 Anna Allué-Guardia
Anna Allué-Guardia Jordi B. Torrelles
Jordi B. Torrelles Alex Sigal
Alex Sigal
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 27 September 2023
Sec. Microbial Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1250198
This article is part of the Research TopicThe Covid-19 and TB Syndemic: Differences and SimilaritiesView all 11 articles
Mycobacterium tuberculosis (M.tb) and SARS-CoV-2 are both infections that can lead to severe disease in the lower lung. However, these two infections are caused by very different pathogens (Mycobacterium vs. virus), they have different mechanisms of pathogenesis and immune response, and differ in how long the infection lasts. Despite the differences, SARS-CoV-2 and M.tb share a common feature, which is also frequently observed in other respiratory infections: the burden of disease in the elderly is greater. Here, we discuss possible reasons for the higher burden in older adults, including the effect of co-morbidities, deterioration of the lung environment, auto-immunity, and a reduced antibody response. While the answer is likely to be multifactorial, understanding the main drivers across different infections may allow us to design broader interventions that increase the health-span of older people.
The older adult population (> 60 years old) is projected to double to 2 billion by 2050 (1, 2). Natural lung aging is associated with progressive changes at both the cellular and organ level, including cellular senescence and chronic inflammation among others (3). This causes a decline in lung function and impaired immunological responses (4–7), which would be expected to influence the response to respiratory infections.
Coronavirus disease 2019 (COVID-19) and tuberculosis (TB) are both predominantly respiratory diseases but do not have a great deal in common beyond that. COVID-19 results from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, a virus that in most people persists for a few weeks or less and is cleared by the adaptive immune response. Protection against symptomatic SARS-CoV-2 infection correlates strongly with the levels of neutralizing antibodies against the virus (8). In support of this, the Omicron variant of SARS-CoV-2 was able to extensively re-infect people with pre-existing immunity (9) because it had high-level escape from neutralizing antibodies elicited by previous infection or vaccination (10). In contrast, TB, caused by Mycobacterium tuberculosis (M.tb), can persist indefinitely in the infected individual (11, 12). Further, TB generally follows a bimodal age pattern, with higher risk of severe disease in children below 5 years of age and adult individuals of > 30 years old (13, 14), while severe COVID-19 is more common in older adults and pediatric COVID-19 deaths are relatively rare (15). These differences may be due to the fact that the immune responses in TB and COVID-19 are different.
Despite the differences, these two infections share common features: first, both are strongly affected by immunosuppression (e.g. during HIV infection), indicating that their control strongly depends on T cell and/or antibody responses, which are compromised by the CD4 T cell depletion and dysregulation during HIV infection (16–24). Indeed, TB is one of the cardinal diseases leading to the death of people living with HIV (PLWH) in the pre-ART era (25, 26). On the other hand, the most striking effect of HIV co-infection in COVID-19 happens in advanced HIV disease (defined as a CD4 T cell counts of less than 200 cells per microliter), where prolonged SARS-CoV-2 infection can last for months (27–33), leading to extensive SARS-CoV-2 genome evolution. A second common feature, which will be the focus of this review, is the remarkably higher disease burden in the elderly population (34). This is also true for most respiratory infections such as respiratory syncytial virus (35, 36), influenza (37, 38), and even rhinovirus, which is usually a mild upper respiratory tract infection, but can become a more severe lower respiratory infection in the elderly, very young children, or immunocompromised people (39).
Globally, COVID-19 has a mortality rate of about 1% (40), although this is influenced and fluctuates depending on many factors, including phenotypic and genotypic host factors, host immunity, and SARS-CoV-2 variants, among others. The elderly are at a higher risk of having more severe disease, which manifests as a lower respiratory tract infection that may require hospitalization, intensive care, and ventilation. It also results in higher mortality (41–43). The increase in the probability to die from COVID-19 as a function of age is dramatic: relative to the under-55 age group, mortality increases 8-fold in the 55-64 age group and 62-fold in the over 65 age group (43).
In contrast to the 1% mortality rate from COVID-19, mortality from TB disease is roughly a quarter of the TB incidence (44). That is, about a quarter of people diagnosed with TB disease will die. However, most people who are exposed to M.tb do not progress to symptomatic disease and instead have subclinical or asymptomatic infection for years. In this case, the infection is controlled by the host immune response (45, 46). In the elderly population, such subclinical or asymptomatic infection has a higher chance to develop into TB disease (47–49). Indeed, more than 90% of TB cases in older individuals result from reactivation of latent TB infection (LTBI) (50). Elderly people that develop TB disease have high mortality, mainly due to treatment failure. A recent report evaluating data from four countries shows that the treatment success rate among people with TB < 65 years old is 82% but decreases among the older age groups to 76% in 65−74 year-olds, 65% in 75−84 year-olds, and 46% in ≥85 year-olds (51).
There are multiple factors that may interact with each other and potentially play a role in the higher disease burden in COVID-19 and TB in the elderly, and their contribution may differ between the two infections. These include age-associated inflammation (inflammaging), a less effective immune response due to immunosenescense, and a highly oxidized lung environment (Figure 1). Although observed less frequently, other factors such as an age-related increase in autoantibodies (autoimmunity) may play a role in higher COVID-19 severity in the elderly. In addition, increasing numbers of people living with comorbidities in the elderly population may be particularly important. These factors tend to arise at different times along the life span (Figure 1). In the next sections, we outline examples for each of these factors, including how they may exacerbate COVID-19 and TB in older individuals.
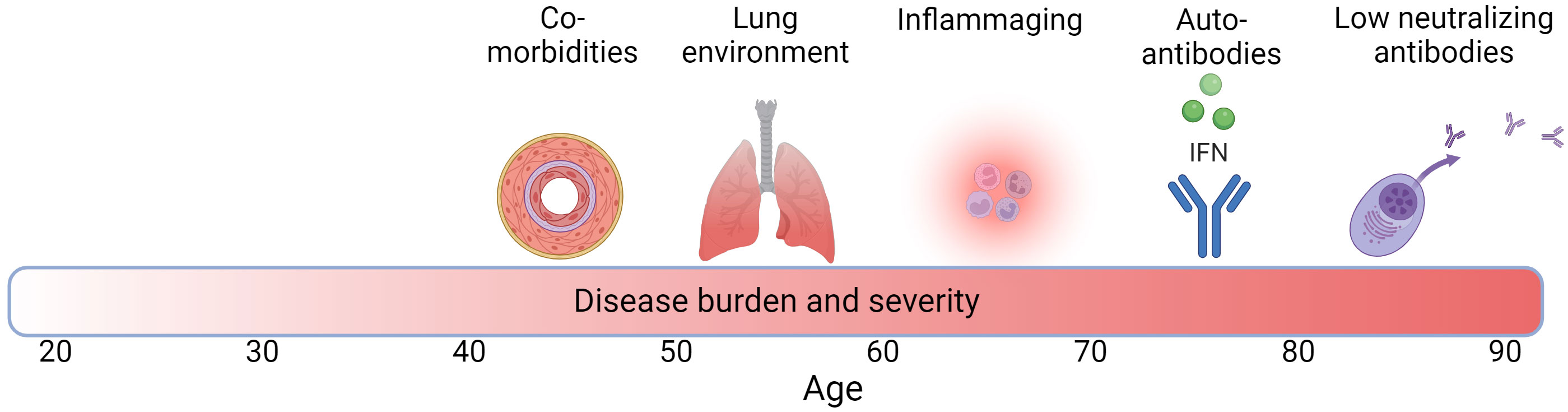
Figure 1 Factors associated with increased TB and COVID-19 disease burden with age across the life span. Darker red in bar denotes higher disease burden and severity and numbers denote age. Created with BioRender.com.
The process of aging is associated with a decline in immune functions marked by immunosenescence, resulting in increased susceptibility to autoimmunity, malignancies, and infectious diseases (4, 5, 52–58). Immunosenescence is relatively well characterized in the adaptive immune system. Age-related adaptive immune dysfunction is related to a lingering level of low-grade inflammation, immune dysfunction, increased number of memory T cells, and the loss ability of T cells to respond to antigens, as well as irreversible T cell loss of proliferation capacity. Interestingly, viral (e.g. SARS-CoV-2) and bacterial (e.g. M.tb) infections can also increase the extent of immune senescence, adding to the increased immune dysfunction and inflammation, especially in the elderly population (reviewed in detail in (59)).
Senescence in the innate immune system, where innate immunity is the first response to infection, is less well-characterized. Evidence for macrophage senescence during aging is supported by decreased pro-inflammatory responses of human (60–64) and mouse phagocytes to lipopolysaccharide (LPS) stimulation (65–70), which could be linked to age-related alterations in Toll-Like Receptor (TLR) expression and/or signaling which recognizes pathogen-associated molecular patterns (65, 66, 71–73).
Still, cellular immunosenescence does not fully explain increased circulating pro-inflammatory cytokines seen in elderly people, non-human primates, and old mice (61, 74–76), or the increased expression of pro-inflammatory genes with aging in several organs (77–81). This has led to a second paradigm, termed inflammaging, in which chronic, low-grade inflammation develops with increasing age in tissues that are frequently exposed to innate immune stimulation and oxidative stress (82, 83). Inflammaging occurs in the human lung, with increased numbers of macrophages and neutrophils in the lung alveolar lining fluid (ALF) of elderly individuals, as well as increased levels of IL-6 and IL-8 (84, 85). Specifically, IL-6 is the commonly used biomarker of inflammaging (86). There is also increased p38 MAPK phosphorylation and nuclear localization of NF-κB (87–89), a critical regulator of inflammation. Resident alveolar macrophages are more activated in the elderly (90–92) and have increased production of pro-inflammatory cytokines in response to TLR stimulation (93). Taken together, there is strong evidence that chronic inflammation occurs in the lungs as we age.
How inflammaging affects M.tb and SARS-CoV-2 infection is not completely understood. However, in both infections, a balanced immune response is thought to be critical both for infection control and to prevent immune system mediated damage. Tumor necrosis factor (TNF), the upstream activator of the NF-κB system, is elevated in inflammaging. High levels of TNF lead to reduced control of M.tb through programmed cell necrosis of activated macrophages via the mitochondrial-lysosomal-endoplasmic reticulum signaling circuit (94–96). Since macrophages are the primary host cells of M.tb as well as the most important line of defense against this pathogen, macrophage death in turn increases M.tb replication since the bacilli are able to robustly grow in the dead infected cells (97).
Immunosenescence and inflammaging are also suspected to contribute to severe COVID-19 in the elderly as well as to persistence of symptoms following acute disease (98, 99). Severe SARS-CoV-2 infection is characterized by a cytokine storm that, combined with a dysfunctional immune response in the elderly, leads to the accumulation of immune cells in the lungs and overproduction of pro-inflammatory molecules such as IL-6 (a marker of inflammaging), resulting in more tissue damage (100–102). High levels of pro-inflammatory molecules (hyperinflammatory syndrome) promote the survival of neutrophils via decreased apoptosis (103); and persistent increased systemic levels of neutrophils and monocytes in COVID-19 patients are associated with increased disease severity (104). Age-associated dysregulation and senescence of T-cells may also influence the immune response to SARS-CoV-2 (105). As seen in HIV infection, CD4 T cell depletion and dysregulation may lead to the inability to clear SARS-CoV-2 infection, most likely due to the inability to generate antibodies which will effectively neutralize the virus (27). This would be expected since CD4 T cells are critical to facilitate the antibody response to infection (106). Lastly, SARS-CoV-2 infection might also increase chronic inflammation in the elderly, resulting in a higher chance of long-term sequelae even after viral clearance (long-COVID) (107). New therapies targeting age-associated pathways may be critical to reduce COVID-19 mortality and/or long-term sequelae in the aging population (108).
Local inflammation and oxidation occur in the aging lung and influences the ALF (109, 110). ALF is a surfactant which reduces surface tension and allows the lung alveoli to expand. It also functions in multiple ways in the innate immune response to lung pathogens. Here we will focus on the ALF as an example of how the lung environment can change with age and its impact on TB and COVID-19.
ALF is generated, secreted, and recycled by alveolar epithelial type II cells (ATII), and is essential for maintaining lung homeostasis (111, 112). ALF in elderly individuals degrades quickly and is not regenerated efficiently because of ATII senescence. In addition, low-grade chronic inflammation in old age is expected to alter ALF component production and activity, due in part to biochemical modifications because of the alveolar oxidation state. We and others have shown that components of human ALF including collectins (which bind pathogen surface oligosaccharides or lipids and mark the pathogen for the innate immune response), surfactant protein (SP)-A and SP-D, homeostatic hydrolytic activities (hydrolases), surfactant lipids, and the complement system are critical elements of the innate immune system during M.tb infection (113–115) and play important roles in M.tb-phagocyte encounters (116–119). Indeed, SP-A upregulates the expression of the mannose receptor in macrophages, which in turn favors M.tb survival within phagocytes. In contrast, SP-D can directly bind M.tb clustering bacteria, favoring recognition and uptake by phagocytes driving better control of the infection (110).
In a study defining the molecular composition of ALF in the aging lung, our findings demonstrate that pro-inflammatory cytokines are increased, SP-A and SP-D and complement components are significantly increased but dysfunctional, ALF hydrolases are decreased, and surfactant lipids are oxidized in both mice and humans (109). Further, a recent quantitative proteomic profiling of the lung environment of human adult vs. elderly ALF investigated molecular fingerprints, pathways, and regulatory networks that characterize the alveolar space in old age compared to younger individuals (120). ALF from elderly individuals had significantly increased production of matrix metalloproteinases, markers of cellular senescence, antimicrobials, and proteins of neutrophilic granule origin, among others, suggesting that neutrophils could be potential contributors to the dysregulated alveolar environment with increasing age.
Consistent with reduced ALF functionality with age, M.tb exposed to human ALF obtained from older adults showed increased intracellular growth in macrophages and ATIIs (121–123), as well as increased bacterial burden and lung tissue damage in mice (121). In addition, M.tb exposed to ALF from healthy 18- to 45-year-old adults upregulated key cell envelope genes associated with amino acid, carbohydrate, and lipid metabolism, as well as genes associated with redox homeostasis and transcriptional regulators, while M.tb exposed to ALF from 60+ year-old individuals showed lower transcriptional responses (124). The changes in ALF in aging support the concept that the pulmonary environment can modify mucosal immune responses, thereby increasing the susceptibility to pulmonary infections in the elderly population.
How ALF influences SARS-CoV-2 infection is mostly unknown. However, patients with severe COVID-19 sometimes harbor IgA autoantibodies against pulmonary SP-B and SP-C, blocking the function of the lung surfactant lipid layer and potentially contributing to alveolar collapse and poor oxygenation (125). Other studies indicate that levels of SP-D in blood could be used as a biomarker for COVID-19 severity as a result of the impairment of the pulmonary barrier caused by prolonged inflammation (126). Still, how the levels, status, and function of ALF components in the alveolar environment determine the outcome of M.tb and SARS-CoV-2 infection and disease severity of TB and COVID-19, respectively, still needs to be elucidated in detail.
The adaptive immune response is essential to control both M.tb and SARS-CoV-2. CD4 T cells are critical in the orchestration of both the antibody and cellular adaptive immune response to infections (106, 127, 128). For SARS-CoV-2, the strongest correlate of protection against symptomatic infection is the level of pre-existing neutralizing antibody immunity (8, 129). Thus, SARS-CoV-2 neutralizing antibody levels are studied extensively as a function of age. A complication of measuring neutralizing antibody levels after infection is that higher disease severity elicits higher antibody levels (130). However, it is possible to distinguish between neutralizing antibody production capacity and disease severity by measuring neutralizing antibody levels to SARS-CoV-2 post-vaccination, with much of the data coming from mRNA vaccines.
One of the first studies examining the neutralizing antibody response to the Pfizer BNT162b2 mRNA vaccine against SARS-CoV-2 found that the fraction of people with a detectable neutralizing antibody response decreased slowly as a function of age up to the age of 80, with almost all individuals responding to the vaccine. After 80, the probability to elicit a neutralizing antibody response plummeted and was close to zero at 90, although the number of individuals in this part of the age range was small in the study (131). A second study used a cutoff of 55 years for who is elderly and showed substantially lower neutralizing antibody levels in the older age group after the first dose of an mRNA vaccine (132). However, this difference decreased with the second dose. A third study done in Singapore also found that people over 60 had lower neutralizing responses with an mRNA vaccine. However, they showed a strong increase in neutralizing antibodies with a third, booster dose (133). The benefit of a booster dose was recapitulated in a group of over-80-year-olds who did not have an antibody response to the first two doses (134).
The role of neutralizing antibodies in the immune response to M.tb is currently unclear. However, the reduced ability to mount an effective neutralizing antibody response may indicate an overall less effective adaptive immune response in the elderly, which would reduce M.tb control.
An essential component of the initial immune response to both M.tb and SARS-CoV-2 is interferon (IFN), which orchestrates the innate immune response to infection. In TB, the type II interferon IFN-γ activates macrophages and enables them to initiate maturation and acidification of the M.tb-containing phagosome, as well as other antimicrobial responses (135). The failure of this process to kill the internalized bacilli leads to macrophage death and M.tb growth in the dead infected cells (97). Mice deficient in IFN-γ quickly succumb to TB (136, 137).
The role of type I interferons during M.tb infection is not completely understood, with some studies reporting a host protective role vs. other studies suggesting a detrimental role under different host-M.tb encounter settings (138, 139). However, type I interferons including IFN-α are an important component of the innate immune response to SARS-CoV-2 (100), and they are rapidly induced in early stages of the infection (140). Multiple SARS-CoV-2 genes attempt to interfere with IFN (141–144). Individuals with inborn errors in type I IFN immunity are much more prone to severe COVID-19 (145) and mice deficient for type I IFN have reduced activation of CD4 and CD8 T cells and reduced recruitment of monocytes and monocyte-derived macrophages to the lung (146).
Anti-IFN antibodies might block IFN binding to IFN receptors, impairing its antiviral effect (147). There have been sporadic case reports of anti-IFN antibodies increasing susceptibility to mycobacterial infections (148–150) or shown to be elevated at the site of infection in advanced TB patients (151). In contrast, SARS-CoV-2 infection is reported to be more severe in individuals with autoantibodies to type I IFN. In one study, 101 of 987 patients with severe COVID-19 have been found to have these autoantibodies, while none of the 663 individuals with asymptomatic or mild SARS-CoV-2 infection had anti-IFN type I antibodies (152). The prevalence of anti-IFN type I antibodies was found to be strongly age dependent (153, 154). Autoantibodies neutralizing high concentrations of IFN-α were present in 0.18% of individuals between 18 and 69 years, 1.1% of individuals between 70 and 79 years, and 3.4% of people >80 years of age (154). Autoantibodies are unlikely to completely explain the higher susceptibility of the elderly population to severe COVID-19. However, such autoimmunity may be a contributing factor in a subset of people (155) and an example of an age dependent affect which is highly variable between people of the same age. Also, antibodies to other host proteins are known to increase with age (156). This may potentially add to disease pathology in a similar way.
An important aspect of the shift towards a global aging population is increasing chronic illness. The top 10 comorbidities associated with the elderly population include hypertension (58%), high cholesterol (47%), arthritis (31%), ischemic heart disease (29%), diabetes (27%), chronic kidney disease (18%), heart failure (14%), depression, Alzheimer disease and dementia (11%) and chronic obstructive pulmonary disease (COPD, 11%) (157). Some of these comorbidities overlap with known risk factors for TB (diabetes) and higher COVID-19 severity (obesity, hypertension, high cholesterol, and diabetes). Thus, a common risk factor for TB and COVID-19 which increases in prevalence in the elderly is diabetes. This is not surprising, as diabetes leads to higher mortality from a range of infectious diseases (158).
Projections suggest that the global incidence of diabetes will double in the next 20 years, with 40% of this estimated to result from the aging population (159, 160). The elderly are at high risk for developing type 2 diabetes due to underlying insulin resistance, impaired pancreatic function, and a higher obesity prevalence linked to changes in body composition and physical inactivity (161, 162). The resulting high blood sugar can cause serious complications such as heart disease, kidney problems, and loss of vision. Furthermore, diabetes in older adults is associated with a higher risk for chronic microvascular and cardiovascular complications and common geriatric syndromes and is linked to higher mortality (163). People with diabetes have altered cytokine release by macrophages and T cells, impaired neutrophil recruitment, and decreased levels of type I interferons as well as reduced numbers of new populations of dendritic cells (DCs) and natural killer (NK) cells (164). Also, the diabetic lung is characterized by structural modifications such as abnormalities in small vessels (alveolar diabetic microangiopathy or microvascular disease) (165), as well as alterations in the interstitial environment (166, 167) and autonomic neuropathy with loss of autonomic innervation in bronchioles (168), which might contribute to adverse outcomes in respiratory diseases (169).
While the interactions between TB and diabetes in the elderly are not completely understood, people with diabetes are 3 times more likely to develop pulmonary TB, especially those with poorly controlled diabetes (170, 171). In vitro and in vivo studies have found reduced association and uptake of M.tb by monocytes from people with diabetes and alveolar macrophages from mice with chronic diabetes, as well as reduced innate immune responses and a persistent systemic hyper-inflammation in TB-diabetic individuals (172–174). Diabetes also promotes TB reactivation due to impaired T cell immunity, specifically because of decreased IFN-γ production by CD4 T cells (175). In addition, cavitary disease (where cavities are abnormal, thick-walled, air-filled spaces in the lung which result when a granuloma encasing M.tb liquifies and ruptures) is more frequently observed in elderly TB patients with diabetes than in non-diabetic elderly patients, suggesting that diabetes promotes cavitation in the aging lung parenchyma (176).
In addition, TB might pose a risk of developing diabetes (177). A persistent inflammatory state in response to TB disease might result in secondary metabolic effects such as “stress hyperglycemia”, defined as temporary hyperglycemia caused by stress during acute illness (178). It has been suggested that stress hyperglycemia may negatively influence TB treatment outcomes, although this relationship is still poorly understood (178).
Individuals with diabetes are at a higher risk for SARS-CoV-2 severe disease and mortality (34, 179–185). According to an analysis done in the South African population, the hazard ratios for mortality range from 3 to 12 for ≥20 years old public-sector patients, with the mortality risk increasing as blood sugar control decreases. Risk may be lower in other populations, perhaps due to better diabetes control: about 3-fold higher for mortality as reported in a meta-analysis (181). While the worse disease outcome of SARS-CoV-2 infection in diabetics is well established, diabetics are not necessarily at higher risk of infection with SARS-CoV-2 (179), indicating that not all aspects of immunity are equally compromised.
Respiratory infections tend to interact in one of two ways. They can synergize, with the cardinal example being Streptococcus pneumoniae bacterial infection after influenza virus infection. This happens because the virus causes damage to the mucosal surface, allowing the bacteria to attach better and invade more easily (186). In addition, the type I interferon response to the virus decreases phagocyte function and therefore control of the bacteria by phagocytosis (186). The other possible interaction is antagonism, and usually happens between viruses. This is called super-infection exclusion and occurs because the type I interferon antiviral response trigged by one virus can inhibit other viruses (187). Two studies by independent groups examined experimental SARS-CoV-2/M.tb co-infection in K18-hACE2 transgenic mice. Both groups found that SARS-CoV-2 infection did not affect M.tb loads or associated pathology. They also observed that M.tb infected mice were more resistant to SARS-CoV-2 infection (188, 189). This is consistent with a report that intravenous administration of BCG, a live attenuated TB vaccine developed from Mycobacterium bovis, protects mice against lethal SARS-CoV-2 challenge (190). Thus, there is currently no mechanistic basis for synergy between SARS-CoV-2 and M.tb. There is still a poor understanding of the pathology and immunological changes associated with M.tb/SARS-CoV-2 co-infection (191), as recently reviewed in (192).
In terms of epidemiology, some studies suggest that the dysregulated immunity during M.tb infection is associated with increased susceptibility and severity of COVID-19 and vice versa (193–198). There is also some evidence suggesting that in the elderly population, TB and COVID-19 may be associated with increased mortality compared to each disease occurring alone (199–201). Mechanisms may include increased lung damage in TB patients with COVID-19, resulting in impaired lung function (202) or higher risk of TB reactivation after COVID-19 infection due to depletion of CD4 T cells and excessive lung fibrosis. Worse outcomes of co-infection may also be because of shared clinical, immunological, and social determinants (203–206), as well as compromised linkage to care for HIV and TB in a pandemic environment (207). In our own South Africa based cohort of SARS-CoV-2 infected individuals, we did not observe a clear enrichment of active TB disease (208) relative to the observed incidence in the South African population (209).
Aging has a negative effect on the outcomes of both SARS-CoV-2 and M.tb infection, and may be considered a subtype of immunosuppression/dysregulation which varies widely between individuals of a similar age. This may be because the effect is multi-factorial and involves age-related inflammation (inflammaging) and senescence of immune cell subsets, as reviewed previously (210). It is particularly damaging to the adaptive arm of the immune response which is critical to control both infections. In addition to that, the lung environment itself also changes with age, and many of the changes are associated with the reduced ability of alveolar fluid to perform its innate immune functions. Aging also increases autoimmunity, and in a subset of individuals this may manifest as autoantibodies to immune mediators such as interferons, with the result that innate immunity becomes less effective at reducing pathogen replication. This is an example of how the effects of aging can be heterogeneous. Lastly, co-morbidities such as type II diabetes increase with age, and such co-morbidities, though they do not necessarily increase the chances of infection, are risk factors for more severe disease if infection does occur.
Conversely, SARS-CoV-2 and M.tb infections may accelerate age-related processes. For example, M.tb infection and TB treatment, as well as long-COVID, might result in cardiovascular complications and induce cardiovascular disease (211), an important comorbidity associated with the older population. Also, SARS-CoV-2 is associated with increased oxidative stress, which also plays a role in the pathogenesis of diabetes (212).
Some of the processes described here are already targets for interventions. For example, the elderly are prioritized for COVID-19 vaccination to compensate for the less effective immune response to SARS-CoV-2 (213). Other interventions, for example better control of diabetes, are available but are not uniformly implemented due to health systems challenges, particularly in low- and middle-income countries (214). Interventions which may increase lung health at a given stage of life are yet little explored, but have the potential to work across pathogens to decrease the effects of infection, which could translate to substantial gains in the health span of aging populations.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was partially supported by the Bill and Melinda Gates award INV-018944 and Wellcome Trust Award 226137/Z/22/Z to AS. This study was also partially supported by the Robert J. Kleberg, Jr and Helen C. Kleberg Foundation to JBT. AAG and JBT are part of the Interdisciplinary NextGen Tuberculosis Research Advancement Center (IN-TRAC) at Texas Biomed, which is supported by the NIAID/NIH under the award number P30AI168439.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The content in this publication is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
2. Schneider JL, Rowe JH, Garcia-de-Alba C, Kim CF, Sharpe AH, Haigis MC. The aging lung: Physiology, disease, and immunity. Cell (2021) 184(8):1990–2019. doi: 10.1016/j.cell.2021.03.005
3. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell (2023) 186(2):243–78. doi: 10.1016/j.cell.2022.11.001
4. Chalise HN. Aging: basic concept. Am J BioMed Sci Res (2019) 1:8–10. doi: 10.34297/AJBSR.2019.01.000503
5. Cho SJ, Stout-Delgado HW. Aging and lung disease. Annu Rev Physiol (2020) 82:433–59. doi: 10.1146/annurev-physiol-021119-034610
6. Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun (2019) 10(1):963. doi: 10.1038/s41467-019-08831-9
7. Kim J, Heise RL, Reynolds AM, Pidaparti RM. Aging effects on airflow dynamics and lung function in human bronchioles. PloS One (2017) 12(8):e0183654. doi: 10.1371/journal.pone.0183654
8. Khoury DS, Cromer D, Reynaldi A, Schlub TE, Wheatley AK, Juno JA, et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat Med (2021) 27:1205–11. doi: 10.1038/s41591-021-01377-8
9. Pulliam JR, van Schalkwyk C, Govender N, von Gottberg A, Cohen C, Groome MJ, et al. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science (2022) 376(6593):eabn4947. doi: 10.1126/science.abn4947
10. Cele S, Jackson L, Khoury DS, Khan K, Moyo-Gwete T, Tegally H, et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature (2021) 602(7898):654–6. doi: 10.1038/s41586-021-04387-1
11. Menzies NA, Swartwood N, Testa C, Malyuta Y, Hill AN, Marks SM, et al. Time since infection and risks of future disease for individuals with mycobacterium tuberculosis infection in the United States. Epidemiology (2021) 32(1):70–8. doi: 10.1097/EDE.0000000000001271
12. Kiazyk S, Ball TB. Latent tuberculosis infection: An overview. Can Commun Dis Rep (2017) 43(3-4):62–6. doi: 10.14745/ccdr.v43i34a01
13. Bunyasi EW, Mulenga H, Luabeya AKK, Shenje J, Mendelsohn SC, Nemes E, et al. Regional changes in tuberculosis disease burden among adolescents in South Africa (2005-2015). PloS One (2020) 15(7):e0235206. doi: 10.1371/journal.pone.0235206
14. Iqbal SA, Winston CA, Bardenheier BH, Armstrong LR, Navin TR. Age-period-cohort analyses of tuberculosis incidence rates by nativity, United States, 1996-2016. Am J Public Health (2018) 108:S315–S20. doi: 10.2105/AJPH.2018.304687
15. Wang JG, Zhong ZJ, Mo YF, Wang LC, Chen R. Epidemiological features of coronavirus disease 2019 in children: a meta-analysis. Eur Rev Med Pharmacol Sci (2021) 25(2):1146–57. doi: 10.26355/eurrev_202101_24685
16. Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science (2014) 343(6169):428–32. doi: 10.1126/science.1243640
17. Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell (2010) 143(5):789–801. doi: 10.1016/j.cell.2010.11.001
18. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505(7484):509–14. doi: 10.1038/nature12940
19. Galloway NL, Doitsh G, Monroe KM, Yang Z, Muñoz-Arias I, Levy DN, et al. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T cells. Cell Rep (2015) 12(10):1555–63. doi: 10.1016/j.celrep.2015.08.011
20. Banda NK, Bernier J, Kurahara DK, Kurrle R, Haigwood N, Sekaly RP, et al. Crosslinking CD4 by human immunodeficiency virus gp120 primes T cells for activation-induced apoptosis. J Exp Med (1992) 176(4):1099–106. doi: 10.1084/jem.176.4.1099
21. Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, et al. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature (1995) 375(6531):497–500. doi: 10.1038/375497a0
22. Westendorp MO, Shatrov VA, Schulze-Osthoff K, Frank R, Kraft M, Los M, et al. HIV-1 Tat potentiates TNF-induced NF-kappa B activation and cytotoxicity by altering the cellular redox state. EMBO J (1995) 14(3):546–54. doi: 10.1002/j.1460-2075.1995.tb07030.x
23. Zeng M, Haase AT, Schacker TW. Lymphoid tissue structure and HIV-1 infection: life or death for T cells. Trends Immunol (2012) 33(6):306–14. doi: 10.1016/j.it.2012.04.002
24. Cooper A, García M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature (2013) 498(7454):376–9. doi: 10.1038/nature12274
25. Sepkowitz KA, Raffalli J, Riley L, Kiehn TE, Armstrong D. Tuberculosis in the AIDS era. Clin Microbiol Rev (1995) 8(2):180–99. doi: 10.1128/CMR.8.2.180
26. Bell LC, Noursadeghi M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat Rev Microbiol (2018) 16(2):80–90. doi: 10.1038/nrmicro.2017.128
27. Cele S, Karim F, Lustig G, San JE, Hermanus T, Tegally H, et al. SARS-CoV-2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host Microbe (2022) 30(2):154–162.e5. doi: 10.1016/j.chom.2022.01.005
28. Lustig G, Ganga Y, Rodel H, Tegally H, Jackson L, Cele S, et al. SARS-CoV-2 evolves increased infection elicited cell death and fusion in an immunosuppressed individual. medRxiv (2022), 22282673. doi: 10.1101/2022.11.23.22282673
29. Riddell AC, Kele B, Harris K, Bible J, Murphy M, Dakshina S, et al. Generation of novel SARS-CoV-2 variants on B.1.1.7 lineage in three patients with advanced HIV disease. Clin Infect Dis (2022) 75(11):2016–8. doi: 10.1093/cid/ciac409
30. Wilkinson SAJ, Richter A, Casey A, Osman H, Mirza JD, Stockton J, et al. Recurrent SARS-CoV-2 mutations in immunodeficient patients. Virus Evol (2022) 8(2):veac050. doi: 10.1093/ve/veac050
31. Maponga TG, Jeffries M, Tegally H, Sutherland A, Wilkinson E, Lessells RJ, et al. Persistent SARS-CoV-2 infection with accumulation of mutations in a patient with poorly controlled HIV infection. Clin Infect Dis (2022). doi: 10.2139/ssrn.4014499
32. Hoffman SA, Costales C, Sahoo MK, Palanisamy S, Yamamoto F, Huang C, et al. SARS-coV-2 neutralization resistance mutations in patient with HIV/AIDS, california, USA. Emerging Infect Diseases (2021) 27(10):2720–3. doi: 10.3201/eid2710.211461
33. Karim F, Moosa MY, Gosnell B, Sandile C, Giandhari J, Pillay S, et al. Persistent SARS-CoV-2 infection and intra-host evolution in association with advanced HIV infection. medRxiv (2021). doi: 10.1101/2021.06.03.21258228
34. Boulle A, Davies M-A, Hussey H, Ismail M, Morden E, Vundle Z. Western cape department of health in collaboration with the national institute for communicable diseases SA. Risk factors for coronavirus disease 2019 (COVID-19) death in a population cohort study from the western cape province, South Africa. Clin Infect Dis (2020) 73(7):e2005–e15. doi: 10.1093/cid/ciaa1198
35. Savic M, Penders Y, Shi T, Branche A, Pircon JY. Respiratory syncytial virus disease burden in adults aged 60 years and older in high-income countries: A systematic literature review and meta-analysis. Influenza Other Respir Viruses (2023) 17(1):e13031. doi: 10.1111/irv.13031
36. Branche AR, Falsey AR. Respiratory syncytial virus infection in older adults: an under-recognized problem. Drugs Aging (2015) 32(4):261–9. doi: 10.1007/s40266-015-0258-9
37. Paget J, Spreeuwenberg P, Charu V, Taylor RJ, Iuliano AD, Bresee J, et al. Global mortality associated with seasonal influenza epidemics: New burden estimates and predictors from the GLaMOR Project. J Glob Health (2019) 9(2):020421. doi: 10.7189/jogh.09.020421
38. Langer J, Welch VL, Moran MM, Cane A, Lopez SMC, Srivastava A, et al. High clinical burden of influenza disease in adults aged >/= 65 years: can we do better? A systematic literature review. Adv Ther (2023) 40(4):1601–27. doi: 10.1007/s12325-023-02432-1
39. Hung IF, Zhang AJ, To KK, Chan JF, Zhu SH, Zhang R, et al. Unexpectedly higher morbidity and mortality of hospitalized elderly patients associated with rhinovirus compared with influenza virus respiratory tract infection. Int J Mol Sci (2017) 18(2):259. doi: 10.3390/ijms18020259
40. WHO. WHO Coronavirus (COVID-19) Dashboard 2023. Available at: https://covid19.who.int/.
41. O’Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature (2021) 590(7844):140–5. doi: 10.1038/s41586-020-2918-0
42. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/S0140-6736(20)30566-3
43. Yanez ND, Weiss NS, Romand JA, Treggiari MM. COVID-19 mortality risk for older men and women. BMC Public Health (2020) 20(1):1742. doi: 10.1186/s12889-020-09826-8
44. Dolin PJ, Raviglione MC, Kochi A. Global tuberculosis incidence and mortality during 1990-2000. Bull World Health Organization (1994) 72(2):213.
45. Wong EB. It is time to focus on asymptomatic tuberculosis. Clin Infect Diseases (2021) 72(12):E1044–E6. doi: 10.1093/cid/ciaa1827
46. Shah M, Dorman SE. Latent tuberculosis infection. New Engl J Med (2021) 385(24):2271–80. doi: 10.1056/NEJMcp2108501
47. Li SJ, Li YF, Song WM, Zhang QY, Liu SQ, Xu TT, et al. Population aging and trends of pulmonary tuberculosis incidence in the elderly. BMC Infect Dis (2021) 21(1):302. doi: 10.1186/s12879-021-05994-z
48. Piergallini TJ, Turner J. Tuberculosis in the elderly: Why inflammation matters. Exp Gerontol (2018) 105:32–9. doi: 10.1016/j.exger.2017.12.021
49. Caraux-Paz P, Diamantis S, de Wazières B, Gallien S. Tuberculosis in the elderly. J Clin Med (2021) 10(24):5888. doi: 10.3390/jcm10245888
50. Kim S, Cohen T, Horsburgh CR, Miller JW, Hill AN, Marks SM, et al. Trends, mechanisms, and racial/ethnic differences of tuberculosis incidence in the US-born population aged 50 years or older in the United States. Clin Infect Dis (2022) 74(9):1594–603. doi: 10.1093/cid/ciab668
51. Teo AKJ, Rahevar K, Morishita F, Ang A, Yoshiyama T, Ohkado A, et al. Tuberculosis in older adults: case studies from four countries with rapidly ageing populations in the western pacific region. BMC Public Health (2023) 23(1):370. doi: 10.1186/s12889-023-15576-0
52. Janssens JP, Pache JC, Nicod LP. Physiological changes in respiratory function associated with ageing. Eur Respir J (1999) 13(1):197–205. doi: 10.1183/09031936.99.14614549
53. Lowery EM, Brubaker AL, Kuhlmann E, Kovacs EJ. The aging lung. Clin Interv Aging (2013) 8:1489–96. doi: 10.2147/CIA.S51152
54. Sharma G, Goodwin J. Effect of aging on respiratory system physiology and immunology. Clin Interv Aging (2006) 1(3):253–60. doi: 10.2147/ciia.2006.1.3.253
55. Miller RA. The aging immune system: primer and prospectus. Science (1996) 273(5271):70–4. doi: 10.1126/science.273.5271.70
56. Pawelec G, Solana R. Immunosenescence. Immunol Today (1997) 18(11):514–6. doi: 10.1016/S0167-5699(97)01145-6
57. Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol (2004) 5(2):133–9. doi: 10.1038/ni1033
58. Pawelec G, Derhovanessian E, Larbi A. Immunosenescence and cancer. Crit Rev Oncol Hematol (2010) 75(2):165–72. doi: 10.1016/j.critrevonc.2010.06.012
59. Lee KA, Flores RR, Jang IH, Saathoff A, Robbins PD. Immune senescence, immunosenescence and aging. Front Aging (2022) 3:900028. doi: 10.3389/fragi.2022.900028
60. Rink L, Cakman I, Kirchner H. Altered cytokine production in the elderly. Mech Ageing Dev (1998) 102(2-3):199–209. doi: 10.1016/S0047-6374(97)00153-X
61. Bruunsgaard H, Andersen-Ranberg K, Hjelmborg J, Pedersen BK, Jeune B. Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am J Med (2003) 115(4):278–83. doi: 10.1016/S0002-9343(03)00329-2
62. van den Biggelaar AH, Huizinga TW, de Craen AJ, Gussekloo J, Heijmans BT, Frolich M, et al. Impaired innate immunity predicts frailty in old age. Leiden 85-plus study Exp Gerontol (2004) 39(9):1407–14. doi: 10.1016/j.exger.2004.06.009
63. van Duin D, Mohanty S, Thomas V, Ginter S, Montgomery RR, Fikrig E, et al. Age-associated defect in human TLR-1/2 function. J Immunol (2007) 178(2):970–5. doi: 10.4049/jimmunol.178.2.970
64. Nyugen J, Agrawal S, Gollapudi S, Gupta S. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J Clin Immunol (2010) 30(6):806–13. doi: 10.1007/s10875-010-9448-8
65. Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol (2002) 169(9):4697–701. doi: 10.4049/jimmunol.169.9.4697
66. Boehmer ED, Goral J, Faunce DE, Kovacs EJ. Age-dependent decrease in Toll-like receptor 4-mediated proinflammatory cytokine production and mitogen-activated protein kinase expression. J Leukoc Biol (2004) 75(2):342–9. doi: 10.1189/jlb.0803389
67. Boehmer ED, Meehan MJ, Cutro BT, Kovacs EJ. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech Ageing Dev (2005) 126(12):1305–13. doi: 10.1016/j.mad.2005.07.009
68. Chelvarajan RL, Collins SM, Van Willigen JM, Bondada S. The unresponsiveness of aged mice to polysaccharide antigens is a result of a defect in macrophage function. J Leukoc Biol (2005) 77(4):503–12. doi: 10.1189/jlb.0804449
69. Chelvarajan RL, Liu Y, Popa D, Getchell ML, Getchell TV, Stromberg AJ, et al. Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leukoc Biol (2006) 79(6):1314–27. doi: 10.1189/jlb.0106024
70. Armstrong JA, Hart PD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med (1971) 134(3 Pt 1):713–40. doi: 10.1084/jem.134.3.713
71. Dunston CR, Griffiths HR. The effect of ageing on macrophage Toll-like receptor-mediated responses in the fight against pathogens. Clin Exp Immunol (2010) 161(3):407–16. doi: 10.1111/j.1365-2249.2010.04213.x
72. Shaw AC, Panda A, Joshi SR, Qian F, Allore HG, Montgomery RR. Dysregulation of human Toll-like receptor function in aging. Ageing Res Rev (2011) 10(3):346–53. doi: 10.1016/j.arr.2010.10.007
73. Hajishengallis G. Too old to fight? Aging and its toll on innate immunity. Mol Oral Microbiol (2010) 25(1):25–37. doi: 10.1111/j.2041-1014.2009.00562.x
74. Bruunsgaard H, Ladelund S, Pedersen AN, Schroll M, Jorgensen T, Pedersen BK. Predicting death from tumour necrosis factor-alpha and interleukin-6 in 80-year-old people. Clin Exp Immunol (2003) 132(1):24–31. doi: 10.1046/j.1365-2249.2003.02137.x
75. Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol (2004) 39(5):687–99. doi: 10.1016/j.exger.2004.01.009
76. Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev (2011) 10(3):319–29. doi: 10.1016/j.arr.2010.11.002
77. Korhonen P, Helenius M, Salminen A. Age-related changes in the regulation of transcription factor NF-kappa B in rat brain. Neurosci Lett (1997) 225(1):61–4. doi: 10.1016/S0304-3940(97)00190-0
78. Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J (2003) 17(9):1183–5. doi: 10.1096/fj.02-1049fje
79. de Magalhaes JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics (2009) 25(7):875–81. doi: 10.1093/bioinformatics/btp073
80. Swindell WR. Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse. BMC Genomics (2009) 10:585. doi: 10.1186/1471-2164-10-585
81. Lumeng CN, Liu J, Geletka L, Delaney C, Delproposto J, Desai A, et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J Immunol (2011) 187(12):6208–16. doi: 10.4049/jimmunol.1102188
82. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging - An evolutionary perspective on immunosenescence. Mol Cell Gerontology (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
83. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol (2018) 15(9):505–22. doi: 10.1038/s41569-018-0064-2
84. Meyer KC, Ershler W, Rosenthal NS, Lu XG, Peterson K. Immune dysregulation in the aging human lung. Am J Respir Crit Care Med (1996) 153(3):1072–9. doi: 10.1164/ajrccm.153.3.8630547
85. Meyer KC, Rosenthal NS, Soergel P, Peterson K. Neutrophils and low-grade inflammation in the seemingly normal aging human lung. Mech Ageing Dev (1998) 104(2):169–81. doi: 10.1016/S0047-6374(98)00065-7
86. Alberro A, Iribarren-Lopez A, Saenz-Cuesta M, Matheu A, Vergara I, Otaegui D. Inflammaging markers characteristic of advanced age show similar levels with frailty and dependency. Sci Rep (2021) 11(1):4358. doi: 10.1038/s41598-021-83991-7
87. Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev (2007) 128(1):92–105. doi: 10.1016/j.mad.2006.11.016
88. Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci (2012) 69(18):2999–3013. doi: 10.1007/s00018-012-0962-0
89. Garcia-Garcia VA, Alameda JP, Page A, Casanova ML. Role of NF-kappaB in ageing and age-related diseases: lessons from genetically modified mouse models. Cells (2021) 10(8):1906. doi: 10.3390/cells10081906
90. Lafuse WP, Rajaram MVS, Wu Q, Moliva JI, Torrelles JB, Turner J, et al. Identification of an increased alveolar macrophage subpopulation in old mice that displays unique inflammatory characteristics and is permissive to mycobacterium tuberculosis infection. J Immunol (2019) 203(8):2252–64. doi: 10.4049/jimmunol.1900495
91. Canan CH, Gokhale NS, Carruthers B, Lafuse WP, Schlesinger LS, Torrelles JB, et al. Characterization of lung inflammation and its impact on macrophage function in aging. J Leukoc Biol (2014) 96(3):473–80. doi: 10.1189/jlb.4A0214-093RR
92. Duong L, Radley HG, Lee B, Dye DE, Pixley FJ, Grounds MD, et al. Macrophage function in the elderly and impact on injury repair and cancer. Immun Ageing (2021) 18(1):4. doi: 10.1186/s12979-021-00215-2
93. Kohut ML, SenChina DS, Madden KS, Martin AE, Felten DL, Moynihan JA. Age effects on macrophage function vary by tissue site, nature of stimulant, and exercise behavior. Exp Gerontol (2004) 39(9):1347–60. doi: 10.1016/j.exger.2004.07.001
94. Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity (2008) 29(2):283–94. doi: 10.1016/j.immuni.2008.06.011
95. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell (2013) 153(3):521–34. doi: 10.1016/j.cell.2013.03.022
96. Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit. Cell (2019) 178(6):1344–61.e11. doi: 10.1016/j.cell.2019.08.004
97. Mahamed D, Boulle M, Ganga Y, Mc Arthur C, Skroch S, Oom L, et al. Intracellular growth of Mycobacterium tuberculosis after macrophage cell death leads to serial killing of host cells. Elife (2017) 6:e22028. doi: 10.7554/eLife.28205
98. Muller L, Di Benedetto S. How immunosenescence and inflammaging may contribute to hyperinflammatory syndrome in COVID-19. Int J Mol Sci (2021) 22(22):12539. doi: 10.3390/ijms222212539
99. Tizazu AM, Mengist HM, Demeke G. Aging, inflammaging and immunosenescence as risk factors of severe COVID-19. Immun Ageing (2022) 19(1):53. doi: 10.1186/s12979-022-00309-5
100. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-coV-2 drives development of COVID-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
101. Brabek J, Jakubek M, Vellieux F, Novotny J, Kolar M, Lacina L, et al. Interleukin-6: molecule in the intersection of cancer, ageing and COVID-19. Int J Mol Sci (2020) 21(21):7937. doi: 10.3390/ijms21217937
102. Mojtabavi H, Saghazadeh A, Rezaei N. Interleukin-6 and severe COVID-19: a systematic review and meta-analysis. Eur Cytokine Netw (2020) 31(2):44–9. doi: 10.1684/ecn.2020.0448
103. Asensi V, Valle E, Meana A, Fierer J, Celada A, Alvarez V, et al. In vivo interleukin-6 protects neutrophils from apoptosis in osteomyelitis. Infect Immun (2004) 72(7):3823–8. doi: 10.1128/IAI.72.7.3823-3828.2004
104. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol (2020) 5(49):eabd7114. doi: 10.1126/sciimmunol.abd7114
105. Pietrobon AJ, Teixeira FME, Sato MN. I mmunosenescence and inflammaging: risk factors of severe COVID-19 in older people. Front Immunol (2020) 11:579220. doi: 10.3389/fimmu.2020.579220
106. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-coV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell (2020) 181(7):1489–501.e15. doi: 10.1016/j.cell.2020.05.015
107. Bektas A, Schurman SH, Franceschi C, Ferrucci L. A public health perspective of aging: do hyper-inflammatory syndromes such as COVID-19, SARS, ARDS, cytokine storm syndrome, and post-ICU syndrome accelerate short- and long-term inflammaging? Immun Ageing (2020) 17(1):23. doi: 10.1186/s12979-020-00196-8
108. Bartleson JM, Radenkovic D, Covarrubias AJ, Furman D, Winer DA, Verdin E. SARS-CoV-2, COVID-19 and the aging immune system. Nat Aging (2021) 1(9):769–82. doi: 10.1038/s43587-021-00114-7
109. Moliva JI, Rajaram MV, Sidiki S, Sasindran SJ, Guirado E, Pan XJ, et al. Molecular composition of the alveolar lining fluid in the aging lung. Age (Dordr) (2014) 36(3):9633. doi: 10.1007/s11357-014-9633-4
110. Torrelles JB, Schlesinger LS. Integrating lung physiology, immunology, and tuberculosis. Trends Microbiol (2017) 25(8):688–97. doi: 10.1016/j.tim.2017.03.007
111. Notter RH. Lung surfactants: Basic science and clinical applications. NY, US: Marcel Dekker (2000) p. 1–444.
112. Canadas O, Olmeda B, Alonso A, Perez-Gil J. Lipid-protein and protein-protein interactions in the pulmonary surfactant system and their role in lung homeostasis. Int J Mol Sci (2020) 21(10):3708. doi: 10.3390/ijms21103708
113. Gaynor CD, McCormack FX, Voelker DR, McGowan SE, Schlesinger LS. Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol (1995) 155(11):5343–51. doi: 10.4049/jimmunol.155.11.5343
114. Ferguson JS, Voelker DR, McCormack FX, Schlesinger LS. Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J Immunol (1999) 163(1):312–21. doi: 10.4049/jimmunol.163.1.312
115. Ferguson JS, Martin JL, Azad AK, McCarthy TR, Kang PB, Voelker DR, et al. Surfactant protein D increases fusion of Mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect Immun (2006) 74(12):7005–9. doi: 10.1128/IAI.01402-06
116. Schlesinger LS, Bellinger-Kawahara CG, Payne NR, Horwitz MA. Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J Immunol (1990) 144(7):2771–80. doi: 10.4049/jimmunol.144.7.2771
117. Ferguson JS, Weis JJ, Martin JL, Schlesinger LS. Complement protein C3 binding to Mycobacterium tuberculosis is initiated by the classical pathway in human bronchoalveolar lavage fluid. Infect Immun (2004) 72(5):2564–73. doi: 10.1128/IAI.72.5.2564-2573.2004
118. Thacker VV, Dhar N, Sharma K, Barrile R, Karalis K. McKinney JD. A lung-on-chip model of early Mycobacterium tuberculosis infection reveals an essential role for alveolar epithelial cells in controlling bacterial growth. Elife (2020) 9:e59961. doi: 10.7554/eLife.59961
119. Hsieh MH, Ou CY, Hsieh WY, Kao HF, Lee SW, Wang JY, et al. Functional analysis of genetic variations in surfactant protein D in mycobacterial infection and their association with tuberculosis. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01543
120. Garcia-Vilanova A, Olmo-Fontanez AM, Moliva JI, Allue-Guardia A, Singh H, Merritt RE, et al. The aging human lung mucosa: A proteomics study. J Gerontol A Biol Sci Med Sci (2022) 77(10):1969–74. doi: 10.1093/gerona/glac091
121. Moliva JI, Duncan MA, Olmo-Fontanez A, Akhter A, Arnett E, Scordo JM, et al. The lung mucosa environment in the elderly increases host susceptibility to mycobacterium tuberculosis infection. J Infect Dis (2019) 220(3):514–23. doi: 10.1093/infdis/jiz138
122. Scordo JM, Olmo-Fontanez AM, Kelley HV, Sidiki S, Arcos J, Akhter A, et al. The human lung mucosa drives differential Mycobacterium tuberculosis infection outcome in the alveolar epithelium. Mucosal Immunol (2019) 12(3):795–804. doi: 10.1038/s41385-019-0156-2
123. Olmo-Fontánez AM, Scordo JM, Garcia-Vilanova A, Maselli DJ, Peters JI, Restrepo BI, et al. Human alveolar lining fluid from the elderly promotes Mycobacterium tuberculosis growth in alveolar epithelial cells and bacterial translocation into the cytosol. bioRxiv (2021), 443884. doi: 10.1101/2021.05.12.443884
124. Allue-Guardia A, Garcia-Vilanova A, Olmo-Fontanez AM, Peters J, Maselli DJ, Wang Y, et al. Host- and Age-Dependent Transcriptional Changes in Mycobacterium tuberculosis Cell Envelope Biosynthesis Genes after Exposure to Human Alveolar Lining Fluid. Int J Mol Sci (2022) 23(2):983. doi: 10.3390/ijms23020983
125. Sinnberg T, Lichtensteiger C, Ali OH, Pop OT, Jochum AK, Risch L, et al. Pulmonary surfactant proteins are inhibited by immunoglobulin A autoantibodies in severe COVID-19. Am J Respir Crit Care Med (2023) 207(1):38–49. doi: 10.1164/rccm.202201-0011OC
126. Salvioni L, Testa F, Sulejmani A, Pepe F, Lovaglio PG, Berta P, et al. (SP-D) as a biomarker of SARS-CoV-2 infection. Clin Chim Acta (2022) 537:140–5. doi: 10.1016/j.cca.2022.10.013
127. Kervevan J, Chakrabarti LA. Role of CD4+ T cells in the control of viral infections: recent advances and open questions. Int J Mol Sci (2021) 22(2):523. doi: 10.3390/ijms22020523
128. Morgan J, Muskat K, Tippalagama R, Sette A, Burel J, Arlehamn CSL. Classical CD4 T cells as the cornerstone of antimycobacterial immunity. Immunol Rev (2021) 301(1):10–29. doi: 10.1111/imr.12963
129. Earle KA, Ambrosino DM, Fiore-Gartland A, Goldblatt D, Gilbert PB, Siber GR, et al. Evidence for antibody as a protective correlate for COVID-19 vaccines. Vaccine (2021) 39(32):4423–8. doi: 10.1016/j.vaccine.2021.05.063
130. Garcia-Beltran WF, Lam EC, Astudillo MG, Yang D, Miller TE, Feldman J, et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell (2021) 184(2):476–88.e11. doi: 10.1016/j.cell.2020.12.015
131. Collier DA, Ferreira I, Kotagiri P, Datir RP, Lim EY, Touizer E, et al. Age-related immune response heterogeneity to SARS-CoV-2 vaccine BNT162b2. Nature (2021) 596(7872):417–22. doi: 10.1038/s41586-021-03739-1
132. Jergović M, Uhrlaub JL, Watanabe M, Bradshaw CM, White LM, LaFleur BJ, et al. Competent immune responses to SARS-CoV-2 variants in older adults following two doses of mRNA vaccination. Nat Commun (2022) 13(1):2891. doi: 10.1038/s41467-022-30617-9
133. Renia L, Goh YS, Rouers A, Le Bert N, Chia WN, Chavatte JM, et al. Lower vaccine-acquired immunity in the elderly population following two-dose BNT162b2 vaccination is alleviated by a third vaccine dose. Nat Commun (2022) 13(1):4615. doi: 10.1038/s41467-022-32312-1
134. Romero-Olmedo AJ, Schulz AR, Hochstätter S, Das Gupta D, Virta I, Hirseland H, et al. Induction of robust cellular and humoral immunity against SARS-CoV-2 after a third dose of BNT162b2 vaccine in previously unresponsive older adults. Nat Microbiol (2022) 7(2):195–9. doi: 10.1038/s41564-021-01046-z
135. Huang L, Nazarova EV, Russell DG. Mycobacterium tuberculosis: bacterial fitness within the host macrophage. Microbiol spectrum (2019) 7(2):7. doi: 10.1128/microbiolspec.BAI-0001-2019
136. Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med (1993) 178(6):2249–54. doi: 10.1084/jem.178.6.2249
137. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med (1993) 178(6):2243–7. doi: 10.1084/jem.178.6.2243
138. Moreira-Teixeira L, Mayer-Barber K, Sher A, O’Garra A. Type I interferons in tuberculosis: Foe and occasionally friend. J Exp Med (2018) 215(5):1273–85. doi: 10.1084/jem.20180325
139. Mundra A, Yegiazaryan A, Karsian H, Alsaigh D, Bonavida V, Frame M, et al. Pathogenicity of type I interferons in mycobacterium tuberculosis. Int J Mol Sci (2023) 24(4):3919. doi: 10.3390/ijms24043919
140. Aiello A, Grossi A, Meschi S, Meledandri M, Vanini V, Petrone L, et al. Coordinated innate and T-cell immune responses in mild COVID-19 patients from household contacts of COVID-19 cases during the first pandemic wave. Front Immunol (2022) 13:920227. doi: 10.3389/fimmu.2022.920227
141. Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res (2020) 179:104811. doi: 10.1016/j.antiviral.2020.104811
142. Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerging Microbes infections (2020) 9(1):1418–28. doi: 10.1080/22221751.2020.1780953
143. Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun (2020) 11(1):3810. doi: 10.1038/s41467-020-17665-9
144. Xia H, Cao Z, Xie X, Zhang X, Chen JY-C, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep (2020) 33(1):108234. doi: 10.1016/j.celrep.2020.108234
145. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
146. Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J Exp Med (2020) 217(12):e20201241. doi: 10.1084/jem.20201241
147. Chen LF, Yang CD, Cheng XB. Anti-interferon autoantibodies in adult-onset immunodeficiency syndrome and severe COVID-19 infection. Front Immunol (2021) 12:788368. doi: 10.3389/fimmu.2021.788368
148. Döffinger R, Helbert MR, Barcenas-Morales G, Yang K, Dupuis S, Ceron-Gutierrez L, et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin Infect Dis (2004) 38(1):e10–4. doi: 10.1086/380453
149. Höflich C, Sabat R, Rosseau S, Temmesfeld B, Slevogt H, Döcke WD, et al. Naturally occurring anti-IFN-gamma autoantibody and severe infections with Mycobacterium cheloneae and Burkholderia cocovenenans. Blood (2004) 103(2):673–5. doi: 10.1182/blood-2003-04-1065
150. Kampmann B, Hemingway C, Stephens A, Davidson R, Goodsall A, Anderson S, et al. Acquired predisposition to mycobacterial disease due to autoantibodies to IFN-gamma. J Clin Invest (2005) 115(9):2480–8. doi: 10.1172/JCI19316
151. Madariaga L, Amurrio C, Martin G, Garcia-Cebrian F, Bicandi J, Lardelli P, et al. Detection of anti-interferon-gamma autoantibodies in subjects infected by Mycobacterium tuberculosis. Int J Tuberc Lung Dis (1998) 2(1):62–8.
152. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
153. Manry J, Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, et al. The risk of COVID-19 death is much greater and age dependent with type I IFN autoantibodies. Proc Natl Acad Sci U S A (2022) 119(21):e2200413119. doi: 10.1073/pnas.2200413119
154. Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci Immunol (2021) 6(62):eabl4340. doi: 10.1126/sciimmunol.abl4340
155. Zhao TV, Sato Y, Goronzy JJ, Weyand CM. T-cell aging-associated phenotypes in autoimmune disease. Front Aging (2022) 3:867950. doi: 10.3389/fragi.2022.867950
156. Wang M, Yin X, Zhang S, Mao C, Cao N, Yang X, et al. Autoantibodies against AT1 receptor contribute to vascular aging and endothelial cell senescence. Aging Dis (2019) 10(5):1012–25. doi: 10.14336/AD.2018.0919
157. Divo MJ, Martinez CH, Mannino DM. Ageing and the epidemiology of multimorbidity. Eur Respir J (2014) 44(4):1055–68. doi: 10.1183/09031936.00059814
158. Zoppini G, Fedeli U, Schievano E, Dauriz M, Targher G, Bonora E, et al. Mortality from infectious diseases in diabetes. Nutrition Metab Cardiovasc Diseases (2018) 28(5):444–50. doi: 10.1016/j.numecd.2017.12.007
159. Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr (2010) 8:29. doi: 10.1186/1478-7954-8-29
161. Batsis JA, Zagaria AB. Addressing obesity in aging patients. Med Clin North Am (2018) 102(1):65–85. doi: 10.1016/j.mcna.2017.08.007
162. Tuduri E, Soriano S, Almagro L, Montanya E, Alonso-Magdalena P, Nadal A, et al. The pancreatic beta-cell in ageing: Implications in age-related diabetes. Ageing Res Rev (2022) 80:101674. doi: 10.1016/j.arr.2022.101674
163. Bellary S, Kyrou I, Brown JE, Bailey CJ. Type 2 diabetes mellitus in older adults: clinical considerations and management. Nat Rev Endocrinol (2021) 17(9):534–48. doi: 10.1038/s41574-021-00512-2
164. Sen S, Chakraborty R, Kalita P, Pathak MP. Diabetes mellitus and COVID-19: Understanding the association in light of current evidence. World J Clin Cases (2021) 9(28):8327–39. doi: 10.12998/wjcc.v9.i28.8327
165. Zhang L, Jiang F, Xie Y, Mo Y, Zhang X, Liu C. Diabetic endothelial microangiopathy and pulmonary dysfunction. Front Endocrinol (Lausanne) (2023) 14:1073878. doi: 10.3389/fendo.2023.1073878
166. Foster DJ, Ravikumar P, Bellotto DJ, Unger RH, Hsia CC. Fatty diabetic lung: altered alveolar structure and surfactant protein expression. Am J Physiol Lung Cell Mol Physiol (2010) 298(3):L392–403. doi: 10.1152/ajplung.00041.2009
167. Marunaka Y. The Proposal of Molecular Mechanisms of Weak Organic Acids Intake-Induced Improvement of Insulin Resistance in Diabetes Mellitus via Elevation of Interstitial Fluid pH. Int J Mol Sci (2018) 19(10):3244. doi: 10.3390/ijms19103244
168. Sharma JK, Rohatgi A, Sharma D. Diabetic autonomic neuropathy: a clinical update. J R Coll Physicians Edinb (2020) 50(3):269–73. doi: 10.4997/jrcpe.2020.310
169. Whyte MB, Vas P, Heiss C, Feher MD. The contribution of diabetic micro-angiopathy to adverse outcomes in COVID-19. Diabetes Res Clin Pract (2020) 164:108217. doi: 10.1016/j.diabres.2020.108217
170. Restrepo BI. Diabetes and tuberculosis. Microbiol Spectr (2016) 4(6):10.1128/microbiolspec.TNMI7-0023-2016. doi: 10.1128/microbiolspec.TNMI7-0023-2016
171. Ngo MD, Bartlett S, Ronacher K. Diabetes-associated susceptibility to tuberculosis: contribution of hyperglycemia vs. Dyslipidemia. Microorganisms (2021) 9(11):2282. doi: 10.3390/microorganisms9112282
172. Vallerskog T, Martens GW, Kornfeld H. Diabetic mice display a delayed adaptive immune response to Mycobacterium tuberculosis. J Immunol (2010) 184(11):6275–82. doi: 10.4049/jimmunol.1000304
173. Kumar NP, Fukutani KF, Shruthi BS, Alves T, Silveira-Mattos PS, Rocha MS, et al. Persistent inflammation during anti-tuberculosis treatment with diabetes comorbidity. Elife (2019) 8:e46477. doi: 10.7554/eLife.46477
174. Kumar Nathella P, Babu S. Influence of diabetes mellitus on immunity to human tuberculosis. Immunology (2017) 152(1):13–24. doi: 10.1111/imm.12762
175. Jali MV, Kavital A, Hiremath MB. Challenges of diabetes in elderly TB patients. Indian J Tuberc (2022) 69 Suppl 2:S264–S6. doi: 10.1016/j.ijtb.2022.10.017
176. Perez-Guzman C, Vargas MH. Diabetes, aging, and tuberculosis. Lung India (2011) 28(3):191–2. doi: 10.4103/0970-2113.83976
177. Menon S, Rossi R, Dusabimana A, Zdraveska N, Bhattacharyya S, Francis J. The epidemiology of tuberculosis-associated hyperglycemia in individuals newly screened for type 2 diabetes mellitus: systematic review and meta-analysis. BMC Infect Dis (2020) 20(1):937. doi: 10.1186/s12879-020-05512-7
178. Magee MJ, Salindri AD, Kyaw NTT, Auld SC, Haw JS, Umpierrez GE. Stress hyperglycemia in patients with tuberculosis disease: epidemiology and clinical implications. Curr Diabetes Rep (2018) 18(9):71. doi: 10.1007/s11892-018-1036-y
179. Fadini G, Morieri M, Longato E, Avogaro DA. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J Endocrinological Invest (2020) 43:867–9. doi: 10.1007/s40618-020-01236-2
180. Zhang Y, Li H, Zhang J, Cao Y, Zhao X, Yu N, et al. The clinical characteristics and outcomes of patients with diabetes and secondary hyperglycaemia with coronavirus disease 2019: A single-centre, retrospective, observational study in Wuhan. Diabetes Obes Metab (2020) 22(8):1443–54. doi: 10.1111/dom.14086
181. Roncon L, Zuin M, Rigatelli G, Zuliani G. Diabetic patients with COVID-19 infection are at higher risk of ICU admission and poor short-term outcome. J Clin Virol (2020) 127:104354. doi: 10.1016/j.jcv.2020.104354
182. Guo L, Shi Z, Zhang Y, Wang C, Do Vale Moreira NC, Zuo H, et al. Comorbid diabetes and the risk of disease severity or death among 8807 COVID-19 patients in China: A meta-analysis. Diabetes Res Clin Pract (2020) 166:108346. doi: 10.1016/j.diabres.2020.108346
183. Ou M, Zhu J, Ji P, Li H, Zhong Z, Li B, et al. Risk factors of severe cases with COVID-19: a meta-analysis. Epidemiol Infect (2020) 148:e175. doi: 10.1017/S095026882000179X
184. Chang MC, Park Y-K, Kim B-O, Park D. Risk factors for disease progression in COVID-19 patients. BMC Infect diseases (2020) 20:1–6. doi: 10.1186/s12879-020-05144-x
185. Singh AK, Gupta R, Ghosh A, Misra A. Diabetes in COVID-19: Prevalence, pathophysiology, prognosis and practical considerations. Diabetes Metab Syndrome: Clin Res Rev (2020) 14(4):303–10. doi: 10.1016/j.dsx.2020.04.004
186. Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D. Viral and bacterial interactions in the upper respiratory tract. PloS Pathog (2013) 9(1):e1003057. doi: 10.1371/journal.ppat.1003057
187. Piret J, Boivin G. Viral interference between respiratory viruses. Emerging Infect Diseases (2022) 28(2):273. doi: 10.3201/eid2802.211727
188. Rosas Mejia O, Gloag ES, Li J, Ruane-Foster M, Claeys TA, Farkas D, et al. Mice infected with Mycobacterium tuberculosis are resistant to acute disease caused by secondary infection with SARS-CoV-2. PloS Pathog (2022) 18(3):e1010093. doi: 10.1371/journal.ppat.1010093
189. Baker PJ, Amaral E P, Castro E, Bohrer AC, Torres-Juárez F, Jordan CM, et al. Co-infection of mice with SARS-CoV-2 and Mycobacterium tuberculosis limits early viral replication but does not affect mycobacterial loads. Front Immunol (2023) 14:1240419. doi: 10.3389/fimmu.2023.1240419
190. Hilligan KL, Namasivayam S, Clancy CS, O’Mard D, Oland SD, Robertson SJ, et al. Intravenous administration of BCG protects mice against lethal SARS-CoV-2 challenge. J Exp Med (2022) 219(2):e20211862. doi: 10.1084/jem.20211862
191. Flores-Lovon K, Ortiz-Saavedra B, Cueva-Chicana LA, Aperrigue-Lira S, Montes-Madariaga ES, Soriano-Moreno DR, et al. Immune responses in COVID-19 and tuberculosis coinfection: A scoping review. Front Immunol (2022) 13:992743. doi: 10.3389/fimmu.2022.992743
192. Aiello A, Najafi-Fard S, Goletti D. Initial immune response after exposure to Mycobacterium tuberculosis or to SARS-COV-2: similarities and differences. Front Immunol (2023) 14. doi: 10.3389/fimmu.2023.1244556
193. Luke E, Swafford K, Shirazi G, Venketaraman V. TB and COVID-19: an exploration of the characteristics and resulting complications of co-infection. Front Biosci (Schol Ed) (2022) 14(1):6. doi: 10.31083/j.fbs1401006
194. Shah T, Shah Z, Yasmeen N, Baloch Z, Xia X. Pathogenesis of SARS-coV-2 and mycobacterium tuberculosis coinfection. Front Immunol (2022) 13:909011. doi: 10.3389/fimmu.2022.909011
195. Petrone L, Petruccioli E, Vanini V, Cuzzi G, Gualano G, Vittozzi P, et al. Coinfection of tuberculosis and COVID-19 limits the ability to in vitro respond to SARS-CoV-2. Int J Infect Dis (2021) 113 Suppl 1:S82–S7. doi: 10.1016/j.ijid.2021.02.090
196. Sheerin D, Abhimanyu, Peton N, Vo W, Allison CC, Wang X. Immunopathogenic overlap between COVID-19 and tuberculosis identified from transcriptomic meta-analysis and human macrophage infection. Iscience (2022) 25(6):104464. doi: 10.1016/j.isci.2022.104464
197. Riou C, du Bruyn E, Stek C, Daroowala R, Goliath RT, Abrahams F, et al. Relationship of SARS-CoV-2-specific CD4 response to COVID-19 severity and impact of HIV-1 and tuberculosis coinfection. J Clin Invest (2021) 131(12):e149125. doi: 10.1172/JCI149125
198. Najafi-Fard S, Aiello A, Navarra A, Cuzzi G, Vanini V, Migliori GB, et al. Characterization of the immune impairment of patients with tuberculosis and COVID-19 coinfection. Int J Infect Dis (2023) 130 Suppl 1:S34–42. doi: 10.1016/j.ijid.2023.03.021
199. Nabity SA, Han E, Lowenthal P, Henry H, Okoye N, Chakrabarty M, et al. Sociodemographic characteristics, comorbidities, and mortality among persons diagnosed with tuberculosis and COVID-19 in close succession in california, 2020. JAMA Network Open (2021) 4(12):e2136853. doi: 10.1001/jamanetworkopen.2021.36853
200. Kim B, Kang YA, Lee J. Heterogeneous impact of Covid-19 response on tuberculosis burden by age group. Sci Rep (2022) 12(1):13773. doi: 10.1038/s41598-022-18135-6
201. Group TC-GS. Tuberculosis and COVID-19 co-infection: description of the global cohort. Eur Respir J (2022) 59(3):2102538. doi: 10.1183/13993003.02538-2021
202. Gupta N, Ish P, Gupta A, Malhotra N, Caminero JA, Singla R, et al. A profile of a retrospective cohort of 22 patients with COVID-19 and active/treated tuberculosis. Eur Respir J (2020) 56(5):2003408. doi: 10.1183/13993003.03408-2020
203. Elziny MM, Ghazy A, Elfert KA, Aboukamar M. Case report: development of miliary pulmonary tuberculosis in a patient with peritoneal tuberculosis after COVID-19 upper respiratory tract infection. Am J Trop Med Hyg (2021) 104(5):1792–5. doi: 10.4269/ajtmh.20-1156
204. Visca D, Ong CWM, Tiberi S, Centis R, D’Ambrosio L, Chen B, et al. Tuberculosis and COVID-19 interaction: A review of biological, clinical and public health effects. Pulmonology (2021) 27(2):151–65. doi: 10.1016/j.pulmoe.2020.12.012
205. Yang H, Lu S. COVID-19 and tuberculosis. J Transl Int Med (2020) 8(2):59–65. doi: 10.2478/jtim-2020-0010
206. Daneshvar P, Hajikhani B, Sameni F, Noorisepehr N, Zare F, Bostanshirin N, et al. COVID-19 and tuberculosis coinfection: An overview of case reports/case series and meta-analysis of prevalence studies. Heliyon (2023) 9(2):e13637. doi: 10.1016/j.heliyon.2023.e13637
207. Norwood J, Kheshti A, Shepherd BE, Rebeiro PF, Ahonkhai A, Kelly S, et al. The impact of COVID-19 on the HIV care continuum in a large urban southern clinic. AIDS Behav (2022) 26(8):2825–9. doi: 10.1007/s10461-022-03615-7
208. Karim F, Gazy I, Cele S, Zungu Y, Krause R, Bernstein M, et al. HIV status alters disease severity and immune cell responses in beta variant SARS-CoV-2 infection wave. Elife (2021) 10:e67397. doi: 10.7554/eLife.67397
209. Conan N, Simons E, Ohler L, Mbatha M, van Cutsem G, Huerga H. Prevalence of TB and health-seeking behaviour. Int J Tuberc Lung Dis (2022) 26(5):463–5. doi: 10.5588/ijtld.22.0001
210. Grifoni A, Alonzi T, Alter G, Noonan DM, Landay AL, Albini A, et al. Impact of aging on immunity in the context of COVID-19, HIV, and tuberculosis. Front Immunol (2023) 14:1146704. doi: 10.3389/fimmu.2023.1146704
211. Marcu DTM, Adam CA, Mitu F, Cumpat C, Aursulesei Onofrei V, Zabara ML, et al. Cardiovascular involvement in tuberculosis: from pathophysiology to diagnosis and complications-A narrative review. Diagnostics (Basel) (2023) 13(3):432. doi: 10.3390/diagnostics13030432
212. Styskal J, Van Remmen H, Richardson A, Salmon AB. Oxidative stress and diabetes: what can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic Biol Med (2012) 52(1):46–58. doi: 10.1016/j.freeradbiomed.2011.10.441
213. Nomoto H, Hayakawa K, Ohmagari N. Impact of prioritized vaccinations for the elderly on the COVID-19 pandemic in Japan. Glob Health Med (2022) 4(2):129–32. doi: 10.35772/ghm.2022.01015
Keywords: SARS-CoV-2, Mycobacterium tuberculosis, COVID-19, TB, elderly, immunity, infectious diseases
Citation: Allué-Guardia A, Torrelles JB and Sigal A (2023) Tuberculosis and COVID-19 in the elderly: factors driving a higher burden of disease. Front. Immunol. 14:1250198. doi: 10.3389/fimmu.2023.1250198
Received: 29 June 2023; Accepted: 11 September 2023;
Published: 27 September 2023.
Edited by:
Subash Babu, International Centers for Excellence in Research (ICER), IndiaReviewed by:
Delia Goletti, National Institute for Infectious Diseases Lazzaro Spallanzani (IRCCS), ItalyCopyright © 2023 Allué-Guardia, Torrelles and Sigal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Allué-Guardia, YWFsbHVlZ3VhcmRpYUB0eGJpb21lZC5vcmc=; Jordi B. Torrelles, anRvcnJlbGxlc0B0eGJpb21lZC5vcmc=; Alex Sigal, YWxleC5zaWdhbEBhaHJpLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.