 Junjie Cheng
Junjie Cheng Hanhai Zeng
Hanhai Zeng Huaijun Chen
Huaijun Chen Linfeng Fan
Linfeng Fan Chaoran Xu2
Chaoran Xu2- 1Intensive Care Unit, The Fourth Affiliated Hospital, School of Medicine, Zhejiang University, Yiwu, China
- 2Department of Neurosurgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Thrombocytopenia, characterized by a decrease in platelet count, is commonly observed in sepsis and COVID-19. In sepsis, thrombocytopenia can result from various mechanisms, including impaired platelet production in the bone marrow, accelerated platelet destruction due to increased inflammation, sequestration of platelets in the spleen, immune-mediated platelet destruction, or dysregulated host responses. Similarly, thrombocytopenia has been reported in COVID-19 patients, but the immune-related mechanisms underlying this association remain unclear. Notably, interventions targeting thrombocytopenia have shown potential for improving outcomes in both sepsis and COVID-19 patients. Understanding these mechanisms is crucial for developing effective treatments.
1 Introduction
Thrombocytopenia refers to a condition characterized by abnormally low levels of blood platelets—a vital component of the body’s clotting system. It can be triggered by various factors such as medications, infections, autoimmune diseases, and other health conditions (1). Thrombocytopenia can lead to symptoms like excessive bruising and bleeding and may result in severe complications such as an increased risk of stroke, heart attack, or death (1). Investigating the causes and exploring effective treatments for thrombocytopenia are essential for improving patient prognosis and quality of life.
Sepsis is a life-threatening condition characterized by excessive inflammation caused by infection. It has seen a significant increase in incidence—8.7% over recent decades—due to the body’s exaggerated response to infections (2). Coagulation disorders play a major role in sepsis-related mortality ranging from mild thrombocytopenia to fatal conditions like disseminated intravascular coagulation (DIC) (3). Platelets are crucial in sepsis, and thrombocytopenia serves as both a prognostic marker and an independent predictor of worse outcomes (4, 5). Notably, thrombocytopenia during sepsis is associated with increased overall 30-day mortality (6). Furthermore, platelets have the potential to be therapeutic targets and modulators of sepsis (7), highlighting the significance of understanding thrombocytopenia in sepsis.
In recent years, the COVID-19 pandemic has profoundly impacted healthcare systems and individuals’ health. Extensive research has shown that SARS-CoV-2—the virus causing COVID-19—affects various tissues and organs beyond the respiratory system. Studies have revealed that SARS-CoV-2 can directly or indirectly affect blood cells such as hematopoietic stem cells, megakaryocytes, and platelets (8, 9). Approximately one-quarter of COVID-19 patients develop thrombocytopenia Ashwell-Morell receptor, particularly within the first week after admission (8, 9). Thrombocytopenia in ICU-admitted COVID-19 patients has been associated with a significantly worse prognosis for severe cases (10), and even patients with normal platelet count at admission but developing thrombocytopenia during ICU stay exhibited lower survival rates compared to those without thrombocytopenia (11). Despite these observations, the mechanisms underlying COVID-19-associated thrombocytopenia remain poorly understood.
Therefore, this review aims to summarize the current knowledge regarding thrombocytopenia in sepsis and its association with COVID-19.
2 Mechanisms of thrombocytopenia in sepsis
Several complex mechanisms contribute to thrombocytopenia during sepsis. Dysregulated host responses, interactions with platelet receptors and complexes, and immune-mediated thrombocytopenia are among the identified mechanisms (12–14). However, the precise mechanisms behind these phenomena require further elucidation. Understanding these mechanisms is crucial for developing appropriate treatment strategies (15, 16).
2.1 Dysregulated host response
Thrombocytopenia can exacerbate the disturbed host response observed in sepsis, as evidenced by animal models and functional data (17). Disease severity strongly influences host response biomarkers in sepsis (12, 18). Interleukin mediators play a role in the host response, with elevated plasma levels of IL-6, IL-8, and IL-10 indicating cytokine network activation. Additionally, vascular endothelial activation (elevated soluble E-selectin and soluble ICAM-1) and compromised vascular integrity are observed. Coagulation abnormalities—prolonged aPTT and PT, increased plasma D-dimer levels, decreased antithrombin and protein C levels—contribute to thrombocytopenia in sepsis (12, 17, 18).
2.2 The interaction between platelet receptors and complexes
Platelet-receptor glycoprotein Ibalpha (GpIbalpha), part of the GP Ib-IX-V complex, and plasma von Willebrand factor (VWF) proteins play a role in hemostasis and the process of platelet attachment to the vascular endothelium (19). In septic shock, platelet inhibition prevents clotting, preserves endothelial function, and reduces tissue damage, potentially leading to improved outcomes (20).
The Toll-like receptors (TLRs) of the innate immune system recognize molecules of microbial origin by interacting with their transmembrane domains (21). TLR2, TLR4, and TLR9 are expressed on the surface of platelets. During sepsis, TLRs are activated, with TLR4 being involved in endotoxemia by recognizing lipopolysaccharide (LPS) proteins. Furthermore, LPS-induced thrombocytopenia is reduced because the expression of TLR4 is significantly decreased in activated platelets (13, 14). Studies have shown that the increase in cGMP in an LPS-induced TLR knockout model is also mediated through the TLR4 pathway and inhibited by anti-TLR4 blocking antibodies. TLR4 interacts with LPS to promote platelet secretion and enhance platelet aggregation (22).
Matrix metalloproteinases (MMPs) are enzymes that modulate extracellular matrix recycling. MMP-2 is a platelet agonist, while MMP-9 is a platelet activation inhibitor (Figure 1). Limited evidence suggests that toll-like receptor 4 (TLR-4) formation and platelet-leukocyte aggregates (PLA) may be associated with the development of sepsis-associated thrombocytopenia. However, current studies have found no difference in levels of MMP-2, MMP-9, and TLR-4 between donors with non-thrombotic and thrombotic sepsis. PLA formation is also increased in patients with thrombocytopenia. MMP-9 in platelets was detected using flow cytometry, gelatin enzyme spectrometry, and ELISA (23). Platelet consumption into the plasma may be the cause of thrombocytopenia in septic shock. The expression of MMP-9 in platelets increases during septic shock, suggesting that MMP-9 could be a potential therapeutic target for thrombocytopenia in sepsis (23).
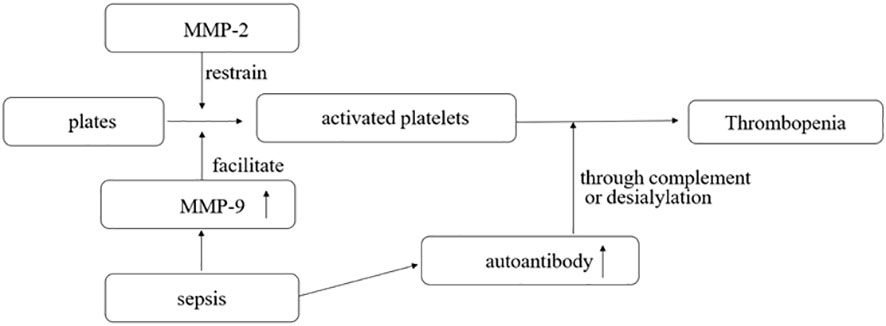
Figure 1 MMP-2 is a platelet agonist, and MMP-9 is a platelet activation inhibitor. In sepsis, the pathogen interacts with the body to produce a large number of antibodies, some of which can destroy activated platelets through complement or desialylation.
2.3 Immune-mediated thrombocytopenia
The mechanism of immune-mediated thrombocytopenia is complex and still under exploration. The classical explanation is that platelets bound to autoantibodies on their surfaces are destroyed in the spleen or liver through interaction with Fcγ receptors (24). Autoantibodies can mediate platelet destruction through complement activation or desialylation (25–27). Aged desialylated platelets are cleared by the liver Ashwell-Morell receptor (AMR) (28). Recent studies have shown that conditioning platelets with anti-GPIB antibodies activate platelets, leading to the translocation of neuraminidase-1 to the surface, which desialylated platelets for Fc-independent hepatic clearance via AMR in the liver (26) (Figure 2). Studies have demonstrated that abnormal T cells, including T-helper (Th) cell differentiation into Th1 and Th17 phenotypes and reduced regulatory T cells, contribute to varying degrees of platelet destruction (24). Additionally, CD8+ T cells, especially cytotoxic CD8+ T cells, cause thrombocytopenia through phagocytosis of splenic macrophages or dendritic cells. Activation of CD8+ T cells in the bone marrow can also lead to damage to megakaryocytes and inhibit platelet production (24). Liver macrophages clear sialic acid-free platelets through macrophage galactolectin (29). Whether macrophage clearance of platelet aggregates affects antibody-induced hepatocellular-mediated platelet clearance remains unclear. Further research is still needed to understand the mechanism of immune-mediated thrombocytopenia.
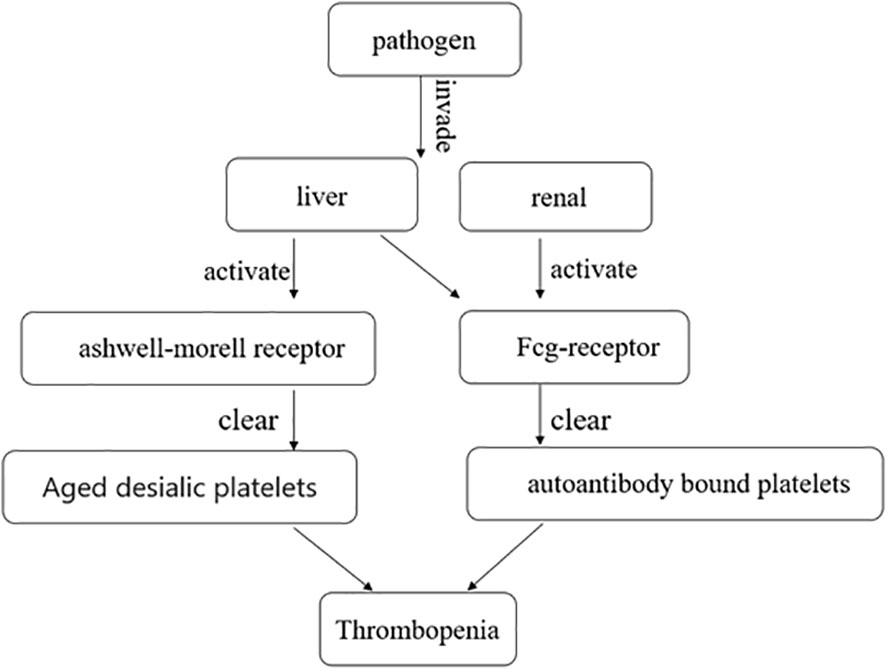
Figure 2 After the pathogen invades the liver and kidney, it clears aged desialic platelets and autoantibody-bound platelets by activating the Ashwell-Morell receptor and Fcg-receptor, respectively, causing thrombocytopenia.
It has been demonstrated that platelet levels of CD63 (LIMP-1), CD62P (P-selectin), and CD31 (platelet-endothelial cell adhesion molecule) are increased, while CD36 (GP IV) levels are significantly decreased during septic shock (30, 31). This suggests that platelet activation is mediated by interactions between platelets and leukocytes, endothelium, and activated endothelium, with platelet adhesion and aggregation being facilitated by these surface antigens (31).
Additionally, heparin-induced thrombocytopenia (HIT) is typically an immune-mediated thrombocytopenia (32) and is mediated by IgG antibodies that have specificity for platelet factor 4 antigen complexes (33). The consensus view is that these immune complexes activate platelets through Fc gamma RIIa receptors, leading to thrombocytopenia and thrombosis (34). Neutrophil extracellular traps (NETs) are complex structures composed of DNA and various proteins, including histones, neutrophil elastase, myeloperoxidase, and antimicrobial proteins. They have been increasingly reported in patients with infections and thrombosis associated with autoimmune and non-immune diseases (35, 36). When activated neutrophils release NETs, they form a mesh-like structure that traps and prevents platelets from binding to their receptors, resulting in thrombocytopenia. HIT immune complexes also activate neutrophils and induce the formation of NETs through MMP (33) (Figure 3).
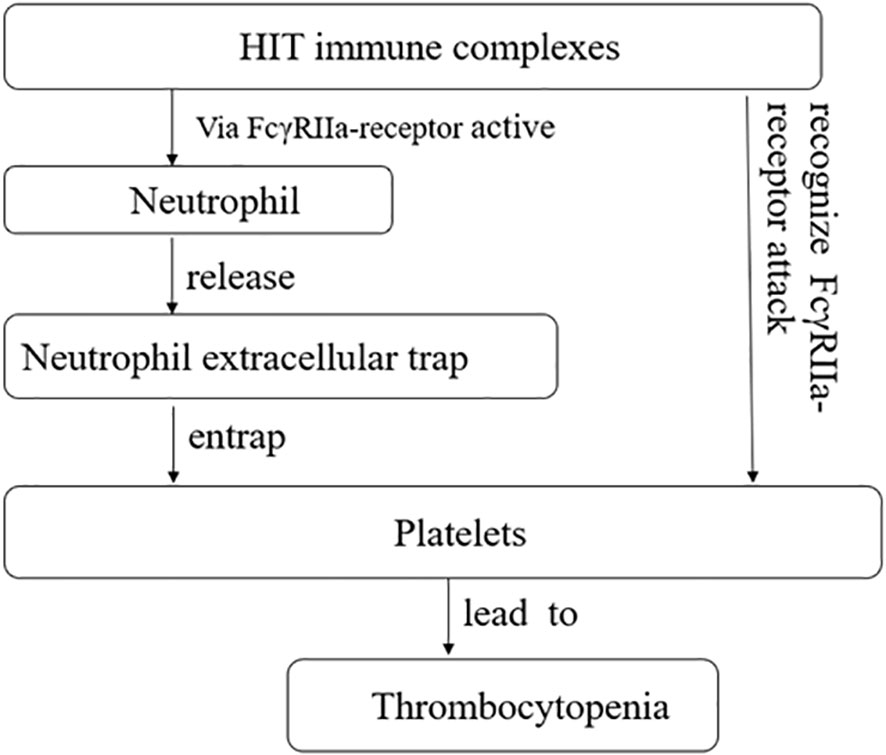
Figure 3 Heparin-induced thrombocytopenia (HIT) immune complexes via FcγRIIa-receptor active neutrophil, and further promotes the release of neutrophil extracellular trap, causing it to seize platelets, causing thrombocytopenia. HIT immune complexes attack plates through FcγRIIa-receptor, causing thrombocytopenia.
Additionally, activated platelet/neutrophil interactions mediated by P-selectin and PSGL-1 also induce NETosis (37). Research has shown that neutropenia eliminates thrombosis. Conversely, neutrophil reconstitution restores thrombus deposition and leads to thrombocytopenia (33). Furthermore, NETs can activate the complement system (38), leading to the generation of anaphylatoxins that can directly lyse platelets and further contribute to the development of thrombocytopenia.
Moreover, studies have found elevated levels of platelet-related IgG in sepsis with thrombocytopenia. Anti-platelet autoantibodies (anti-GP IIb/IX) have also been detected in a small number of patients, such as idiopathic thrombocytopenic purpura, suggesting a potential immune-related process for thrombocytopenia in sepsis (15). In mouse models of immune thrombocytopenia (ITP), monoclonal antibodies IgG, which bind to cell surface-associated antigens, prevent the development of immune thrombocytopenia (39, 40). It has been suggested that phagocytosis of reticuloendothelial cells in the bone marrow may also cause thrombocytopenia (41).
2.4 Disseminated intravascular coagulation
Disseminated intravascular coagulation (DIC) often occurs in patients with sepsis. It is mediated by pathogen-related molecules, leading to upregulated expression of tissue factors and inhibition of anticoagulation and fibrinolysis mechanisms (42). Platelet activation and white blood cell involvement are widely reported (43). The underlying disease process, such as trauma-induced mechanical endothelial injury, sepsis-related inflammation, obstetric complications, or cancer, results in excessive release of tissue factor, leading to overproduction of thrombin. Thrombin increases its production by converting soluble fibrinogen into insoluble fibrin chains and activating other thrombin and factors. Thrombin activates platelets as a key component of the primary hemostatic mechanism through the TF/VIIa axis and the involvement of Factor XIIa. Additionally, complement-mediated responses can affect lytic cells and/or bacterial pathogens through the release of damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) and other cellular components that promote blood coagulation (3).
Once a fibrinolytic clot begins to form, the fibrinolytic cascade is activated to counteract increased fibrin deposition and aggregation in microvessels. However, this fibrinolytic activity is impaired by anti-fibrinolytic components such as thrombin-activated fibrinolytic inhibitors (TAFI), plasminogen activator inhibitors (PAI-1), and other prethrombotic mediators (44, 45). Similarly, damage to the physiological anticoagulant pathway, including tissue factor pathway inhibitors (TFPI), antithrombin (AT), and activated protein C (APC), fails to inhibit the progressive procoagulant state to some extent. Currently, there is a bidirectional cross-talk between abnormal coagulation and inflammatory mediators, where inflammation effectively induces the coagulation cascade while abnormal coagulation profoundly alters and perpetuates inflammation (46, 47). In sepsis, major pro-inflammatory factors include IL-1, IL-6, TNF-α, elastase, and cathepsin (48). Recent research has elucidated the role of other thrombogenic inflammatory response factors such as NETs, extracellular vesicles, and shedding of endothelial calyx (49). DIC involves various mechanisms, including endothelial dysfunction and vascular endothelial injury, initiation of the coagulation pathway, platelet aggregation, dysfunction of the anticoagulation system, impaired fibrinolysis, activation of the complement system, upregulation of the inflammatory response, and many other factors that contribute to thrombocytopenia. Furthermore, it is assumed that all patients with sepsis have certain disorders of coagulation and clot activation regardless of obvious symptoms of disseminated intravascular coagulation (DIC). Moreover, DIC also correlates with disease severity (50).
2.5 Increased destruction of platelets
There are specific diseases that, when they cause sepsis, are also accompanied by thrombocytopenia. The mechanisms underlying this increased platelet destruction are not well understood. One example is hemolytic uremic syndrome, a thrombotic microvascular disease primarily caused by endothelial cell injury, leading to a series of syndromes (51). These factors collectively contribute to peripheral thrombocytopenia (51). Additionally, there are articles describing other causes of thrombocytopenia in sepsis, such as hypersplenism, bone marrow failure, heparin-induced thrombocytopenia (HIT), drug-induced thrombocytopenia (DIT), and blood dilution (42, 52, 53). Further research is needed to elucidate the mechanisms underlying these specific diseases.
3 Mechanisms of thrombocytopenia in COVID-19
Based on previous studies, thrombocytopenia has been identified as one of the most common symptoms of COVID-19 (54). Furthermore, it has been observed that thrombocytopenia is associated with a threefold increased risk for severe COVID-19 and an elevated risk of mortality (55). Although the exact mechanisms of thrombocytopenia in COVID-19 are still being researched, several potential causes have been suggested. One prominent cause is the cytokine storm, which occurs when the immune system produces an excessive amount of cytokines that can destroy platelets and contribute to thrombocytopenia. Additionally, other causes include direct viral-induced cytopenias and immune-mediated destruction of platelets (56–58) (Figure 4).
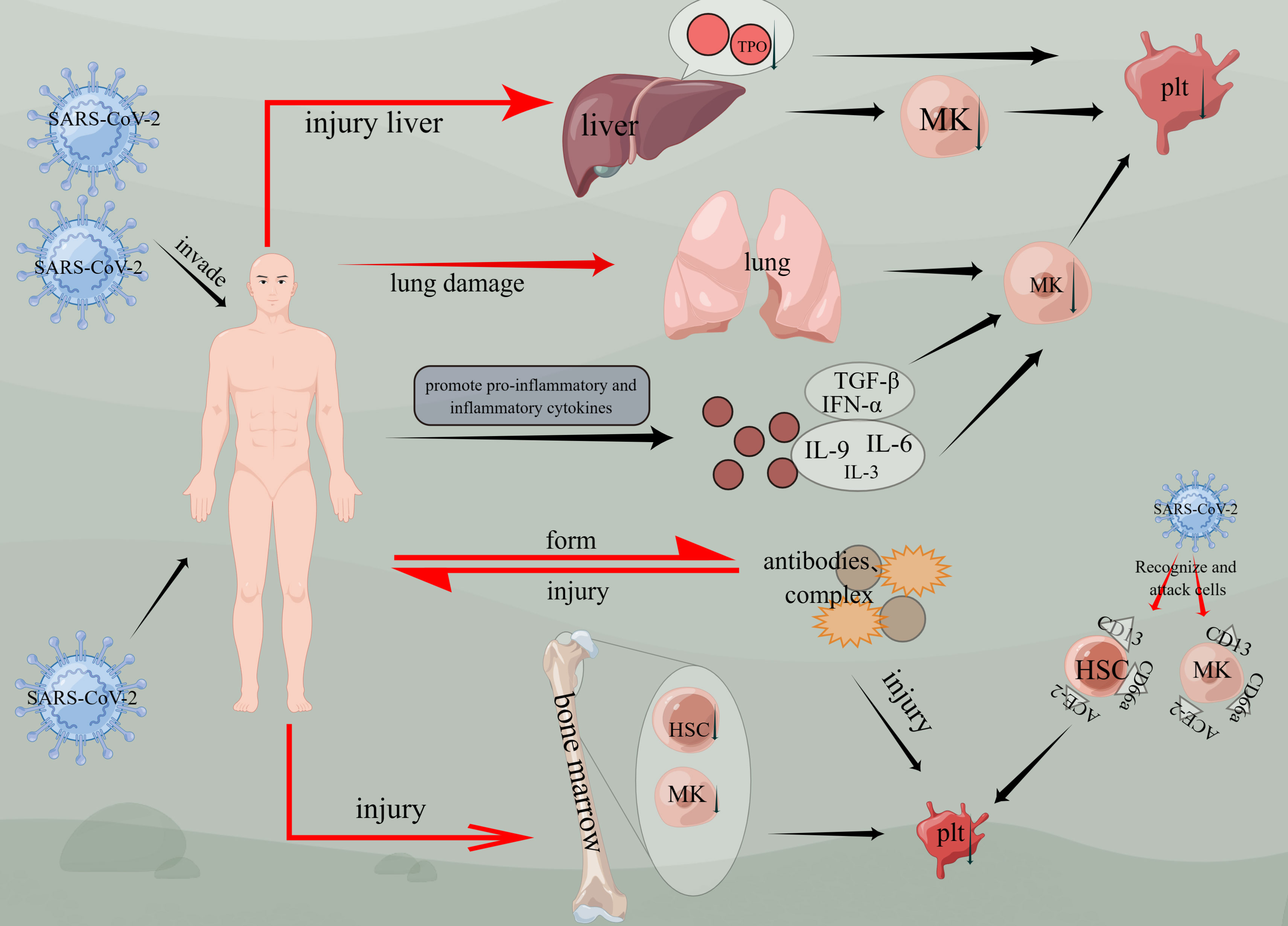
Figure 4 Mechanisms of thrombocytopenia caused by COVID-19. Viruses invade the body, cause liver and lung damage, and then damage megakaryocytes, causing thrombocytopenia. Viruses affect bone marrow hematopoietic function, affect hematopoietic stem cells and megakaryocyte generation, and cause thrombocytopenia. Thrombocytopenia is induced by CD13, ACE-2, and CD66a on hematopoietic stem cells or megakaryocytes. Inflammatory factors such as TGF-β, IFN-α, IL-3, IL-6, and IL-9 are activated, thus affecting the generation of megakaryocytes and the production of platelets. The virus enters the body and forms antibodies or compounds, and damages the body again. (figure by figdraw).
3.1 Thrombocytopenia by affecting platelet production in COVID-19
Platelets are produced from megakaryocytes (MK), which derive from hematopoietic stem cells (59, 60). Hematopoietic tissue expresses ACE2 receptors, which are utilized by SARS-CoV-2 and SARS-CoV to invade host cells and tissues (61). CD34+ stem cells, platelets, and MK cell lines express CD13 and EACAM1a (CD66a) (62, 63). Studies have shown that SARS-CoV infects human MK progenitor cells and CD34+ cells (58), exhibiting similar antigenic characteristics to human HCoV-229E (64, 65). HCoV-229E enters monocytes and macrophages via CD13 and CEACAM1a (CD66a) receptors, inducing cell apoptosis (62, 66, 67). Therefore, CD13 and CD66a are potential receptors for viral entry. The interaction between viruses and host cells in the production of specific antibodies (68) can trigger autoimmune antibodies that lead to specific cell death. Consequently, SARS-CoV-2 infection may induce the production of autoantibodies and immune complexes or directly attack hematopoietic stem cells (HSCs) or hematopoietic progenitor cells, resulting in thrombocytopenia. For instance, individuals with thrombocytopenia infected with HIV-1 produce antibodies that cross-react with HIV-1 gp160/120 antigen, leading to increased levels of circulating immune complexes (69, 70). The immune system identifies and targets platelets packaged by antibodies and immune complexes via cells of the reticuloendothelial system, thereby attacking platelets. Hematopoietic cells expressing similar antigens are also vulnerable to damage (63). In summary, viral infections generate antibodies and immune complexes that are recognized by the body’s immune system, causing thrombocytopenia by attacking blood-forming cells. Thus, it is plausible that SARS-CoV-2 might induce the production of autoantibodies and immune complexes or directly target HSCs or hematopoietic progenitor cells, ultimately resulting in thrombocytopenia.
In addition, dysfunction of the local renin-angiotensin system can result in an abnormal bone marrow microenvironment (71, 72). SARS-CoV granules and inflammatory cytokines such as IL-1β and TNF-α promote ACE2 removal from the cell surface. This weakens ACE2 function, leading to renin-angiotensin system dysfunction and increased inflammation (68). Megakaryocytes are also present in the blood vessels of lung tissue, where they contribute to platelet release (73). When the virus attacks lung tissue, it damages lung epithelial cells and endothelial cells, causing vascular leakage and releasing large amounts of pro-inflammatory cytokines and chemokines (68). SARS patients often exhibit diffuse alveolar injury characterized by pulmonary tissue congestion, pulmonary edema, alveolar hyaline membrane formation, and fibrosis (62, 64). Extensive damage to the alveoli leads to the destruction of lung capillaries and subsequent lysis of megakaryocytes, resulting in thrombocytopenia (73, 74). Consequently, elevated levels of chemokines, inflammatory factors, growth factors, and anti-inflammatory factors affect the hematopoietic microenvironment. Moreover, an imbalance in the bone marrow microenvironment may impair thrombopoietin production as well as megakaryocyte differentiation and maturation to some extent, thereby contributing to thrombocytopenia.
3.2 Cytokine storm and immune system-mediated thrombocytopenia in COVID-19
During the COVID-19 pandemic, it has been observed that COVID-19 is a highly coagulant disorder associated with inflammation commonly seen in ICU settings. This phenomenon is often referred to as “thromboinflammation” and is linked to cytokine storms (75). In the case of COVID-19, the cytokines involved in cytokine storm mainly include tumor necrosis factor-alpha (TNF-α), interleukin 1β (IL-1β), interleukin 6 (IL-6), interleukin 8 (IL-8), interleukin 10 (IL-10), interferon alpha (IFN-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (56). When these cytokines are stimulated and activated, they cause inflammation, damage tissues, and lead to organ dysfunction. Additionally, many cytokines such as IL-3, IL-6, IL-9, IL-1, and stem cell factors can stimulate MK production (76–79). However, tumor growth factor-beta (TGF-β), platelet factor 4, and interferon-alpha (IFN-α) inhibit MK production (80). Elevated TGF-β levels have been observed in SARS patients. Plasma from active SARS patients inhibited the formation of MK colony-forming units, which could be neutralized by anti-TGF-β antibodies. This suggests that virus-induced TGF-β-mediated cytokine storms inhibit megakaryocyte generation, resulting in thrombocytopenia (58). IFN-α induces the production of cytokine signal transduction inhibitors that inhibit the expression of megakaryocyte-regulating transcription factors to some extent. This directly inhibits thrombopoietin (TPO)-mediated MK growth. Ultrastructural research supports this mechanism (81, 82), further indicating insufficient platelet production.
In summary, the specific mechanism of cytokine storm causing thrombocytopenia in COVID-19 is still being investigated. However, it is evident that when infected with COVID-19, a cytokine storm can lead to platelet disorders or thrombocytopenia by either directly destroying platelets or stimulating the production of antibodies that bind and destroy platelets.
Additionally, cases of thrombus formation with thrombocytopenia have been reported following administration of the AstraZeneca recombinant adenovirus vector vaccine (ChAdOx1 nCov-19) (83). This phenomenon is referred to as vaccine-induced immune thrombotic thrombocytopenia (VITT) or thrombosis with thrombocytopenia syndrome (TTS) (84). Studies have shown that some mechanisms of VITT are similar to those of heparin-induced thrombocytopenia (HIT), particularly involving platelet-activating anti-platelet factor 4 (anti-PF4) antibodies (85). Although pathogenic platelet-activating antibodies in VITT caused by vaccination are not common (86), it is important to actively explore the relevant mechanisms and implement effective prevention measures.
In conclusion, there are multiple mechanisms contributing to thrombocytopenia in critically ill patients and those infected with COVID-19, often involving a combination of several factors. It is crucial to consider different pathophysiological mechanisms when treating thrombocytopenia to effectively address the condition.
4 Treatments
4.1 Treatments of thrombocytopenia in sepsis
In the context of sepsis, several potential therapeutic targets have been identified for managing thrombocytopenia. Interleukin-11 (IL-11) has been shown to effectively prevent and treat chemotherapy-associated thrombocytopenia by increasing the production and differentiation of megakaryocytes. Studies have demonstrated that sepsis patients treated with IL-11 have a lower mortality rate (87). However, the use of IL-11 is limited due to serious side effects and is no longer used in clinical practice.
Another potential treatment option is recombinant human thrombopoietin, which has shown promising results in improving platelet counts more rapidly to normal levels and reducing the need for platelet transfusions in sepsis patients (88).
An animal study revealed that platelet granase B-mediated apoptosis occurs in the spleen and lung during sepsis. The progression of sepsis was found to be slowed down by inhibiting granase B using the platelet GPIIb/IIIa receptor inhibitor etibatitide both in vitro and in vivo. Inhibitors of GPIIb/IIIa receptors and other antiplatelet drugs delayed survival in mice with sepsis (89).
TAK-242, a compound that binds to TLR4 and inhibits lipopolysaccharide (LPS) activation, has shown potential as a toll-like receptor 4 (TLR4) inhibitor for treating immune factor-induced thrombocytopenia in sepsis (90, 91). Clinical studies using anti-TLR4 antibodies have demonstrated increased survival rates in mice with LPS endotoxemia (92). Additionally, TLR3, TLR5 antagonists, or TLR9 agonists have also improved survival rates in mice with LPS endotoxemia, suggesting their potential use in treating sepsis-associated thrombocytopenia (93, 94).
4.2 Treatments of thrombocytopenia in COVID-19
In the context of COVID-19, several treatment options have shown promise for managing thrombocytopenia. Tocilizumab (TCZ), a recombinant humanized monoclonal antibody against the interleukin-6 (IL-6) receptor, has demonstrated significant efficacy in treating cytokine release syndrome in COVID-19 patients. It binds to membranous IL-6R (mIL-6R) and soluble IL-6R (sIL-6R) (95). An Italian study reported that intravenous tocilizumab significantly improved the prognosis of ICU patients (96). Anakinra, an antagonist of IL-1R, has been suggested to block the secretion of IL-1β by macrophages, preventing tissue damage and inhibiting excessive platelet accumulation by blocking endothelial cell exposure and coagulation cascade propagation. Notably, patients treated with IL-1R inhibitors have shown better outcomes compared to those treated with IL-6R inhibitors (97).
Complement inhibitors have also shown potential for managing thrombocytopenia in COVID-19. Inhibition of the complement system can help inhibit abnormal activation of the complement cascade and thrombotic microangiopathies. Studies using eculizumab, a C5-blocking agent, demonstrated a rapid reduction in lactic acid levels, improvement in hypoxia, restoration of platelet counts, and improved prothrombin time (PT) within 15 days (98, 99). AMY-101 has been suggested as another complement inhibitor that follows a similar action to eculizumab in inhibiting hyperinflammatory states (100).
Inhibiting chemokines and chemokine receptors may contribute to the recovery of platelet numbers in COVID-19 patients. The anti-CCR5 antibody leronlimab has shown efficacy in reducing IL-6 levels, restoring T cell populations, reducing inflammatory responses, and indirectly mitigating virus-induced damage (101). Further exploration is needed to determine if leronlimab can effectively treat thrombocytopenia caused by COVID-19.
Thrombopoietin receptor agonists (TPO-RAs) are often used to treat COVID-19-related immune thrombocytopenias (ITPs). However, their use carries a risk of thrombosis (102, 103). Steroids, such as corticosteroids, have been used to treat COVID-19-related ITPs and have shown positive effects in reducing systemic inflammatory response and improving patient outcomes (104). A combination therapy using dexamethasone and intravenous immunoglobulin (IVIG) has demonstrated increased platelet counts within 12 hours after treatment and improved bleeding control and oxygenation in severe COVID-19 patients with ITP (105).
Recombinant human ACE2 (hrsACE2), which binds to viral spike proteins, has been explored as a therapeutic option for preventing tissue damage. Intravenous administration of hrsACE2 for seven days in severe COVID-19 patients has been shown to significantly reduce angiotensin II levels, inhibit IL-6 and IL-8-mediated inflammatory response, alleviate organ damage caused by SARS-CoV-2, and indirectly improve thrombocytopenia (106).
By targeting these receptors and utilizing specific drugs, it is possible to inhibit the process of thrombocytopenia in sepsis and COVID-19. Understanding these mechanisms can aid in the development of effective prevention strategies that improve outcomes for sepsis and COVID-19 patients.
5 Conclusion
Thrombocytopenia, characterized by low platelet count, is a common symptom observed in both sepsis and COVID-19 patients. The exact mechanisms underlying thrombocytopenia in these conditions are still being researched, but several potential causes have been identified. In sepsis, cytokine storm plays a significant role in inducing thrombocytopenia. Excessive production of cytokines can directly destroy platelets or stimulate the production of antibodies that bind and destroy platelets. Therapeutic targets for managing sepsis-associated thrombocytopenia include IL-11 and recombinant human thrombopoietin, which have shown promising results in improving platelet counts. Additionally, inhibitors targeting TLR4 and GPIIb/IIIa receptors have demonstrated potential in animal studies. In this context, cytokine release syndrome contributes to thrombocytopenia. Drugs like tocilizumab (TCZ) and anakinra have shown efficacy in managing cytokine release syndrome by targeting IL-6 and IL-1 pathways. Complement inhibitors such as eculizumab have demonstrated positive outcomes in reducing inflammation and improving platelet counts. Chemokine inhibitors and thrombopoietin receptor agonists are also being explored as treatment options. However, it is important to maintain a balanced view when considering these treatments. Some therapeutic options come with limitations or risks. For example, IL-11 has serious side effects that restrict its clinical use, while TPO-RAs carry a risk of thrombosis. Furthermore, the effectiveness of certain drugs like leronlimab in treating thrombocytopenia caused by COVID-19 requires further investigation. And looking ahead, continued research is necessary to gain a comprehensive understanding of the mechanisms underlying thrombocytopenia in sepsis and COVID-19. This will enable the development of more targeted and effective treatments. It is crucial to consider the potential benefits and risks associated with each therapeutic approach, taking into account individual patient characteristics and disease severity. While progress has been made in identifying therapeutic targets for thrombocytopenia in sepsis and COVID-19, further research is needed to optimize treatment strategies. A critical evaluation of available options will help ensure that interventions are balanced, comprehensive, and tailored to the specific needs of patients. Prospective studies should focus on identifying biomarkers for early detection, elucidating the interplay between platelets and immune responses, and evaluating the efficacy of targeted therapies.
Author contributions
ML and HZ contributed to the conception and design of the study. HC, LF, CX, HH, and TT have conducted a literature review and analysis. JC wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet (2018) 392:75–87. doi: 10.1016/S0140-6736(18)30696-2
3. Giustozzi M, Ehrlinder H, Bongiovanni D, Borovac JA, Guerreiro RA, Gasecka A, et al. Coagulopathy and sepsis: Pathophysiology, clinical manifestations, and treatment. Blood Rev (2021) 50:100864. doi: 10.1016/j.blre.2021.100864
4. Vardon-Bounes F, Ruiz S, Gratacap MP, Garcia C, Payrastre B, Minville V. Platelets are critical key players in sepsis. Int J Mol Sci (2019) 20(14):8. doi: 10.3390/ijms20143494
5. Thiery-Antier N, Binquet C, Vinault S, Meziani F, Boisrame-Helms J, Quenot JP, et al. Is thrombocytopenia an early prognostic marker in septic shock? Crit Care Med (2016) 44:764–72. doi: 10.1097/CCM.0000000000001520
6. Schupp T, Weidner K, Rusnak J, Jawhar S, Forner J, Dulatahu F, et al. Diagnostic and prognostic role of platelets in patients with sepsis and septic shock. Platelets (2023) 34:2131753. doi: 10.1080/09537104.2022.2131753
7. Greco E, Lupia E, Bosco O, Vizio B, Montrucchio G. Platelets and multi-organ failure in sepsis. Int J Mol Sci (2017) 18(10):5. doi: 10.3390/ijms18102200
8. Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, et al. China med treatment expert, clinical characteristics of coronavirus disease 2019 in China. New Engl J Med (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
9. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
10. Rampotas A, Pavord S. Platelet aggregates, a marker of severe COVID-19 disease. J Clin Pathol (2021) 74:750–1. doi: 10.1136/jclinpath-2020-206933
11. Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PloS Biol (2021) 19(2):5. doi: 10.1371/journal.pbio.3001109
12. Parlato M, Cavaillon JM. Host response biomarkers in the diagnosis of sepsis: a general overview. Methods Mol Biol (2015) 1237:149–211. doi: 10.1007/978-1-4939-1776-1_15
13. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol (2005) 83:196–8. doi: 10.1111/j.1440-1711.2005.01314.x
14. Andonegui G, Kerfoot SM, McNagny K, Ebbert KVJ, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood (2005) 106:2417–23. doi: 10.1182/blood-2005-03-0916
15. Stéphan F CM, Kaplan C, Maillet J, Novara A, Fagon J, Bonnet F. Autoantibodies against platelet glycoproteins in critically ill patients with thrombocytopenia. Am J Med (2000) 108(7):554–60. doi: 10.1016/s0002-9343(00)00332-6
16. Bedet A, Razazi K, Boissier F, Surenaud M, Hue S, Giraudier S, et al. Mechanisms of thrombocytopenia during septic shock: A multiplex cluster analysis of endogenous sepsis mediators. Shock (2018) 49:641–8. doi: 10.1097/SHK.0000000000001015
17. Claushuis TA, van Vught LA, Scicluna BP, Wiewel MA, Klein Klouwenberg PM, Hoogendijk AJ, et al. Risk Stratification of Sepsis, Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood (2016) 127:3062–72. doi: 10.1182/blood-2015-11-680744
18. Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care (2010) 14:9–11. doi: 10.1186/cc8872
19. Huizinga EG, Tsuji S, Romijn RAP, Schiphorst ME, de Groot PG, Sixma JJ, et al. Structures of glycoprotein Ib alpha and its complex with von Willebrand factor A1 domain. Science (2002) 297:1176–9. doi: 10.1126/science.107355
20. Pu Q, Wiel E, Corseaux D, Bordet R, Azrin MA, Ezekowitz MD, et al. Beneficial effect of glycoprotein IIb/IIIa inhibitor (AZ-1) on endothelium in Escherichia coli endotoxin-induced shock. Crit Care Med (2001) 29:1181–8. doi: 10.1097/00003246-200106000-00019
21. Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun (2009) 388:621–5. doi: 10.1016/j.bbrc.2009.08.062
22. Zhang GY, Han JY, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the cGMP-Dependent Protein Kinase Pathway. J Immunol (2009) 182:7997–8004. doi: 10.4049/jimmunol.0802884
23. Larkin CM, Hante NK, Breen EP, Tomaszewski KA, Eisele S, Radomski MW, et al. Role of matrix metalloproteinases 2 and 9, toll-like receptor 4 and platelet-leukocyte aggregate formation in sepsis-associated thrombocytopenia. PloS One (2018) 13:e0196478. doi: 10.1371/journal.pone.0196478
24. Zufferey A, Kapur R, Semple JW. Pathogenesis and therapeutic mechanisms in immune thrombocytopenia (ITP). J Clin Med (2017) 6(2):3–4. doi: 10.3390/jcm6020016
25. Wang W, Irani RA, Zhang YJ, Ramin SM, Blackwell SC, Tao LJ, et al. Autoantibody-mediated complement C3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension (2012) 60:712. doi: 10.1161/HYPERTENSIONAHA.112.191817
26. Li J, van der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun (2015) 6:2. doi: 10.1038/ncomms8737
27. Li J, Callum JL, Lin YL, Zhou Y, Zhu GH, Ni HY. Severe platelet desialylation in a patient with glycoprotein Ib/IX antibody-mediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica (2014) 99:E61–3. doi: 10.3324/haematol.2013.102897
28. Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falai H, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med (2015) 21:47–54. doi: 10.1038/nm.3770
29. Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira F, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med (2020) 217(4):2. doi: 10.1084/jem.20190723
30. van Velzen JF, Laros-van Gorkom BAP, Pop GAM, van Heerde WL. Multicolor flow cytometry for evaluation of platelet surface antigens and activation markers. Thromb Res (2012) 130:92–8. doi: 10.1016/j.thromres.2012.02.041
31. Salat A, Bodingbauer G, Boehm D, Murabito M, Tochkow E, Sautner T, et al. Changes of platelet surface antigens in patients suffering from abdominal septic shock. Thromb Res (1999) 95:289–94. doi: 10.1016/S0049-3848(99)00046-8
32. Fahey VA. Heparin-induced thrombocytopenia. J Vasc Nurs Off Publ Soc Peripheral Vasc Nurs (1995) 13:112–6. doi: 10.1016/S1062-0303(05)80003-2
33. Perdomo J, Leung HHL, Ahmadi Z, Feng Y, Passam FH, Chong BH. Neutrophil activation and netosis are the key drivers of thrombosis in heparin-induced thrombocytopenia. Blood (2018) 132:378–8. doi: 10.1182/blood-2018-99-116421
34. Warkentin TE. HIT: still stringing us along. Blood (2020) 135:1193–4. doi: 10.1182/blood.2020005157
35. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The emerging role of neutrophil extracellular traps (NETs) in tumor progression and metastasis. Front Immunol (2020) 11:1749. doi: 10.3389/fimmu.2020.01749
36. Wigerblad G, Kaplan MJ. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat Rev Immunol (2022) 23(5):274–88. doi: 10.1038/s41577-022-00787-0
37. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
38. Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol (2015) 181:518–27. doi: 10.1111/cei.12654
39. Crow AR, Song S, Semple JW, Freedman J, Lazarus AH. IVIg inhibits reticuloendothelial system function and ameliorates murine passive-immune thrombocytopenia independent of anti-idiotype reactivity. Br J Haematology (2001) 115:679–86. doi: 10.1046/j.1365-2141.2001.03136.x
40. Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science (2001) 291:484–6. doi: 10.1126/science.291.5503.484
41. Francois B, Trimoreau F, Vignon P, Fixe P, Praloran V, Gastinne H. Thrombocytopenia in the sepsis syndrome: Role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med (1997) 103:114–20. doi: 10.1016/S0002-9343(97)00136-8
42. Bibas M. Bleeding and coagulopathies in critical care. New Engl J Med (2014) 370:2152–2. doi: 10.1056/NEJMc1403768
43. Johansson D, Shannon O, Rasmussen M. Platelet and neutrophil responses to gram positive pathogens in patients with bacteremic infection. PloS One (2011) 6(11):4–5. doi: 10.1371/journal.pone.0026928
44. Hayakawa M, Sawamura A, Gando S, Jesmin S, Naito S, Ieko M, et al. and insufficient activation of fibrinolysis by both plasmin and neutrophil elastase promote organ dysfunction in disseminated intravascular coagulation associated with sepsis. Thromb Res (2012) 130:906–13. doi: 10.1016/j.thromres.2012.01.015
45. Gando S. Role of fibrinolysis in sepsis. Semin Thromb Hemostasis (2013) 39:392–9. doi: 10.1055/s-0033-1334140
46. Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation (2004) 109:2698–704. doi: 10.1161/01.CIR.0000131660.51520.9A
47. Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res (2016) 118:1392–408. doi: 10.1161/CIRCRESAHA.116.306853
48. Lupu F, Keshari RS, Lambris JD, Coggeshall KM. Crosstalk between the coagulation and complement systems in sepsis. Thromb Res (2014) 133:S28–31. doi: 10.1016/j.thromres.2014.03.014
49. Iba T, Levy JH, Raj A, Warkentin TE. Advance in the management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J Clin Med (2019) 8(5):2–5. doi: 10.3390/jcm8050728
50. Kinasewitz GT, Yan SB, Basson B, Comp P, Russell JA, Cariou A, et al. Universal changes in biomarkers of coagulation and inflammation occur in patients with severe sepsis, regardless of causative micro-organism ISRCTN74215569. Crit Care (2004) 8:R82–90. doi: 10.1186/cc2459
51. Fakhouri F, Zuber J, Fremeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet (2017) 390:681–96. doi: 10.1016/S0140-6736(17)30062-4
52. Lee KH, Hui KP, Tan WC. Thrombocytopenia in sepsis: a predictor of mortality in the intensive care unit. Singapore Med J (1993) 34(3):245–6.
53. Stephan F, Hollande J, Richard O, Cheffi A, Maier-Redelsperger M, Flahault A. Thrombocytopenia in a surgical ICU. Chest (1999) 115:1363–70. doi: 10.1378/chest.115.5.1363
54. Al-Tawfiq JA, Hinedi K, Abbasi S, Babiker M, Sunji A, Eltigani M. Hematologic, hepatic, and renal function changes in hospitalized patients with Middle East respiratory syndrome coronavirus. Int J Lab Hematol (2017) 39:272–8. doi: 10.1111/ijlh.12620
55. Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin Chim Acta (2020) 506:145–8. doi: 10.1016/j.cca.2020.03.022
56. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
57. Yang M, Ng MH, Li CK. Thrombocytopenia in patients with severe acute respiratory syndrome (review). Hematol (Amsterdam Netherlands) (2005) 10:101–5. doi: 10.1080/10245330400026170
58. Yang M, Ng MH, Li CK, Chan PK, Liu C, Ye JY, et al. Thrombopoietin levels increased in patients with severe acute respiratory syndrome. Thromb Res (2008) 122:473–7. doi: 10.1016/j.thromres.2007.12.021
59. Drissen R, Buza-Vidas N, Woll P, Thongjuea S, Gambardella A, Giustacchini A, et al. Distinct myeloid progenitor-differentiation pathways identified through single-cell RNA sequencing. Nat Immunol (2016) 17:666. doi: 10.1038/ni.3412
60. Kaushansky K. Historical review: megakaryopoiesis and thrombopoiesis. Blood (2008) 111:981–6. doi: 10.1182/blood-2007-05-088500
61. Mercurio I, Tragni V, Busto F, De Grassi A, Pierri CL. Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. Cell Mol Life Sci (2021) 78:1501–22. doi: 10.1007/s00018-020-03580-1
62. Yang M, Li CK, Li K, Hon KLE, Ng MHL, Chan PKS, et al. Hematological findings in SARS patients and possible mechanisms (Review). Int J Mol Med (2004) 14:311–5. doi: 10.3892/ijmm.14.2.311
63. Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci United States America (2004) 101:15748–53. doi: 10.1073/pnas.0403812101
64. Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet (2003) 361:1319–25. doi: 10.1016/S0140-6736(03)13077-2
65. Yu CJ, Chen YC, Hsiao CH, Kuo TC, Chang SC, Lu CY, et al. Identification of a novel protein 3a from severe acute respiratory syndrome coronavirus. FEBS Lett (2004) 565:111–6. doi: 10.1016/j.febslet.2004.03.086
66. Nomura R, Kiyota A, Suzaki E, Kataoka K, Senda T, Fujimoto T. Human coronavirus 229E binds to CD13 in raft and enters the cell through caveolae. Anatomical Sci Int (2004) 79:287. doi: 10.1128/JVI.78.16.8701-8708.2004
67. Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and Malignant tissues. Semin Cancer Biol (1999) 9:67–81. doi: 10.1006/scbi.1998.0119
68. Fu YJ, Cheng YX, Wu YT. Understanding SARS-coV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virologica Sin (2020) 35:266–71. doi: 10.1007/s12250-020-00207-4
69. Scaradavou A. HIV-related thrombocytopenia. Blood Rev (2002) 16:73–6. doi: 10.1054/blre.2001.0188
70. Nardi M, Tomlinson S, Greco MA, Karpatkin S. Complement-independent, peroxide-induced antibody lysis of platelets in HIV-1-related immune thrombocytopenia. Cell (2001) 106:551–61. doi: 10.1016/S0092-8674(01)00477-9
71. Strawn WB, Richmond RS, Tallant EA, Gallagher PE, Ferrario CM. Renin-angiotensin system expression in rat bone marrow haematopoietic and stromal cells. Br J Haematology (2004) 126:120–6. doi: 10.1111/j.1365-2141.2004.04998.x
72. Haznedaroglu IC, Malkan UY. Local bone marrow renin-angiotensin system in the genesis of leukemia and other malignancies. Eur Rev Med Pharmacol Sci (2016) 20(19):4089–111.
73. Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature (2017) 544:105–9. doi: 10.1038/nature21706
74. Weibel ER. Oxygen effect on lung cells. Arch Internal Med (1971) 128:54–6. doi: 10.1001/archinte.1971.00310190058005
75. Bikdeli B, Madhavan MV, Gupta A, Jimenez D, Burton JR, Nigoghossian CD, et al. Pharmacological agents targeting thromboinflammation in COVID-19: review and implications for future research. Thromb Haemostasis (2020) 120:1004–24. doi: 10.1055/s-0040-1713152
76. Sugimoto N, Eto K. Platelet production from induced pluripotent stem cells. J Thromb Haemost (2017) 15:1717–27. doi: 10.1111/jth.13736
77. Monroy RL, Davis TA, Donahue RE, MacVittie TJ. In vivo stimulation of platelet production in a primate model using IL-1 and IL-3. Exp Hematol (1991) 19(7):629–35.
78. Williams N, Bertoncello I, Jackson H, Arnold J, Kavnoudias H. The role of interleukin 6 in megakaryocyte formation, megakaryocyte development and platelet production. Ciba Foundation symposium (1992) 167:160–70; discussion 170-3. doi: 10.1002/9780470514269.ch10
79. Cortin V, Garnier A, Pineault N, Lemieux R, Boyer L, Proulx C. Efficient in vitro megakaryocyte maturation using cytokine cocktails optimized by statistical experimental design. Exp Hematol (2005) 33:1182–91. doi: 10.1016/j.exphem.2005.06.020
80. Behrens K, Alexander WS. Cytokine control of megakaryopoiesis. Growth Factors (2018) 36:89–103. doi: 10.1080/08977194.2018.1498487
81. Yamane A, Nakamura T, Suzuki H, Ito M, Ohnishi Y, Ikeda Y, et al. Interferon-alpha 2b-induced thrombocytopenia is caused by inhibition of platelet production but not proliferation and endomitosis in human megakaryocytes. Blood (2008) 112:542–50. doi: 10.1182/blood-2007-12-125906
82. Wang Q, Fox NE, Kaushansky K. Interferon-alpha directly inhibits thrombopoietin-induced megakaryocyte proliferation and differentiation. Zhonghua Xue Ye Xue Za Zhi (2001) 22(6):296–9.
83. Sadoff J, Davis K, Douoguih M. Thrombotic thrombocytopenia after ad26.COV2.S vaccination - response from the manufacturer. New Engl J Med (2021) 384:1965–6. doi: 10.1056/NEJMc2106075
84. Elsheikh S, Lip GYH. Editorial commentary: COVID-19 and COVID-19 vaccination: Observations on thrombosis and thrombocytopenia. Trends Cardiovasc Med (2022) 32:257–8. doi: 10.1016/j.tcm.2022.03.002
85. Iba T, Levy JH. Thrombosis and thrombocytopenia in COVID-19 and after COVID-19 vaccination. Trends Cardiovasc Med (2022) 32:249–56. doi: 10.1016/j.tcm.2022.02.008
86. Thiele T, Ulm L, Holtfreter S, Schonborn L, Kuhn SO, Scheer C, et al. Frequency of positive anti-PF4/polyanion antibody tests after COVID-19 vaccination with ChAdOx1 nCoV-19 and BNT162b2. Blood (2021) 138:299–303. doi: 10.1182/blood.2021012217
87. Wan B, Zhang H, Fu HY, Chen YK, Yang LP, Yin JT, et al. Recombinant human interleukin-11 (IL-11) is a protective factor in severe sepsis with thrombocytopenia: A case-control study. Cytokine (2015) 76:138–43. doi: 10.1016/j.cyto.2015.08.001
88. Wu Q, Ren JA, Wu XW, Wang GF, Gu GS, Liu S, et al. Recombinant human thrombopoietin improves platelet counts and reduces platelet transfusion possibility among patients with severe sepsis and thrombocytopenia: A prospective study. J Crit Care (2014) 29:362–6. doi: 10.1016/j.jcrc.2013.11.023
89. Sharron M, Hoptay CE, Wiles AA, Garvin LM, Geha M, Benton AS, et al. Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PloS One (2012) 7(7):6. doi: 10.1371/journal.pone.0041549
90. Samarpita S, Kim JY, Rasool MK, Kim KS. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res Ther (2020) 22(1):8–9. doi: 10.1186/s13075-020-2097-2
91. Wittebole X, Castanares-Zapatero D, Laterre PF. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediators Inflammation (2010) 2010:3–4. doi: 10.1155/2010/568396
92. Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, et al. Protection from lethal Gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci United States America (2009) 106:2348–52. doi: 10.1073/pnas.0808146106
93. Ranjith-Kumar CT, Duffy KE, Jordan JL, Eaton-Bassiri A, Vaughan R, Hoose SA, et al. Single-stranded oligonucleotides can inhibit cytokine production induced by human toll-like receptor 3. Mol Cell Biol (2008) 28:4507–19. doi: 10.1128/MCB.00308-08
94. Lahiri A, Lahiri A, Das P, Vani J, Shaila MS, Chakravortty D. TLR 9 activation in dendritic cells enhances salmonella killing and antigen presentation via involvement of the reactive oxygen species. PloS One (2010) 5(10):2–3. doi: 10.1371/journal.pone.0013772
95. Zhang S, Li L, Shen A, Chen Y, Qi Z. Rational use of tocilizumab in the treatment of novel coronavirus pneumonia. Clin Drug Invest (2020) 40:511–8. doi: 10.1007/s40261-020-00917-3
96. Toniati P, Piva S, Cattalini M, Garrafa E, Regola F, Castelli F, et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun Rev (2020) 19(7):4. doi: 10.1016/j.autrev.2020.102568
97. Nemchand P, Tahir H, Mediwake R, Lee J. Cytokine storm and use of anakinra in a patient with COVID-19. BMJ Case Rep (2020) 13(9):2. doi: 10.1136/bcr-2020-237525
98. Annane D, Heming N, Grimaldi-Bensouda L, Fremeaux-Bacchi V, Vigan M, Roux A-L, et al. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: A proof-of-concept study. Eclinicalmedicine (2020) 28:4–5. doi: 10.1016/j.eclinm.2020.100590
99. Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol (2020) 215:3. doi: 10.1016/j.clim.2020.108450
100. Mastellos DC, da Silva B, Fonseca BAL, Fonseca NP, Auxiliadora-Martins M, Mastaglio S, et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin Immunol (2020) 220:8. doi: 10.1016/j.clim.2020.108598
101. Patterson BK, Seethamraju H, Dhody K, Corley MJ, Kazempour K, Lalezari J, et al. CCR5 inhibition in critical COVID-19 patients decreases inflammatory cytokines, increases CD8 T-cells, and decreases SARS-CoV2 RNA in plasma by day 14. Int J Infect Dis (2021) 103:25–32. doi: 10.1016/j.ijid.2020.10.101
102. Siegal D, Crowther M, Cuker A. Thrombopoietin receptor agonists in primary immune thrombocytopenia. Semin Hematol (2013) 50(Suppl 1):S18–21. doi: 10.1053/j.seminhematol.2013.03.005
103. Alonso-Beato R, Morales-Ortega A, Fernandez F, Moron AIP, Rios-Fernandez R, Rubio JLC, et al. Immune thrombocytopenia and COVID-19: Case report and review of literature. Lupus (2021) 30:1515–21. doi: 10.1177/09612033211021161
104. Albani F, Fusina F, Granato E, Capotosto C, Ceracchi C, Gargaruti R, et al. Corticosteroid treatment has no effect on hospital mortality in COVID-19 patients. Sci Rep (2021) 11:2–4. doi: 10.1038/s41598-020-80654-x
105. Martincic Z, Skopec B, Rener K, Mavric M, Vovko T, Jereb M, et al. Severe immune thrombocytopenia in a critically ill COVID-19 patient. Int J Infect Dis (2020) 99:269–71. doi: 10.1016/j.ijid.2020.08.002
Keywords: platelet count, sepsis, thrombocytopenia, COVID-19, platelet
Citation: Cheng J, Zeng H, Chen H, Fan L, Xu C, Huang H, Tang T and Li M (2023) Current knowledge of thrombocytopenia in sepsis and COVID-19. Front. Immunol. 14:1213510. doi: 10.3389/fimmu.2023.1213510
Received: 28 April 2023; Accepted: 12 September 2023;
Published: 28 September 2023.
Edited by:
Madhumita Chatterjee, Experimental Therapy and Toxicology, GermanyReviewed by:
Emilia Barreto-Duran, Jagiellonian University, PolandCarsten Deppermann, Johannes Gutenberg University Mainz, Germany
Copyright © 2023 Cheng, Zeng, Chen, Fan, Xu, Huang, Tang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Li, bWlubGl6anVAemp1LmVkdS5jbg==