with implications in haplo-identical hematopoietic stem cell transplantation. Cancers (Basel) (2020) 12(12):3595. doi: 10.3390/cancers12123595
41. Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature (2011) 476(7358):96–100. doi: 10.1038/nature10237
42. Hölzemer A, Thobakgale CF, Jimenez Cruz CA, Garcia-Beltran WF, Carlson JM, van Teijlingen NH, et al. Selection of an HLA-C*03:04-restricted HIV-1 p24 Gag sequence variant is associated with viral escape from KIR2DL3+ Natural killer cells: data from an observational cohort in South Africa. PloS Med (2015) 12(11):e1001900. doi: 10.1371/journal.pmed.1001900
43. Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science (2004) 305(5685):872–4. doi: 10.1126/science.1097670
44. Littera R, Chessa L, Deidda S, Angioni G, Campagna M, Lai S, et al. Natural killer-cell immunoglobulin-like receptors trigger differences in immune response to SARS-CoV-2 infection. PloS One (2021) 16(8):e0255608. doi: 10.1371/journal.pone.0255608
45. Wauquier N, Padilla C, Becquart P, Leroy E, Vieillard V. Association of KIR2DS1 and KIR2DS3 with fatal outcome in Ebola virus infection. Immunogenetics (2010) 62(11-12):767–71. doi: 10.1007/s00251-010-0480-x
46. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res (2006) 66(1):563–70. doi: 10.1158/0008-5472.CAN-05-2776
47. Hara R, Onizuka M, Matsusita E, Kikkawa E, Nakamura Y, Matsushita H, et al. NKG2D gene polymorphisms are associated with disease control of chronic myeloid leukemia by dasatinib. Int J Hematol (2017) 106(5):666–74. doi: 10.1007/s12185-017-2294-1
48. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “One for all, all for one”. Front Immunol (2018) 9:476. doi: 10.3389/fimmu.2018.00476
49. Khor CC, Chau TN, Pang J, Davila S, Long HT, Ong RT, et al. Genome-wide association study identifies susceptibility loci for dengue shock syndrome at MICB and PLCE1. Nat Genet (2011) 43(11):1139–41. doi: 10.1038/ng.960
50. Dang TN, Naka I, Sa-Ngasang A, Anantapreecha S, Chanama S, Wichukchinda N, et al. A replication study confirms the association of GWAS-identified SNPs at MICB and PLCE1 in Thai patients with dengue shock syndrome. BMC Med Genet (2014) 15:58. doi: 10.1186/1471-2350-15-58
51. Ding W, Ma Y, Zhu W, Pu W, Zhang J, Qian F, et al. Allele facilitates the metastasis of KRAS-mutant colorectal cancer. Front Genet (2020) 11:511. doi: 10.3389/fgene.2020.00511
52. Tamaki S, Sanefuzi N, Ohgi K, Imai Y, Kawakami M, Yamamoto K, et al. An association between the MICA-A5.1 allele and an increased susceptibility to oral squamous cell carcinoma in Japanese patients. J Oral Pathol Med (2007) 36(6):351–6. doi: 10.1111/j.1600-0714.2007.00539.x
53. Tamaki S, Kawakami M, Yamanaka Y, Shimomura H, Imai Y, Ishida J, et al. Relationship between soluble MICA and the MICA A5.1 homozygous genotype in patients with oral squamous cell carcinoma. Clin Immunol (2009) 130(3):331–7. doi: 10.1016/j.clim.2008.09.004
54. Lavado-Valenzuela R, Benavides M, Carabantes F, Alonso A, Caballero A. MHC class I chain-related gene A transmembrane polymorphism in Spanish women with breast cancer. Tissue Antigens (2009) 74(1):46–9. doi: 10.1111/j.1399-0039.2009.01254.x
55. Jumnainsong A, Romphruk AV, Jearanaikoon P, Klumkrathok K, Romphruk A, Luanrattanakorn S, et al. Association of polymorphic extracellular domains of MICA with cervical cancer in northeastern Thai population. Tissue Antigens (2007) 69(4):326–33. doi: 10.1111/j.1399-0039.2006.00754.x
56. Kared H, Martelli S, Ng TP, Pender SL, Larbi A. CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol Immunother (2016) 65(4):441–52. doi: 10.1007/s00262-016-1803-z
57. Borrego F, Robertson MJ, Ritz J, Peña J, Solana R. CD69 is a stimulatory receptor for natural killer cell and its cytotoxic effect is blocked by CD94 inhibitory receptor. Immunology (1999) 97(1):159–65. doi: 10.1046/j.1365-2567.1999.00738.x
58. Marden CM, North J, Anderson R, Bakhsh IA, Addison E, Pittman H, et al. CD69 is required for activated NK cell-mediated killing of resistant targets. Blood (2005) 106(11):3322–. doi: 10.1182/blood.V106.11.3322.3322
59. Hashemi E, Malarkannan S. Tissue-resident NK cells: development, maturation, and clinical relevance. Cancers (Basel) (2020) 12(6):1553. doi: 10.3390/cancers12061553
60. Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods (2004) 294(1-2):15–22. doi: 10.1016/j.jim.2004.08.008
61. Velotti F, Barchetta I, Cimini FA, Cavallo MG. Granzyme B in inflammatory diseases: apoptosis, inflammation, extracellular matrix remodeling, epithelial-to-mesenchymal transition and fibrosis. Front Immunol (2020) 11:587581. doi: 10.3389/fimmu.2020.587581
62. Osińska I, Popko K, Demkow U. Perforin: an important player in immune response. Cent Eur J Immunol (2014) 39(1):109–15. doi: 10.5114/ceji.2014.42135
63. Yoon SR, Kim TD, Choi I. Understanding of molecular mechanisms in natural killer cell therapy. Exp Mol Med (2015) 47(2):e141. doi: 10.1038/emm.2014.114
64. Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med (2019) 7(5):105. doi: 10.21037/atm.2019.01.42
65. Engelman A, Cherepanov P. The structural biology of HIV-1: mechanistic and therapeutic insights. Nat Rev Microbiol (2012) 10(4):279–90. doi: 10.1038/nrmicro2747
66. Deeks SG, Overbaugh J, Phillips A, Buchbinder S. HIV infection. Nat Rev Dis Primers (2015) 1:15035. doi: 10.1038/nrdp.2015.35
67. Cohn LB, Chomont N, Deeks SG. The biology of the HIV-1 latent reservoir and implications for cure strategies. Cell Host Microbe (2020) 27(4):519–30. doi: 10.1016/j.chom.2020.03.014
68. Douglas SD, Durako SJ, Tustin NB, Houser J, Muenz L, Starr SE, et al. Natural killer cell enumeration and function in HIV-infected and high-risk uninfected adolescents. AIDS Res Hum Retroviruses (2001) 17(6):543–52. doi: 10.1089/08892220151126643
69. Tarazona R, Casado JG, Delarosa O, Torre-Cisneros J, Villanueva JL, Sanchez B, et al. Selective depletion of CD56(dim) NK cell subsets and maintenance of CD56(bright) NK cells in treatment-naive HIV-1-seropositive individuals. J Clin Immunol (2002) 22(3):176–83. doi: 10.1023/A:1015476114409
70. Hong HS, Eberhard JM, Keudel P, Bollmann BA, Ballmaier M, Bhatnagar N, et al. HIV infection is associated with a preferential decline in less-differentiated CD56dim CD16+ NK cells. J Virol (2010) 84(2):1183–8. doi: 10.1128/JVI.01675-09
71. Slyker JA, Lohman-Payne B, John-Stewart GC, Dong T, Mbori-Ngacha D, Tapia K, et al. The impact of HIV-1 infection and exposure on natural killer (NK) cell phenotype in Kenyan infants during the first year of life. Front Immunol (2012) 3:399. doi: 10.3389/fimmu.2012.00399
72. Hong HS, Ahmad F, Eberhard JM, Bhatnagar N, Bollmann BA, Keudel P, et al. Loss of CCR7 expression on CD56(bright) NK cells is associated with a CD56(dim)CD16(+) NK cell-like phenotype and correlates with HIV viral load. PloS One (2012) 7(9):e44820. doi: 10.1371/journal.pone.0044820
73. Dierks P, Wroblewski R, Eberhard JM, Martrus G, Degen O, Hertling S, et al. Brief report: increased frequency of CD39+ CD56bright natural killer cells in HIV-1 infection correlates with immune activation and disease progression. J Acquir Immune Defic Syndr (2017) 74(4):467–72. doi: 10.1097/QAI.0000000000001266
74. Jiang Y, Yang M, Sun X, Chen X, Ma M, Yin X, et al. IL-10(+) NK and TGF-beta(+) NK cells play negative regulatory roles in HIV infection. BMC Infect Dis (2018) 18(1):80. doi: 10.1186/s12879-018-2991-2
75. Cao WJ, Zhang XC, Wan LY, Li QY, Mu XY, Guo AL, et al. Immune dysfunctions of CD56(neg) NK cells are associated with HIV-1 disease progression. Front Immunol (2021) 12:811091. doi: 10.3389/fimmu.2021.811091
76. Milush JM, López-Vergès S, York VA, Deeks SG, Martin JN, Hecht FM, et al. CD56negCD16+ NK cells are activated mature NK cells with impaired effector function during HIV-1 infection. Retrovirology (2013) 10:158. doi: 10.1186/1742-4690-10-158
77. Cocker ATH, Liu F, Djaoud Z, Guethlein LA, Parham P. CD56-negative NK cells: Frequency in peripheral blood, expansion during HIV-1 infection, functional capacity, and KIR expression. Front Immunol (2022) 13:992723. doi: 10.3389/fimmu.2022.992723
78. Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, et al. Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci USA (2005) 102(8):2886–91. doi: 10.1073/pnas.0409872102
79. Ma M, Yin X, Zhao X, Guo C, Zhu X, Liu T, et al. CD56(-) CD16(+) NK cells from HIV-infected individuals negatively regulate IFN-gamma production by autologous CD8(+) T cells. J Leukoc Biol (2019) 106(6):1313–23. doi: 10.1002/JLB.3A0819-171RR
80. Cocker ATH, Guethlein LA, Parham P. The CD56-CD16+ NK cell subset in chronic infections. Biochem Soc Trans (2023) 51(3):1201–12. doi: 10.1042/BST20221374
81. Zhang Z, Zhou Y, Lu J, Chen YF, Hu HY, Xu XQ, et al. Changes in NK cell subsets and receptor expressions in HIV-1 infected chronic patients and HIV controllers. Front Immunol (2021) 12:792775. doi: 10.3389/fimmu.2021.792775
82. Mavilio D, Benjamin J, Daucher M, Lombardo G, Kottilil S, Planta MA, et al. Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc Natl Acad Sci USA (2003) 100(25):15011–6. doi: 10.1073/pnas.2336091100
83. Mahapatra S, Shearer WT, Minard CG, Mace E, Paul M, Orange JS. NK cells in treated HIV-infected children display altered phenotype and function. J Allergy Clin Immunol (2019) 144(1):294–303 e13. doi: 10.1016/j.jaci.2018.11.052
84. Dillon SM, Lee EJ, Bramante JM, Barker E, Wilson CC. The natural killer cell interferon-gamma response to bacteria is diminished in untreated HIV-1 infection and defects persist despite viral suppression. J Acquir Immune Defic Syndr (2014) 65(3):259–67. doi: 10.1097/01.qai.0000435603.50598.2b
85. Kaczmarek DJ, Kokordelis P, Kramer B, Glassner A, Wolter F, Goeser F, et al. Alterations of the NK cell pool in HIV/HCV co-infection. PloS One (2017) 12(4):e0174465. doi: 10.1371/journal.pone.0174465
86. Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity (1999) 10(6):661–71. doi: 10.1016/S1074-7613(00)80065-5
87. Schwartz O, Maréchal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med (1996) 2(3):338–42. doi: 10.1038/nm0396-338
88. Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, et al. HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe (2016) 19(5):686–95. doi: 10.1016/j.chom.2016.04.005
89. Bachtel ND, Umviligihozo G, Pickering S, Mota TM, Liang H, Del Prete GQ, et al. HLA-C downregulation by HIV-1 adapts to host HLA genotype. PloS Pathog (2018) 14(9):e1007257. doi: 10.1371/journal.ppat.1007257
90. Ward J, Davis Z, DeHart J, Zimmerman E, Bosque A, Brunetta E, et al. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PloS Pathog (2009) 5(10):e1000613. doi: 10.1371/journal.ppat.1000613
91. Richard J, Sindhu S, Pham TN, Belzile JP, Cohen EA. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood (2010) 115(7):1354–63. doi: 10.1182/blood-2009-08-237370
92. Sakai K, Dimas J, Lenardo MJ. The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc Natl Acad Sci USA (2006) 103(9):3369–74. doi: 10.1073/pnas.0509417103
93. Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell EM, Planelles V, et al. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe (2010) 8(5):397–409. doi: 10.1016/j.chom.2010.10.008
94. Davis ZB, Cogswell A, Scott H, Mertsching A, Boucau J, Wambua D, et al. A conserved HIV-1-derived peptide presented by HLA-E renders infected T-cells highly susceptible to attack by NKG2A/CD94-bearing natural killer cells. PloS Pathog (2016) 12(2):e1005421. doi: 10.1371/journal.ppat.1005421
95. Bonaparte MI, Barker E. Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood (2004) 104(7):2087–94. doi: 10.1182/blood-2004-02-0696
96. Luteijn R, Sciaranghella G, van Lunzen J, Nolting A, Dugast AS, Ghebremichael MS, et al. Early viral replication in lymph nodes provides HIV with a means by which to escape NK-cell-mediated control. Eur J Immunol (2011) 41(9):2729–40. doi: 10.1002/eji.201040886
97. Guo AL, Jiao YM, Zhao QW, Huang HH, Deng JN, Zhang C, et al. Implications of the accumulation of CXCR5(+) NK cells in lymph nodes of HIV-1 infected patients. EBioMedicine (2022) 75:103794. doi: 10.1016/j.ebiom.2021.103794
98. George AF, Luo X, Neidleman J, Hoh R, Vohra P, Thomas R, et al. Deep phenotypic analysis of blood and lymphoid T and NK cells from HIV+ Controllers and ART-suppressed individuals. Front Immunol (2022) 13:803417. doi: 10.3389/fimmu.2022.803417
99. Sungur CM, Wang Q, Ozanturk AN, Gao H, Schmitz AJ, Cella M, et al. Human NK cells confer protection against HIV-1 infection in humanized mice. J Clin Invest (2022) 132(24):e162694. doi: 10.1172/JCI162694
100. Sips M, Sciaranghella G, Diefenbach T, Dugast AS, Berger CT, Liu Q, et al. Altered distribution of mucosal NK cells during HIV infection. Mucosal Immunol (2012) 5(1):30–40. doi: 10.1038/mi.2011.40
101. Vollmers S, Lobermeyer A, Niehrs A, Fittje P, Indenbirken D, Nakel J, et al. Host KIR/HLA-C genotypes determine HIV-mediated changes of the NK cell repertoire and are associated with vpu sequence variations impacting downmodulation of HLA-C. Front Immunol (2022) 13:922252. doi: 10.3389/fimmu.2022.922252
102. Olvera A, Pérez-Álvarez S, Ibarrondo J, Ganoza C, Lama JR, Lucchetti A, et al. The HLA-C*04: 01/KIR2DS4 gene combination and human leukocyte antigen alleles with high population frequency drive rate of HIV disease progression. AIDS (2015) 29(5):507–17. doi: 10.1097/QAD.0000000000000574
103. Merino AM, Dugast AS, Wilson CM, Goepfert PA, Alter G, Kaslow RA, et al. KIR2DS4 promotes HIV-1 pathogenesis: new evidence from analyses of immunogenetic data and natural killer cell function. PloS One (2014) 9(6):e99353. doi: 10.1371/journal.pone.0099353
104. Mhandire K, Zijenah LS, Tshabalala M, Yindom LM, Mlambo T, Mhandire DZ, et al. KIR and HLA-C genetic polymorphisms influence plasma IP-10 concentration in antiretroviral therapy-naive HIV-infected adult Zimbabweans. OMICS (2019) 23(2):111–8. doi: 10.1089/omi.2018.0147
105. Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med (2007) 204(12):3027–36. doi: 10.1084/jem.20070695
106. Pelak K, Need AC, Fellay J, Shianna KV, Feng S, Urban TJ, et al. Copy number variation of KIR genes influences HIV-1 control. PloS Biol (2011) 9(11):e1001208. doi: 10.1371/annotation/7e17b146-a69c-4e83-9230-7340486d9dc8
107. Florez-Alvarez L, Hernandez JC, Zapata W. NK Cells in HIV-1 Infection: From Basic Science to Vaccine Strategies. Front Immunol (2018) 9:2290.
108. Reeves RK, Li H, Jost S, Blass E, Li H, Schafer JL, et al. Antigen-specific NK cell memory in rhesus macaques. Nat Immunol (2015) 16(9):927–32. doi: 10.1038/ni.3227
109. Tiemessen CT, Shalekoff S, Meddows-Taylor S, Schramm DB, Papathanasopoulos MA, Gray GE, et al. Cutting Edge: Unusual NK cell responses to HIV-1 peptides are associated with protection against maternal-infant transmission of HIV-1. J Immunol (2009) 182(10):5914–8. doi: 10.4049/jimmunol.0900419
110. Tiemessen CT, Shalekoff S, Meddows-Taylor S, Schramm DB, Papathanasopoulos M, Gray G, et al. Natural killer cells that respond to human immunodeficiency virus type 1 (HIV-1) peptides are associated with control of HIV-1 infection. J Infect Dis (2010) 202(9):1444–53. doi: 10.1086/656535
111. Gondois-Rey F, Cheret A, Granjeaud S, Mallet F, Bidaut G, Lecuroux C, et al. NKG2C(+) memory-like NK cells contribute to the control of HIV viremia during primary infection: Optiprim-ANRS 147. Clin Transl Immunol (2017) 6(7):e150. doi: 10.1038/cti.2017.22
112. Jensen SS, Hartling HJ, Tingstedt JL, Larsen TK, Nielsen SD, Pedersen C, et al. HIV-specific ADCC improves after antiretroviral therapy and correlates with normalization of the NK cell phenotype. J Acquir Immune Defic Syndr (2015) 68(2):103–11. doi: 10.1097/QAI.0000000000000429
113. Nabatanzi R, Bayigga L, Cose S, Rowland-Jones S, Canderan G, Joloba M, et al. Aberrant natural killer (NK) cell activation and dysfunction among ART-treated HIV-infected adults in an African cohort. Clin Immunol (2019) 201:55–60. doi: 10.1016/j.clim.2019.02.010
114. Lichtfuss GF, Cheng WJ, Farsakoglu Y, Paukovics G, Rajasuriar R, Velayudham P, et al. Virologically suppressed HIV patients show activation of NK cells and persistent innate immune activation. J Immunol (2012) 189(3):1491–9. doi: 10.4049/jimmunol.1200458
115. Giuliani E, Vassena L, Di Cesare S, Malagnino V, Desimio MG, Andreoni M, et al. NK cells of HIV-1-infected patients with poor CD4(+) T-cell reconstitution despite suppressive HAART show reduced IFN-gamma production and high frequency of autoreactive CD56(bright) cells. Immunol Lett (2017) 190:185–93. doi: 10.1016/j.imlet.2017.08.014
116. Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S. An overview of HCV molecular biology, replication and immune responses. Virol J (2011) 8:161. doi: 10.1186/1743-422X-8-161
117. Revie D, Salahuddin SZ. Human cell types important for hepatitis C virus replication in vivo and in vitro: old assertions and current evidence. Virol J (2011) 8:346. doi: 10.1186/1743-422X-8-346
118. Millman AJ, Nelson NP, Vellozzi C. Hepatitis C: review of the epidemiology, clinical care, and continued challenges in the direct acting antiviral era. Curr Epidemiol Rep (2017) 4(2):174–85. doi: 10.1007/s40471-017-0108-x
119. Amadei B, Urbani S, Cazaly A, Fisicaro P, Zerbini A, Ahmed P, et al. Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology (2010) 138(4):1536–45. doi: 10.1053/j.gastro.2010.01.006
120. Grebely J, Prins M, Hellard M, Cox AL, Osburn WO, Lauer G, et al. Hepatitis C virus clearance, reinfection, and persistence, with insights from studies of injecting drug users: towards a vaccine. Lancet Infect Dis (2012) 12(5):408–14. doi: 10.1016/S1473-3099(12)70010-5
121. de Groen RA, Groothuismink ZMA, van Oord G, Kootstra NA, Janssen HLA, Prins M, et al. NK cells in self-limited HCV infection exhibit a more extensively differentiated, but not memory-like, repertoire. J Viral Hepat (2017) 24(11):917–26. doi: 10.1111/jvh.12716
122. Golden-Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology (2010) 52(5):1581–9. doi: 10.1002/hep.23896
123. Harrison RJ, Ettorre A, Little AM, Khakoo SI. Association of NKG2A with treatment for chronic hepatitis C virus infection. Clin Exp Immunol (2010) 161(2):306–14. doi: 10.1111/j.1365-2249.2010.04169.x
124. Ji HF, Wang J, Yu L, Niu JQ, Ayana DA, Jiang YF. High frequencies of CD158b+ NK cells are associated with persistent hepatitis C virus infections. Ann Hepatol (2013) 12(4):539–47. doi: 10.1016/S1665-2681(19)31337-7
125. Frias M, Rivero-Juárez A, Rodriguez-Cano D, Camacho Á, López-López P, Risalde M, et al. HLA-B, HLA-C and KIR improve the predictive value of IFNL3 for Hepatitis C spontaneous clearance. Sci Rep (2018) 8(1):659. doi: 10.1038/s41598-017-17531-7
126. Morishima C, Paschal DM, Wang CC, Yoshihara CS, Wood BL, Yeo AE, et al. Decreased NK cell frequency in chronic hepatitis C does not affect ex vivo cytolytic killing. Hepatology (2006) 43(3):573–80. doi: 10.1002/hep.21073
127. Dessouki O, Kamiya Y, Nagahama H, Tanaka M, Suzu S, Sasaki Y, et al. Chronic hepatitis C viral infection reduces NK cell frequency and suppresses cytokine secretion: Reversion by anti-viral treatment. Biochem Biophys Res Commun (2010) 393(2):331–7. doi: 10.1016/j.bbrc.2010.02.008
128. Golden-Mason L, Madrigal-Estebas L, McGrath E, Conroy MJ, Ryan EJ, Hegarty JE, et al. Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut (2008) 57(8):1121–8. doi: 10.1136/gut.2007.130963
129. Zarife MA, Reis EA, Carmo TM, Lopes GB, Brandao EC, Silva HR, et al. Increased frequency of CD56Bright NK-cells, CD3-CD16+CD56- NK-cells and activated CD4+T-cells or B-cells in parallel with CD4+CDC25High T-cells control potentially viremia in blood donors with HCV. J Med Virol (2009) 81(1):49–59. doi: 10.1002/jmv.21340
130. Golden-Mason L, Hahn YS, Strong M, Cheng L, Rosen HR. Extracellular HCV-core protein induces an immature regulatory phenotype in NK cells: implications for outcome of acute infection. PloS One (2014) 9(7):e103219. doi: 10.1371/journal.pone.0103219
131. Cosgrove C, Berger CT, Kroy DC, Cheney PC, Ghebremichael M, Aneja J, et al. Chronic HCV infection affects the NK cell phenotype in the blood more than in the liver. PloS One (2014) 9(8):e105950. doi: 10.1371/journal.pone.0105950
132. Miot C, Beaumont E, Duluc D, Le Guillou-Guillemette H, Preisser L, Garo E, et al. IL-26 is overexpressed in chronically HCV-infected patients and enhances TRAIL-mediated cytotoxicity and interferon production by human NK cells. Gut (2015) 64(9):1466–75. doi: 10.1136/gutjnl-2013-306604
133. Larochette V, Miot C, Poli C, Beaumont E, Roingeard P, Fickenscher H, et al. IL-26, a cytokine with roles in extracellular DNA-induced inflammation and microbial defense. Front Immunol (2019) 10:204. doi: 10.3389/fimmu.2019.00204
134. Nel I, Lucar O, Petitdemange C, Beziat V, Lapalus M, Bedossa P, et al. Accumulation of intrahepatic TNF-alpha-producing NKp44+ NK cells correlates with liver fibrosis and viral load in chronic HCV infection. Med (Baltimore) (2016) 95(19):e3678. doi: 10.1097/MD.0000000000003678
135. Maretti-Mira AC, Salomon MP, Hsu AM, Dara L, Golden-Mason L. Etiology of end-stage liver cirrhosis impacts hepatic natural killer cell heterogenicity. Front Immunol (2023) 14:1137034. doi: 10.3389/fimmu.2023.1137034
136. Lunemann S, Malone DF, Hengst J, Port K, Grabowski J, Deterding K, et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J Infect Dis (2014) 209(9):1362–73. doi: 10.1093/infdis/jit561
137. Alter G, Jost S, Rihn S, Reyor LL, Nolan BE, Ghebremichael M, et al. Reduced frequencies of NKp30+NKp46+, CD161+, and NKG2D+ NK cells in acute HCV infection may predict viral clearance. J Hepatol (2011) 55(2):278–88. doi: 10.1016/j.jhep.2010.11.030
138. Zhang C, Wang XM, Li SR, Twelkmeyer T, Wang WH, Zhang SY, et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat Commun (2019) 10(1):1507. doi: 10.1038/s41467-019-09212-y
139. Spaan M, van Oord G, Kreefft K, Hou J, Hansen BE, Janssen HL, et al. Immunological analysis during interferon-free therapy for chronic hepatitis C virus infection reveals modulation of the natural killer cell compartment. J Infect Dis (2016) 213(2):216–23. doi: 10.1093/infdis/jiv391
140. Zhang X, Jiang Y, Li S, Bian D, Liu M, Kong M, et al. Direct-acting antiviral-induced transient recovery of NK cells in early-stage treatment of chronic hepatitis C patients. J Clin Transl Hepatol (2022) 10(6):1117–24. doi: 10.14218/JCTH.2021.00427
141. Mele D, Oliviero B, Mantovani S, Ludovisi S, Lombardi A, Genco F, et al. Adaptive natural killer cell functional recovery in hepatitis C virus cured patients. Hepatology (2021) 73(1):79–90. doi: 10.1002/hep.31273
142. Cunningham C, Gatherer D, Hilfrich B, Baluchova K, Dargan DJ, Thomson M, et al. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J Gen Virol (2010) 91(Pt 3):605–15. doi: 10.1099/vir.0.015891-0
143. Wang YQ, Zhao XY. Human cytomegalovirus primary infection and reactivation: insights from virion-carried molecules. Front Microbiol (2020) 11:1511. doi: 10.3389/fmicb.2020.01511
144. Lopez-Vergès S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci USA (2011) 108(36):14725–32. doi: 10.1073/pnas.1110900108
145. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood (2012) 119(11):2665–74. doi: 10.1182/blood-2011-10-386995
146. Rolle A, Brodin P. Immune adaptation to environmental influence: the case of NK cells and HCMV. Trends Immunol (2016) 37(3):233–43. doi: 10.1016/j.it.2016.01.005
147. Bayard C, Lepetitcorps H, Roux A, Larsen M, Fastenackels S, Salle V, et al. Coordinated expansion of both memory T cells and NK cells in response to CMV infection in humans. Eur J Immunol (2016) 46(5):1168–79. doi: 10.1002/eji.201546179
148. Hendricks DW, Balfour HH Jr., Dunmire SK, Schmeling DO, Hogquist KA, Lanier LL. Cutting edge: NKG2C(hi)CD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein-Barr virus. J Immunol (2014) 192(10):4492–6. doi: 10.4049/jimmunol.1303211
149. Djaoud Z, David G, Bressollette C, Willem C, Rettman P, Gagne K, et al. Amplified NKG2C+ NK cells in cytomegalovirus (CMV) infection preferentially express killer cell Ig-like receptor 2DL: functional impact in controlling CMV-infected dendritic cells. J Immunol (2013) 191(5):2708–16. doi: 10.4049/jimmunol.1301138
150. Campos C, Lopez N, Pera A, Gordillo JJ, Hassouneh F, Tarazona R, et al. Expression of NKp30, NKp46 and DNAM-1 activating receptors on resting and IL-2 activated NK cells from healthy donors according to CMV-serostatus and age. Biogerontology (2015) 16(5):671–83. doi: 10.1007/s10522-015-9581-0
151. Beziat V, Dalgard O, Asselah T, Halfon P, Bedossa P, Boudifa A, et al. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur J Immunol (2012) 42(2):447–57. doi: 10.1002/eji.201141826
152. Costa-Garcia M, Vera A, Moraru M, Vilches C, Lopez-Botet M, Muntasell A. Antibody-mediated response of NKG2Cbright NK cells against human cytomegalovirus. J Immunol (2015) 194(6):2715–24. doi: 10.4049/jimmunol.1402281
153. Barnes S, Schilizzi O, Audsley KM, Newnes HV, Foley B. Deciphering the immunological phenomenon of adaptive natural killer (NK) cells and cytomegalovirus (CMV). Int J Mol Sci (2020) 21(22):8864. doi: 10.3390/ijms21228864
154. Cichocki F, Miller JS, Anderson SK, Bryceson YT. Epigenetic regulation of NK cell differentiation and effector functions. Front Immunol (2013) 4:55. doi: 10.3389/fimmu.2013.00055
155. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity (2015) 42(3):443–56. doi: 10.1016/j.immuni.2015.02.008
156. Della Chiesa M, Muccio L, Moretta A. CMV induces rapid NK cell maturation in HSCT recipients. Immunol Lett (2013) 155(1-2):11–3. doi: 10.1016/j.imlet.2013.09.020
157. Hassan N, Eldershaw S, Stephens C, Kinsella F, Craddock C, Malladi R, et al. CMV reactivation initiates long-term expansion and differentiation of the NK cell repertoire. Front Immunol (2022) 13:935949. doi: 10.3389/fimmu.2022.935949
158. Storek J, Dawson MA, Storer B, Stevens-Ayers T, Maloney DG, Marr KA, et al. Immune reconstitution after allogeneic marrow transplantation compared with blood stem cell transplantation. Blood (2001) 97(11):3380–9. doi: 10.1182/blood.V97.11.3380
159. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol (2012) 189(10):5082–8. doi: 10.4049/jimmunol.1201964
160. Steinhauer DA, Skehel JJ. Genetics of influenza viruses. Annu Rev Genet (2002) 36:305–32. doi: 10.1146/annurev.genet.36.052402.152757
161. Bouvier NM, Palese P. The biology of influenza viruses. Vaccine (2008) 26 Suppl 4(Suppl 4):D49–53. doi: 10.1016/j.vaccine.2008.07.039
162. Kalil AC, Thomas PG. Influenza virus-related critical illness: pathophysiology and epidemiology. Crit Care (2019) 23(1):258. doi: 10.1186/s13054-019-2539-x
163. Brownlie D, Rodahl I, Varnaite R, Asgeirsson H, Glans H, Falck-Jones S, et al. Comparison of lung-homing receptor expression and activation profiles on NK cell and T cell subsets in COVID-19 and influenza. Front Immunol (2022) 13:834862. doi: 10.3389/fimmu.2022.834862
164. Carlin LE, Hemann EA, Zacharias ZR, Heusel JW, Legge KL. Natural killer cell recruitment to the lung during influenza A virus infection is dependent on CXCR3, CCR5, and virus exposure dose. Front Immunol (2018) 9:781. doi: 10.3389/fimmu.2018.00781
165. Verbist KC, Rose DL, Cole CJ, Field MB, Klonowski KD. IL-15 participates in the respiratory innate immune response to influenza virus infection. PloS One (2012) 7(5):e37539. doi: 10.1371/journal.pone.0037539
166. Fox A, Le NM, Horby P, van Doorn HR, Nguyen VT, Nguyen HH, et al. Severe pandemic H1N1 2009 infection is associated with transient NK and T deficiency and aberrant CD8 responses. PloS One (2012) 7(2):e31535. doi: 10.1371/journal.pone.0031535
167. Jost S, Quillay H, Reardon J, Peterson E, Simmons RP, Parry BA, et al. Changes in cytokine levels and NK cell activation associated with influenza. PloS One (2011) 6(9):e25060. doi: 10.1371/journal.pone.0025060
168. Jost S, Reardon J, Peterson E, Poole D, Bosch R, Alter G, et al. Expansion of 2B4+ natural killer (NK) cells and decrease in NKp46+ NK cells in response to influenza. Immunology (2011) 132(4):516–26. doi: 10.1111/j.1365-2567.2010.03394.x
169. Juarez-Reyes A, Noyola DE, Monsivais-Urenda A, Alvarez-Quiroga C, Gonzalez-Amaro R. Influenza virus infection but not H1N1 influenza virus immunization is associated with changes in peripheral blood NK cell subset levels. Clin Vaccine Immunol (2013) 20(8):1291–7. doi: 10.1128/CVI.00194-13
170. Zheng J, Wen L, Yen HL, Liu M, Liu Y, Teng O, et al. Phenotypic and functional characteristics of a novel influenza virus hemagglutinin-specific memory NK cell. J Virol (2021) 95(12):e00165–21. doi: 10.1128/JVI.00165-21
171. Mao H, Tu W, Qin G, Law HK, Sia SF, Chan PL, et al. Influenza virus directly infects human natural killer cells and induces cell apoptosis. J Virol (2009) 83(18):9215–22. doi: 10.1128/JVI.00805-09
172. Mao H, Liu Y, Sia SF, Peiris JSM, Lau YL, Tu W. Avian influenza virus directly infects human natural killer cells and inhibits cell activity. Virol Sin (2017) 32(2):122–9. doi: 10.1007/s12250-016-3918-y
173. Guo H, Kumar P, Moran TM, Garcia-Sastre A, Zhou Y, Malarkannan S. The functional impairment of natural killer cells during influenza virus infection. Immunol Cell Biol (2009) 87(8):579–89. doi: 10.1038/icb.2009.60
174. Cooper GE, Ostridge K, Khakoo SI, Wilkinson TMA, Staples KJ. Human CD49a(+) lung natural killer cell cytotoxicity in response to influenza A virus. Front Immunol (2018) 9:1671. doi: 10.3389/fimmu.2018.01671
175. Du N, Zhou J, Lin X, Zhang Y, Yang X, Wang Y, et al. Differential activation of NK cells by influenza A pseudotype H5N1 and 1918 and 2009 pandemic H1N1 viruses. J Virol (2010) 84(15):7822–31. doi: 10.1128/JVI.00069-10
176. Scharenberg M, Vangeti S, Kekalainen E, Bergman P, Al-Ameri M, Johansson N, et al. Influenza A virus infection induces hyperresponsiveness in human lung tissue-resident and peripheral blood NK cells. Front Immunol (2019) 10:1116. doi: 10.3389/fimmu.2019.01116
177. Zhou G, Juang SW, Kane KP. NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol (2013) 43(4):929–38. doi: 10.1002/eji.201242620
178. Jansen CA, de Geus ED, van Haarlem DA, van de Haar PM, Löndt BZ, Graham SP, et al. Differential lung NK cell responses in avian influenza virus infected chickens correlate with pathogenicity. Sci Rep (2013) 3:2478. doi: 10.1038/srep02478
179. Bongen E, Vallania F, Utz PJ, Khatri P. KLRD1-expressing natural killer cells predict influenza susceptibility. Genome Med (2018) 10(1):45. doi: 10.1186/s13073-018-0554-1
180. Liu Y, Zheng J, Liu Y, Wen L, Huang L, Xiang Z, et al. Uncompromised NK cell activation is essential for virus-specific CTL activity during acute influenza virus infection. Cell Mol Immunol (2018) 15(9):827–37. doi: 10.1038/cmi.2017.10
181. Ishikawa H, Tanaka K, Kutsukake E, Fukui T, Sasaki H, Hata A, et al. IFN-gamma production downstream of NKT cell activation in mice infected with influenza virus enhances the cytolytic activities of both NK cells and viral antigen-specific CD8+ T cells. Virology (2010) 407(2):325–32. doi: 10.1016/j.virol.2010.08.030
182. Duan X, Lu J, Zhou K, Wang J, Wu J, Fu Gao G, et al. NK-cells are involved in thymic atrophy induced by influenza A virus infection. J Gen Virol (2015) 96(11):3223–35. doi: 10.1099/jgv.0.000276
183. Wagstaffe HR, Pickering H, Houghton J, Mooney JP, Wolf AS, Prevatt N, et al. Influenza vaccination primes human myeloid cell cytokine secretion and NK cell function. J Immunol (2019) 203(6):1609–18. doi: 10.4049/jimmunol.1801648
184. Long BR, Michaelsson J, Loo CP, Ballan WM, Vu BA, Hecht FM, et al. Elevated frequency of gamma interferon-producing NK cells in healthy adults vaccinated against influenza virus. Clin Vaccine Immunol (2008) 15(1):120–30. doi: 10.1128/CVI.00357-07
185. Dou Y, Fu B, Sun R, Li W, Hu W, Tian Z, et al. Influenza vaccine induces intracellular immune memory of human NK cells. PloS One (2015) 10(3):e0121258. doi: 10.1371/journal.pone.0121258
186. Park J, Fong Legaspi SL, Schwartzman LM, Gygli SM, Sheng ZM, Freeman AD, et al. An inactivated multivalent influenza A virus vaccine is broadly protective in mice and ferrets. Sci Transl Med (2022) 14(653):eabo2167. doi: 10.1126/scitranslmed.abo2167
187. Naqvi AAT, Fatima K, Mohammad T, Fatima U, Singh IK, Singh A, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys Acta Mol Basis Dis (2020) 1866(10):165878. doi: 10.1016/j.bbadis.2020.165878
188. Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol (2022) 20(5):270–84. doi: 10.1038/s41579-022-00713-0
189. Jiang Y, Wei X, Guan J, Qin S, Wang Z, Lu H, et al. COVID-19 pneumonia: CD8(+) T and NK cells are decreased in number but compensatory increased in cytotoxic potential. Clin Immunol (2020) 218:108516. doi: 10.1016/j.clim.2020.108516
190. Li M, Guo W, Dong Y, Wang X, Dai D, Liu X, et al. Elevated exhaustion levels of NK and CD8(+) T cells as indicators for progression and prognosis of COVID-19 disease. Front Immunol (2020) 11:580237. doi: 10.3389/fimmu.2020.580237
191. Xu G, Qi F, Li H, Yang Q, Wang H, Wang X, et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov (2020) 6:73. doi: 10.1038/s41421-020-00225-2
192. Maucourant C, Filipovic I, Ponzetta A, Aleman S, Cornillet M, Hertwig L, et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci Immunol (2020) 5(50):eabd6832. doi: 10.1126/sciimmunol.abd6832
193. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med (2020) 26(7):1070–6. doi: 10.1038/s41591-020-0944-y
194. Witkowski M, Tizian C, Ferreira-Gomes M, Niemeyer D, Jones TC, Heinrich F, et al. Untimely TGFbeta responses in COVID-19 limit antiviral functions of NK cells. Nature (2021) 600(7888):295–301. doi: 10.1038/s41586-021-04142-6
195. Galán M, Vigón L, Fuertes D, Murciano-Antón MA, Casado-Fernández G, Domínguez-Mateos S, et al. Persistent overactive cytotoxic immune response in a spanish cohort of individuals with long-COVID: identification of diagnostic biomarkers. Front Immunol (2022) 13:848886. doi: 10.3389/fimmu.2022.848886
196. Beckmann ND, Comella PH, Cheng E, Lepow L, Beckmann AG, Tyler SR, et al. Downregulation of exhausted cytotoxic T cells in gene expression networks of multisystem inflammatory syndrome in children. Nat Commun (2021) 12(1):4854. doi: 10.1038/s41467-021-24981-1
197. Huang L, Shi Y, Gong B, Jiang L, Zhang Z, Liu X, et al. Dynamic blood single-cell immune responses in patients with COVID-19. Signal Transduct Target Ther (2021) 6(1):110. doi: 10.1038/s41392-021-00526-2
198. Herrera L, Martin-Inaraja M, Santos S, Ingles-Ferrandiz M, Azkarate A, Perez-Vaquero MA, et al. Identifying SARS-CoV-2 ‘memory’ NK cells from COVID-19 convalescent donors for adoptive cell therapy. Immunology (2022) 165(2):234–49. doi: 10.1111/imm.13432
199. Malengier-Devlies B, Filtjens J, Ahmadzadeh K, Boeckx B, Vandenhaute J, De Visscher A, et al. Severe COVID-19 patients display hyper-activated NK cells and NK cell-platelet aggregates. Front Immunol (2022) 13:861251. doi: 10.3389/fimmu.2022.861251
200. Hoste L, Roels L, Naesens L, Bosteels V, Vanhee S, Dupont S, et al. TIM3+ TRBV11-2 T cells and IFNγ signature in patrolling monocytes and CD16+ NK cells delineate MIS-C. J Exp Med (2022) 219(2):e20211381. doi: 10.1084/jem.20211381
201. Bozzano F, Dentone C, Perrone C, Di Biagio A, Fenoglio D, Parodi A, et al. Extensive activation, tissue trafficking, turnover and functional impairment of NK cells in COVID-19 patients at disease onset associates with subsequent disease severity. PloS Pathog (2021) 17(4):e1009448. doi: 10.1371/journal.ppat.1009448
202. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest (2020) 130(9):4694–703. doi: 10.1172/JCI138554
203. Varchetta S, Mele D, Oliviero B, Mantovani S, Ludovisi S, Cerino A, et al. Unique immunological profile in patients with COVID-19. Cell Mol Immunol (2021) 18(3):604–12. doi: 10.1038/s41423-020-00557-9
204. Hutanu A, Manu D, Gabor MR, Vasiesiu AM, Andrejkovits AV, Dobreanu M. Dynamic evaluation of natural killer cells subpopulations in COVID-19 patients. Int J Mol Sci (2022) 23(19):11875. doi: 10.3390/ijms231911875
205. Yaqinuddin A, Kashir J. Innate immunity in COVID-19 patients mediated by NKG2A receptors, and potential treatment using Monalizumab, Cholroquine, and antiviral agents. Med Hypotheses (2020) 140:109777. doi: 10.1016/j.mehy.2020.109777
206. Casado JL, Moraga E, Vizcarra P, Velasco H, Martin-Hondarza A, Haemmerle J, et al. Expansion of CD56(dim)CD16(neg) NK cell subset and increased inhibitory KIRs in hospitalized COVID-19 patients. Viruses (2021) 14(1):46. doi: 10.3390/v14010046
207. Hsieh WC, Lai EY, Liu YT, Wang YF, Tzeng YS, Cui L, et al. NK cell receptor and ligand composition influences the clearance of SARS-CoV-2. J Clin Invest (2021) 131(21):e146408. doi: 10.1172/JCI146408
208. Leem G, Cheon S, Lee H, Choi SJ, Jeong S, Kim ES, et al. Abnormality in the NK-cell population is prolonged in severe COVID-19 patients. J Allergy Clin Immunol (2021) 148(4):996–1006 e18. doi: 10.1016/j.jaci.2021.07.022
209. Wilk AJ, Lee MJ, Wei B, Parks B, Pi R, Martinez-Colon GJ, et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J Exp Med (2021) 218(8):e20210582. doi: 10.1101/2020.12.18.423363
210. Vigón L, García-Pérez J, Rodríguez-Mora S, Torres M, Mateos E, Castillo de la Osa M, et al. Impaired antibody-dependent cellular cytotoxicity in a spanish cohort of patients with COVID-19 admitted to the ICU. Front Immunol (2021) 12:742631. doi: 10.3389/fimmu.2021.742631
211. Fionda C, Ruggeri S, Sciume G, Laffranchi M, Quinti I, Milito C, et al. Age-dependent NK cell dysfunctions in severe COVID-19 patients. Front Immunol (2022) 13:1039120. doi: 10.3389/fimmu.2022.1039120
212. Huang CY, Chung CL, Hu TH, Chen JJ, Liu PF, Chen CL. Recent progress in TGF-β inhibitors for cancer therapy. BioMed Pharmacother (2021) 134:111046. doi: 10.1016/j.biopha.2020.111046
213. Wang H, Chen M, Sang X, You X, Wang Y, Paterson IC, et al. Development of small molecule inhibitors targeting TGF-β ligand and receptor: Structures, mechanism, preclinical studies and clinical usage. Eur J Med Chem (2020) 191:112154. doi: 10.1016/j.ejmech.2020.112154
214. Chinnapaiyan S, Dutta RK, Nair M, Chand HS, Rahman I, Unwalla HJ. TGF-beta1 increases viral burden and promotes HIV-1 latency in primary differentiated human bronchial epithelial cells. Sci Rep (2019) 9(1):12552. doi: 10.1038/s41598-019-49056-6
215. Kaczmarek Michaels K, Natarajan M, Euler Z, Alter G, Viglianti G, Henderson AJ. Blimp-1, an intrinsic factor that represses HIV-1 proviral transcription in memory CD4+ T cells. J Immunol (2015) 194(7):3267–74. doi: 10.4049/jimmunol.1402581
216. Samer S, Thomas Y, Arainga M, Carter C, Shirreff LM, Arif MS, et al. Blockade of TGF-beta signaling reactivates HIV-1/SIV reservoirs and immune responses in vivo. JCI Insight (2022) 7(21):e162290. doi: 10.1172/jci.insight.162290
217. Barros-Martins J, Forster R, Bosnjak B. NK cell dysfunction in severe COVID-19: TGF-beta-induced downregulation of integrin beta-2 restricts NK cell cytotoxicity. Signal Transduct Target Ther (2022) 7(1):32. doi: 10.1038/s41392-022-00892-5
218. Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal (2016) 9(415):ra19. doi: 10.1126/scisignal.aad1884
219. Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol (2020) 11:167. doi: 10.3389/fimmu.2020.00167
220. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell (2018) 175(7):1731–43 e13. doi: 10.1016/j.cell.2018.10.014
221. Ducoin K, Oger R, Bilonda Mutala L, Deleine C, Jouand N, Desfrancois J, et al. Targeting NKG2A to boost anti-tumor CD8 T-cell responses in human colorectal cancer. Oncoimmunology (2022) 11(1):2046931. doi: 10.1080/2162402X.2022.2046931
222. Ruggeri L, Urbani E, Andre P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica (2016) 101(5):626–33. doi: 10.3324/haematol.2015.135301
223. Li F, Wei H, Wei H, Gao Y, Xu L, Yin W, et al. Blocking the natural killer cell inhibitory receptor NKG2A increases activity of human natural killer cells and clears hepatitis B virus infection in mice. Gastroenterology (2013) 144(2):392–40. doi: 10.1053/j.gastro.2012.10.039
224. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol (2020) 17(5):533–5. doi: 10.1038/s41423-020-0402-2
225. Sarwar Z, Ahmad T, Kakar S. Potential approaches to combat COVID-19: a mini-review. Mol Biol Rep (2020) 47(12):9939–49. doi: 10.1007/s11033-020-05988-1
226. Benson DM, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120(22):4324–33. doi: 10.1182/blood-2012-06-438028
227. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res (2016) 22(21):5211–22. doi: 10.1158/1078-0432.CCR-16-1108
228. Vey N, Goncalves A, Karlin L, Lebouvier-Sadot S, Broussais F, Marie D, et al. A phase 1 dose-escalation study of IPH2102 (lirilumab, BMS-986015, LIRI), a fully human anti KIR monoclonal antibody (mAb) in patients (pts) with various hematologic (HEM) or solid Malignancies (SOL). J Clin Oncol (2015) 33(15_suppl):3065. doi: 10.1200/jco.2015.33.15_suppl.3065
229. Benson DM, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood (2011) 118(24):6387–91. doi: 10.1182/blood-2011-06-360255
230. Benson DM, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res (2015) 21(18):4055–61. doi: 10.1158/1078-0432.CCR-15-0304
231. Vey N, Bourhis J-H, Dombret H, Bordessoule D, Prébet T, Charbonnier A, et al. A phase I study of the anti-natural killer inhibitory receptor (KIR) monoclonal antibody (1-7F9, IPH2101) in elderly patients with acute myeloid leukemia (AML): clinical and immunological effects of a single dose followed by repeated dosing. Blood (2009) 114(22):632–. doi: 10.1182/blood.V114.22.632.632
232. Bagot M, Porcu P, Marie-Cardine A, Battistella M, William BM, Vermeer M, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol (2019) 20(8):1160–70. doi: 10.1016/S1470-2045(19)30320-1
233. Liu J, Chen Z, Li Y, Zhao W, Wu J, Zhang Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Front Pharmacol (2021) 12:731798. doi: 10.3389/fphar.2021.731798
234. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol (2017) 8:561. doi: 10.3389/fphar.2017.00561
235. Benson DM, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116(13):2286–94. doi: 10.1182/blood-2010-02-271874
236. MacFarlane AW, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res (2014) 2(4):320–31. doi: 10.1158/2326-6066.CIR-13-0133
237. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget (2016) 7(45):72961–77. doi: 10.18632/oncotarget.12150
238. Ai L, Chen J, Yan H, He Q, Luo P, Xu Z, et al. Research status and outlook of PD-1/PD-L1 inhibitors for cancer therapy. Drug Des Devel Ther (2020) 14:3625–49. doi: 10.2147/DDDT.S267433
239. Norris S, Coleman A, Kuri-Cervantes L, Bower M, Nelson M, Goodier MR. PD-1 expression on natural killer cells and CD8(+) T cells during chronic HIV-1 infection. Viral Immunol (2012) 25(4):329–32. doi: 10.1089/vim.2011.0096
240. Bertoletti A, Gehring AJ. The immune response during hepatitis B virus infection. J Gen Virol (2006) 87(6):1439–49. doi: 10.1099/vir.0.81920-0
241. Salem ML, El-Badawy A. Programmed death-1/programmed death-L1 signaling pathway and its blockade in hepatitis C virus immunotherapy. World J Hepatol (2015) 7(23):2449–58. doi: 10.4254/wjh.v7.i23.2449
242. Pauken KE, Godec J, Odorizzi PM, Brown KE, Yates KB, Ngiow SF, et al. The PD-1 pathway regulates development and function of memory CD8. Cell Rep (2020) 31(13):107827. doi: 10.1016/j.celrep.2020.107827
243. Aghbash PS, Eslami N, Shamekh A, Entezari-Maleki T, Baghi HB. SARS-CoV-2 infection: The role of PD-1/PD-L1 and CTLA-4 axis. Life Sci (2021) 270:119124. doi: 10.1016/j.lfs.2021.119124
244. McNally B, Ye F, Willette M, Flaño E. Local blockade of epithelial PDL-1 in the airways enhances T cell function and viral clearance during influenza virus infection. J Virol (2013) 87(23):12916–24. doi: 10.1128/JVI.02423-13
245. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature (2009) 458(7235):206–10. doi: 10.1038/nature07662
246. Palmer BE, Neff CP, Lecureux J, Ehler A, Dsouza M, Remling-Mulder L, et al. In vivo blockade of the PD-1 receptor suppresses HIV-1 viral loads and improves CD4+ T cell levels in humanized mice. J Immunol (2013) 190(1):211–9. doi: 10.4049/jimmunol.1201108
247. Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology (2010) 138(2):682–93, 93.e1-4. doi: 10.1053/j.gastro.2009.09.052
248. Moebius J, Guha R, Peterson M, Abdi K, Skinner J, Li S, et al. PD-1 expression on NK cells in malaria-exposed individuals is associated with diminished natural cytotoxicity and enhanced antibody-dependent cellular cytotoxicity. Infect Immun (2020) 88(3):e00711–19. doi: 10.1128/IAI.00711-19
249. Rao M, Valentini D, Dodoo E, Zumla A, Maeurer M. Anti-PD-1/PD-L1 therapy for infectious diseases: learning from the cancer paradigm. Int J Infect Dis (2017) 56:221–8. doi: 10.1016/j.ijid.2017.01.028
250. Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J (2015) 21(6):486–91. doi: 10.1097/PPO.0000000000000156
251. Iliopoulou EG, Kountourakis P, Karamouzis MV, Doufexis D, Ardavanis A, Baxevanis CN, et al. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol Immunother (2010) 59(12):1781–9. doi: 10.1007/s00262-010-0904-3
252. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13(1):98–107. doi: 10.3109/14653249.2010.515582
253. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy (2008) 10(6):625–32. doi: 10.1080/14653240802301872
254. Woan KV, Kim H, Bjordahl R, Davis ZB, Gaidarova S, Goulding J, et al. Harnessing features of adaptive NK cells to generate iPSC-derived NK cells for enhanced immunotherapy. Cell Stem Cell (2021) 28(12):2062–75 e5. doi: 10.1016/j.stem.2021.08.013
255. Ni Z, Knorr DA, Bendzick L, Allred J, Kaufman DS. Expression of chimeric receptor CD4zeta by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo. Stem Cells (2014) 32(4):1021–31. doi: 10.1002/stem.1611
256. Kim JT, Zhang TH, Carmona C, Lee B, Seet CS, Kostelny M, et al. Latency reversal plus natural killer cells diminish HIV reservoir in vivo. Nat Commun (2022) 13(1):121. doi: 10.1038/s41467-021-27647-0
257. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med (2009) 206(1):25–34. doi: 10.1084/jem.20082013
258. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi: 10.1200/JCO.2014.57.3329
259. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med (2016) 8(357):357ra123. doi: 10.1126/scitranslmed.aaf2341
260. Wagstaffe HR, Nielsen CM, Riley EM, Goodier MR. IL-15 promotes polyfunctional NK cell responses to influenza by boosting IL-12 production. J Immunol (2018) 200(8):2738–47. doi: 10.4049/jimmunol.1701614
261. Garrido C, Abad-Fernandez M, Tuyishime M, Pollara JJ, Ferrari G, Soriano-Sarabia N, et al. Interleukin-15-stimulated natural killer cells clear HIV-1-infected cells following latency reversal. J Virol (2018) 92(12):262. doi: 10.1128/JVI.00235-18
262. Terrén I, Orrantia A, Astarloa-Pando G, Amarilla-Irusta A, Zenarruzabeitia O, Borrego F. Cytokine-induced memory-like NK cells: from the basics to clinical applications. Front Immunol (2022) 13:884648. doi: 10.3389/fimmu.2022.884648
263. Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood (2012) 120(24):4751–60. doi: 10.1182/blood-2012-04-419283
264. Berrien-Elliott MM, Wagner JA, Fehniger TA. Human cytokine-induced memory-like natural killer cells. J Innate Immun (2015) 7(6):563–71. doi: 10.1159/000382019
265. Shapiro RM, Birch GC, Hu G, Vergara Cadavid J, Nikiforow S, Baginska J, et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for posttransplant relapse. J Clin Invest (2022) 132(11):e154334. doi: 10.1172/JCI154334
266. Bednarski JJ, Zimmerman C, Berrien-Elliott MM, Foltz JA, Becker-Hapak M, Neal CC, et al. Donor memory-like NK cells persist and induce remissions in pediatric patients with relapsed AML after transplant. Blood (2022) 139(11):1670–83. doi: 10.1182/blood.2021013972
267. Berrien-Elliott MM, Foltz JA, Russler-Germain DA, Neal CC, Tran J, Gang M, et al. Hematopoietic cell transplantation donor-derived memory-like NK cells functionally persist after transfer into patients with leukemia. Sci Transl Med (2022) 14(633):eabm1375. doi: 10.1126/scitranslmed.abm1375
268. Ellis-Connell AL, Balgeman AJ, Zarbock KR, Barry G, Weiler A, Egan JO, et al. ALT-803 transiently reduces simian immunodeficiency virus replication in the absence of antiretroviral treatment. J Virol (2018) 92(3):e01748–17. doi: 10.1128/JVI.01748-17
269. Webb GM, Molden J, Busman-Sahay K, Abdulhaqq S, Wu HL, Weber WC, et al. The human IL-15 superagonist N-803 promotes migration of virus-specific CD8+ T and NK cells to B cell follicles but does not reverse latency in ART-suppressed, SHIV-infected macaques. PloS Pathog (2020) 16(3):e1008339. doi: 10.1371/journal.ppat.1008339
270. Miller JS, Davis ZB, Helgeson E, Reilly C, Thorkelson A, Anderson J, et al. Safety and virologic impact of the IL-15 superagonist N-803 in people living with HIV: a phase 1 trial. Nat Med (2022) 28(2):392–400. doi: 10.1038/s41591-021-01651-9
271. Lynch RM, Boritz E, Coates EE, DeZure A, Madden P, Costner P, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med (2015) 7(319):319ra206. doi: 10.1126/scitranslmed.aad5752
272. Caskey M. Broadly neutralizing antibodies for the treatment and prevention of HIV infection. Curr Opin HIV AIDS (2020) 15(1):49–55. doi: 10.1097/COH.0000000000000600
273. Haynes BF, Wiehe K, Borrow P, Saunders KO, Korber B, Wagh K, et al. Strategies for HIV-1 vaccines that induce broadly neutralizing antibodies. Nat Rev Immunol (2023) 23(3):142–58. doi: 10.1038/s41577-022-00753-w
274. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med (2009) 361(23):2209–20. doi: 10.1056/NEJMoa0908492
275. Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med (2012) 366(14):1275–86. doi: 10.1056/NEJMoa1113425
276. Karasavvas N, Billings E, Rao M, Williams C, Zolla-Pazner S, Bailer RT, et al. The Thai Phase III HIV Type 1 Vaccine trial (RV144) regimen induces antibodies that target conserved regions within the V2 loop of gp120. AIDS Res Hum Retroviruses (2012) 28(11):1444–57. doi: 10.1089/aid.2012.0103
277. Yates NL, Liao HX, Fong Y, deCamp A, Vandergrift NA, Williams WT, et al. Vaccine-induced Env V1-V2 IgG3 correlates with lower HIV-1 infection risk and declines soon after vaccination. Sci Transl Med (2014) 6(228):228ra39. doi: 10.1126/scitranslmed.3007730
278. Bonsignori M, Pollara J, Moody MA, Alpert MD, Chen X, Hwang KK, et al. Antibody-dependent cellular cytotoxicity-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J Virol (2012) 86(21):11521–32. doi: 10.1128/JVI.01023-12
279. Li SS, Gilbert PB, Tomaras GD, Kijak G, Ferrari G, Thomas R, et al. FCGR2C polymorphisms associate with HIV-1 vaccine protection in RV144 trial. J Clin Invest (2014) 124(9):3879–90. doi: 10.1172/JCI75539
280. Alpert MD, Harvey JD, Lauer WA, Reeves RK, Piatak M, Carville A, et al. ADCC develops over time during persistent infection with live-attenuated SIV and is associated with complete protection against SIV(mac)251 challenge. PloS Pathog (2012) 8(8):e1002890. doi: 10.1371/journal.ppat.1002890
281. Forthal DN, Gilbert PB, Landucci G, Phan T. Recombinant gp120 vaccine-induced antibodies inhibit clinical strains of HIV-1 in the presence of Fc receptor-bearing effector cells and correlate inversely with HIV infection rate. J Immunol (2007) 178(10):6596–603. doi: 10.4049/jimmunol.178.10.6596
282. Nag P, Kim J, Sapiega V, Landay AL, Bremer JW, Mestecky J, et al. Women with cervicovaginal antibody-dependent cell-mediated cytotoxicity have lower genital HIV-1 RNA loads. J Infect Dis (2004) 190(11):1970–8. doi: 10.1086/425582
283. Wren LH, Chung AW, Isitman G, Kelleher AD, Parsons MS, Amin J, et al. Specific antibody-dependent cellular cytotoxicity responses associated with slow progression of HIV infection. Immunology (2013) 138(2):116–23. doi: 10.1111/imm.12016
284. Lu CL, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science (2016) 352(6288):1001–4. doi: 10.1126/science.aaf1279
285. Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, et al. Antibody neutralization and escape by HIV-1. Nature (2003) 422(6929):307–12. doi: 10.1038/nature01470
286. Meijers M, Vanshylla K, Gruell H, Klein F, Lässig M. Predicting in vivo escape dynamics of HIV-1 from a broadly neutralizing antibody. Proc Natl Acad Sci USA (2021) 118(30):e2104651118. doi: 10.1073/pnas.2104651118
287. Dingens AS, Arenz D, Weight H, Overbaugh J, Bloom JD. An antigenic atlas of HIV-1 escape from broadly neutralizing antibodies distinguishes functional and structural epitopes. Immunity (2019) 50(2):520–32.e3. doi: 10.1016/j.immuni.2018.12.017
288. Isitman G, Stratov I, Kent SJ. Antibody-dependent cellular cytotoxicity and NK cell-driven immune escape in HIV infection: implications for HIV vaccine development. Adv Virol (2012) 2012:637208. doi: 10.1155/2012/637208
289. Tudor D, Yu H, Maupetit J, Drillet AS, Bouceba T, Schwartz-Cornil I, et al. Isotype modulates epitope specificity, affinity, and antiviral activities of anti-HIV-1 human broadly neutralizing 2F5 antibody. Proc Natl Acad Sci USA (2012) 109(31):12680–5. doi: 10.1073/pnas.1200024109
290. Richardson SI, Lambson BE, Crowley AR, Bashirova A, Scheepers C, Garrett N, et al. IgG3 enhances neutralization potency and Fc effector function of an HIV V2-specific broadly neutralizing antibody. PloS Pathog (2019) 15(12):e1008064. doi: 10.1371/journal.ppat.1008064
291. de Taeye SW, Bentlage AEH, Mebius MM, Meesters JI, Lissenberg-Thunnissen S, Falck D, et al. FcγR binding and ADCC activity of human IgG allotypes. Front Immunol (2020) 11:740. doi: 10.3389/fimmu.2020.00740
292. Demaria O, Gauthier L, Debroas G, Vivier E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur J Immunol (2021) 51(8):1934–42. doi: 10.1002/eji.202048953
293. Pinto S, Pahl J, Schottelius A, Carter PJ, Koch J. Reimagining antibody-dependent cellular cytotoxicity in cancer: the potential of natural killer cell engagers. Trends Immunol (2022) 43(11):932–46. doi: 10.1016/j.it.2022.09.007
294. Au KM, Park SI, Wang AZ. Trispecific natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci Adv (2020) 6(27):eaba8564. doi: 10.1126/sciadv.aba8564
295. Mei Y, Xiang B, Yang J. Novel strategies for redirecting NK cells in cancer immunotherapy. J Nutr Oncol (2017) 2(2):66–72. doi: 10.34175/jno201702003
296. Ma J, Mo Y, Tang M, Shen J, Qi Y, Zhao W, et al. Bispecific antibodies: from research to clinical application. Front Immunol (2021) 12:626616. doi: 10.3389/fimmu.2021.626616
297. Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol (2020) 17(5):451–61. doi: 10.1038/s41423-020-0417-8
298. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ Targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res (2016) 22(14):3440–50. doi: 10.1158/1078-0432.CCR-15-2710
299. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol (2015) 93(3):290–6. doi: 10.1038/icb.2014.93
300. Sung JA, Pickeral J, Liu L, Stanfield-Oakley SA, Lam CY, Garrido C, et al. Dual-Affinity Re-Targeting proteins direct T cell-mediated cytolysis of latently HIV-infected cells. J Clin Invest (2015) 125(11):4077–90. doi: 10.1172/JCI82314
301. Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood (2011) 117(17):4542–51. doi: 10.1182/blood-2010-09-306449
302. Phung SK, Miller JS, Felices M. Bi-specific and tri-specific NK cell engagers: the new avenue of targeted NK cell immunotherapy. Mol Diagn Ther (2021) 25(5):577–92. doi: 10.1007/s40291-021-00550-6
303. Oboshi W, Watanabe T, Matsuyama Y, Kobara A, Yukimasa N, Ueno I, et al. The influence of NK cell-mediated ADCC: Structure and expression of the CD16 molecule differ among FcγRIIIa-V158F genotypes in healthy Japanese subjects. Hum Immunol (2016) 77(2):165–71. doi: 10.1016/j.humimm.2015.11.001
304. Coënon L, Villalba M. From CD16a biology to antibody-dependent cell-mediated cytotoxicity improvement. Front Immunol (2022) 13:913215. doi: 10.3389/fimmu.2022.913215
305. Talathi SP, Shaikh NN, Pandey SS, Saxena VA, Mamulwar MS, Thakar MR. FcγRIIIa receptor polymorphism influences NK cell mediated ADCC activity against HIV. BMC Infect Dis (2019) 19(1):1053. doi: 10.1186/s12879-019-4674-z
306. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of biKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol (2016) 1441:333–46. doi: 10.1007/978-1-4939-3684-7_28
307. Tay SS, Carol H, Biro M. TriKEs and BiKEs join CARs on the cancer immunotherapy highway. Hum Vaccin Immunother (2016) 12(11):2790–6. doi: 10.1080/21645515.2016.1198455
308. Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol (2017) 31:64–75. doi: 10.1016/j.smim.2017.07.011
309. Davis ZB, Lenvik T, Hansen L, Felices M, Cooley S, Vallera D, et al. A novel HIV envelope bi-specific killer engager enhances natural killer cell mediated ADCC responses against HIV-infected cells. Blood (2016) 128(22):2517–. doi: 10.1182/blood.V128.22.2517.2517
310. Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med (2012) 2(8):a006866. doi: 10.1101/cshperspect.a006866
311. Chen W, Feng Y, Gong R, Zhu Z, Wang Y, Zhao Q, et al. Engineered single human CD4 domains as potent HIV-1 inhibitors and components of vaccine immunogens. J Virol (2011) 85(18):9395–405. doi: 10.1128/JVI.05119-11
312. Sakihama T, Smolyar A, Reinherz EL. Oligomerization of CD4 is required for stable binding to class II major histocompatibility complex proteins but not for interaction with human immunodeficiency virus gp120. Proc Natl Acad Sci USA (1995) 92(14):6444–8. doi: 10.1073/pnas.92.14.6444
313. Lamarre D, Capon DJ, Karp DR, Gregory T, Long EO, Sékaly RP. Class II MHC molecules and the HIV gp 120 envelope protein interact with functionally distinct regions of the CD4 molecule. EMBO J (1989) 8(11):3271–7. doi: 10.1002/j.1460-2075.1989.tb08487.x
314. Chen W, Feng Y, Prabakaran P, Ying T, Wang Y, Sun J, et al. Exceptionally potent and broadly cross-reactive, bispecific multivalent HIV-1 inhibitors based on single human CD4 and antibody domains. J Virol (2014) 88(2):1125–39. doi: 10.1128/JVI.02566-13
315. Li W, Wu Y, Kong D, Yang H, Wang Y, Shao J, et al. One-domain CD4 fused to human anti-CD16 antibody domain mediates effective killing of HIV-1-infected cells. Sci Rep (2017) 7(1):9130. doi: 10.1038/s41598-017-07966-3
316. Li H, Song W, Li Z, Zhang M. Preclinical and clinical studies of CAR-NK-cell therapies for Malignancies. Front Immunol (2022) 13:992232. doi: 10.3389/fimmu.2022.992232
317. Lu T, Ma R, Dong W, Teng KY, Kollath DS, Li Z, et al. Off-the-shelf CAR natural killer cells secreting IL-15 target spike in treating COVID-19. Nat Commun (2022) 13(1):2576. doi: 10.1038/s41467-022-30216-8
318. Mazarzaei A, Vafaei M, Ghasemian A, Mirforughi SA, Rajabi Vardanjani H, Alwan NAS. Memory and CAR-NK cell-based novel approaches for HIV vaccination and eradication. J Cell Physiol (2019) 234(9):14812–7. doi: 10.1002/jcp.28280
Keywords: NK cells, NK cell dysfunction, HIV, hepatitis C, CMV, influenza, SARS-CoV-2, immunotherapy
Citation: Bjorgen JC, Dick JK, Cromarty R, Hart GT and Rhein J (2023) NK cell subsets and dysfunction during viral infection: a new avenue for therapeutics? Front. Immunol. 14:1267774. doi: 10.3389/fimmu.2023.1267774
Received: 27 July 2023; Accepted: 25 September 2023;
Published: 19 October 2023.
Edited by:
Niels A. W. Lemmermann, University Hospital Bonn, GermanyReviewed by:
Silvana Gaudieri, University of Western Australia, AustraliaCopyright © 2023 Bjorgen, Dick, Cromarty, Hart and Rhein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacob C. Bjorgen, YmpvcmcwOTFAdW1uLmVkdQ==
†Present address: Jacob C. Bjorgen, Viral Pathogenesis and Evolution Section, Laboratory of Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.