 Yujian Teng
Yujian Teng Licheng Xu
Licheng Xu Linli Tian
Linli Tian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 July 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1224443
This article is part of the Research Topic Reactive Oxygen Species (ROS) Signaling and Immune Diseases. View all 9 articles
Tumor-associated macrophages (TAMs) are significant immunocytes infiltrating the tumor microenvironment(TME). Recent research has shown that TAMs exhibit diversity in terms of their phenotype, function, time, and spatial distribution, which allows for further classification of TAM subtypes. The metabolic efficiency of fatty acid oxidation (FAO) varies among TAM subtypes. FAO is closely linked to the production of reactive oxygen species (ROS), which play a role in processes such as oxidative stress. Current evidence demonstrates that FAO and ROS can influence TAMs’ recruitment, polarization, and phagocytosis ability either individually or in combination, thereby impacting tumor progression. But the specific mechanisms associated with these relationships still require further investigation. We will review the current status of research on the relationship between TAMs and tumor development from three aspects: ROS and TAMs, FAO and TAMs, and the interconnectedness of FAO, ROS, and TAMs.
Various components of the tumor microenvironment(TME) play a crucial role in tumorigenesis and progression. Among these components, tumor-associated macrophages (TAMs) can regulate the TME through different infiltration levels and polarization characteristics (1, 2). TAMs in TME mainly originate from peripheral blood mononuclear cells (PBMCs) and tissue-resident macrophages (TRMs) (3). PBMCs are recruited to TME primarily mediated by chemokine-mediated pathways involving CCR2/CCL2 (4, 5), CSF1/M-CSF (6–8), and others. TRMs are present in tissues during embryonic development with tissue-specific and self-renewal abilities. In the past, TAMs were often classified into two distinct phenotypes, M1 and M2, with lipopolysaccharides and interferon-gamma (IFN-gamma) or interleukin (IL)-4 and IL-13 as representative inducers, respectively (9). M1 promotes the formation of an inflammatory environment and possess tumor-killing capabilities. M2 assists in constructing an immunosuppressive microenvironment by secreting IL-10, transforming growth factor beta (TGF-beta), or prostaglandin E2 (PGE2) to promote tissue repair and tumorigenic development. However, researchers have revealed that in most cases, TAMs exhibit a transitional phenotype between M1 and M2 (10–12). The emergence of this transitional subtype has expanded the previous classification criteria for TAMs. It has been demonstrated that TAMs can regulate tumor progression by modulating their own phenotype (13–15).
Reactive oxygen species (ROS), as a kind of redox byproduct, mainly includes superoxide, hydrogen peroxide (H2O2), and hydroxyl radical (HO-) (16). Mitochondria, endoplasmic reticulum,and peroxisomes are the primary sites of ROS production (17). ROS possess high chemical reactivity, and the oxidative stress effects will occur when the cell’s antioxidant capacity cannot coordinate the excess ROS (18, 19). For tumors, such ROS and oxidative stress effect are both necessary and lethal, and they regulate TAM-related mechanisms through multiple signaling pathways (20) (as shown in Figure 1).
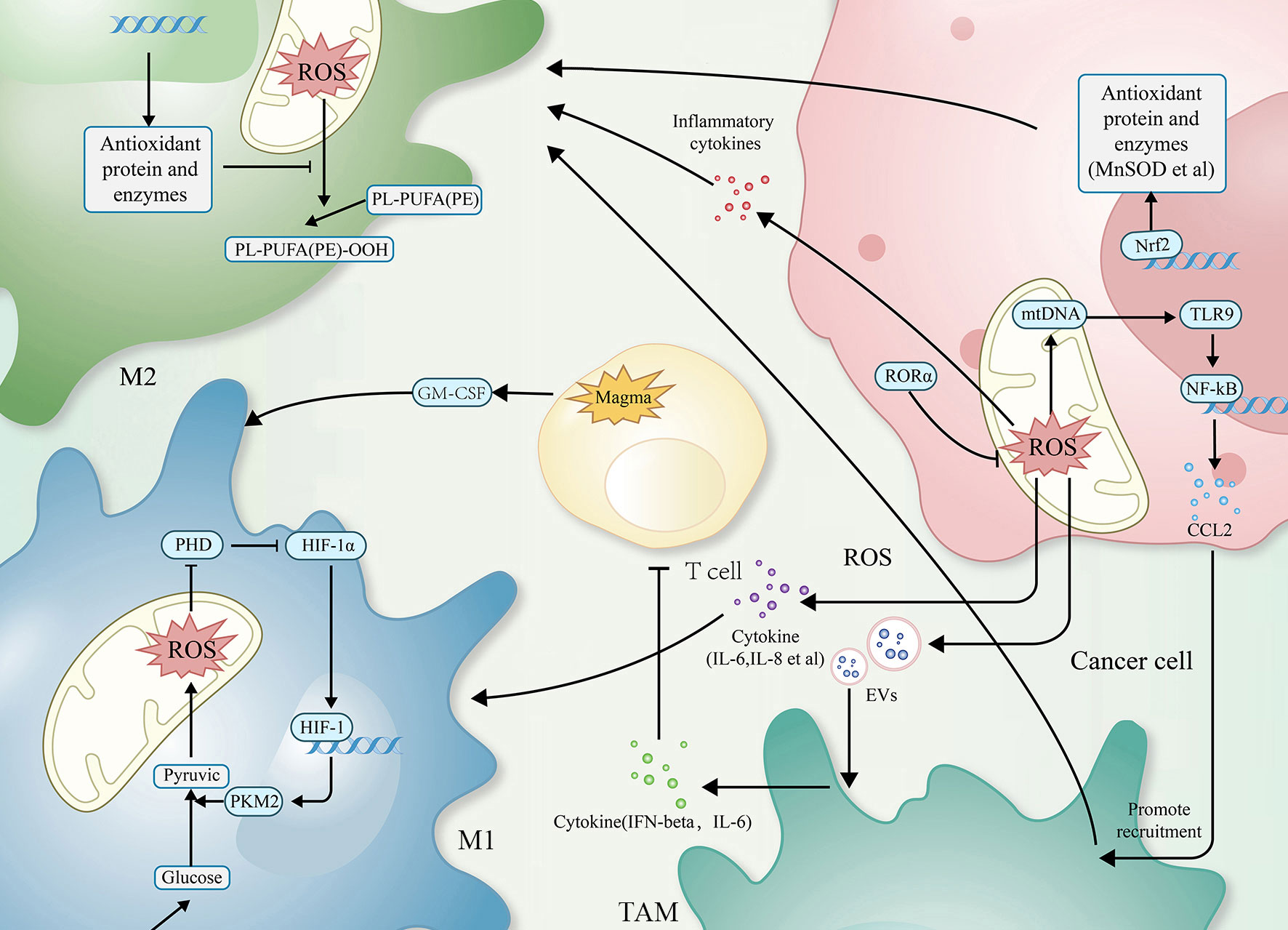
Figure 1 The relationship between ROS and TAMs. Simplified figure depicting various possible mechanisms of TAMs associated with ROS at a cellular-level affect oncogenesis and development of tumor in TME.
Lipids, including triglycerides, cholesterol, and phospholipids, play crucial roles in cellular function. Fatty acids and glycerol constitute the triglycerides. Moreover, both tumor cells and immune cells undergo lipid metabolism, including fatty acid oxidation (FAO), reprogramming to survive the harsh environment (21). TAMs, known for their high plasticity (22), exhibit differences in FAO efficiency between M1 and M2 phenotypes. The metabolic reprogramming associated with it, especially FAO, plays an essential role in regulating tumor progression (23–25), and this article will describe how FAO synergistically influences tumor progression in conjunction with TAMs (as shown in Figure 2).
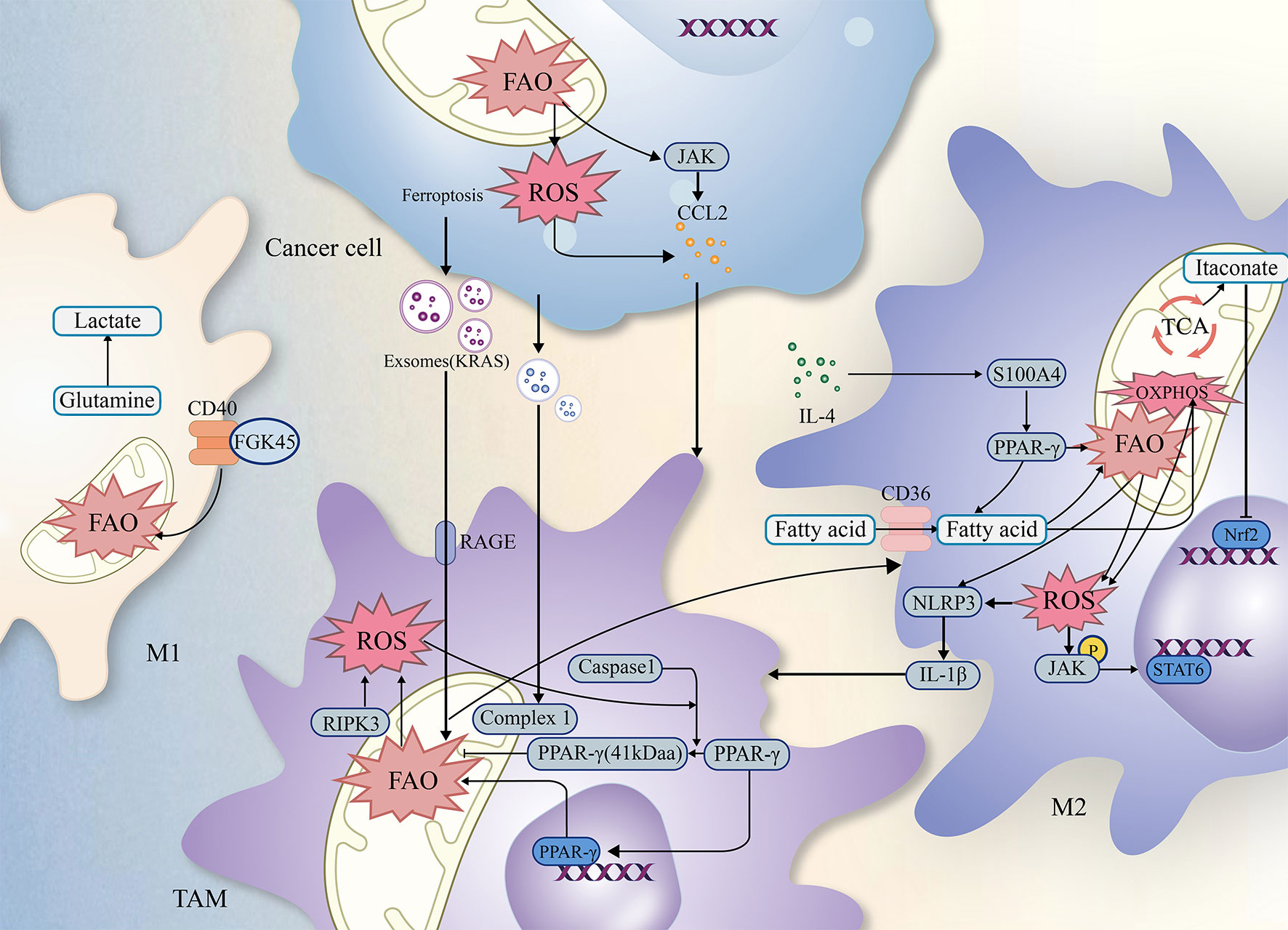
Figure 2 The relationship of ROS FAO and TAMs. Simplified figure depicting various possible mechanisms of TAMs associated with ROS and FAO at a cellular-level affect oncogenesis and development of tumor in TME.
Previous studies have revealed complex interactions among TAMs, ROS, FAO, which may influence the biological functions of tumors through multiple factors (as shown in Figure 2). As a result, integrating the critical signaling pathways involving TAMs, FAO, and ROS establishes a theoretical foundation and offers research implications for further investigation into tumor treatment modalities.
In studies investigating the regulation of tumor progression by immune cells, researchers have observed that ROS at different levels can either promote or inhibit tumor growth depending on various molecular signaling pathways. The generation of ROS involves multiple mechanisms, with mitochondria being the primary site of ROS production. Electron leakage in the electron transport chain (ETC) is the primary source of ROS (26). Electron leaking from complex I and III of ETC react with O2 to generate ROS in the inner mitochondrial membrane, influenced by the redox state of the ETC, the proton dynamics and the local O2 concentration (27). ROS can also generate in other organelles, such as endoplasmic reticulum or peroxisome (17). In addition, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) (28), xanthine oxidase (XO) (29), cytochrome p450 (30), nitric oxide synthase (NOS) are also in ROS generation (31). The generated ROS are further involved in various molecular signaling pathways that regulate tumor cell proliferation, differentiation and apoptosis (32–34).
Growing evidence demonstrating that ROS play a role in modulating the tumor microenvironment through multiple mechanisms. ROS can directly induce cell death in tumor cells (35) and also regulate tumor progression by acting on the recruitment and polarization of TAMs (36, 37). We will discuss the relationship between ROS and TAMs and summarize the current status of research on the relevant molecular signaling pathways (as shown in Table 1).
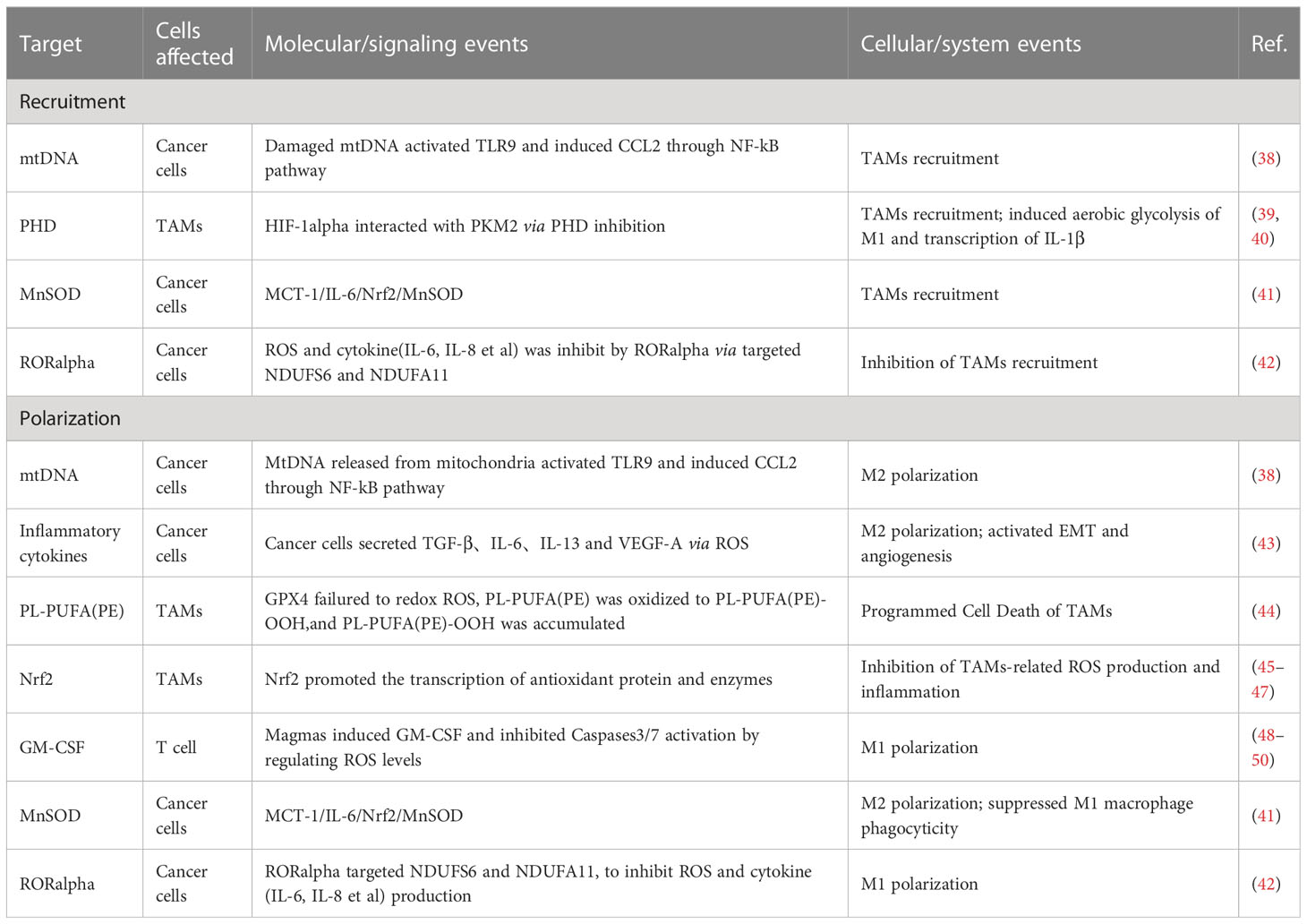
Table 1 Targets of ROS associated with TAMs.
ROS are involved in regulating the infiltration of TAMs through a mechanism of action related to influencing the macrophage recruitment, which is one of the primary ways to affect TAMs. In related studies, by activating T cells and natural killer (NK) cells, ROS recruits both neutrophils and macrophages into the TME and, in this way, kills cancer cells and inhibits tumor progression (51, 52); TAMs can also secrete ROS after being recruited to the TME, which contributes to reduces the activity of T cells and NK cells (53). Excessive ROS can damage mitochondrial DNA (mtDNA). Mitochondria of hepatocellular carcinoma cells release mtDNA into the cytoplasm in response to ROS, activating Toll-like receptors (TLRs), of which TLR9 can induce CCL2 to recruit macrophages to the TME.Additionally, TLR9 activation can also contribute to the maintenance of the M2 phenotype of TAMs (38).
In the M1 phenotype of TAMs, via inhibiting the prolyl hydroxylase domain (PHD), ROS-induced the generation of hypoxia-inducible factor 1alpha (HIF-1alpha). HIF-1alpha can interact with pyruvate kinase M2 (PKM2), increasing the transcriptional levels of macrophage glycolysis-related enzymes and sustaining aerobic glycolysis. It can also induce angiogenesis and participate in the recruitment of TAMs (39, 40).
The recruitment of TAMs provides favourable conditions for tumor development. As research has progressed, the interconversion mechanism of between M1 and M2 phenotypes of macrophages, involving ROS, has been recognized as a critical factor in regulating the function of TAMs. In most cases, elevated ROS promotes M2 polarization, a process in which ROS often plays a pro-tumor role. Mitochondrial Lon, a chaperonin, can induce ROS production and participate in M2 polarization, while M2 can induce Lon production, forming a positive feedback loop (43, 54). In a study of specific mechanisms, researchers found Lon can release ROS-dependent inflammatory cytokines such as TGF-beta, IL-6, IL-13, and vascular endothelial-derived growth factor-A (VEGF-A) through p38 and NF-κB signaling pathways to promote epithelial-mesenchymal transition (EMT), angiogenesis, and M2 polarization (43). In addition to the passive TAMs regulation by ROS, TAMs actively modulate ROS levels through NOX2, leading to high levels of ROS production. This, in turn, recruits and regulates other immune cells in the TME, including myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Treg). Together, TAMs and these immune cells work to effectively regulate the immune function of TME, establishing an immunosuppressive microenvironment (55–59).
High levels of ROS can also limit tumor progression by activating cell death pathways. In a 2012 paper by Brent R. Stockwell, it was described that cells can Interact with excess ROS through mechanisms such as the Fenton reaction. The combination of excess ROS with a dysregulated antioxidant system resulting in the reaction of intracellular lipids with ROS, producing lipid peroxide (LOOH). And the accumulation of LOOH drives a novel form of programmed cell death called ferroptosis (60–64), which also occurs in TAMs (44). Extracellular vesicles (EVs) are also essential participants in the mechanisms involved. These exosomes can carry immunosuppressive components and chemokines (65). TAMs exposed to tumor cell-derived exosomes exhibit a metabolic profile similar to the M2 phenotype, enhancing FAO, oxidative phosphorylation (OXPHOS), and oxygen consumption rates (66–68). EVs can also down-regulate T cell immune function by inducing TAMs to produce IFN-beta and IL-6. In this way, EVs disrupt the immune function of immune cells (65).
Mammals have evolved various complex antioxidant systems in vivo to scavenge ROS and mitigate the harmful effects of oxidative stress, including superoxide dismutase (SOD) (69), catalase (CAT) (70), peroxidase (PRDX) (71, 72), glutathione peroxidase (GPX) (48), and mitochondrial autophagy (73, 74).Additionally, a central regulator of antioxidant genes known as Nrf2 can be dissociated from (KEAP1) under conditions of oxidative stress to exert antioxidant functions (45–47). Recently research has shown that beta-glucan with antioxidant properties can modulate ROS production in LPS-induced RAW264.7 mouse macrophages. Beta-glucan achieves this by regulating Nrf2 through the activation of the scavenger receptor Dectin-1 (75). Activation of Dectin-1 enhances the expression of the antioxidant enzyme heme oxygenase-1 (HO-1) within macrophages, thereby reducing ROS levels and oxidative stress (76). In studying the relationship between GPX and macrophages, researchers observed that dysfunctional GPX4 induced ferroptosis in macrophages by accumulating lipid peroxides (63, 64). In addition, the mammalian protein translocation mechanism, Magmas, can be involved in regulating ROS levels to maintain redox homeostasis, in which Magmas protects cells with oxidative stress damage by inhibiting the activation of Caspases3/7 (48), induces granulocyte-macrophage colony-stimulating factor (GM-CSF) to promote M1 polarization, and enhances the antigen-presenting effect of macrophage (49, 50). When targeting Mn-superoxide dismutase (MnSOD) or MnSOD-related genes to inhibit MnSOD levels, researchers observed that TAMs infiltration and M2 polarization processes were inhibited (41, 48). Retinoid orphan nuclear receptor alpha (RORalpha), which is also involved in the complex process of the antioxidant system, can significantly (42) reduce ROS levels, decreases macrophage infiltration, and enhances M1 polarization (42). All of these antioxidant mechanisms involved in the regulation of TAMs further reveal the feasibility of targeting the anti-ROS oxidative system to impede tumor progression.
To date, we have identified several tumor molecular signaling pathways regarding how ROS affects phenotypes of TAMs (as shown in Table 1). Directly targeting these pathways to reduce the recruitment and function of TAMs in TME and reversing M2-like TAMs to M1,or modulating macrophages phagocytosis has emerged as an extremely promising strategy for antitumor immunotherapy.
The researchers observed that TAMs predominantly exhibited an M1 phenotype during the initiation stage of tumor development. As the tumor progressed to an advanced stage, TAMs primarily expressed an M2 phenotype (21, 77). In the early stage of tumor development, TAMs preferentially utilize glycolysis metabolism for energy generation (78). However, as the tumor progressed, FAO and OXPHOS gradually became the predominant modes of TAMs’ metabolism (79). The metabolic reprogramming from M0 to M1 is achieved by inhibiting macrophage mitochondrial function and improving substrate utilization, regulated by HIF-1α and its downstream proteins (80–82). Conversely, when macrophages transitioned from M0 and M1 phenotypes to M2 phenotypes, their FAO and OXPHOS metabolic efficiency increased, accompanied by activation of the tricarboxylic acid cycle (TCA) cycle (77, 83, 84).
In studies examining the influence of lipids on tumor progression, we have observed that TAMs infiltrate more in lipid-rich droplets of TME. Furthermore, a short-term high-fat diet can activate the macrophages in adipose tissue of patients with colorectal cancer, and reduce the risk of cancer metastasis, tentatively suggesting a potential correlation between lipid metabolism and TAMs (85). Examination of the lipid metabolic profile of TAMs has revealed heterogeneity in their fatty acid metabolic profile under different phenotypes, leading to the speculation that FAO metabolic activity is associated with the TAMs’ phenotypes (77, 83). In most cases, cells undergo three steps from fatty acid oxidation to the final production of energy: FAO, conversion of acetyl-coenzyme A (Acetyl-CoA) by TCA, and OXPHOS (86). Beta-oxidation of fatty acids is the primary metabolic pathway of FAO, and when the efficiency of beta-oxidation metabolism is enhanced in TAMs, tumor-invasive abilities become stronger (87).
Further study of specific mechanisms, researchers found that the metabolic efficiency of FAO plays a crucial role in regulating mitochondrial function and polarization of TAMs. Beta-oxidation is closely related to the phenotype of TAMs (77, 83). Peroxisome proliferator-activated receptor (PPAR) system is an essential regulator of fatty acid metabolism and is involved in the metabolic reprogramming of TAMs to M2 phenotypic polarization (88, 89). The PPAR system mediated through signal transducer and activator of transcription 6 (STAT6) and PPARgamma coactivator 1-beta (PGC-1beta) elevated the metabolic efficiency of FAO in TAMs (23, 90, 91). In further studies, the expression levels of PPAR-gamma and its downstream CD36 were upregulated by the action of the upstream S100A4 protein, which induced M2 polarization responses in the form of enhanced fatty acid absorption and FAO (88). Researchers have observed that intact structure is a prerequisite for the regulation of FAO by PPARs, but receptor-interacting protein kinase 3 (RIPK3)-mediated Caspase-1 can disrupt the integrity of PPAR-gamma in TAMs, leading to the generation of a PPAR-gamma 41 kDa fragment that can move into the mitochondria. This fragment prevents the buildup of lipid droplets and the promotion of cancerous M2 cells by inhibiting the function of FAO and MCAD enzymes in a time-dependent manner (92–94). In another study, ovarian cancer stem cells can also promote the M2 polarization of TAMs with the PPARγ/NF-κB pathway (95). In addition to the PPAR system, the researchers observed that ubiquitin-specific protease14 (USP14)-mediated deubiquitination of SIRT1 can elevate its downstream fatty acid oxidation-related pathway, SIRT1/PGC-1α, in IL-4+IL-10-induced M2 macrophages. We can find the levels of M2 marker CD206 and SIRT1/PGC-1α expressed higher in USP14+ macrophages compared to USP14- macrophages. And this alteration did not affect the expression of other key proteins involved in FAO, such as PPARs, as confirmed by qRT-PCR. However, the elevated USP14 alone did not lead to M2 polarization, which further suggests that there may not be only one single FAO-related signaling pathway during the polarization schedule of TAMs (96).
Exosomes are also involved in FAO-mediated phenotypic regulation of TAMs. Kirsten rat sarcoma viral oncogene homolog (KRASG12D) protein, carried by the exosome, can polarize M2-type macrophages by regulating STAT3-dependent FAO after internalized by TAMs. Moreover, macrophages with high expression of KRASG12D showed increased expression of FAO-related genes such as carnitine palmitoyl-transferase 1A (CPT1A) and acyl-CoA dehydrogenase short chain (ACADS) (97). However, unlike the results obtained by testing FAO-related genes in TAMs, the activity of the rate-limiting enzyme ACADS in TME was positively correlated with M1 and Treg infiltration levels but negatively correlated with and M2 (98). Further studies identified methylation sites of ACADs and differences in the expression of methylation levels of ACADs between cancer and normal tissues, suggesting that epigenetic alterations in ACADS may be involved in forming this phenomenon (98).
The specific signaling pathways have not been sufficiently studied. Previous studies have shown that the IFN-gamma, GM-CSF and LPS are important influences in the inducement of M1 polarization (99). Unlike the mechanisms associated with the oxidative decomposition of fatty acids alone, the secretion of IFN-gamma can inhibit the srebp1-mediated fatty acid synthesis pathway in immunosuppressed (M2-like) TAMs and stimulate FAO (100). Perhaps we can target the crosstalk between IFN-gamma and FAO to regulate the phenotype of TAMs by regulatting the secretion of IFN-gamma (101–104). However, we should not overlook the combined efficacy of the treatments. For example, inhibiting CD8+ T cells by Treg cells may enhance the secretion level of IFN-gamma, but we should also consider the anti-tumor effects of CD8+ T cells (99, 105). Therefore, a comprehensive approach is needed to select the ideal target for treatment.
In an attempt to investigate the link between metabolic reprogramming and TAMs, some metabolites are also involved in the mechanisms regulating the phenotype of TAMs (106, 107). One study found that CD40 activation altered the NAD+/NADH ratio through lactate production and enhanced M1 polarization, which relied on glutamine-lactate conversion. However, unlike lipopolysaccharide (LPS)-activated M1, CD40-activated M1 exhibited elevated activity of FAO and TCA cycle. The researchers speculate that a combination of CD40 activation and type I interferons (IFN-I) deficiency may contribute to this alteration (78, 108). Additionally, alpha-ketoglutarate (AKG), an essential intermediate in the TCA cycle, can regulate both FAO and Jumonji domain-containing protein-3(JMJD3)-dependent epigenetic modifications of the M2 genes, thereby increasing the ratio of AKG’s downstream product succinate to AKG and inducing M2 polarization (106, 109–111).
In conclusion, signaling pathways related to fatty acid metabolism exert some influence on TAMs regulation, either by directly altering FAO efficiency or by affecting the production of related metabolites (as shown in Table 2). The discovery of this phenomenon provides a new theoretical basis for regulating phenotypic alteration and polarization of TAMs through FAO.

Table 2 Targets of FAO associated with TAMs.
Previous studies have demonstrated that ROS and FAO can separately regulate TAMs (38, 77, 83). For instance, modulating cellular oxidative stress levels by targeting ROS can interfere with the tumor microenvironment and modulate the phenotype of TAMs (19, 112). In addition, targeting and regulating FAO-related signaling pathways can inhibit the growth and survival of cancer cells and TAMs (79, 87, 113). In some studies, we observed that ROS could affect mitochondrial FAO (114, 115) by disrupting mitochondrial DNA (mtDNA) due to the proximity of ROS production sites to mtDNA (65, 114, 116), and such a spatial relationship provides an opportunity to study the interplay among ROS, FAO, and TAMs.
Several potential signaling pathways suggest that ROS and FAO jointly regulate the level of TAMs and tumor progression (as shown in Table 3). Intact mitochondrial structures support the proper functioning of FAO (38, 77, 83), and FAO-related mechanisms can regulate the phenotype of TAMs (115, 122). In contrast, NADH and FADH2 produced by FAO contribute to electron leakage processes in the ETC collectively participate in the mitochondrial generation of ROS, which further influences phenotypic changes in TAMs (123). It is due to these interconnected mechanisms that researchers have attempted to establish links between TAMs, ROS, and FAO to overcome current limitations in diagnosis and treatment.

Table 3 Targets involving TAMs, ROS, and FAO.
As mentioned, PPARs play a crucial role in regulating FAO. The deficiency of RIPK3 reduces ROS levels through the ROS/Caspase-1/PPAR pathway, which inhibits Caspase-1-mediated PPAR-gamma catabolic processes. Maintaining PPAR function and integrity improves FAO efficiency, leading to M2 polarization and TAMs recruitment (92). Scavenger receptors (SRs) are a group of endocytic receptors involved in various processes such as apoptosis, autoimmunity, inflammation, and lipid metabolism. CD36 protein is a member of SRs localized on the cell surface in adipose tissue, gastrointestinal tract, heart, skeletal muscle, and macrophages (124). In 2007, Nada A. Abumrad’s team first identified the critical role of CD36 in fatty acid uptake and lipid accumulation (125), which enables cells to generate energy through FAO instead of glycolysis (24). Current studies have revealed that CD36 can enhance the effectiveness of FAO but also promotes the generation of ROS (126). And the generated ROS levels can promote Janus kinase 1 (JAK1) phosphorylation and Src homology region 2 (SH-2) domain-containing phosphatase 1 (SHP1) dephosphorylation in response to oxidative stress, which regulates the transcription of TAM genes (24) and the polarization towards the M2 phenotype (117). Complex I (NADH: ubiquinone oxidoreductase) and III (ubiquinone: cytochrome c oxidoreductase) have been shown to have a crucial role in ROS production. Furthermore, complex IV, one of the regulatory sites of oxidative phosphorylation, is closely related to the final generation of ATP from FAO (127, 128). Complex IV (cytochrome c oxidase) expresses at higher levels in M2 compared to M1 (p < 0.05), contrasting with the difference of complex I, in which the complex I was lower in M2 than in M1 (p < 0.05) (118). The study did not reveal the specific mechanism leading to this phenomenon. However, it also enhances the possibility of the mitochondrial complexes as potential therapeutic target that involves the interplay of ROS, FAO, and TAMs. In a study on hepatocellular carcinoma cells, researchers found that changes in FAO efficiency in TAMs can regulate tumor cell migration. Further studies showed that M2-type macrophages upregulate IL-1β secretion levels by regulating FAO in a NLRP3- and ROS-dependent manner (120). The secreted IL-1β then enhances cell migration by activating the NF-κB pathway in tumor cells (129). To investigate the mechanism related to the oncogene retinoblastoma gene (RB) in malignancies, researchers knocked out the RB gene in mouse sarcoma and breast cancer models. As a result, AMPK was activated, which increased FAO by inhibiting ACC activity, thereby promoting mitochondrial ROS production and JNK activation. This activation led to the involvement of the CCL2/CCR2 axis in a mitochondrial ROS and JNK-dependent manner, recruiting immune cells, including TAMs and MDSCs, into TME (121).
In addition, some potential mechanisms that link TAMs, ROS, and FAO. Researchers have found that macrophages with the M1 phenotype are more resistant to ferroptosis compared to M2 (44), and several studies on ferroptosis suggest the interplay among TAMs, ROS, and FAO (44, 130–132). The occurrence of ferroptosis is dependent on the oxidation of ROS, and one study recently demonstrated that inducing cellular ferroptosis lead to mitochondria shrink, or even disappearance of mitochondrial ridges, impairing the function of FAO (61, 64, 115, 122). In one study, ROS and ferroptosis mediated the release of KRASG12D-containing exosomes. Scavenger receptors, specifically receptor for advanced glycation end products (RAGE) on tumor-associated macrophages (TAMs), mediate the uptake of KRASG12D. This uptake promotes the M2 phenotype through STAT3-dependent FAO and is positively correlated with survival rates (97).
As a critical transcription factor regulating the expression of antioxidant genes (133, 134), can reduce ROS generation through increased transcription. Nrf2 has been shown to control the efficiency of FAO by acting on OXPHOS and regulating the production of TAM-related ROS (133). Apart from its direct impact on redox processes, Nrf2 also regulates macrophages by inhibiting the transcriptional function of pro-inflammatory factors (135). In a study using itaconate to target the inhibition of KEAP1-NRF2 complex degradation, undamaged Nrf2 was released and translocated to the nucleus, activating the transcription of downstream genes. This resulted in the inhibition of both lipid peroxidation and ferroptosis in macrophages (132, 136). However, the exact mechanism of Nrf2 in TAMs still needs to be thoroughly investigated. As a macrophage-specific metabolite generated in the presence of Immunoresponsive gene 1 (IRG1), itaconate production is increased in M2 (137, 138) and correlated with beta-oxidation efficiency in TAMs (119, 139). Itaconate is one of the highly upregulated metabolites in peritoneal tissue-resident macrophages in B16 melanoma cells or ID8 ovarian cancer cells (119). Knockdown of IRG1 lead to the downregulation of itaconate and reduced FAO, OXPHOS, and ROS levels in TAMs. This significantly inhibited tumor progression, although the studies did not indicate what mechanism led to this phenomenon (119, 140). However, these influences can be considered potential clinical biomarkers while altering TAMs polarization.
Considering the significance of TAMs in regulating tumor progression and the complex interactions among ROS, FAO, and TAMs, some studies have explored the inclusion of TAMs with ROS or FAO to identify more effective anti-cancer therapies (141).
Studies focusing on ROS have demonstrated that downregulation of ROS in TAMs often leads to a skewed phenotype towards M1 polarization, which provides a theoretical basis for therapeutic modalities that target NOX2, Lon proteins, RORα to alter ROS levels and thus reconfigure the phenotype of TAMs (42, 54, 56–59). In recent years, several signaling pathways between ROS and PD-L1 in immunotherapy-related studies involving TAMs have been identified (19, 56). For instance, in a triple-negative breast cancer (TNBC)-related study, induced generation of ROS in a manner that activates NF-κB signaling to promote PD-L1 expression on the surface of TAMs (142). In contrast, in the field of therapeutics combined with nanotechnology, iron oxide nanoparticles (IONPs) were found to reprogram TAMs toward an immunogenic phenotype in a manner that modulates changes in ROS production levels through the activation of Caspase-3, which is closely related to apoptosis-reduced cell survival in mouse mammary tumors (142). Additionally, researchers have observed that different levels of ROS may indicate variable tumor sensitivity to chemotherapy. Thus, it is crucial to closely monitor the dynamics of ROS during patient treatment (142). Elevated expression of FAO-related genes and increased FAO efficiency are metabolic characteristics of macrophages skewed towards the M2 phenotype (24), Inhibiting or enhancing FAO metabolic efficiency in macrophages can induce polarization towards the M1 or M2 phenotype, respectively (24). Macrophages will exhibit anti tumor effects with the M1 phenotype and promote tumors with the M2 phenotype. Therefore, shifting the balance of TAMs to the M1 phenotype by altering FAO will inhibit tumor progression. Since PPARs are critical transcription factor for FAO promotion in TAMs and is involved in regulating the polarization of TAMs, they are considered potential target for cancer therapy. In related studies, modulation of PPAR-gamma in TAMs by S100A4 and others has also emerged as a potential cancer treatment modality (88). In addition, by mediating fatty acids uptake, CD36 can regulate the metabolic efficiency of FAO and OXPHOS to influence the phenotype of TAMs. Currently, there are fewer studies specifically targeting FAO in TAMs. Most studies reprogram the phenotype of TAMs by targeting mitochondrial function or OXPHOS alone or in combination with treatment (143–145). This part of the study also provides a basis for studying FAO as a therapy target.
Previous studies show that cancer cells are susceptible to developing resistance to single treatments (146). Combining different therapeutic modalities to reduce cancer resistance and improve treatment efficacy has also been a significant challenge for researchers. Therapies that modulate the phenotype of TAMs by targeting FAO or ROS have shown effects on tumorigenesis and progression, respectively. Given the close interaction between FAO and ROS, targeting both pathways provides valuable intervention points. However, the specific mechanisms through which they influence tumorigenesis and progression are still being elucidated. Therefore, a combination therapies targeting different molecular key pathways have been selected to achieve more potent anti-cancer effects.
We found combined targeting both FAO and ROS play an crutial role in inhibiting tumor progression. Targeting RIPK3, CD36, RIPK3 can directly regulate TAMs polarization toward an anti-tumor M1 phenotype. Methods that affect CCL2 secretion (121) or the use of etomoxir and siRNA to modulate IL-1β secretion in TAMs capacity to reduce TAMs recruitment (120). Numerous therapeutic approaches targeting FAO and ROS in TAMs are currently under development. For instance, a potential therapeutic involving two targets was identified. Decitabine, a DNA methyltransferase inhibitor, was found to have an impact on the hypomethylation of RIPK3. This inhibits the FAO process in TAMs through the ROS/Caspase-1/PPAR-gamma signaling pathway and leads to a reversal of the pro-tumor phenotype of TAMs (92). An agent called α-T-K nanoemulsions, prepared with the combination of KIRA673-75 (IRE1-XBP1 inhibitor) and α-tocopherol (ROS inhibitor), had a dual inhibitory effect. Under α-T-K intervention, macrophages showed increased expression of CD86, a marker of M1-type cells, and decreased expression of CD206, a marker of M2-type cells. Simultaneously, the IRE1-XBP1 pathway, which upregulated FAO, was inhibited, resulting in decreased levels of ROS and FAO (147). This intervention led to reduced tumor cell survival and improved efficacy of immunotherapy for lung cancer. Importantly, in experimental settings, simultaneous inhibition of both ROS and FAO showed superior antitumor effects compared to either drug alone (147). And α-T-K demonstrated fewer adverse effects in a mouse model (147), so we believe this work will provide a valuable reference for cancer treatment, bringing hope for more effective tumor therapy by combining the targeting of ROS with FAO.
We review the signaling pathways involving ROS, FAO, and TAMs, which present new opportunities for therapeutic interventions in tumors. ROS can influence the biological function of tumors by regulating the recruitment, polarization, and phagocytosis ability of TAMs, in which ROS production and the antioxidant system in vivo play an important role. The efficiency of FAO metabolism and the regulation of related metabolites also impact the function of TAMs. Meanwhile, several signaling pathways that affect the biological processes of tumors have been identified, which efficiently modulate tumor progression through regulatory mechanisms involving TAMs, ROS, and FAO. However, the current research findings are still far from sufficient, and further investigations are still needed to gain a deeper understanding and explore the intricate relationship between TAMs, oxidative stress, and nutrient metabolism for potential therapeutic targets.
Conceptualization, YT, LT, and ML; Investigation, LX, YT, WL, and PL; Visualization, YT and PL; Writing - Original Draft, YT, LX, and WL; Writing - Review & Editing, YT, LT, and LX; Funding Acquisition, LT and ML; Supervision, YT, WL, LT, and ML. All authors contributed to the article and approved the submitted version.
National Natural Science Foundation of China (Grant No. 82072987), National Natural Science Foundation of China (Grant No. 82273332) and National Natural Science Foundation of China (Grant No. 82173209).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol (2022) 23(8):1148–56. doi: 10.1038/s41590-022-01267-2
2. Kowal J, Kornete M, Joyce JA. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy (2019) 11(8):677–89. doi: 10.2217/imt-2018-0156
3. Min J, Dong F, Wu P, Xu X, Wu Y, Tan Y, et al. A radiomic approach to access tumor immune status by cd8(+)Trms on surgically resected non-small-cell lung cancer. OncoTargets Ther (2021) 14:4921–31. doi: 10.2147/ott.S316994
4. Schmall A, Al-Tamari HM, Herold S, Kampschulte M, Weigert A, Wietelmann A, et al. Macrophage and cancer cell cross-talk via ccr2 and cx3cr1 is a fundamental mechanism driving lung cancer. Am J Respir Crit Care Med (2015) 191(4):437–47. doi: 10.1164/rccm.201406-1137OC
5. Xu M, Wang Y, Xia R, Wei Y, Wei X. Role of the ccl2-ccr2 signalling axis in cancer: mechanisms and therapeutic targeting. Cell proliferation (2021) 54(10):e13115. doi: 10.1111/cpr.13115
6. Ahmed N, Kadife E, Raza A, Short M, Jubinsky PT, Kannourakis G. Ovarian cancer, cancer stem cells and current treatment strategies: A potential role of magmas in the current treatment methods. Cells (2020) 9(3):719. doi: 10.3390/cells9030719
7. Guan W, Li F, Zhao Z, Zhang Z, Hu J, Zhang Y. Tumor-associated macrophage promotes the survival of cancer cells upon docetaxel chemotherapy via the csf1/csf1r-cxcl12/cxcr4 axis in castration-resistant prostate cancer. Genes (2021) 12(5):773. doi: 10.3390/genes12050773
8. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol (2019) 12(1):76. doi: 10.1186/s13045-019-0760-3
9. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. Il-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev (2010) 24(3):241–55. doi: 10.1101/gad.1874010
10. Muraille E, Leo O, Moser M. Th1/th2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front Immunol (2014) 5:603. doi: 10.3389/fimmu.2014.00603
11. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell (2018) 174(5):1293–308.e36. doi: 10.1016/j.cell.2018.05.060
12. Degboé Y, Poupot R, Poupot M. Repolarization of unbalanced macrophages: unmet medical need in chronic inflammation and cancer. Int J Mol Sci (2022) 23(3):1496. doi: 10.3390/ijms23031496
13. Kato Y, Tabata K, Kimura T, Yachie-Kinoshita A, Ozawa Y, Yamada K, et al. Lenvatinib plus anti-pd-1 antibody combination treatment activates cd8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PloS One (2019) 14(2):e0212513. doi: 10.1371/journal.pone.0212513
14. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via ahr and cd39. Nat Neurosci (2019) 22(5):729–40. doi: 10.1038/s41593-019-0370-y
15. Zhu Q, Wu X, Wu Y, Wang X. Interaction between treg cells and tumor-associated macrophages in the tumor microenvironment of epithelial ovarian cancer. Oncol Rep (2016) 36(6):3472–8. doi: 10.3892/or.2016.5136
16. Cheung EC, Vousden KH. The role of ros in tumour development and progression. Nat Rev Cancer (2022) 22(5):280–97. doi: 10.1038/s41568-021-00435-0
17. Sarniak A, Lipińska J, Tytman K, Lipińska S. Endogenous mechanisms of reactive oxygen species (Ros) generation. Postepy higieny i medycyny doswiadczalnej (Online) (2016) 70(0):1150–65. doi: 10.5604/17322693.1224259
18. Hajam YA, Rani R, Ganie SY, Sheikh TA, Javaid D, Qadri SS, et al. Oxidative stress in human pathology and aging: molecular mechanisms and perspectives. Cells (2022) 11(3):552. doi: 10.3390/cells11030552
19. Kuo CL, Ponneri Babuharisankar A, Lin YC, Lien HW, Lo YK, Chou HY, et al. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: foe or friend? J Biomed Sci (2022) 29(1):74. doi: 10.1186/s12929-022-00859-2
20. Covarrubias A, Byles V, Horng T. Ros sets the stage for macrophage differentiation. Cell Res (2013) 23(8):984–5. doi: 10.1038/cr.2013.88
21. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell (2022) 13(12):877–919. doi: 10.1007/s13238-021-00846-7
22. Anagnostakis F, Piperi C. Targeting options of tumor-associated macrophages (Tam) activity in gliomas. Curr neuropharmacol (2023) 21(3):457–70. doi: 10.2174/1570159x20666220120120203
23. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab (2019) 30(1):36–50. doi: 10.1016/j.cmet.2019.06.001
24. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res (2020) 80(7):1438–50. doi: 10.1158/0008-5472.Can-19-2994
25. Li S, Yu J, Huber A, Kryczek I, Wang Z, Jiang L, et al. Metabolism drives macrophage heterogeneity in the tumor microenvironment. Cell Rep (2022) 39(1):110609. doi: 10.1016/j.celrep.2022.110609
26. Olsen RK, Cornelius N, Gregersen N. Genetic and cellular modifiers of oxidative stress: what can we learn from fatty acid oxidation defects? Mol Genet Metab (2013) 110 Suppl:S31–9. doi: 10.1016/j.ymgme.2013.10.007
27. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417(1):1–13. doi: 10.1042/bj20081386
28. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
29. Elsayed S, Elsaid KA. Protein phosphatase 2a regulates xanthine oxidase-derived ros production in macrophages and influx of inflammatory monocytes in a murine gout model. Front Pharmacol (2022) 13:1033520. doi: 10.3389/fphar.2022.1033520
30. Contreras-Zentella ML, Villalobos-García D, Hernández-Muñoz R. Ethanol metabolism in the liver, the induction of oxidant stress, and the antioxidant defense system. Antioxidants (Basel Switzerland) (2022) 11(7):1258 doi: 10.3390/antiox11071258
31. Byrne NJ, Rajasekaran NS, Abel ED, Bugger H. Therapeutic potential of targeting oxidative stress in diabetic cardiomyopathy. Free Radical Biol Med (2021) 169:317–42. doi: 10.1016/j.freeradbiomed.2021.03.046
32. Chen CL, Zhang L, Jin Z, Kasumov T, Chen YR. Mitochondrial redox regulation and myocardial ischemia-reperfusion injury. Am J Physiol Cell Physiol (2022) 322(1):C12–c23. doi: 10.1152/ajpcell.00131.2021
33. Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J, et al. Mitochondria and mitochondrial ros in cancer: novel targets for anticancer therapy. J Cell Physiol (2016) 231(12):2570–81. doi: 10.1002/jcp.25349
34. Williamson J, Hughes CM, Cobley JN, Davison GW. The mitochondria-targeted antioxidant mitoq, attenuates exercise-induced mitochondrial DNA damage. Redox Biol (2020) 36:101673. doi: 10.1016/j.redox.2020.101673
35. Winterbourn CC. Hydrogen peroxide reactivity and specificity in thiol-based cell signalling. Biochem Soc Trans (2020) 48(3):745–54. doi: 10.1042/bst20190049
36. Moloney JN, Cotter TG. Ros signalling in the biology of cancer. Semin Cell Dev Biol (2018) 80:50–64. doi: 10.1016/j.semcdb.2017.05.023
37. Jelic MD, Mandic AD, Maricic SM, Srdjenovic BU. Oxidative stress and its role in cancer. J Cancer Res Ther (2021) 17(1):22–8. doi: 10.4103/jcrt.JCRT_862_16
38. Bao D, Zhao J, Zhou X, Yang Q, Chen Y, Zhu J, et al. Mitochondrial fission-induced mtdna stress promotes tumor-associated macrophage infiltration and hcc progression. Oncogene (2019) 38(25):5007–20. doi: 10.1038/s41388-019-0772-z
39. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates hif-1α Activity and il-1β Induction and is a critical determinant of the warburg effect in lps-activated macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2014.12.005
40. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. Pyruvate kinase M2 is a phd3-stimulated coactivator for hypoxia-inducible factor 1. Cell (2011) 145(5):732–44. doi: 10.1016/j.cell.2011.03.054
41. Al Haq AT, Tseng HY, Chen LM, Wang CC, Hsu HL. Targeting prooxidant mnsod effect inhibits triple-negative breast cancer (Tnbc) progression and M2 macrophage functions under the oncogenic stress. Cell Death Dis (2022) 13(1):49. doi: 10.1038/s41419-021-04486-x
42. Mao W, Xiong G, Wu Y, Wang C, St Clair D, Li JD, et al. Rorα Suppresses cancer-associated inflammation by repressing respiratory complex I-dependent ros generation. Int J Mol Sci (2021) 22(19):10665. doi: 10.3390/ijms221910665
43. Kuo CL, Chou HY, Chiu YC, Cheng AN, Fan CC, Chang YN, et al. Mitochondrial oxidative stress by lon-pycr1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett (2020) 474:138–50. doi: 10.1016/j.canlet.2020.01.019
44. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16(3):278–90. doi: 10.1038/s41589-019-0462-8
45. Zhu H, Jia Z, Li YR. Nrf2 signaling in macrophages. Reactive oxygen species (Apex NC) (2016) 2(6):417–20. doi: 10.20455/ros.2016.875
46. Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel nrf2-regulated genes by chip-seq: influence on retinoid X receptor alpha. Nucleic Acids Res (2012) 40(15):7416–29. doi: 10.1093/nar/gks409
47. Crisman E, Duarte P, Dauden E, Cuadrado A, Rodríguez-Franco MI, López MG, et al. Keap1-nrf2 protein-protein interaction inhibitors: design, pharmacological properties and therapeutic potential. Medicinal Res Rev (2023) 43(1):237–87. doi: 10.1002/med.21925
48. Srivastava S, Sinha D, Saha PP, Marthala H, D'Silva P. Magmas functions as a ros regulator and provides cytoprotection against oxidative stress-mediated damages. Cell Death Dis (2014) 5(8):e1394. doi: 10.1038/cddis.2014.355
49. Jubinsky PT, Messer A, Bender J, Morris RE, Ciraolo GM, Witte DP, et al. Identification and characterization of magmas, a novel mitochondria-associated protein involved in granulocyte-macrophage colony-stimulating factor signal transduction. Exp Hematol (2001) 29(12):1392–402. doi: 10.1016/s0301-472x(01)00749-4
50. Trus E, Basta S, Gee K. Who's in charge here? Macrophage colony stimulating factor and granulocyte macrophage colony stimulating factor: competing factors in macrophage polarization. Cytokine (2020) 127:154939. doi: 10.1016/j.cyto.2019.154939
51. Darcy PK, Neeson P, Yong CS, Kershaw MH. Manipulating immune cells for adoptive immunotherapy of cancer. Curr Opin Immunol (2014) 27:46–52. doi: 10.1016/j.coi.2014.01.008
52. Seledtsov VI, Goncharov AG, Seledtsova GV. Clinically feasible approaches to potentiating cancer cell-based immunotherapies. Hum Vaccines immunotherapeutics (2015) 11(4):851–69. doi: 10.1080/21645515.2015.1009814
53. Kono K, Salazar-Onfray F, Petersson M, Hansson J, Masucci G, Wasserman K, et al. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur J Immunol (1996) 26(6):1308–13. doi: 10.1002/eji.1830260620
54. Cheng CW, Kuo CY, Fan CC, Fang WC, Jiang SS, Lo YK, et al. Overexpression of lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis (2013) 4(6):e681. doi: 10.1038/cddis.2013.204
55. Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, et al. Nadph oxidase deficiency underlies dysfunction of aged cd8+ Tregs. J Clin Invest (2016) 126(5):1953–67. doi: 10.1172/jci84181
56. Bailly C. Regulation of pd-L1 expression on cancer cells with ros-modulating drugs. Life Sci (2020) 246:117403. doi: 10.1016/j.lfs.2020.117403
57. Yu X, Lao Y, Teng XL, Li S, Zhou Y, Wang F, et al. Senp3 maintains the stability and function of regulatory T cells via bach2 desumoylation. Nat Commun (2018) 9(1):3157. doi: 10.1038/s41467-018-05676-6
58. Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci United States America (2010) 107(41):17686–91. doi: 10.1073/pnas.1012016107
59. Guo Z, Wang G, Wu B, Chou WC, Cheng L, Zhou C, et al. Dcaf1 regulates treg senescence via the ros axis during immunological aging. J Clin Invest (2020) 130(11):5893–908. doi: 10.1172/jci136466
60. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
61. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell (2017) 168(3):344–61. doi: 10.1016/j.cell.2016.12.034
62. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci (2016) 41(3):274–86. doi: 10.1016/j.tibs.2015.11.012
63. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death differentiation (2016) 23(3):369–79. doi: 10.1038/cdd.2015.158
64. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
65. Cheng AN, Cheng LC, Kuo CL, Lo YK, Chou HY, Chen CH, et al. Mitochondrial lon-induced mtdna leakage contributes to pd-L1-mediated immunoescape via sting-ifn signaling and extracellular vesicles. J immunother Cancer (2020) 8(2):e001372. doi: 10.1136/jitc-2020-001372
66. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microrna-mediated metabolic shift. Oncogene (2019) 38(26):5158–73. doi: 10.1038/s41388-019-0782-x
67. Baig MS, Roy A, Rajpoot S, Liu D, Savai R, Banerjee S, et al. Tumor-derived exosomes in the regulation of macrophage polarization. Inflammation Res (2020) 69(5):435–51. doi: 10.1007/s00011-020-01318-0
68. Gregory CD, Dransfield I. Apoptotic tumor cell-derived extracellular vesicles as important regulators of the onco-regenerative niche. Front Immunol (2018) 9:1111. doi: 10.3389/fimmu.2018.01111
69. Wang Y, Branicky R, Noë A, Hekimi S. Superoxide dismutases: dual roles in controlling ros damage and regulating ros signaling. J Cell Biol (2018) 217(6):1915–28. doi: 10.1083/jcb.201708007
70. Xu D, Wu L, Yao H, Zhao L. Catalase-like nanozymes: classification, catalytic mechanisms, and their applications. Small (Weinheim an der Bergstrasse Germany) (2022) 18(37):e2203400. doi: 10.1002/smll.202203400
71. Lee YJ. Knockout mouse models for peroxiredoxins. Antioxidants (Basel Switzerland) (2020) 9(2):182. doi: 10.3390/antiox9020182
72. Szeliga M. Comprehensive analysis of the expression levels and prognostic values of prdx family genes in glioma. Neurochemistry Int (2022) 153:105256. doi: 10.1016/j.neuint.2021.105256
73. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ros: physiology and pathology. Trends Biochem Sci (2011) 36(1):30–8. doi: 10.1016/j.tibs.2010.07.007
74. Magalhaes-Novais S, Blecha J, Naraine R, Mikesova J, Abaffy P, Pecinova A, et al. Mitochondrial respiration supports autophagy to provide stress resistance during quiescence. Autophagy (2022) 18(10):2409–26. doi: 10.1080/15548627.2022.2038898
75. Kazakova E, Iamshchikov P, Larionova I, Kzhyshkowska J. Macrophage scavenger receptors: tumor support and tumor inhibition. Front Oncol (2022) 12:1096897. doi: 10.3389/fonc.2022.1096897
76. Yu C, Chen H, Du D, Lv W, Li S, Li D, et al. B-glucan from saccharomyces cerevisiae alleviates oxidative stress in lps-stimulated raw264.7 cells via dectin-1/nrf2/ho-1 signaling pathway. Cell Stress chaperones (2021) 26(4):629–37. doi: 10.1007/s12192-021-01205-5
77. Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, et al. Metabolic reprogramming in macrophage responses. biomark Res (2021) 9(1):1. doi: 10.1186/s40364-020-00251-y
78. Wang Y, Wang D, Yang L, Zhang Y. Metabolic reprogramming in the immunosuppression of tumor-associated macrophages. Chin Med J (2022) 135(20):2405–16. doi: 10.1097/cm9.0000000000002426
79. Muri J, Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol (2021) 21(6):363–81. doi: 10.1038/s41577-020-00478-8
80. Blouin CC, Pagé EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha. Blood (2004) 103(3):1124–30. doi: 10.1182/blood-2003-07-2427
81. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep (2016) 17(3):684–96. doi: 10.1016/j.celrep.2016.09.008
82. Liu S, Liu J, Ma Q, Cao L, Fattah RJ, Yu Z, et al. Solid tumor therapy by selectively targeting stromal endothelial cells. Proc Natl Acad Sci United States America (2016) 113(28):E4079–87. doi: 10.1073/pnas.1600982113
83. Kumar S, Mittal S, Gupta P, Singh M, Chaluvally-Raghavan P, Pradeep S. Metabolic reprogramming in tumor-associated macrophages in the ovarian tumor microenvironment. Cancers (2022) 14(21):5224. doi: 10.3390/cancers14215224
84. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res (2015) 3(11):1236–47. doi: 10.1158/2326-6066.Cir-15-0036
85. Xiang W, Shi R, Zhang D, Kang X, Zhang L, Yuan J, et al. Dietary fats suppress the peritoneal seeding of colorectal cancer cells through the tlr4/cxcl10 axis in adipose tissue macrophages. Signal transduction targeted Ther (2020) 5(1):239. doi: 10.1038/s41392-020-00327-z
86. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab (2013) 18(2):153–61. doi: 10.1016/j.cmet.2013.05.017
87. Xiang Y, Miao H. Lipid metabolism in tumor-associated macrophages. Adv Exp Med Biol (2021) 1316:87–101. doi: 10.1007/978-981-33-6785-2_6
88. Liu S, Zhang H, Li Y, Zhang Y, Bian Y, Zeng Y, et al. S100a4 enhances protumor macrophage polarization by control of ppar-Γ-dependent induction of fatty acid oxidation. J immunother Cancer (2021) 9(6):e002548. doi: 10.1136/jitc-2021-002548
89. Zhou D, Ji L, Chen Y. Tspo modulates il-4-induced microglia/macrophage M2 polarization via ppar-Γ Pathway. J Mol Neurosci MN (2020) 70(4):542–9. doi: 10.1007/s12031-019-01454-1
90. Puthenveetil A, Dubey S. Metabolic reprograming of tumor-associated macrophages. Ann Trans Med (2020) 8(16):1030. doi: 10.21037/atm-20-2037
91. Dubey S, Ghosh S, Goswami D, Ghatak D, De R. Immunometabolic attributes and mitochondria-associated signaling of tumor-associated macrophages in tumor microenvironment modulate cancer progression. Biochem Pharmacol (2023) 208:115369. doi: 10.1016/j.bcp.2022.115369
92. Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. Ripk3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res (2020) 8(5):710–21. doi: 10.1158/2326-6066.Cir-19-0261
93. Mojsilovic SS, Mojsilovic S, Villar VH, Santibanez JF. The metabolic features of tumor-associated macrophages: opportunities for immunotherapy? Analytical Cell Pathol (Amsterdam) (2021) 2021:5523055. doi: 10.1155/2021/5523055
94. Niu Z, Shi Q, Zhang W, Shu Y, Yang N, Chen B, et al. Caspase-1 cleaves pparγ for potentiating the pro-tumor action of tams. Nat Commun (2017) 8(1):766. doi: 10.1038/s41467-017-00523-6
95. Deng X, Zhang P, Liang T, Deng S, Chen X, Zhu L. Ovarian cancer stem cells induce the M2 polarization of macrophages through the pparγ and nf-Kb pathways. Int J Mol Med (2015) 36(2):449–54. doi: 10.3892/ijmm.2015.2230
96. He F, Chen Y, He D, He S. Usp14-mediated deubiquitination of sirt1 in macrophage promotes fatty acid oxidation amplification and M2 phenotype polarization. Biochem Biophys Res Commun (2023) 646:19–29. doi: 10.1016/j.bbrc.2022.12.076
97. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic kras protein. Autophagy (2020) 16(11):2069–83. doi: 10.1080/15548627.2020.1714209
98. Wu Q, Yan T, Chen Y, Chang J, Jiang Y, Zhu D, et al. Integrated analysis of expression and prognostic values of acyl-coa dehydrogenase short-chain in colorectal cancer. Int J Med Sci (2021) 18(16):3631–43. doi: 10.7150/ijms.63953
99. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
100. Lee LY, Oldham WM, He H, Wang R, Mulhern R, Handy DE, et al. Interferon-Γ Impairs human coronary artery endothelial glucose metabolism by tryptophan catabolism and activates fatty acid oxidation. Circulation (2021) 144(20):1612–28. doi: 10.1161/circulationaha.121.053960
101. Yerrapragada RM, Mampallil D. Interferon-Γ Detection in point of care diagnostics: short review. Talanta (2022) 245:123428. doi: 10.1016/j.talanta.2022.123428
102. Lin CY, Chen WL, Huang YC, Lim CL, Yang CH. Gum arabic in combination with ifn-Γ Promotes the M1 polarization in macrophage. Int J Biol macromolecules (2022) 209(Pt A):506–12. doi: 10.1016/j.ijbiomac.2022.04.024
103. Abdi K, Laky K, Abshari M, Hill EM, Lantz L, Singh NJ, et al. Dendritic cells trigger ifn-Γ Secretion by nk cells independent of il-12 and il-18. Eur J Immunol (2022) 52(9):1431–40. doi: 10.1002/eji.202149733
104. Zhao X, Peng T, Cao X, Hou Y, Li R, Han T, et al. In vivo G-csf treatment activates the gr-socs1 axis to suppress ifn-Γ Secretion by natural killer cells. Cell Rep (2022) 40(11):111342. doi: 10.1016/j.celrep.2022.111342
105. Friedmann Angeli JP, Xavier da Silva TN, Schilling B. Cd8(+) T cells puf(a)Ing the flames of cancer ferroptotic cell death. Cancer Cell (2022) 40(4):346–8. doi: 10.1016/j.ccell.2022.03.003
106. Chen D, Zhang X, Li Z, Zhu B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics (2021) 11(3):1016–30. doi: 10.7150/thno.51777
107. Hezaveh K, Shinde RS, Klötgen A, Halaby MJ, Lamorte S, Ciudad MT, et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity (2022) 55(2):324–40.e8. doi: 10.1016/j.immuni.2022.01.006
108. Liu PS, Chen YT, Li X, Hsueh PC, Tzeng SF, Chen H, et al. Cd40 signal rewires fatty acid and glutamine metabolism for stimulating macrophage anti-tumorigenic functions. Nat Immunol (2023) 24(3):452–62. doi: 10.1038/s41590-023-01430-3
109. Sun Y, Zheng Z, Zhang H, Yu Y, Ma J, Tang K, et al. Chemotherapeutic tumor microparticles combining low-dose irradiation reprogram tumor-promoting macrophages through a tumor-repopulating cell-curtailing pathway. Oncoimmunology (2017) 6(6):e1309487. doi: 10.1080/2162402x.2017.1309487
110. Altman BJ, Stine ZE, Dang CV. From krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(11):749. doi: 10.1038/nrc.2016.114
111. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. A-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol (2017) 18(9):985–94. doi: 10.1038/ni.3796
112. Cuschieri J, Maier RV. Oxidative stress, lipid rafts, and macrophage reprogramming. Antioxidants Redox Signaling (2007) 9(9):1485–97. doi: 10.1089/ars.2007.1670
113. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg cells promote the srebp1-dependent metabolic fitness of tumor-promoting macrophages via repression of cd8(+) T cell-derived interferon-Γ. Immunity (2019) 51(2):381–97.e6. doi: 10.1016/j.immuni.2019.06.017
114. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circulatory Physiol (2011) 301(6):H2181–90. doi: 10.1152/ajpheart.00554.2011
115. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ros promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting tfam-mediated mtdna maintenance. Theranostics (2021) 11(4):1845–63. doi: 10.7150/thno.50905
116. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett (2018) 592(5):728–42. doi: 10.1002/1873-3468.12956
117. Larionova I, Kazakova E, Patysheva M, Kzhyshkowska J. Transcriptional, epigenetic and metabolic programming of tumor-associated macrophages. Cancers (2020) 12(6):1411. doi: 10.3390/cancers12061411
118. Pritchard A, Tousif S, Wang Y, Hough K, Khan S, Strenkowski J, et al. Lung tumor cell-derived exosomes promote M2 macrophage polarization. Cells (2020) 9(5):1303. doi: 10.3390/cells9051303
119. Weiss JM, Davies LC, Karwan M, Ileva L, Ozaki MK, Cheng RY, et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest (2018) 128(9):3794–805. doi: 10.1172/jci99169
120. Zhang Q, Wang H, Mao C, Sun M, Dominah G, Chen L, et al. Fatty acid oxidation contributes to il-1β Secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol Immunol (2018) 94:27–35. doi: 10.1016/j.molimm.2017.12.011
121. Li F, Kitajima S, Kohno S, Yoshida A, Tange S, Sasaki S, et al. Retinoblastoma inactivation induces a protumoral microenvironment via enhanced ccl2 secretion. Cancer Res (2019) 79(15):3903–15. doi: 10.1158/0008-5472.Can-18-3604
122. Feng J, Chen Z, Ma Y, Yang X, Zhu Z, Zhang Z, et al. Akap1 contributes to impaired mtdna replication and mitochondrial dysfunction in podocytes of diabetic kidney disease. Int J Biol Sci (2022) 18(10):4026–42. doi: 10.7150/ijbs.73493
123. Yang Z, Min Z, Yu B. Reactive oxygen species and immune regulation. Int Rev Immunol (2020) 39(6):292–8. doi: 10.1080/08830185.2020.1768251
124. Maréchal L, Laviolette M, Rodrigue-Way A, Sow B, Brochu M, Caron V, et al. The cd36-pparγ Pathway in metabolic disorders. Int J Mol Sci (2018) 19(5):1529. doi: 10.3390/ijms19051529
125. Nassir F, Wilson B, Han X, Gross RW, Abumrad NA. Cd36 is important for fatty acid and cholesterol uptake by the proximal but not distal intestine. J Biol Chem (2007) 282(27):19493–501. doi: 10.1074/jbc.M703330200
126. Pillai SS, Pereira DG, Zhang J, Huang W, Beg MA, Knaack DA, et al. Contribution of adipocyte na/K-atpase A1/cd36 signaling induced exosome secretion in response to oxidized ldl. Front Cardiovasc Med (2023) 10:1046495. doi: 10.3389/fcvm.2023.1046495
127. Kadenbach B. Complex iv - the regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion (2021) 58:296–302. doi: 10.1016/j.mito.2020.10.004
128. Lapuente-Brun E, Moreno-Loshuertos R, Acín-Pérez R, Latorre-Pellicer A, Colás C, Balsa E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Sci (New York NY) (2013) 340(6140):1567–70. doi: 10.1126/science.1230381
129. Zhou J, Zheng S, Liu T, Liu Q, Chen Y, Tan D, et al. Il-1β from M2 macrophages promotes migration and invasion of escc cells enhancing epithelial-mesenchymal transition and activating nf-Kb signaling pathway. J Cell Biochem (2018) 119(8):7040–52. doi: 10.1002/jcb.26918
130. Youssef LA, Rebbaa A, Pampou S, Weisberg SP, Stockwell BR, Hod EA, et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood (2018) 131(23):2581–93. doi: 10.1182/blood-2017-12-822619
131. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatol (Baltimore Md) (2017) 66(2):449–65. doi: 10.1002/hep.29117
132. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discovery (2022) 8(1):43. doi: 10.1038/s41420-021-00807-3
133. Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J (2014) 457(3):415–24. doi: 10.1042/bj20130863
134. Zhao J, Lin X, Meng D, Zeng L, Zhuang R, Huang S, et al. Nrf2 mediates metabolic reprogramming in non-small cell lung cancer. Front Oncol (2020) 10:578315. doi: 10.3389/fonc.2020.578315
135. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun (2016) 7:11624. doi: 10.1038/ncomms11624
136. Samec M, Mazurakova A, Lucansky V, Koklesova L, Pecova R, Pec M, et al. Flavonoids attenuate cancer metabolism by modulating lipid metabolism, amino acids, ketone bodies and redox state mediated by nrf2. Eur J Pharmacol (2023) 949:175655. doi: 10.1016/j.ejphar.2023.175655
137. Nelson VL, Nguyen HCB, Garcìa-Cañaveras JC, Briggs ER, Ho WY, DiSpirito JR, et al. Pparγ Is a nexus controlling alternative activation of macrophages. Via Glutamine Metab Genes Dev (2018) 32(15-16):1035–44. doi: 10.1101/gad.312355.118
138. Ganta VC, Choi MH, Kutateladze A, Fox TE, Farber CR, Annex BH. A microrna93-interferon regulatory factor-9-immunoresponsive gene-1-itaconic acid pathway modulates M2-like macrophage polarization to revascularize ischemic muscle. Circulation (2017) 135(24):2403–25. doi: 10.1161/circulationaha.116.025490
139. Weiss JM, Palmieri EM, Gonzalez-Cotto M, Bettencourt IA, Megill EL, Snyder NW, et al. Itaconic acid underpins hepatocyte lipid metabolism in non-alcoholic fatty liver disease in male mice. Nat Metab (2023) 5(6):981–95. doi: 10.1038/s42255-023-00801-2
140. Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, et al. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating B-oxidation-dependent mitochondrial ros production. Cell Metab (2013) 18(2):265–78. doi: 10.1016/j.cmet.2013.06.018
141. Myers KV, Amend SR, Pienta KJ. Targeting tyro3, axl and mertk (Tam receptors): implications for macrophages in the tumor microenvironment. Mol Cancer (2019) 18(1):94. doi: 10.1186/s12943-019-1022-2
142. Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive Oxygen Species Modulate Macrophage Immunosuppressive Phenotype through the up-Regulation of Pd-L1. Proc Natl Acad Sci United States America (2019) 116(10):4326–35. doi: 10.1073/pnas.1819473116
143. Wu L, Chen F, Chang X, Li L, Yin X, Li C, et al. Combined cellular thermometry reveals that salmonella typhimurium warms macrophages by inducing a pyroptosis-like phenotype. J Am Chem Soc (2022) 144(42):19396–409. doi: 10.1021/jacs.2c07287
144. Dahlem C, Siow WX, Lopatniuk M, Tse WKF, Kessler SM, Kirsch SH, et al. Thioholgamide a, a new anti-proliferative anti-tumor agent, modulates macrophage polarization and metabolism. Cancers (2020) 12(5):1288. doi: 10.3390/cancers12051288
145. Hasan MN, Luo L, Ding D, Song S, Bhuiyan MIH, Liu R, et al. Blocking nhe1 stimulates glioma tumor immunity by restoring oxphos function of myeloid cells. Theranostics (2021) 11(3):1295–309. doi: 10.7150/thno.50150
146. Weiss F, Lauffenburger D, Friedl P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer (2022) 22(3):157–73. doi: 10.1038/s41568-021-00427-0
Keywords: tumor-associated macrophages, reactive oxygen species, fatty acid oxidation, tumor microenvironment, recruitment, polarization, antioxidant systems, metabolism
Citation: Teng Y, Xu L, Li W, Liu P, Tian L and Liu M (2023) Targeting reactive oxygen species and fat acid oxidation for the modulation of tumor-associated macrophages: a narrative review. Front. Immunol. 14:1224443. doi: 10.3389/fimmu.2023.1224443
Received: 17 May 2023; Accepted: 06 July 2023;
Published: 21 July 2023.
Edited by:
Sukh Mahendra Singh, Banaras Hindu University, IndiaReviewed by:
Armando Rojas, Catholic University of the Maule, ChileCopyright © 2023 Teng, Xu, Li, Liu, Tian and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linli Tian, dGlhbmxpbmxpNzhAMTYzLmNvbQ==; Ming Liu, bGl1bWluZ2VudEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.