 Viktor Rydberg
Viktor Rydberg Sigridur Sunna Aradottir
Sigridur Sunna Aradottir Ann-Charlotte Kristoffersson
Ann-Charlotte Kristoffersson Naila Svitacheva
Naila Svitacheva Diana Karpman
Diana Karpman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 07 September 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1254759
Background: Complement activation in atypical hemolytic uremic syndrome (aHUS), C3 glomerulonephropathy (C3G) and immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) may be associated with rare genetic variants. Here we describe gene variants in the Swedish and Norwegian populations.
Methods: Patients with these diagnoses (N=141) were referred for genetic screening. Sanger or next-generation sequencing were performed to identify genetic variants in 16 genes associated with these conditions. Nonsynonymous genetic variants are described when they have a minor allele frequency of <1% or were previously reported as being disease-associated.
Results: In patients with aHUS (n=94, one also had IC-MPGN) 68 different genetic variants or deletions were identified in 60 patients, of which 18 were novel. Thirty-two patients had more than one genetic variant. In patients with C3G (n=40) 29 genetic variants, deletions or duplications were identified in 15 patients, of which 9 were novel. Eight patients had more than one variant. In patients with IC-MPGN (n=7) five genetic variants were identified in five patients. Factor H variants were the most frequent in aHUS and C3 variants in C3G. Seventeen variants occurred in more than one condition.
Conclusion: Genetic screening of patients with aHUS, C3G and IC-MPGN is of paramount importance for diagnostics and treatment. In this study, we describe genetic assessment of Nordic patients in which 26 novel variants were found.
Rare genetic variants have been associated with the ultra-rare complement-mediated kidney diseases atypical hemolytic uremic syndrome (aHUS) and C3 glomerulopathy (C3G) as well as the rare kidney disease immune complex-associated membranoproliferative glomerulonephritis (IC-MPGN). HUS is characterized by the simultaneous development of acute kidney injury, non-immune hemolytic anemia, and thrombocytopenia (1). The most common cause of HUS is gastrointestinal infection with Shiga toxin-producing enterohemorrhagic E. coli (EHEC) (1). A less common form is aHUS, associated with overactivation of the alternative pathway of complement due to heterozygous variants or the presence of circulating auto-antibodies (2, 3).
In aHUS overactivation of complement has been associated with gain-of-function mutations in complement components such as C3 (4) and factor B (5) or loss-of-function mutations in complement regulators such as factor H (6), factor I (7), and membrane-cofactor protein (MCP or CD46) (8). Rare genetic variants have also been described in factor H-related 5 (9), the terminal complement pathway regulator clusterin (10), thrombomodulin (11), diacylglycerol kinase epsilon (DGKE), often associated with development of HUS in infancy (12), and the fibrinolytic factor plasminogen (13). Variants affecting components of the classical pathway of complement, such as C2 and C4 binding-protein, have also been described (14–16). In addition to specific genetic variants, alone or in combination (17), certain haplotypes in factor H (18) and MCP (19) have been described as being risk-associated for the development of disease. Some patients have antibodies to factor H (3) which may occur in combination with deletions or hybrid genes in factor H-related proteins (20).
aHUS is associated with both familial and sporadic cases (1). The presence of a heterozygous mutation is not necessarily associated with disease. In addition to an underlying genetic variant, found in about 60-70% of cases, a trigger, such as infection or pregnancy, can contribute to the development of aHUS (2). Disease recurrences are common, and the natural course of disease is characterized by the development of end-stage kidney disease which may also recur after transplantation (17).
C3G is a chronic form of glomerulonephritis that can lead to end-stage kidney disease and is further subdivided into C3 glomerulonephritis (C3GN) and Dense Deposit Disease (DDD) based on ultramorphology (21, 22). Complement activation via the alternative pathway occurs primarily in the fluid phase (21, 22) either due to mutations or auto-antibodies. This leads to increased C3 consumption, low serum C3 and increased complement deposition in glomeruli (22). Immunofluorescence of kidney biopsies shows predominant C3 deposition and little or no immunoglobulin deposits (23). Patients with C3G carry mutations in C3 and complement factor B (CFB), as well as in complement factor H (CFH), complement factor I (CFI) and complement factor H-related 5 (CFHR5) (24). C3 nephritic factors are auto-antibodies that bind the C3 convertase prolonging its half-life (21, 24) and are found in in many patients (25). Some patients with C3G may have an underlying monoclonal gammopathy (23).
IC-MPGN is also a chronic form of glomerulonephritis that can progress to end-stage renal disease (26). It cannot be differentiated from C3GN based on renal symptoms and ultramorphology but immunofluorescence exhibits deposits of immunoglobulins as well as complement (23, 26). Although IC-MPGN can be associated with infections, such as viral hepatitis, or monoclonal gammopathy (23), cluster analysis revealed that some patients have excessive complement activation via the alternative pathway (27), properdin-dependent C3 nephritic factors (28) as well as genetic variants in complement (26). This suggests an overlapping spectrum of disease between C3G and IC-MPGN with regard to etiology and clinical course (26, 29).
Genetic variants have been found in these complement-mediated kidney diseases in several population studies (24, 30–37). The aim of this paper is to describe genetic variants associated with aHUS, C3G and IC-MPGN in patients from Sweden and Norway. Sequencing encompassed the genes encoding complement factor H, C3, factor I, factor B, MCP, C5, factor H-related proteins 1-5, clusterin, DGKE, thrombomodulin, plasminogen, and properdin. Twenty-six novel genetic variants were found, and certain variants were found in more than one condition.
Patients with suspected complement-mediated renal diseases or thrombotic thrombocytopenic purpura (TTP) are referred to the Dept of Pediatrics, Lund University, for genetic analysis. In this study patients with suspected TTP were not included. A total of 141 patients, both children and adults, living in Sweden or Norway, were included, 94 patients with a clinical diagnosis of aHUS, 40 patients with C3G and 8 patients with IC-MPGN. One patient had both aHUS and IC-MPGN. The diagnosis of aHUS was made by the referring physician based on the simultaneous presentation of non-immune hemolytic anemia, thrombocytopenia, and acute kidney injury as well as negative testing for enterohemorrhagic E. coli. Patients with documented defects in cobalamin metabolism were excluded. The diagnosis of C3G and IC-MPGN was based on kidney biopsy results. Patient data consisted of age of disease debut, laboratory data (such as complement levels and the presence of auto-antibodies), course of disease including kidney failure and familial cases. Data regarding treatment was not uniformly available as most samples were collected upon initial diagnosis. Patient data are presented in Table 1. Informed consent was obtained for diagnostic genetic analysis and the project was approved by the Swedish Ethical Review Authority, approval no. 2021-04438. The Swedish Ethical Review Authority waived the requirement for written consent from patients included retrospectively in this study. All patients included after October 2021 gave informed written consent.
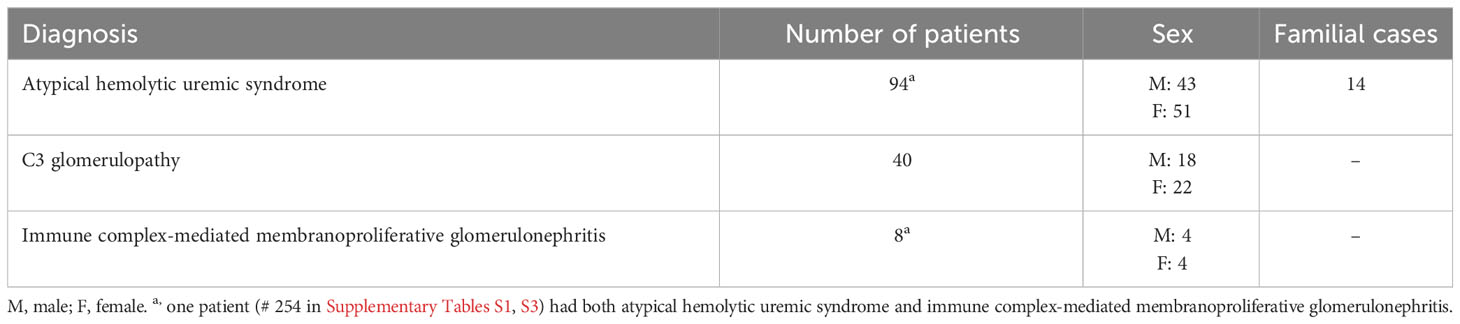
Table 1 Patients investigated in this study.
Genomic DNA was extracted and analyzed by Sanger sequencing until the end of 2016. These samples were assayed for variants in genes encoding factor H, factor I, membrane cofactor protein CD46, C3 and factor B. From 2017 samples were assayed by next generation sequencing first using whole exome sequencing and from 2020 by whole genome sequencing. A panel of 17 genes was assayed including complement factor H (CFH), factor I (CFI), membrane cofactor protein (MCP, CD46), C3, factor B (CFB), properdin (CFP), clusterin (CLU), factor H-related proteins 1-5 (CFHR1-CFHR5), ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), thrombomodulin (THBD), DGKE, C5 and plasminogen (PLG). In this study variants in ADAMTS13 were not included.
For Sanger sequencing genomic DNA was combined with DNA polymerase, primers and nucleotides. The PCR product was sequenced bidirectionally using fluorescent chain-terminating nucleotides (38) from Big dye terminator kit (Applied Biosystems, Foster City, CA) and analyzed on an Applied Biosystems DNA Analyzer, model 3730.
Next generation sequencing was performed in collaboration with the Center for Molecular Diagnostics, Skåne University Hospital, Lund, using either whole exome or whole genome sequencing. Whole-exome sequencing (WES) libraries were generated using Agilent SureSelect Clinical Research Exome v2. Whole-genome sequencing libraries were prepared using Illumina TruSeq PCR-Free. Sequencing was done on either Next Seq 500 (WES) or Novaseq (WES and NGS), with 2 x 150 bp paired end reads and a target depth of at least 30x. Resulting reads were analyzed using the Broad Institute best practices (https://www.broadinstitute.org/gatk/guide/best-practices) as implemented in the Sentieon software suite (https://www.sentieon.com/). Briefly, reads were mapped to the human genome (build hg19) with BWA MEM and variants were identified using DNAscope as implemented in Sentieon. Structural variants were detected with CNVnator and Manta for WGS, and with CNVkit for WES. Variants were annotated using Ensembl Variant Effect Predictor (VEP, https://www.ensembl.org/info/docs/tools/vep/) and filtered for the following genes: CFH, CFHR1-5, CFI, MCP, CFB, C3, C5, CFP, DGKE, PLG, THBD and CLU. All relevant variants were verified in Integrative Genomics Viewer (IGV, https://software.broadinstitute.org/software/igv). Interpretation of variants was performed using Scout software (Similarities from COntinUous Traits https://clinical-genomics.github.io/scout) and prediction was performed using Mastermind (mastermind.genomenon.com). Certain DNA samples were also sequenced at Centogene, Rostock Germany.
Sequencing data included nucleotide shift and amino acid alterations as well as zygosity. This information was analyzed using databases describing mutations and polymorphisms in the included genes such as www.complement-db.org, https://gnomad.broadinstitute.org/ and www.ncbi.nlm.nih.gov/snp for variant calling. Minor allele frequency was defined by the frequency of the second most common allele. Minor allele frequency less than 1% was defined as a possible mutation. Variants that were previously described as associated with disease were included even if the minor allele frequency was > 1%. Variants were considered novel if not previously published in the medical literature. Variants that were previously reported in the ClinVar database in association with complement-mediated diseases such as age-related macular degeneration, aHUS or C3G are mentioned.
C3 was analyzed by nephelometry and C3d by double-decker rocket immunoelectrophoresis according to hospital routines. Low and high levels were defined as below or above the laboratory reference values. Antibodies against factor H and C3 or C4 nephritic factor were detected at the Department of Clinical Immunology, Skåne University Hospital in Lund as per hospital routines. Factor H antibodies were detected as previously described with minor modifications (3). C3 nephritic factor was detected using three methods, by ELISA (39), by hemolytic assay (40) and by crossed immunoelectrophoresis (41) and (42). If any of these assays were positive the patient was defined as having C3 nephritic factor. C4 nephritic factor was detected as previously described (43) with minor modifications.
Of all 141 patients with aHUS, C3G and IC-MPGN 80 patients were found to have genetic variants. Of 94 aHUS patients, 60 patients had genetic variants. Of these 32 patients had more than one variant and a total of 68 different aHUS-related variants were identified, considering deletions in CFHR1 and CFHR3 as one variant, as these genes are adjacent to each other. The genetic variants in aHUS patients are summarized in Table 2 and Supplementary Table 1. Deletions in CFHR1 and CFHR3 are only reported when homozygous except for one patient with antibodies to factor H (patient 17). Eighteen variants were novel to this study and have not been described before. Two of these was previously reported in ClinVar in association with complement-mediated disease. The association between the genetic variants and kidney function, if known, is summarized in Supplementary Table 1.
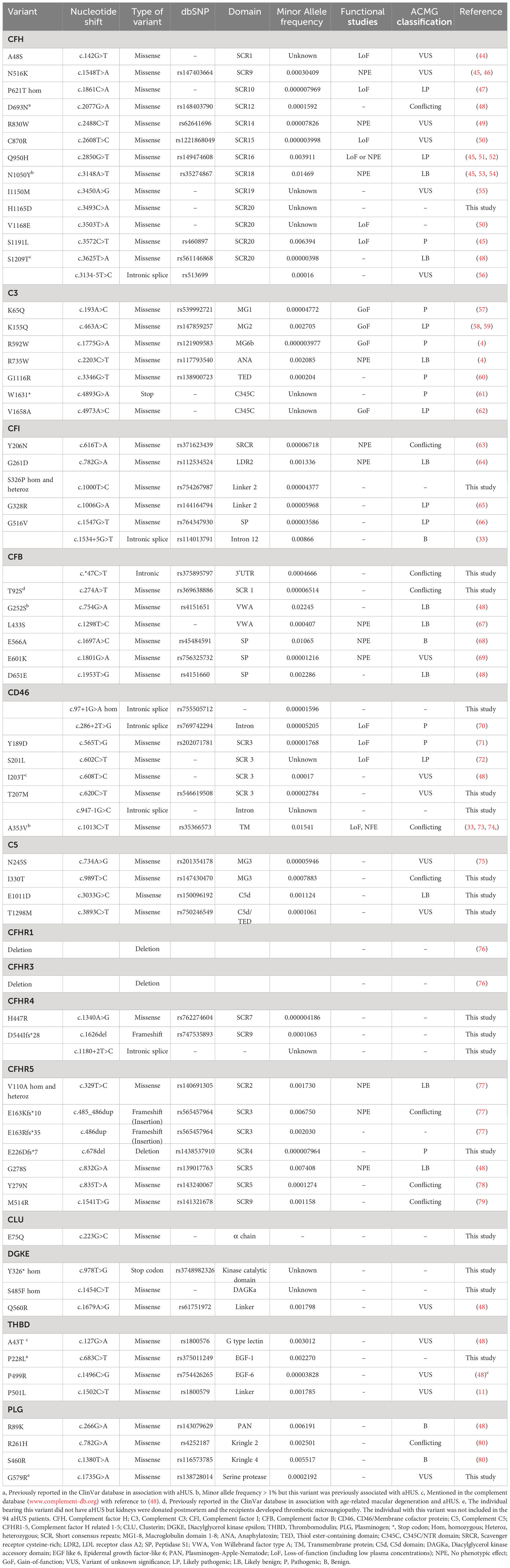
Table 2 Variants found in aHUS patients included in this study.
In 40 C3G patients 15 patients were found to have 29 genetic variants which could be associated with disease (considering homozygous deletions in CFHR1 and CFHR3 as one variant). Of these 8 patients had more than one variant. Nine variants in C3G were novel to this study and have not been described before (two of these were previously reported in ClinVar in association with complement-mediated diseases). The genetic variants are summarized in Table 3. The association between the genetic variants and kidney function, if known, is summarized in Supplementary Table 2.
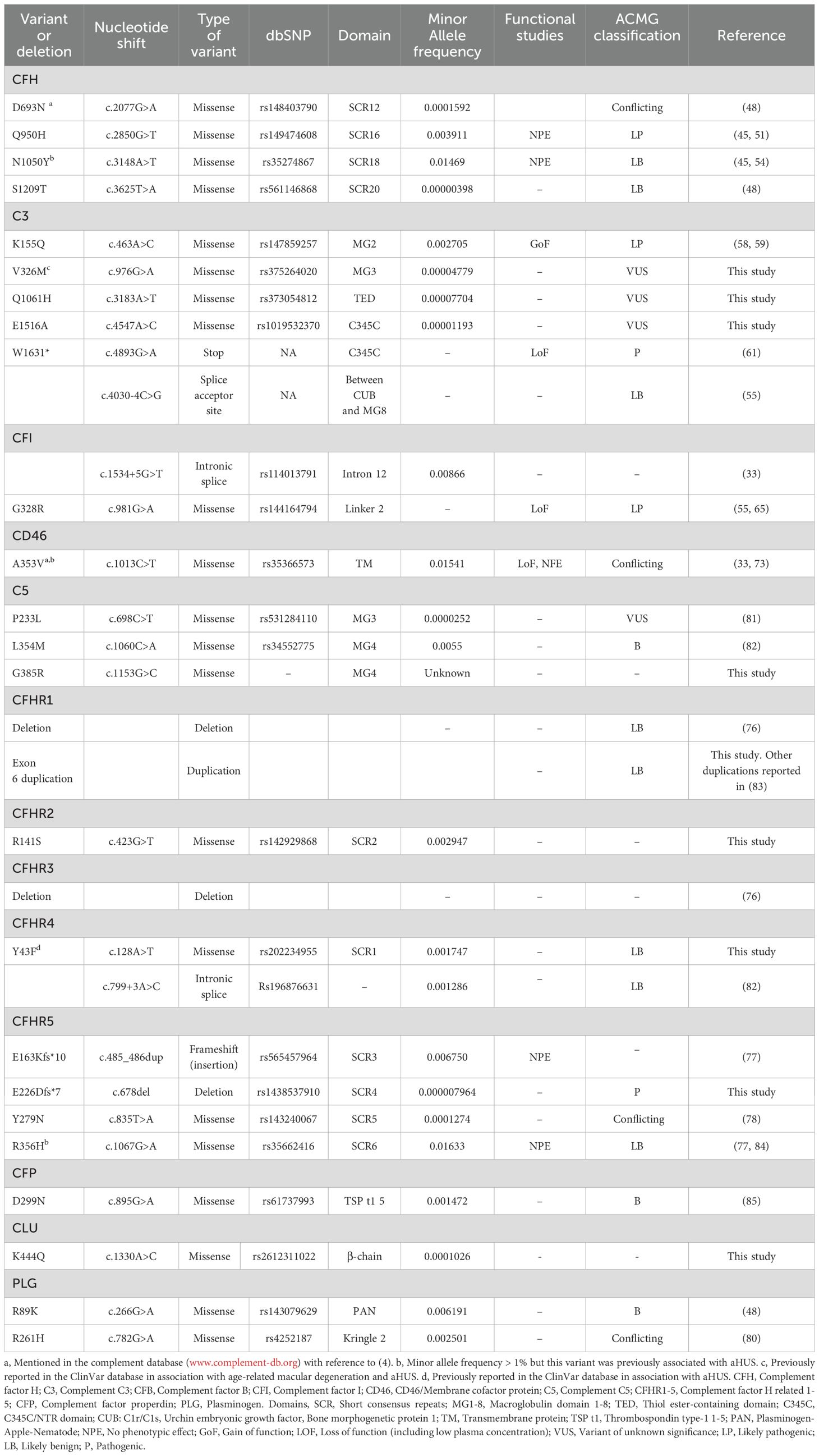
Table 3 Variants in C3G patients included in this study.
Eight patients with IC-MPGN were investigated but one of these also had aHUS (patient 254 in Supplementary Tables 1, 3). In the remaining seven patients, four patients had five genetic variants. The genetic variants are summarized in Table 4. The association between the genetic variants and kidney function, if known, is summarized in Supplementary Table 3.
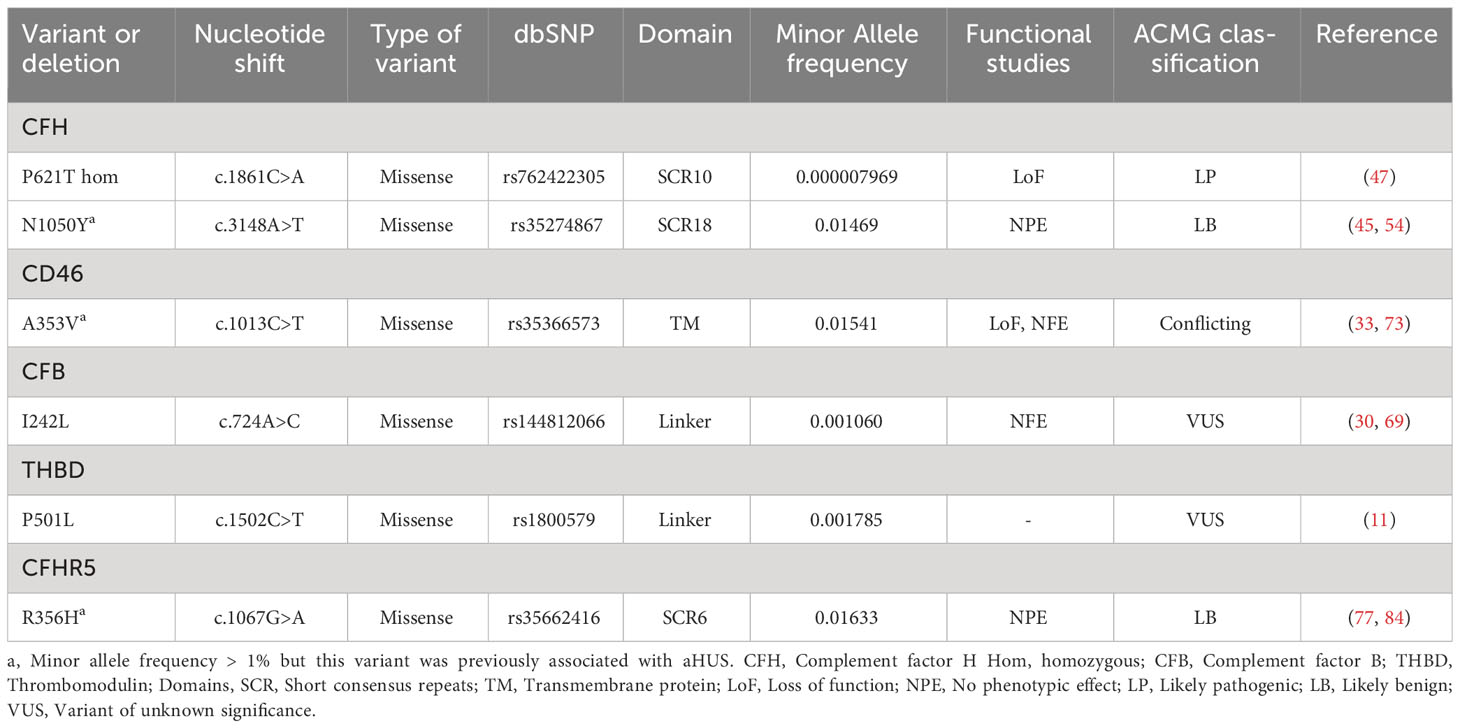
Table 4 Variants in MPGN-IC patients included in this study.
The number of variants found in patients whose DNA underwent Sanger sequencing but did not undergo next generation sequencing may represent an underestimation as only five genes were sequenced encoding CFH, C3, CFI, MCP and CFB.
The number of genetic variants detected in patients with aHUS and C3G is summarized in Tables 5, 6, respectively. For patients with IC-MPGN only five variants were detected. In aHUS most variants were detected in the gene encoding CFH followed by C3 and in C3G the reverse was found.
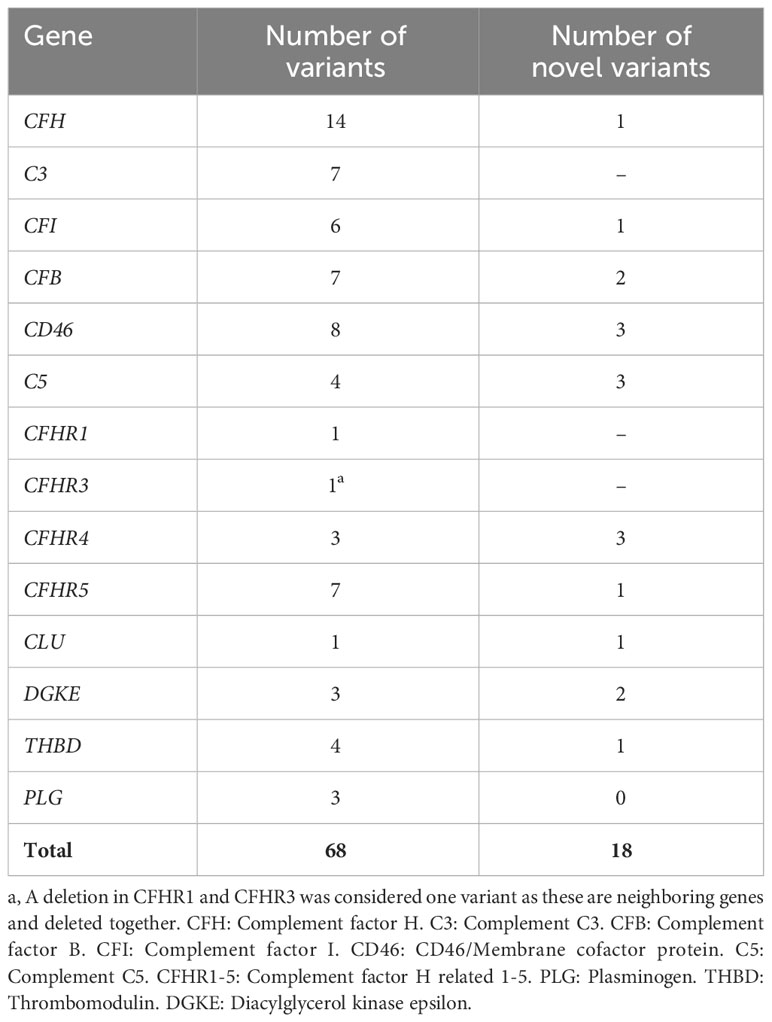
Table 5 Prevalence of variants in aHUS patients included in this study (n=94).
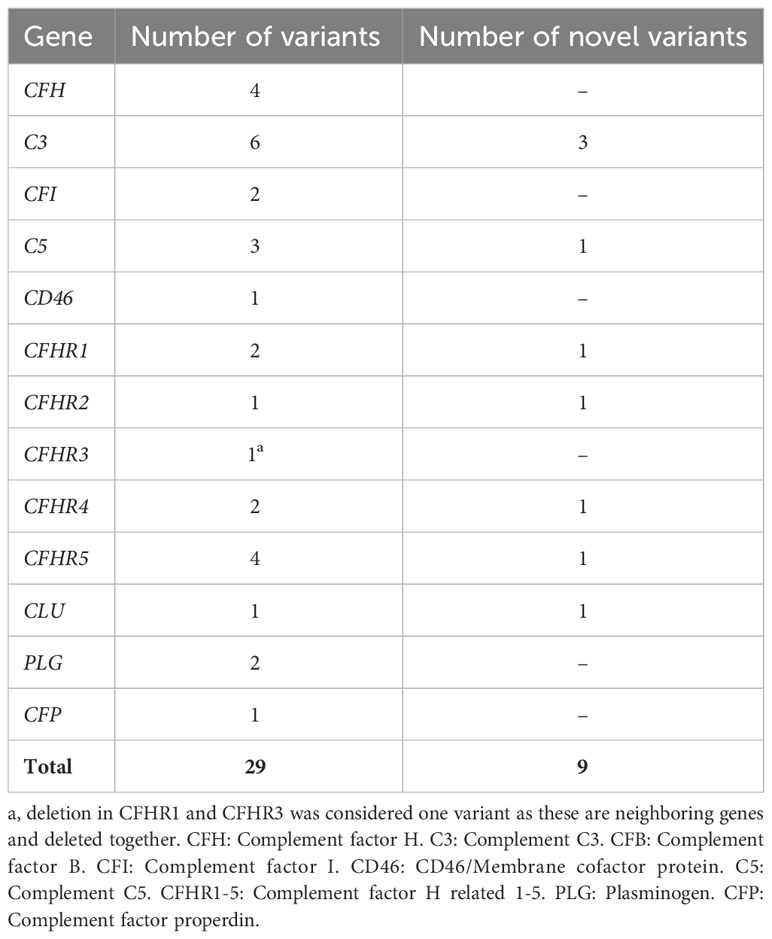
Table 6 Prevalence of variants in C3G patients included in this study (n=40).
The location of all genetic variants detected is presented in Figure 1 which also depicts which variants are novel and which were found in more than one condition. Most variants were heterozygous, however, homozygous variants were found in CFH (P621T), CFI (S326P), CD46 (c.97 + 1G>A), CFHR5 (V110A), DGKE (Y326* and S485F), in patients with aHUS (the patient with the homozygous CFH variant P621T also had IC-MPGN). In both aHUS and C3G homozygous deletions in CFHR1 and CFHR3 were detected.
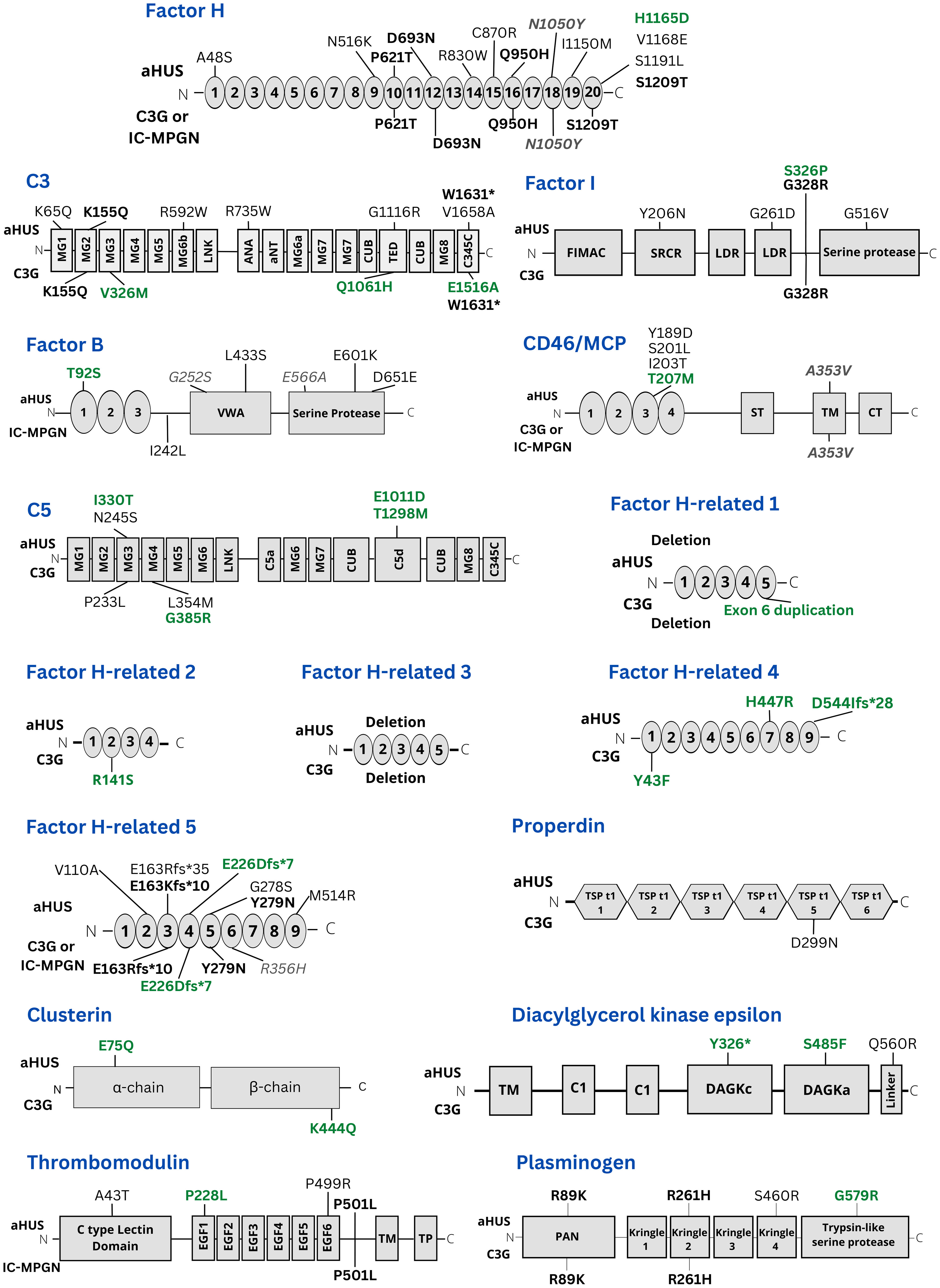
Figure 1 Presentation of all genetic variants found in this study. The location of variants in the genes studied herein is presented. Variants shown above the gene domains were found in aHUS patients. Variants presented under gene domains were found in C3G cases. Variants labeled in black were previously known. Variants labelled in green are novel. Variants in bold were present in both aHUS and C3G patients. Variants in grey italics have a minor allele frequency > 1% but have been associated with complement-mediated kidney diseases. MG1-8: Macroglobulin domain 1-8. ANA: Anaphylatoxin. CUB, C1r/C1s, Urchin embryonic growth factor, Bone morphogenetic protein 1; LNK, Linker; aNT, alpha N-terminal; TED, Thiol ester-containing domain; C345C, C345C/NTR domain; FIMAC, Factor I membrane attack complex; SRCR, Scavenger receptor cysteine-rich; LDR, LDL receptor class A2; VWA, Von Willebrand factor type A; TM, Transmembrane; ST, Ser/Thr-rich; CT, Cytoplasmic domain; EGF 1-6, Epidermal growth factor-like 1-6; PAN, Plasminogen-Apple-Nematode; DAGKc, Diacylglycerol kinase catalytic domain; DAGKa, Diacylglycerol kinase accessory domain; Int, Intron; TSP t1, Thrombospondin type-1 1-5.
Certain variants were found in both aHUS and C3G or IC-MPGN patients. These included CFH (P621T in the same patient, D693N, Q950H, N1050Y (the latter with MAF>1%) and S1209T), C3 (K155Q and W1631*), CFI (G328R, c.1534 + 5G>T), CD46 (A353V, MAF >1%), CFHR5 (E163Kfs*10, E226DFS*7, Y279N), THBD (P501L), PLG (R89K and R261H) and homozygous deletions of CFHR1 and CFHR3. Of these CFHR5 E226DFS*7 was novel and found in both aHUS and C3G patients.
Upon referral for genetic testing, we had access to clinical information describing the course of disease in most patients. When these data were available, we correlated the genetic findings to the presence of kidney failure, as presented in Supplementary Tables 1-3. The genetic variants were correlated to levels of C3, C3d and the presence of antibodies to factor H in aHUS (Supplementary Table 1) or nephritic factors in C3G or IC-MPGN (Supplementary Tables 2, 3).
In complement-mediated kidney diseases aHUS, C3G, and IC-MPGN understanding a patient’s genotype and its correlation to disease phenotype is of paramount importance for diagnostics and choice of treatment. Genetic investigation is also crucial for determining the risk of disease recurrence, the suitability of kidney transplantation and the choice of donor, as well as evaluation of the risk of disease development in family members bearing the same variant, including family members considered as kidney donors. This study investigated the genotype of a large cohort of patients with these diseases using a panel of disease-associated genes. In aHUS 68 different variants were identified and in C3G 29 different variants were identified, most variants were heterozygous and 26 were novel. Importantly, 40 patients with aHUS and C3G had more than one genetic variant, exemplifying the complexity of interpreting disease heredity.
Seventeen genetic variants, novel as well as previously reported, had a dual disease phenotype occurring in both patients with aHUS and C3G. The presence of a disease-associated genetic variant is not tantamount to the development of symptoms, as it is known that disease penetrance is incomplete (67, 86) and we show here that identical genetic alterations may be associated with different disease phenotypes. This suggests that disease expression may be related to additional genetic factors (complement genes as well as others), the exposome such as triggering factors and, as yet, undefined environmental, lifestyle or epigenetic factors.
In aHUS and C3G genetic variants were previously identified in complement components and complement regulatory proteins (24, 30–37). The variants may cause a loss-of-function in regulators or a gain-of-function in complement factors. Variants in CFH have been shown to lead to a loss-of-function by reducing cofactor activity, as shown for Q950H (52) and V1168E (50), impairing C3b binding, demonstrated for S1191L (87), or by deficiency associated with decreased protein secretion, as shown for P621T (47). Additionally, variant S1191L exhibited impaired ability to regulate complement activation on cell surfaces (87). Genetic variants in factor I can lead to quantitative deficiency or functional defects in protease activity rendering the enzyme incapable of inactivating C3b (59). This was, however, not specifically demonstrated for the variants presented herein, as, for example, the aHUS-associated variant G261D did not exhibit complement dysregulation (64). CD46 variants may also cause loss-of-function by reduced expression on cell surfaces, as shown for c.286 + 2T>G and S201L (70), decreased cofactor activity, or affect C3b/C4b binding capacity (72, 86). Likewise, thrombomodulin inhibits complement activation by promoting C3b inactivation and mutated variants have exhibited less C3b inactivation to iC3b on cell surfaces, as shown for the P501L variant (11).
C3 and CFB variants may exhibit gain-of-function properties in the respective encoded proteins. The variant V1658A in C3 results in increased C3 convertase formation (62) and K155Q confers increased hemolytic activity (58). Additionally, the C3 variant K65Q is associated with reduced binding to factor H (57), and variant R592W exhibits impaired binding to the complement regulator CD46/MCP (4). CFB variants may increase the affinity to C3b, thus stabilizing the convertase, as shown for variant D371G (69) and by increased resistance to factor H-mediated decay (32, 88). However, not all variants show dysfunction in in vitro models, as reported for L433S, I242L and E601K (67, 69).
Variants in DGKE cause the development of aHUS in young children, often in combination with proteinuria. It has been suggested that homozygous DGKE variants lead to loss-of-function and consequently a prothrombotic state (12), however the mechanism by which the variants Y326* and S485F lead to disease has yet to be deciphered. Likewise, plasminogen deficiency or dysfunction is associated with reduced proteolytic activity in growing thrombi which may contribute to a thrombosis (89).
Many patients had several variants in more than one gene. In these cases, the individual variants themselves may not lead to disease development, but when combined may result in complement over-activation (67, 90). As previously described in an anephric aHUS patient, with a CFH disease-associated haplotype as well as CFI and CFB variants, the patient had evidence of complement activation, developed carotid artery stenosis, and was successfully treated with eculizumab (67). Some genetic variants are distinctly pathological, even without the presence of other variants, such as the C3 mutation V1636A (62). On the contrary, some variants that in vitro exhibit a dysfunctional protein, may be present in unaffected carriers that remain disease-free, such as the CFH variant V1168E (50) and the CFB variant D371G (91).
For the variants presented herein we provide prediction as to their possible pathogenicity, when available. Prediction models can efficiently assess if a genetic alteration is benign or pathogenic (92), however, they are not always accurate and may require combining various prediction scores (93), thus interpretation can be challenging. Functional tests, using mutant compared to wild-type proteins, can more accurately demonstrate protein dysfunction. Considering the rarity and complexity of disease expression, we recommend performing mutagenesis to predict the effect of genetic variants on complement activation.
This study found that 72% of aHUS and 38% of C3G patients had genetic variants, therefore not all patients who develop disease carry variants in the screened genes. In aHUS this may, in part, be due to the presence of antibodies to factor H and in C3G to the presence of nephritic factors which may cause disease. Additionally, 47 patients were sequenced using only the Sanger method. In these patients, all the known disease-related genes were not sequenced and the percentage of patients with genetic variants may be higher. Even though the frequency of variants in this cohort may be underestimated, other cohorts have identified patients with no known cause for both aHUS and C3G. Previous studies show that about 45-60% of aHUS patients (13, 30, 94) and 30-40% of C3G patients carry mutations (24, 35, 36). Additionally, in C3G and IC-MPGN 50% of patients included herein had C3NeF (or C4NeF) which is comparable to previous reports regarding C3NeF (35, 36). Clearly, all disease-related mechanisms for aHUS and C3G have not been found and future research may uncover new disease-related genes which are not routinely screened for at present.
Patient samples included in this study were collected over a 20-year period. Thus, many patients were included before complement-inhibitory therapy with eculizumab was available. A further limitation of the study is that data regarding treatments was not fully available in referrals, mostly because DNA samples were submitted as part of a diagnostic work-up and, in certain cases, before a diagnosis was made and treatment was initiated. For these reasons, this study could not associate specific genetic variants with the need for complement-inhibitory therapy and outcome.
To conclude, this study presents genetic variants found in Swedish and Norwegian patients with aHUS, C3G and IC-MPGN, 26 of which were novel. Some patients had multiple variants in genes encoding complement proteins. Bearing a genetic variant does not necessarily lead to occurrence of disease as there is incomplete penetrance of the disease phenotype. Furthermore, certain variants were found in both aHUS and C3G suggesting that factors additional to genetic composition can dictate the phenotype. Further investigations are required to better understand the impact variants have on protein functionality, and how they lead to disease expression.
All data are available within the article and its supplements. Datasets were deposited in Zenodo, doi: 10.5281/zenodo.8124309.
Informed consent was obtained for diagnostic genetic analysis and the project was approved by the Swedish Ethical Review Authority, approval no. 2021-04438. The Swedish Ethical Review Authority waived the requirement for written consent from patients included retrospectively in this study. All patients included after October 2021 gave informed written consent. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants or their legal guardians/next of kin.
VR: Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. SA: Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. A-CK: Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. NS: Investigation, Writing – original draft. DK: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
The Swedish Research Council (2022-00415 (EJPRD2022-016 CompRare), 2020-02200, 2017-01920), The Knut and Alice Wallenberg Foundation (Wallenberg Clinical Scholar 2015.0320), Skåne Centre of Excellence in Health, The Inga-Britt and Arne Lundberg’s Research Foundation, Kidneeds Fund, Olle Engkvist Byggmästare Foundation and The Swedish Freemason’s Foundation for Children’s Welfare (all to DK).
The authors thank all referring physicians and their patients, with a special thanks to Professor Anna Bjerre, Oslo University Hospital, Oslo Norway. Associate Professor Lillemor Skattum is acknowledged for providing methods used at the Department of Clinical Immunology, Skåne University Hospital in Lund and Dr. Petter Storm, Lund University, Sweden for providing methods used at the Center for Molecular Diagnostics, Skåne University Hospital. Professor Zoltan Prohászka, Department of Medicine and Hematology, Semmelweis University, Budapest, Hungary, is acknowledged for sequencing the sample from patient #348. This study, in preliminary form, was included in the masters’ thesis of VR.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1254759/full#supplementary-material
1. Karpman D, Loos S, Tati R, Arvidsson I. Haemolytic uraemic syndrome. J Intern Med (2017) 281(2):123–48. doi: 10.1111/joim.12546
2. Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol (2016) 31(1):15–39. doi: 10.1007/s00467-015-3076-8
3. Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol (2005) 16(2):555–63. doi: 10.1681/ASN.2004050380
4. Frémeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood (2008) 112(13):4948–52. doi: 10.1182/blood-2008-01-133702
5. Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA (2007) 104(1):240–5. doi: 10.1073/pnas.0603420103
6. Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int (1998) 53(4):836–44. doi: 10.1111/j.1523-1755.1998.00824.x
7. Kavanagh D, Kemp EJ, Mayland E, Winney RJ, Duffield JS, Warwick G, et al. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol (2005) 16(7):2150–5. doi: 10.1681/ASN.2005010103
8. Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA (2003) 100(22):12966–71. doi: 10.1073/pnas.2135497100
9. Westra D, Vernon KA, Volokhina EB, Pickering MC, van de Kar NC, van den Heuvel LP. Atypical hemolytic uremic syndrome and genetic aberrations in the complement factor H-related 5 gene. J Hum Genet (2012) 57(7):459–64. doi: 10.1038/jhg.2012.57
10. Ståhl AL, Kristoffersson A, Olin AI, Olsson ML, Roodhooft AM, Proesmans W, et al. A novel mutation in the complement regulator clusterin in recurrent hemolytic uremic syndrome. Mol Immunol (2009) 46(11-12):2236–43. doi: 10.1016/j.molimm.2009.04.012
11. Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med (2009) 361(4):345–57. doi: 10.1056/NEJMoa0810739
12. Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet (2013) 45(5):531–6. doi: 10.1038/ng.2590
13. Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol (2014) 25(1):55–64. doi: 10.1681/ASN.2013050453
14. Urban A, Volokhina E, Felberg A, Stasiłojć G, Blom AM, Jongerius I, et al. Gain-of-function mutation in complement C2 protein identified in a patient with aHUS. J Allergy Clin Immunol (2020) 146(4):916–9.e11. doi: 10.1016/j.jaci.2020.02.014
15. Urban A, Kowalska D, Stasiłojć G, Kuźniewska A, Skrobińska A, Arjona E, et al. Gain-of-function mutations R249C and S250C in complement C2 protein increase C3 deposition in the presence of C-reactive protein. Front Immunol (2021) 12:724361. doi: 10.3389/fimmu.2021.724361
16. Blom AM, Bergström F, Edey M, Diaz-Torres M, Kavanagh D, Lampe A, et al. A novel non-synonymous polymorphism (p.Arg240His) in C4b-binding protein is associated with atypical hemolytic uremic syndrome and leads to impaired alternative pathway cofactor activity. J Immunol (2008) 180(9):6385–91. doi: 10.4049/jimmunol.180.9.6385
17. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol (2013) 24(3):475–86. doi: 10.1681/ASN.2012090884
18. de Córdoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol (2008) 151(1):1–13. doi: 10.1111/j.1365-2249.2007.03552.x
19. Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, López-Trascasa M, Sánchez-Corral P, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet (2005) 14(5):703–12. doi: 10.1093/hmg/ddi066
20. Zipfel PF, Mache C, Müller D, Licht C, Wigger M, Skerka C. DEAP-HUS: deficiency of CFHR plasma proteins and autoantibody-positive form of hemolytic uremic syndrome. Pediatr Nephrol (2010) 25(10):2009–19. doi: 10.1007/s00467-010-1446-9
21. Wong EKS, Kavanagh D. Diseases of complement dysregulation-an overview. Semin Immunopathol (2018) 40(1):49–64. doi: 10.1007/s00281-017-0663-8
22. Smith RJH, Appel GB, Blom AM, Cook HT, D'Agati VD, Fakhouri F, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol (2019) 15(3):129–43. doi: 10.1038/s41581-018-0107-2
23. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis–a new look at an old entity. N Engl J Med (2012) 366(12):1119–31. doi: 10.1056/NEJMra1108178
24. Bu F, Borsa NG, Jones MB, Takanami E, Nishimura C, Hauer JJ, et al. High-throughput genetic testing for thrombotic microangiopathies and C3 glomerulopathies. J Am Soc Nephrol (2016) 27(4):1245–53. doi: 10.1681/ASN.2015040385
25. Corvillo F, Okrój M, Nozal P, Melgosa M, Sánchez-Corral P, López-Trascasa M. Nephritic factors: an overview of classification, diagnostic tools and clinical associations. Front Immunol (2019) 10:886. doi: 10.3389/fimmu.2019.00886
26. Noris M, Daina E, Remuzzi G. Membranoproliferative glomerulonephritis: no longer the same disease and may need very different treatment. Nephrol Dial Transpl (2023) 38(2):283–90. doi: 10.1093/ndt/gfab281
27. Iatropoulos P, Daina E, Curreri M, Piras R, Valoti E, Mele C, et al. Cluster analysis identifies distinct pathogenetic patterns in C3 glomerulopathies/immune complex-mediated membranoproliferative GN. J Am Soc Nephrol (2018) 29(1):283–94. doi: 10.1681/ASN.2017030258
28. Donadelli R, Pulieri P, Piras R, Iatropoulos P, Valoti E, Benigni A, et al. Unraveling the molecular mechanisms underlying complement dysregulation by nephritic factors in C3G and IC-MPGN. Front Immunol (2018) 9:2329. doi: 10.3389/fimmu.2018.02329
29. Kovala M, Seppälä M, Räisänen-Sokolowski A, Meri S, Honkanen E, Kaartinen K. Diagnostic and prognostic comparison of immune-complex-mediated membranoproliferative glomerulonephritis and C3 glomerulopathy. Cells (2023) 12(5):712. doi: 10.3390/cells12050712
30. Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat (2010) 31(6):E1445–60. doi: 10.1002/humu.21256
31. Fan X, Yoshida Y, Honda S, Matsumoto M, Sawada Y, Hattori M, et al. Analysis of genetic and predisposing factors in Japanese patients with atypical hemolytic uremic syndrome. Mol Immunol (2013) 54(2):238–46. doi: 10.1016/j.molimm.2012.12.006
32. Zhang T, Lu J, Liang S, Chen D, Zhang H, Zeng C, et al. Comprehensive analysis of complement genes in patients with atypical hemolytic uremic syndrome. Am J Nephrol (2016) 43(3):160–9. doi: 10.1159/000445127
33. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood (2006) 108(4):1267–79. doi: 10.1182/blood-2005-10-007252
34. Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis (2015) 66(2):359–75. doi: 10.1053/j.ajkd.2015.03.040
35. Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnorMalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int (2012) 82(4):454–64. doi: 10.1038/ki.2012.63
36. Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, et al. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol (2016) 71:131–42. doi: 10.1016/j.molimm.2016.01.010
37. Szarvas N, Szilágyi Á, Csuka D, Takács B, Rusai K, Müller T, et al. Genetic analysis and functional characterization of novel mutations in a series of patients with atypical hemolytic uremic syndrome. Mol Immunol (2016) 71:10–22. doi: 10.1016/j.molimm.2016.01.003
38. Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet (2011) 52(4):413–35. doi: 10.1007/s13353-011-0057-x
39. Skattum L. Analysis of C3 nephritic factors by ELISA. Methods Mol Biol (2019) 1901:177–82. doi: 10.1007/978-1-4939-8949-2_13
40. Rother U. A new screening test for C3 nephritis factor based on a stable cell bound convertase on sheep erythrocytes. J Immunol Methods (1982) 51(1):101–7. doi: 10.1016/0022-1759(82)90386-6
41. Peters DK, Martin A, Weinstein A, Cameron JS, Barratt TM, Ogg CS, et al. Complement studies in membrano-proliferative glomerulonephritis. Clin Exp Immunol (1972) 11(3):311–20.
42. Sjöholm AG. Complement components and complement activation in acute poststreptococcal glomerulonephritis. Int Arch Allergy Appl Immunol (1979) 58(3):274–84. doi: 10.1159/000232203
43. Blom AM, Corvillo F, Magda M, Stasiłojć G, Nozal P, Pérez-Valdivia M, et al. Testing the activity of complement convertases in serum/plasma for diagnosis of C4NeF-mediated C3 glomerulonephritis. J Clin Immunol (2016) 36(5):517–27. doi: 10.1007/s10875-016-0290-5
44. Schmitt R, Krmar RT, Kristoffersson A, Söderberg M, Karpman D. IgA nephropathy associated with a novel N-terminal mutation in factor H. Eur J Pediatr (2011) 170(1):107–10. doi: 10.1007/s00431-010-1279-3
45. Martín Merinero H, Zhang Y, Arjona E, Del Angel G, Goodfellow R, Gomez-Rubio E, et al. Functional characterization of 105 factor H variants associated with aHUS: lessons for variant classification. Blood (2021) 138(22):2185–201. doi: 10.1182/blood.2021012037
46. Le Quintrec M, Lionet A, Kamar N, Karras A, Barbier S, Buchler M, et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am J Transpl (2008) 8(8):1694–701. doi: 10.1111/j.1600-6143.2008.02297.x
47. Vaziri-Sani F, Holmberg L, Sjöholm AG, Kristoffersson AC, Manea M, Frémeaux-Bacchi V, et al. Phenotypic expression of factor H mutations in patients with atypical hemolytic uremic syndrome. Kidney Int (2006) 69(6):981–8. doi: 10.1038/sj.ki.5000155
48. Osborne AJ, Breno M, Borsa NG, Bu F, Frémeaux-Bacchi V, Gale DP, et al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol (2018) 200(7):2464–78. doi: 10.4049/jimmunol.1701695
49. Merinero HM, García SP, García-Fernández J, Arjona E, Tortajada A, Rodríguez de Córdoba S. Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int (2018) 93(2):470–81. doi: 10.1016/j.kint.2017.07.015
50. Ståhl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood (2008) 111(11):5307–15. doi: 10.1182/blood-2007-08-106153
51. Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet (2003) 12(24):3385–95. doi: 10.1093/hmg/ddg363
52. Mohlin FC, Nilsson SC, Levart TK, Golubovic E, Rusai K, Müller-Sacherer T, et al. Functional characterization of two novel non-synonymous alterations in CD46 and a Q950H change in factor H found in atypical hemolytic uremic syndrome patients. Mol Immunol (2015) 65(2):367–76. doi: 10.1016/j.molimm.2015.02.013
53. Abrera-Abeleda MA, Nishimura C, Smith JL, Sethi S, McRae JL, Murphy BF, et al. Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease). J Med Genet (2006) 43(7):582–9. doi: 10.1136/jmg.2005.038315
54. Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell recognition. Am J Hum Genet (2001) 68(2):485–90. doi: 10.1086/318203
55. Åkesson A, Martin M, Blom AM, Rossing M, Gabrielaite M, Zetterberg E, et al. Clinical characterization and identification of rare genetic variants in atypical hemolytic uremic syndrome: A Swedish retrospective observational study. Ther Apher Dial (2021) 25(6):988–1000. doi: 10.1111/1744-9987.13634
56. Larsen CP, Wilson JD, Best-Rocha A, Beggs ML, Hennigar RA. Genetic testing of complement and coagulation pathways in patients with severe hypertension and renal microangiopathy. Mod Pathol (2018) 31(3):488–94. doi: 10.1038/modpathol.2017.154
57. Volokhina E, Westra D, Xue X, Gros P, van de Kar N, van den Heuvel L. Novel C3 mutation p.Lys65Gln in aHUS affects complement factor H binding. Pediatr Nephrol (2012) 27(9):1519–24. doi: 10.1007/s00467-012-2183-z
58. Mohlin FC, Gros P, Mercier E, Gris JR, Blom AM. Analysis of C3 gene variants in patients with idiopathic recurrent spontaneous pregnancy loss. Front Immunol (2018) 9:1813. doi: 10.3389/fimmu.2018.01813
59. Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Int (2010) 77(4):339–49. doi: 10.1038/ki.2009.472
60. Brackman D, Sartz L, Leh S, Kristoffersson AC, Bjerre A, Tati R, et al. Thrombotic microangiopathy mimicking membranoproliferative glomerulonephritis. Nephrol Dial Transpl (2011) 26(10):3399–403. doi: 10.1093/ndt/gfr422
61. El Sissy C, Rosain J, Vieira-Martins P, Bordereau P, Gruber A, Devriese M, et al. Clinical and genetic spectrum of a large cohort with total and sub-total complement deficiencies. Front Immunol (2019) 10:1936. doi: 10.3389/fimmu.2019.01936
62. Sartz L, Olin AI, Kristoffersson AC, Ståhl AL, Johansson ME, Westman K, et al. A novel C3 mutation causing increased formation of the C3 convertase in familial atypical hemolytic uremic syndrome. J Immunol (2012) 188(4):2030–7. doi: 10.4049/jimmunol.1100319
63. de Jong S, Volokhina EB, de Breuk A, Nilsson SC, de Jong EK, van der Kar N, et al. Effect of rare coding variants in the CFI gene on Factor I expression levels. Hum Mol Genet (2020) 29(14):2313–24. doi: 10.1093/hmg/ddaa114
64. Nilsson SC, Karpman D, Vaziri-Sani F, Kristoffersson AC, Salomon R, Provot F, et al. A mutation in factor I that is associated with atypical hemolytic uremic syndrome does not affect the function of factor I in complement regulation. Mol Immunol (2007) 44(8):1835–44. doi: 10.1016/j.molimm.2006.10.005
65. de Jong S, de Breuk A, Bakker B, Katti S, Hoyng CB, Nilsson SC, et al. Functional analysis of variants in complement factor I identified in age-related macular degeneration and atypical hemolytic uremic syndrome. Front Immunol (2021) 12:789897. doi: 10.3389/fimmu.2021.789897
66. Zhao W, Ding Y, Lu J, Zhang T, Chen D, Zhang H, et al. Genetic analysis of the complement pathway in C3 glomerulopathy. Nephrol Dial Transpl (2018) 33(11):1919–27. doi: 10.1093/ndt/gfy033
67. Békássy ZD, Kristoffersson AC, Cronqvist M, Roumenina LT, Rybkine T, Vergoz L, et al. Eculizumab in an anephric patient with atypical haemolytic uraemic syndrome and advanced vascular lesions. Nephrol Dial Transpl (2013) 28(11):2899–907. doi: 10.1093/ndt/gft340
68. Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, et al. Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol (2014) 25(9):2053–65. doi: 10.1681/ASN.2013070796
69. Aradottir SS, Kristoffersson AC, Roumenina LT, Bjerre A, Kashioulis P, Palsson R, et al. Factor D inhibition blocks complement activation induced by mutant factor B associated with atypical hemolytic uremic syndrome and membranoproliferative glomerulonephritis. Front Immunol (2021) 12:690821. doi: 10.3389/fimmu.2021.690821
70. Frémeaux-Bacchi V, Arzouk N, Ferlicot S, Charpentier B, Snanoudj R, Dürrbach A. Recurrence of HUS due to CD46/MCP mutation after renal transplantation: a role for endothelial microchimerism. Am J Transpl (2007) 7(8):2047–51. doi: 10.1111/j.1600-6143.2007.01888.x
71. Fremeaux-Bacchi V, Moulton EA, Kavanagh D, Dragon-Durey MA, Blouin J, Caudy A, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol (2006) 17(7):2017–25. doi: 10.1681/ASN.2005101051
72. Schack VR, Herlin MK, Pedersen H, Jensen JMB, Faerch M, Bundgaard B, et al. Novel homozygous CD46 variant with C-isoform expression affects C3b inactivation in atypical hemolytic uremic syndrome. Eur J Immunol (2022) 52(10):1610–9. doi: 10.1002/eji.202249838
73. Fang CJ, Fremeaux-Bacchi V, Liszewski MK, Pianetti G, Noris M, Goodship TH, et al. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood (2008) 111(2):624–32. doi: 10.1182/blood-2007-04-084533
74. Bu F, Zhang Y, Wang K, Borsa NG, Jones MB, Taylor AO, et al. Genetic analysis of 400 patients refines understanding and implicates a new gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol (2018) 29(12):2809–19. doi: 10.1681/ASN.2018070759
75. Ravindran A, Fervenza FC, Smith RJH, De Vriese AS, Sethi S. C3 glomerulopathy: ten years' Experience at mayo clinic. Mayo Clin Proc (2018) 93(8):991–1008. doi: 10.1016/j.mayocp.2018.05.019
76. Zipfel PF, Edey M, Heinen S, Józsi M, Richter H, Misselwitz J, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PloS Genet (2007) 3(3):e41. doi: 10.1371/journal.pgen.0030041
77. Garam N, Cserhalmi M, Prohászka Z, Szilágyi Á, Veszeli N, Szabó E, et al. FHR-5 serum levels and CFHR5 genetic variations in patients with immune complex-mediated membranoproliferative glomerulonephritis and C3-glomerulopathy. Front Immunol (2021) 12:720183. doi: 10.3389/fimmu.2021.720183
78. Vaught AJ, Braunstein EM, Jasem J, Yuan X, Makhlin I, Eloundou S, et al. Germline mutations in the alternative pathway of complement predispose to HELLP syndrome. JCI Insight (2018) 3(6):e99128. doi: 10.1172/jci.insight.99128
79. Hughes AE, Bridgett S, Meng W, Li M, Curcio CA, Stambolian D, et al. Sequence and expression of complement factor H gene cluster variants and their roles in age-related macular degeneration risk. Invest Ophthalmol Vis Sci (2016) 57(6):2763–9. doi: 10.1167/iovs.15-18744
80. Sadovnick AD, Traboulsee AL, Bernales CQ, Ross JP, Forwell AL, Yee IM, et al. Analysis of plasminogen genetic variants in multiple sclerosis patients. G3 (Bethesda) (2016) 6(7):2073–9. doi: 10.1534/g3.116.030841
81. Avasare RS, Canetta PA, Bomback AS, Marasa M, Caliskan Y, Ozluk Y, et al. Mycophenolate mofetil in combination with steroids for treatment of C3 glomerulopathy: A case series. Clin J Am Soc Nephrol (2018) 13(3):406–13. doi: 10.2215/CJN.09080817
82. Gastoldi S, Aiello S, Galbusera M, Breno M, Alberti M, Bresin E, et al. An ex vivo test to investigate genetic factors conferring susceptibility to atypical haemolytic uremic syndrome. Front Immunol (2023) 14:1112257. doi: 10.3389/fimmu.2023.1112257
83. Piras R, Valoti E, Alberti M, Bresin E, Mele C, Breno M, et al. CFH and CFHR structural variants in atypical Hemolytic Uremic Syndrome: Prevalence, genomic characterization and impact on outcome. Front Immunol (2022) 13:1011580. doi: 10.3389/fimmu.2022.1011580
84. Monteferrante G, Brioschi S, Caprioli J, Pianetti G, Bettinaglio P, Bresin E, et al. Genetic analysis of the complement factor H related 5 gene in haemolytic uraemic syndrome. Mol Immunol (2007) 44(7):1704–8. doi: 10.1016/j.molimm.2006.08.004
85. Seitsonen S, Onkamo P, Torniainen S, Ihalainen M, Immonen I, Meri S, et al. Screening of DNA-variants in the properdin gene (CFP) in age-related macular degeneration (AMD). Mol Immunol (2010) 47(6):1334–6. doi: 10.1016/j.molimm.2010.01.001
86. Bhatia D, Khandelwal P, Sinha A, Hari P, Cheong HI, Bagga A. Incomplete penetrance of CD46 mutation causing familial atypical hemolytic uremic syndrome. Pediatr Nephrol (2015) 30(12):2215–20. doi: 10.1007/s00467-015-3189-0
87. Heinen S, Sanchez-Corral P, Jackson MS, Strain L, Goodship JA, Kemp EJ, et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat (2006) 27(3):292–3. doi: 10.1002/humu.9408
88. Roumenina LT, Jablonski M, Hue C, Blouin J, Dimitrov JD, Dragon-Durey MA, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood (2009) 114(13):2837–45. doi: 10.1182/blood-2009-01-197640
89. Hyvärinen S, JokIranta TS. Minor role of plasminogen in complement activation on cell surfaces. PloS One (2015) 10(12):e0143707. doi: 10.1371/journal.pone.0143707
90. Rodríguez de Córdoba S. Genetic variability shapes the alternative pathway complement activity and predisposition to complement-related diseases. Immunol Rev (2023) 313(1):71–90. doi: 10.1111/imr.13131
91. Aradottir SS, Kristoffersson AC, Jensson BO, Sulem P, Gong H, Palsson R, et al. Factor B mutation in monozygotic twins discordant for atypical hemolytic uremic syndrome. Kidney Int Rep (2023) 8(5):1097–101. doi: 10.1016/j.ekir.2023.02.1069
92. Pandurangan AP, Blundell TL. Prediction of impacts of mutations on protein structure and interactions: SDM, a statistical approach, and mCSM, using machine learning. Protein Sci (2020) 29(1):247–57. doi: 10.1002/pro.3774
93. Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet (2015) 24(8):2125–37. doi: 10.1093/hmg/ddu733
Keywords: complement, atypical hemolytic uremic syndrome, C3 glomerulopathy, membranoproliferative glomerulonephritis, genes
Citation: Rydberg V, Aradottir SS, Kristoffersson A-C, Svitacheva N and Karpman D (2023) Genetic investigation of Nordic patients with complement-mediated kidney diseases. Front. Immunol. 14:1254759. doi: 10.3389/fimmu.2023.1254759
Received: 07 July 2023; Accepted: 21 August 2023;
Published: 07 September 2023.
Edited by:
Christoph Daniel, University Hospital Erlangen, GermanyReviewed by:
Agnes Szilagyi, Semmelweis University, HungaryCopyright © 2023 Rydberg, Aradottir, Kristoffersson, Svitacheva and Karpman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Karpman, ZGlhbmEua2FycG1hbkBtZWQubHUuc2U=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.