 Hui Zhao
Hui Zhao Cheng Tang
Cheng Tang Miao Wang1
Miao Wang1- 1The Geriatrics, Graduate School of Anhui University of Chinese Medicine, Hefei, China
- 2Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3The Geriatrics, The Second Affiliated Hospital of Anhui University of Chinese Medicine, Hefei, China
Rheumatoid arthritis (RA) is an autoimmune disease of unknown etiology. Due to the rise in the incidence rate of RA and the limitations of existing therapies, the search for new treatment strategies for RA has become a global focus. Ferroptosis is a novel programmed cell death characterized by iron-dependent lipid peroxidation, with distinct differences from apoptosis, autophagy, and necrosis. Under the conditions of iron accumulation and the glutathione peroxidase 4 (GPX4) activity loss, the lethal accumulation of lipid peroxide is the direct cause of ferroptosis. Ferroptosis mediates inflammation, oxidative stress, and lipid oxidative damage processes, and also participates in the occurrence and pathological progression of inflammatory joint diseases including RA. This review provides insight into the role and mechanism of ferroptosis in RA and discusses the potential and challenges of ferroptosis as a new therapeutic strategy for RA, with an effort to provide new targets for RA prevention and treatment.
1 Introduction
Rheumatoid arthritis (RA) is a multifactorial autoimmune disease of unknown etiology mainly characterized by synovial hyperplasia, pannus formation, as well as cartilage and bone destruction, resulting in joint pain, stiffness, and swelling. In RA pathogenesis, activated RA fibroblast-like synoviocytes (RA-FLSs) exhibit proliferative features similar to tumor cells and subsequently cause cartilage erosion, which will eventually lead to joint destruction (1, 2). RA patients initially present with the main complaint of finger and/or wrist joint pain, but as the disease progresses, RA may also involve large joints such as the knee, shoulder, and hip joints, resulting in limited joint activity, irreversible joint deformities, and even disability. Apart from joint symptoms, RA may also give rise to systemic multi-system injury and extra-articular manifestations (3). Due to the intensification of population aging, prolongation of life expectancy, and deterioration of the global environment, the morbidity of RA is ascending annually. Statistics estimate that approximately 1% of the global population suffers from RA, and the incidence rate of RA in women is three to four times higher than that in men (4, 5). The current treatment strategies for RA mainly focus on regulating immune inflammatory responses to alleviate arthritis symptoms (6). However, these existing treatment methods still have insurmountable shortcomings such as various adverse drug reactions, limited individualized treatment regimens, significant differences in treatment effectiveness, and drug resistance. Effective therapies that can delay or reverse the pathological progression of RA are urgently needed. Therefore, further elucidating the pathogenesis of RA and identifying new therapeutic targets will help develop more personalized, effective, and safe treatment approaches, thereby improving the quality of life of RA patients and reducing the socioeconomic burden of the disease.
It has been hypothesized that iron is excessively accumulated in the synovium of RA patients to perpetuate inflammation (7, 8). Ferroptosis is a type of regulatory cell death that depends on intracellular iron accumulation. The fundamental features of ferroptosis, such as iron deposition, lipid peroxidation, glutathione (GSH) depletion, and glutathione peroxidase 4 (GPX4) inactivation, have been gradually emphasized in the pathogenesis of RA (9). As a significant regulatory factor in inflammatory responses, ferroptosis has recently been reported to be strongly associated with the initiation and progression of various inflammatory arthritis including RA (10). This review provides an in-depth study of the role and mechanisms of ferroptosis in RA, discusses the potential and challenges of ferroptosis as a novel therapeutic strategy for RA, and highlights the potential of ferroptosis as a promising treatment target for RA.
2 Overview of ferroptosis
Cell death is a fundamental physiological process in all aspects of life. In the past, most studies focused on the diverse roles of apoptosis, pyroptosis, autophagy, and necrosis in diseases. In 2012, Dixon et al. first reported a novel form of regulated cell death with distinct morphological, biochemical, and genetic properties from the aforementioned cell death types, named “ferroptosis” (11). Ferroptosis morphologically exhibits the same features as necrosis, including loss of plasma membrane integrity, cytoplasm and organelle swelling, increased mitochondrial membrane density, reduced or absent cristae, and outer membrane rupture (12, 13). The exact pathogenesis of ferroptosis has not yet been clarified. Current studies indicate iron accumulation, lipid peroxidation, and redox system dysregulation as central biochemical events leading to ferroptosis (14). During ferroptosis, excessive accumulation of iron ions can catalyze the production of reactive oxygen species (ROS) through the Fenton reaction to promote lipid peroxidation, causing oxidative damage to the cell membrane (15). Subsequently, oxidative stress further damages the cell membrane and important intracellular biomolecules due to the disruption of antioxidant defense mechanisms, and this imbalance between oxidative damage and antioxidant defense is a crucial driver of ferroptosis (16, 17). GPX4 inactivation based on GSH depletion is a central regulator in the antioxidant defense system, and GPX4 can utilize the substrate GSH to reduce oxidation to scavenge free radicals and other harmful substances (18). Therefore, it can be assumed that the core mechanism of ferroptosis is the insufficient ability of GPX4 to scavenge peroxides and the excessive accumulation of lipid peroxides. Since the mechanisms of ferroptosis are exceptionally complex and involve multiple signaling pathways, an in-depth study of these mechanisms can provide new ideas and approaches for the treatment and prevention of ferroptosis-related diseases.
3 Ferroptosis mechanisms
3.1 Iron metabolism
As one of the abundant trace elements in the crust, iron plays an imperative role in life activities such as DNA synthesis, redox reactions, enzymatic processes, cellular respiration, and metabolism (19, 20). Therefore, precise iron metabolism is required for the homeostasis of intracellular iron ions. Ferroptosis, as a response to abnormal cellular iron metabolism, typically occurs in a state of iron overload. After the circulating iron ions (Fe3+) bind to transferrin (Tf) on the plasma membrane, the complex is transferred into the cell by binding to the specific transferrin receptor 1 (TfR1) and localized in the endosome (21). The acidic environment in the endosome promotes the shedding of Fe3+and reduces it to Fe2+ via the six-transmembrane epithelial antigen of prostate 3 (STEAP3), which is then exported to the cytoplasm via divalent metal transporter protein 1 (DMT1) or ZRT/IRT-like protein 8/14 (ZIP8/14). In the cytoplasm, a majority of Fe2+ is stored in ferritin and a small fraction of Fe2+ is stored in the labile iron pool (LIP). Excess Fe2+ is oxidized to Fe3+, which is transported out of the cell and re-entered into the circulation via ferroportin (FPN). Ferritin, a nanocage composed of ferritin light chain (FTL) and ferritin heavy chain (FTH), reduces Fe2+ to non-toxic Fe3+ (22). In response to the autophagic degradation by nuclear receptor coactivator 4 (NCOA4), ferritin releases active Fe2+ (23, 24) to catalyze lipid peroxidation through the Fenton reaction (25) (Figure 1).
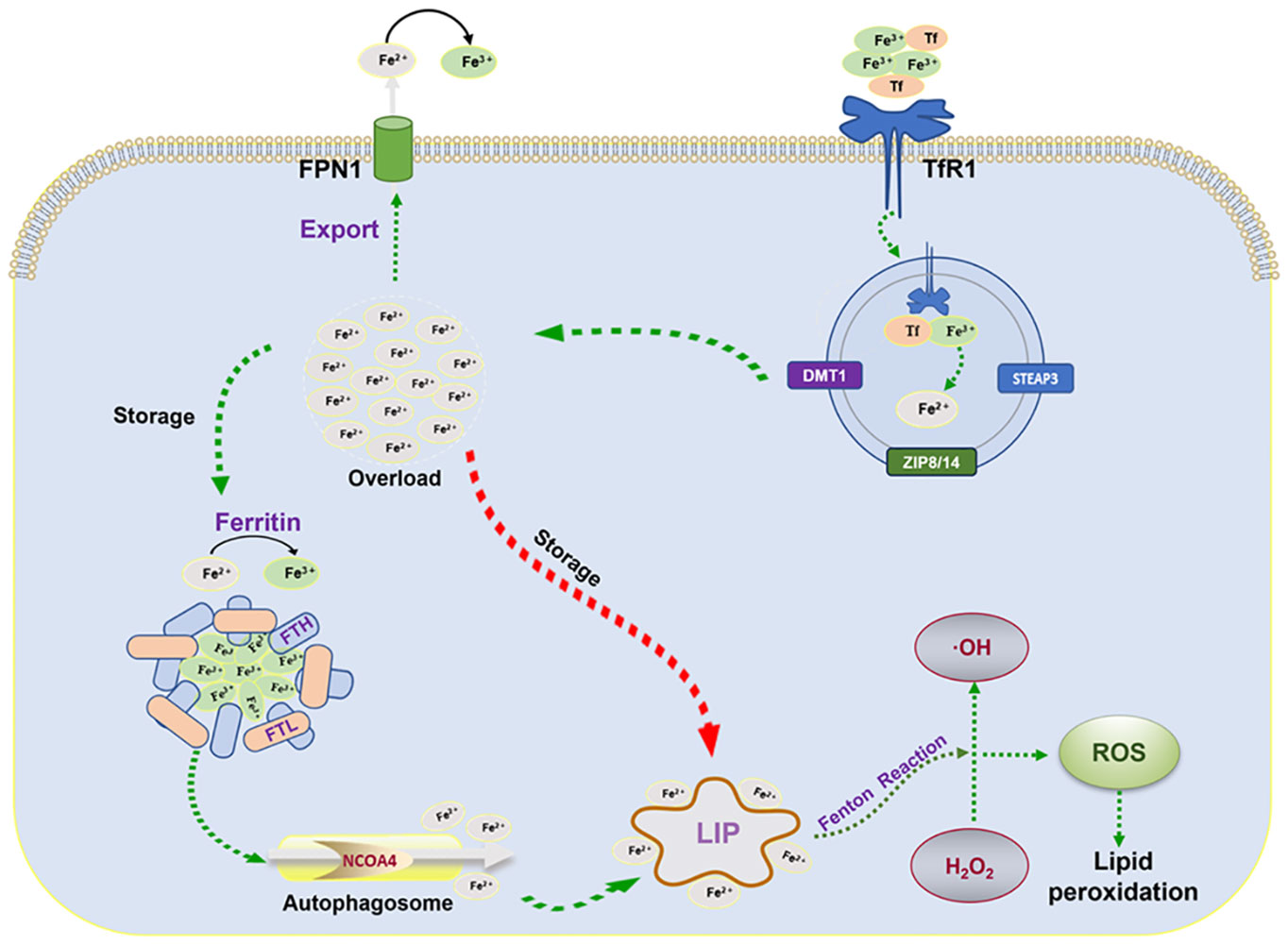
Figure 1 Mechanisms of iron metabolism: FPN1, iron transporter protein; TfR1, transferrin receptor 1; FTL, ferritin light chain; FTH, ferritin heavy chain; NCOA4, nuclear receptor coactivator 4; LIP, labile iron pool; ROS, reactive oxygen species; STEAP3, six-transmembrane epithelial antigen of the prostate 3; DMT1, divalent metal transporter protein 1.
3.2 Lipid peroxidation and oxidative stress
Ferroptosis is ultimately driven by ROS-mediated lipid peroxidation. Polyunsaturated fatty acids (PUFAs), represented by arachidonic acid (AA) and adrenaline (AdA), are most susceptible to peroxidation (15). Free intracellular PUFAs form esterification products through the binding of coenzyme A (CoA) and acyl-coenzyme A synthase long-chain family member 4 (ACSL4), which are subsequently transported to phospholipid-like lipids (PL) of the cell membrane via lysophosphatidylcholine acyltransferase 3 (LPCAT3) (26, 27). In the presence of lipoxygenase (Lox) peroxidation, PUFA-PL is oxidized, inducing the accumulation of PL hydrogen peroxide on the plasma membrane and thus affecting membrane function (28) (Figure 2).
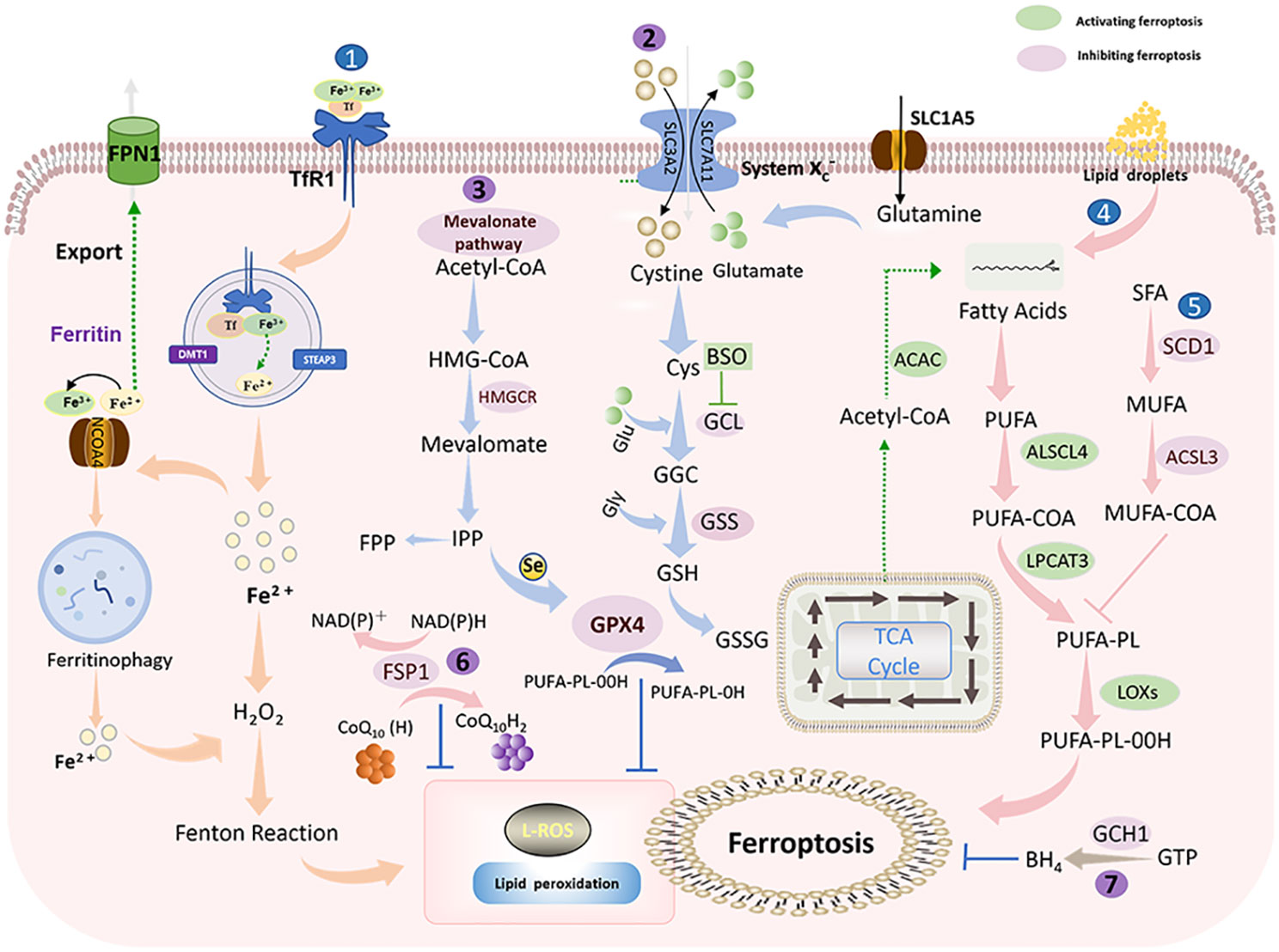
Figure 2 Ferroptosis mechanism: Ferroptosis is mainly caused by iron-dependent lipid peroxidation. It is mainly divided into ferroptosis promotion pathway (blue) and ferroptosis inhibition pathway (purple) (1). Iron metabolic pathway (2). Cystine/glutamate (also known as Xc-system)-GSH-GPX4 pathway: Cystine is imported into cells via the Xc-system, where it is oxidized to cysteine (Cys), followed by the synthesis of glutathione (GSH) in the presence of glutamate-cysteine ligase (GCL) and glutathione synthase (GSS). GSH is a potent reducing agent. GPX4 inhibits ferroptosis by using GSH as a reducing cofactor to reduce lipid hydroperoxides to lipid alcohols (3). Mevalonate pathway: Acetyl coenzyme A is first converted to HMG-CoA, which is then reduced to mevalonate by HMGCR, and mevalonate is converted to IPP. Finally, selenocysteine residues are added to the catalytic center of GPX4 to activate GPX4. At the same time, IPP can also produce coenzyme Q10 and then enter the FSP1 pathway (4). Lipid metabolic pathway: PUFAs are metabolized by ACSL4 and LPCAT3 and then oxidized by lipoxygenase HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; IPP, isopentenyl pyrophosphate; GPX4, glutathione peroxidase 4; SLC1A5, solute carrier family 1 member 5; GLS, glutaminase; SLC3A2, solute carrier family 3 member 2. SLC7A11, solute carrier family 7 member 11; GSH, glutathione; Xc- system: glutamate reverse transporter protein, GPX4: glutathione peroxidase 4.
3.3 Antioxidant defense
Lipid peroxidation can be countered by antioxidant systems, in which the cystine/glutamate-GSH-GPX4 is the main antioxidant pathway (29). Cystine/glutamate (also known as the Xc- system) is a system composed of light chain solute carrier family 7 member 11 (SLC7A11, xCT) and heavy chain solute carrier family 3 member 2 (SLC3A2). Cystine is reduced to cysteine intracellularly via Xc- to participate in GSH synthesis (30). Glutathione peroxide GPX4 is a primary factor responsible for the scavenging of lipid peroxides, which can convert GSH to oxidized glutathione (GSSG) and simultaneously reduce toxic lipid peroxides to non-toxic alcohols (31). Moreover, FSP1-CoQ10-NADPH (32) and GTP-BH4 (33) are two antioxidant systems independent of GPX4 that effectively protect cells from lipid peroxidation damage.
4 Association between ferroptosis and RA
Recent studies have found that the basic features of ferroptosis such as iron deposition, lipid peroxidation, GSH depletion, and GPX4 inactivation are implicated in RA pathogenesis. Immune inflammatory dysregulation and intestinal microbial imbalance in RA are confirmed to be associated with ferroptosis. These findings suggest that ferroptosis is closely related to the occurrence and development of RA.
4.1 Disorders of iron metabolism in RA
Disturbances in iron metabolism lead to the development of RA through a variety of mechanisms, including promoting oxidative stress, inducing inflammatory responses, and impairing immune cell functions. RA patients commonly present with elevated iron metabolism indicators like serum iron and ferritin, and the concentration of iron is positively correlated with the severity of joint inflammation (34, 35). FTL, FTH, and non-characteristic resistance-associated macrophage proteins such as Nramp2 and DMT1 can also be detected in fibroblast-like synoviocytes (FLSs) and macrophages isolated from synovial tissues of RA patients (36). In addition, animal experiments have also demonstrated that intravenous iron injection can exacerbate RA synovial inflammation (37). Iron acts as an important regulatory factor in immune responses, and its metabolism is of great significance to autoimmune diseases including RA (38, 39). Iron overload has profound effects on the immune system, including suppressing the phagocytosis of monocytes and macrophages, enhancing the number and viability of suppressor T cells, impairing the proliferative capacity of Th cells, and altering the distribution of lymphocytes in different compartments of the immune system (40). Moreover, iron overload may also trigger immune inflammatory responses by activating the NF-κB signaling pathway and stimulating the secretion of inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-β (20). In a recent mouse experiment (41), the application of iron chelators effectively reduces iron deposition and ameliorates iron overload-induced oxidative damage and immune dysfunction.
4.2 Lipid peroxidation and oxidative stress in RA
Oxidative stress as a major pathogenic hallmark of RA promotes the local microenvironment at the RA lesion sites, enhances abnormal synoviocytes proliferation, and exacerbates inflammatory infiltration (42, 43). Oxidative stress is the result of an imbalance between the generation of reactive oxygen species (ROS) and the antioxidant defense systems. ROS-mediated mitochondrial DNA (mtDNA) tends to be recognized as a pathogen-associated molecular pattern (PAMP) by the innate immune system, leading to the activation of multiple inflammatory pathways (44). A higher ROS concentration and a lower antioxidant potential have been observed in the plasma of patients with active RA (45). Furthermore, emerging studies have confirmed that there exists a mutually reinforcing positive feedback mechanism between oxidative stress and inflammation, where ROS and mitochondrial damage products are good activators of inflammatory responses (46, 47).
In RA, excessive ROS accumulation and sustained inflammatory activation promote synovial, vascular, and joint damage. FLSs are the main effector cells of RA inflammation. The activated FLSs recruit peripheral monocytes/macrophages to synovial tissues by releasing chemokines and inflammatory mediators (48). Moreover, RA-FLSs can secrete macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor κB ligand (RANKL) to promote bone destruction (49). Related studies have found (50, 51) that ROS can promote the abnormal proliferation of RA-FLSs and mediate multiple pathological processes including erosion, migration, inflammation, and joint damage. As a second messenger, ROS can upregulate hypoxia-inducible factor-1α (HIF-1α), activate the JAK3/STAT4 pathway, and promote RANKL expression (52). Moreover, Notch-1 is known to be strongly expressed in the perivascular area of synovial tissues and ROS can also induce the formation of vascular opacities by upregulating the expression of vascular endothelial growth factor (VEGF) and Notch-1 (53). Of note, ROS accumulation underlies RA, and the accumulated ROS has different effects over different cell types. For example, excessive ROS is toxic to non-FSLs and triggers ferroptosis in these cells; however, ROS accumulation has a pro-proliferative and pro-survival role in FSLs. Several studies have attempted to explore alternative methods to assess the disease activity and prognosis of RA by monitoring the levels of ROS, mtDNA, and other markers of oxidative stress (46, 54, 55). However, further studies and explorations are still needed to address the use of oxidative stress markers in the treatment of RA.
4.3 Antioxidant defense in RA
RA patients commonly present with lower levels of antioxidant markers such as GSH and GSH peroxidase (GSH Px) compared to healthy individuals and systemic lupus erythematosus (SLE) patients, and methotrexate treatment can notably alter the oxidative stress parameters in RA patients (56). The use of antioxidants, such as vitamin E, vitamin C, and selenium, has shown promising efficacy in alleviating RA disease conditions (57–59). These findings suggest that antioxidants and oxidative stress markers may be potential therapeutic targets for RA. However, further studies and clinical trials are required to evaluate the efficacy and safety of these potential targets.
4.4 Immunity and inflammation in RA
Dysregulation of the immune-inflammatory response is accepted as the predominant pathogenic mechanism of RA (5). A previous study has summed up (60) the effects of ferroptosis on immune cells in two aspects. On the one hand, ferroptosis affects the number and function of immune cells. On the other hand, ferroptotic cells can be recognized by immune cells and subsequently trigger a series of inflammatory immune responses. Thus, we report the association between ferroptosis and immune cells, especially macrophages, neutrophils, and T cells upstream of RA.
Macrophages are immune cells with antigen-presenting and phagocytic properties and have critical functions in the initiation and perpetuation of RA synovitis. Iron overload promotes the polarization of M1 macrophages and increases the levels of M1 macrophage markers (IL-6, TNF-α, and IL-1β) (61). Meanwhile, TNF-α released from M1 macrophages upregulates the expressions of acyl-CoA synthetases (ACSL3 and ACSL57) (62), promotes L-ROS accumulation, and eventually triggers ferroptosis. More significantly, M1 macrophages also elevate the ROS concentration via the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) pathway (63). Furthermore, erythrocytes phagocytized by macrophages are first catabolized to heme and then to iron under the action of heme oxygenase, and iron is deposited in RA macrophages to provide suitable conditions and environment for the occurrence of ferroptosis (64, 65).
As the first line of host defense against invading pathogens, neutrophils can be the first type of innate immune cells reaching the RA synovial joints to drive inflammation by producing chemokines, ROS, and neutrophil extracellular traps (NETs) (66). Neutrophil ferroptosis prevalently occurs in SLE patients (67), but whether neutrophil ferroptosis is associated with RA is not yet clear. Notably, there is an imbalance between oxidative stress and antioxidant defense in neutrophils of RA patients (68), which is exactly the key to driving ferroptosis. Besides, mitochondrial formyl peptides (mtNFPs) are known to induce the release of ROS from neutrophils, and significantly elevated levels of mtNFPs have been found in RA patients (69), suggesting that neutrophil ferroptosis is most likely present in RA. However, mechanisms regarding neutrophil ferroptosis in RA still require further scientific studies.
T cells are the leading inflammatory cells in RA synovial tissues. Ferroptosis is found to regulate the activity of cytotoxic T cells (CD8) and helper T cells (CD4). T cell activation is dependent on the production of cysteine (Cys) by antigen-presenting cells. Ferroptosis-related proteins, Xc- anti-transport proteins, and neutral amino acid transporter proteins are available to provide T cells with the required Cys (70). An in vitro experiment has revealed that ferroptosis inhibitor FSP1 or GPX4 overexpression protects CD8 T cells from ferroptosis, while the utilization of GPX4 inhibitors significantly enhances the sensitivity of T cells to ferroptosis (71). Further investigation (72) suggests that T cells from RA patients can store lipid droplets, leading to excess fatty acids and thus providing substrates for lipid peroxidation and ferroptosis.
4.5 Intestinal microorganisms in ferroptosis
With the intensive investigation of the pathogenesis of RA, there is increasing evidence supporting the association between intestinal flora and RA. Evidence points out (73) that dysbacteriosis is likely to initiate hyperactivity of the intestinal mucosal immune system, activate both innate and adaptive immunity, and then overtake the synovial joints, a process known as the “gut-joint axis”. More crucially, the intestinal microbiota also participates in ferroptosis by facilitating oxidative stress balance and reducing ROS accumulation. Excessive oral intake of iron causes intestinal iron accumulation and, in some cases, promotes ROS accumulation and interferes with intestinal microbiota homeostasis, eventually triggering RA development (74). Free iron may play a role in inflammatory diseases such as RA through the mediation of microbial activation rather than through Fenton response and oxidative stress mechanisms, implying that iron metabolism is closely related to the regulation of microbiota and immune response (75). Further, intestinal microbiota metabolites mediate ferroptosis by regulating the transferrin-long-chain acyl coenzyme A synthase 4 (TFR-ACSL4) pathway and inducing subsequent lipid metabolism disorders and inflammatory responses, while the use of iron chelators can effectively mitigate this process (76).
5 Potential role of ferroptosis in RA inflammation and tissue damage
5.1 Ferroptosis products and derivatives can act as inflammatory mediators
Ferroptosis is an immunogenic form of cell death, and its metabolites and derivatives can also act as inflammatory mediators to further exacerbate RA inflammatory responses. Ferroptosis can induce the release of damage-associated molecular patterns (DAMPs), thereby promoting immune cell activation and enhancing inflammatory responses (77). On the one hand, DAMPs promote the maturation and antigen presentation of immune cells such as macrophages and dendritic cells to enhance immune responses. On the other hand, DAMPs are well-recognized triggers that result in the elevation of inflammatory cytokine levels and then initiate inflammatory signaling pathways by binding to corresponding receptors. The execution of ferroptosis is driven by lipid peroxidation. The main products of lipid peroxidation derivatives, such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), may lead to cartilage degeneration and subchondral bone remodeling, and 4-HNE has also been demonstrated to activate inflammatory signaling pathways (78).
PUFAs are responsible for ferroptosis-inducing lipid peroxidation. PUFA-derived lipid mediators, AA and docosahexaenoic acid (DHA), serve as inflammatory activators in RA (79). COX-2, an enzyme involved in the AA oxidation reaction, rapidly induces inflammatory responses at the site of inflammation and accelerates the inflammatory process under specific conditions. In the presence of PPARγ (peroxisome proliferator-activated receptor γ)-dependent mechanisms, the expressions of COX-2 and prostaglandin D synthase 2 (PGD2) is promptly upregulated, resulting in a vicious cycle that enhances the inflammatory response in a positive feedback manner (80, 81). Although the role of ferroptosis and associated inflammatory mediators in RA has been supported to some extent, further in-depth scientific studies are still necessary to fully understand this complex process (Figure 3).
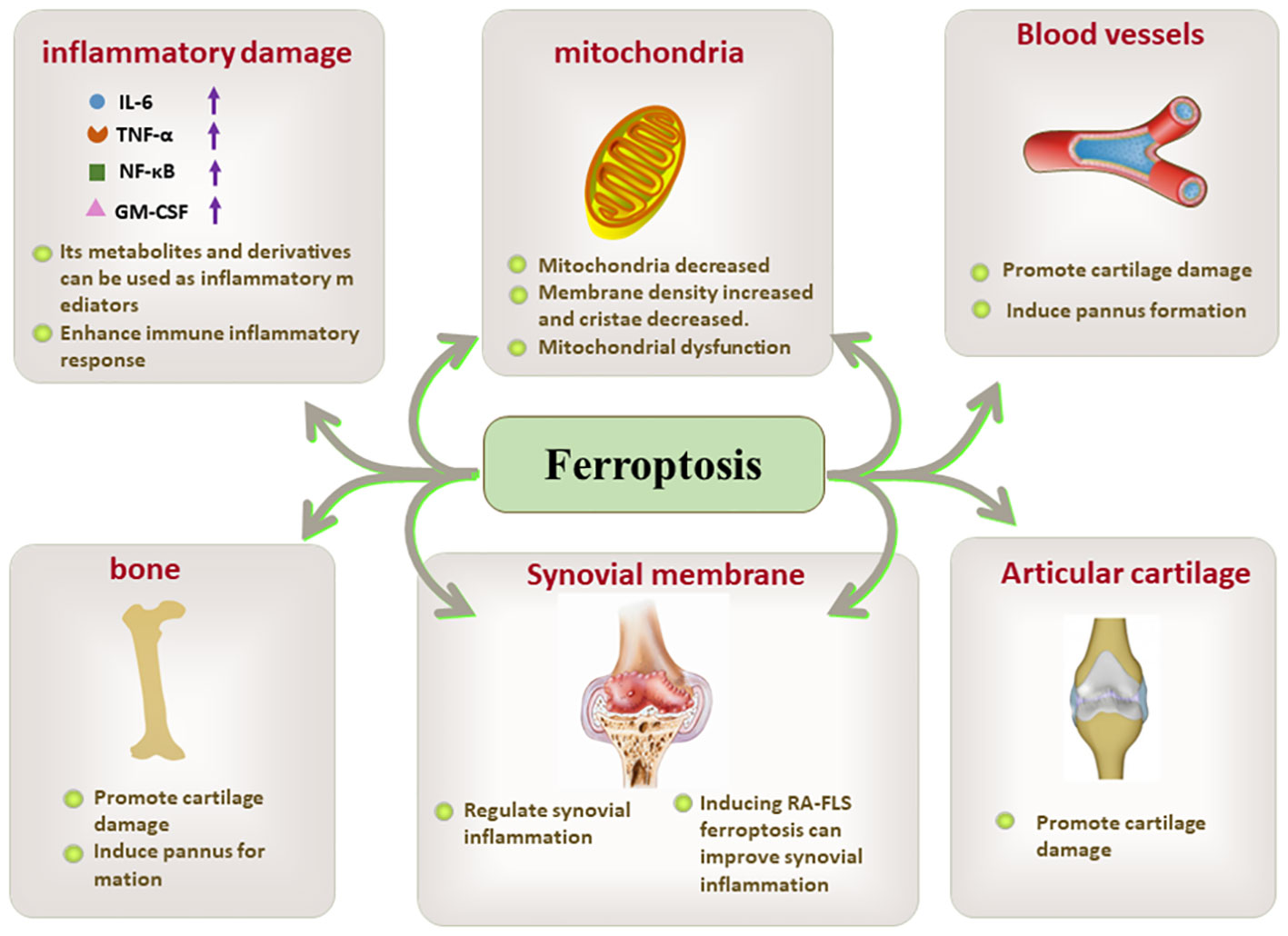
Figure 3 The key role of ferroptosis in inflammatory damage, mitochondria, blood vessels. bone, synovial membrane, and articular cartilage.
5.2 Ferroptosis regulates RA synovial inflammation
Synovial inflammation is a central event in the pathogenesis of RA, as well as a major trigger for bone and cartilage destruction and vascular opacification formation. Synovium is mainly composed of FLSs and immune cells. FLSs, the dominant non-immune cells of the synovium, develop abnormal proliferative capacity and aggressive characteristics under oxidative stress conditions (82, 83). Immune cells including macrophages, lymphocytes, and T cells play immune surveillance and inflammatory regulatory roles in the synovium. Activated T cells and macrophages in the synovium form an environment conducive to ferroptosis and inflammation, and also induce ROS production in a positive feedback manner, thus promoting the release of pro-inflammatory factors into the synovium and synovial fluid and further amplifying synovial inflammation (84). Moreover, compared with osteoarthritis, both RA synovial tissues and synovial fluids present ferroptosis-related characteristics such as lipid peroxidation and iron overload, as well as massive cascading proliferation and “tumor-like” growth of RA-FLSs (85) (Figure 4).
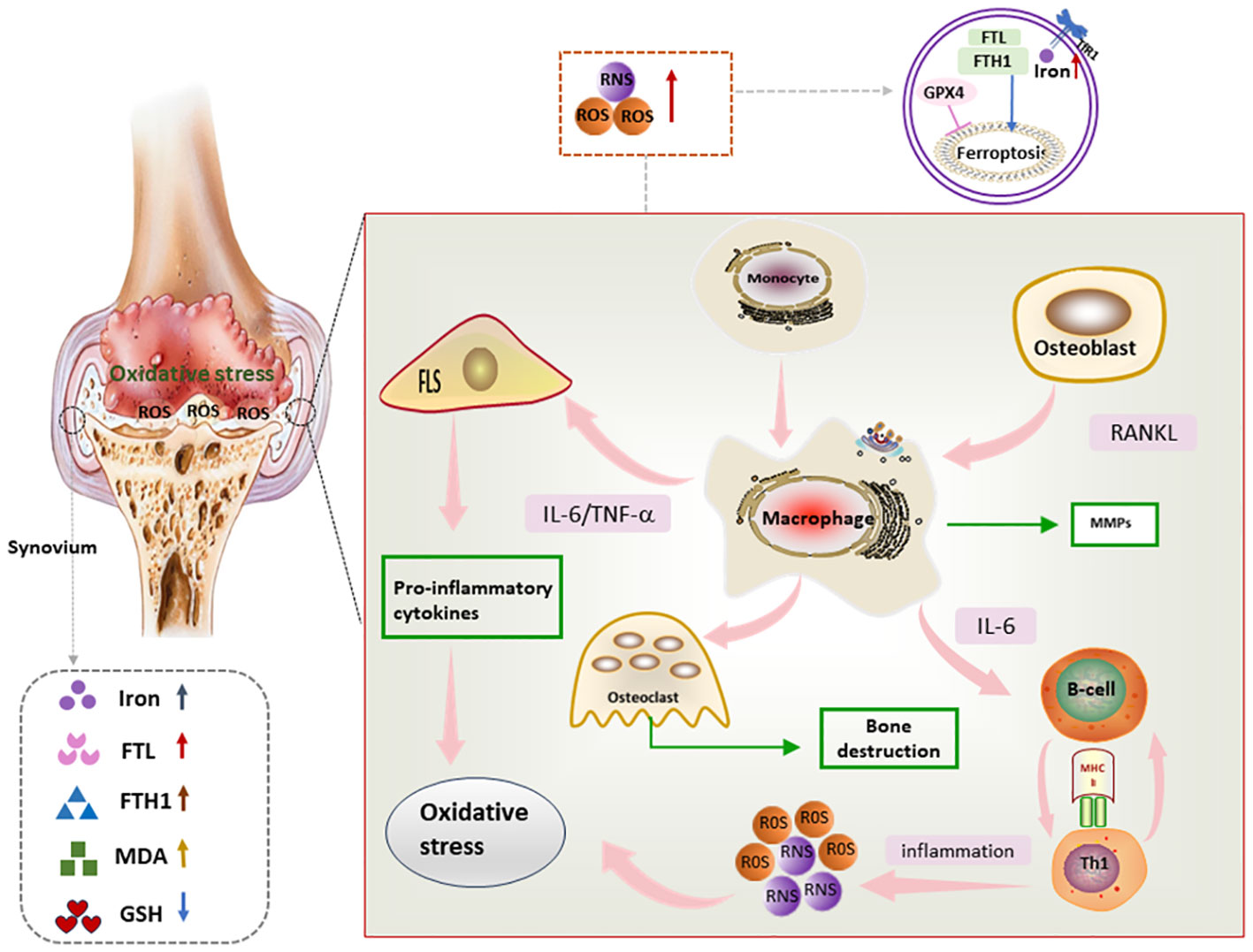
Figure 4 Mechanism of ferroptosis in RA synovium Lipid peroxides as well as iron deposition can be seen in RA synovium. In addition, proinflammatory factors secreted by immune cells and FLS in RA synovium promote oxidative stress, which in turn further exacerbates the inflammatory response, and through this vicious circle, ferroptosis and synovial inflammation are promoted, FLS, fibroblast-like synoviocyte; IL-6, interleukin 6; RNS, reactive nitrogen species; ROS, reactive oxygen species; TNF, tumor necrosis factor; RANKL, Receptor Activator for Nuclear Factor-kB Ligand; MMPs, matrix metalloproteinases.
FLSs are considered to be dominant drivers of RA pathogenesis. Fibroblast activating protein-α (FAPα) is a RA fibroblast marker located on the synovial cell surface. According to a previous study (85), the number of FAPα+ fibroblasts is significantly increased in the inflammatory synovium of collagen-induced arthritis (CIA) mice. The TNF antagonist etanercept can significantly enhance the killing effect of the ferroptosis inducer imidazole ketone erastin (IKE) on RA-FLSs, and the combination of the two restores synovial homeostasis by promoting FAPα+ fibroblasts ferroptosis and reducing the number of FAPα+ fibroblasts. Thus, inducing the ferroptosis of RA-FLSs may be a potential target for the treatment of RA. In contrast, treatment with the GPX4 inhibitor RSL3 is observed to specifically induce cell death in FAPα+ fibroblasts. Interestingly, RSL3 does not increase cell death in macrophages, endothelial cells, T cells, or B cells (85). The possible reasons for this may be as follows. Firstly, RA-FLSs proliferate abnormally in response to the elevation of ROS and lipid oxidation, and the GPX4 inhibitor RSL3 specifically scavenges ROS accumulation in the synovial membrane and downregulates the number of FLSs. Therefore, FLSs are sensitive to RSL3. Secondly, FAPa+ fibroblasts are almost undetectable in non-inflammatory conditions, while they significantly increase in inflamed synovial membranes, and the synovial inflammatory milieu tends to promote ferroptosis and lipid ROS production, and induction of ferroptosis is effective in reducing the number of FLSs and thereby improving RA. It can thus be concluded that GPX4 inhibitors have the potential to selectively affect the cell survival in specific cell types. However, further studies are still needed to comprehensively validate and understand the therapeutic potential of GPX4 inhibitors and their effects on different cell types. Not only that, future studies are expected to identify surface proteins more specific to fibroblasts, which will facilitate the development of fibroblast ferroptosis targeting therapy.
Glycolysis is an essential energy production and metabolic process, and its regulatory mechanism involves several signaling pathways such as Akt/mTOR/HIF-1α (86). Overexpression of SLC2A3 activates glycolysis and HIF-1 signaling pathways (87). In the meantime, the ferroptosis inducer RSL3 can trigger ferroptosis of RA-FLSs by reducing the expression of SLC2A3 and suppressing the glycolytic metabolism of RA-FLSs, which further reveals the intrinsic mechanism of RSL3-induced ferroptosis of RA-FLSs.
The nuclear factor E2-related factor 2 (Nrf2)-related anti-oxidative stress is strongly associated with ferroptosis suppression. Nrf2 activation can prevent oxidative stress and inflammatory damage in RA synovium by transcribing various antioxidant enzymes including superoxide dismutase (SOD), heme oxygenase-1 (HO-1), and GSH (88). It is discovered (89) that activation of Nrf2 not only regulates a series of inflammation-related signaling molecules but also inhibits the production of ROS and represses the proliferation and migration of RA-FLSs, thereby effectively alleviating RA synovial inflammation.
5.3 Ferroptosis exacerbates bone and cartilage damage in RA
In addition to synovial inflammation, progressive bone and cartilage destruction is another catastrophic pathological change in RA. The imbalance between bone resorption by osteoclasts and bone formation by osteoblasts is the major contributor to the development of bone damage in RA, and ferroptosis further exacerbates this process.
Osteoclasts are multinucleated giant cells formed by the fusion of monocyte/macrophage precursor cells. M-CSF and RANKL are crucial for osteoclast differentiation and development. A typical osteoclast phenotype on the surface of multinucleated giant cells can be observed at the bone-pannus joint interface in CIA rats, suggesting the involvement of osteoclasts in RA-associated bone destruction (90). Furthermore, cytokines such as IL-17 and IL-6 in RA arthritis induce RANKL expression in osteoblasts and RA-FLSs, indirectly stimulating osteoclast formation (91, 92). ROS, a principal feature of ferroptosis, also interacts with RNAKL and M-CSF to promote osteoclast differentiation. In addition, activation of the Janus kinase (JAK)2/STAT3 pathway effectively promotes RANKL expression and induces osteoclast differentiation, and ROS can activate the JAK2/STAT3 pathway by upregulating HIF-1α expression (49, 52). An interesting experiment shows (93) that iron overload induces osteoblast apoptosis and osteoclast differentiation, and that icariin can inhibit the function of osteoclasts by regulating ROS and mitochondrial membrane potential (MMP) homeostasis while attenuating iron overload-induced oxidative damage in osteoblasts.
As articular cartilage is an important structural and functional unit of the joint, cartilage degeneration and damage can cause impairment and loss of joint function in RA patients (94). On the one hand, IL-7 and TNF in RA can degrade cartilage by stimulating chondrocytes to secrete cartilage-degrading metalloproteinases (MMP). On the other hand, the enhanced sensitivity of chondrocytes to IL-1 and TNF in synovial fluid in turn accelerates cartilage degradation (95). Not merely that, knockdown of GPX4 is found to increase chondrocyte sensitivity to oxidative stress and also activate the MAPK/NF-κB pathway to promote extracellular matrix (ECM) degradation, while this process can be effectively mitigated by the use of Fer-1, a ferroptosis inhibitor (96). However, the research on ferroptosis and bone and cartilage damage in RA is still in its infancy and still needs more supporting evidence.
5.4 Effect of ferroptosis on osteoporosis
RA patients have both disease-specific risk factors for OP and fractures. OP is a common clinical disorder mainly characterized by bone mass loss and bone microarchitectural deterioration (97). During the course of RA, inflammatory factors induce osteoclast differentiation but inhibit osteoblast maturation. The aggravation of bone loss caused by this imbalance in bone metabolism is the main reason for RA complicated with OP (98, 99). Moreover, the long-term clinical use of mainstream drugs such as glucocorticoids and immunosuppressants can also induce the development of OP. Epidemiological surveys have shown (100) that approximately 60-80% of RA patients are complicated with OP.
Iron metabolism disorder is identified as an independent risk factor for OP. Iron overload can disrupt bone homeostasis by significant inhibition of osteoblast differentiation and stimulation of osteoclastogenesis, consequently leading to OP (101). In an iron overload rat model constructed by intraperitoneal injection of iron dextrose, iron overload causes thinning of bone trabeculae and cortex, and increase of bone resorption. Further findings (102) reveal that iron overload leads to the increase of MMP and the accumulation of lipid peroxide by affecting GSH and fatty acid cycle, which further promotes the activation of osteoclasts and apoptosis of osteoblasts, leading to increased bone resorption and decreased bone formation.
Furthermore, the hypoxic microenvironment of RA is known to inhibit RANKL-induced ferritin phagocytosis and protect osteoclasts from ferroptosis. Conversely, the HIF-1α inhibitor 2ME2 effectively promotes osteoclast ferroptosis and prevents ovariectomy-induced OP in rats (52). Not only that, targeting osteoblast ferroptosis is also a potential therapeutic strategy for OP. One study has found (103) that the expression of NADPH oxidase 4 (NOX4) is elevated in OP mice and patients, and NOX4 overexpression drives osteoblast ferroptosis and subsequently causes bone loss in OP mice.
5.5 Ferroptosis modulates RA pannus formation and angiogenesis
Pannus formation in the synovial cavity is a major pathological factor in RA and can cause irreversible damage to the joint and cartilage. Synovial pannus is composed of neoplastic microvessels, proliferating hypertrophic synovial cells, inflammatory cells, and mechanized fibrin, exhibiting tumor-like tissue properties (104). Angiogenesis is an early event in the pathogenesis of RA, which is crucial for the proliferation of synovial tissue and the formation of pannus (105, 106).
RA-FLSs exhibit tumor-like biological characteristics that facilitate pannus formation. On the one hand, RA-FLSs secrete reactive substances, such as IL-6, MMP, and TNF-α, which not only aggravate the process of bone erosion but also exacerbate the development of pannus (107). On the other hand, FLSs produce MMP to digest a variety of proteins in cartilage and supporting structures, further promoting the invasion and expansion of pannus (108). In addition, VEGF secreted by RA-FLSs is an important factor in promoting angiogenesis (109, 110). Thus, it is evident that inducing ferroptosis of RA-FLSs not only regulates synovial homeostasis but also has important implications for the inhibition of angiogenesis.
An appropriate ROS concentration is conducive to normal angiogenesis and vascular homeostasis, while a high concentration of ROS chronically induces ferroptosis and consequently causes pathological damage to blood vessels. However, it must be emphasized that ROS as a trigger for RA and FLS proliferation is not an inevitable consequence. A recent study (111) has indicated the involvement of ROS in VEGF-dependent signaling processes in endothelial cells. Specifically, ROS transcribes VEGF genes by activating the NF-κB pathway to participate in key pathological processes such as angiogenesis and proliferation. In addition, ROS also functions in other cell types, such as macrophages, fibroblasts, endothelial cells, and keratin-forming cells. These cells produce VEGF in response to ROS stimulation and regulate angiogenesis through upstream and downstream effects of VEGF/VEGFR2 signaling (112, 113).
The Notch signaling pathway responds to the regulation of ROS and is closely associated with RA angiogenesis (53). Its mechanism may be related to the induction of angiogenesis and endothelial cell migration by activating FLSs. Due to insufficient research results, it is necessary to further explore the relationship between ferroptosis and RA angiogenesis, which may help to reveal new mechanisms underlying the pathological process of RA and provide potential targets for the development of new therapeutic strategies.
6 Ferroptosis may be a key target in controlling RA
Compelling evidence supports the potential of ferroptosis as a prospective target for the prevention and treatment of RA (114–116): using ferroptosis inducers to induce ferroptosis of RA-FLSs or using ferroptosis inhibitors to reduce inflammation and joint damage. Searching for ferroptosis inhibitors or inducers has become a hot research topic in recent years, but due to the intricate regulatory mechanisms of ferroptosis and individual differences of patients, the targeted therapy of ferroptosis still stays in the preliminary stage.
Consistent with previous findings that promoting ferroptosis of RA-FLSs improves RA, the results of a recent study also indicate (117) that glycine drives ferroptosis of RA-FLSs through methylation of the S-adenosylmethionine (SAM)-associated GPX4 promoter and further enhances this effect by reducing FTH1 expression. This study provides important insights into the molecular mechanisms of ferroptosis in RA and the development of new therapeutic strategies. However, further studies are still needed to validate and extend these results.
Another study has also found (118) that the bioactive peptide G1dP3 promotes RA-FLS ferroptosis via a p53/SLC7A11 axis-dependent manner and has a potential therapeutic role in RA. It is speculated that somatic mutations of p53 in RA-FLSs may be the direct cause of synovial hyperplasia and subsequent pannus formation (119). p53 is one of the most commonly mutated tumor suppressor genes, and p53 expression is elevated in activated FLSs (120). Studies have confirmed (121, 122) that p53 can induce ferroptosis not only by inhibiting SLC7A11 activity but also by increasing CDKN1A expression or decreasing DPP4 activity, suggesting the dual regulatory role of p53 in the regulation of ferroptosis. In addition, ferroptosis induced by salazosulfapyridine likewise exerts dual effects (123). On the one hand, salazosulfapyridine inhibits ferroptosis by inhibiting systemic Xc- and downregulating GSH and GPX4. On the other hand, salazosulfapyridine induces the Fenton reaction by upregulating the ferric ion levels, thus generating excessive lipid ROS and inducing ferroptosis.
Antioxidants also have a regulatory effect on ferroptosis. ROS promotes Th17 differentiation and IL-17 production through activation of the RORγt and STAT3 pathways. CoQ10, a ROS scavenger and anti-inflammatory substance, has been shown to regulate Th17 and IL-17 through the STAT3 pathway, thereby inhibiting ferroptosis and ameliorating joint inflammation in CIA mice (124). In addition, the antioxidant and anti-inflammatory effects of natural extract polyphenols (like resveratrol, quercetin, rutin, curcumin, etc.) have been widely reported (125). Sirtuin 1 (SIRT1) prevents inflammatory responses in articular chondrocytes, and resveratrol, a natural SIRT1 activator, can exert a protective effect on chondrocytes by inhibiting NF-κB and MMP-13 expression (126). Green tea polyphenol EGCG has been shown to activate the antioxidant defense system in chondrocytes by inhibiting ERK and p12 MAPK activation while blocking pro-inflammatory signaling pathways, thereby alleviating RA (127). Xanthine oxidase (XO) is an essential generator of free radicals, and quercetin has been proven to stifle XO and prevent oxidative damage. In addition, one study reports that icariin can inhibit ferroptosis of synovial cells and exert protective effects by activating the Xc-/GPX4 axis (128).
As described in a previous study (129), TRPM7-mediated ferroptosis of chondrocytes is likely to be a promising target for the prevention and treatment of RA. TRPM7 is highly expressed in the articular cartilage of adjuvant arthritis rats and knockdown of TRPM7 alleviates articular cartilage destruction in RA (130). Further, inhibition of TRPM7 activity attenuates RA articular cartilage destruction and chondrocyte ferroptosis by suppressing the PKCα-NOX4 axis (129). Moreover, another study has discovered (131) that glycyrrhizin, a natural flavonoid extracted from Ural licorice root, can effectively ameliorate RA inflammation by blocking the MAPK signaling and inhibiting angiogenesis.
Despite the promising results on ferroptosis modulators revealed by preclinical studies, the effects of sustained ferroptosis modulation on normal tissues and organs remain unresolved. Therefore, it is essential to conduct comprehensive toxicity studies and monitor potential off-target effects to ensure the safety of ferroptosis modulation in RA treatment.
7 Discussion
Ferroptosis can be utilized as an interventional target for RA treatment. However, the therapeutic effects of ferroptosis regulation may vary from tissue to site since ferroptosis is an extremely complex process. For instance, although ferroptosis inducers can effectively suppress RA synovial inflammation by inducing FLSs ferroptosis, the use of ferroptosis inducers may also exacerbate inflammatory responses given the pro-inflammatory nature of ferroptosis, thus promoting bone erosion and bone destruction in RA, and further inducing OP development and angiogenesis. Specifically, two issues need to be considered for the targeted therapy of ferroptosis in RA. One is the off-target effect of non-FLS cell types, as ferroptosis induction may affect other cell populations in the synovium. Another consideration is the delicate balance between promoting ferroptosis of target cells and minimizing excessive cell death, which may lead to tissue damage and healing impairment. Therefore, the underlying mechanisms of ferroptosis events and autoimmune diseases still need more scientific exploration, and further studies are required to optimize the specificity and efficacy of ferroptosis-targeted drugs in RA.
Except for ferroptosis, there are other cell death modalities in RA, such as apoptosis, pyroptosis, autophagy, and necrosis, which coexist in a mixed form and play a synergistic role in the pathological progression of RA. The interactions and regulatory dynamics among these cell death pathways are not fully understood. Further research on the interactions and specific functions of these cell death pathways in RA contributes to a deeper understanding of the pathological mechanisms and targeted therapeutic strategies of RA. In addition, the current understanding remains limited regarding the cellular mechanisms of different cell death isoforms to maintain the stability of the internal environment, respond to external stresses, and defend against pathogenic threats. There are a lot of unknown aspects in this field that require further in-depth studies.
Furthermore, no consensus has been reached regarding the potential deleterious or protective effects of ferroptosis in RA. A key question that remains to be addressed is the identification of the optimal window for modulating ferroptosis and achieving therapeutic balance. In particular, the optimal timing and duration of ferroptosis induction in RA-FLSs are unclear. How to select and when to apply the best inducer or inhibitor applicable to ferroptosis in RA also demands in-depth studies. Another critical issue is the lack of specific biomarkers for monitoring ferroptosis activity in RA. The development of reliable biomarkers not only contributes to patient stratification and treatment response monitoring, but also helps to assess the efficacy of iron ferroptosis-targeted therapies in clinical trials.
In conclusion, ferroptosis in RA represents an exciting research field with important research and application prospects. in-depth research on the regulatory mechanisms of ferroptosis and the role of ferroptosis in RA is expected to lead to the development of more precise and efficient ferroptosis-targeted therapeutic strategies for RA.
Author contributions
HuZ: Writing – original draft. CT: Writing – review & editing. MW: Writing – review & editing. HoZ: Writing – original draft. YZ: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (No 81403484); Anhui Province University Natural Science Fund Project of China (No KJ2019A0448); National Project Cultivation Fund Project Plan (No. 2019py01) Anhui Province Clinical Medical Research Center [Anhui Provincial Science and Technology Department Anhui Social Science (2020) No. 41]; Anhui Province Traditional Chinese Medicine Inheritance Innovation Scientific Research Project (No 2020ccyb13); The training Program of Outstanding talents in Colleges and Universities (No. gxgnfx2021122); Anhui Province Medical and Health Key Specialist Construction Project (Anhui Health Letter [2021] No.273).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis-immune cell-fibroblast-bone interactions. Nat Rev Rheumatol (2022) 18(7):415–29. doi: 10.1038/s41584-022-00793-5
2. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388(10055):2023–38. doi: 10.1016/S0140-6736(16)30173-8
3. Osipova D, Janssen R, Martens HA. Rheumatoid arthritis: more than a joint disease. Ned Tijdschr Geneeskd (2020) 164:D4166.
4. Scublinsky D, Gonzalez CD. Quantifying disease in challenging conditions: incidence and prevalence of rheumatoid arthritis. J Rheumatol (2016) 43(7):1263–4. doi: 10.3899/jrheum.160522
5. Radu AF, Bungau SG. Management of rheumatoid arthritis: an overview. Cells (2021) 10(11):2857. doi: 10.3390/cells10112857
6. Singh JA. Treatment guidelines in rheumatoid arthritis. Rheum Dis Clin North Am (2022) 48(3):679–89. doi: 10.1016/j.rdc.2022.03.005
7. Baker JF, Ghio AJ. Iron homoeostasis in rheumatic disease. Rheumatol (Oxford) (2009) 48(11):1339–44. doi: 10.1093/rheumatology/kep221
8. Telfer JF, Brock JH. Proinflammatory cytokines increase iron uptake into human monocytes and synovial fibroblasts from patients with rheumatoid arthritis. Med Sci Monit (2004) 10(4):BR91–5. doi: 10.1051/medsci/2004204487
9. Xie Z, Hou H, Luo D, An R, Zhao Y, Qiu C. ROS-dependent lipid peroxidation and reliant antioxidant ferroptosis-suppressor-protein 1 in rheumatoid arthritis: a covert clue for potential therapy. Inflammation (2021) 44(1):35–47. doi: 10.1007/s10753-020-01338-2
10. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell (2022) 185(14):2401–21. doi: 10.1016/j.cell.2022.06.003
11. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
12. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447(7146):864–8. doi: 10.1038/nature05859
13. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
14. Stockwell BR, Friedmann AJ, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
15. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U.S.A. (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
16. Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med (2019) 23(8):4900–12. doi: 10.1111/jcmm.14511
17. Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol (2020) 8:586578. doi: 10.3389/fcell.2020.586578
18. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPX4. Free Radic Biol Med (2020) 152:175–85. doi: 10.1016/j.freeradbiomed
19. Lelièvre P, Sancey L, Coll JL, Deniaud A, Busser B. Iron dysregulation in human cancer: altered metabolism, biomarkers for diagnosis, prognosis, monitoring and rationale for therapy. Cancers (Basel) (2020) 12(12):3524. doi: 10.3390/cancers12123524
20. Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry (2012) 51(29):5705–24. doi: 10.1021/bi300752r
21. Lawen A, Lane DJ. Mammalian iron homeostasis in health and disease: uptake, storage, transport, and molecular mechanisms of action. Antioxid Redox Signal (2013) 18(18):2473–507. doi: 10.1089/ars.2011.4271
22. Aversa I, Chirillo R, Chiarella E, Zolea F, Di Sanzo M, Biamonte F, et al. Chemoresistance in H-ferritin silenced cells: the role of NF-kappaB. Int J Mol Sci (2018) 19(10):2969. doi: 10.3390/ijms19102969
23. Biamonte F, Battaglia AM, Zolea F, Oliveira DM, Aversa I, Santamaria G, et al. Ferritin heavy subunit enhances apoptosis of non-small cell lung cancer cells through modulation of miR-125b/p53 axis. Cell Death Dis (2018) 9(12):1174. doi: 10.1038/s41419-018-1216-3
24. Salatino A, Aversa I, Battaglia AM, Sacco A, Di Vito A, Santamaria G. H-ferritin affects cisplatin-induced cytotoxicity in ovarian cancer cells through the modulation of ROS. Oxid Med Cell Longev (2019) 2019:3461251. doi: 10.1155/2019/3461251
25. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 509(7498):105–9. doi: 10.1038/nature13148
26. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
27. Cui J, Wang Y, Tian X, Miao Y, Ma L, Zhang C. LPCAT3 is transcriptionally regulated by YAP/ZEB/EP300 and collaborates with ACSL4 and YAP to determine ferroptosis sensitivity. Antioxid Redox Signal (2023) 39(7-9):491–511. doi: 10.1089/ars.2023.0237
28. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
29. Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis (2017) 8(3):e2716. doi: 10.1038/cddis.2017.133
30. Lin W, Wang C, Liu G, Bi C, Wang X, Zhou Q, et al. SLC7A11/xCT in cancer: biological functions and therapeutic implications. Am J Cancer Res (2020) 10(10):3106–26.
31. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal (2018) 29(1):61–74. doi: 10.1089/ars.2017.7115
32. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
33. Kraft V, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci (2020) 6(1):41–53. doi: 10.1021/acscentsci.9b01063
34. Ahmadzadeh N, Shingu M, Nobunaga M. Iron-binding proteins and free iron in synovial fluids of rheumatoid arthritis patients. Clin Rheumatol (1989) 8(3):345–51. doi: 10.1007/BF02030347
35. Yazar M, Sarban S, Kocyigit A, Isikan UE. Synovial fluid and plasma selenium, copper, zinc, and iron concentrations in patients with rheumatoid arthritis and osteoarthritis. Biol Trace Elem Res (2005) 106(2):123–32. doi: 10.1385/BTER:106:2:123
36. Telfer JF, Brock JH. Expression of ferritin, transferrin receptor, and non-specific resistance associated macrophage proteins 1 and 2 (Nramp1 and Nramp2) in the human rheumatoid synovium. Ann Rheum Dis (2002) 61(8):741–4. doi: 10.1136/ard.61.8.741
37. Dabbagh AJ, Blake DR, Morris CJ. Effect of iron complexes on adjuvant arthritis in rats. Ann Rheum Dis (1992) 51(4):516–21. doi: 10.1136/ard.51.4.516
38. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood (2003) 102(3):783–8. doi: 10.1182/blood-2003-03-0672
39. Ni S, Yuan Y, Kuang Y, Li X. Iron metabolism and immune regulation. Front Immunol (2022) 13:816282. doi: 10.3389/fimmu.2022.816282
40. Walker EJ, Walker SM. Effects of iron overload on the immune system. Ann Clin Lab Sci (2000) 30(4):354–65. doi: 10.1007/BF00314208
41. Ba T, Zhao D, Chen Y, Zeng C, Zhang C, Niu S. L-citrulline supplementation restrains ferritinophagy-mediated ferroptosis to alleviate iron overload-induced thymus oxidative damage and immune dysfunction. Nutrients (2022) 14(21):4549. doi: 10.3390/nu14214549
42. Mateen S, Moin S, Khan AQ, Zafar A, Fatima N. Increased reactive oxygen species formation and oxidative stress in rheumatoid arthritis. PloS One (2016) 11(4):e0152925. doi: 10.1371/journal.pone.0152925
43. Zamudio-Cuevas Y, Martínez-Flores K, Martínez-Nava GA, Clavijo-Cornejo D, Fernández-Torres J, Sánchez-Sánchez R. Rheumatoid arthritis and oxidative stress. Cell Mol Biol (Noisy-le-grand) (2022) 68(6):174–84. doi: 10.14715/cmb/2022.68.6.28
44. Jing W, Liu C, Su C, Liu L, Chen P, Li X. Role of reactive oxygen species and mitochondrial damage in rheumatoid arthritis and targeted drugs. Front Immunol (2023) 14:1107670. doi: 10.3389/fimmu.2023.1107670
45. Fonseca LJS, Nunes-Souza V, Goulart MOF, Rabelo LA. Oxidative stress in rheumatoid arthritis: what the future might hold regarding novel biomarkers and add-on therapies. Oxid Med Cell Longev (2019) 2019:7536805. doi: 10.1155/2019/7536805
46. Veselinovic M, Barudzic N, Vuletic M, Zivkovic V, Tomic-Lucic A, Djuric D. Oxidative stress in rheumatoid arthritis patients: relationship to diseases activity. Mol Cell Biochem (2014) 391(1-2):225–32. doi: 10.1007/s11010-014-2006-6
47. Garcia-Gonzalez A, Gaxiola-Robles R, Zenteno-Savin T. Oxidative stress in patients with rheumatoid arthritis. Rev Invest Clin (2015) 67(1):46–53. doi: 10.3310/hta19410
48. Huang QQ, Doyle R, Chen SY, Sheng Q, Misharin AV, Mao Q, et al. Critical role of synovial tissue-resident macrophage niche in joint homeostasis and suppression of chronic inflammation. Sci Adv (2021) 7(2):eabd0515. doi: 10.1126/sciadv.abd0515
49. Danks L, Komatsu N, Guerrini MM, Sawa S, Armaka M, Kollias G, et al. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann Rheum Dis (2016) 75(6):1187–95. doi: 10.1136/annrheumdis-2014-207137
50. Feng LJ, Jiang TC, Zhou CY, Yu CL, Shen YJ, Li J, et al. Activated macrophage-like synoviocytes are resistant to endoplasmic reticulum stress-induced apoptosis in antigen-induced arthritis. Inflammation Res (2014) 63(5):335–46. doi: 10.1007/s00011-013-0705-1
51. Masoumi M, Bashiri H, Khorramdelazad H, Barzaman K, Hashemi N, Sereshki HA, et al. Destructive roles of fibroblast-like synoviocytes in chronic inflammation and joint damage in rheumatoid arthritis. Inflammation (2021) 44(2):466–79. doi: 10.1007/s10753-020-01371-1
52. Zhu J, Tang Y, Wu Q, Ji YC, Feng ZF, Kang FW. HIF-1 alpha facilitates osteocyte-mediated osteoclastogenesis by activating JAK2/STAT3 pathway in vitro. J Cell Physiol (2019) 234(11):21182–92. doi: 10.1002/jcp.28721
53. Gao W, Sweeney C, Connolly M, Kennedy A, Ng CT, McCormick J. Notch-1 mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum (2012) 64(7):2104–13. doi: 10.1002/art.34397
54. Mukhopadhyay K, De S, Kundu S, Ghosh P, Chatterjee S, Chatterjee M. Evaluation of levels of oxidative stress asa potential biomarker in patients with rheumatoid arthritis. J Family Med Prim Care (2021) 10(5):1981–6. doi: 10.4103/jfmpc.jfmpc_2412_20
55. Quiñonez-Flores CM, González-Chávez SA, Del Río Nájera D, Pacheco-Tena C. Oxidative stress relevance in the pathogenesis of the rheumatoid arthritis: A systematic review. BioMed Res Int (2016) 2016:6097417. doi: 10.1155/2016/6097417
56. Hassan SZ, Gheita TA, Kenawy SA, Fahim AT, El-Sorougy IM, Abdou MS. Oxidative stress in systemic lupus erythematosus and rheumatoid arthritis patients: relationship to disease manifestations and activity. Int J Rheum Dis (2011) 14(4):325–31. doi: 10.1111/j.1756-185X.2011.01630.x
57. Bjorklund G, Shanaida M, Lysiuk R, Antonyak H, Klishch I, Shanaida V, et al. Selenium: an antioxidant with a critical role in anti-aging. Molecules (2022) 27(19):6613. doi: 10.3390/molecules2719661
58. Rosenbaum CC, O’Mathúna DP, Chavez M, Shields K. Antioxidants and antiinflammatory dietary supplements for osteoarthritis and rheumatoid arthritis. Altern Ther Health Med (2010) 16(2):32–40.
59. Zhou X, Mi J, Liu Z. Causal association of diet-derived circulating antioxidants with the risk of rheumatoid arthritis: A Mendelian randomization study. Semin Arthritis Rheum (2022) 56:152079. doi: 10.1016/j.semarthrit.2022.152079
60. Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med (2021) 218(6):e20210518. doi: 10.1084/jem.20210518
61. Handa P, Thomas S, Morgan-Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol (2019) 105(5):1015–26. doi: 10.1002/JLB.3A0318-108R
62. Jung HS, Shimizu-Albergine M, Shen X, Kramer F, Shao D, Vivekanandan-Giri A, et al. TNF-alpha induces acyl-CoA synthetase 3 to promote lipid droplet formation in human endothelial cells. J Lipid Res (2020) 61(1):33–44. doi: 10.1194/jlr.RA119000256
63. Yang Y, Wang Y, Guo L, Gao W, Tang TL, Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis (2022) 13(4):355. doi: 10.1038/s41419-022-04775-z
64. Soe-Lin S, Sheftel AD, Wasyluk B, Ponka P. Nramp1 equips macrophages for efficient iron recycling. Exp Hematol (2008) 36(8):929–37. doi: 10.1016/j.exphem
65. Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Body iron metabolism and pathophysiology of iron overload. Int J Hematol (2008) 88(1):7–15. doi: 10.1007/s12185-008-0120-5
66. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol (2020) 11:584116. doi: 10.1101/2020.07.16.20155291
67. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol (2021) 22(9):1107–17. doi: 10.1038/s41590-021-00993-3
68. Kaushal J, Kamboj A, Anupam K, Tandon A, Sharma A, Bhatnagar A. Interplay of redox imbalance with matrix gelatinases in neutrophils and their association with disease severity in rheumatoid arthritis patients. Clin Immunol (2022) 237:108965. doi: 10.1016/j.clim.2022.108965
69. Duvvuri B, Baddour AA, Deane KD, Feser ML, Nelson JL, Demoruelle MK, et al. Mitochondrial N-formyl methionine peptides associate with disease activity as well as contribute to neutrophil activation in patients with rheumatoid arthritis. J Autoimmun (2021) 119:102630. doi: 10.1016/j.jaut.2021.102630
70. Levring TB, Hansen AK, Nielsen BL, Kongsbak M, von Essen MR, Woetmann A, et al. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci Rep (2012) 2:266. doi: 10.1038/srep00266
71. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8(+) T cells. Cancer Immunol Res (2021) 9(2):184–99. doi: 10.1158/2326-6066.CIR-20-0384
72. Weyand CM, Wu B, Goronzy JJ. The metabolic signature of T cells in rheumatoid arthritis. Curr Opin Rheumatol (2020) 32(2):159–67. doi: 10.1097/BOR.0000000000000683
73. Zaiss MM, Joyce Wu HJ, Mauro D, Schett G, Ciccia F. The gut-joint axis in rheumatoid arthritis. Nat Rev Rheumatol (2021) 17(4):224–37. doi: 10.1038/s41584-021-00585-3
74. Seyoum Y, Baye K, Humblot C. Iron homeostasis in host and gut bacteria - a complex interrelationship. Gut Microbes (2021) 13(1):1–19. doi: 10.1080/19490976.2021.1874855
75. Kell DB, Pretorius E. No effects without causes: the Iron Dysregulation and Dormant Microbes hypothesis for chronic, inflammatory diseases. Biol Rev Camb Philos Soc (2018) 93(3):1518–57. doi: 10.1111/brv.12407
76. Liu S, Gao Z, He W, Wu Y, Liu J, Zhang S, et al. The gut microbiota metabolite glycochenodeoxycholate activates TFR-ACSL4-mediated ferroptosis to promote the development of environmental toxin-linked MAFLD. Free Radic Biol Med (2022) 193(Pt 1):213–26. doi: 10.1016/j.freeradbiomed.2022.10.270
77. Mao H, Zhao Y, Li H, Lei L. Ferroptosis as an emerging target in inflammatory diseases. Prog Biophys Mol Biol (2020) 155:20–8. doi: 10.1016/j.pbiomolbio.2020.04.001
78. Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem (2015) 30(1):11–26. doi: 10.1007/s12291-014-0446-0
79. Ajabnoor SM, Thorpe G, Abdelhamid A, Hooper L. Long-term effects of increasing omega-3, omega-6 and total polyunsaturated fats on inflammatory bowel disease and markers of inflammation: a systematic review and meta-analysis of randomized controlled trials. Eur J Nutr (2021) 60(5):2293–316. doi: 10.1007/s00394-020-02413-y
80. Raphael W, Sordillo LM. Dietary polyunsaturated fatty acids and inflammation: the role of phospholipid biosynthesis. Int J Mol Sci (2013) 14(10):21167–88. doi: 10.3390/ijms141021167
81. Deng L, He S, Guo N, Tian W, Zhang W, Luo L. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflammation Res (2023) 72(2):281–99. doi: 10.1007/s00011-022-01672-1
82. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233(1):233–55. doi: 10.1111/j.0105-2896.2009.00859.x
83. Tsaltskan V, Firestein GS. Targeting fibroblast-like synoviocytes in rheumatoid arthritis. Curr Opin Pharmacol (2022) 67:102304. doi: 10.1016/j.coph.2022.102304
84. Hitchon CA, El-Gabalawy HS. Oxidation in rheumatoid arthritis. Arthritis Res Ther (2004) 6(6):265–78. doi: 10.1186/ar1447
85. Wu J, Feng Z, Chen L, Li Y, Bian H, Geng J, et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat Commun (2022) 13(1):676. doi: 10.1038/s41467-021-27948-4
86. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
87. Xiang J, Chen H, Lin Z, Chen J, Luo L. Identification and experimental validation of ferroptosis-related gene SLC2A3 is involved in rheumatoid arthritis. Eur J Pharmacol (2023) 943:175568. doi: 10.1016/j.ejphar.2023.175568
88. Chadha S, Behl T, Kumar A, Khullar G, Arora S. Role of Nrf2 in rheumatoid arthritis. Curr Res Transl Med (2020) 68(4):171–81. doi: 10.1016/j.retram.2020.05.002
89. Zhang Y, Wang G, Wang T, Cao W, Zhang L, Chen X. Nrf2-Keap1 pathway-mediated effects of resveratrol on oxidative stress and apoptosis in hydrogen peroxide-treated rheumatoid arthritis fibroblast-like synoviocytes. Ann N Y Acad Sci (2019) 1457(1):166–78. doi: 10.1111/nyas.14196
90. Suzuki Y, Nishikaku F, Nakatuka M, Koga Y. Osteoclast-like cells in murine collagen induced arthritis. J Rheumatol (1998) 25(6):1154–60. doi: 10.1097/00124743-199806000-00017
91. Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatol (Oxford) (2008) 47(11):1635–40. doi: 10.1093/rheumatology/ken363
92. Tanaka S, Tanaka Y. RANKL as a therapeutic target of rheumatoid arthritis. J Bone Miner Metab (2021) 39(1):106–12. doi: 10.1007/s00774-020-01159-1
93. Jing X, Du T, Chen K, Guo J, Xiang W, Yao X. Icariin protects against iron overload-induced bone loss via suppressing oxidative stress. J Cell Physiol (2019) 234(7):10123–37. doi: 10.1002/jcp.27678
94. Komatsu N, Takayanagi H. Bone and cartilage destruction in rheumatoid arthritis. Clin Calcium (2012) 22(2):179–85.
95. Ostrowska M, Maśliński W, Prochorec-Sobieszek M, Nieciecki M, Sudoł-Szopińska I. Cartilage and bone damage in rheumatoid arthritis. Reumatologia (2018) 56(2):111–20. doi: 10.5114/reum.2018.75523
96. Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao J, et al. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine (2022) 76:103847. doi: 10.1016/j.ebiom.2022.103847
97. Wysham KD, Baker JF, Shoback DM. Osteoporosis and fractures in rheumatoid arthritis. Curr Opin Rheumatol (2021) 33(3):270–6. doi: 10.1097/BOR.0000000000000789
98. Matuszewska A, Szechinski J. Mechanisms of osteoporosis development in patients with rheumatoid arthritis. Postepy Hig Med Dosw (Online) (2014) 68:145–52. doi: 10.5604/17322693.1088339
99. Baker R, Narla R, Baker JF, Wysham KD. Risk factors for osteoporosis and fractures in rheumatoid arthritis. Best Pract Res Clin Rheumatol (2022) 36(3):101773. doi: 10.1016/j.berh.2022.101773
100. Lodder MC, de Jong Z, Kostense PJ, Molenaar ET, Staal K, Voskuyl AE, et al. Bone mineral density in patients with rheumatoid arthritis: relation between disease severity and low bone mineral density. Ann Rheum Dis (2004) 63(12):1576–80. doi: 10.1136/ard.2003.016253
101. Tsay J, Yang Z, Ross FP, Cunningham-Rundles S, Lin H, Coleman R, et al. Bone loss caused by iron overload in a murine model: importance of oxidative stress. Blood (2010) 116(14):2582–9. doi: 10.1182/blood-2009-12-260083
102. Liu X, Wang T, Wang W, Liang X, Mu Y, Xu Y, et al. Emerging potential therapeutic targets of ferroptosis in skeletal diseases. Oxid Med Cell Longev (2022) 2022:3112388. doi: 10.1155/2022/3112388
103. Zhang H, Wang A, Li G, Zhai Q, Huang Z, Wang X, et al. Osteoporotic bone loss from excess iron accumulation is driven by NOX4-triggered ferroptosis in osteoblasts. Free Radic Biol Med (2023) 198:123–36. doi: 10.1016/j.freeradbiomed.2023.01.026
104. Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D. Angiogenesis in rheumatoid arthritis. Histol Histopathol (2006) 21(5):557–66. doi: 10.14670/HH-21.557
105. Elshabrawy HA, Chen Z, Volin MV, Ravella S, Virupannavar S, Shahrara S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis (2015) 18(4):433–48. doi: 10.1007/s10456-015-9477-2
106. Avouac J, Pezet S, Vandebeuque E, Orvain C, Gonzalez V, Marin G, et al. Semaphorins: from angiogenesis to inflammation in rheumatoid arthritis. Arthritis Rheumatol (2021) 73(9):1579–88. doi: 10.1002/art.41701
107. Ji M, Ryu HJ, Baek HM, Shin DM, Hong JH. Dynamic synovial fibroblasts are modulated by NBCn1 as a potential target in rheumatoid arthritis. Exp Mol Med (2022) 54(4):503–17. doi: 10.1038/s12276-022-00756-6
108. Hattori Y, Kida D, Kaneko A. Normal serum matrix metalloproteinase-3 levels can be used to predict clinical remission and normal physical function in patients with rheumatoid arthritis. Clin Rheumatol (2019) 38(1):181–7. doi: 10.1007/s10067-017-3829-9
109. Micheroli R, Elhai M, Edalat S, Frank-Bertoncelj M, Bürki K, Ciurea A, et al. Role of synovial fibroblast subsets across synovial pathotypes in rheumatoid arthritis: a deconvolution analysis. RMD Open (2022) 8(1):e001949. doi: 10.1136/rmdopen-2021-001949
110. Long L, Guo H, Chen X, Liu Y, Wang R, Zheng X, et al. Advancement in understanding the role of ferroptosis in rheumatoid arthritis. Front Physiol (2022) 13:1036515. doi: 10.3389/fphys.2022.1036515
111. Huang YJ, Nan GX. Oxidative stress-induced angiogenesis. J Clin Neurosci (2019) 63:13–6. doi: 10.1016/j.jocn.2019.02.019
112. Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood (2014) 123(5):625–31. doi: 10.1182/blood-2013-09-512749
113. Hopf HW, Gibson JJ, Angeles AP, Constant JS, Feng JJ, Rollins MD, et al. Hyperoxia and angiogenesis. Wound Repair Regener (2005) 13(6):558–64. doi: 10.1111/j.1524-475X.2005.00078.x
114. Zhao T, Yang Q, Xi Y, Xie Z, Shen J, Li Z, et al. Ferroptosis in rheumatoid arthritis: A potential therapeutic strategy. Front Immunol (2022) 13:779585. doi: 10.3389/fimmu.2022.779585
115. Chang S, Tang M, Zhang B, Xiang D, Li F. Ferroptosis in inflammatory arthritis: A promising future. Front Immunol (2022) 13:955069. doi: 10.3389/fimmu.2022.955069
116. Lai B, Wu CH, Wu CY, Luo SF, Lai JH. Ferroptosis and autoimmune diseases. Front Immunol (2022) 13:916664. doi: 10.3389/fimmu.2022.916664
117. Ling H, Li M, Yang C, Sun S, Zhang W, Zhao L, et al. Glycine increased ferroptosis via SAM-mediated GPX4 promoter methylation in rheumatoid arthritis. Rheumatol (Oxford) (2022) 61(11):4521–34. doi: 10.1093/rheumatology/keac069
118. Hu J, Zhang R, Chang Q, Ji M, Zhang H, Geng R, et al. p53: A regulator of ferroptosis induced by galectin-1 derived peptide 3 in MH7A cells. Front Genet (2022) 13:920273. doi: 10.3389/fgene.2022.920273
119. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol (2020) 16(6):316–33. doi: 10.1038/s41584-020-0413-5
120. Taghadosi M, Adib M, Jamshidi A, Mahmoudi M, Farhadi E. The p53 status in rheumatoid arthritis with focus on fibroblast-like synoviocytes. Immunol Res (2021) 69(3):225–38. doi: 10.1007/s12026-021-09202-7
121. Jiang L, Hickman JH, Wang SJ, Gu W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle (2015) 14(18):2881–5. doi: 10.1080/15384101.2015.1068479
122. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep (2017) 20(7):1692–704. doi: 10.1016/j.celrep.2017.07.055
123. Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv H, et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett (2020) 483:127–36. doi: 10.1016/j.canlet.2020.02.015
124. Jhun J, Moon J, Ryu J, Shin Y, Lee S, Cho KH, et al. Liposome/gold hybrid nanoparticle encoded with CoQ10 (LGNP-CoQ10) suppressed rheumatoid arthritis via STAT3/Th17 targeting. PloS One (2020) 15(11):e241080. doi: 10.1371/journal.pone.0241080
125. Behl T, Upadhyay T, Singh S, Chigurupati S, Alsubayiel AM, Mani V, et al. Polyphenols targeting MAPK mediated oxidative stress and inflammation in rheumatoid arthritis. Molecules (2021) 26(21):6570. doi: 10.3390/molecules26216570
126. Ravalli S, Szychlinska MA, Leonardi RM, Musumeci G. Recently highlighted nutraceuticals for preventive management of osteoarthritis. World J Orthop (2018) 9(11):255–61. doi: 10.5312/wjo.v9.i11.255
127. Jean-Gilles D, Li L, Vaidyanathan VG, King R, Cho B, Worthen DR, et al. Inhibitory effects of polyphenol punicalagin on type-II collagen degradation in vitro and inflammation in vivo. Chem Biol Interact (2013) 205(2):90–9. doi: 10.1016/j.cbi.2013.06.018
128. Luo H, Zhang R. Icariin enhances cell survival in lipopolysaccharide-induced synoviocytes by suppressing ferroptosis via the Xc-/GPX4 axis. Exp Ther Med (2021) 21(1):72. doi: 10.3892/etm.2020.9504
129. Zhou R, Chen Y, Li S, Wei X, Hu W, Tang S, et al. TRPM7 channel inhibition attenuates rheumatoid arthritis articular chondrocyte ferroptosis by suppression of the PKCalpha-NOX4 axis. Redox Biol (2022) 55:102411. doi: 10.1016/j.redox.2022.102411
130. Ma G, Yang Y, Chen Y, Wei X, Ding J, Zhou RP, et al. Blockade of TRPM7 alleviates chondrocyte apoptosis and articular cartilage damage in the adjuvant arthritis rat model through regulation of the Indian hedgehog signaling pathway. Front Pharmacol (2021) 12:655551. doi: 10.3389/fphar.2021.655551
Keywords: ferroptosis, rheumatoid arthritis, lipid peroxidation, iron metabolism, emerging target
Citation: Zhao H, Tang C, Wang M, Zhao H and Zhu Y (2023) Ferroptosis as an emerging target in rheumatoid arthritis. Front. Immunol. 14:1260839. doi: 10.3389/fimmu.2023.1260839
Received: 18 July 2023; Accepted: 22 September 2023;
Published: 19 October 2023.
Edited by:
Alessandra Bettiol, University of Florence, ItalyReviewed by:
Changzheng Chen, Renmin Hospital of Wuhan University, ChinaZhiqiang Lin, Masonic Medical Research Institute (MMRI), United States
Copyright © 2023 Zhao, Tang, Wang, Zhao and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zhu, enlkemYyMDA4QDE2My5jb20=