 Alessandra Aiello
Alessandra Aiello- Translational Research Unit, National Institute for Infectious Diseases Lazzaro Spallanzani- Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb) and Coronavirus disease-2019 (COVID-19), whose etiologic agent is severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), are currently the two deadliest infectious diseases in humans, which together have caused about more than 11 million deaths worldwide in the past 3 years. TB and COVID-19 share several aspects including the droplet- and aerosol-borne transmissibility, the lungs as primary target, some symptoms, and diagnostic tools. However, these two infectious diseases differ in other aspects as their incubation period, immune cells involved, persistence and the immunopathological response. In this review, we highlight the similarities and differences between TB and COVID-19 focusing on the innate and adaptive immune response induced after the exposure to Mtb and SARS-CoV-2 and the pathological pathways linking the two infections. Moreover, we provide a brief overview of the immune response in case of TB-COVID-19 co-infection highlighting the similarities and differences of each individual infection. A comprehensive understanding of the immune response involved in TB and COVID-19 is of utmost importance for the design of effective therapeutic strategies and vaccines for both diseases.
Introduction
Coronavirus disease-2019 (COVID-19), whose etiologic agent is severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) and tuberculosis (TB), that is caused by the bacterial pathogen Mycobacterium tuberculosis (Mtb), are the two-leading causes of death from a single infectious agent in humans. In the past 3 years, SARS-CoV-2 has been responsible for more than 7 million deaths, and Mtb for 4.5 million worldwide (1, 2).
SARS-CoV-2 is an enveloped RNA-based single-stranded virus recently emerged belonging to the Betacoronavirus genus. The first case of COVID-19 dates back to 2019 in Wuhan, China, and it is thought to be the result of a zoonotic spill-over event that likely occurred from bats and humans and finally caused the global pandemic (3). More than 700 million SARS-CoV-2 infections have been reported worldwide (to date, as of June 2023) (1). According to WHO, the largest number of confirmed cases are in Europe, Western Pacific and Americas (Table 1) (1). The spread of the virus was probably aided also by the onset of highly mutated forms of SARS-CoV-2, defined as “variants of concern” (VOCs), with enhanced transmission rate and with relatively lower morbidity and mortality compared to the ancestral strain (94, 95).
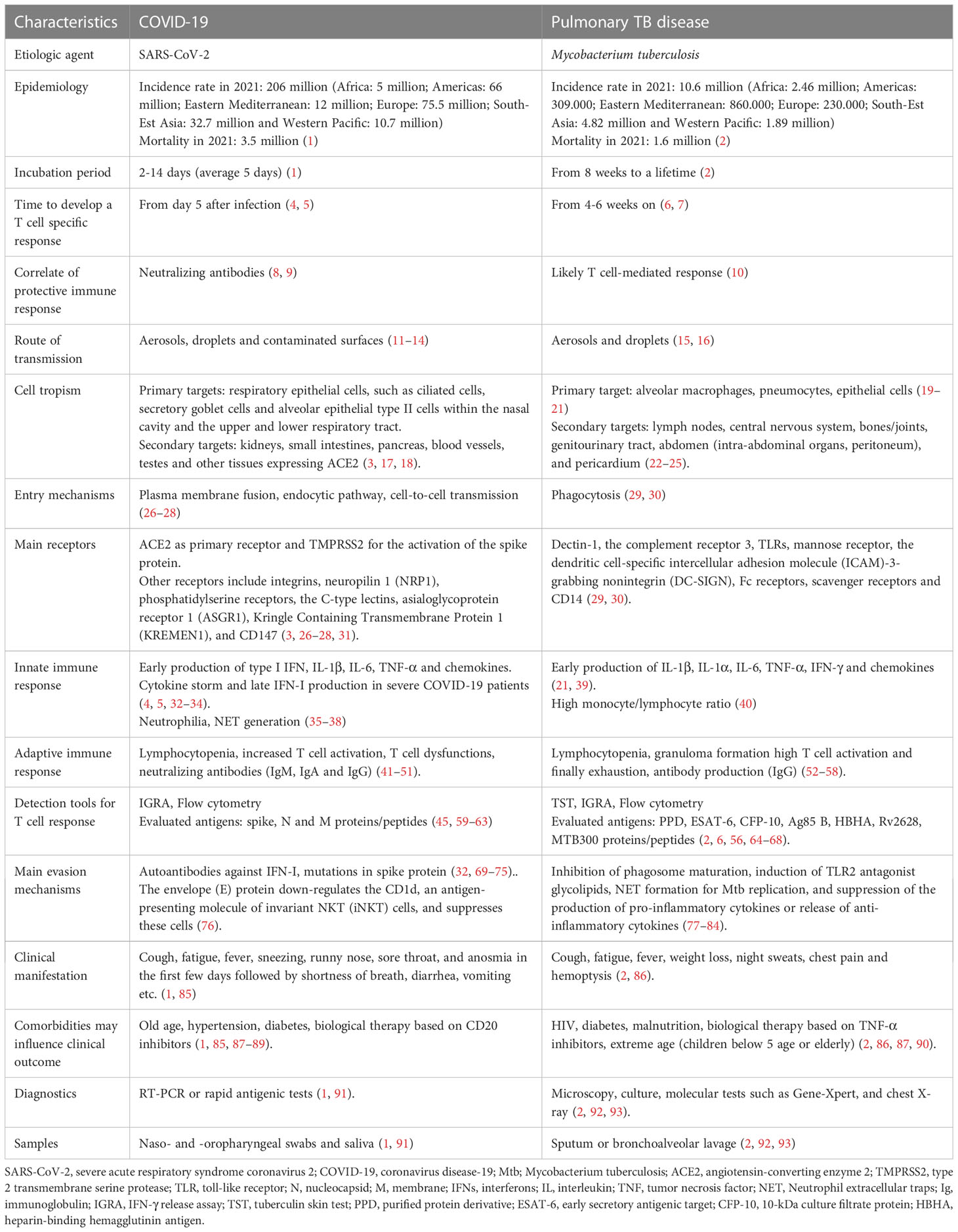
Table 1 Comparison of the features of SARS-CoV-2 and M. tuberculosis in terms of cell tropism, disease development and diagnosis.
On the contrary, Mtb is an ancient slow growing bacterium that has plagued the human population for thousand years. To date, it is estimated that one third of the world population is infected with Mtb (2), and about 5-10% of the Mtb-exposed and -infected individuals will progress to TB disease. In most of them bacilli are detectable in the sputum (15). According to WHO, the largest number of confirmed cases are in Africa and South-Est Asia (Table 1) (2).
Although Mtb and SARS-CoV-2 are distinct pathogens, they share several features summarized in Table 1. The main transmission route for both pathogens is via droplets (> 100 µm particles) or aerosols (< 100 µm particles) that are expelled by an ill individual by coughing, sneezing, talking, and breathing (11, 16). These particles can travel short distances in the air before being inhaled (12). However, for SARS-CoV-2, the infection can also occur as a result of contact with contaminated surfaces or objects on which virions can persist even for 72 hours (13, 14). Regarding Mtb, infection can also occur during autopsies (96) or during the spill of caseus material, i.e. from a scrofula when the cervical tuberculous lymphadenitis drains the material outside (97–99).
While SARS-CoV-2 shows a short incubation period (2-14 days) before symptoms onset, in Mtb infection it can range from eight weeks to a lifetime (1, 2) (Table 1 and Figure 1).
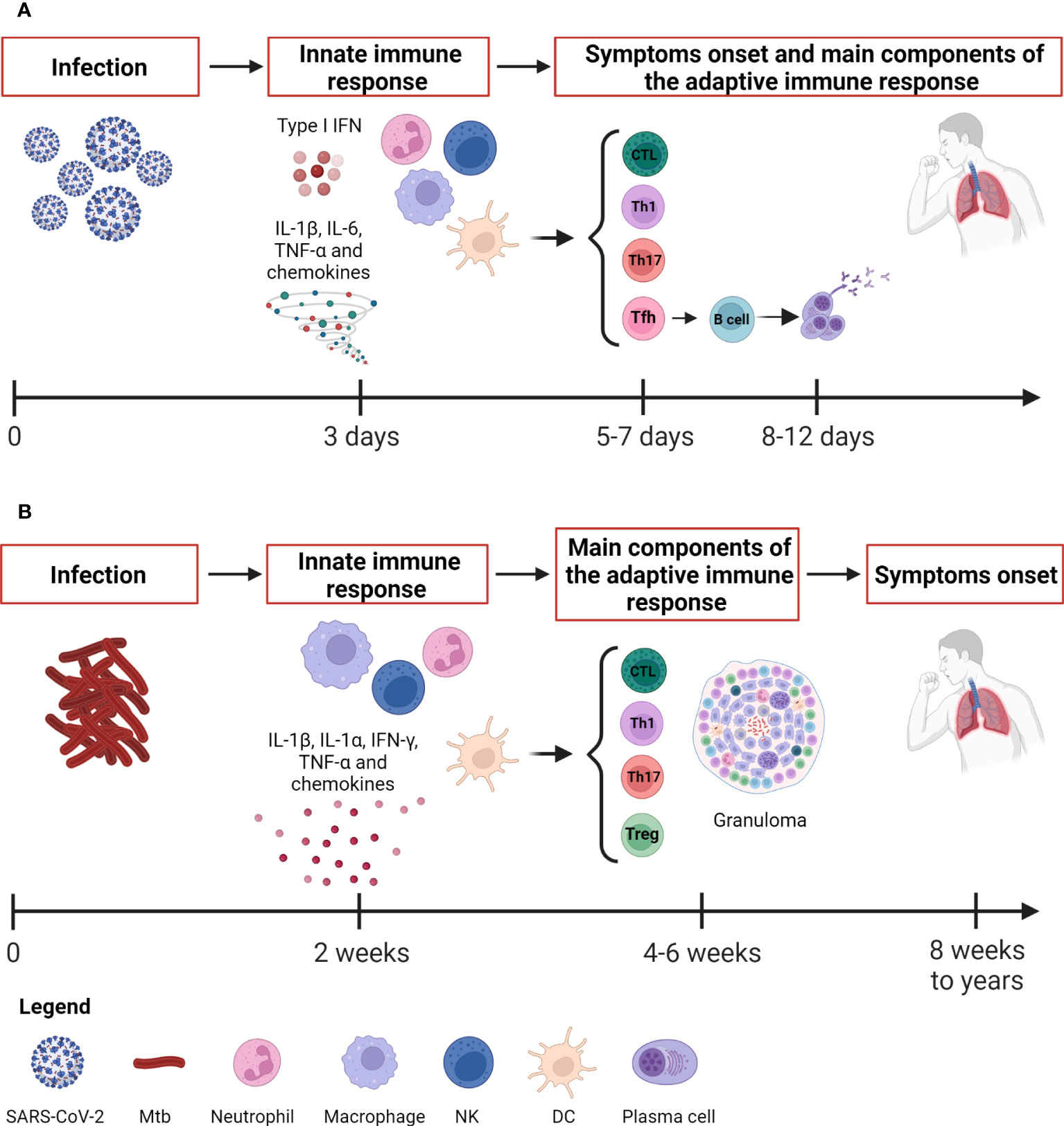
Figure 1 Kinetic of the immune response to SARS-CoV-2 and Mtb. (A) SARS-CoV-2 infection evolves rapidly. The innate immune response occurs after about 3 days and is detectable through immunoenzymatic assays and flow cytometry. The antigen-specific T cell response appears around 5-7 days concurrently also with the onset of symptoms, whereas the antibody response appears later around 8-12 days. The adaptive immune response is detectable by immunoenzymatic assays, flow cytometry and IGRA. (B) Mtb causes a slow-progressing infection that might result in the development of TB disease even after many years. The innate immune response occurs after about 2 weeks and is detectable through immunoenzymatic assays and flow cytometry as for SARS-CoV-2. The antigen-specific T cell response is detectable around 4-6 weeks by means IGRA, TST, immunoenzymatic assays and flow cytometry. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; Mtb, Mycobacterium tuberculosis; IFNs, interferons; DCs, dendritic cells; NK, natural killer; Tfh, T follicular helper lymphocytes; Th, T helper; IGRA, IFN-γ release assay; TST, tuberculin skin test. Created with BioRender.com.
Considering the route of transmission, it is not surprising that both SARS-CoV-2 and Mtb firstly infect the respiratory system causing symptoms such as cough, fatigue and fever. In addition, SARS-CoV-2-infected subjects also experience sneezing, runny nose, sore throat, and anosmia in the first few days followed by shortness of breath, diarrhea, vomiting, etc. (85), whereas in TB patients weight loss, night sweats, chest pain and coughing up of blood were reported (86). This similarity in symptoms might make the diagnosis difficult; however, in most cases the COVID-19 symptoms are short-lived compared to those of TB, which has a long incubation with long-lasting symptoms duration.
Both agents can be detected in respiratory samples such as nasopharyngeal swab or saliva for SARS-CoV-2, and sputum or bronchoalveolar lavage (BAL) for Mtb.
The diagnosis can require different tools. For SARS-CoV-2 infection, molecular swab is the first choice in case of suspected symptomatic individuals, contacts of confirmed cases with symptoms and for the screening of health workers. In other contexts, it is recommended to use rapid antigenic tests that are less labor-intensive and costly and can provide results in less than half an hour (91) (Table 1).
Regarding Mtb, two main types of tests are used to determine the traditionally called latent infection, now defined “tuberculosis infection” (2): the tuberculin skin test (TST) and interferon (IFN)-γ release assays (IGRA). For patients with suspected pulmonary TB, the Center for Disease Control (CDC) recommends performing an acid-fast-bacilli smear on three different sputum specimens (92). Moreover, Gene-Xpert (Cepheid, Sunnyvale, CA, USA) is a widely accepted diagnostic test for TB detection in direct smear negative cases (93).
Notably, SARS-CoV-2 and Mtb-infected individuals show a diverse spectrum of clinical manifestations. Patients infected with SARS-CoV-2 can experience a clinical outcome ranging from asymptomatic to mild/moderate infection up to severe disease (particularly with Wuhan strain and in those not vaccinated), which can also progress to acute respiratory distress syndrome (ARDS) (1). Indeed, SARS-CoV-2 can interfere with the host immune system leading to hyperinflammatory state, immune dysregulation, and extensive lung damage (100, 101).
Differently, Mtb-exposed individuals remain clinically asymptomatic due to the development of an immune response that controls Mtb replication (102, 103). It has been shown that some individuals heavily exposed to Mtb can clear the infection early before the emergence of the adaptive immune response, can keep a negative score to the TST and IGRA, and therefore do not show any evidence of infection (104). The lack of a detectable adaptive immune response in these resistant individuals suggests the key role mediated by the local innate immunity. The difficulty of treating and eradicating Mtb is related to the ability of the mycobacteria to survive and replicate within human cells.
In both infections, the clinical manifestations may be more severe in presence of comorbidities. In this regard, they share similar risk factors in terms of comorbidities as advanced age (87), and diabetes (90), although they have specific peculiarities as hypertension and biological therapy with CD20 inhibitors for COVID-19 and HIV infection, malnourishment and biological therapy based on TNF-α inhibitors for TB (90) (Table 1).
An effective and timely immune response plays a pivotal role in affecting the clinical course of both COVID-19 and TB. This review aims to provide an overview of innate and adaptive immune responses induced after the exposure to Mtb and SARS-CoV-2 highlighting the similarities and differences of each individual infection and their crosstalk in TB-COVID-19 co-infection.
Cell tropism and entry mechanisms
Viral entry is the first and pivotal step for the viral life cycle. Not surprisingly, blocking virus entry is a primary target of several therapeutic strategies to prevent the subsequent steps and inhibit viral replication and host cell pathology (3). Although both SARS-CoV-2 and Mtb are airborne pathogen entering via droplets, and primarily infect the human respiratory system, they differ by cellular tropism and entry mechanisms.
SARS-CoV-2 and cell tropism and entry mechanisms
SARS-CoV-2 has a broad spectrum of tropism. The human angiotensin-converting enzyme 2 (ACE2) represents the major cellular entry point for the virus, thus the expression of ACE2 defines which tissues can be potentially infected by SARS-CoV-2 (3, 26). The epithelial cells such as subset of ciliated cells, secretory goblet cells and alveolar epithelial type II cells within the nasal cavity and the upper and lower respiratory tract, represent the primary targets for the initial infection and spread of SARS-CoV-2. In this regard, the higher amount of viral RNA was found in ciliated and epithelial progenitors (105). Interestingly, although the human respiratory tract is the main target for the virus due to its airborne transmissibility, ACE2 expression in kidneys and gastrointestinal tract is even higher than the lungs (17). Notably, extrapulmonary organs such as the kidneys, small intestines, pancreas, blood vessels, testes and other tissues can be additional targets for SARS-CoV-2, thus explaining the variety of symptoms associated to the infection (17, 18).
SARS-CoV-2 gains access to cells mainly through two possible routes, the plasma membrane fusion and the endocytic pathway. The entry route used by the virus is dependent on the expression of cell surface proteases, which are needed for the activation of the viral protein (27, 28), and it is primarily mediated by the structural protein spike, a trimeric glycoprotein that binds to the ACE2 (26). After binding, spike undergoes a conformational change that allows the proteolytic cleavage before membrane fusion (Figure 2).
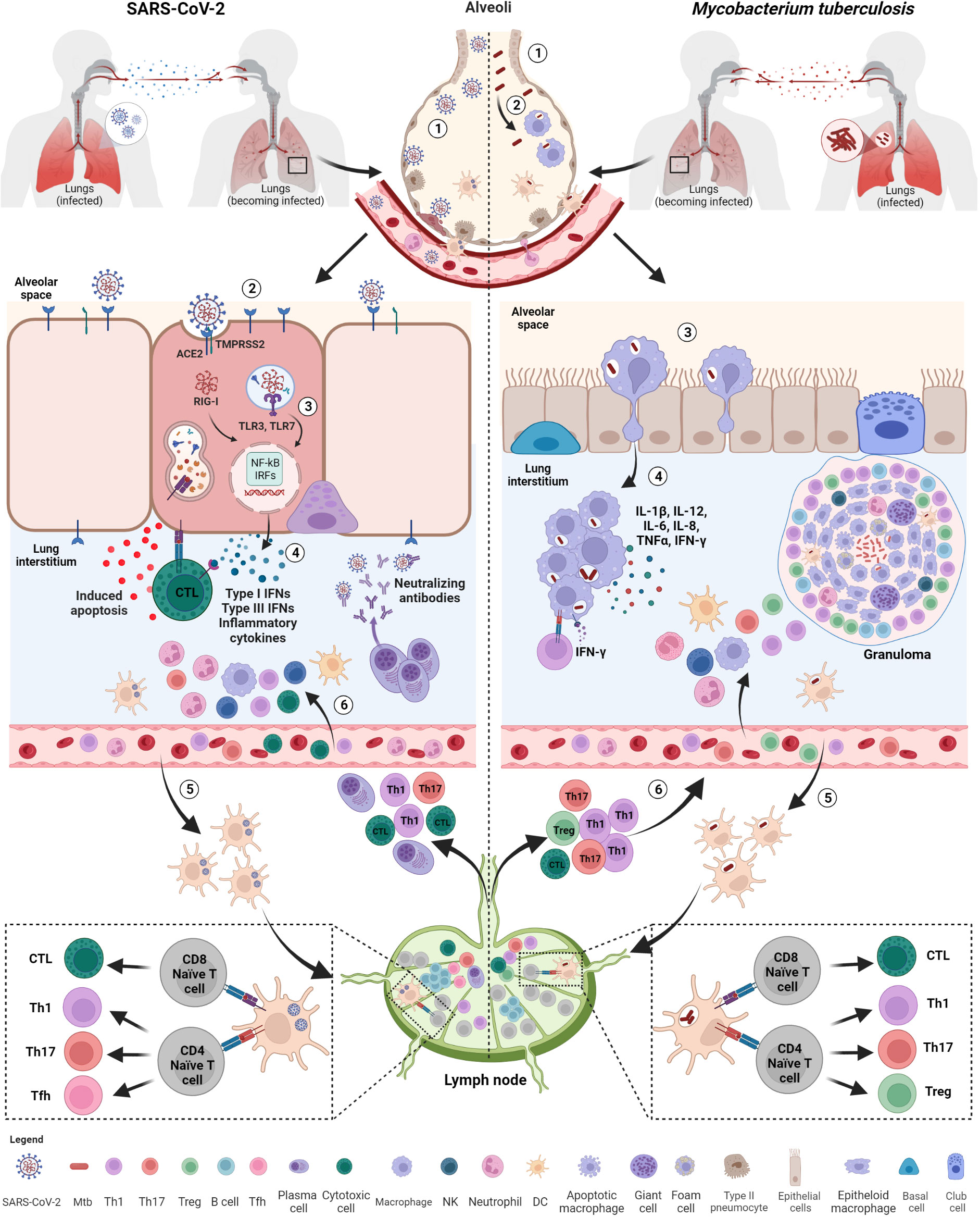
Figure 2 Initial immune response after exposure to SARS-CoV-2 and Mtb. Both SARS-CoV-2 and M. tuberculosis (Mtb) are transmitted by aerosols or droplets. SARS-CoV-2 infection (1): virions enter into the airways and (2), once arrived in the lung, infect epithelial lung cells via recognition and binding of the spike protein to the ACE2 cell receptor. (3) Viral RNA, once released inside the cells, is recognized by endosomal (TLR3, TLR7) or cytosolic (RIG-I) receptors and activate downstream signaling pathways (NF-kB and IRFs) (4) leading to the release of IFNs, pro-inflammatory cytokines and chemokines favoring immune cell recruitment, including neutrophils and DCs. (5) Infected DCs migrate to the lymph nodes for T and B cell priming. (6) Primed T cells and plasma cells go back to the infection site via blood where they exert their functions, including apoptosis induced by cytotoxic T cells and viral neutralization. Mtb infection: (1) Mtb bacilli enter into the airways and (2) are phagocytosed by alveolar macrophages. (3) Alveolar macrophages migrate to lung interstitium, where they form aggregates and (4) release cytokines promoting the recruitment of immune cells, such neutrophils, macrophages and DCs. (5) Infected DCs migrate to lymph nodes to prime T cells that are recruited at the infection sites where they contribute to the formation of the organized granuloma. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; Mtb, Mycobacterium tuberculosis; ACE2, angiotensin-converting enzyme 2; TMPRSS2, type 2 transmembrane serine protease; TLR, toll-like receptor; IFNs, interferons; RIG, retinoic acid-inducible gene-I; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; IRFs, interferon regulatory factors; DCs, dendritic cells; NK, natural killer; Tfh, T follicular helper lymphocytes; Th, T helper. Created with BioRender.com.
Spike activation can occur either at the cell surface or in endosomes and consists of two different proteolytic events. The first proteolytic event occurs during spike biosynthesis and it is mediated by the host pro-protein convertase furin that cleaves the polybasic S1/S2 junction (106) generating the two subunits S1 and S2 non-covalently linked and with different roles in the viral entry (107). The amino-terminal S1 subunit includes a receptor-binding domain (RBD) that is involved in the initial recognition of ACE2 receptor (108), whereas the carboxy-terminal S2 presents highly conserved regions that catalyse the fusion between viral and host cell membranes, crucial to release the viral RNA genome and start the replication in the target cell. A further cleavage at the S2’ site is needed to expose the S2’ fragment, a highly hydrophobic fusion peptide that starts the fusion of membranes (109, 110).
Interestingly, TMPRSS2, which is a type 2 transmembrane serine protease (TTSPs) expressed in the human upper and lower respiratory tract, heart, prostate and gastrointestinal tracts (111–113), has been shown to prime spikes on cell surface thus allowing the entry via membrane fusion (26). In the absence or insufficient availability of cell surface proteases, in particular TMPRSS2, SARS-CoV-2 prefers to enter via clathrin-mediated endocytosis (114). In this case, the conformational modifications of the spike occur in the acidic environment of endosomes and its cleavage is mediated by the members of the cathepsin family (e.g. B and L). While the virus takes 10 minutes to enter the cells via cell surface membrane fusion, the pH-dependent endocytosis process needs about 40–60 minutes after infection (28).
The cleavage of S1/S2 can have an impact on viral fitness and transmission, thus affecting viral infectivity (115). Notably, during the COVID-19 pandemic, several mutations have accumulated in S1 and S2 subunits of the spike causing the emergence of several SARS-CoV-2 VOCs capable of escaping the immune system, while preserving the steps of activation of the spike protein. The different infectivity rate in the epithelial cells of the nose, bronchi, and lung by SARS-CoV-2 VOC is correlated with the different protease expression, subsequent transmissibility, and severity of disease (18, 87, 116–118).
Emerged Omicron subvariants are less dependent on TMPRSS2-mediated spike activation at the plasma membrane, showing a reduced replication of the virus in the lung and intestinal cultures, while a similar replication rate was observed in the nasal epithelia compared to the Delta variant (117, 119, 120). Likely, this modified tropism allowed a major air transmission of the virus, in accordance with the highest rate of spread observed in the latest variants compared with the ancestral one (69). Moreover, the different spike protease tropism resulted in the diminished pathogenesis in the lung.
Besides TMPRSS2, other TTSPs or metalloproteases can mediate SARS-CoV-2 entry. For instance, TMPRSS2 and TMPRSS4 promote viral entry into human enterocytes of the proximal digestive tract (121), and matrix metalloproteases (MMPs), such as ADAM10 and ADAM17, seem to be involved in the cleavage at the S2 site in cells lacking TMPRSS2 (122–124). Moreover, coagulation factors, such as factor Xa and thrombin, can directly cleave spike protein at both cleavage sites and thus further contributing to infection at the stage of viral entry (125, 126).
Furthermore, other molecules have been suggested as alternative receptors for the SARS-CoV-2 entry process including integrins, neuropilin 1 (NRP1), phosphatidylserine receptors, the C-type lectins, asialoglycoprotein receptor 1 (ASGR1), Kringle Containing Transmembrane Protein 1 (KREMEN1), and CD147, as reviewed by Jackson and colleagues (27, 31).
Notably, SARS-CoV-2 could also infect cells through other mechanisms that allow the virus to escape the immune recognition favoring its spread in the host. In this regard, SARS-CoV-2-infected cells can directly fuse with adjacent cells expressing ACE2 through S1/S2 cleaved SARS-CoV-2 spikes resulting in the formation of multinucleated cells or syncytia (127, 128). The syncytia formation favors a cell-to-cell transmission of the virus without even the need to assemble viral particles or to release the virus in the extracellular environment (129). SARS-CoV-2-induced multinucleated pneumocytes and syncytia formation is a feature of severe COVID-19 patients, suggesting their involvement in the COVID-19 pathogenesis (130–132). Moreover, these structures might cause direct cytopathic effects to lymphocytes. In this regard, Zhang and colleagues reported that lymphocytes could be internalized by syncytia by forming cell-in-cell structures and leading to cell death (133).
Another possible mechanism for viral entry is mediated by extracellular vesicles (EVs) containing particles or viral components well documented in SARS-CoV-2-infected cells (134).
Regardless of the mechanism and molecules involved in SARS-CoV-2 entry, the virus replicates triggering the host immune response.
M. tuberculosis and cell tropism and entry mechanisms
As for SARS-CoV-2, the first interactions between bacteria and host occur in the lungs after the inhalation of the aerosolized Mtb. The size of Mtb droplets (2–5 µm particles) is important to ensure the passage through the upper respiratory tract into the alveolar space, where bacilli primarily encounter pneumocytes, epithelial cells (AEC), and alveolar macrophages (AMs) with anti-bacterial capacities (19–21). On the other hand, larger droplets can be stuck in the upper airways or oropharynx probably explaining the onset of the extrapulmonary forms of TB localized in the oropharynx but lacking evidence of concurrent pulmonary disease (135).
Once entered into the airways, Mtb is phagocytosed by AMs, which are permissive for infection establishment. In the upper airway, Mtb invades the specialized epithelial cells called microfold cell (M cell) through the binding to the scavenger receptor B1 in both mouse and human tissue (136, 137). Similar to SARS-CoV-2, Mtb can disseminate to other organs including the lymphatics and lymph nodes that are the main sites of extrapulmonary TB (22). Lymphatic endothelial cells, the adipose tissue and the bone marrow have been identified as extrapulmonary niches where Mtb may persist for long time (23–25).
The receptors involved in the Mtb entry into cells have not been fully demonstrated. Phagocytosis of Mtb by macrophages seems not occur via a single receptor-mediated pathway, but rather it seems to be mediated by multiple receptors including dectin-1, the complement receptor 3, mannose receptor, the dendritic cell-specific intercellular adhesion molecule (ICAM)-3-grabbing nonintegrin (DC-SIGN), Fc receptors, scavenger receptors and CD14 (29). Other receptors, such as Toll-like receptors (TLRs) are involved in the recognition of mycobacteria. To enable their entrance into AMs, mycobacteria exploit a group of pathogen-associated molecular patterns (PAMPs) expressed on its surface, including mycobacterial lipoproteins such as the 19 kDa surface antigen LpqH, which acts as an adhesin playing a crucial role in both host-pathogen interactions and pleiotropic immune regulation through the engagement of the TLR1/TLR2 (30). The downstream signaling and the phagosomal fate depend on the type of receptor engaged during the phagocytosis.
Macrophages containing Mtb then migrate from the air space to the lung interstitium in an IL1-R signaling- and ESX-1 secretion system-dependent manner (138, 139). This is the first step preceding the formation of the granuloma, the pathologic hallmark of TB (Figure 2).
Innate immune response
Whereas Mtb causes a slow-progressing infection that might result in the development of TB disease even after many years, the SARS-CoV-2 infection evolves rapidly causing COVID-19 (Figure 1). Within the immunological response to Mtb and SARS-CoV-2, both the innate and adaptive responses play an important role. The innate immune response is a nonspecific response that serves as initial defense against pathogens. It consists of humoral components (cytokines, chemokines, interferons, complement and coagulation-fibrinolysis systems, and naturally occurring antibodies) and cellular components (natural killer cells, macrophages, dendritic cells and other innate lymphocytes). Innate immunity aids in controlling the infection, in the identification and eradication of infected cells as well as in the development of the adaptive immunity (59, 140).
Innate immune response to SARS-CoV-2
The heterogeneous course of SARS-CoV-2 infection depends on the immune response at the early stages of infection (141). Considering the rapid course of COVID-19, the capability of patients with asymptomatic or mild disease to control the infection is likely due to the innate immune response since the adaptive response occurs days later, with the T cell immunity preceding the B cell response occurring after 2 weeks (Figure 1).
Early on, an effective control of SARS-CoV-2 spread depends on the induction of a robust antiviral response and on the ability of alveolar macrophages to eliminate the virus and the infected cells through phagocytosis.
Immune cells resident within the lung recognize SARS-CoV-2 through several pathogen-recognition receptors (PRRs), such as TLRs (TL3 and TLR7), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and inflammasomes. As a result, downstream signaling pathways involving nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and interferon regulatory factors (IRFs) are activated inducing the production of multiple pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, several chemokines (CCL20, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL8 and CXCL16) (32, 33) and antiviral IFNs resulting in the initial inflammation. The local innate immune response attracts and activates into the site of infection further innate immune cells such as neutrophils, monocytes, dendritic cells (DCs), natural killer (NK), and innate lymphoid cells aimed to promote viral clearance (142) (Figure 2). Consequently, the combined action of innate immune cells, cytokines, and chemokines may have an impact on the outcome of SARS-CoV-2 infection (143).
Although SARS-CoV-2 induces a pro-inflammatory state, there are reports of reduced IFN release (70, 144); in fact, SARS-CoV-2 is more effective at suppressing IFN responses compared to other respiratory viruses (71). Type I IFN, which includes IFN-α and IFN-β, represents the primary defensive response against viral infections by the induction of antiviral effector molecules encoded by IFN-stimulated genes (ISGs) and immunomodulatory responses (145). In SARS-CoV-2-infected individuals, the presence of a quick type I IFN production soon after infection contributes to protection against critical illness as observed in studies conducted in individuals exposed to COVID-19 cases (4, 5, 34).
On the contrary, if a strong and rapid antiviral response is lacking, the ongoing infection can lead to an exuberant release of cytokines and chemokines that is amplified by the further infiltration of circulating immune cells, finally provoking the so-called “cytokine storm”, which can be caused by infectious and non-infectious agents, and which in COVID-19 is responsible for the immunopathology associated with its severe presentation (141). Based on the evidence, individuals with highly compromised IFN-I response, which means no IFN-β and low IFN-α production and activity due to neutralizing auto-antibodies or inherited errors of type I IFN immunity, do not control the primary SARS-CoV-2 infection and they are more at risk of fatal COVID-19 (70, 146–148). Moreover, a low number and an impaired functionality of plasmacytoid dendritic cells (pDCs), which are the main IFN producers, have been found in bronchoalveolar lavage fluid (BALF) from severe or critical patients compared to the moderate ones (149). Also, a lower frequency of circulating pDCs was found in samples from SARS-CoV-2-infected individuals than in controls (150).
In vitro studies have shown the presence of a huge amount of NF-kB-dependent proinflammatory mediators in BALF (CCL2, CCL3, CCL4, and CXCL10) (151) and in circulation (IP-10, IL-6, and IL-8, IL-1, IFN-γ, IL-17, TNF-α, MCP-1, G-CSF, GM-CSF, IL-1RA, CCL2, CCL3, CCL5, CCL8, CXCL2, CXCL8, CXCL9, and CXCL16) (32, 60, 70, 152–155).
Patients with COVID-19 generally show migration of neutrophils and monocytes into the nasopharyngeal mucosa in response to chemokines released by infected epithelial cells (e.g. CXCL1, CXCL3, CXCL6, CXCL15, CXCL16, and CXCL17) (156).
Once reached the lung, neutrophils as phagocytes may exert a protective role in the clearance of the infection by secreting leukotrienes, reactive oxygen species (ROS), and forming neutrophil extracellular traps (NET), which are aggregates of extracellular DNA, histones, microbicidal proteins and proteases aimed to entrap and kill pathogens. However, neutrophils are known to be implicated in COVID-19 pathology as hyperinflammation drivers through increased cytokine production and cell degranulation (35). Indeed, their extensive and prolonged activation causes an hyperinflammatory environment and cellular infiltrations that may result in the tissue damage observed in the ARDS and increased mortality (36, 37). Indeed, a high neutrophil-to-lymphocyte ratio (NLR), that is a marker of inflammation and infection, and NET DNA complexes have been found in severe COVID-19 compared with mild/moderate cases or healthy controls (38).
In addition to NET generation, another source of hyperinflammation associated with COVID-19 is the activation of the NLRP3-inflammasome due to the interaction of the nucleoprotein (N) with NLRP3 (157). In this regard, a study conducted in an ACE2 humanized mouse model of COVID-19 showed that, in response to infection, macrophages activate inflammasomes causing the release of IL-1β and IL-18 and undergo pyroptosis, thus favoring the pathogenesis of acute lung injury (158).
During SARS-CoV-2 infection, monocytes/macrophages are involved either as virus target or as producer of inflammatory cytokines and undergo phenotypical changes (159). Alterations in the phenotype of monocytes consisting of reduced antigenic presentation and dysregulated immune response have been observed (35). In the peripheral blood of COVID-19 patients there are cell subsets of mixed M1/M2 macrophages secreting IL-6, TNF-α and IL-10 and characterized by higher expression of CD80 and CD86 (35, 160–162).
NK cells are innate lymphocytes that are recruited along with macrophages and neutrophils in the lungs as confirmed by the analysis on BALF samples of COVID-19 patients (163). NK cells usually exert an antiviral activity through the production of the effector cytokines IFN-γ and TNF-α and limit tissue fibrosis (164). Regarding the protective role of NK cells against infection, Witkowski and colleagues reported that SARS-CoV-2-infected individuals with a higher NK cell number at hospitalization showed a more rapid clearance of viral load (165). Although during early stages of infection NK cells may contribute to control viral replication and dissemination, their migration in affected tissue may favor the enhancement of inflammation. In this context, a reduced peripheral cell count and functional impairment of NK cells with an enhanced expression of the cytolytic proteins perforin and granzyme B have been found in patients with severe COVID-19 (166–168).
CD1d-restricted NKT cells are other types of innate lymphocytes that are involved in antiviral immunity (169). To counteract their function, the envelope (E) protein of SARS-CoV-2 reduces the expression of the antigen-presenting molecule CD1d thus inhibiting the activation of innate NKT cells and enhancing SARS-CoV-2 virulence (76).
The activation of the innate immune system is essential to mount an effective adaptive immune response. In this regard, DCs, as professional antigen presenting cells (APCs), represent a point of junction between innate and adaptive immune response as they migrate to lymph nodes to activate naïve T lymphocytes (170).
Innate immune response to M. tuberculosis
The innate immune response to Mtb infection is multifaceted with several different cell types and functions involved. Upon pattern recognition, a variety of cellular functions, including phagocytosis, autophagy, and apoptosis will be launched by the host to clear or control Mtb (171–173). In particular, macrophages with antimicrobial mechanisms such as nitric oxide synthesis and antimicrobial peptides such as cathelicidin represent the first defense line against Mtb infection (174).
The investigation of the early events and host responses against Mtb in humans is very challenging and difficult as the progression of infection is generally slow and individuals often do not know the exact time of exposure or infection (175). Therefore, a validated model that recapitulates TB in human lungs is critical to support TB research. In this regard, a number of in vitro systems (176), spheroids (177), human airway organoids (178), and experimental animal models of TB such as zebrafish (179), mouse (180), guinea pig (181), rabbit (182) and rat (183) have provided new insights into the local events that occur during few days and weeks post Mtb infection. In particular, Mtb infection in nonhuman primates closely recapitulates human TB and these models can be used to study the full spectrum of infection outcome and pathology of TB (184).
Early in infection, the infected cells are activated and start to release some early mediators of inflammation such as TNF-α, IL-1α, IL-1β, IFN-γ and chemo-attractant molecules (e.g. CXCL5, CXCL8), some of which also characterize the early stages of SARS-CoV-2 infection (Figure 2). These soluble factors mediate the recruitment to the site of infection of different blood cell types including neutrophils, monocytes, macrophages and DCs (21, 39), which are necessary for starting early granuloma formation (139). These innate granulomas include cells that are not yet fully activated, thus favoring the dissemination of mycobacteria from infected macrophages to uninfected cells.
Notably, the EVs released from infected cells containing mycobacterial components, including lipoarabinomannan, the Ag85 complex and lipoproteins, have been shown to contribute to the migration of immune cells to the lungs (185). Moreover, EVs can modulate immune response by promoting the release of proinflammatory cytokines and by increasing autophagy and superoxide production (185, 186).
During the first 10 days post-infection, Mtb almost exclusively resides and replicates inside AMs, suggesting that these cells provide an early niche for Mtb growth (139, 187, 188). In a murine model of TB, Mtb was reported to be equally distributed between AMs, DCs and neutrophils 14 days post-aerosol challenge (189).
As already mentioned for SARS-CoV-2 and also known for other infections including Mtb, DCs play a crucial role by transporting bacteria from the site of infection to the draining lymph nodes (64) in order to prime naïve T cells and start an adaptive immune response (190, 191). An involvement of CCR2+ inflammatory monocytes in the Mtb delivery to pulmonary lymph nodes has also been reported (192). Notably, a higher monocyte/lymphocyte ratio is observed in Mtb-infected patients (40).
Neutrophils are other professional phagocytes that have been shown to be involved in the early innate immune response against Mtb through a direct antimicrobial activity and chemokines/cytokines production (193). They readily phagocytose Mtb and can destroy it via ROS, proteases and antimicrobial peptides (AMPs). They can also undergo apoptosis and microbe-containing apoptotic neutrophils can be phagocytosed by macrophages and DCs and then transported to the lymph nodes (194, 195).
In addition, mucosal-associated invariant T cells (MAITs) are a group of T cells restricted to a nonclassical molecule MR-1 and not to the classical major histocompatibility complex (MHC) molecules. MAITs are also involved in the early responses to Mtb by producing IFN-γ and TNF-α, and showing cytotoxic activity upon recognition of microbe-derived riboflavin metabolites (196).
Moreover, there is evidence for a role of NK cells in controlling Mtb infection, by killing the pathogen through antibody-dependent cellular cytotoxicity, directly targeting the Mtb by binding to cell wall components such as mycolic acid, arabinogalactan, peptidoglycan through receptors including TLR-2, NKp44, NKp46, and NK group 2D (NKG2D), promoting the maturation of phagolysosome and phagocytosis by producing cytokines such as IFN-γ and TNF-α and by killing Mtb-infected macrophages through the release of granules (perforin, granulysin, and granzyme) (175, 197–199). However, it is not well known whether the role of NK cells is as important as that of macrophages or cytokines such as IFN-γ or TNF-α.
Nonetheless, Mtb has evolved several strategies to evade the host’s immune system through its unique cell wall structure, intracellular survival, dormancy and the ability to modulate immune response. Mtb has adapted to survive and replicate in macrophages by inhibiting phagosome maturation (77–80) and promoting necrosis over apoptosis (200). Several types of programmed necrosis in response to Mtb infection, such as inflammasome-mediated pyroptosis and NET-associated NETosis have been identified (201–203). However, NETosis may facilitate the interactions between neutrophils and other immune cells rather than killing Mtb directly (81). Moreover, the formation of NETs can be induced via type I IFN signaling to favor MTB replication (82). Mtb inhibits also innate immune response by induction of TLR2 antagonist glycolipids (83). It also modulates the immune response through the release of molecules that suppress the production of proinflammatory cytokines or even by inducing the production of anti-inflammatory cytokines (84).
As for SARS-CoV-2, the control of Mtb infection requires a timely innate response as well as an effective adaptive response.
Adaptive immune response
The adaptive immune response comprises antibody and cell-mediated responses and takes approximately 2 to 3 weeks before we can measure it (59). It is involved in the specific recognition of pathogens and in the establishment of the immunological memory. Notwithstanding the importance of innate responses, a coordinated cellular immunity is crucial for disease control in both SARS-CoV-2 and Mtb infection.
T cell response to SARS-CoV-2
In the majority of cases, SARS-CoV-2 infection induces adaptive antigen-specific responses, viral clearance and immunological memory finally resulting in an asymptomatic or mild disease. However, a failure of the first line defense mechanisms, particularly of innate IFN, may act as triggering factor for viral proliferation and immune dysregulation. Indeed, the delayed/ineffective adaptive responses and exaggerated inflammatory response can promote immunopathogenesis of COVID-19, particularly ARDS (204–207).
Several lines of evidence from both human studies and animal model systems have shown that an effective T cell response is required to control and eradicate SARS-CoV-2 infection by releasing cytokines and other anti-inflammatory factors (208).
During the infection, subepithelial DCs present SARS-CoV-2-specific peptides through MHC class I and II molecules on the cell surface, thus promoting the activation of CD8+ and CD4+ T cells, respectively, which migrate to the lung after antigen exposure. Indeed, the lung is characterized by the presence of tissue-resident T cells with a memory phenotype (CD69+, CD103+/-, CD45RA,CCR7-) originated from the priming of naïve T cells (209). Interestingly, an involvement of EVs in the regulation of antigen presentation and T cell activation has also been reported (210).
While CD8+ T cells recognize and kill the infected cells, CD4+ T cells contribute to activate B cells for antibody secretion and CD8+ T cells to exert the cytotoxic activity, and to produce cytokines that favor immune cell migration at the site of infection (143).
Initial studies conducted by Grifoni and colleagues, and subsequently confirmed by others showed CD4+ and CD8+ viral specific T cell responses in most infected individuals mainly against spike antigen, although present also against other structural (nucleocapsid and membrane proteins) and non-structural SARS-CoV-2 antigens (61–63). Since spike protein has been identified as the most immunogenic antigen, it has been employed for many of the currently used SARS-CoV-2 vaccines (61, 62).
Unlike Mtb infection, the early development of antigen-specific T cell responses is generally observed within 7 days after the onset of COVID-19 symptoms, peaks at 14 days and may be detectable even if SARS-CoV-2 specific antibodies are lacking (5) (Figure 1). Several studies have shown that asymptomatic or pauci-symptomatic individuals are characterized by a strong SARS-CoV-2-specific CD4+ T cell response (41, 211–213). Surprisingly, CD4+ T cell responses were also observed in 40% to 60% of unexposed individuals likely because of the cross-recognition between SARS-CoV-2 and other “common cold” coronaviruses (63).
T cell activity has been associated with a less disease severity (8, 59). The critical role played by T cells in the protection against the severe disease has been highlighted also with the occurrence of different VOCs with an increased ability to escape neutralizing antibodies (214–216). Indeed, the spike-specific T cell response induced by both vaccination and natural infection seems to be not affected by the amino acid mutations that characterize the VOCs, including Omicron, in healthy subjects and in the vulnerable populations (217–221). Indeed, the availability of thousands of SARS-CoV-2 epitopes that may be recognized by T cells makes unlikely that the virus may successfully escape the T cell response by mutating the epitopes.
SARS-CoV-2 infection mainly support the differentiation of CD4+ T lymphocytes toward T helper 1 (Th1), T helper 17 (Th17) and T follicular helper (Tfh) cells (Figure 2).
An appropriate Th1 immune response is necessary for protection against COVID-19, as an early and rapid expansion of IFN-γ-secreting SARS-CoV-2-specific T cells was detected over the course of acute infection and was associated with viral clearance (42, 222) and mild disease (43, 223, 224).
Chauss and colleagues showed that asymptomatic SARS-CoV-2-infected individuals present in the BALF CD4+ T cells switched from a predominantly pro-inflammatory Th1 phenotype toward an IFN-γ and IL-10-producing phenotype that enable them the viral control without causing pathology (225). The mechanism behind the switching phenotype is triggered by cell-intrinsic complement that orchestrates an autocrine/paracrine autoregulatory vitamin D (VitD) loop to initiate Th1 shutdown. During this process, Vitamin D induces epigenetic changes in the CD4+ T cells and recruits transcriptional factors, including c-JUN, STAT3 and BACH2 finally resulting in the switch off of Th1 programs and in the IL-10 induction (225). In patients with severe COVID-19 these regulatory processes are lacking and thus exacerbated Th1 cytokine profiles are prevail (226).
The lack of a fine-tuned Th1 immune response can cause an exacerbated reaction that precedes cytokine storm promoting the differentiation of Th2 cells that are related to a poor prognosis (227). In this regard, Gil-Etayo and colleagues observed in COVID-19 patients a significant reduction in the percentage of Th1 and Th17 cells whereas a higher frequency of activated Th2 cells. Moreover, a higher number of senescent Th2 cells together with higher levels of IL-15 were observed in patients with a fatal outcome (227).
In addition, Th17 cells are strongly activated in severe COVID-19, thus favoring cell-mediated immunopathology through the production of IL-17 and GM-CSF (44). IL-17 released by Th17 cells induces the activation of monocytes/macrophages, DCs, and neutrophils which, in turn, increases the release of cytokines (IL-1, IL-6, IL-8, IL-21, TNF-α, and MCP-1), thus promoting the cytokine storm (44).
It has been reported that the polarization of CD4+ T cells toward Th17 instead of Th1 can be promoted by neutrophils as well as by the up-regulation of pro-inflammatory cytokines IL-1β, IL-6 and IL-23 (228).
Tfh cells are localized within the germinal centers of the secondary lymphoid organs and they are primarily involved in the activation and proliferation of B cells, and the production of high affinity antibodies (59) as well as in the assistance of CD8+ T cell functions (229).
In rhesus macaques CD8+ T cells are crucial for viral clearance especially when a reduced humoral response is present (230). In this regard, a weak CD8+ T cell response has been associated with a poor prognosis (45, 231). Indeed, a delayed or lacking CD8+ T cell response was found in patients with severe or fatal outcomes probably due to the inability of T cells to rapidly limit viral replication (59).
Besides CD4+ and CD8+ T cells, regulatory T cells (Treg) have been shown to play a critical role in SARS-CoV-2 infection, particularly as regulators of the inflammatory response. Perturbations in Treg phenotype, such as the reduced expression of Foxp3 and cytokines including IL-10 and TGF-β, have been associated with disease severity (232).
Quantitative and/or functional deficiency of T cells is associated with pathological processes responsible for tissue damage. Indeed, a characteristic hallmark of severe COVID-19 is the peripheral lymphopenia accompanied by a reduced count of monocytes, eosinophils, basophils, but not neutrophils (46). Possible explanations for T cell depletion is the SARS-CoV-2 infection of T cells through the binding of the spike protein to the CD147 or CD26 expressed on cell surface (233), their recruitment to infected site, or their apoptosis via Fas/Fas ligand or TNF (234–237). Furthermore, increased levels of IL-6, IL-10 and TNF-α may contribute to lymphopenia (47, 238). The prolonged peripheral lymphocytopenia increases the risk of secondary bacterial infections (88). Also, an immunosuppression following hyperinflammation in COVID-19 disease has been described, in particular NLRs and TLRs were shown to be associated to immunosuppression (239).
A reduced number of peripheral Treg cells has also been observed in severe cases of COVID-19, likely leading to the development of lung pathology (232).
As COVID-19 progresses, a different T cell functionality has also been observed. Early during the acute phase of SARS-CoV-2 infection, T lymphocytes are characterized by a highly activated cytotoxic phenotype, whereas in convalescent individuals they show a polyfunctional and memory phenotype (41, 44, 47, 240). CD8+ T cells expressing markers of exhaustion such as PD-1+ TIM3+ increase over the infection and this scenario seems to be related to IL-10 blood levels. The hyper-activation of T cells along with the dysfunctionality of DCs and Tregs may increase the overwhelming alveoli inflammation and cytokine storm in COVID-19 (241).
In light of what is reported in literature, an efficient T cell response is fundamental for viral clearance.
Antibody response to SARS-CoV-2
The antibody response usually appears by 1-2 weeks later than SARS-CoV-2 specific T cell response that is detectable 5-6 days post-infection (4, 5) (Figure 1). Within few days post-infection, B cells are rapidly activated in extrafollicular foci to differentiate in short-lived plasma cells that predominantly produce IgM antibodies but also IgG or IgA-switched to initially stem viral infection, while waiting for the production of antibodies with higher affinity. The first IgM, IgA and IgG are measurable in the sera between 8 and 12 days after symptom onset (48). Subsequently, within the germinal centers in the secondary lymphoid organs, antigen-specific B cells undergo somatic hypermutation and isotype-switching resulting in the production of high-affinity IgG antibodies that mainly recognize nucleocapsid and spike proteins (242, 243). Cross-sectional and longitudinal studies showed that Enzyme-linked immunosorbent assay (ELISA) titers and neutralizing antibodies are detectable around 14 days after symptom onset, peak in 3 to 4 weeks, and decline subsequently causing a reduction of protection and increasing the risk of SARS-CoV-2 re-infection (9, 49, 50, 244).
However, it has been observed that anti-RBD antibodies, neutralizing activity and RBD-specific memory B cells are mostly stable between 6 and 12 months after infection (245, 246), likely owing to the presence of a long-lived plasma cell compartment located in the bone marrow (247–249).
The protective role of the antibodies is limited to those specific for the viral spike protein because they neutralize the virus by hindering the binding between spike and ACE2 receptor and thus blocking its entry, and by promoting effector functions via the binding to the complement and Fc receptors (250).
In the case of neutralizing antibodies, the engagement of Fc receptors can potentiate neutralization (251, 252). Non-neutralizing antibodies may promote antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). In this regard, high ADCC activities are detected mainly in hospitalized patients and showed a kinetic similar to antibody titers with a peak at 2-4 weeks post-infection followed by a gradual decline (253–255).
Most of the antibodies are directed against epitopes localized in the receptor-binding motif (RBM) within the RBD of spike, whereas a minority is directed against the N-terminal domain (NTD) (256–258). Anti-NTD antibodies have less neutralizing activity than anti-RBD antibodies and they may act by interfering with the conformational changes necessary for fusion or binding to receptors such as transmembrane lectins DC-SIGN, L-SIGN and SIGLEC1 (259, 260).
The antibody response, either qualitative and quantitative, is dependent on the amount of the antigen and on the activity of the germinal centers. In this regard, patients with severe COVID-19 show higher titers of total and neutralizing antibodies than mild or asymptomatic patients, likely due to the stronger antigen response (261, 262). On the other hand, individuals undergoing B-cell depleting therapies, such as anti-CD20, show an impaired antibody response that is associated with a more severe course of COVID-19 (263).
While circulating antibodies may help to control viral dissemination within the host, mucosal antibodies such as the dimeric form of IgA that is secreted in the upper respiratory tract, play an important role in preventing the transmission of SARS-CoV-2, present a stronger neutralizing activity than circulating antibodies, and contribute to protection against re-infection (51, 264). Indeed, SARS-CoV-2 specific IgA have been found in saliva samples collected from infected individuals (249).
During SARS-CoV-2 infection, also autoantibodies targeting self-antigens, including type I IFN, were identified in some COVID-19 patients, particularly in those with a severe disease that are characterized by a reduced IFN production, as mentioned above (32, 70, 71). COVID-19 patients are also characterized by changes in B-cell subpopulations. In particular, increased number of proliferating, metabolically hyperactive plasma blasts and reduction of memory B cells have been found in patients with severe disease, whereas they disappeared with convalescence (261, 265, 266).
Nonetheless, SARS-CoV-2 has evolved different strategies to escape the immune response. Unlike bacteria such as Mtb, RNA viruses are usually characterized by high mutation rates. SARS-CoV-2 exploits this ability to accumulate mutations in the spike protein in order to avoid the immune recognition by neutralizing antibodies and to increase its transmissibility (69). In particular, the emerging VOCs has accumulated mutations mainly located in the RBM, in part due to the pressure exerted by the host immune system. It has been proposed that the concurrent onset of multiple mutations in the spike protein might occur during the prolonged infection in immunocompromised patients resulting in the emergence of variant strains (72, 73). These mutations increased affinity of the virus for the ACE2 receptor and improved its ability to evade the neutralizing antibody response induced by natural infection or following vaccination with the spike protein derived from the ancestral strain (74, 75).
Altogether, the humoral response has been shown to play a crucial role in the host immune protection against SARS-CoV-2 together with the T cell response.
T cell response to M. tuberculosis
The infected monocytes, macrophages and DCs are thought to be key elements leading to Mtb dissemination and granuloma formation (39, 267). The infected professional antigen-presenting DCs travel to the lung draining lymph nodes where priming of naïve CD4+ and CD8+ T cells is initiated (52, 64, 190, 191, 268). Priming is a critical step for the initiation of the adaptive immunity that is crucial to hinder bacilli dissemination and control the infection. However, the adaptive (T cell) response takes longer to appear in infected hosts because Mtb or its antigens are transported late into the lymph nodes for T cell priming (269). In mice this occurs within 2-3 weeks post-infection (64, 65, 270), but in humans and non-human primates Mtb-specific T cell response in the periphery, measured as a response to TST, or IGRA, is usually not detectable until 4–6 weeks post-infection (6, 7).
It was found that Mtb-infected DCs in the lymph node are capable to release soluble and intact Mtb antigens that can be caught by uninfected DCs and efficiently presented to naïve CD4+ T cells to optimize CD4+ T cell priming and to initiate the adaptive immune response (271). Surprisingly, the capacity of Mtb-infected DCs in activation and proliferation of naïve Mtb-specific CD4+ T cells in the murine lymph node was found to be impaired likely due to lower MHC class II-peptide presentation by these infected APCs (189).
The primed T (and likely B) lymphocytes can then move to the site of infection and contribute to the formation of the organized granuloma that consists of modified macrophages as epithelioid cells and multinucleated giant cells accompanied by neutrophils and DCs in the center, infiltrated immune cells including granulocytes, antigen-specific T cells and few B cells in the periphery, with variable degrees of fibrosis or central caseous necrosis (Figure 2) (272, 273). Although the mechanisms driving protection and pathology within the granuloma microenvironments are still poorly understood, such mechanisms can be very important for the prognosis, and outcome of the disease (52).
Notably, granuloma structure and function protect the host from the dissemination of the infection, but it is also a way to facilitate the persistency of the infection (274). In fact, sterilizing immunity following Mtb infection is rare and even in the presence of a robust adaptive immune response to Mtb, the nature of the granulomas as well as the immune escape mechanisms of Mtb can restrict the host immune response to reliably eliminate the infection. This leads to develop a controlled infection, traditionally called latent infection, in most infected individuals. Mtb can survive in a dormant (non-replicating) state favored by hypoxic conditions inside solid granulomas that makes it difficult to be detected by the immune system (53, 275).
Within the granuloma, Mtb antigens persistently stimulate immune cells leading to immune activation, chronic inflammation, and finally cell exhaustion (54). Different T cell types and functions can exert a beneficial or even detrimental role. The peripheral localization of T cells restricts their access to the central core of the granuloma, where Mtb-infected macrophages reside, and this can limit the interactions between macrophages and lymphocytes. Moreover, a Mtb-induced immunosuppressive environment has been indicated in the granuloma in which IL-10 impairs Th1 activity and lysis of infected macrophages (276).
The important role for T-cell immunity and particularly IFN-γ-producing Th1 in controlling Mtb infection has been demonstrated in humans (10) and animal models (277, 278). IFN-γ is a key factor involved in CD4+ T cell-mediated protection by increasing autophagy and promoting phagosome maturation in macrophages (79) inducing the production of antimicrobial peptides (279), and limits the accumulation of non-protective CD4+ T cells in the lung vasculature (280).
In humans, HIV infection appears to be an important risk factor for TB disease progression likely due to CD4+ T cell depletion (10, 90). Also, depletion of CD4+ T cells in cynomolgus macaques with acute Mtb infection leads to exacerbated disease in most animals (278). Moreover, TB disease increases HIV replication, in vivo and in vitro through a mechanism of immune activation (281, 282).
The activation and proliferation of antigen-specific naïve CD4+ T cell subsets strongly depends on the cytokine milieu released by APCs. Particularly, macrophages are the main source of IL-1β, IL-6, IL-18, TNF-α, IL-10, and TGF-β, while DCs are the main producers of IL-12, IL-23, IL-27 and IFN-β (39). For instance, IL-12 produced by DCs differentiates naïve CD4+ T cells to Th1 which promote activation of the cell-mediated immunity needed to counteract intracellular pathogens (55). These cells secrete pro-inflammatory cytokines such as IL-2, IFN-γ and TNF-α to activate macrophages and cytotoxic CD8+ T cells (283). TNF-α is known to be necessary for the formation of a well-organized granuloma and host protection, as confirmed by the higher risk of developing TB disease and disseminated infection in subjects who underwent anti-TNF-α treatment (284, 285).
Activated cytotoxic CD8+ T cells and macrophages kill and eliminate pathogens and infected host cells by cytotoxic effector molecules such as perforin, granzymes and granulysin and by death receptor/ligand ligation (286).
Furthermore, IL-23 produced by DCs drives differentiation and functionality of Th17 cells that produce IL-17 which is a cytokine involved in neutrophil recruitment (287). IL-17 signaling appears to be essential for recruiting neutrophils to the site of infection early after Mtb infection in murine models (288), but a dysregulated production of this cytokine was also found to be associated with immunopathology driven by excess neutrophil recruitment and inflammation (289, 290).
Although inflammation is required for an effective immune response against harmful pathogens, the balance between pro- and anti-inflammatory cytokines is critical to control the disease and lung damage during Mtb infection (56, 291). Anti-inflammatory cytokines such as IL-4, IL-5, IL-13 released by Th2 cells and IL-10 and TGF-β by regulatory T cells are needed to suppress inflammation during immune response. However, these cells may promote long-term persistence of Mtb by favoring active immunosuppression rather than the expected tissue repair response (292).
In patients with TB disease, TST positive, in vitro PPD stimulation induced the production of IL-10, IFN-γ, and cell proliferation, whereas in those TST-negative PPD induced IL-10 but not IFN-γ release, without cell proliferation (293).
Altogether, a better understanding of the dynamically balanced immune response is fundamental for therapeutic strategies and subsequently for vaccine development.
The role of B cells and antibodies in TB
Although Mtb infection induces strong antibody responses, the role of antibodies and B cells in TB has not been fully elucidated. Previous studies on B cell depletion have failed to definitively establish a role for these cells or antibodies in Mtb infection and control, although recent studies have demonstrated potentially protective roles of antibodies in humans and non-human primates (NHPs) after intravenous bacille Calmette-Guérin (BCG) vaccination (294, 295).
It has been shown that TB disease is associated with decreased B cell count and function compared with individuals who are infected with Mtb but without any clinical symptoms, suggesting that TB patients may be less able to develop successful antibody responses against Mtb (296–298).
Moreover, distinct glycosylation patterns on the Fc part of the antibodies (296), and isotype skewing to less potently immune-activating variants like IgG4 have been considered for this altered functional response (298, 299).
Surprisingly, heavily Mtb-exposed individuals who “resisted” to infection showed higher antibody functionality compared to those with TB infection, indicating an important role of antibodies in early protective immunity (300, 301).
Studies have shown that the interaction of Mtb with macrophages can be affected by antibodies in a variety of ways (57, 58). For instance, bacterial opsonization may alter vesicular trafficking and macrophage signaling. Moreover, the binding of antibodies to Fc receptors (activator or inhibitory) on macrophages can modulate their function (58).
Together, data suggest that B cells and antibodies may play an important role in protective immunity against mycobacterial infections; however, the diversity of antibody functions, the heterogeneity of the humoral immune response to Mtb, as well as the complexity of the interactions between B cells and other immune cells have been indicated as the major challenges to understand the impact of the humoral immune system in the immune protection at each stage of Mtb infection (58).
M. tuberculosis and SARS-CoV-2 co-infection
Information on TB-COVID-19 co-infection in humans is still limited. Co-infection was reported around 1% in the Philippines (302), 5% in South Africa (303), and between 0.37% and 4.47% in China (304). Recent works suggest that TB-COVID-19 co-infection is associated with elevated risk of unfavorable clinical outcome, with a longer time to recovery, treatment failure, loss to follow-up rates, and higher rates of mortality compared to patients with COVID-19 alone (89, 305–308).
However, mechanistic studies are needed to understand the interactions during Mtb and SARS-CoV-2 dual infections, their effect on the host immune response and clinical outcomes. Understanding the early events and pathophysiology of TB-COVID-19 co-infection is warranted to find better ways to manage such cases, particularly in the high TB endemic areas. The dysregulated immune response induced by each pathogen can lead to an unbalanced inflammatory response, which can promote the progression and worsening of both diseases.
To date, the immune response for each pathogen has been well studied, whereas the impact of Mtb and SARS-CoV-2 co-infection on the innate and adaptive immune response, their crosstalk and cumulative impact on disease outcome in humans still need to be delineated (309–313).
In fact, the studies available have mostly focused on the clinical features of co-infected patients, characterizing a marked lymphopenia and increased levels of some markers of inflammation, such as C-reactive protein (CRP), D-dimer, ferritin, and describing the lung tissue damages (308, 312, 314, 315).
There are few published studies either in vitro, ex-vivo using human samples from co-infected individuals or animal models evaluating the immune response and immunopathology in the context of co-infection (Table 2).
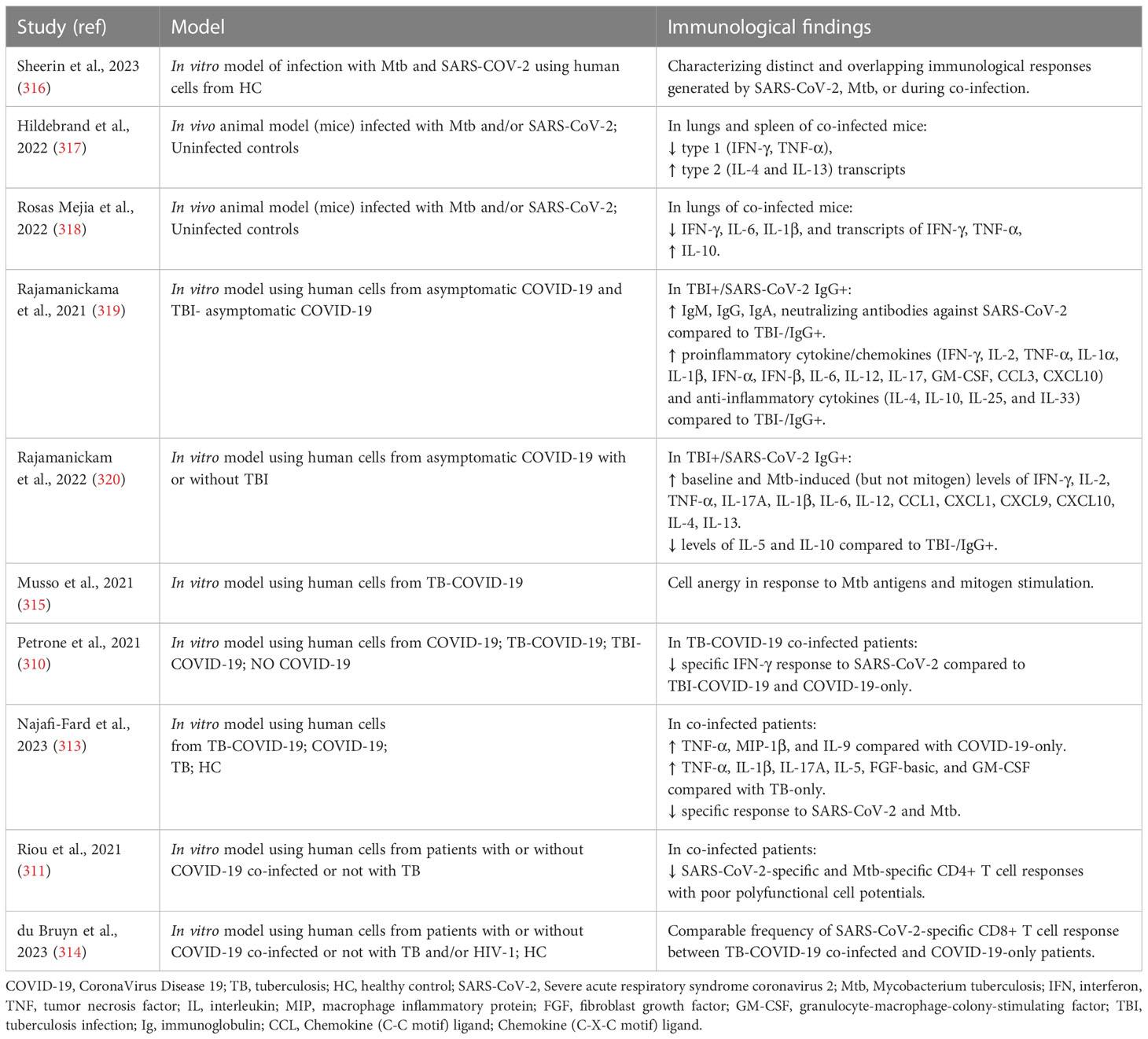
Table 2 Studies that evaluated the immunopathology and the immune response in the context of M. tuberculosis and SARS-CoV-2 co-infection.
In vitro studies were recently performed by Sheerin and colleagues using a single-cell RNA-seq (scRNA-seq) approach to analyze the results from a co-infection performed using a whole blood platform (24 or 96 hours) from healthy adults. The authors characterized different and overlapping immunological responses generated by SARS-CoV-2 (ancestral strain) and Mtb (lineage 4 laboratory strain H37Rv) when a single infection or co-infection occurs. Based on marker gene expression, they identified 13 distinct clusters of cells showing diverse proportions of monocytes, T cells and neutrophils between different conditions and timepoints. The co-infected condition showed the major immune activation effect early (24h) post-infection with 238 immunological pathways uniquely enriched, including IFN-γ and TNF production, while 182 shared pathways were overlapping at 96h post-infection among different conditions. In contrast to SARS-CoV-2-only infection that caused extensive cell death by 96h post-infection, Mtb-only and co-infected conditions maintained monocyte, T cell and NK cell signatures, and negative regulation of the signaling of extrinsic apoptosis (316).
Interesting animal studies evaluating the impact of aerosol Mtb and SARS-CoV-2 co-infection in transgenic (K18-hACE2) C57BL/6 mice showed that pre-infection with Mtb resulted in lower SARS-CoV-2 viral loads at the lung tissue level, likely mediated by the heightened immune microenvironment of the lungs. In addition, after SARS-CoV-2 superinfection, increased bacterial loads in Mtb-infected tissues and decreased histiocytic inflammation were found. Moreover, SARS-CoV-2 caused a decreasing trend in type 1 (IFN-γ and TNF-α) and an increasing trend in type 2 (IL-4 and IL-13) cytokine transcript levels in Mtb-infected mice. These findings, which are usually associated with disseminated Mtb infection, suggest that SARS-CoV-2 may have a deleterious effect on TB outcome (317) through the immune dysregulation, potentially resulting in granuloma collapse and the subsequent Mtb dissemination (311).
Using two concomitant murine models of COVID-19 (SARS-CoV-2 infection of K18-hACE2 mice and mouse-adapted SARS-CoV-2 [MACoV2] infection of C57BL/6 mice) it was shown that chronically Mtb H37Rv-infected mice were resistant to the pathological consequences of secondary SARS-CoV-2 infection, and SARS-CoV-2 infection did not affect Mtb burdens. Single-cell RNA sequencing of the lungs of the co-infected animals showed that resistance could be due to T and B cells expansion upon viral challenge. Interestingly, lower lung protein levels of IFN-γ, IL-6 and IL-1β as well as mRNA levels of IFN-γ and TNF-α and higher levels of IL-10 were found in co-infection than in Mtb-monoinfection at the 30 days post-infection (318), similar to Hildebrand and colleagues (317)
Regarding the evaluation of the immune responses in co-infected humans, two studies have demonstrated that Mtb infection can modulate humoral (antibody) and cytokine responses to SARS-CoV-2 infection (319) and vice versa (320) in investigations conducted in TB endemic countries. Rajamanickam and colleagues demonstrated that individuals seropositive (IgG+) for SARS-CoV-2 infection and with TB infection (TBI+/SARS-CoV-2 IgG+) were characterized by higher levels of specific antibodies (IgM, IgG and IgA) and neutralizing antibodies against SARS-CoV-2 compared to individuals with only SARS-CoV-2 infection. Moreover, elevated plasma levels of proinflammatory cytokine/chemokine responses including IFN-γ, IL-2, TNF-α, IL-1α, IL-1β, IFN-α, IFN-β, IL-6, IL-12, IL-17, GM-CSF, CCL3, CXCL10 and anti-inflammatory cytokines such as IL-4, IL-10, IL-25 and IL-33 were found in TBI+/SARS-CoV-2 IgG+ subjects. These results show that Mtb infection can modulate the immune responses in asymptomatic SARS-CoV-2-infected individuals (319). In an additional study, it was shown that TBI+/SARS-CoV-2 IgG+ individuals have higher baseline and Mtb-induced (but not mitogen) levels of several pro- and anti-inflammatory cytokines/chemokines including IFN-γ, IL-2, TNF-α, IL-17A, IL-1β, IL-6, IL-12, CCL1, CXCL1, CXCL9, CXCL10, IL-4, IL-13 and reduced levels of IL-5 and IL-10 compared to TBI-/SARS-CoV-2 IgG+ individuals. These findings suggest modulating effects of SARS-CoV-2 infection on the immune responses of individuals with Mtb infection (320). However, these results were obtained in TB-infected individuals with only asymptomatic SARS-CoV-2 infection and the influence of each pathogen on the disease severity and the outcome of each infection were not evaluated.
Differently, clinical outcome was assessed in a case of multidrug-resistant (MDR)/TB-COVID-19 co-infected patient affected by bilateral cavitary pulmonary TB, that subsequently developed COVID-19-associated pneumonia which led to a fatal outcome. Death was probably due to the immuno-suppressed state of the patient, as shown by the low lymphocyte count and by the lack of response to Mtb antigens and mitogen (315).
In addition, a cohort of TB-COVID-19 co-infected patients with different severity of COVID-19 showed a reduced ability to mount a specific immune response to SARS-CoV-2 stimulation compared to patients with TBI and COVID-19 (TBI-COVID-19) or with COVID-19 only (310). In particular, in TB-COVID-19 co-infected patients TNF-α, MIP-1β, and IL-9 showed significant elevated levels compared to COVID-19 only, and TNF-α had the highest discriminant power. Moreover, TNF-α, IL-1β, IL-17A, IL-5, FGF-basic, and GM-CSF were increased in co-infected compared to patients with TB-only. Importantly, co-infection was associated with an impairment of SARS-CoV-2-specific and a reduced Mtb-specific immune response (313).
In agreement with these results, Riou and colleagues demonstrated in TB-COVID-19 co-infection impaired SARS-CoV-2-specific and Mtb-specific CD4+ T cells with reduced polyfunctional cell potentials, proliferation cell capacity, and augmented cell activation markers (311). However, the frequency of SARS-CoV-2 specific CD8+ T cell response to peptides spanning the M, N and S sequences in TB-COVID-19 co-infected patients was found to be comparable with patients with COVID-19 only (314).
Furthermore, several recent case studies have raised concerns regarding the Mtb reactivation in TB-infected subjects following SARS-CoV-2 co-infection. These reports suggest that since the control of both Mtb and SARS-CoV-2 replication depends on cellular immunity, it is possible that the immune dysregulation caused by SARS-CoV-2 or the immunomodulatory therapies used for COVID-19 treatment may increase the risk for TB reactivation (321–326).
Both SARS-CoV-2 and Mtb have immunomodulating potentials to change the outcome of the course of each disease in co-infected patients: SARS-CoV-2 may cause immunosuppression and cytokine storm, which can contribute to the Mtb reactivation (327) and lung tissue damage; Mtb may cause T-cell exhaustion and uncontrolled release of proinflammatory cytokines resulting in lung damage (328, 329), thus potentially contributing to the susceptibility to SARS-CoV-2 infection and to a more severe COVID-19.
In TB-infected individuals, T cells are responsible for Mtb control via the granuloma formation. Co-infection with SARS-CoV-2 in these individuals may negatively affect immune regulation in the granuloma leading to Mtb reactivation (322, 330). This alteration of the immune system has been reported using a large-scale meta-analysis of transcriptomic data showing that some immune genes are enriched in COVID-19 and TB diseases (309). The findings from case reports indicate the presence of similarities in the immunopathogenesis of the two diseases, which may exacerbate disease severity during co-infection. Subclinical and clinical TB disease may increase the risk of severe COVID-19 disease and also SARS-CoV-2 co-infection may induce the progression to TB disease (309), as reported above (321–326). In this regard, IFN-I which is strongly induced by viral infection may be detrimental in the context of Mtb by inhibiting B cell responses, inducing the release of immunosuppressive molecules or reducing the macrophagic activation induced by IFN-γ (145), Also, the hyperinflammatory milieu caused by Mtb may raise the risk of severe COVID-19 and vice versa (331). Mtb spread or reactivation might be favored by inflammatory molecules released from the SARS-CoV-2-induced necroptosis, whereas the apoptosis might mitigate it (332). Moreover, while COVID-19 therapies targeting pro-inflammatory cytokines may limit the acute immunopathology, they may also repress the responses needed to control Mtb containment (308).
Altogether, these studies suggest that co-infection alters the capacity of the host to respond to and control Mtb and/or SARS-CoV-2, indicating the need for further investigation of the underlying immunological pathways.
Final remarks
SARS-CoV-2 and Mtb are currently the two deadliest infectious diseases in humans. While the route of infection and the target organ are similar, the time to disease manifestation and the pathways driving immunopathology differ significantly (Figure 3).
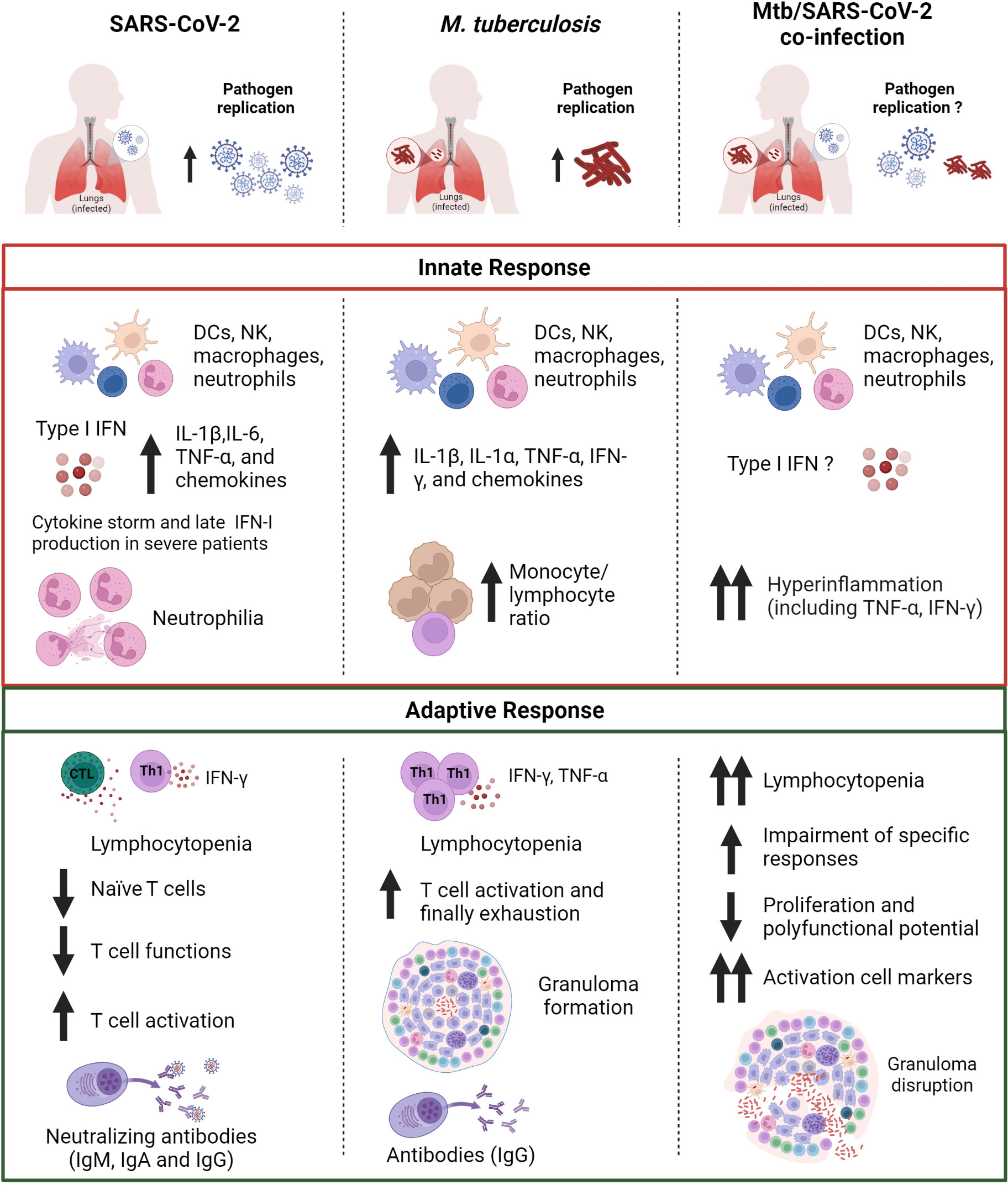
Figure 3 Comparison of the immune response in SARS-CoV-2, Mtb or Mtb/SARS-CoV-2 infection. The innate immune response induced after exposure to SARS-CoV-2 or Mtb is characterized by the production of pro-inflammatory cytokines including IL-1β and TNF-α. In Mtb/SARS-CoV-2 co-infection there is an overproduction of pro-inflammatory cytokines. SARS-CoV-2 infection presents also an early type I IFN production, which is absent or delayed in severe COVID-19 patients. SARS-CoV-2 infection is also characterized by a higher neutrophil count, whereas a higher monocyte/lymphocyte ratio is observed in Mtb-infected patients. Both SARS-CoV-2 and Mtb infected subjects show lymphocytopenia and T cell activation, which are even more prominent in case of co-infection. In co-infected individuals a major impairment of antigen-specific response to Mtb and SARS-CoV-2, and granuloma disruption is present. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; Mtb, Mycobacterium tuberculosis; IFNs, interferons; DCs, dendritic cells; NK, natural killer; Th, T helper; Ig, immunoglobulin. Created with BioRender.com.
Evidence reported here show that both innate and adaptive immune response are critical components for the protection against SARS-CoV-2 and Mtb. The immune response to both SARS-CoV-2 and Mtb is complex and multifaceted, and there are still many aspects that are not well understood. However, it is known that an appropriate activation of the innate immunity in the early stages of infection followed by adaptive immunity is necessary to curb the pathogen dissemination in the host.
The comparison of these two pathogens highlights how the innate immune response induced after exposure to SARS-CoV-2 or Mtb share the production of some pro-inflammatory cytokines including IL-1β and TNF-α. Similar results were found in Mtb/SARS-CoV-2 co-infection. For SARS-CoV-2 infection, the early and robust IFN-I production as well as neutralizing antibodies have an outmost importance for guarantee an efficient control of viral spread and to determine the clinical outcome of COVID-19. On the other hand, in Mtb infection a central role is played by the alveolar macrophages and the cytokines they release as TNF-α and IL-1β.
Although the infections caused by the individual pathogens have been intensively studied, there are still many unanswered questions about the influence of these pathogens on each other, the immune response, and clinical outcome in the context of co-infection. Recent data has raised concerns regarding the Mtb reactivation following SARS-CoV-2 infection likely due to immune dysregulation caused by SARS-CoV-2 or immunomodulatory COVID-19 therapies. Further clinical and scientific research is needed to better understand the interaction and outcome of the co-infection.
Author contributions
AA contributed to the writing of introduction, immune response to SARS-CoV-2, final remarks and created the figures. SN-F was responsible for immune response to M. tuberculosis, co-infection and tables. DG conceived the review, contributed to the first draft and revised the whole manuscript. All the authors approved the final version of the manuscript.
Funding
This work was supported by INMI ‘Lazzaro Spallanzani’ Ricerca Corrente Linea 1 and Linea 4 funded by the Italian Ministry of Health and by generous liberal donation/funding for COVID-19 research from Camera di Commercio, Industria e Artigianato di Roma (resolution number 395 on 25 May 2021).
Conflict of interest
Author DG has been a member of the advisory board of Biomerieux and Eli Lilly in 2020 and 2021 and is currently scientific advisor of PDB Biotec. She received fees for educational training or consultancy from Almirall, Biogen, Celgene, Diasorin, Janssen, Qiagen and Quidel.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. WHO. Coronavirus disease (COVID-19) – World Health Organization (2023). Available at: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (Accessed June 13, 2023).
2. Global tuberculosis report 2022 . Available at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (Accessed June 19, 2023).
3. Najafi Fard S, Petrone L, Petruccioli E, Alonzi T, Matusali G, Colavita F, et al. In vitro models for studying entry, tissue tropism, and therapeutic approaches of highly pathogenic coronaviruses. BioMed Res Int (2021) 2021:8856018. doi: 10.1155/2021/8856018
4. Chandran A, Rosenheim J, Nageswaran G, Swadling L, Pollara G, Gupta RK, et al. Rapid synchronous type 1 IFN and virus-specific T cell responses characterize first wave non-severe SARS-CoV-2 infections. Cell Rep Med (2022) 3:100557. doi: 10.1016/j.xcrm.2022.100557
5. Aiello A, Grossi A, Meschi S, Meledandri M, Vanini V, Petrone L, et al. Coordinated innate and T-cell immune responses in mild COVID-19 patients from household contacts of COVID-19 cases during the first pandemic wave. Front Immunol (2022) 13:920227. doi: 10.3389/fimmu.2022.920227
6. Capuano SV, Croix DA, Pawar S, Zinovik A, Myers A, Lin PL, et al. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect Immun (2003) 71:5831–44. doi: 10.1128/IAI.71.10.5831-5844.2003
7. Lin PL, Rodgers M, Smith L, Bigbee M, Myers A, Bigbee C, et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect Immun (2009) 77:4631–42. doi: 10.1128/IAI.00592-09
8. Goldblatt D, Alter G, Crotty S, Plotkin SA. Correlates of protection against SARS-CoV-2 infection and COVID-19 disease. Immunol Rev (2022) 310:6–26. doi: 10.1111/imr.13091
9. Regev-Yochay G, Lustig Y, Joseph G, Gilboa M, Barda N, Gens I, et al. Correlates of protection against COVID-19 infection and intensity of symptomatic disease in vaccinated individuals exposed to SARS-CoV-2 in households in Israel (ICoFS): a prospective cohort study. Lancet Microbe (2023) 4:e309–18. doi: 10.1016/S2666-5247(23)00012-5
10. Lawn SD, Myer L, Edwards D, Bekker L-G, Wood R. Short-term and long-term risk of tuberculosis associated with CD4 cell recovery during antiretroviral therapy in South Africa. AIDS (2009) 23:1717–25. doi: 10.1097/QAD.0b013e32832d3b6d
11. Wang J, Du G. COVID-19 may transmit through aerosol. Ir J Med Sci (2020) 189:1143–4. doi: 10.1007/s11845-020-02218-2
12. Zhou L, Ayeh SK, Chidambaram V, Karakousis PC. Modes of transmission of SARS-CoV-2 and evidence for preventive behavioral interventions. BMC Infect Dis (2021) 21:496. doi: 10.1186/s12879-021-06222-4
13. Baker CA, Gibson KE. Persistence of SARS-CoV-2 on surfaces and relevance to the food industry. Curr Opin Food Sci (2022) 47:100875. doi: 10.1016/j.cofs.2022.100875
14. Chin AWH, Chu JTS, Perera MRA, Hui KPY, Yen H-L, Chan MCW, et al. Stability of SARS-CoV-2 in different environmental conditions. Lancet Microbe (2020) 1:e10. doi: 10.1016/S2666-5247(20)30003-3
15. Hobby GL, Holman AP, Iseman MD, Jones JM. Enumeration of tubercle bacilli in sputum of patients with pulmonary tuberculosis. Antimicrob Agents Chemother (1973) 4:94–104. doi: 10.1128/AAC.4.2.94
16. Patterson B, Bryden W, Call C, McKerry A, Leonard B, Seldon R, et al. Cough-independent production of viable Mycobacterium tuberculosis in bioaerosol. Tuberculosis (2021) 126:102038. doi: 10.1016/j.tube.2020.102038
17. Liu J, Li Y, Liu Q, Yao Q, Wang X, Zhang H, et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discovery (2021) 7:1–4. doi: 10.1038/s41421-021-00249-2
18. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
19. Scordo JM, Knoell DL, Torrelles JB. Alveolar epithelial cells in mycobacterium tuberculosis infection: active players or innocent bystanders? J Innate Immun (2016) 8:3–14. doi: 10.1159/000439275
20. Corleis B, Dorhoi A. Early dynamics of innate immunity during pulmonary tuberculosis. Immunol Lett (2020) 221:56–60. doi: 10.1016/j.imlet.2020.02.010
21. de Waal AM, Hiemstra PS, Ottenhoff TH, Joosten SA, van der Does AM. Lung epithelial cells interact with immune cells and bacteria to shape the microenvironment in tuberculosis. Thorax (2022) 77:408–16. doi: 10.1136/thoraxjnl-2021-217997
22. Behr MA, Waters WR. Is tuberculosis a lymphatic disease with a pulmonary portal? Lancet Infect Dis (2014) 14:250–5. doi: 10.1016/S1473-3099(13)70253-6
23. Lerner TR, de Souza Carvalho-Wodarz C, Repnik U, Russell MRG, Borel S, Diedrich CR, et al. Lymphatic endothelial cells are a replicative niche for Mycobacterium tuberculosis. J Clin Invest (2016) 126:1093–108. doi: 10.1172/JCI83379
24. Beigier-Bompadre M, Montagna GN, Kühl AA, Lozza L, Iii JW, Kupz A, et al. Mycobacterium tuberculosis infection modulates adipose tissue biology. PloS Pathog (2017) 13:e1006676. doi: 10.1371/journal.ppat.1006676
25. Mayito J, Andia I, Belay M, Jolliffe DA, Kateete DP, Reece ST, et al. Anatomic and cellular niches for mycobacterium tuberculosis in latent tuberculosis infection. J Infect Dis (2019) 219:685–94. doi: 10.1093/infdis/jiy579
26. Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-coV-2 is essential for infection of human lung cells. Mol Cell (2020) 78:779–784.e5. doi: 10.1016/j.molcel.2020.04.022
27. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
28. Koch J, Uckeley ZM, Doldan P, Stanifer M, Boulant S, Lozach P-Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J (2021) 40:e107821. doi: 10.15252/embj.2021107821
29. Schäfer G, Jacobs M, Wilkinson RJ, Brown GD. Non-opsonic recognition of Mycobacterium tuberculosis by phagocytes. J Innate Immun (2009) 1:231–43. doi: 10.1159/000173703
30. Chatterjee S, Kundapura SV, Basak AJ, Mukherjee D, Dash S, Ganguli N, et al. High-resolution crystal structure of LpqH, an immunomodulatory surface lipoprotein of Mycobacterium tuberculosis reveals a distinct fold and a conserved cleft on its surface. Int J Biol Macromol (2022) 210:494–503. doi: 10.1016/j.ijbiomac.2022.04.196
31. Eslami N, Aghbash PS, Shamekh A, Entezari-Maleki T, Nahand JS, Sales AJ, et al. SARS-coV-2: receptor and co-receptor tropism probability. Curr Microbiol (2022) 79:133. doi: 10.1007/s00284-022-02807-7
32. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-coV-2 drives development of COVID-19. Cell (2020) 181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026
33. Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
34. Severa M, Diotti RA, Etna MP, Rizzo F, Fiore S, Ricci D, et al. Differential plasmacytoid dendritic cell phenotype and type I Interferon response in asymptomatic and severe COVID-19 infection. PloS Pathog (2021) 17:e1009878. doi: 10.1371/journal.ppat.1009878
35. Parackova Z, Zentsova I, Bloomfield M, Vrabcova P, Smetanova J, Klocperk A, et al. Disharmonic inflammatory signatures in COVID-19: augmented neutrophils’ but impaired monocytes’ and dendritic cells’ Responsiveness. Cells (2020) 9:2206. doi: 10.3390/cells9102206
36. Al-Kuraishy HM, Al-Gareeb AI, Al-Hussaniy HA, Al-Harcan NAH, Alexiou A, Batiha GE-S. Neutrophil Extracellular Traps (NETs) and Covid-19: A new frontiers for therapeutic modality. Int Immunopharmacol (2022) 104:108516. doi: 10.1016/j.intimp.2021.108516
37. Veras FP, Gomes GF, Silva BMS, Caetité DB, Almeida CJLR, Silva CMS, et al. Targeting neutrophils extracellular traps (NETs) reduces multiple organ injury in a COVID-19 mouse model. Respir Res (2023) 24:66. doi: 10.1186/s12931-023-02336-2
38. Masso-Silva JA, Moshensky A, Lam MTY, Odish MF, Patel A, Xu L, et al. Increased peripheral blood neutrophil activation phenotypes and neutrophil extracellular trap formation in critically ill coronavirus disease 2019 (COVID-19) patients: A case series and review of the literature. Clin Infect Dis (2022) 74:479–89. doi: 10.1093/cid/ciab437
39. Etna MP, Giacomini E, Severa M, Coccia EM. Pro- and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin Immunol (2014) 26:543–51. doi: 10.1016/j.smim.2014.09.011
40. Adane T, Melku M, Ayalew G, Bewket G, Aynalem M, Getawa S. Accuracy of monocyte to lymphocyte ratio for tuberculosis diagnosis and its role in monitoring anti-tuberculosis treatment: Systematic review and meta-analysis. Med (Baltimore) (2022) 101:e31539. doi: 10.1097/MD.0000000000031539
41. Sekine T, Perez-Potti A, Rivera-Ballesteros O, Strålin K, Gorin J-B, Olsson A, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell (2020) 183:158–168.e14. doi: 10.1016/j.cell.2020.08.017
42. Notarbartolo S, Ranzani V, Bandera A, Gruarin P, Bevilacqua V, Putignano AR, et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci Immunol (2021) 6:eabg5021. doi: 10.1126/sciimmunol.abg5021
43. Oja AE, Saris A, Ghandour CA, Kragten NAM, Hogema BM, Nossent EJ, et al. Divergent SARS-CoV-2-specific T- and B-cell responses in severe but not mild COVID-19 patients. Eur J Immunol (2020) 50:1998–2012. doi: 10.1002/eji.202048908
44. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
45. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-coV-2 in acute COVID-19 and associations with age and disease severity. Cell (2020) 183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038
46. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in wuhan, China. Clin Infect Dis (2020) 71:762–8. doi: 10.1093/cid/ciaa248
47. Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L, et al. Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J Infect Dis (2020) 221:1762–9. doi: 10.1093/infdis/jiaa150
48. Long Q-X, Liu B-Z, Deng H-J, Wu G-C, Deng K, Chen Y-K, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med (2020) 26:845–8. doi: 10.1038/s41591-020-0897-1
49. Seow J, Graham C, Merrick B, Acors S, Pickering S, Steel KJA, et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat Microbiol (2020) 5:1598–607. doi: 10.1038/s41564-020-00813-8
50. Wang K, Long Q-X, Deng H-J, Hu J, Gao Q-Z, Zhang G-J, et al. Longitudinal dynamics of the neutralizing antibody response to severe acute respiratory syndrome coronavirus 2 (SARS-coV-2) infection. Clin Infect Dis (2021) 73:e531–9. doi: 10.1093/cid/ciaa1143
51. Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Viant C, Gaebler C, et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci Transl Med (2021) 13:eabf1555. doi: 10.1126/scitranslmed.abf1555
52. Flynn JL, Chan J. Immune cell interactions in tuberculosis. Cell (2022) 185:4682–702. doi: 10.1016/j.cell.2022.10.025
53. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol (2012) 3:411. doi: 10.3389/fimmu.2012.00411
54. Ashenafi S, Brighenti S. Reinventing the human tuberculosis (TB) granuloma: Learning from the cancer field. Front Immunol (2022) 13:1059725. doi: 10.3389/fimmu.2022.1059725
55. Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology (2008) 123:326–38. doi: 10.1111/j.1365-2567.2007.02719.x
56. Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PloS Pathog (2015) 11:e1004603. doi: 10.1371/journal.ppat.1004603
57. Chan J, Mehta S, Bharrhan S, Chen Y, Achkar JM, Casadevall A, et al. The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Semin Immunol (2014) 26:588–600. doi: 10.1016/j.smim.2014.10.005
58. Achkar JM, Chan J, Casadevall A. B cells and antibodies in the defense against Mycobacterium tuberculosis infection. Immunol Rev (2015) 264:167–81. doi: 10.1111/imr.12276
59. Sette A, Crotty S. Adaptive immunity to SARS-coV-2 and COVID-19. Cell (2021) 184:861–80. doi: 10.1016/j.cell.2021.01.007
60. Petrone L, Petruccioli E, Vanini V, Cuzzi G, Najafi Fard S, Alonzi T, et al. A whole blood test to measure SARS-CoV-2-specific response in COVID-19 patients. Clin Microbiol Infect (2021) 27:286.e7–286.e13. doi: 10.1016/j.cmi.2020.09.051
61. Aiello A, Najafi Fard S, Petruccioli E, Petrone L, Vanini V, Farroni C, et al. Spike is the most recognized antigen in the whole-blood platform in both acute and convalescent COVID-19 patients. Int J Infect Dis (2021) 106:338–47. doi: 10.1016/j.ijid.2021.04.034
62. Petruccioli E, Najafi Fard S, Navarra A, Petrone L, Vanini V, Cuzzi G, et al. Exploratory analysis to identify the best antigen and the best immune biomarkers to study SARS-CoV-2 infection. J Transl Med (2021) 19:272. doi: 10.1186/s12967-021-02938-8
63. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-coV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell (2020) 181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015
64. Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med (2008) 205:105–15. doi: 10.1084/jem.20071367
65. Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, Fountain JJ, et al. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci U.S.A. (2008) 105:10961–6. doi: 10.1073/pnas.0801496105
66. Kauffman KD, Sallin MA, Hoft SG, Sakai S, Moore R, Wilder-Kofie T, et al. Limited Pulmonary Mucosal-Associated Invariant T Cell Accumulation and Activation during Mycobacterium tuberculosis Infection in Rhesus Macaques. Infect Immun (2018) 86:e00431–18. doi: 10.1128/IAI.00431-18
67. Goletti D, Butera O, Vanini V, Lauria FN, Lange C, Franken KLMC, et al. Response to Rv2628 latency antigen associates with cured tuberculosis and remote infection. Eur Respir J (2010) 36:135–42. doi: 10.1183/09031936.00140009
68. Sali M, Buonsenso D, D’Alfonso P, De Maio F, Ceccarelli M, Battah B, et al. Combined use of Quantiferon and HBHA-based IGRA supports tuberculosis diagnosis and therapy management in children. J Infect (2018) 77:526–33. doi: 10.1016/j.jinf.2018.09.011
69. Bálint G, Vörös-Horváth B, Széchenyi A. Omicron: increased transmissibility and decreased pathogenicity. Sig Transduct Target Ther (2022) 7:1–3. doi: 10.1038/s41392-022-01009-8
70. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science (2020) 369:718–24. doi: 10.1126/science.abc6027
71. Galani I-E, Rovina N, Lampropoulou V, Triantafyllia V, Manioudaki M, Pavlos E, et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol (2021) 22:32–40. doi: 10.1038/s41590-020-00840-x
72. McCarthy KR, Rennick LJ, Nambulli S, Robinson-McCarthy LR, Bain WG, Haidar G, et al. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science (2021) 371:1139–42. doi: 10.1126/science.abf6950
73. Kemp SA, Collier DA, Datir RP, Ferreira IATM, Gayed S, Jahun A, et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature (2021) 592:277–82. doi: 10.1038/s41586-021-03291-y
74. Collier DA, De Marco A, Ferreira IATM, Meng B, Datir RP, Walls AC, et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature (2021) 593:136–41. doi: 10.1038/s41586-021-03412-7
75. Zhou D, Dejnirattisai W, Supasa P, Liu C, Mentzer AJ, Ginn HM, et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell (2021) 184:2348–2361.e6. doi: 10.1016/j.cell.2021.02.037
76. Lu H, Liu Z, Deng X, Chen S, Zhou R, Zhao R, et al. Potent NKT cell ligands overcome SARS-CoV-2 immune evasion to mitigate viral pathogenesis in mouse models. PloS Pathog (2023) 19:e1011240. doi: 10.1371/journal.ppat.1011240
77. Carranza C, Chavez-Galan L. Several routes to the same destination: inhibition of phagosome-lysosome fusion by mycobacterium tuberculosis. Am J Med Sci (2019) 357:184–94. doi: 10.1016/j.amjms.2018.12.003
78. Hayakawa E, Tokumasu F, Nardone GA, Jin AJ, Hackley VA, Dvorak JA. A Mycobacterium tuberculosis-derived lipid inhibits membrane fusion by modulating lipid membrane domains. Biophys J (2007) 93:4018–30. doi: 10.1529/biophysj.107.104075
79. Schnettger L, Rodgers A, Repnik U, Lai RP, Pei G, Verdoes M, et al. A rab20-dependent membrane trafficking pathway controls M. tuberculosis replication by regulating phagosome spaciousness and integrity. Cell Host Microbe (2017) 21:619–628.e5. doi: 10.1016/j.chom.2017.04.004
80. Jayachandran R, Sundaramurthy V, Combaluzier B, Mueller P, Korf H, Huygen K, et al. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell (2007) 130:37–50. doi: 10.1016/j.cell.2007.04.043
81. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis- induced neutrophil extracellular traps activate human macrophages. J Innate Immun (2013) 5:591–602. doi: 10.1159/000348676
82. Chowdhury CS, Kinsella RL, Nehls EM, Naik SK, Lane DS, Talukdar P, et al. Type I IFN signaling mediates NET release to promote Mycobacterium tuberculosis replication and granuloma caseation. (2022) 2022:. doi: 10.1101/2022.11.29.518376
83. Blanc L, Gilleron M, Prandi J, Song O-R, Jang M-S, Gicquel B, et al. Mycobacterium tuberculosis inhibits human innate immune responses via the production of TLR2 antagonist glycolipids. Proc Natl Acad Sci U.S.A. (2017) 114:11205–10. doi: 10.1073/pnas.1707840114
84. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in mycobacterium tuberculosis infection. Microbiol Spectr (2016) 4. doi: 10.1128/microbiolspec.TBTB2-0018-2016
85. CDC. COVID-19 and your health, in: Centers for disease control and prevention (2020). Available at: https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms (Accessed June 13, 2023).
86. Fact sheets | General | Tuberculosis: general information | TB | CDC (2022). Available at: https://www.cdc.gov/tb/publications/factsheets/general/tb.htm (Accessed June 13, 2023).
87. Grifoni A, Alonzi T, Alter G, Noonan DM, Landay AL, Albini A, et al. Impact of aging on immunity in the context of COVID-19, HIV, and tuberculosis. Front Immunol (2023) 14:1146704. doi: 10.3389/fimmu.2023.1146704
88. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
89. Western Cape Department of Health in collaboration with the National Institute for Communicable Diseases, South Africa. Risk factors for coronavirus disease 2019 (COVID-19) death in a population cohort study from the western cape province, South Africa. Clin Infect Dis (2021) 73:e2005–15. doi: 10.1093/cid/ciaa1198
90. Goletti D, Pisapia R, Fusco FM, Aiello A, Van Crevel R. Epidemiology, pathogenesis, clinical presentation and management of TB in patients with HIV and diabetes. Int J Tuberc Lung Dis (2023) 27:284–90. doi: 10.5588/ijtld.22.0685
91. ISS. Test di laboratorio per SARS-CoV-2 e loro uso in sanità pubblica, online la nota tecnica ad interim . Available at: https://www.inail.it/cs/internet/comunicazione/news-ed-eventi/news/news-nota-tecnica-test-sars-cov-2-2020.html (Accessed June 15, 2023).
92. Lewinsohn DM, Leonard MK, LoBue PA, Cohn DL, Daley CL, Desmond E, et al. Official american thoracic society/infectious diseases society of america/centers for disease control and prevention clinical practice guidelines: diagnosis of tuberculosis in adults and children. Clin Infect Dis (2017) 64:111–5. doi: 10.1093/cid/ciw778
93. Diriba K, Churiso G. The prevalence of Mycobacterium tuberculosis using Gene Xpert among tuberculosis suspected patients in Gedeo Zone, Southern Ethiopia. Eur J Med Res (2022) 27:24. doi: 10.1186/s40001-022-00650-x
94. Liu X, Mostafavi H, Ng WH, Freitas JR, King NJC, Zaid A, et al. The delta SARS-coV-2 variant of concern induces distinct pathogenic patterns of respiratory disease in K18-hACE2 transgenic mice compared to the ancestral strain from wuhan. mBio (2022) 13:e0068322. doi: 10.1128/mbio.00683-22
95. Lyngse FP, Mortensen LH, Denwood MJ, Christiansen LE, Møller CH, Skov RL, et al. Household transmission of the SARS-CoV-2 Omicron variant in Denmark. Nat Commun (2022) 13:5573. doi: 10.1038/s41467-022-33328-3
96. Flavin RJ, Gibbons N, O’Briain DS. Mycobacterium tuberculosis at autopsy–exposure and protection: an old adversary revisited. J Clin Pathol (2007) 60:487–91. doi: 10.1136/jcp.2005.032276
97. Grosset J. Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary. Antimicrob Agents Chemother (2003) 47:833–6. doi: 10.1128/AAC.47.3.833-836.2003
98. Canetti G DM. Thé Tubercle bacillus in the pulmonary lesion of man, in: Thé Tubercle bacillus in the pulmonary lesion of man (1955). Available at: https://www.cabdirect.org/cabdirect/abstract/19562700285 (Accessed June 19, 2023).
99. Opie, Aronson. Tubercle bacilli in latent tuberculous lesions in lung tissue without tuberculous lesions. Am J Med Sci (1928) 176:460. doi: 10.1097/00000441-192809000-00064
100. Silva MJA, Ribeiro LR, Gouveia MIM, Marcelino B dos R, dos Santos CS, Lima KVB, et al. Hyperinflammatory response in COVID-19: A systematic review. Viruses (2023) 15:553. doi: 10.3390/v15020553
101. Valdebenito S, Bessis S, Annane D, Lorin de la Grandmaison G, Cramer–Bordé E, Prideaux B, et al. COVID-19 lung pathogenesis in SARS-coV-2 autopsy cases(2021) (Accessed July 27, 2023).
102. O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MPR. The immune response in tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
103. Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol (2009) 27:393–422. doi: 10.1146/annurev.immunol.021908.132703
104. Kroon EE, Kinnear CJ, Orlova M, Fischinger S, Shin S, Boolay S, et al. An observational study identifying highly tuberculosis-exposed, HIV-1-positive but persistently TB, tuberculin and IGRA negative persons with M. tuberculosis specific antibodies in Cape Town, South Africa. EBioMedicine (2020) 61:103053. doi: 10.1016/j.ebiom.2020.103053
105. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell (2020) 181:1475–1488.e12. doi: 10.1016/j.cell.2020.05.006
106. Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-coV-2 spike glycoprotein. Cell (2020) 181:281–292.e6. doi: 10.1016/j.cell.2020.02.058
107. Yu S, Hu H, Ai Q, Bai R, Ma K, Zhou M, et al. SARS-coV-2 spike-mediated entry and its regulation by host innate immunity. Viruses (2023) 15:639. doi: 10.3390/v15030639
108. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature (2020) 581:215–20. doi: 10.1038/s41586-020-2180-5
109. Benton DJ, Wrobel AG, Xu P, Roustan C, Martin SR, Rosenthal PB, et al. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature (2020) 588:327–30. doi: 10.1038/s41586-020-2772-0
110. Yu S, Zheng X, Zhou B, Li J, Chen M, Deng R, et al. SARS-CoV-2 spike engagement of ACE2 primes S2’ site cleavage and fusion initiation. Proc Natl Acad Sci U.S.A. (2022) 119:e2111199119. doi: 10.1073/pnas.2111199119
111. Wettstein L, Kirchhoff F, Münch J. The transmembrane protease TMPRSS2 as a therapeutic target for COVID-19 treatment. Int J Mol Sci (2022) 23:1351. doi: 10.3390/ijms23031351
112. Vaarala MH, Porvari KS, Kellokumpu S, Kyllönen AP, Vihko PT. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J Pathol (2001) 193:134–40. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH743>3.0.CO;2-T
113. Liu Y, Qu H-Q, Qu J, Tian L, Hakonarson H. Expression pattern of the SARS-coV-2 entry genes ACE2 and TMPRSS2 in the respiratory tract. Viruses (2020) 12:1174. doi: 10.3390/v12101174
114. Yang H, Yuan H, Zhao X, Xun M, Guo S, Wang N, et al. Cytoplasmic domain and enzymatic activity of ACE2 are not required for PI4KB dependent endocytosis entry of SARS-CoV-2 into host cells. Virol Sin (2022) 37:380–9. doi: 10.1016/j.virs.2022.03.003
115. Lau S-Y, Wang P, Mok BW-Y, Zhang AJ, Chu H, Lee AC-Y, et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg Microbes Infect (2020) 9:837–42. doi: 10.1080/22221751.2020.1756700
116. Hui KPY, Ho JCW, Cheung M-C, Ng K-C, Ching RHH, Lai K-L, et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature (2022) 603:715–20. doi: 10.1038/s41586-022-04479-6
117. Meng B, Abdullahi A, Ferreira IATM, Goonawardane N, Saito A, Kimura I, et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature (2022) 603:706–14. doi: 10.1038/s41586-022-04474-x
118. Alonzi T, Aiello A, Repele F, Falasca L, Francalancia M, Garbuglia AR, et al. Cysteamine exerts in vitro antiviral activity against the SARS-CoV-2 Delta and Omicron variants. Cell Death Discovery (2022) 8:288. doi: 10.1038/s41420-022-01080-8
119. Halfmann PJ, Iida S, Iwatsuki-Horimoto K, Maemura T, Kiso M, Scheaffer SM, et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature (2022) 603:687–92. doi: 10.1038/s41586-022-04441-6
120. Suzuki R, Yamasoba D, Kimura I, Wang L, Kishimoto M, Ito J, et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature (2022) 603:700–5. doi: 10.1038/s41586-022-04462-1
121. Zang R, Gomez Castro MF, McCune BT, Zeng Q, Rothlauf PW, Sonnek NM, et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci Immunol (2020) 5:eabc3582. doi: 10.1126/sciimmunol.abc3582
122. Jocher G, Grass V, Tschirner SK, Riepler L, Breimann S, Kaya T, et al. ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion. EMBO Rep (2022) 23:e54305. doi: 10.15252/embr.202154305
123. Harte JV, Wakerlin SL, Lindsay AJ, McCarthy JV, Coleman-Vaughan C. Metalloprotease-dependent S2’-activation promotes cell-cell fusion and syncytiation of SARS-coV-2. Viruses (2022) 14:2094. doi: 10.3390/v14102094
124. Yamamoto M, Gohda J, Kobayashi A, Tomita K, Hirayama Y, Koshikawa N, et al. Metalloproteinase-dependent and TMPRSS2-independent cell surface entry pathway of SARS-coV-2 requires the furin cleavage site and the S2 domain of spike protein. mBio (2022) 13:e0051922. doi: 10.1128/mbio.00519-22
125. Kastenhuber ER, Mercadante M, Nilsson-Payant B, Johnson JL, Jaimes JA, Muecksch F, et al. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. Elife (2022) 11:e77444. doi: 10.7554/eLife.77444
126. Wettstein L, Immenschuh P, Weil T, Conzelmann C, Almeida-Hernández Y, Hoffmann M, et al. Native and activated antithrombin inhibits TMPRSS2 activity and SARS-CoV-2 infection. J Med Virol (2023) 95:e28124. doi: 10.1002/jmv.28124
127. Buchrieser J, Dufloo J, Hubert M, Monel B, Planas D, Rajah MM, et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J (2021) 40:e107405. doi: 10.15252/embj.2020107405
128. Rajah MM, Bernier A, Buchrieser J, Schwartz O. The mechanism and consequences of SARS-coV-2 spike-mediated fusion and syncytia formation. J Mol Biol (2022) 434:167280. doi: 10.1016/j.jmb.2021.167280
129. Zeng C, Evans JP, King T, Zheng Y-M, Oltz EM, Whelan SPJ, et al. SARS-CoV-2 spreads through cell-to-cell transmission. Proc Natl Acad Sci (2022) 119:e2111400119. doi: 10.1073/pnas.2111400119
130. Bussani R, Schneider E, Zentilin L, Collesi C, Ali H, Braga L, et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine (2020) 61:103104. doi: 10.1016/j.ebiom.2020.103104
131. Sanders DW, Jumper CC, Ackerman PJ, Bracha D, Donlic A, Kim H, et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. Elife (2021) 10:e65962. doi: 10.7554/eLife.65962
132. Wang H, Guo S, Yang H. Rapid quantitative monitoring of SARS-CoV-2 spike protein-mediated syncytia formation using split NanoLuc. J Med Virol (2022) 94:6073–7. doi: 10.1002/jmv.28053
133. Zhang Z, Zheng Y, Niu Z, Zhang B, Wang C, Yao X, et al. SARS-CoV-2 spike protein dictates syncytium-mediated lymphocyte elimination. Cell Death Differ (2021) 28:2765–77. doi: 10.1038/s41418-021-00782-3
134. Mendonça L, Howe A, Gilchrist JB, Sheng Y, Sun D, Knight ML, et al. Correlative multi-scale cryo-imaging unveils SARS-CoV-2 assembly and egress. Nat Commun (2021) 12:4629. doi: 10.1038/s41467-021-24887-y
135. Fennelly KP, Jones-López EC. Quantity and quality of inhaled dose predicts immunopathology in tuberculosis. Front Immunol (2015) 6:313. doi: 10.3389/fimmu.2015.00313
136. Khan HS, Nair VR, Ruhl CR, Alvarez-Arguedas S, Galvan Rendiz JL, Franco LH, et al. Identification of scavenger receptor B1 as the airway microfold cell receptor for Mycobacterium tuberculosis. eLife (2020) 9:e52551. doi: 10.7554/eLife.52551
137. Nair VR, Franco LH, Zacharia VM, Khan HS, Stamm CE, You W, et al. Microfold cells actively translocate mycobacterium tuberculosis to initiate infection. Cell Rep (2016) 16:1253–8. doi: 10.1016/j.celrep.2016.06.080
138. Lovey A, Verma S, Kaipilyawar V, Ribeiro-Rodrigues R, Husain S, Palaci M, et al. Early alveolar macrophage response and IL-1R-dependent T cell priming determine transmissibility of Mycobacterium tuberculosis strains. Nat Commun (2022) 13:884. doi: 10.1038/s41467-022-28506-2
139. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe (2018) 24:439–446.e4. doi: 10.1016/j.chom.2018.08.001
140. Boechat JL, Chora I, Morais A, Delgado L. The immune response to SARS-CoV-2 and COVID-19 immunopathology - Current perspectives. Pulmonology (2021) 27:423–37. doi: 10.1016/j.pulmoe.2021.03.008
141. Schultze JL, Aschenbrenner AC. COVID-19 and the human innate immune system. Cell (2021) 184:1671–92. doi: 10.1016/j.cell.2021.02.029
142. Roukens AHE, Pothast CR, König M, Huisman W, Dalebout T, Tak T, et al. Prolonged activation of nasal immune cell populations and development of tissue-resident SARS-CoV-2-specific CD8+ T cell responses following COVID-19. Nat Immunol (2022) 23:23–32. doi: 10.1038/s41590-021-01095-w
143. Schiuma G, Beltrami S, Bortolotti D, Rizzo S, Rizzo R. Innate immune response in SARS-coV-2 infection. Microorganisms (2022) 10:501. doi: 10.3390/microorganisms10030501
144. Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science (2020) 369:1210–20. doi: 10.1126/science.abc6261
145. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi: 10.1038/nri3787
146. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 370:eabd4585. doi: 10.1126/science.abd4585
147. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020) 370:eabd4570. doi: 10.1126/science.abd4570
148. Contoli M, Papi A, Tomassetti L, Rizzo P, Vieceli Dalla Sega F, Fortini F, et al. Blood interferon-α Levels and severity, outcomes, and inflammatory profiles in hospitalized COVID-19 patients. Front Immunol (2021) 12:648004. doi: 10.3389/fimmu.2021.648004
149. Venet M, Ribeiro MS, Décembre E, Bellomo A, Joshi G, Nuovo C, et al. Severe COVID-19 patients have impaired plasmacytoid dendritic cell-mediated control of SARS-CoV-2. Nat Commun (2023) 14:694. doi: 10.1038/s41467-023-36140-9
150. Zhou R, To KK-W, Wong Y-C, Liu L, Zhou B, Li X, et al. Acute SARS-coV-2 infection impairs dendritic cell and T cell responses. Immunity (2020) 53:864–877.e5. doi: 10.1016/j.immuni.2020.07.026
151. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect (2020) 9:761–70. doi: 10.1080/22221751.2020.1747363
152. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
153. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukocyte Biol (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
154. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J Microbiology Immunol Infection (2020) 53:368–70. doi: 10.1016/j.jmii.2020.03.005
155. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol (2020) 146:119–127.e4. doi: 10.1016/j.jaci.2020.04.027
156. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol (2020) 38:970–9. doi: 10.1038/s41587-020-0602-4
157. Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun (2021) 12:4664. doi: 10.1038/s41467-021-25015-6
158. Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature (2022) 606:585–93. doi: 10.1038/s41586-022-04802-1
159. Qin S, Jiang Y, Wei X, Liu X, Guan J, Chen Y, et al. Dynamic changes in monocytes subsets in COVID-19 patients. Hum Immunol (2021) 82:170–6. doi: 10.1016/j.humimm.2020.12.010
160. Toor D, Jain A, Kalhan S, Manocha H, Sharma VK, Jain P, et al. Tempering macrophage plasticity for controlling SARS-coV-2 infection for managing COVID-19 disease. Front Pharmacol (2020) 11:570698. doi: 10.3389/fphar.2020.570698
161. Lian Q, Zhang K, Zhang Z, Duan F, Guo L, Luo W, et al. Differential effects of macrophage subtypes on SARS-CoV-2 infection in a human pluripotent stem cell-derived model. Nat Commun (2022) 13:2028. doi: 10.1038/s41467-022-29731-5
162. Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, et al. Frontline Science: COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J Leukocyte Biol (2021) 109:13–22. doi: 10.1002/JLB.4HI0720-470R
163. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
164. Björkström NK, Strunz B, Ljunggren H-G. Natural killer cells in antiviral immunity. Nat Rev Immunol (2022) 22:112–23. doi: 10.1038/s41577-021-00558-3
165. Witkowski M, Tizian C, Ferreira-Gomes M, Niemeyer D, Jones TC, Heinrich F, et al. Untimely TGFβ responses in COVID-19 limit antiviral functions of NK cells. Nature (2021) 600:295–301. doi: 10.1038/s41586-021-04142-6
166. Bi J. NK cell dysfunction in patients with COVID-19. Cell Mol Immunol (2022) 19:127–9. doi: 10.1038/s41423-021-00825-2
167. Krämer B, Knoll R, Bonaguro L, ToVinh M, Raabe J, Astaburuaga-García R, et al. Early IFN-α signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID-19. Immunity (2021) 54:2650–2669.e14. doi: 10.1016/j.immuni.2021.09.002
168. Lenart M, Górecka M, Bochenek M, Barreto-Duran E, Szczepański A, Gałuszka-Bulaga A, et al. SARS-CoV-2 infection impairs NK cell functions via activation of the LLT1-CD161 axis. Front Immunol (2023) 14:1123155. doi: 10.3389/fimmu.2023.1123155
169. Crosby CM, Kronenberg M. Invariant natural killer T cells: front line fighters in the war against pathogenic microbes. Immunogenetics (2016) 68:639–48. doi: 10.1007/s00251-016-0933-y
170. Wang X, Guan F, Miller H, Byazrova MG, Candotti F, Benlagha K, et al. The role of dendritic cells in COVID-19 infection. Emerg Microbes Infect (2023) 12:2195019. doi: 10.1080/22221751.2023.2195019
171. Lerner TR, Borel S, Gutierrez MG. The innate immune response in human tuberculosis. Cell Microbiol (2015) 17:1277–85. doi: 10.1111/cmi.12480
172. Khan N, Vidyarthi A, Javed S, Agrewala JN. Innate Immunity Holding the Flanks until Reinforced by Adaptive Immunity against Mycobacterium tuberculosis Infection. Front Microbiol (2016) 7:328. doi: 10.3389/fmicb.2016.00328
173. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U.S.A. (2012) 109:E3168–3176. doi: 10.1073/pnas.1210500109
174. Schön T, Elmberger G, Negesse Y, Pando RH, Sundqvist T, Britton S. Local production of nitric oxide in patients with tuberculosis. Int J Tuberc Lung Dis (2004) 8:1134–7.
175. Cadena AM, Flynn JL, Fortune SM. The importance of first impressions: early events in mycobacterium tuberculosis infection influence outcome. mBio (2016) 7:e00342–00316. doi: 10.1128/mBio.00342-16
176. Reuschl A-K, Edwards MR, Parker R, Connell DW, Hoang L, Halliday A, et al. Innate activation of human primary epithelial cells broadens the host response to Mycobacterium tuberculosis in the airways. PloS Pathog (2017) 13:e1006577. doi: 10.1371/journal.ppat.1006577
177. Mukundan S, Singh P, Shah A, Kumar R, O’Neill KC, Carter CL, et al. In vitro miniaturized tuberculosis spheroid model. Biomedicines (2021) 9:1209. doi: 10.3390/biomedicines9091209
178. Iakobachvili N, Leon-Icaza SA, Knoops K, Sachs N, Mazères S, Simeone R, et al. Mycobacteria-host interactions in human bronchiolar airway organoids. Mol Microbiol (2022) 117:682–92. doi: 10.1111/mmi.14824
179. Clay H, Davis JM, Beery D, Huttenlocher A, Lyons SE, Ramakrishnan L. Dichotomous role of the macrophage in early Mycobacterium marinum infection of the zebrafish. Cell Host Microbe (2007) 2:29–39. doi: 10.1016/j.chom.2007.06.004
180. Gupta UD, Katoch VM. Animal models of tuberculosis. Tuberculosis (Edinb) (2005) 85:277–93. doi: 10.1016/j.tube.2005.08.008
181. Kashino SS, Napolitano DR, Skobe Z, Campos-Neto A. Guinea pig model of Mycobacterium tuberculosis latent/dormant infection. Microbes Infect (2008) 10:1469–76. doi: 10.1016/j.micinf.2008.08.010
182. Subbian S, Tsenova L, O’Brien P, Yang G, Koo M-S, Peixoto B, et al. Phosphodiesterase-4 inhibition alters gene expression and improves isoniazid-mediated clearance of Mycobacterium tuberculosis in rabbit lungs. PloS Pathog (2011) 7:e1002262. doi: 10.1371/journal.ppat.1002262
183. Singhal A, Aliouat EM, Hervé M, Mathys V, Kiass M, Creusy C, et al. Experimental tuberculosis in the Wistar rat: a model for protective immunity and control of infection. PloS One (2011) 6:e18632. doi: 10.1371/journal.pone.0018632
184. Lin PL, Myers A, Smith L, Bigbee C, Bigbee M, Fuhrman C, et al. Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum (2010) 62:340–50. doi: 10.1002/art.27271
185. Singh PP, Smith VL, Karakousis PC, Schorey JS. Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J Immunol (2012) 189:777–85. doi: 10.4049/jimmunol.1103638
186. Alvarez-Jiménez VD, Leyva-Paredes K, García-Martínez M, Vázquez-Flores L, García-Paredes VG, Campillo-Navarro M, et al. Extracellular vesicles released from mycobacterium tuberculosis-infected neutrophils promote macrophage autophagy and decrease intracellular mycobacterial survival. Front Immunol (2018) 9:272. doi: 10.3389/fimmu.2018.00272
187. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response to Mycobacterium tuberculosis in vivo. Sci Immunol (2019) 4:eaaw6693. doi: 10.1126/sciimmunol.aaw6693
188. Repasy T, Lee J, Marino S, Martinez N, Kirschner DE, Hendricks G, et al. Intracellular bacillary burden reflects a burst size for Mycobacterium tuberculosis in vivo. PloS Pathog (2013) 9:e1003190. doi: 10.1371/journal.ppat.1003190
189. Wolf AJ, Linas B, Trevejo-Nuñez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol (2007) 179:2509–19. doi: 10.4049/jimmunol.179.4.2509
190. Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, et al. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med (2006) 203:1805–15. doi: 10.1084/jem.20052545
191. Tian T, Woodworth J, Sköld M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J Immunol (2005) 175:3268–72. doi: 10.4049/jimmunol.175.5.3268
192. Samstein M, Schreiber HA, Leiner IM, Sušac B, Glickman MS, Pamer EG. Essential yet limited role for CCR2+ inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. eLife (2013) 2:e01086. doi: 10.7554/eLife.01086
193. Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol (2011) 186:7110–9. doi: 10.4049/jimmunol.1100001
194. Zheng L, He M, Long M, Blomgran R, Stendahl O. Pathogen-induced apoptotic neutrophils express heat shock proteins and elicit activation of human macrophages. J Immunol (2004) 173:6319–26. doi: 10.4049/jimmunol.173.10.6319
195. Alemán M, de la Barrera S, Schierloh P, Yokobori N, Baldini M, Musella R, et al. Spontaneous or Mycobacterium tuberculosis-induced apoptotic neutrophils exert opposite effects on the dendritic cell-mediated immune response. Eur J Immunol (2007) 37:1524–37. doi: 10.1002/eji.200636771
196. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature (2012) 491:717–23. doi: 10.1038/nature11605
197. Sia JK, Georgieva M, Rengarajan J. Innate Immune Defenses in Human Tuberculosis: An Overview of the Interactions between Mycobacterium tuberculosis and Innate Immune Cells. J Immunol Res (2015) 2015:747543. doi: 10.1155/2015/747543
198. Portevin D, Via LE, Eum S, Young D. Natural killer cells are recruited during pulmonary tuberculosis and their ex vivo responses to mycobacteria vary between healthy human donors in association with KIR haplotype. Cell Microbiol (2012) 14:1734–44. doi: 10.1111/j.1462-5822.2012.01834.x
199. Abebe F. Immunological basis of early clearance of Mycobacterium tuberculosis infection: the role of natural killer cells. Clin Exp Immunol (2021) 204:32–40. doi: 10.1111/cei.13565
200. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol (2009) 10:899–906. doi: 10.1038/ni.1758
201. Mohareer K, Asalla S, Banerjee S. Cell death at the cross roads of host-pathogen interaction in Mycobacterium tuberculosis infection. Tuberculosis (Edinb) (2018) 113:99–121. doi: 10.1016/j.tube.2018.09.007
202. Filio-Rodríguez G, Estrada-García I, Arce-Paredes P, Moreno-AltamIrano MM, Islas-Trujillo S, Ponce-Regalado MD, et al. In vivo induction of neutrophil extracellular traps by Mycobacterium tuberculosis in a Guinea pig model. Innate Immun (2017) 23:625–37. doi: 10.1177/1753425917732406
203. Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun (2020) 11:2270. doi: 10.1038/s41467-020-16143-6
204. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
205. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
206. Wong L-YR, Perlman S. Author Correction: Immune dysregulation and immunopathology induced by SARS-CoV-2 and related coronaviruses - are we our own worst enemy? Nat Rev Immunol (2022) 22:200. doi: 10.1038/s41577-021-00673-1
207. Zhang Q, Bastard P, COVID Human Genetic Effor, Cobat A, Casanova J-L. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature (2022) 603:587–98. doi: 10.1038/s41586-022-04447-0
208. Kudlay D, Kofiadi I, Khaitov M. Peculiarities of the T cell immune response in COVID-19. Vaccines (Basel) (2022) 10:242. doi: 10.3390/vaccines10020242
209. Snyder ME, Farber DL. Human lung tissue resident memory T cells in health and disease. Curr Opin Immunol (2019) 59:101–8. doi: 10.1016/j.coi.2019.05.011
210. Smith VL, Cheng Y, Bryant BR, Schorey JS. Exosomes function in antigen presentation during an in vivo Mycobacterium tuberculosis infection. Sci Rep (2017) 7:43578. doi: 10.1038/srep43578
211. Le Bert N, Clapham HE, Tan AT, Chia WN, Tham CYL, Lim JM, et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J Exp Med (2021) 218:e20202617. doi: 10.1084/jem.20202617
212. Boyton RJ, Altmann DM. The immunology of asymptomatic SARS-CoV-2 infection: what are the key questions? Nat Rev Immunol (2021) 21:762–8. doi: 10.1038/s41577-021-00631-x
213. Reynolds CJ, Swadling L, Gibbons JM, Pade C, Jensen MP, Diniz MO, et al. Discordant neutralizing antibody and T cell responses in asymptomatic and mild SARS-CoV-2 infection. Sci Immunol (2020) 5:eabf3698. doi: 10.1126/sciimmunol.abf3698
214. Cao Y, Wang J, Jian F, Xiao T, Song W, Yisimayi A, et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature (2022) 602:657–63. doi: 10.1038/s41586-021-04385-3
215. van Dorp L, Houldcroft CJ, Richard D, Balloux F. COVID-19, the first pandemic in the post-genomic era. Curr Opin Virol (2021) 50:40–8. doi: 10.1016/j.coviro.2021.07.002
216. Liu L, Iketani S, Guo Y, Chan JF-W, Wang M, Liu L, et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature (2022) 602:676–81. doi: 10.1038/s41586-021-04388-0
217. De Marco L, D’Orso S, Pirronello M, Verdiani A, Termine A, Fabrizio C, et al. Assessment of T-cell reactivity to the SARS-coV-2 omicron variant by immunized individuals. JAMA Netw Open (2022) 5:e2210871. doi: 10.1001/jamanetworkopen.2022.10871
218. Gao Y, Cai C, Grifoni A, Müller TR, Niessl J, Olofsson A, et al. Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat Med (2022) 28:472–6. doi: 10.1038/s41591-022-01700-x
219. Tarke A, Coelho CH, Zhang Z, Dan JM, Yu ED, Methot N, et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell (2022) 185:847–859.e11. doi: 10.1016/j.cell.2022.01.015
220. Petrone L, Picchianti-Diamanti A, Sebastiani GD, Aiello A, Laganà B, Cuzzi G, et al. Humoral and cellular responses to spike of δ SARS-CoV-2 variant in vaccinated patients with immune-mediated inflammatory diseases. Int J Infect Dis (2022) 121:24–30. doi: 10.1016/j.ijid.2022.04.027
221. Petrone L, Tortorella C, Aiello A, Farroni C, Ruggieri S, Castilletti C, et al. Humoral and cellular response to spike of delta SARS-coV-2 variant in vaccinated patients with multiple sclerosis. Front Neurol (2022) 13:881988. doi: 10.3389/fneur.2022.881988
222. Tan AT, Linster M, Tan CW, Le Bert N, Chia WN, Kunasegaran K, et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep (2021) 34:108728. doi: 10.1016/j.celrep.2021.108728
223. Bergamaschi L, Mescia F, Turner L, Hanson AL, Kotagiri P, Dunmore BJ, et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity (2021) 54:1257–1275.e8. doi: 10.1016/j.immuni.2021.05.010
224. Tarke A, Potesta M, Varchetta S, Fenoglio D, Iannetta M, Sarmati L, et al. Early and polyantigenic CD4 T cell responses correlate with mild disease in acute COVID-19 donors. Int J Mol Sci (2022) 23:7155. doi: 10.3390/ijms23137155
225. Chauss D, Freiwald T, McGregor R, Yan B, Wang L, Nova-Lamperti E, et al. Autocrine vitamin D signaling switches off pro-inflammatory programs of TH1 cells. Nat Immunol (2022) 23:62–74. doi: 10.1038/s41590-021-01080-3
226. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
227. Gil-Etayo FJ, Suàrez-Fernández P, Cabrera-Marante O, Arroyo D, Garcinuño S, Naranjo L, et al. T-helper cell subset response is a determining factor in COVID-19 progression. Front Cell Infect Microbiol (2021) 11:624483. doi: 10.3389/fcimb.2021.624483
228. Pourgholaminejad A, Aghdami N, Baharvand H, Moazzeni SM. Is TGFβ as an anti-inflammatory cytokine required for differentiation of inflammatory TH17 cells? J Immunotoxicol (2016) 13:775–83. doi: 10.1080/1547691X.2016.1193574
229. Zander R, Kasmani MY, Chen Y, Topchyan P, Shen J, Zheng S, et al. Tfh-cell-derived interleukin 21 sustains effector CD8+ T cell responses during chronic viral infection. Immunity (2022) 55:475–493.e5. doi: 10.1016/j.immuni.2022.01.018
230. McMahan K, Yu J, Mercado NB, Loos C, Tostanoski LH, Chandrashekar A, et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature (2021) 590:630–4. doi: 10.1038/s41586-020-03041-6
231. Mallajosyula V, Ganjavi C, Chakraborty S, McSween AM, Pavlovitch-Bedzyk AJ, Wilhelmy J, et al. CD8+ T cells specific for conserved coronavirus epitopes correlate with milder disease in COVID-19 patients. Sci Immunol (2021) 6:eabg5669. doi: 10.1126/sciimmunol.abg5669
232. Dhawan M, Rabaan AA, Alwarthan S, Alhajri M, Halwani MA, Alshengeti A, et al. Regulatory T cells (Tregs) and COVID-19: unveiling the mechanisms, and therapeutic potentialities with a special focus on long COVID. Vaccines (2023) 11:699. doi: 10.3390/vaccines11030699
233. Wang K, Chen W, Zhang Z, Deng Y, Lian J-Q, Du P, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther (2020) 5:283. doi: 10.1038/s41392-020-00426-x
234. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
235. Li H, Liu L, Zhang D, Xu J, Dai H, Tang N, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet (2020) 395:1517–20. doi: 10.1016/S0140-6736(20)30920-X
236. Peteranderl C, Herold S. The impact of the interferon/TNF-related apoptosis-inducing ligand signaling axis on disease progression in respiratory viral infection and beyond. Front Immunol (2017) 8:313. doi: 10.3389/fimmu.2017.00313
237. André S, Picard M, Cezar R, Roux-Dalvai F, Alleaume-Butaux A, Soundaramourty C, et al. T cell apoptosis characterizes severe Covid-19 disease. Cell Death Differ (2022) 29:1486–99. doi: 10.1038/s41418-022-00936-x
238. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
239. Carreto-Binaghi LE, Herrera MT, Guzmán-Beltrán S, Juárez E, Sarabia C, Salgado-Cantú MG, et al. Reduced IL-8 secretion by NOD-like and toll-like receptors in blood cells from COVID-19 patients. Biomedicines (2023) 11:1078. doi: 10.3390/biomedicines11041078
240. Jiang Y, Wei X, Guan J, Qin S, Wang Z, Lu H, et al. COVID-19 pneumonia: CD8+ T and NK cells are decreased in number but compensatory increased in cytotoxic potential. Clin Immunol (2020) 218:108516. doi: 10.1016/j.clim.2020.108516
241. Alahdal M, Elkord E. Exhaustion and over-activation of immune cells in COVID-19: Challenges and therapeutic opportunities. Clin Immunol (2022) 245:109177. doi: 10.1016/j.clim.2022.109177
242. Suthar MS, Zimmerman MG, Kauffman RC, Mantus G, Linderman SL, Hudson WH, et al. Rapid generation of neutralizing antibody responses in COVID-19 patients. Cell Rep Med (2020) 1:100040. doi: 10.1016/j.xcrm.2020.100040
243. Premkumar L, Segovia-Chumbez B, Jadi R, Martinez DR, Raut R, Markmann A, et al. The receptor binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Sci Immunol (2020) 5:eabc8413. doi: 10.1126/sciimmunol.abc8413
244. Muecksch F, Wise H, Batchelor B, Squires M, Semple E, Richardson C, et al. Longitudinal serological analysis and neutralizing antibody levels in coronavirus disease 2019 convalescent patients. J Infect Dis (2021) 223:389–98. doi: 10.1093/infdis/jiaa659
245. Wang Z, Muecksch F, Schaefer-Babajew D, Finkin S, Viant C, Gaebler C, et al. Naturally enhanced neutralizing breadth against SARS-CoV-2 one year after infection. Nature (2021) 595:426–31. doi: 10.1038/s41586-021-03696-9
246. Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science (2021) 371:eabf4063. doi: 10.1126/science.abf4063
247. Wajnberg A, Amanat F, Firpo A, Altman DR, Bailey MJ, Mansour M, et al. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science (2020) 370:1227–30. doi: 10.1126/science.abd7728
248. Guo L, Ren L, Yang S, Xiao M, Chang D, Yang F, et al. Profiling early humoral response to diagnose novel coronavirus disease (COVID-19). Clin Infect Dis (2020) 71:778–85. doi: 10.1093/cid/ciaa310
249. Isho B, Abe KT, Zuo M, Jamal AJ, Rathod B, Wang JH, et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci Immunol (2020) 5:eabe5511. doi: 10.1126/sciimmunol.abe5511
250. Zohar T, Alter G. Dissecting antibody-mediated protection against SARS-CoV-2. Nat Rev Immunol (2020) 20:392–4. doi: 10.1038/s41577-020-0359-5
251. Winkler ES, Gilchuk P, Yu J, Bailey AL, Chen RE, Chong Z, et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell (2021) 184:1804–1820.e16. doi: 10.1016/j.cell.2021.02.026
252. Yamin R, Jones AT, Hoffmann H-H, Schäfer A, Kao KS, Francis RL, et al. Fc-engineered antibody therapeutics with improved anti-SARS-CoV-2 efficacy. Nature (2021) 599:465–70. doi: 10.1038/s41586-021-04017-w
253. Lee WS, Selva KJ, Davis SK, Wines BD, Reynaldi A, Esterbauer R, et al. Decay of Fc-dependent antibody functions after mild to moderate COVID-19. Cell Rep Med (2021) 2:100296. doi: 10.1016/j.xcrm.2021.100296
254. Anand SP, Prévost J, Nayrac M, Beaudoin-Bussières G, Benlarbi M, Gasser R, et al. Longitudinal analysis of humoral immunity against SARS-CoV-2 Spike in convalescent individuals up to 8 months post-symptom onset. Cell Rep Med (2021) 2:100290. doi: 10.1016/j.xcrm.2021.100290
255. Yu Y, Wang M, Zhang X, Li S, Lu Q, Zeng H, et al. Antibody-dependent cellular cytotoxicity response to SARS-CoV-2 in COVID-19 patients. Signal Transduct Target Ther (2021) 6:346. doi: 10.1038/s41392-021-00759-1
256. Piccoli L, Park Y-J, Tortorici MA, Czudnochowski N, Walls AC, Beltramello M, et al. Mapping neutralizing and immunodominant sites on the SARS-coV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell (2020) 183:1024–1042.e21. doi: 10.1016/j.cell.2020.09.037
257. Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, et al. Deep mutational scanning of SARS-coV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell (2020) 182:1295–1310.e20. doi: 10.1016/j.cell.2020.08.012
258. McCallum M, Bassi J, De Marco A, Chen A, Walls AC, Di Iulio J, et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science (2021) 373:648–54. doi: 10.1126/science.abi7994
259. McCallum M, De Marco A, Lempp FA, Tortorici MA, Pinto D, Walls AC, et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell (2021) 184:2332–2347.e16. doi: 10.1016/j.cell.2021.03.028
260. Lempp FA, Soriaga LB, Montiel-Ruiz M, Benigni F, Noack J, Park Y-J, et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature (2021) 598:342–7. doi: 10.1038/s41586-021-03925-1
261. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol (2020) 5:eabd7114. doi: 10.1126/sciimmunol.abd7114
262. Long Q-X, Tang X-J, Shi Q-L, Li Q, Deng H-J, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med (2020) 26:1200–4. doi: 10.1038/s41591-020-0965-6
263. Burgener S, Rochat P, Dollenmaier G, Benz G, Kistler AD, Fulchini R. Progression of COVID-19 in a patient on anti-CD20 antibody treatment: case report and literature review. Case Rep Infect Dis (2022) 2022:8712424. doi: 10.1155/2022/8712424
264. Russell MW, Moldoveanu Z, Ogra PL, Mestecky J. Mucosal immunity in COVID-19: A neglected but critical aspect of SARS-coV-2 infection. Front Immunol (2020) 11:611337. doi: 10.3389/fimmu.2020.611337
265. De Biasi S, Lo Tartaro D, Meschiari M, Gibellini L, Bellinazzi C, Borella R, et al. Expansion of plasmablasts and loss of memory B cells in peripheral blood from COVID-19 patients with pneumonia. Eur J Immunol (2020) 50:1283–94. doi: 10.1002/eji.202048838
266. Bernardes JP, Mishra N, Tran F, Bahmer T, Best L, Blase JI, et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity (2020) 53:1296–1314.e9. doi: 10.1016/j.immuni.2020.11.017
267. Slight SR, Khader SA. Chemokines shape the immune responses to tuberculosis. Cytokine Growth Factor Rev (2013) 24:105–13. doi: 10.1016/j.cytogfr.2012.10.002
268. Olmos S, Stukes S, Ernst JD. Ectopic activation of Mycobacterium tuberculosis-specific CD4+ T cells in lungs of CCR7-/- mice. J Immunol (2010) 184:895–901. doi: 10.4049/jimmunol.0901230
269. Lin PL, Pawar S, Myers A, Pegu A, Fuhrman C, Reinhart TA, et al. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun (2006) 74:3790–803. doi: 10.1128/IAI.00064-06
270. Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun (2002) 70:4501–9. doi: 10.1128/IAI.70.8.4501-4509.2002
271. Srivastava S, Ernst JD. Cell-to-cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell Host Microbe (2014) 15:741–52. doi: 10.1016/j.chom.2014.05.007
272. Gideon HP, Hughes TK, Tzouanas CN, Wadsworth MH, Tu AA, Gierahn TM, et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity (2022) 55:827–846.e10. doi: 10.1016/j.immuni.2022.04.004
273. Donovan ML, Bielefeldt-Ohmann H, Rollo RF, McPherson SJ, Schultz TE, Mori G, et al. Distinct contributions of the innate immune receptors TLR2 and RP105 to formation and architecture of structured lung granulomas in mice infected with Mycobacterium tuberculosis. Immunology (2023) 169:13–26. doi: 10.1111/imm.13606
274. Guirado E, Mbawuike U, Keiser TL, Arcos J, Azad AK, Wang S-H, et al. Characterization of host and microbial determinants in individuals with latent tuberculosis infection using a human granuloma model. mBio (2015) 6:e02537–02514. doi: 10.1128/mBio.02537-14
275. Wayne LG, Lin KY. Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect Immun (1982) 37:1042–9. doi: 10.1128/iai.37.3.1042-1049.1982
276. de la Barrera S, Aleman M, Musella R, Schierloh P, Pasquinelli V, Garcia V, et al. IL-10 down-regulates costimulatory molecules on Mycobacterium tuberculosis-pulsed macrophages and impairs the lytic activity of CD4 and CD8 CTL in tuberculosis patients. Clin Exp Immunol (2004) 138:128–38. doi: 10.1111/j.1365-2249.2004.02577.x
277. Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med (2001) 193:271–80. doi: 10.1084/jem.193.3.271
278. Lin PL, Rutledge T, Green AM, Bigbee M, Fuhrman C, Klein E, et al. CD4 T cell depletion exacerbates acute Mycobacterium tuberculosis while reactivation of latent infection is dependent on severity of tissue depletion in cynomolgus macaques. AIDS Res Hum Retroviruses (2012) 28:1693–702. doi: 10.1089/AID.2012.0028
279. Fabri M, Stenger S, Shin D-M, Yuk J-M, Liu PT, Realegeno S, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med (2011) 3:104ra102. doi: 10.1126/scitranslmed.3003045
280. Sallin MA, Sakai S, Kauffman KD, Young HA, Zhu J, Barber DL. Th1 differentiation drives the accumulation of intravascular, non-protective CD4 T cells during tuberculosis. Cell Rep (2017) 18:3091–104. doi: 10.1016/j.celrep.2017.03.007
281. Goletti D, Weissman D, Jackson RW, Graham NM, Vlahov D, Klein RS, et al. Effect of Mycobacterium tuberculosis on HIV replication. Role Immune activation. J Immunol (1996) 157:1271–8. doi: 10.4049/jimmunol.157.3.1271
282. Goletti D, Weissman D, Jackson RW, Collins F, Kinter A, Fauci AS. The in vitro induction of human immunodeficiency virus (HIV) replication in purified protein derivative-positive HIV-infected persons by recall antigen response to Mycobacterium tuberculosis is the result of a balance of the effects of endogenous interleukin-2 and proinflammatory and antiinflammatory cytokines. J Infect Dis (1998) 177:1332–8. doi: 10.1086/515276
283. Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol Rev (2015) 264:74–87. doi: 10.1111/imr.12274
284. Boom WH, Schaible UE, Achkar JM. The knowns and unknowns of latent Mycobacterium tuberculosis infection. J Clin Invest (2021) 131:e136222. doi: 10.1172/JCI136222
285. Cantini F, Niccoli L, Capone A, Petrone L, Goletti D. Risk of tuberculosis reactivation associated with traditional disease modifying anti-rheumatic drugs and non-anti-tumor necrosis factor biologics in patients with rheumatic disorders and suggestion for clinical practice. Expert Opin Drug Saf (2019) 18:415–25. doi: 10.1080/14740338.2019.1612872
286. Chen CY, Huang D, Wang RC, Shen L, Zeng G, Yao S, et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PloS Pathog (2009) 5:e1000392. doi: 10.1371/journal.ppat.1000392
287. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol (2014) 14:585–600. doi: 10.1038/nri3707
288. Freches D, Korf H, Denis O, Havaux X, Huygen K, ROmano M. Mice genetically inactivated in interleukin-17A receptor are defective in long-term control of Mycobacterium tuberculosis infection. Immunology (2013) 140:220–31. doi: 10.1111/imm.12130
289. Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, et al. Cutting edge: IFN-gamma regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol (2006) 177:1416–20. doi: 10.4049/jimmunol.177.3.1416
290. Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity (2009) 31:974–85. doi: 10.1016/j.immuni.2009.10.007
291. Najafi-Fard S, Petruccioli E, Farroni C, Petrone L, Vanini V, Cuzzi G, et al. Evaluation of the immunomodulatory effects of interleukin-10 on peripheral blood immune cells of COVID-19 patients: Implication for COVID-19 therapy. Front Immunol (2022) 13:984098. doi: 10.3389/fimmu.2022.984098
292. Cicchese JM, Evans S, Hult C, Joslyn LR, Wessler T, Millar JA, et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol Rev (2018) 285:147–67. doi: 10.1111/imr.12671
293. Boussiotis VA, Tsai EY, Yunis EJ, Thim S, Delgado JC, Dascher CC, et al. IL-10-producing T cells suppress immune responses in anergic tuberculosis patients. J Clin Invest (2000) 105:1317–25. doi: 10.1172/JCI9918
294. Irvine EB, O’Neil A, Darrah PA, Shin S, Choudhary A, Li W, et al. Robust IgM responses following intravenous vaccination with Bacille Calmette-Guérin associate with prevention of Mycobacterium tuberculosis infection in macaques. Nat Immunol (2021) 22:1515–23. doi: 10.1038/s41590-021-01066-1
295. Watson A, Li H, Ma B, Weiss R, Bendayan D, Abramovitz L, et al. Human antibodies targeting a Mycobacterium transporter protein mediate protection against tuberculosis. Nat Commun (2021) 12:602. doi: 10.1038/s41467-021-20930-0
296. Alter G, Ottenhoff THM, Joosten SA. Antibody glycosylation in inflammation, disease and vaccination. Semin Immunol (2018) 39:102–10. doi: 10.1016/j.smim.2018.05.003
297. Roy Chowdhury R, Vallania F, Yang Q, Lopez Angel CJ, Darboe F, Penn-Nicholson A, et al. A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature (2018) 560:644–8. doi: 10.1038/s41586-018-0439-x
298. Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, et al. A functional role for antibodies in tuberculosis. Cell (2016) 167:433–443.e14. doi: 10.1016/j.cell.2016.08.072
299. Grace PS, Dolatshahi S, Lu LL, Cain A, Palmieri F, Petrone L, et al. Antibody subclass and glycosylation shift following effective TB treatment. Front Immunol (2021) 12:679973. doi: 10.3389/fimmu.2021.679973
300. Lu LL, Smith MT, Yu KKQ, Luedemann C, Suscovich TJ, Grace PS, et al. IFN-γ-independent immune markers of Mycobacterium tuberculosis exposure. Nat Med (2019) 25:977–87. doi: 10.1038/s41591-019-0441-3
301. Melkie ST, Arias L, Farroni C, Jankovic Makek M, Goletti D, Vilaplana C. The role of antibodies in tuberculosis diagnosis, prophylaxis and therapy: a review from the ESGMYC study group. Eur Respir Rev (2022) 31:210218. doi: 10.1183/16000617.0218-2021
302. Sy KTL, Haw NJL, Uy J. Previous and active tuberculosis increases risk of death and prolongs recovery in patients with COVID-19. Infect Dis (Lond) (2020) 52:902–7. doi: 10.1080/23744235.2020.1806353
303. Dheda K, Perumal T, Moultrie H, Perumal R, Esmail A, Scott AJ, et al. The intersecting pandemics of tuberculosis and COVID-19: population-level and patient-level impact, clinical presentation, and corrective interventions. Lancet Respir Med (2022) 10:603–22. doi: 10.1016/S2213-2600(22)00092-3
304. Gao Y, Liu M, Chen Y, Shi S, Geng J, Tian J. Association between tuberculosis and COVID-19 severity and mortality: A rapid systematic review and meta-analysis. J Med Virol (2021) 93:194–6. doi: 10.1002/jmv.26311
305. TB/COVID-19 Global Study Group. Tuberculosis and COVID-19 co-infection: description of the global cohort. Eur Respir J (2022) 59:2102538. doi: 10.1183/13993003.02538-2021
306. Jassat W, Mudara C, Ozougwu L, Tempia S, Blumberg L, Davies M-A, et al. Difference in mortality among individuals admitted to hospital with COVID-19 during the first and second waves in South Africa: a cohort study. Lancet Glob Health (2021) 9:e1216–25. doi: 10.1016/S2214-109X(21)00289-8
307. Tadolini M, García-García J-M, Blanc F-X, Borisov S, Goletti D, Motta I, et al. On tuberculosis and COVID-19 co-infection. Eur Respir J (2020) 56:2002328. doi: 10.1183/13993003.02328-2020
308. Tadolini M, Codecasa LR, García-García J-M, Blanc F-X, Borisov S, Alffenaar J-W, et al. Active tuberculosis, sequelae and COVID-19 co-infection: first cohort of 49 cases. Eur Respir J (2020) 56:2001398. doi: 10.1183/13993003.01398-2020
309. Sheerin D, Abhimanyu n, Peton N, Vo W, Allison CC, Wang X, et al. Immunopathogenic overlap between COVID-19 and tuberculosis identified from transcriptomic meta-analysis and human macrophage infection. iScience (2022) 25:104464. doi: 10.1016/j.isci.2022.104464
310. Petrone L, Petruccioli E, Vanini V, Cuzzi G, Gualano G, Vittozzi P, et al. Coinfection of tuberculosis and COVID-19 limits the ability to in vitro respond to SARS-CoV-2. Int J Infect Dis (2021) 113 Suppl 1:S82–7. doi: 10.1016/j.ijid.2021.02.090
311. Riou C, du Bruyn E, Stek C, Daroowala R, Goliath RT, Abrahams F, et al. Relationship of SARS-CoV-2-specific CD4 response to COVID-19 severity and impact of HIV-1 and tuberculosis coinfection. J Clin Invest (2021) 131:e149125. doi: 10.1172/JCI149125
312. Stochino C, Villa S, Zucchi P, Parravicini P, Gori A, Raviglione MC. Clinical characteristics of COVID-19 and active tuberculosis co-infection in an Italian reference hospital. Eur Respir J (2020) 56:2001708. doi: 10.1183/13993003.01708-2020
313. Najafi-Fard S, Aiello A, Navarra A, Cuzzi G, Vanini V, Migliori GB, et al. Characterization of the immune impairment of tuberculosis and COVID-19 coinfected patients. Int J Infect Dis (2023) 130, Supplement 1, S34–S42. doi: 10.1016/j.ijid.2023.03.021
314. du Bruyn E, Stek C, Daroowala R, Said-Hartley Q, Hsiao M, Schafer G, et al. Effects of tuberculosis and/or HIV-1 infection on COVID-19 presentation and immune response in Africa. Nat Commun (2023) 14:188. doi: 10.1038/s41467-022-35689-1
315. Musso M, Di Gennaro F, Gualano G, Mosti S, Cerva C, Fard SN, et al. Concurrent cavitary pulmonary tuberculosis and COVID-19 pneumonia with in vitro immune cell anergy. Infection (2021) 49:1061–4. doi: 10.1007/s15010-021-01576-y
316. Sheerin D, Phan TK, Eriksson EM, Consortium CP, Coussens AK. Distinct and overlapping immunological responses to SARS-CoV-2 and Mycobacterium tuberculosis identified by single-cell RNA-seq of co-infected whole blood. (2023) 2023:. doi: 10.1101/2023.05.24.23290499
317. Hildebrand RE, Chandrasekar SS, Riel M, Touray BJB, Aschenbroich SA, Talaat AM. Superinfection with SARS-CoV-2 Has Deleterious Effects on Mycobacterium bovis BCG Immunity and Promotes Dissemination of Mycobacterium tuberculosis. Microbiol Spectr (2022) 10:e0307522. doi: 10.1128/spectrum.03075-22
318. Rosas Mejia O, Gloag ES, Li J, Ruane-Foster M, Claeys TA, Farkas D, et al. Mice infected with Mycobacterium tuberculosis are resistant to acute disease caused by secondary infection with SARS-CoV-2. PloS Pathog (2022) 18:e1010093. doi: 10.1371/journal.ppat.1010093
319. Rajamanickam A, Kumar NP, Padmapriyadarsini C, Nancy A, Selvaraj N, Karunanithi K, et al. Latent tuberculosis co-infection is associated with heightened levels of humoral, cytokine and acute phase responses in seropositive SARS-CoV-2 infection. J Infect (2021) 83:339–46. doi: 10.1016/j.jinf.2021.07.029
320. Rajamanickam A, Pavan Kumar N, Chandrasekaran P, Nancy A, Bhavani PK, Selvaraj N, et al. Effect of SARS-CoV-2 seropositivity on antigen - specific cytokine and chemokine responses in latent tuberculosis. Cytokine (2022) 150:155785. doi: 10.1016/j.cyto.2021.155785
321. Pozdnyakov A, Jin A, Bader M. Reactivation of pulmonary tuberculosis in a patient with COVID-19: case report and review of literature. Infect Dis Clin Pract (Baltim Md) (2021) 29:e468–70. doi: 10.1097/IPC.0000000000001032
322. Leonso A-A, Brown K, Prol R, Rawat S, Khunger A, Bromberg R. A rare case of latent tuberculosis reactivation secondary to a COVID-19 infection. Infect Dis Rep (2022) 14:446–52. doi: 10.3390/idr14030048
323. Garg N, Lee YI. REACTIVATION TB WITH SEVERE COVID-19. Chest (2020) 158:A777. doi: 10.1016/j.chest.2020.08.724
324. Khayat M, Fan H, Vali Y. COVID-19 promoting the development of active tuberculosis in a patient with latent tuberculosis infection: A case report. Respir Med Case Rep (2021) 32:101344. doi: 10.1016/j.rmcr.2021.101344
325. COLBY S, SHAH R. TB REACTIVATION FOLLOWING COVID-19 INFECTION. Chest (2022) 162:A329. doi: 10.1016/j.chest.2022.08.255
326. Tang W, Leonhardt L, Pervez A, Sarvepalli S. A case of pleural tuberculosis vs latent tuberculosis reactivation as a result of COVID-19 infection and treatment. J Community Hosp Intern Med Perspect (2022) 12:89–93. doi: 10.55729/2000-9666.1078
327. Azkur AK, Akdis M, Azkur D, Sokolowska M, van de Veen W, Brüggen M-C, et al. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy (2020) 75:1564–81. doi: 10.1111/all.14364
328. Khan N, Vidyarthi A, Amir M, Mushtaq K, Agrewala JN. T-cell exhaustion in tuberculosis: pitfalls and prospects. Crit Rev Microbiol (2017) 43:133–41. doi: 10.1080/1040841X.2016.1185603
329. Kumar NP, Moideen K, Banurekha VV, Nair D, Babu S. Plasma proinflammatory cytokines are markers of disease severity and bacterial burden in pulmonary tuberculosis. Open Forum Infect Dis (2019) 6:ofz257. doi: 10.1093/ofid/ofz257
330. Srivastava S, Jaggi N. TB positive cases go up in ongoing COVID-19 pandemic despite lower testing of TB: An observational study from a hospital from Northern India. Indian J Tuberc (2022) 69:157–60. doi: 10.1016/j.ijtb.2021.04.014
331. Luke E, Swafford K, Shirazi G, Venketaraman V. TB and COVID-19: an exploration of the characteristics and resulting complications of co-infection. Front Biosci (Schol Ed) (2022) 14:6. doi: 10.31083/j.fbs1401006
Keywords: SARS-CoV-2, M. tuberculosis, COVID-19, tuberculosis, innate response, T cell response, antibody response, co-infection
Citation: Aiello A, Najafi-Fard S and Goletti D (2023) Initial immune response after exposure to Mycobacterium tuberculosis or to SARS-COV-2: similarities and differences. Front. Immunol. 14:1244556. doi: 10.3389/fimmu.2023.1244556
Received: 22 June 2023; Accepted: 31 July 2023;
Published: 17 August 2023.
Edited by:
Stefan H. E. Kaufmann, Max Planck Institute for Infection Biology, GermanyReviewed by:
Luis Horacio Gutiérrez-González, National Institute of Respiratory Diseases-Mexico (INER), MexicoKerry L. Hilligan, National Institute of Allergy and Infectious Diseases (NIH), United States
Copyright © 2023 Aiello, Najafi-Fard and Goletti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Delia Goletti, delia.goletti@inmi.it
†These authors have contributed equally to this work and share first authorship