 Muhammad Babar Khawar
Muhammad Babar Khawar Fei Ge
Fei Ge Ali Afzal
Ali Afzal Haibo Sun
Haibo Sun- 1Institute of Translational Medicine, Medical College, Yangzhou University, Yangzhou, China
- 2Jiangsu Key Laboratory of Experimental & Translational Non-Coding RNA Research Yangzhou, Yangzhou, China
- 3Applied Molecular Biology and Biomedicine Lab, Department of Zoology, University of Narowal, Narowal, Pakistan
- 4Haian Hospital of Traditional Chinese Medicine Affiliated to Nanjing University of Chinese Medicine, Nantong, Jiangsu, China
Chimeric antigen receptor (CAR) T cell therapy for solid tumors shows promise, but several hurdles remain. Strategies to overcome barriers such as CAR T therapy-related toxicities (CTT), immunosuppression, and immune checkpoints through research and technology are needed to put the last nail to the coffin and offer hope for previously incurable malignancies. Herein we review current literature and infer novel strategies for the mitigation of CTT while impeding immune suppression, stromal barriers, tumor heterogeneity, on-target/off-tumor toxicities, and better transfection strategies with an emphasis on clinical research and prospects.
Introduction
Malignant tumors, despite the potency of chemotherapy, remain a resilient adversary and are adept at evading traditional treatment methods, much like a wily fox. Regarding the fact, immunotherapy has been propelled to the vanguard of experimental research owing to its remarkable success in hematological malignancies by utilizing T cells with an expression of chimeric antigen receptors (CARs), albeit solid tumors again outstand owing to cytotoxicity, immunosuppression, heterogenetic nature, an anomaly in metabolism, and resistant tumor territory. Interestingly, CAR T cell therapy appears promising in conquering such obstacles being obliged to nano-immunoengineering (1), gene editing (2), armoring with cytokines (3), dual targeting CARs (4), and switchable CARs (5). The initial development of CARs aimed to augment the efficacy as a single unit; however, later generations focused on the optimization of various aspects of CARs as mentioned earlier. Generally, each generation (as summarized in Table 1) has different co-stimulatory domains added, resulting in improved response to tumor cells and longer shelf-life. Exclusively, gen-4-CAR T cells contain constitutive or inducible transgenic sequences to avoid systemic toxicity, and the gen-5-CAR T cells have the addition of an IL-2 receptor for JAK/STAT pathway activation in an antigen-dependent manner. Regardless of such expansion, the actual potential, particularly for solid tumors, is yet to be determined. Moreover, in-depth understandings of the mechanisms of loss of CAR activity, such as CAR T cell exhaustion, antigen escape, and resistance mechanisms, need to be further investigated. Further improvement of transfection strategies is promising to overcome the barriers of the solid tumor microenvironment (TME). Hitherto we have reviewed some novel applications of nanotechnology in immunoengineering of CAR T cells (6), limitations and their possible solutions for improving different generations of CARs (7), and some novel approaches to enhance the efficacy of natural killer cells (NK) with CARs (8). Herein we discuss promising strategies to mitigate CAR T cell-related toxicities. Looking forward, we provide promising insights into the improved delivery of CARs in solid TME and discuss the strategies for better transfection of CAR T cells with an emphasis on translational and clinical research.

Table 1 Comparison of the CAR T cell generations.
CAR T cell therapy-related toxicities
CAR T cell therapy-related toxicities (CTT) are owed to various challenges—for example, chance expression of target antigens in non-tumoral cells and heterogenous inflammatory responses (9). The latter fact is duly supported by a recent study which reveal that 79% of patients showed poor prognosis following the CAR T cell therapy, though a fraction of patients showed prolonged remissions upon reinfusion of CARs and salvage treatment (10). In a cohort of 110 patients (NCT03173417), the majority likewise had cytokine release syndrome (CRS) and neurotoxicity (11). In addition, a systematic analysis reported worse CRS (26%) and neurotoxicity (12%) within the cohort, without any significant difference in patients achieving complete remission or those experiencing inflammatory responses (12). Nonetheless, reinfusion of CARs following the failure of prior CAR T cell therapy showed prolonged remission rates (13) and, combined with a high dose of cyclophosphamide, resulted in CAR T cell expansion in peripheral blood (NCT01860937) (14). Apart from this, studies have reported various forms of CTT—for example, hemophagocytic lymphohistiocytosis was observed in approximately 14.8% of pediatric and young adults who underwent CD19-specific CAR T cell therapy (15). Moreover, tumor lysis syndrome (16), hypogammaglobulinemia (17), and febrile neutropenia (NCT02414269) (18) have been reported less frequently in association with CTT. The fact is attributed to several factors, such as the variable nature of targeting antigens and individual patient factors. Additional research is imperative to enhance our comprehension and mitigate the potential risks and CTT beyond CRS and neurotoxicity. A comprehensive exploration of these aspects will enable the refinement and optimization of current strategies, thereby enhancing the safety and efficacy of CAR T cell therapy.
Mechanism of CAR T cell therapy-related toxicities
Henceforward, two kinds of CTT are common: CRS and neurotoxicity. The exact mechanism of the pathophysiology of CTT is yet to be demarcated. However, studies have identified a significant mechanism involving an interaction between macrophages and T cells, leading to the release of multiple cytokines and oxides in murine models (19). Subsequently, it was suggested that the infusion of CAR T cells following macrophage depletion could be a promising approach to eliminate CRS (20). Although this depletion increases the risk of infections, the complications can be managed using IL-6 or corticosteroids or a combination of both (21). Recently, another study showed a positive correlation between CRS and hematological toxicities (22). Endothelial activation is a potent member in CRS pathology (23) as supported by a cohort of 133 B cell malignant patients who received CD19 CAR T cell infusion in a dose-dependent manner. In total, 70% of patients developed CRS with variable severity indices (24). Not just this—endothelial activation also disrupts the blood–brain barrier (BBB) in patients with neurotoxicity as warranted by the magnetic resonance imaging results of severe cases (23). A mechanistic approach was developed in a study that showed that CAR T cell products such as tisagenlecleucel responded by the expansion of proliferative memory-like CD8 clones, while another product, axicabtagene ciloleucel responders, displayed more heterogeneous populations. The latter non-responders had elevations in CAR T-regulatory cells, which suppressed conventional CAR T cell expansion and drove late relapses in an in vivo model (25). One of the recommendations, in the context explained above, is reinfusion of CAR T cells along with a combinatorial use of IL-6 and steroids inclusive of former macrophage depletion. Nevertheless, there are various other mechanisms of pathology of CTT, which are potent to grab the attention of researchers, yet careful monitoring and personalized treatment plans are warranted to manage the potential adverse reactions and ensure the safety of the patients.
Mitigation of CAR T cell therapy-related toxicities
Currently, some strategies have been reported for the mitigation of CTT; however, further understanding of the exact approach will lead to the precise development of CARs. Researchers are focusing on the preemptive mitigation of CTT in combination with various immunosuppressors which have been proven to be effective devoid of any attenuation of CAR T efficiency—for instance, a clinical study confirms no negative impact on the antitumor activity of CAR T cell therapy when combined with tocilizumab and/or steroid administration and supports no expansion and persistence of CD19-targeted CAR T cells (26). Following the conclusion, a prospective study evaluated the efficacy of tocilizumab to mitigate CRS in 70 patients divided into two groups of high and low tumor burden. The overall response rates (87% in high-tumor-burden and 100% in low-tumor-burden patients) were good enough to support the very strategy as an effective one. The clinical trial successfully met the primary and final goals of the study without any attenuation of the antitumor activity of CAR T cell therapy (27). Dexamethasone—a glucocorticoid, has also been of interest in the mitigation of neurological toxicities due to its exceptional penetration into the central nervous system as per the guidelines of the National Cancer Institute. IL-7 receptor alpha is a well-recognized facet needed for CAR T cell persistence and memory T cell formation (28). In this case, dexamethasone has been shown to upregulate IL-7 receptors to enhance CAR T cell persistence and antitumor activity (29). Urak and colleagues suggest that the effect of dexamethasone is not only restricted to specific T cell subsets while the same upregulation was also observed in peripheral blood mononuclear cells (PBMCs), naïve cells, memory T cell-derived CAR T cells, and untransduced T cells. The study does not affect the efficacy of CAR T cells in vivo or in vitro. These facts are promising in understanding the mechanism of dexamethasone in CAR T cell therapy. When combined with ruxolitinib, dexamethasone synergistically reduced the disease symptoms in a murine model. Mechanistically, JAK-dependent cytokines IL-2 and IL-12 conferred resistance to dexamethasone, but not etoposide, and ruxolitinib attenuated STAT5 activation to enhance dexamethasone-mediated cell death in the presence of IL-2 or IL-12 (30). Other various drugs are under investigation to be used in combination with CAR T cell therapy. In this text, we advocated the inhibition of CTT utilizing reported cytokines and combining various immunosuppressants and steroids. These drugs reveal an in-depth understanding of the mechanism of mitigation of CTT, including CRS and neurotoxicity. Further research in this area is de rigueur to proceed with the mitigation of CTT for effective CAR T cell therapy.
Improving the barriers for better delivery in tumor microenvironment
Following a pertinent success in hematological malignancies where CAR T cells find fewer disturbances for the effective killing of tumor cells, solid TME presents several physical and immune barriers that impede CAR T cell trafficking in solid TME. These include heterogeneity of tumor antigens, nutrient-poor milieu, T cell exhaustion, and recruitment of immunosuppressor cells.
Immunosuppression
Mechanistically, chemokine-transforming growth factor β (TGF-β) activates tumor-associated fibroblasts, which, in return, upregulates the extracellular matrix proteins to T cell infiltration into solid TME (31). Interestingly, a direct restriction to CAR T cell infiltration by TGF-β has also been observed, whereby TGF-β not only downregulates CXCR3 expression but also increases the binding of Smad2 to CXCR3 promoter (32). The search for effective and safer therapeutics targeting the TGF-β pathway is further complicated by various functions that TGF-β performs in normal tissues (33). We accordingly advocate further extensive research to investigate the contextual role of TGF-β for immunosuppression in TME. Rather than TGF-β, several other cytokine and chemokine profiles recruit T-reg cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages and thus limit CAR T cell infiltration. T-reg cells secrete immunosuppressive cytokines and downregulate antigen-presenting cells via cytotoxic T lymphocyte antigen 4, thus preventing T cell activation (34). The immune-suppressive effect of MDSCs also has an unfavorable effect on CAR T cell therapy (35). Krishnamoorthy et al. reviewed a detailed mechanism of suppression of T cells mediated by MDSCs (36). They stated that, generally, MDSCs suppress the immune response by inhibiting the functions of T cells in four distinct pathways as shown in Figure 1A. In this way, we recommend utilizing the drug which could be capable of blocking the MDSC functions within the solid TME. In this aspect, the delivery systems must be capable of targeted unloading of cargo. We also suggest that CAR T cells should be engineered genetically to express molecules that specifically target and eliminate MDSCs within the TME. This recommendation is interestingly supported by a study in which the authors developed CAR T cells with co-expression of receptors for MDSCs. The study provides an evidence of superior anti-tumoral activity of CAR T cells which also improved T cell proliferation in solid TME (38).
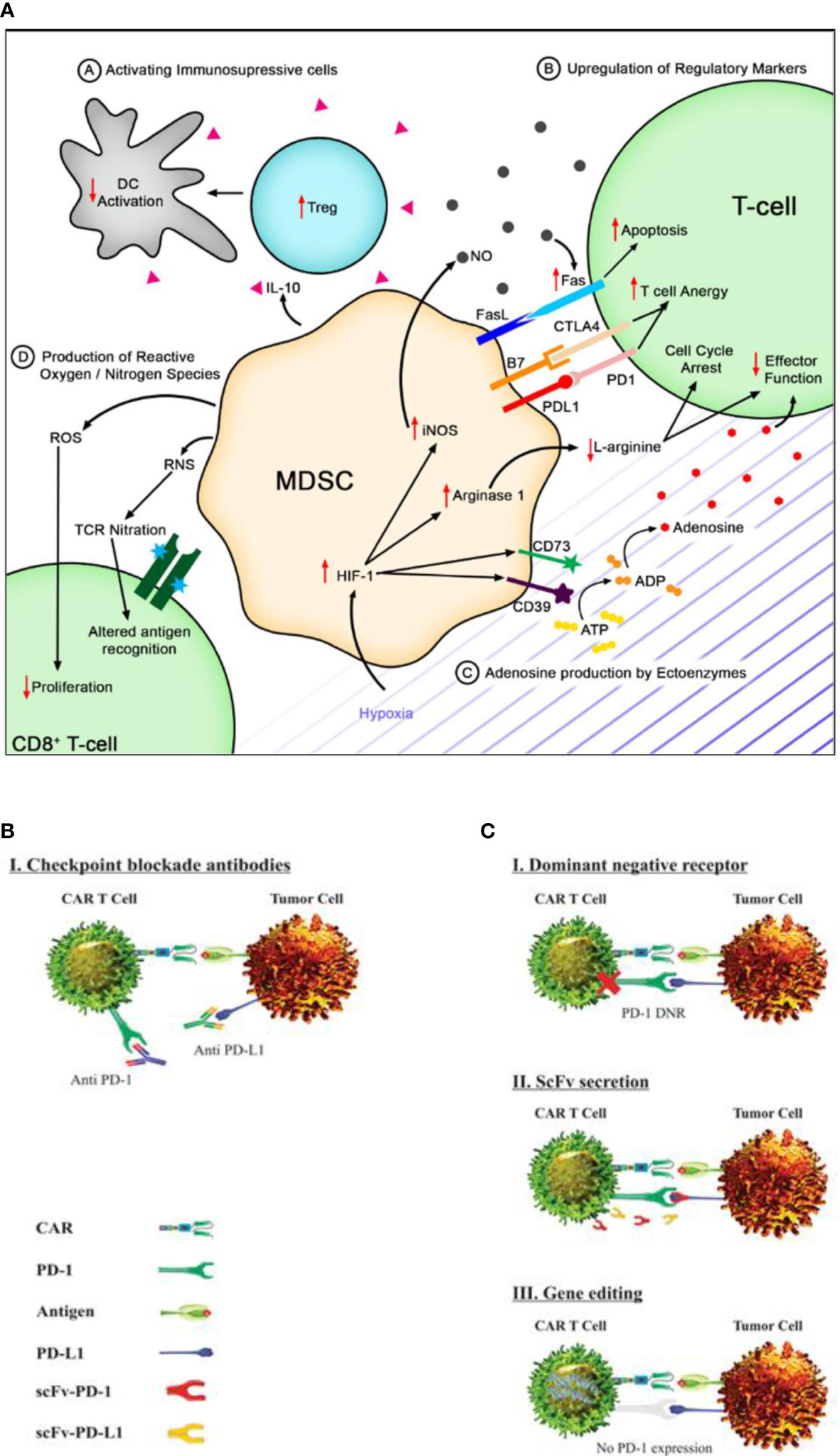
Figure 1 Immune suppression and checkpoint inhibition in solid tumor microenvironment. (A) Myeloid-derived suppressor cells (MDSCs) secrete IL-10 to activate immunosuppressive cells like T-regs, leading to reduced T cell activation. They can also induce the upregulation of checkpoint molecules on T cells, inducing anergy or apoptosis. Hypoxia in the tumor microenvironment can upregulate CD73 and CD39, leading to increased adenosine. MDSCs produce ROS and RNS that can decrease T cell proliferation and alter antigen recognition [reproduced from (36)]. (B) Immune checkpoint blockade using monoclonal antibodies targeting PD-1 and PD L1 has shown promise in restoring anti-tumor immune responses by rescuing T cell function and enhancing T cell-mediated killing of cancer cells [reproduced from (37)]. (C) Genetically engineered chimeric antigen receptor T cells can locally inhibit immune checkpoint molecules by expressing dominant-negative receptors, secreting blocking scFv, or disrupting gene expression to interfere with intracellular signaling [reproduced from (37)].
Tumor heterogeneity
Once the minute, safe, and appropriate tumor-associated antigens (TAAs) are determined in the development of CAR T cell therapy, it becomes crucial to address tumor antigen heterogeneity in the subsequent step, as it may be expressed variably in both tumor and normal cells (39, 40). Dana H et al., in 2021, systematically summarized various antigens, targeted in different cancers (41), showing promise in the mitigation of tumor heterogeneity. An interesting study employing a combinatorial therapy demonstrated a compatibility between CARs, T cell receptors (TCRs), and hnCD16 in alleviating tumor heterogeneity (42). This technique is likewise effective for resistant tumors owing to antigen escape. By computational modeling, cellular interactions were examined, and it was thus concluded with a suggestion to employ combinatorial therapy for effective treatment (43). As mentioned above in the mitigation of CTT, the presence of IL-12 enhances the synergistic effect of steroid therapy in combination with ruxolitinib. This is an evident boon that it is present in solid TME (44). We hereof recommend a tumor-specific condition employing the IL-12-mediated expression of CARs. Nevertheless, investigations are warranted to inspect off-tumor toxicity along with IL-12 dosages. A similar study was conducted aiming to augment tumor specificity (45). NK cells detect tumor ligands through NK receptors, including NKp30. A subset of CD8+ T cells expressing NKp30 recognizes B7H6, a tumor ligand frequently overexpressed in cancers (46). Apropos of this research, Correia et al. (2021) latterly engineered a dual recognition strategy in the form of innate-like NKp30+CD8+ T cells with a combined expression of TCRs and CARs, thus successfully targeting tumor heterogeneity (47). Furthermore, novel criteria for the conscription of CAR T cell therapy, including preselection for the intensity and proportion of targeted antigen, are warranted. Although prescribing the definite criteria could be tricky, hereto we suggest concurrent or in combination targeting of different tumor antigens, which is promising in leading to a more effective antitumoral effect and possibly reducing the emergence of tumor variants that lack the targeted antigen.
Immune checkpoint inhibition
Generally, upon tumor initiation, normal and tumor cells upregulate inhibitory signaling pathways, driving the tumor progression and its immune escape. Blockade therapy has shown to reverse this suppression and has shown promising effectiveness in hematological malignancies (48). Henceforth, the use of immune checkpoint inhibitors, specifically PD-1 blockade, has been under investigation. Chronic antigen stimulation in tumor sites leads to inhibitory receptor upregulation and CAR T cell exhaustion in a PDL1-dependent manner (49). Blocking the PD-1:PDL1 interaction using mAbs can rescue exhausted CAR T cells as shown in Figure 1B, but multiple administrations are required due to the short half-life of antibodies and immune-related adverse events (50). Localized immune checkpoint inhibition at the tumor site using modified CAR T cells secreting PD-1 blocking scFv can increase the killing activity and restore the cytotoxic activity in solid tumors. The very strategy has shown a positive response in CAR T cell exhaustion in vivo that indicates a promising synergistic effect with improved efficacy (51–53). Genetically modified CAR T cells are also under investigation for local inhibition of immune checkpoints incorporating specific mechanisms—for example, engineered CAR T cells expressing a dominant-negative receptor can disrupt the signaling of immune checkpoint molecules. Genetically modified CAR T cells also secrete blocking single-chain variable fragments, which can prevent immune checkpoint molecules from binding to their targets. Lastly, CAR T cells can be engineered to interfere with intracellular signaling pathways by disrupting the gene expression as depicted in Figure 1C (54). By implementing these innovative techniques, it is anticipated that CAR T cell therapy can overcome immune checkpoint-mediated suppression and offers enhanced therapeutic outcomes for cancer patients.
MDSCs can upregulate checkpoint molecules like CTLA4, T cell immunoglobulin, and mucin domain-containing protein 3 (TIM3) on T cells, leading to T cell apoptosis through Fas signaling (55). Another study focuses on targeting B7-H3 immune checkpoint in non-small cell lung cancer which shows promise in T cell activation and target cell death. The study also finds that anti-B7-H3 blockade has a role in altering glucose metabolism through reactive oxygen species-mediated pathways (56). Moreover, rebalancing the TGF-β1/BMP signaling pathways shows promise in exhausted T cells and induces higher antitumoral targeting while synergizing the immune checkpoint blockade (57). This data may provide better insights into solid TME for effective tumor-killing activity.
On-target/off-tumor toxicities
A careful selection of tumor-specific antigens is warranted as they are rare and require precise identification following the use of TAAs. The main issue is the heterogeneity of tumor-specific antigens and their expression on normal, non-tumoral cells vulnerating them for off-tumor toxicity. Generally, the antigens targeted by CAR T cells are tumor-associated, rather than tumor specific. While these antigens may be highly expressed within the solid tumor microenvironment, it is important to note that normal cells also express a small amount of these antigens (58).
Recent developments in the field of CAR T cell therapy have led to the emergence of novel technologies which hold significant potential in addressing the challenges of on-target/off-tumor toxicity. Researchers have developed novel technologies like ZAP-70-based logic-gated intracellular network CARs through the application of Boolean logic gating and the engineering of intracellular T cell signaling molecules (59). This fact demonstrates superior efficacy and safety while bypassing upstream signaling proteins. The utilization of logic gating circuits and synthetic biology in CAR T cell engineering has shown promising specificity in preclinical mouse models (60). However, the translation of these approaches into clinical studies is yet to be fully explored and evaluated. Furthermore, SynNotch CAR utilizes synthetic Notch receptors in mitigating the toxicity by enhancing the specificity and functionality (61). It allows enhanced selectivity via improved discrimination between tumor cells and healthy tissues, minimizing off-target toxicity (62). The activation of SynNotch CAR is likewise programmable by enabling downstream signaling pathways and expressing effector molecules to offer enhanced flexibility (63). Notably, it also allows fine-tuning of immune responses by incorporating inducible signaling domains or regulatory elements that modulate CAR-T cell activity, promoting controlled and adaptable immune responses (64). Another important development is the T cell redirected for universal cytokine killing (TRUCK) CAR which combines the killing ability of CAR with the production of therapeutic cytokines (65) such as inducible IL-18 (66). There are conditional cytokine expression systems that enable controlled cytokine release, reducing the risk of off-target effects and cytokine-related toxicities (67, 68). Importantly, TRUCK CARs allow antigen-specific targeting minimizing off-target toxicity (69, 70).
Specifically, apart from the above-mentioned developments, gene editing practice is more promising in this research area. To enhance T cell responses to antigens and decrease exhaustion rates, the very technology has been employed (71). Furthermore, the use of such technologies may allow for the development of T cell populations that can be used in allogeneic applications. Exploiting clustered regularly interspaced short palindromic repeat (CRISPR) in mitigating on-target/off-tumor toxicities is itself another research arena whereby it is used to create CAR resistance to immune suppression (72). Modifying CAR T cells to target tumor-specific antigens using CRISPR is another promising strategy as it has been demonstrated that CRISPR has been used to knock out the expression of the CD19 antigen on normal B cells, allowing CAR T cells to target only tumor cells that express CD19 (72). This approach has the potential to reduce the risk of CTT without targeting normal cells. An extensive review of such modifications via CRISPR technology for CARs (as shown in Figure 2) presents encouraging outcomes in clinical and preclinical trials (73). Further research into all these insights is necessary to develop effective strategies for mitigating the risks associated with CAR T cell therapy.
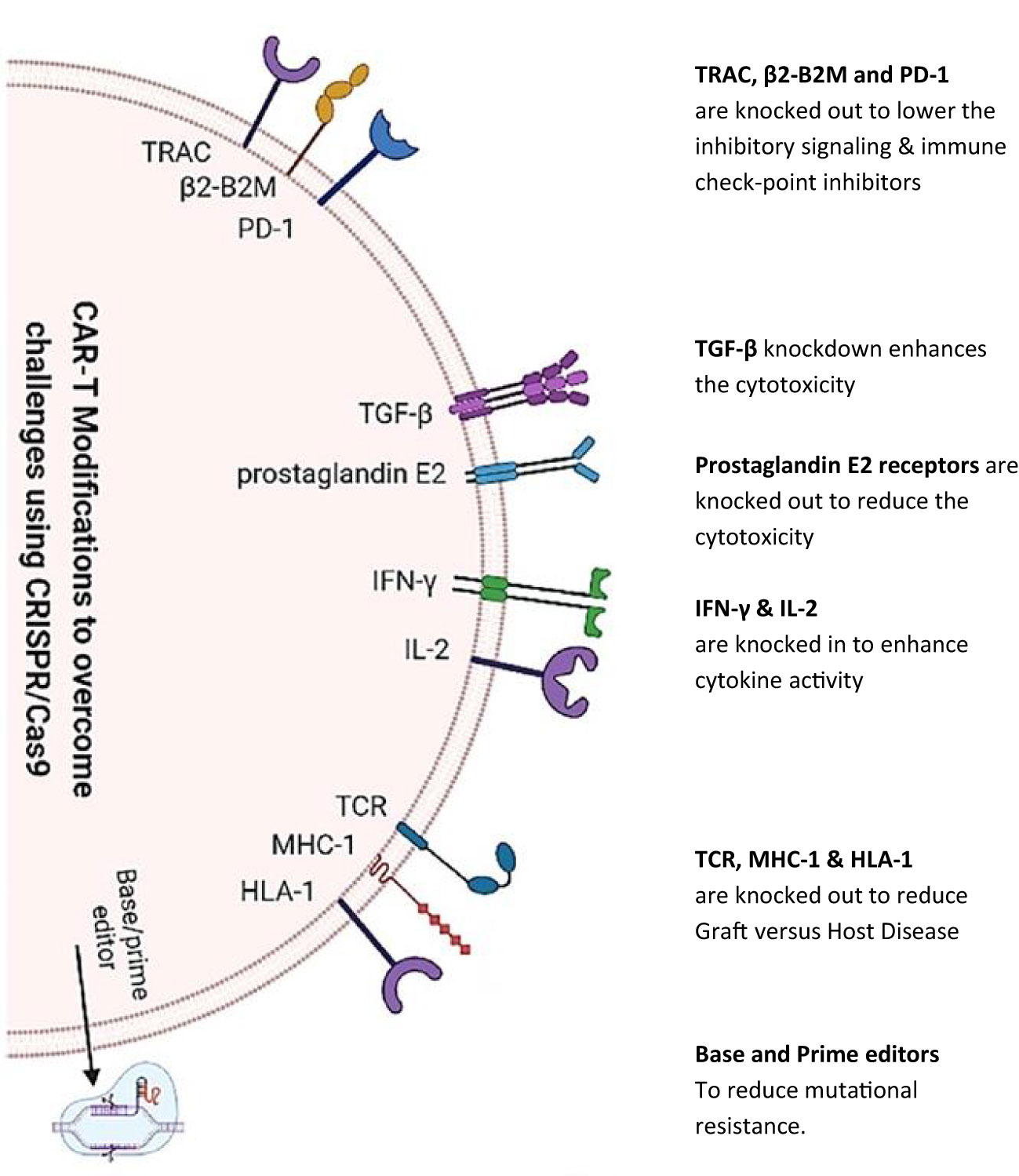
Figure 2 CRISPR/Cas9 knock in/out of various targeted genes to modify chimeric antigen receptor T cell therapy. CRISPR/Cas9 technology [as depicted by Khan et al. (2022)] is being utilized to reduce inhibitory signaling, knock in/out genes for toxicity reduction, and boost cytokine secretion. Furthermore, gene editing has been used to reduce adverse host reactions by knocking out TCR, major histocompatibility complex class 1 (MHC-1), and HLA-1 genes.
Tumor stromal barrier
Herein the aforementioned barriers have been on the bull’s eye to steer CARs toward promising efficacy. However, newly emergent evidence suggests novel roadblocks in the form of tumor parenchyma and stromal cells in solid TME (74). These two distinct yet synergistic elements engage in a dynamic cross-talk, thereby facilitating and sustaining a resistant solid TME. Notwithstanding, the very notion not only initiates and promotes tumor growth but also augments the metastatic potential of tumors (75), presenting new research areas in overcoming the roadblocks to CAR T cell therapy. The promising candidates comprise tumor-associated fibroblasts (TAFs) secreting fibronectin and collagen-like proteins. Further cellular entities such as MDSCs, epithelial, endothelial, or mesenchymal cells, adipose cells, and bone marrow-derived mesenchymal cells have also been observed to transit to TAFs (75, 76) as depicted in Figure 3. Noticing all these entities, CAR T cell therapy acquires a giant barrier to incapacitate. Consequently, the development of innovative strategies and combinatorial strategies with other established treatment modalities are warranted as a promising gateway to CAR T cell therapy research arena.
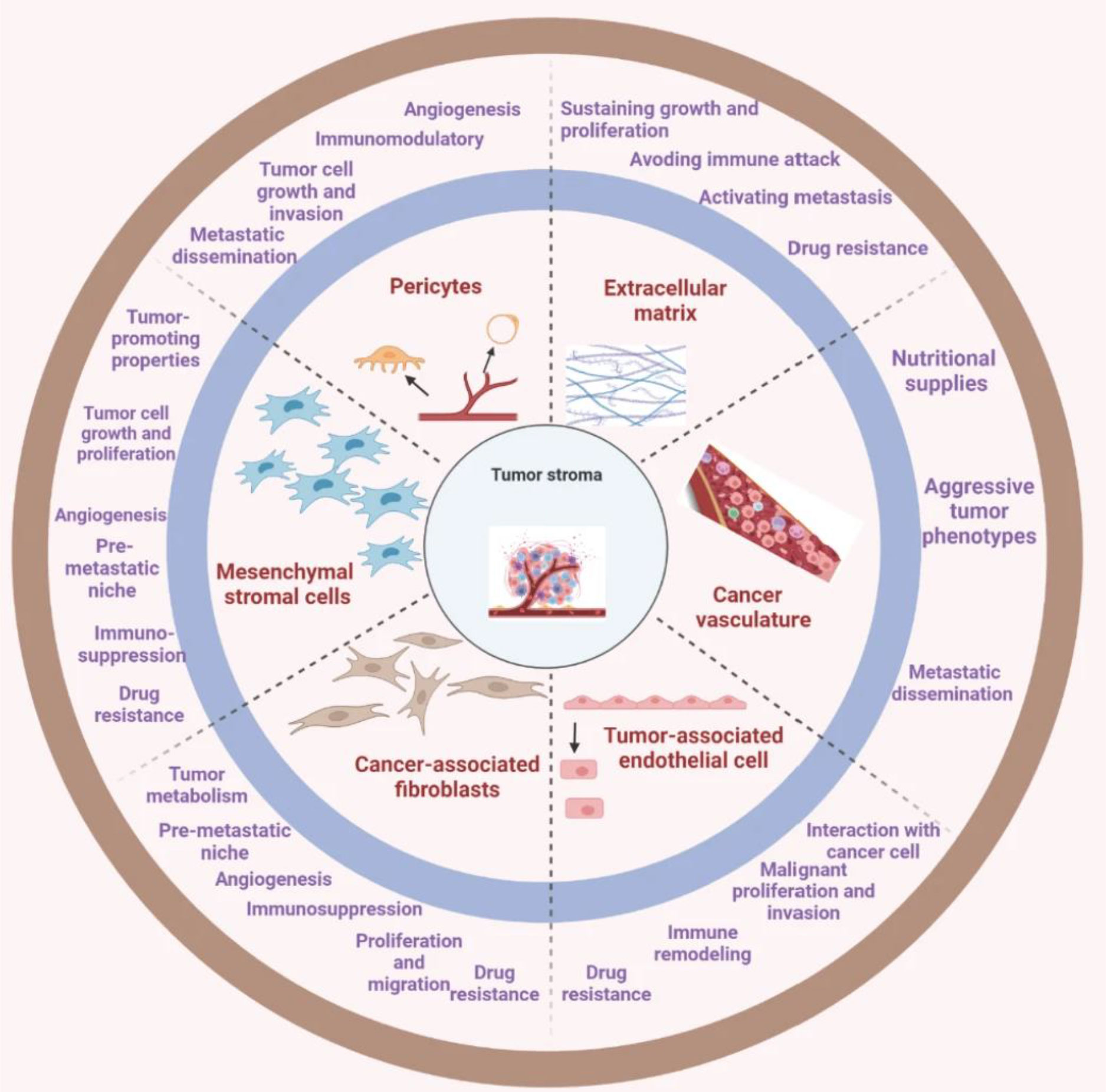
Figure 3 The tumor stroma comprises various key entities. The deep understandings present a comprehensive mechanism of the interactions between tumor and stromal cells and how they contribute to the development and progression of cancer. This can be crucial in the development of targeted therapies that specifically address the tumor microenvironment to improve treatment outcomes for cancer patients [reproduced from (75)].
Accordingly, modulation of TME in overcoming stromal barriers has shown promise. Geng F. et al., in 2019, examined the effectiveness of a DNA vaccine expressing fibroblast activation protein-α (FAP-α) in altering the solid TME in breast cancer (77). In this aspect, CARs that should target FAP-α to mitigate the barrier are needed to be developed, allowing more infiltration and persistence, or else CAR T cell therapy may be combined with the pre-infusion of nanoparticles (NPs) with FAP-specific antibodies cargo as evidenced by Zhen et al. in 2017 (78). There are a lot more options; one of that is targeting pathways of stromal interactions which may be employed as a pre-emptive strategy or combinatorial agents as extensively reviewed by van der Spek et al. in 2020 (76).
By and large, the text herein highlights the potential of CAR T cell therapy and underscores a prerequisite for sustained research and collaboration to optimize its clinical application and offer hope for patients with solid tumors.
Better transfection strategies in the development of CAR T cells
There is a multitude of methods for transfecting the CAR T gene, most notably electroporation, viral transduction to RNA electroporation, and even newer means of improving the overall efficiency of CAR T cell therapies, such as the manipulation genes which have produced valuable desired nucleases: transcription activator-like effector nucleases (TALEN) and the widely known CRISPR (79).
However, even with the large success and even more so the novelty, there are many limitations to its administration. As mentioned earlier, electroporation, one of the most commonly used means of transfection—has shown to have many limitations: non-specific delivery and undesired targets, accompanied with certain cell death rates (80). Studies have also indicated a risk of cytotoxicity (81). The transfection through electroporation correspondently results in membrane disruption that affects expressions. Another limitation faced by electroporation is its inadequacy in its application for in vivo purposes (82).
These limitations have been and can be tackled in many ways, one being by incorporating conventional anti-cancer drugs (83). The findings suggest approaching certain targeted tumors by combining specific mathematical and statistical computer models that have shown to improve the desired result and efficacy of transfecting CAR T through electroporation (84, 85). Perfusing the targeted organ or tissue or the surrounding vessels could also enhance the outcome. A study suggests that CAR Ts may be produced via plasmids and IL15-IL15R fusion proteins, resulting in effectiveness and superior endurance in in vivo environments (86).
CAR T cells are often challenged in forming proper resistance, of which the expression of the desired targeted antigen has been shown to be either partly or completely lost by many tumors (87). Several studies have indicated that certain undesired downregulation of expression was observed. With the viral transfection methods, the limitations are more inclined to expense viability. The production of high-grade and clinically safe viral CAR T cells is quite expensive (88). With that, the general viral CAR T cells are highly complex structures and require intense observations in their completion. The prolonged expression of these CARs has led to tonic signaling and cell death via activation induction of T cells. Even in the case of the rather novel CARs, such as the aforementioned TALEN, the authors discussed how receptor downregulation may mute the activation of natural killer cells caused by the lack of MHC-I (89, 90).
As mentioned earlier, another prevailing setback of CAR T cell therapy may result in neurotoxicity, which is a result of the CAR T cells prompting CRS, which has been observed in many patients treated with CAR T cell therapy (91). A way to improve this setback is by developing a method that can alter the switching ability of CAR T cells, and we advocate that switchable CARs may provide temporal control over neurotoxicity and effector performance. When toxicity first manifests, turning down the CAR T cell effector activity with medication can be helpful and life-saving. However, after the toxicities are gone, they will need to be switched back on to prevent impairing the antitumor activity. This is well elaborated by a recent study via the use of protease-regulated signal neutralization by an inhibitable protease (SNIP) (92). Comparatively to constitutively active CAR T cells, SNIP CAR T cells displayed higher cytotoxicity against tumors, however an in vivo durability and more likely a memory phenotype. The expression of markers linked to T cell exhaustion was significantly reduced in SNIP CAR T cells, perhaps as a result of avoiding prolonged T cell activation.
Lastly, the overall cost of CAR T has oftentimes been the issue in developing and improving the therapy. Hence, to lower the overall cost of CAR T cell therapy, better and more affordable gene transfer methods are required. Although lentiviral/retroviral vector-mediated gene transfer is more expensive than non-viral methods, there is a growing body of clinical data supporting the latter method (93).
Translational and clinical research
Notwithstanding significant clinical successes, there remain challenges in the translation of CAR T cell therapy to routine clinical practice. Currently, CTT mitigation, improving barriers for better trafficking into the TME, and transfection approaches are of major focus in clinical research as shown in Table 2. A preclinical study focusing on checkpoint blockade demonstrated that the co-expression of PD-1-CD28 in TRuC T cells enhances cytokine production and supports anti-tumor efficacy in xenograft mouse models. The results suggest that PD-1-CD28 co-expression could have therapeutic potential in preventing PD-L1-induced T cell hypofunction (94).
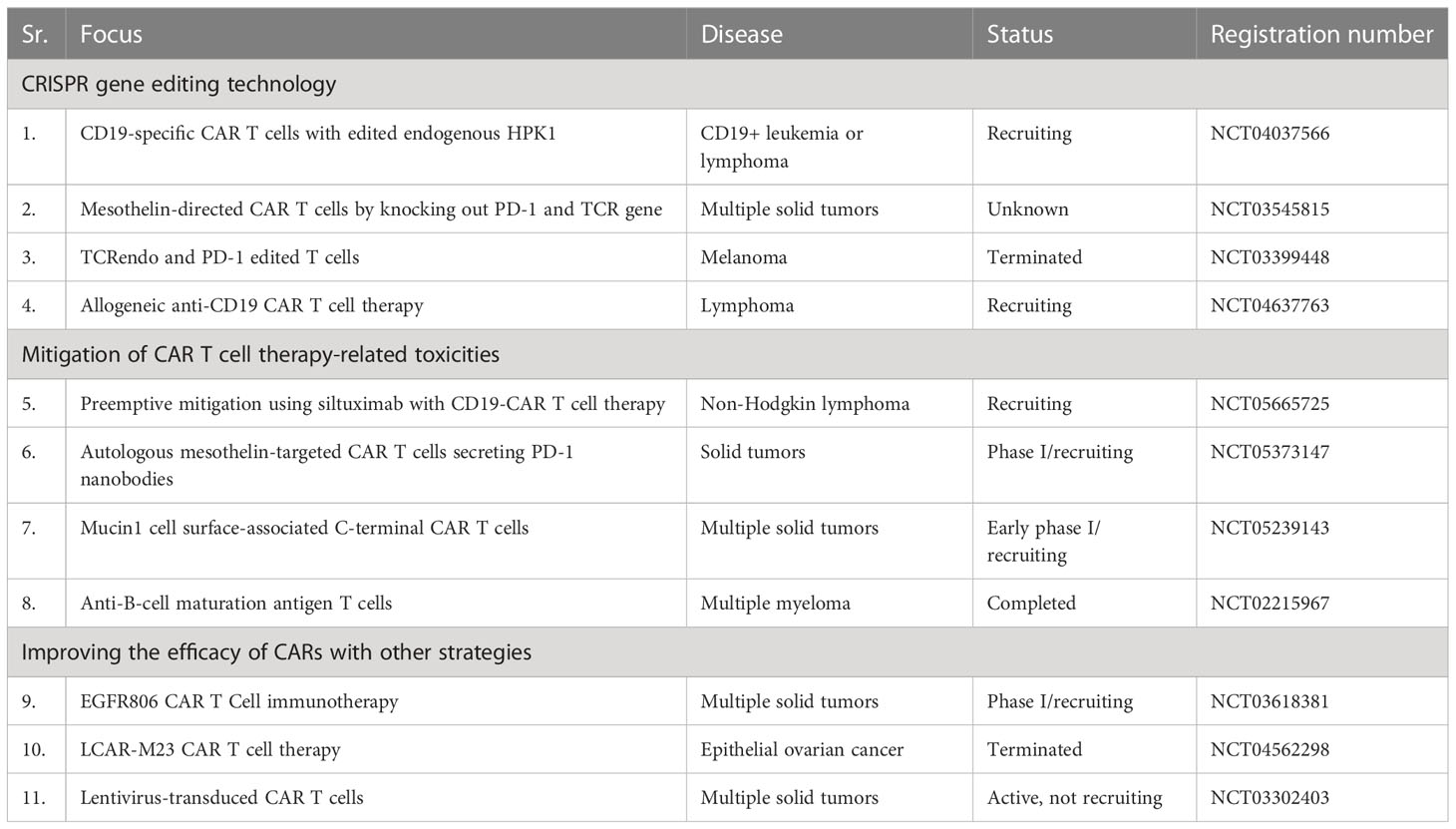
Table 2 Summary of CAR T cell therapeutic approaches to clinical research in improving the barriers.
Tumor-specific antigens have also been targeted in several clinical trials reducing off-tumor toxicities—for instance, NCT04489862 is an early phase I study which aims to target α-PD-1 and MSLN in non-small cell lung cancer at Wuhan Union Hospital, China. Another phase I study (NCT03198052) is recruiting patients. The study aims to target various antigens such as mesothelin, MUC1, PD-L1, and EGFR. Several studies have successfully obtained higher response rates for solid tumors using monodrug synergized with CAR T cell therapy—for example, a study (NCT03548207) enrolled 97 patients and used cyclophosphamide and fludarabine as an autologous CAR T cell monodrug and obtained a 95% overall response rate.
For CRISPR technology in CAR T cell therapy, there are a limited number of clinical studies for solid malignancies—for instance, phase I study (NCT03545815) establishes a groundwork utilizing PD-1 and TCR disruption for solid TME (95). None of the participants displayed any unexpected adverse events or dose-limiting toxicity. The post-infusion detection of predominantly TCR-positive CAR T cells was observed in the peripheral blood of merely three patients. Preclinical experiments suggested a decrease in the persistence of TCR-deficient CAR T cells, encouraging further investigation in human clinical trials. Various other clinical studies such as NCT04037566 are currently underway to investigate the effectiveness of CD19-CAR T cells modified with endogenous HPK1 for treating lymphoma.
Conclusion and future prospects
CAR T cell therapy for solid TME is presenting encouraging results subsequent to its success in hematological malignancies. The incapacitation of various barriers for CAR T cells is under extensive investigation to achieve therapy with mitigated CTT, improved delivery to solid TME impeding blockades, and better transfection strategies. While improved outcomes have been observed, CARs still face numerous hurdles that need to be overcome. In this way, this review presents strong recommendations and suggestions highlighting the gaps and areas of focus. There is an urgent need to focus on immunosuppression due to cytokines and immunosuppressive cells. Investigations for CAR T reinfusion with the combination of cytokine/steroids or other immunosuppressors for preemptive mitigation of toxicities are warranted. While immune suppression has been observed with chemokine inhibition, such as TGF-β, the beneficial effects of these molecules should not be overlooked under normal circumstances. Therefore, we recommend comprehensive research in this area, taking into account their contextual roles. Additionally, CRISPR technology can effectively overcome immune checkpoint inhibition and other barriers.
Even though significant progress has been made in the development of CAR T cell therapy, further research is required to address the remaining challenges and optimize its clinical application. With ongoing advancements and collaborations in the field, CAR T cell therapy may provide new hope for patients with previously incurable malignancies. Finally, improvements in CAR T cell therapy vis-à-vis to herewith discussed prospects may prove to be promising in refining the current research in fighting against solid tumors, which can steer CARs toward another all-embracing victory in solid tumors.
Author contributions
MK, FG and AA proposed the idea, collected the data, and wrote the manuscript. HS proposed the idea and modified, supervised, and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded in part by the Startup Foundation for Advanced Talents and Science and Technology Innovation Foundation at Yangzhou University (HS), the Innovative Training Grant of College Students in Jiangsu Province (202111117097Y, FG and HS), and Postgraduate Research and Practice Innovation Program of Jiangsu Province (SJCX21_1655). This study was funded in part by The Medical Research Project of Jiangsu Provincial Health Commission (M2022024); Scientific Research Project of Nantong Municipal Health Commission (MS2022102) and Natural Science Foundation of Nanjing University of Traditional Chinese Medicine (no.XZR2020084).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nawaz W, Xu S, Li Y, Huang B, Wu X, Wu Z. Nanotechnology and immunoengineering: how nanotechnology can boost CAR-T therapy. Acta Biomaterialia (2020) 109:21–36. doi: 10.1016/j.actbio.2020.04.015
2. Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J Hematol Oncol (2020) 13(1):1–16. doi: 10.1186/s13045-020-00890-6
3. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
4. Hirabayashi K, Du H, Xu Y, Shou P, Zhou X, Fucá G, et al. Dual-targeting CAR-T cells with optimal co-stimulation and metabolic fitness enhance antitumor activity and prevent escape in solid tumors. Nat Cancer (2021) 2(9):904–18. doi: 10.1038/s43018-021-00244-2
5. Stepanov AV, Kalinin RS, Shipunova VO, Zhang D, Xie J, Rubtsov YP, et al. Switchable targeting of solid tumors by BsCAR T cells. Proc Natl Acad Sci (2022) 119(46):e2210562119. doi: 10.1073/pnas.2210562119
6. Khawar MB, Afzal A, Abbasi MH, Sheikh N, Sun H. Nano-immunoengineering of CAR-T cell therapy against tumor microenvironment: the way forward in combating cancer. OpenNano (2023) 10:100124. doi: 10.1016/j.onano.2023.100124
7. Abbasi MH, Riaz A, Khawar MB, Farooq A, Majid A, Sheikh N. CAR-T-Cell therapy: present progress and future strategies. BioMed Res Ther (2022) 9(2):4920–9. doi: 10.15419/bmrat.v9i2.726
8. Khawar MB, Sun H. CAR-NK cells: from natural basis to design for kill. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.707542
9. Fischer JW, Bhattarai N. CAR-T cell therapy: mechanism, management, and mitigation of inflammatory toxicities. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.693016
10. Wudhikarn K, Flynn JR, Rivière I, Gönen M, Wang X, Senechal B, et al. Interventions and outcomes of adult patients with b-ALL progressing after CD19 chimeric antigen receptor T-cell therapy. Blood (2021) 138(7):531–43. doi: 10.1182/blood.2020009515
11. Zhang X, Lu X-A, Yang J, Zhang G, Li J, Song L, et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with b-cell acute lymphoblastic leukemia with high-risk features. Blood Adv (2020) 4(10):2325–38. doi: 10.1182/bloodadvances.2020001466
12. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol (2020) 7(11):e816–26. doi: 10.1016/S2352-3026(20)30277-5
13. Myers RM, Li Y, Barz Leahy A, Barrett DM, Teachey DT, Callahan C, et al. Humanized CD19-targeted chimeric antigen receptor (CAR) T cells in CAR-naive and CAR-exposed children and young adults with relapsed or refractory acute lymphoblastic leukemia. J Clin Oncol (2021) 39(27):3044–55. doi: 10.1200/JCO.20.03458
14. Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory b-ALL. Blood (2019) 134(26):2361–8. doi: 10.1182/blood.2019001641
15. Hines MR, Keenan C, Maron Alfaro G, Cheng C, Zhou Y, Sharma A, et al. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br J Haematol (2021) 194(4):701–7. doi: 10.1111/bjh.17662
16. Miao L, Zhang Z, Ren Z, Li Y. Reactions related to CAR-T cell therapy. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.663201
17. Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Müller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol (2021) 32(1):34–48. doi: 10.1016/j.annonc.2020.10.478
18. Adusumilli PS, Zauderer MG, Rusch VW, O'Cearbhaill RE, Zhu A, Ngai DA, et al. Abstract CT036: a phase I clinical trial of malignant pleural disease treated with regionally delivered autologous mesothelin-targeted CAR T cells: safety and efficacy. Cancer Res (2019) 79(13_Supplement):CT036–6. doi: 10.1158/1538-7445.AM2019-CT036
19. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med (2018) 24(6):731–8. doi: 10.1038/s41591-018-0041-7
20. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med (2018) 24(6):739–48. doi: 10.1038/s41591-018-0036-4
21. Gudiol C, Lewis RE, Strati P, Kontoyiannis DP. Chimeric antigen receptor T-cell therapy for the treatment of lymphoid malignancies: is there an excess risk for infection? Lancet Haematol (2021) 8(3):e216–28. doi: 10.1016/S2352-3026(20)30376-8
22. Juluri KR, Wu QV, Voutsinas J, Hou J, Hirayama AV, Mullane E, et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Adv (2022) 6(7):2055–68. doi: 10.1182/bloodadvances.2020004142
23. Chou CK, Turtle CJ. Insight into mechanisms associated with cytokine release syndrome and neurotoxicity after CD19 CAR-T cell immunotherapy. Bone Marrow Transplant (2019) 54(Suppl 2):780–4. doi: 10.1038/s41409-019-0602-5
24. Hay KA, Hanafi L-A, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood (2017) 130(21):2295–306. doi: 10.1182/blood-2017-06-793141
25. Haradhvala NJ, Leick MB, Maurer K, Gohil SH, Larson RC, Yao N, et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory b cell lymphoma. Nat Med (2022) 28(9):1848–59. doi: 10.1038/s41591-022-01959-0
26. Summers C, Annesley C, Bleakley M, Dahlberg A, Jensen MC, Gardner R. Long term follow-up after SCRI-CAR19v1 reveals late recurrences as well as a survival advantage to consolidation with HCT after CAR T cell induced remission. Blood (2018) 132(Supplement 1):967–7. doi: 10.1182/blood-2018-99-115599
27. Kadauke S, Myers RM, Li Y, Aplenc R, Baniewicz D, Barrett DM, et al. Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric b-cell acute lymphoblastic leukemia: a prospective clinical trial. J Clin Oncol (2021) 39(8):920. doi: 10.1200/JCO.20.02477
28. Swan SL, Mehta N, Ilich E, Shen SH, Wilkinson DS, Anderson AR, et al. IL7 and IL7 Flt3L co-expressing CAR T cells improve therapeutic efficacy in mouse EGFRvIII heterogeneous glioblastoma. Front Immunol (2023) 14. doi: 10.3389/fimmu.2023.1085547
29. Urak R, Munoz A, Hsieh H-J, Taus E, Forman SJ, Wang X. Dexamethasone enhanced CAR T cell persistence and function through upregulation of interleukin 7 receptor. Blood (2021) 138(Supplement 1):1715–5. doi: 10.1182/blood-2021-153507
30. Meyer LK, Verbist K, Albeituni S, Bassett R, Hermiston ML, Nichols KE. The combination of dexamethasone and ruxolitinib synergistically attenuates disease manifestations in a preclinical model of hemophagocytic lymphohistiocytosis. Blood (2019) 134(Supplement_1):81–1. doi: 10.1182/blood-2019-129112
31. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T-cell therapy and the tumor microenvironment: current challenges and opportunities. Mol Ther - Oncolytics (2022) 25:69–77. doi: 10.1016/j.omto.2022.03.009
32. Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A, et al. TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat Commun (2020) 11(1):1749. doi: 10.1038/s41467-020-15404-8
33. Nixon BG, Gao S, Wang X, Li MO. TGFβ control of immune responses in cancer: a holistic immuno-oncology perspective. Nat Rev Immunol (2022) 23(6):346–62. doi: 10.1038/s41577-022-00796-z
34. Li C, Zhou W, Li C. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19(1):116. doi: 10.1158/1557-3125.HIPPO19-B11
35. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther (2021) 12(1):1–16. doi: 10.1186/s13287-020-02128-1
36. Krishnamoorthy M, Gerhardt L, Maleki Vareki S. Immunosuppressive effects of myeloid-derived suppressor cells in cancer and immunotherapy. Cells (2021) 10(5):1170. doi: 10.3390/cells10051170
37. Marofi F, Achmad H, Bokov D, Abdelbasset WK, Alsadoon Z, Chupradit S, et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem Cell Res Ther (2022) 13(1):140. doi: 10.1186/s13287-022-02819-x
38. Nalawade SA, Shafer P, Bajgain P, McKenna MK, Ali A, Kelly L, et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J ImmunoTher Cancer (2021) 9(11):e003237. doi: 10.1136/jitc-2021-003237
39. Khan JF, Khan AS, Brentjens RJ. Application of CAR T cells for the treatment of solid tumors. Prog Mol Biol Trans Sci (2019) 164:293–327. doi: 10.1016/bs.pmbts.2019.07.004
40. Abbott RC, Cross RS, Jenkins MR. Finding the keys to the CAR: identifying novel target antigens for T cell redirection immunotherapies. Int J Mol Sci (2020) 21(2):515. doi: 10.3390/ijms21020515
41. Dana H, Chalbatani GM, Jalali SA, Mirzaei HR, Grupp SA, Suarez ER, et al. CAR-T cells: early successes in blood cancer and challenges in solid tumors. Acta Pharm Sin B (2021) 11(5):1129–47. doi: 10.1016/j.apsb.2020.10.020
42. Yang B-H, Lin Y-SE, Shirinbak S, Yeh W-I, Pribadi M, Chu H-Y, et al. Combination of three unique anti-tumor modalities engineered into iPSC-derived T cells demonstrate a synergistic effect in overcoming tumor heterogeneity and cancer escape. Blood (2021) 138:2793. doi: 10.1182/blood-2021-153268
43. Giorgadze T, Fischel H, Tessier A, Norton K-A. Investigating two modes of cancer-associated antigen heterogeneity in an agent-based model of chimeric antigen receptor T-cell therapy. Cells (2022) 11(19), 3165. doi: 10.3390/cells11193165
44. Ham S, Lima LG, Lek E, Möller A. The impact of the cancer microenvironment on macrophage phenotypes. Front Immunol (2020) 11:1308. doi: 10.3389/fimmu.2020.01308
45. Khanali J, Azangou-Khyavy M, Boroomand-Saboor M, Ghasemi M, Niknejad H. JAK/STAT-dependent chimeric antigen receptor (CAR) expression: a design benefiting from a dual AND/OR gate aiming to increase specificity, reduce tumor escape and affect tumor microenvironment. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.638639
46. Correia MP, Stojanovic A, Bauer K, Juraeva D, Tykocinski L-O, Lorenz H-M, et al. Distinct human circulating NKp30+ FcϵRIγ+ CD8+ T cell population exhibiting high natural killer-like antitumor potential. Proc Natl Acad Sci (2018) 115(26):E5980–9. doi: 10.1073/pnas.1720564115
47. Correia MP, Stojanovic A, Wels WS, Cerwenka A. Innate-like NKp30+CD8+ T cells armed with TCR/CAR target tumor heterogeneity. OncoImmunology (2021) 10(1):1973783. doi: 10.1080/2162402X.2021.1973783
48. Song W, Zhang M. Use of CAR-T cell therapy, PD-1 blockade, and their combination for the treatment of hematological malignancies. Clin Immunol (2020) 214:108382. doi: 10.1016/j.clim.2020.108382
49. Wang J, Zhang R, Lin Z, Zhang S, Chen Y, Tang J, et al. CDK7 inhibitor THZ1 enhances antiPD-1 therapy efficacy via the p38α/MYC/PD-L1 signaling in non-small cell lung cancer. J Hematol Oncol (2020) 13:1–16. doi: 10.1186/s13045-020-00926-x
50. Wang D-R, Wu X-L, Sun Y-L. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduction Targeted Ther (2022) 7(1):331. doi: 10.1038/s41392-022-01136-2
51. Zhang Z-Z, Wang T, Wang X-F, Zhang Y-Q, Song S-X, Ma C-Q. Improving the ability of CAR-T cells to hit solid tumors: challenges and strategies. Pharmacol Res (2022) 175:106036. doi: 10.1016/j.phrs.2021.106036
52. Xia L, Du WL, Chen XY, Zhang YN. Targeting triple-negative breast cancer with combination therapy of EGFR CAR T cells and CDK7 InhibitionTargeting TNBC with EGFR CAR T cells and CDK7 inhibition. Cancer Immunol Res (2021) 9(6):707–22. doi: 10.1158/2326-6066.CIR-20-0405
53. Ping Y, Li F, Nan S, Zhang D, Shi X, Shan J, et al. Augmenting the effectiveness of CAR-T cells by enhanced self-delivery of PD-1-neutralizing scFv. Front Cell Dev Biol (2020) 8:803. doi: 10.3389/fcell.2020.00803
54. Zhu H, You Y, Shen Z, Shi L. EGFRvIII-CAR-T cells with PD-1 knockout have improved anti-glioma activity. Pathol Oncol Res (2020) 26(4):2135–41. doi: 10.1007/s12253-019-00759-1
55. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer (2019) 120(1):16–25. doi: 10.1038/s41416-018-0333-1
56. Liu J, Yang S, Cao B, Zhou G, Zhang F, Wang Y, et al. Targeting B7-H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. J Hematol Oncol (2021) 14(1):21. doi: 10.1186/s13045-020-01024-8
57. Saadey AA, Yousif A, Osborne N, Shahinfar R, Chen Y-L, Laster B, et al. Rebalancing TGFβ1/BMP signals in exhausted T cells unlocks responsiveness to immune checkpoint blockade therapy. Nat Immunol (2023) 24(2):280–94. doi: 10.1038/s41590-022-01384-y
58. Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol (2019) 12:1–10. doi: 10.1186/s13045-019-0813-7
59. Tousley AM, Rotiroti MC, Labanieh L, Rysavy LW, Kim W-J, Lareau C, et al. Co-Opting signalling molecules enables logic-gated control of CAR T cells. Nature (2023) 615(7952):507–16. doi: 10.1038/s41586-023-05778-2
60. Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol (2023) 20(1):49–62. doi: 10.1038/s41571-022-00704-3
61. Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Trans Med (2021) 13(591):eabd8836. doi: 10.1126/scitranslmed.abd8836
62. Labanieh L, Majzner RG, Mackall CL. Programming CAR-T cells to kill cancer. Nat Biomed Eng (2018) 2(6):377–91. doi: 10.1038/s41551-018-0235-9
63. Celichowski P, Turi M, Charvátová S, Radhakrishnan D, Feizi N, Chyra Z, et al. Tuning CARs: recent advances in modulating chimeric antigen receptor (CAR) T cell activity for improved safety, efficacy, and flexibility. J Trans Med (2023) 21(1):197. doi: 10.1186/s12967-023-04041-6
64. Miao L, Zhang J, Huang B, Zhang Z, Wang S, Tang F, et al. Special chimeric antigen receptor (CAR) modifications of T cells: a review. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.832765
65. Chmielewski M, Abken H. TRUCKS, the fourth-generation CAR T cells: current developments and clinical translation. Adv IN Cell AND Gene Ther (2020) 3(3):e84. doi: 10.1002/acg2.84
66. Glienke W, Dragon AC, Zimmermann K, Martyniszyn-Eiben A, Mertens M, Abken H, et al. GMP-compliant manufacturing of TRUCKs: CAR T cells targeting GD2 and releasing inducible IL-18. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.839783
67. Huang Z, Chavda VP, Bezbaruah R, Dhamne H, Yang D-H, Zhao H-B. CAR T-cell therapy for the management of mantle cell lymphoma. Mol Cancer (2023) 22(1):67. doi: 10.1186/s12943-023-01755-5
68. Silveira CRF, Corveloni AC, Caruso SR, Macêdo NA, Brussolo NM, Haddad F, et al. Cytokines as an important player in the context of CAR-T cell therapy for cancer: their role in tumor immunomodulation, manufacture, and clinical implications. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.947648
69. Hanssens H, Meeus F, De Veirman K, Breckpot K, Devoogdt N. The antigen-binding moiety in the driver's seat of CARs. Medicinal Res Rev (2022) 42(1):306–42. doi: 10.1002/med.21818
70. Liu B, Yan L, Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res (2019) 9(2):228.
71. Atsavapranee ES, Billingsley MM, Mitchell MJ. Delivery technologies for T cell gene editing: applications in cancer immunotherapy. eBioMedicine (2021) 67:103354. doi: 10.1016/j.ebiom.2021.103354
72. Razeghian E, Nasution MKM, Rahman HS, Gardanova ZR, Abdelbasset WK, Aravindhan S, et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther (2021) 12(1):428. doi: 10.1186/s13287-021-02510-7
73. Khan A, Sarkar E. CRISPR/Cas9 encouraged CAR-T cell immunotherapy reporting efficient and safe clinical results towards cancer. Cancer Treat Res Commun (2022) 33:100641. doi: 10.1016/j.ctarc.2022.100641
74. Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev (2013) 32(1):303–15. doi: 10.1007/s10555-012-9415-3
75. Xu M, Zhang T, Xia R, Wei Y, Wei X. Targeting the tumor stroma for cancer therapy. Mol Cancer (2022) 21(1):208. doi: 10.1186/s12943-022-01670-1
76. van der Spek YM, Kroep JR, Tollenaar RAEM, Mesker WE. Chemotherapy resistance and stromal targets in breast cancer treatment: a review. Mol Biol Rep (2020) 47(10):8169–77. doi: 10.1007/s11033-020-05853-1
77. Geng F, Guo J, Guo Q-Q, Xie Y, Dong L, Zhou Y, et al. A DNA vaccine expressing an optimized secreted FAPα induces enhanced anti-tumor activity by altering the tumor microenvironment in a murine model of breast cancer. Vaccine (2019) 37(31):4382–91. doi: 10.1016/j.vaccine.2019.06.012
78. Zhen Z, Tang W, Wang M, Zhou S, Wang H, Wu Z, et al. Protein nanocage mediated fibroblast-activation protein targeted photoimmunotherapy to enhance cytotoxic T cell infiltration and tumor control. Nano Lett (2017) 17(2):862–9. doi: 10.1021/acs.nanolett.6b04150
79. Yang M, Tkach D, Boyne A, Kazancioglu S, Duclert A, Poirot L, et al. Optimized two-step electroporation process to achieve efficient nonviral-mediated gene insertion into primary T cells. FEBS Open Bio (2022) 12(1):38–50. doi: 10.1002/2211-5463.13292
80. Lukjanov V, Koutná I, Šimara P. CAR T-cell production using nonviral approaches. J Immunol Res (2021) 2021, 6644685. doi: 10.1155/2021/6644685
81. Harris E, Elmer JJ. Optimization of electroporation and other non-viral gene delivery strategies for T cells. Biotechnol Prog (2021) 37(1):e3066. doi: 10.1002/btpr.3066
82. Pinto IS, Cordeiro RA, Faneca H. Polymer- and lipid-based gene delivery technology for CAR T cell therapy. J Controlled Release (2023) 353:196–215. doi: 10.1016/j.jconrel.2022.11.038
83. Gajewska-Naryniecka A, Szwedowicz U, Łapińska Z, Rudno-Rudzińska J, Kielan W, Kulbacka J. Irreversible electroporation in pancreatic Cancer—An evolving experimental and clinical method. Int J Mol Sci (2023) 24, 4381. doi: 10.3390/ijms24054381
84. Han X, Zhang N, Zhang Y, Li Z, Wang Y, Mao L, et al. Survival model database of human digestive system cells exposed to electroporation pulses: an in vitro and in silico study. Front Public Health (2022) 10:2863. doi: 10.3389/fpubh.2022.948562
85. Lee EW, Shahrouki P, Peterson S, Tafti BA, Ding P-X, Kee ST. Safety of irreversible electroporation ablation of the pancreas. Pancreas (2021) 50(9):1281–6. doi: 10.1097/MPA.0000000000001916
86. Sun Y, Su Y, Wang Y, Liu N, Li Y, Chen J, et al. CD19 CAR-T cells with membrane-bound IL-15 for b-cell acute lymphoblastic leukemia after failure of CD19 and CD22 CAR-T cells: case report. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.728962
87. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
88. Kath J, Du W, Pruene A, Braun T, Thommandru B, Turk R, et al. Pharmacological interventions enhance virus-free generation of TRAC-replaced CAR T cells. Mol Ther - Methods Clin Dev (2022) 25:311–30. doi: 10.1016/j.omtm.2022.03.018
89. Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat Biotechnol (2021) 39(1):56–63. doi: 10.1038/s41587-020-0601-5
90. Jo S, Das S, Williams A, Chretien A-S, Pagliardini T, Le Roy A, et al. Endowing universal CAR T-cell with immune-evasive properties using TALEN-gene editing. Nat Commun (2022) 13(1):3453. doi: 10.1038/s41467-022-30896-2
91. Pant A, Lim M. CAR-T therapy in GBM: current challenges and avenues for improvement. Cancers (2023) 15:1249. doi: 10.3390/cancers15041249
92. Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell (2022) 185(10):1745–63. doi: 10.1016/j.cell.2022.03.041
93. Jogalekar MP, Rajendran RL, Khan F, Dmello C, Gangadaran P, Ahn B-C. CAR T-Cell-Based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.925985
94. Lesch S, Nottebrock A, Rataj F, Heise C, Endres S, Kobold S. PD-1-CD28 fusion protein strengthens mesothelin-specific TRuC T cells in preclinical solid tumor models. Cell Oncol (2023) 46(1):227–35. doi: 10.1007/s13402-022-00747-9
Keywords: CAR T, immunosuppression, immune checkpoints, stromal barrier, transfection strategies
Citation: Khawar MB, Ge F, Afzal A and Sun H (2023) From barriers to novel strategies: smarter CAR T therapy hits hard to tumors. Front. Immunol. 14:1203230. doi: 10.3389/fimmu.2023.1203230
Received: 10 April 2023; Accepted: 22 June 2023;
Published: 14 July 2023.
Edited by:
Jiakai Hou, University of Houston, United StatesReviewed by:
Anliang Xia, Nanjing University, ChinaXiaoming Feng, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2023 Khawar, Ge, Afzal and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haibo Sun, ZnJlbmtzdW5AMTI2LmNvbQ==
†These authors have contributed equally to this work