 Yang Zhao
Yang Zhao Junfeng Du
Junfeng Du Xiaofei Shen
Xiaofei Shen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 March 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1157537
This article is part of the Research Topic New insights into Innate Immune cell-based immunotherapies in cancer View all 19 articles
Myeloid-derived suppressor cells (MDSCs) are one of the major negative regulators in tumor microenvironment (TME) due to their potent immunosuppressive capacity. MDSCs are the products of myeloid progenitor abnormal differentiation in bone marrow, which inhibits the immune response mediated by T cells, natural killer cells and dendritic cells; promotes the generation of regulatory T cells and tumor-associated macrophages; drives the immune escape; and finally leads to tumor progression and metastasis. In this review, we highlight key features of MDSCs biology in TME that are being explored as potential targets for tumor immunotherapy. We discuss the therapies and approaches that aim to reprogram TME from immunosuppressive to immunostimulatory circumstance, which prevents MDSC immunosuppression activity; promotes MDSC differentiation; and impacts MDSC recruitment and abundance in tumor site. We also summarize current advances in the identification of rational combinatorial strategies to improve clinical efficacy and outcomes of cancer patients, via deeply understanding and pursuing the mechanisms and characterization of MDSCs generation and suppression in TME.
Accumulating evidence shows that the formation of the tumor immune microenvironment (TIME) is closely related to tumor malignant development and metastasis. The occurrence and progression of tumors is a complex pathophysiological process. Most researchers believe that the TIME includes: 1) secretion of immunosuppressive factors, including interleukins, chemokines, growth factors, and other cytokines, inducing inflammatory responses and forming a local milieu conductive to tumor propagation (1, 2); and 2) many immune cell components, such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T (Treg) cells, tumor-associated dendritic cells, mast cells, and type 2 natural killer T (NKT) cells, are involved in tumor immunosuppression (3–6). Due to their contribution to tumorigenesis and progression, MDSCs are recognized as the critical factor in TIME and their function exacerbates the disease. MDSCs may be the basis of tumor immunosuppression. First, although in tumor patients, one or several of the above-mentioned immunosuppressive cells are detected, MDSCs can be detected in most patients. Second, MDSCs suppress the innate immune response and delay the adaptive immune response. Third. MDSCs can induce expansion of immunosuppressive cells (TAMs and Treg cells).
MDSCs, have high heterogeneity, were first referred around 30 years ago and have unique characteristics and an important place in many diseases, especially cancer (7). MDSCs are a group of innate immune cells derived from myeloid lineage at different developmental stages with strong heterogeneity, which can differentiate into dendritic cells (DCs), macrophages and granulocytes under physiological conditions. However, under pathological conditions, like inflammation, trauma, tumors and autoimmune diseases, release of immunosuppressive factors block the differentiation of myeloid progenitors, promote their expansion, and further recruit them to the blood, spleen, liver, and tumor tissue (7–9). MDSCs play a pivotal and central role in governing and maintaining the TIME in solid tumors (10). MDSCs are composed of the myeloid cells with similar biological activities, but distinct phenotypes. Unlike monocytes, macrophages and DCs, which express specific molecular markers on the cell surface, MDSCs are composed of a mixture of granulocytes and monocytes, without clear and specific markers on their surface (11). In mice, MDSCs are defined as cells that co-express myeloid antigens Gr-1 and CD11b (CD11b+Gr-1+). In addition, according to the expression of Ly6C and Ly6G, CD11b+Gr-1+ cells can be further divided into granulocytic MDSCs (CD11b+Ly6G+Ly6Clow, G-MDSC) and monocytic MDSCs (CD11b+Ly6G-Ly6Chigh, M-MDSC) subtypes (9, 12). MDSCs induced by human solid tumors were divided into two subgroups: CD33+HLA-DRlowHIF1α+/STAT3+ and CD11b+HLA-DRlowC/EBPβ+, according to their phenotypes and molecular mechanisms impeding other immune cells (13). Human monocytic MDSCs (M-MDSCs) were defined as CD11b+CD14+HLADR-/lowCD15-, while granulocytic MDSCs (G-MDSCs) were defined as CD11b+CD14-CD15+ or CD11b+CD14-CD66b+ (14, 15).
In recent years, it has been discovered that MDSCs directly participate in the promotion of tumor progression and metastasis and are closely related to the clinical treatment of malignant tumors. In this review, we describe the functional and regulatory mechanism of MDSCs within TME. Notable clinical success in tumor immunotherapies, such as immune checkpoint blockade (ICB), and adoptive cell therapy (ACT), have reinvigorated our interest in the field of immunotherapy and established it as a mode of tumor therapy along with traditional strategies, like surgery, chemotherapy and radiotherapy (16, 17). Therefore, we will discuss the emerging data associated with the therapeutic strategies that targeting MDSCs. Furtherly, we also highlight what the aspects of MDSCs requires an in-depth understanding to discriminate and evaluate reasonable and sensitive combinatorial strategies to increase the efficacy of tumor immunotherapy for cancer patients.
In the setting of cancer, MDSCs can be generated by common myeloid progenitor (CMP) in bone marrow, recruited to tumor site and expand massively by tumor-derived factors or inflammatory signals, including inflammatory cytokines, chemokines, growth factors, and other pathological mediators accelerate the expansion and recruitment of immature myeloid cells to tumor site to suppress the host antitumor response (Figure 1). Classical ideas propose that the direct immunosuppressive function of MDSCs depends on secreting inhibitory factors, including production of nitric oxide (NO), elimination of key nutritional factors by depleting L-arginine (via arginase1), sequestering L-cysteine, or decreasing local tryptophan levels due to the activity of indole amine 2,3 dioxygenase (IDO) (18–21) (Figure 1). The mechanisms of immunosuppression by G-MDSCs and M-MDSCs are distinct to the tumor site. Tumor-mediated G-MDSC mainly inhibited T cells via reactive oxygen species (ROS), whereas M-MDSC inhibited T cell mainly through arginase and inducible NO synthase (iNOS) (22–25). The deprivation of L-Arginine, catabolized by MDSC-secreted Arg-1, restrained T cells proliferation via disrupting the expression of CD3ξ chain (19, 26). Nitric oxide (NO) is synthesized by NOSs, which are ubiquitously expressed in MDSCs, and induce T cell apoptosis by blocking JAK/STAT/nuclear factor kappa-B (NF-κB) signaling pathway (27). Peroxide nitrate (PNT) is produced by MDSCs and inhibits CD8+ T cells migration through reducing the integration of MHC I molecules with antigenic peptides on tumor cells and nitrate chemokines (28, 29). In response to a variety of growth factors and cytokines, the progenitor of MDSCs drive a complex transcription network, allowing for their expansion and preventing the further differentiation. The abovementioned factors trigger multiple signaling pathways in MDSCs (11, 30), and most of them converge on the activation of signal transducer and activator of transcription Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling, which upregulates immunosuppressive mediators such as iNOS, ROS, and arginase (31). STAT3 activation promoted the accumulation of MDSCs in melanoma (32), and STAT3 inhibition weakened the suppressive function of MDSCs (33). The MDSCs amplification and function are related to the downstream signals of STAT3. The calcium-binding pro-inflammatory protein factors S100A9 and S100A8, upregulated by STAT3 activation, could block the differentiation and maturation of dendritic cells (DC) and promote MDSCs accumulation (34). The exact mechanism of this process remains to be explored, but some scholars have pointed out that this may be related to the S100A9 and S100A8 heterodimers participating in the formation of the nicotinamide adenine dinucleotide phosphoric acid (NADPH) oxidase complex, which increasing the production of ROS in myeloid cells. STAT1 is the main transcription factor under IFN-γ or IL-1β stimulation, which is believed to have an important relationship with the activity of iNOS and arginase. Some studies point out that MDSCs lacking STAT1 are unable to suppress T cell function due to decreased secretion of iNOS and lower expression of arginase (35). STAT6 activated by IL-4 and IL-13 enhances the activity of arginase and inducing transforming growth factor β (TGFβ) production by MDSCs through IL-4Rα (36). Besides, Nuclear factor (NF-κB), prostaglandin E2 (PGE2)/cyclooxygenase 2 (COX2), and Ras were also of the great significance in the molecular mechanism of suppressing T cell activity mediated by specific subgroups of MDSCs (37, 38). MDSCs are affected by both novel anti-cancer immune therapies, as well as the conventional treatments such as radiotherapy. Following radiotherapy, cytoplasmic double stranded DNA stimulates the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway, resulting in type I interferon production (39). cGAS/STING signaling becomes a key factor in inhibiting MDSC function after radiotherapy via multiple mechanisms. The treatment of cGAMP, the STING agonist, prevented MDSC immunosuppressive function via reducing NO in B16 melanoma tumor-bearing mice (40). Furthermore, STING agonist treatment combined with the STAT3 inhibitor and markedly regressed tumor growth in syngeneic mice by increasing CD8+ T cells and Tregs and MDSCs in TME (41). Collectively, cGAS/STING and JAK/STAT pathway are both recognized as the central signaling pathway in controlling MDSC generation, accumulations and function in tumor progression. The rationale combinatorial treatment of STAT inhibitors and STING agonists will be potential therapeutic strategy and make advances in tumor immunotherapy.
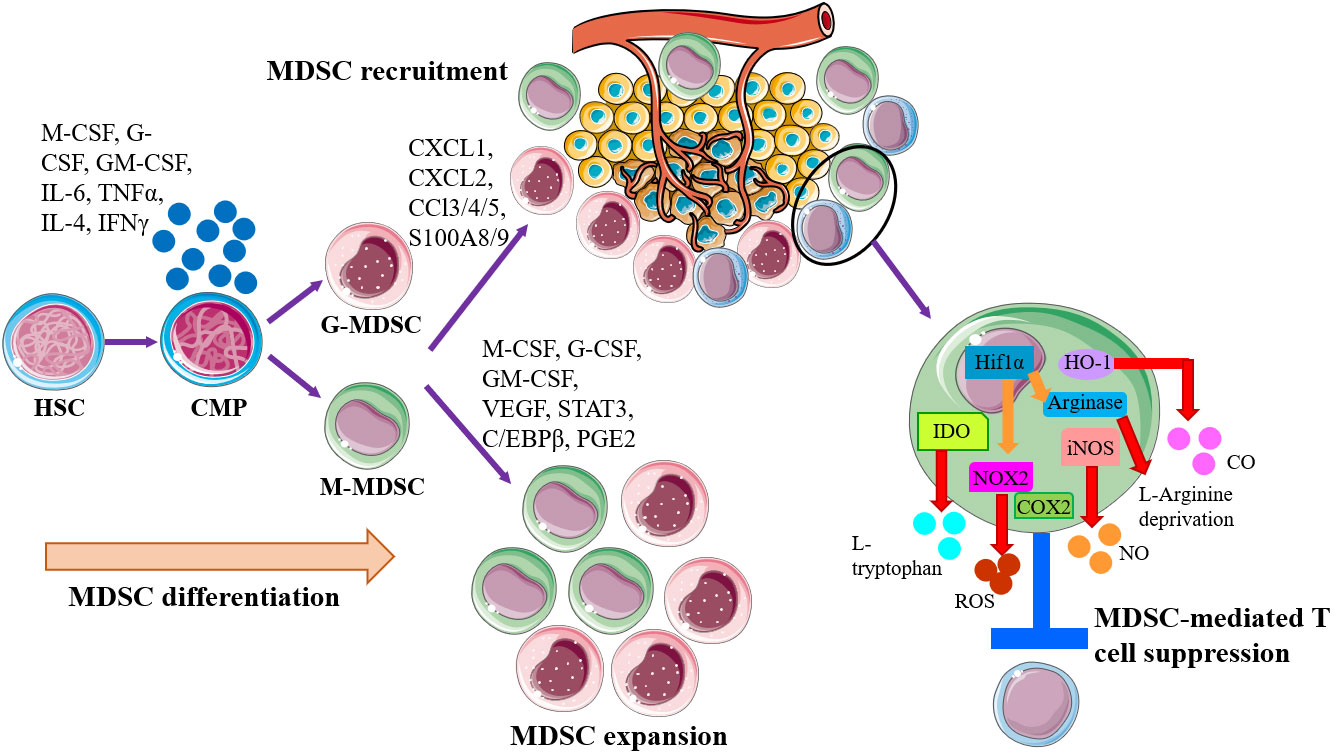
Figure 1 Schematic of MDSC generation, expansion, recruitment, and role in the establishment of tumor immune inhibitory microenvironment. MDSCs are generally generated by common myeloid progenitor (CMP) in bone marrow, governed by the abnormal or pathological signals, especially proinflammatory factors. Then MDSCs are recruited to tumor site by tumor-derived factors or inflammatory signals for establishing the microenvironments that promoting tumor cell escape. Within the tumor microenvironment (TME), both monocytic-MDSC (M-MDSC) and granulocytic-MDSC (G-MDSC) will expand and exert immunosuppressive functions to induce T cell suppression and anergy via multiple mechanisms, like arginase1, iNOS, IDO, HO-1, and NOX2.
Another major mechanism mediating immunosuppression is the induction and recruitment of other regulatory cells, like Treg cells (42). The characteristic of the TME enable crosstalk between MDSCs and Tregs that allows them to modulate each other mutually. MDSCs in TIME selectively facilitated expansion and induction of Treg cells via a TGFβ-dependent manner (43), or dependent on MDSC-secreted IL-10 and IFN-γ (23). Furthermore, MDSCs can provide additional signals for Treg cell induction and development via upregulating ligands expressed on the surface of MDSCs for several costimulatory molecules, such as CD86, programmed death ligand (PD-L1), and leukocyte immunoglobulinlike receptor subfamily B (LILRB4). MDSCs promoted the induction and expansion of tumor-specific Treg cells via taking in tumor antigens and presenting them to T cells, also converted T cells in other differentiated states into Treg cells to assist tumor evasion (44, 45). The correlation of arginase-1 expression with increasing expression of immune checkpoint receptor and ligands results in more potent suppressive activity of MDSCs (46). Twofold repression caused by MDSCs and Treg cells will create strong immune tolerance and promote tumor progression and propagation.
Thus, there is a consensus that the TME can induce MDSCs with the more potent suppressive activity via increasing the expression of a series of immunosuppressive molecules. Exploring more suitable approaches to blockade the immunosuppressive molecules expressed by MDSCs will be hopeful to disrupt the immunosuppression mediated by MDSCs in TME.
The TME plays an important role in supporting and promoting tumor growth and metastasis, where MDSCs have an important role in immuno-suppression. More studies are trying to explore and achieve tumor therapy by changing the TME (soil) to prevent the activity of tumor cells (seeds). Targeting the TIME has become a new approach for tumor therapy in recent years. In view of the important role of MDSCs in the TIME, therapeutic strategies targeting MDSCs are being explored (Table 1): 1) promoting the differentiation and maturation of MDSCs; 2) inhibition of the expansion and accumulation of MDSCs; 3) elimination of MDSCs in TME; 4) abolition of MDSCs immunosuppression (Figure 2).

Table 1 The therapeutic strategies of targeting MDSCs in preclinical cancer trails.
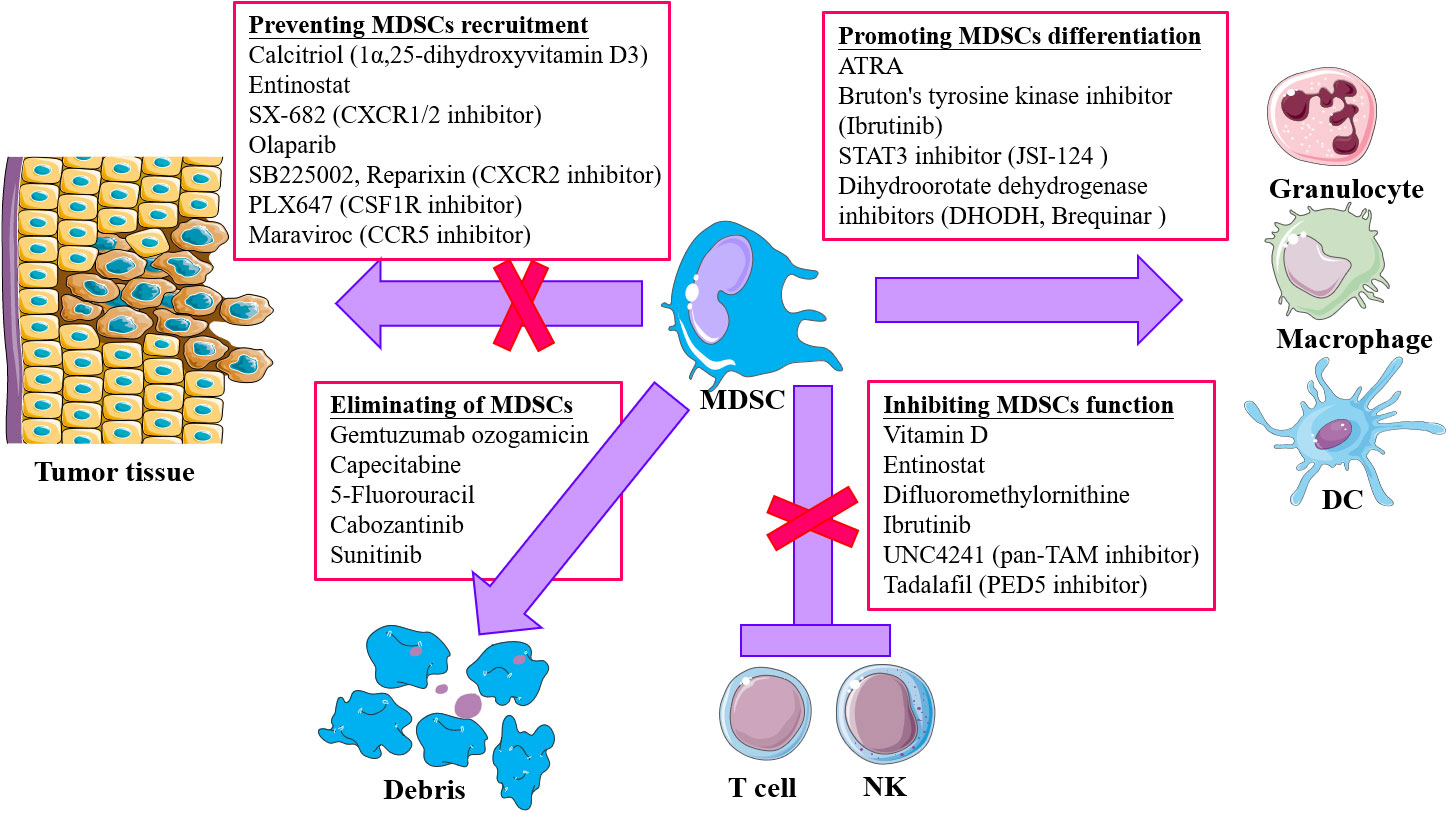
Figure 2 Strategies for targeting MDSCs in tumor immunotherapy. Four main approaches are included: 1) Accelerating and promoting differentiation and maturation via multiple agents, like all-trans retinoic acid (ATRA), STAT3 inhibitor, dihydroorotate dehydrogenase inhibitors, and Bruton’s tyrosine kinase (RTK) inhibitor; 2) blocking MDSCs recruitment and infiltration into tumor microenvironment (TME) through chemokine receptor inhibitors targeting chemokine receptors responsible for migration of MDSCs to TME; 3) depletion of MDSCs by low-dose chemotherapy and tyrosine kinase inhibitor (TKi); 4) attenuating the suppressive activity of MDSCs via targeting and inhibiting the effector molecules, like iNOS, COX2.
MDSCs are a mixture of immature myeloid cell populations with high heterogeneity and immunosuppressive activity. All-trans retinoic acid (ARTA) could promote the differentiation of MDSCs into granulocytes, macrophages and DC, improve the host anti-tumor immune response via neutralizing the production of ROS (79–81). For example, the administration of formic acid receptor (RAR) antagonist (which does not affect retinol X receptor (RXR)) in mice can cause the accumulation of granulocytes in various hematopoietic organs, including bone marrow, suggesting that the RAR pathway blocks the differentiation and maturation of granulocyte precursors. 1,25-dihydroxy vitamin D3 (1,25(OH)2D3), which is the active metabolite of vitamin D3, has been identified as a potent natural modulator of innate and adaptive immunity. Vitamin D3 combined with various cytokines induced the differentiation of CD34+ progenitors isolated from patients with head and neck squamous cell carcinoma (HNSCC), resulting in increased numbers of cells phenotypically similar to mature DCs (82). Recent studies have provided important insights that primitive myeloid leukemic cell lines can be driven to differentiate into monocyte-like cells by 1,25(OH)2D3, which may be useful in differentiation therapy of myeloid leukemia and myelodysplastic syndromes (MDS) (83, 84). However, the role of vitamin D3 in myeloid cell differentiation remains controversial. A recent study showed that DCs treated with 1α,25(OH)2D3 (calcitriol) did not differentiate or mature, locking the cells in a tolerogenic/immature state (85). Vitamin D3-induced tolerogenic DCs are thought to develop their regulatory properties through a semimature profile, inhibition or reduction of T-cell responses, and switching the immune response to a Th2 profile (86–89). Vitamin-D3-induced tolerogenic DCs with the semimature phenotype, anti-inflammatory profile, and low capacity to induce T-cell proliferation, can be used clinical for inducing immunotolerance (90). Calcitriol attenuated the recruitment of MDSCs and increased infiltration of cytotoxic T cells following radiotherapy in hepatocellular carcinoma and prostate cancer (82, 91). Thus, the role of vitamin D3 in tumor therapy is complex and the application of vitamin D3 for clinical use by targeting MDSCs still needs more study. By promoting the development of MDSCs into normal monocytes and granulocytes, not only reduces MDSCs, but also increases the mature myeloid cells in TIME, thereby inhibiting tumor growth.
As mentioned above, the expansion of MDSCs is regulated by tumor-derived suppressive factors secreted by tumor cells and released by the TME. It mainly includes IL-6, GM-CSF, G-CSF, VEGF, COX-2 and other cytokines, which can trigger a variety of different signal transduction and signal activation pathways in MDSC. The STAT3 signaling pathway is an important regulator for MDSC amplification mediated by these factors and could be the ideal target. STAT protein has an N-terminal DNA-binding domain and C-terminal protein-binding domain. Tumor-derived suppressive factors binding to corresponding receptors leads to continuous activation of STAT3, which then upregulates expression of STAT3-related genes and produces proteins (survivin and cyclin) and matrix metalloproteinase-9 (MMP-9) that promote MDSCs expansion. Targeting the STAT3 signaling pathway has become a research hotspot in the inhibition of MDSC expansion (92). MMP-9 is an important target for tumor therapy via inhibiting amplification of MDSCs and facilitating formation of the TME. MMP-9 inhibitors promote the normalization of hematopoietic function, thus reducing the production of MDSCs in tumor-bearing BALB-neuT mice expressing an activated rat c-erbB-2/neu transgene model (93). VEGF is currently recognized as the most powerful angiogenic factor, which can specifically act on vascular endothelial cells and promote vascular endothelial hyperplasia. Studies have confirmed that VEGF in tumor tissues can promote the generation of neovascularization, inhibit the development of DCs and induce the generation of MDSCs (94, 95). Therefore, blockade of VEGF could be another approach for tumor therapy, by removing immunosuppression. In a tumor-bearing mouse model, tumor growth was significantly inhibited after administration of anti-VEGF antibody, and there was a reduced number of MDSCs in tumor tissue and peripheral blood. However, the mechanism by which anti-VEGF monoclonal antibody (mAb) inhibits the expansion of MDSCs remains to be elucidated (96, 97). Bevacizumab was the first FDA-approved monoclonal antibody against VEGF, which brings hope to patients with advanced tumors by anti-tumor microangiogenesis and inhibiting the progression of metastatic lesions. Although, the application of anti-VEGF mAb has been verified and evaluated in a multitude of clinical trials for tumor therapy, because of the multiple effects mediated by blocking VEGF, the efficacy could not be only attributed to MDSC reduction. Sunitinib, as the anti-angiogenic drug, is a receptor tyrosine-kinase inhibitor and immunomodulator, that potently prevents MDSC accumulation and restores normal T-cell function in tumor-bearing mice, independent of its capacity to inhibit tumor progression, as well as reverses MDSC accumulation and T-cell inhibition even in the blood of non-responder renal cell carcinoma (RCC) patients (98).
Chemokines as key mediators of MDSCs recruitment have been extensively studied in many tumor models and cancer patients (Table 1). The recruitment of MDSCs from bone marrow and spleen to tumor tissues is mainly through multiple signaling pathways. A pivotal role of CCL2-CCR2 signaling in MDSC recruitment and tumor progression has been demonstrated in melanoma and hepatocellular carcinoma (HCC) mouse models (99, 100). Thus, blocking CCL2 with soluble CCR2 fragment or inhibition of CCR2 with blocking antibody could decrease tumor accumulation of MDSCs in the tumor bone metastases model by injecting prostate cancer cells directly into murine tibiae, making it a potential target for anti-tumor therapy (101). mCCR5-Ig fusion protein, anti-CCR5 antibody, or even CCL5-neutralizing mAb were all found that could reduce the number and suppressive capacity of tumor infiltration MDSCs, prevent the tumor metastasis, promote the survival of B16 tumor-bearing mouse and even improve the efficacy of anti-PD-1 tumor therapy (102, 103). Chemokine (C-X-C motif) ligand 8, also known as IL-8, highly expressed in various tumors, including colon, ovarian, breast, pancreatic, prostate, and hematological malignancies (104–106), has been demonstrated that could recruit MDSCs to tumor sites via CXCR1/CXCR2 (107). The treatment of Reparixin, the pharmacological inhibitor of CXCR1 and CXCR2, caused the significant reduction of G-MDSCs numbers, in colon adenocarcinoma HT29 xenograft tumor and colon carcinoma CT26-GM-derived subcutaneous tumor models (108–110). Furthermore, inhibition of MDSC trafficking by SX-682, a CXCR1/2 inhibitor, enhanced NK-Cell immunotherapy in head and neck cancer models (55). HuMax-IL8, an anti-IL-8 mAb, reduced the number of G-MDSCs in MDA-MB-231 breast cancer xenografts (111). CXCL8 levels determine the efficacy of sunitinib treatment, which was demonstrated to effectively target MDSCs, suggesting that CXCL8 acts as a potential target in anti-tumor therapy (112). Blocking MDSC recruitment to tumor tissues may be an effective approach for disrupting the formation of TIME and improving the efficacy of anti-tumor therapy.
MDSCs mainly express reactive ROS, arginase1, NOS and peroxynitrite to exert their immunosuppressive function (28, 113). Therefore, appropriate inhibition of those factors, serving as important potential therapeutic targets, can eliminate the immunosuppression of MDSCs. ROS, as part of the major mechanism by which MDSCs suppress T-cell responses, activate anti-oxidative pathways and induce transcriptional programs that regulate the fate and function of MDSCs. Nuclear factor erythroid 2-related factor 2 (Nrf2) activation in regulating the constitutive activation and availability of antioxidant enzymes, including NADPH, NADPH quinone oxidoreductase 1 (NQO1), hem oxygenase (HO), might be a central mechanism enabling cells to increase mitochondrial ATP production by simultaneously counteracting subsequent high ROS levels (114). Selective activation of Nrf2 can decrease the intracellular ROS production, inhibit the immunosuppression of MDSCs, prevent tumor metastasis, and induce tumor regression. Furthermore, the synthetic triterpenoid C-28 methyl ester of 2-cyano-3,12-dioxooleana-1,9,-dien-28-oic acid (CDDO-Me) completely abolished the immunosuppressive activity of MDSCs by reducing ROS production in mouse tumor models (115). Moreover, the treatment of pancreatic cancer patients with CDDO-Me did not affect the number of MDSCs in peripheral blood of patients but significantly improved the immune response (115). Agents that target MDSCs, such as sanguinarine (SNG), are now being considered for treatment of lung cancer. SNG was found to inhibit the immunosuppressive activity of MDSCs via decreasing the expression of Arg-1, iNOS, and ROS, as well as inducing the differentiation of MDSCs into macrophages and DCs through the NF-κB pathway in vitro from Lewis lung cancer mouse model (116). The type I interferons pathway is well known to promote anti-tumor immunity by diverse mechanisms. Emerging evidence shows that the downregulation of the IFNAR1 chain is found in MDSC from cancer patients and mouse tumor models. The decrease in IFNAR1 depends on the activation of the p38 protein kinase and is required for activation of the immunosuppressive phenotype (117). Stabilizing IFNAR1 using p38 inhibitor combined with IFN induction therapy elicits a robust anti-tumor effect via undermining suppressive activity of MDSCs in tumor bearing mice (117). The JAK/STAT signaling pathway is one of the well-known pathways induces immune escape of tumors via cytokines and growth factors to control MDSC generation and differentiation (118, 119). Blockade of STAT3, STAT5 or even NF-κB by the selective inhibitors can inhibit the immunosuppression of MDSCs (120). AMP-activated protein kinase α (AMPKα) signaling was increased in tumor-MDSCs from tumor-bearing mice and patients with ovarian cancer, which was induced by tumor-derived GM-CSF and occurred in a STAT5-dependent manner (121). In addition, genetic deletion of ampkα1-coding gene, prkaa1 antagonized M-MDSC differentiation to macrophages and re-routed M-MDSC, but not G-MDSC, into cells that elicited direct antitumor cytotoxic effects through NOS2-mediated actions, suggesting the therapeutic use of AMPK inhibitors to overcome MDSC-induced T-cell dysfunction and AMPK inhibition as a potential therapeutic strategy to restore protective myelopoiesis in cancer. G-MDSCs in the TME spontaneously die by ferroptosis, inducing the release of oxygenated lipids and limiting the activity of human and mouse T cells, although decreasing the presence of G-MDSCs (122). Thus, genetic and pharmacological inhibition of ferroptosis by liproxstatin-1, abolishes suppressive activity of G-MDSCs, reduces tumor progression and synergizes with immune checkpoint blockade (ICB) to suppress tumor growth in immunocompetent mice (122). However, induction of ferroptosis in immunocompetent mice promotes tumor growth. Therefore, ferroptosis is a unique and targetable immunosuppressive mechanism of G-MDSCs in TME that can be pharmacologically modulated to limit tumor progression. In human hepatocellular carcinoma (HCC), the tumor-surrounding fibrotic livers were markedly enriched with M-MDSCs, along with the poor survival rates. Mechanistically, activated hematopoietic stem cell (HSC) induced monocyte-intrinsic p38 mitogen-activated protein kinase (MAPK) signaling to trigger enhancer reprogramming for M-MDSC development and immunosuppression (123). Treatment with p38 inhibitor inhibited HSC-M-MDSC crosstalk to prevent HCC growth (123). Concomitant with patient-derived M-MDSC suppression by i-BET762, combined treatment with anti-PD-L1 synergistically enhanced tumor-infiltrating lymphocytes, resulting in tumor eradication and prolonging survival in the fibrotic-HCC mouse model (123). It has been reported that mouse and human G-MDSCs exclusively upregulate fatty acid transport protein 2 (FATP2), which was controlled by GM-CSF, through activation of the STAT5 (124). The selective pharmacological inhibition of FATP2 abrogated the suppressive activity of G-MDSCs and substantially delayed tumor progression (124). In combination with immune checkpoint inhibitors (ICIs), FATP2 inhibition blocked tumor progression in mice and has the potential to improve the efficacy of cancer therapy.
An initial attempt was made to clear the MDSCs with antibodies against Gr-1, but since Gr-1 is not specifically expressed by MDSCs, which is also expressed by mature granulocytes. Therefore, the elimination of MDSCs may also lead to a decline in normal immune cells. In addition, once the plasma concentration of the antibody in plasma decreases or the immune system responds to the antibody, the number of MDSCs increases rapidly, which enhances the immunosuppressive function of MDSCs in the TME. Low dose of chemotherapy, such as 5-fluorouracil (5FU), paclitaxel, cisplatin and gemcitabine, has been shown to effectively eliminate MDSC in tumor-bearing mice, and enhanced anti-tumor immunity (72, 73, 125–127). The number of MDSCs in the spleen was significantly reduced, although DCs, T cells, NK cells, macrophages and B cells were not significantly affected. The mechanism may be that the chemotherapy drugs belong to base analogs, which can prolong and block the DNA synthesis in the cell cycle and induce cell death. However, the mechanism of selective killing of MDSCs needs to be further clarified. Additionally, subclinical doses of platinum-based drugs, such as cisplatin, prevented the generation and suppressive activity of M-MDSCs by inhibiting STAT3-COX2 signaling pathway, along with decreasing COX2 and arginase1 expression in M-MDSCs of melanoma and head and neck squamous cell carcinoma (HNSCC) patients (128). Therefore, some chemotherapy drugs can play an active role in anti-tumor immunotherapy by targeting MDSCs in a certain dosage and course of treatment.
The remodeling of metabolic states also contributes to the shape of the TIME and plays an important role in regulating MDSCs in the TME. The radiotherapy-augmented Warburg effect helps myeloid cells to acquire an immunosuppressive phenotype, resulting in limited treatment efficacy for pancreatic ductal adenocarcinoma (PDAC) (129). Sustained increase in lactate secretion, resulting from the radiation augmented Warburg effect, was responsible for the enhanced immunosuppressive phenotype of MDSCs after radiotherapy (129). Thus, targeting lactate derived from tumor cells and the hypoxia-inducible factor-1α (HIF-1α) signaling in MDSCs could reinstate antitumor T-cell responses and inhibit tumor progression after radiotherapy in pancreatic cancer, indicating distinct promise for clinical therapies to alleviate radio resistance in PDAC. Glutamine metabolism is a crucial element of cancer cell metabolism. Glutamine is important for nucleotide synthesis, amino acid production, redox balance, glycosylation, extracellular matrix production, autophagy, and epigenetics (130, 131). Emerging evidence shows that targeting tumor glutamine metabolism leads to a decrease in G-CSF and hence recruitment and generation of MDSCs as well as immunogenic cell death, leading to an increase in inflammatory TAMs (132). Alternatively, inhibiting glutamine metabolism of the MDSCs themselves not only led to activation-induced cell death and conversion of MDSCs to inflammatory macrophages, also impaired suppressive function of MDSCs via inhibiting IDO expression in the tumor and MDSCs, that resulted in a marked decrease in kynurenine levels, and rendered checkpoint blockade-resistant tumors susceptible to immunotherapy in tumor-bearing mice (132). Therefore, the application of glutamine antagonism in synergistic targeting inhibition of tumor and MDSCs may hold promise for clinical therapy to inhibit tumor growth and metastasis. Therapeutic liver-X nuclear receptor (LXR) agonism was also found to reduce MDSC abundance in murine models and in patients treated in a first-in-human dose escalation phase 1 trial, accompanied with the activation of cytotoxic T lymphocyte (CTL) responses in mice and patients (133).
Tumor immunotherapy, such as ICB and adoptive cell therapy, has attracted much attention in recent years due to its remarkable efficacy. However, preliminary and limited success is achieved in hematological malignancies and in certain solid tumors, owing to the limitations in curative effect, or in technology. Combined treatment strategy is suggested which may improve the efficacy of mono-immunotherapy and compensate for the deficiency of monotherapy. MDSCs mediate tumor metastasis and are implicated in immune evasion through shaping the TME, and are referred as the “queen bee” of the TME (134). Strategies to reverse the suppressive TME should also attract and activate immune effectors with antitumor activity (Table 2). Cytokine-induced killer (CIK) cell-based immunotherapy is effective as adjuvant therapy in HCC with early stage but lacks efficacy in advanced HCC. MDSCs are increased in response to CIK cell therapy and subsequently may be targeted to provide an additional therapeutic benefit. A study on immunosuppressive mechanisms focusing on CIKs found that combination treatment with a PDE5 inhibitor reversed the MDSC suppressor function via arginase-1 and iNOS blockade and systemic treatment with a PDE5 inhibitor prevented MDSC accumulation in the TME of the tumor bearing mice (135). Similarly, treatment with a PDE5 inhibitor suppressed CD14+HLA-DR−/low MDSCs immunosuppressive activity and enhanced CIK activity against human HCC cell lines in vitro, suggesting targeting MDSCs is an efficient strategy to enhance the antitumor efficacy of CIKs for the treatment of patients with HCC. The possible combination of olaparib with EGFRvIII-targeting CAR (806-28Z CAR) T cells has been explored (57). The hostile TME is also one of the major obstacles to the efficacy of chimeric antigen receptor modified T (CAR-T) cells, and the recruitment of MDSCs within the TME may contribute to the unsatisfactory performance of CAR-T cells in solid tumors. Olaparib might suppress the recruitment of MDSCs to improve the TIME, which contributes to the infiltration and survival of CAR-T cells on breast cancer in mice (57). The additional mechanistic rationale for combining the third-generation PARPi (olaparib) with CAR-T therapy for the treatment of breast cancer was supported. GPC3-CAR T cell treatment together with C1632, the inhibition of Lin28, which targets IDO1 and PDL1, led to enhanced anti-tumor activity in a HCC xenograft mouse model (136). Combination of targeting IDO1 and PDL1 with CAR-T cells serves as a dual targeting agent against tumor cells and MDSCs in TME and enhances immunotherapeutic potential of CAR-T cells against tumor.
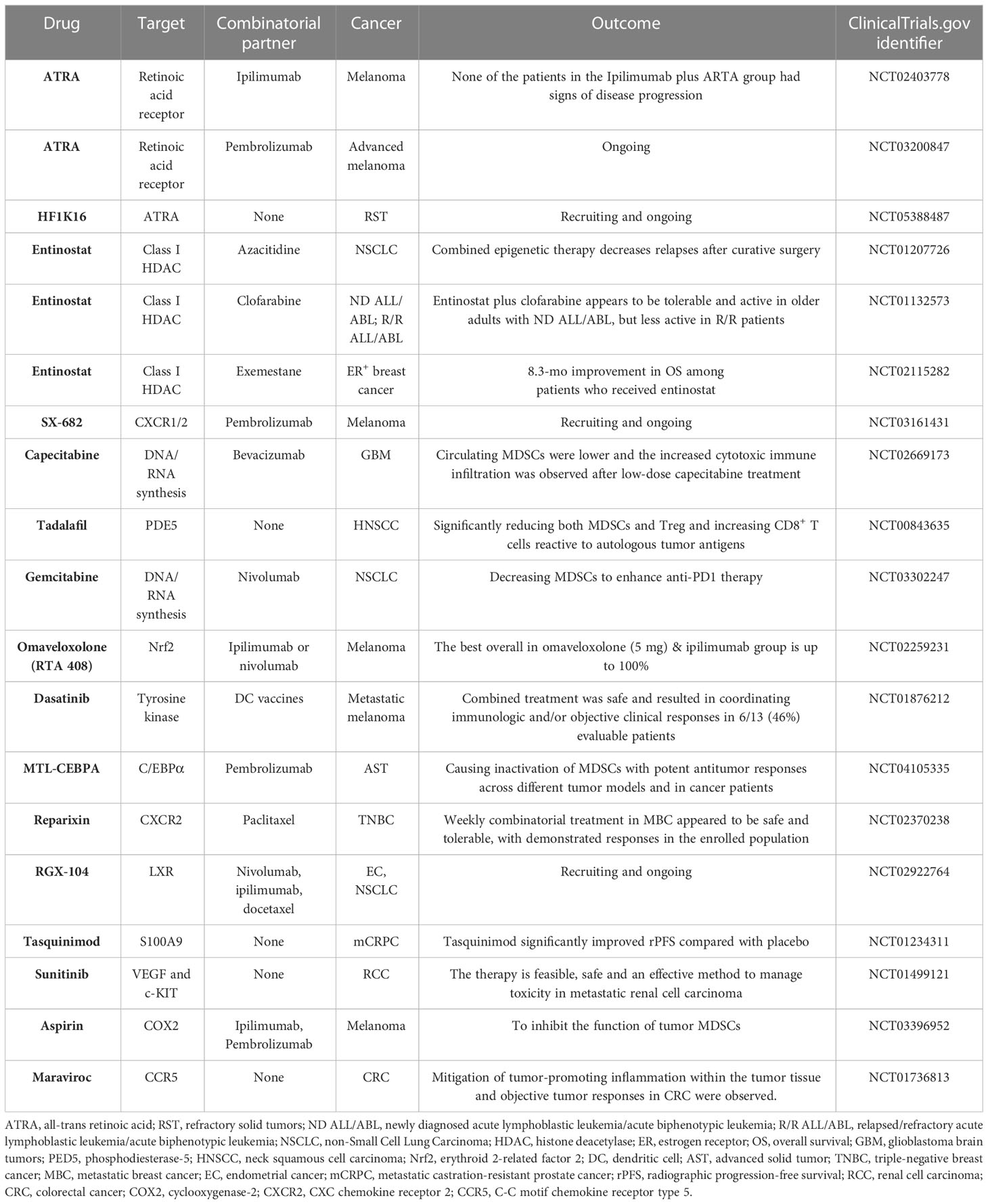
Table 2 Summary of clinical trials targeting MDSCs in cancer.
Although ICB therapy has made remarkable achievements in tumor immunotherapy, there is still a large proportion of patients that do not respond to ICIs or develop resistance (137, 138). Furthermore, ICB therapy is disappointing with a response rate < 10% in cancers with a poorly immunogenic or “cold” TIME, requiring further strategies for effective immunotherapy (139, 140). Immunotherapy non-responders often harbor high levels of circulating MDSCs which can predict the response to cancer immunotherapies, which is an important factor in developing resistance to ICB therapy and mediates immunosuppression in TME, hindering the efficacy of such therapy (141). In particular, the levels of MDSCs indicate whether the patients will respond to ICIs, which the close association between MDSCs level in patients with the efficacy of anti-PD1 or anti-CTLA4 therapy has been observed (142–144). PD-L1 is usually expressed in the majority of cancers, and PD-L1 expression by host myeloid cells is more effective than that on cancer cells in suppressing CTL function (145–149). MDSCs may also suppress CTL activity by PD-L1-dependent and -independent mechanisms (29). Therefore, combining TAM and G-MDSC inhibitors reduced both populations in the tumor site, and dramatically enhanced the effect of ICB with anti-PD-1 in our preclinical model of cholangiocarcinoma (CCA) (150). Combining ICI treatment with MDSC depletion has been successful and has been investigated in some pre-clinical studies. Combined treatment using entinostat and 5-azacytidine, epigenetic modulatory drugs, with ICB antibodies (anti-PD1 and anti-CTLA4), led to complete tumor regression and metastatic progression in the aggressive triple-negative breast cancer (TNBC) model 4T1, with >80% survival rate 100 days after tumor implantation (54, 151). IL-6 plays a major role in the accumulation and activation of MDSCs during tumor development. Importantly, increased IL-6 levels positively correlate with disease progression and MDSC enrichment in cancer patients (152). Preclinical studies of IL-6/IL-6R blockade to target MDSCs in cancer have been conducted. IL-6 blockade or anti-IL-6R monoclonal antibody reversed the effect of ICIs in HCC, colorectal cancer, melanoma, triple negative breast cancer, and squamous cell carcinoma, along with marked reduction of MDSCs, decreased suppressive activity of MDSCs, or an increase in tumor infiltrating CD8+ effector T cells (153–157). Only combination therapy in targeting MDSCs and immune checkpoints was more effective for anti-tumor therapy, while only using the epigenetic modulatory drugs did not mediate the anti-tumor immunity (66). Mechanistic insight into the reversibility of epigenetic modification through small-molecule inhibitors has unlocked the possibility of targeting specific epigenetic pathways to reprogramme the MDSC population into an immunostimulatory phenotype. The histone deacetylases inhibitor, entinostat, was shown to block the formation of the premetastatic niche via promoting MDSCs differentiation into pro-inflammatory macrophages and the therapeutic use of entinostat has been observed limited efficacy in some clinical trials (NCT01207726, NCT01132573, NCT02115282) (54). However, entinostat-driven inhibition of MDSC activity combined with ICI resulted in the tumor regression and longer tumor-free survival by improving the infiltration and function of granzymeB+CD8+ T cells in mouse models of HER2 transgenic breast cancer and the Panc02 metastatic pancreatic cancer mouse models (3, 67). The m6A demethylase Alkbh5 has effects on m6A density and splicing events in tumors during ICB therapy and modulates MDSCs accumulation in TME by regulating Mct4/Slc16a3 expression and lactate content of the TME in the employed melanoma and colon syngeneic mouse models (158). Thus, a small-molecule Alkbh5 inhibitor enhanced the efficacy of ICB cancer immunotherapy. ATRA can have positive effects on anti-tumor therapy by reprogramming MDSCs state within the TME. Some clinical trials have also demonstrated the potential significance of ATRA alone or in combination with ICB or target- orientated anticancer drug in anti-tumor therapy (NCT02403778, NCT03200847). It has been reported that addition of ATRA, which reduces expression of immunosuppressive genes including PD-L1, IL-10, and IDO by MDSCs, to standard of care ipilimumab appeared safe (159). Finally, ATRA significantly decreased the frequency of circulating MDSCs compared to ipilimumab alone in advanced-stage melanoma (159). Additionally, ATRA has been demonstrated to increase the efficacy of anti-VEGFR2 antibodies alone and in combination with chemotherapy in preclinical breast cancer models, reverse the anti-VEGFR2-induced accumulation of intratumoral MDSCs, alleviate hypoxia, and counteract the disorganization of tumor microvessels (47). Although, the clinical efficacy of ATRA has been evaluated, the more effective combination treatment needs to be further explored. We highlight the current clinical trials ongoing and testing the combination of targeting-MDSC with ICB, chemotherapy in Table 2.
Genomic and epigenomic editing provides more opportunities for immunotherapies to create and alter properties. Furthermore, gene editing for immune cell therapies saves the cost and labor participating in the manufacture of the cell products. Gene-modified T cell therapy has been developed as a way to deliver T cells targeting different targets of tumor. However, the immunosuppressive tumor microenvironment is still one of multiple barriers existing in solid tumors that continue to hinder the efficacy of CAR-T cells. Thus, gene modification may enable CAR-T cells acting as a dual targeting agent against tumor cells and MDSCs. One study has developed a modified CAR T cells with IL15, targeting the receptor IL15 receptor alpha (IL15Rα) expressed on MDSC in human and murine glioblastomas (GBMs) (160). The fusion of IL15 to the antibody part of CAR T cells generates a dual targeting system that diminishes the frequency MDSC and tumor cells and improved the survival of mice in two GBM models (160). Another study engineered CAR-T cells to deliver RN7SL1, an endogenous RNA that activates RIG-I/MDA5 signaling (161). RN7SL1 promotes expansion and effector-memory differentiation of CAR-T cells, and transferred RN7SL1 restricts MDSC development, decreases TGFβ in myeloid cells, and fosters DC subsets with costimulatory features, which enables CAR-T cells to enhance autonomous and endogenous immune function (161). To reverse the suppressive tumor microenvironment, some study developed gene modified T-cells bearing a chimeric receptor in which activating receptor NKG2D fused to intracellular domains of 4-1BB and CD3z (NKG2D CAR) (162). The NKG2D CAR-T cells target MDSCs, which overexpress Rae1 (NKG2D ligands) within the TME. NKG2D CAR-T cells eliminated MDSCs and improved antitumor activity of subsequently infused CAR-T cells in a novel orthotopic implantation of syngeneic pancreatic ductal adenocarcinoma (PDAC) tissue slices mice model (162). Similar results were observed in the study that used gene-modified NK cells bearing a chimeric receptor in which the activating receptor NKG2D was fused to the cytotoxic ζ-chain of the T-cell receptor (NKG2D.ζ), and targeted MDSCs that overexpressed NKG2D ligands within the TME (163, 164). Confirmed in the clinical trial, NKG2D.ζ-NK cells generated from patients with neuroblastoma killed autologous intra-tumoral MDSCs capable of suppressing CAR-T function (NCT03373097) (164). Combination therapy with NKG2D.ζ-NK cells and CAR-T cells for solid tumors may provide superior efficacy compared to CAR-T cells therapy alone. PD-L1 axis is a key immunosuppressive signal provided by tumor cells and MDSCs in TME, which limits CAR-T cell function. Some studies designed the CAR-T cells secreting anti-PD-L1 single-chain variable fragment (scFv) or generated a novel PD-L1-targeting chimeric switch receptor (PD-L1.BB CSR) (165–167). The former CAR-T cells secreting anti-PD-L1 scFv which could bind to PD-L1 on PD-L1high tumor cells and MDSCs competitively and block their binding with anti-PD-L1 monoclonal antibodies, leading to increased efficacy (165). The latter CAR-T cells can bind to PD-L1, switching the inhibitory signal into an additional 4-1BB signal, displayed superior fitness and enhanced functions in culture medium, causing rapid and durable eradication of pleural and peritoneal metastatic tumors in xenograft models (166). Furthermore, a phase I clinical trial related to this study, was initiated in patients with pleural or peritoneal metastasis (NCT04684459). Thus, those studies open the opportunity for investigating other targeting moieties on the surface of MDSCs, specifically those enriched in cells of TME, and applying these modifications to CAR T cells for their direct dual functions against glioma cells and immunosuppressive MDSC.
Recent report showed that a vaccine based on heat-killed pathogens induced spleen M-MDSCs that can be activated to kill dendritic cells (DCs), an additional mechanism that may help to explain the difficulties found to develop a very successful anti-pathogen vaccine (168). Tumor vaccines harness the tumor as the source of antigens and implement sequential immunomodulation to generate systemic and lasting antitumor immunity. Mechanism accounting for these is based on isolated patient-derived DCs, through pulsing them with tumor-associated antigens (TAAs) or neoantigens and maturation signals, followed by their reinfusion, or directly inject the antigens subcutaneously by activating DCs in vivo in patients (169). A major challenge facing the future of tumor vaccines for cancer treatments is to persist the cytotoxic T cell responses and overcome inhibitory signals from MDSCs in TME, understand the mechanisms of resistance to vaccines and to develop combination therapies that enhance antitumor immunity and durable responses. In a syngeneic B16F0 melanoma model and using tyrosinase related protein 1 (TRP1) as a vaccine antigen, it has been found that simultaneous delivery of IL-12 and a PD-L1-silencing shRNA was the only combination that exhibited therapeutically relevant anti-melanoma activities (170). Interestingly, the lentivector co-expressing IL-12 and the PD-L1 silencing shRNA was the only one that counteracted MDSC suppressive activities, potentially underlying the observed anti-melanoma therapeutic benefit (170). Prospective evaluation of candidate cancer treatment using ex vivo differentiated MDSCs highlights therapies with significant therapeutic potential and the therapy of the IL12-encoding combined with PD-L1 silencing lentivector vaccines demonstrated promising anti-melanoma activities. A prophylactic vaccine by employing exosomes derived from murine ESCs engineered to produce GM-CSF (ES-exo/GM-CSF vaccine) successfully protects mice from the outgrowth of an implantable form of murine lung cancer and provides protection against metastasized pulmonary tumors, by decreasing the frequencies of tumor infiltrating immunosuppressive immune cells, including Treg cells and MDSCs (171). Similar to this idea, treatment with sunitinib, which inhibits G-MDSCs prior to the vaccine composed of peptides of the tumor antigen survivin (SVX vaccine), could magnify the vaccine-mediated immune responses in a colorectal carcinoma mouse model (172). IFNa and 5-Aza-2’-deoxycytidine combined with a DC targeting DNA vaccine (a MIP3a fused vaccine targeting two common melanoma antigens, gp100 and trp2) exhibited greater tumor infiltration of DCs, and NK cells, as well as reduced levels of MDSCs in vaccinated groups in the B16F10 melanoma model (173). The combination therapy alters the tumor immune cell infiltration and elicits protective immune responses, but the underlying mechanisms needs to be explored. The advent of vaccines in multiple solid tumors has prompted the development of new therapeutic combinations that target MDSCs, modulating the TIME and the systemic antitumor response. Moreover, to explore new strategies to optimize the efficacy of standard immunotherapies, it is essential to find approaches that target MDSCs in antitumor immunization.
Tumor immunotherapy has undergone remarkable advances in recent years and has shown great potential for cancer patients. For most cancer patients, a favorable initial response to immunotherapy, is followed by limited responses and cancer relapse and recurrence, due to the multiple mechanisms inducing tumor immunosuppression (174). It also becomes more urgent and possible to reinforce the immune responses against tumors by ICB, adoptive cell transfer (ACT) therapy or tumor vaccines, and abolish TIME. Many studies found nonresponses to those therapies, mostly due to ignorance of the shape of the TIME after treatment. Thus, exploration of combination immunotherapeutic strategies coupled with other immunotherapy with reprogramming the TIME will be the top priority. It should be noted that MDSCs are known to suppress the anti-tumor immune response to induce host tolerance, support cancer stem cells and increase tumor angiogenesis and vascular maturation (175–178), suggesting that MDSC-targeted therapeutic approaches have broad implications in a wide range of cancer therapies in addition to immunotherapy. A growing number of studies have demonstrated that circulating MDSCs in cancer patients are a negative prognostic biomarker in predicting disease course, tumor stage, or metastatic spread (179). Thus, MDSCs have been recognized as a promising therapeutic target and prognostic biomarker for cancer patients, while the diversity, complexity, and heterogeneity of human MDSCs make it difficult to define their phenotypes accurately and uniformly in cancer. Therefore, it is necessary to investigate the phenotypes and characterizations of MDSCs in different types of tumors to establish the precise means to eliminate MDSCs. Although, the phenotypes and suppressive mechanisms seem to be shared among tumor MDSC subsets, it is necessary to identify and distinguish the differences in detail, in order to make more use of accurate individualized treatment regimens. We have reported here that numerous preclinical trails in mouse tumor models, have exhibited favorable efficacy by targeting MDSCs (61, 180).
The field of MDSC research still has more questions than answers. Better characterization of human MDSCs and a clearer understanding of whether MDSC-targeted cues will be of clinical significance are main priorities in this field. We do not yet know the outcomes of the ongoing clinical trials, including inhibition of MDSC immunosuppressive activity, blockade of MDSC recruitment and expansion, and promotion of MDSC differentiation into mature non-suppressive cells. However, reprogramming MDSCs in tumors, combined with newly developing immunotherapy, like ICB or ACT, seems to be a potential new approach to improve antitumor immunity, although adverse events of the treatment strategies should be taken into consideration. Removing the negative or suppressive immune response and improving the positive immune response is a theoretically ideal scheme to mediate anti-tumor immunity. However, the systemic depletion of suppressive cells, like Treg cells, also causes serious immune-related adverse events (181, 182). Hence, how to realize the immunoregulation in or around the tumor sites to augment antitumor immunity will be challenging. Therefore, developing therapeutic strategies targeting MDSC subpopulation is of paramount importance to improve the effectiveness of tumor therapy. We are at an interesting point in the translation of cancer immunotherapies where an improved knowledge of targeting MDSCs will be critical.
All authors contributed to the article and approved the submitted version. XS and JD was responsible for the overall conceptualization, supervision and writing of the review. YZ was responsible for the draft of the original article, figures and tables.
This work was supported by grants from the National Natural Science Foundation of China (81970500, XF Shen; 81802846, Y Zhao), the Natural Science Foundation of Jiangsu Province (BK20180116, Y Zhao), and the China Postdoctoral Science Foundation (2021M693162, Y Zhao).
We gratefully acknowledge Dr. Xingzhou Wang for his review of our manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Brown JM. Tumor microenvironment and the response to anticancer therapy. Cancer Biol Ther (2002) 1(5):453–8. doi: 10.4161/cbt.1.5.157
2. Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol (2020) 88:106939. doi: 10.1016/j.intimp.2020.106939
3. Dysthe M, Parihar R. Myeloid-derived suppressor cells in the tumor microenvironment. Adv Exp Med Biol (2020) 1224:117–40. doi: 10.1007/978-3-030-35723-8_8
4. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
5. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19(1):116. doi: 10.1158/1557-3125.HIPPO19-B11
6. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
7. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425. doi: 10.1158/0008-5472.CAN-06-3037
8. Rodrigues JC, Gonzalez GC, Zhang L, Ibrahim G, Kelly JJ, Gustafson MP, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol (2010) 12(4):351–65. doi: 10.1093/neuonc/nop023
9. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi: 10.1182/blood-2007-07-099226
10. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest (2015) 125(9):3356–64. doi: 10.1172/JCI80005
11. Zhao Y, Wu TT, Shao S, Shi BY, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology (2016) 5(2):e1004983. doi: 10.1080/2162402X.2015.1004983
12. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791
13. Lechner MG, Megiel C, Russell SM, Bingham B, Arger N, Woo T, et al. Functional characterization of human Cd33+ and Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J Transl Med (2011) 9:90. doi: 10.1186/1479-5876-9-90
14. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
15. Elliott LA, Doherty GA, Sheahan K, Ryan EJ. Human tumor-infiltrating myeloid cells: Phenotypic and functional diversity. Front Immunol (2017) 8:86. doi: 10.3389/fimmu.2017.00086
16. Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao JJ, et al. The next decade of immune checkpoint therapy. Cancer Discovery (2021) 11(4):838–57. doi: 10.1158/2159-8290.CD-20-1680
17. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi: 10.1126/science.aaa4967
18. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer (2014) 134(12):2853–64. doi: 10.1002/ijc.28622
19. Raber P, Ochoa AC, Rodriguez PC. Metabolism of l-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest (2012) 41(6-7):614–34. doi: 10.3109/08820139.2012.680634
20. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi: 10.1158/0008-5472.CAN-09-2587
21. Yu JP, Du WJ, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190(7):3783–97. doi: 10.4049/jimmunol.1201449
22. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8(+) T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol (2004) 172(7), 4647. doi: 10.4049/jimmunol.172.2.989
23. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
24. Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest (2006) 116(10):2777–90. doi: 10.1172/JCI28828
25. Kusmartsev SA, Li Y, Chen SB. Gr-1(+) myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol (2000) 165(2):779–85. doi: 10.4049/jimmunol.165.2.779
26. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
27. Ghorpade DS, Holla S, Sinha AY, Alagesan SK, Balaji KN. Nitric oxide and KLF4 protein epigenetically modify class II transactivator to repress major histocompatibility complex II expression during mycobacterium bovis bacillus calmette-guerin infection. J Biol Chem (2013) 288(28):20592–606. doi: 10.1074/jbc.M113.472183
28. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182(8):4499–506. doi: 10.4049/jimmunol.0802740
29. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
30. Wu T, Zhao Y, Wang H, Li Y, Shao L, Wang R, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep (2016) 6:20250. doi: 10.1038/srep20250
31. Xu W, Li S, Li M, Yang X, Xie S, Lin L, et al. Targeted elimination of myeloid-derived suppressor cells via regulation of the STAT pathway alleviates tumor immunosuppression in neuroblastoma. Immunol Lett (2021) 240:31–40. doi: 10.1016/j.imlet.2021.09.011
32. Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14(+)HLA-DR-/low cells in melanoma patients are Stat3(hi) and overexpress CD80, CD83, and DC-sign. Cancer Res (2010) 70(11):4335–45. doi: 10.1158/0008-5472.CAN-09-3767
33. Dufait I, Van Valckenborgh E, Menu E, Escors D, De Ridder M, Breckpot K. Signal transducer and activator of transcription 3 in myeloid-derived suppressor cells: an opportunity for cancer therapy. Oncotarget (2016) 7(27):42698–715. doi: 10.18632/oncotarget.8311
34. Cheng PY, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med (2008) 205(10):2235–49. doi: 10.1084/jem.20080132
35. Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol (2005) 174(8):4880–91. doi: 10.4049/jimmunol.174.8.4880
36. Munera V, Popovic PJ, Bryk J, Pribis J, Caba D, Matta BM, et al. Stat 6-dependent induction of myeloid derived suppressor cells after physical injury regulates nitric oxide response to endotoxin. Ann Surg (2010) 251(1):120–6. doi: 10.1097/SLA.0b013e3181bfda1c
37. Trikha P, Carson WE. Signaling pathways involved in MDSC regulation. Bba-Rev Cancer (2014) 1846(1):55–65. doi: 10.1016/j.bbcan.2014.04.003
38. Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, et al. Tumor-derived prostaglandin E2 promotes p50 NF-kappaB-Dependent differentiation of monocytic MDSCs. Cancer Res (2020) 80(13):2874–88. doi: 10.1158/0008-5472.CAN-19-2843
39. Kho VM, Mekers VE, Span PN, Bussink J, Adema GJ. Radiotherapy and cGAS/STING signaling: Impact on MDSCs in the tumor microenvironment. Cell Immunol (2021) 362:104298. doi: 10.1016/j.cellimm.2021.104298
40. Cheng H, Xu Q, Lu X, Yuan H, Li T, Zhang Y, et al. Activation of STING by cGAMP regulates MDSCs to suppress tumor metastasis via reversing epithelial-mesenchymal transition. Front Oncol (2020) 10:896. doi: 10.3389/fonc.2020.00896
41. Pei J, Zhang Y, Luo Q, Zheng W, Li W, Zeng X, et al. STAT3 inhibition enhances CDN-induced STING signaling and antitumor immunity. Cancer Lett (2019) 450:110–22. doi: 10.1016/j.canlet.2019.02.029
42. Zhao Y, Shen XF, Cao K, Ding J, Kang X, Guan WX, et al. Dexamethasone-induced myeloid-derived suppressor cells prolong allo cardiac graft survival through iNOS- and glucocorticoid receptor-dependent mechanism. Front Immunol (2018) 9:282. doi: 10.3389/fimmu.2018.00282
43. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4(+)CD25(+) regulatory T cell proliferation. J Exp Med (2005) 202(7):919–29. doi: 10.1084/jem.20050463
44. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in b-cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68(13):5439–49. doi: 10.1158/0008-5472.CAN-07-6621
45. Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood (2011) 117(24):6532–41. doi: 10.1182/blood-2010-11-317321
46. Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH, et al. Population alterations of l-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14(-)/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol (2010) 136(1):35–45. doi: 10.1007/s00432-009-0634-0
47. Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res (2018) 78(12):3220–32. doi: 10.1158/0008-5472.CAN-17-3415
48. Varikuti S, Singh B, Volpedo G, Ahirwar DK, Jha BK, Saljoughian N, et al. Ibrutinib treatment inhibits breast cancer progression and metastasis by inducing conversion of myeloid-derived suppressor cells to dendritic cells. Br J Cancer (2020) 122(7):1005–13. doi: 10.1038/s41416-020-0743-8
49. Colligan SH, Amitrano AM, Zollo RA, Peresie J, Kramer ED, Morreale B, et al. Inhibiting the biogenesis of myeloid-derived suppressor cells enhances immunotherapy efficacy against mammary tumor progression. J Clin Invest (2022) 132(23):e158661. doi: 10.1172/JCI158661
50. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity (2016) 44(2):303–15. doi: 10.1016/j.immuni.2016.01.014
51. Fernandez A, Oliver L, Alvarez R, Hernandez A, Raymond J, Fernandez LE, et al. Very small size proteoliposomes abrogate cross-presentation of tumor antigens by myeloid-derived suppressor cells and induce their differentiation to dendritic cells. J Immunother Cancer (2014) 2:5. doi: 10.1186/2051-1426-2-5
52. Oliver L, Alvarez R, Diaz R, Valdes A, Colligan SH, Nemeth MJ, et al. Mitigating the prevalence and function of myeloid-derived suppressor cells by redirecting myeloid differentiation using a novel immune modulator. J Immunother Cancer (2022) 10(9):e004710. doi: 10.1136/jitc-2022-004710
53. Chen PT, Hsieh CC, Wu CT, Yen TC, Lin PY, Chen WC, et al. 1alpha,25-dihydroxyvitamin D3 inhibits esophageal squamous cell carcinoma progression by reducing IL6 signaling. Mol Cancer Ther (2015) 14(6):1365–75. doi: 10.1158/1535-7163.MCT-14-0952
54. Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature (2020) 579(7798):284–90. doi: 10.1038/s41586-020-2054-x
55. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin Cancer Res (2020) 26(6):1420–31. doi: 10.1158/1078-0432.CCR-19-2625
56. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight (2019) 4(7). doi: 10.1172/jci.insight.126853
57. Sun R, Luo H, Su J, Di S, Zhou M, Shi B, et al. Olaparib suppresses MDSC recruitment via SDF1alpha/CXCR4 axis to improve the anti-tumor efficacy of CAR-T cells on breast cancer in mice. Mol Ther (2021) 29(1):60–74. doi: 10.1016/j.ymthe.2020.09.034
58. Li Y, Yang B, Miao H, Liu L, Wang Z, Jiang C, et al. Nicotinamide n-methyltransferase promotes M2 macrophage polarization by IL6 and MDSC conversion by GM-CSF in gallbladder carcinoma. Hepatology (2023). doi: 10.1097/HEP.0000000000000028
59. Kong Q, Ma M, Zhang L, Liu S, He S, Wu J, et al. Icariside II potentiates the anti-PD-1 antitumor effect by reducing chemotactic infiltration of myeloid-derived suppressor cells into the tumor microenvironment via ROS-mediated inactivation of the SRC/ERK/STAT3 signaling pathways. Phytomedicine (2022) 110:154638. doi: 10.1016/j.phymed.2022.154638
60. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell (2017) 32(5):654–68.e5. doi: 10.1016/j.ccell.2017.10.005
61. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. Ebiomedicine (2016) 6:50–8. doi: 10.1016/j.ebiom.2016.02.024
62. Ban Y, Mai J, Li X, Mitchell-Flack M, Zhang T, Zhang L, et al. Targeting autocrine CCL5-CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res (2017) 77(11):2857–68. doi: 10.1158/0008-5472.CAN-16-2913
63. Yang L, Wang B, Qin J, Zhou H, Majumdar APN, Peng F. Blockade of CCR5-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy in gastric cancer. Immunopharmacol Immunotoxicol (2018) 40(1):91–7. doi: 10.1080/08923973.2017.1417997
64. Prasad M, Zorea J, Jagadeeshan S, Shnerb AB, Mathukkada S, Bouaoud J, et al. MEK1/2 inhibition transiently alters the tumor immune microenvironment to enhance immunotherapy efficacy against head and neck cancer. J Immunother Cancer (2022) 10(3):e003917. doi: 10.1136/jitc-2021-003917
65. Bruns H, Bottcher M, Qorraj M, Fabri M, Jitschin S, Dindorf J, et al. CLL-cell-mediated MDSC induction by exosomal miR-155 transfer is disrupted by vitamin d. Leukemia (2017) 31(4):985–8. doi: 10.1038/leu.2016.378
66. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res (2017) 23(17):5187–201. doi: 10.1158/1078-0432.CCR-17-0741
67. Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res (2018) 6(12):1561–77. doi: 10.1158/2326-6066.CIR-18-0070
68. Holtzhausen A, Harris W, Ubil E, Hunter DM, Zhao J, Zhang Y, et al. TAM family receptor kinase inhibition reverses MDSC-mediated suppression and augments anti-PD-1 therapy in melanoma. Cancer Immunol Res (2019) 7(10):1672–86. doi: 10.1158/2326-6066.CIR-19-0008
69. Ye C, Geng Z, Dominguez D, Chen S, Fan J, Qin L, et al. Targeting ornithine decarboxylase by alpha-difluoromethylornithine inhibits tumor growth by impairing myeloid-derived suppressor cells. J Immunol (2016) 196(2):915–23. doi: 10.4049/jimmunol.1500729
70. Ishfaq M, Pham T, Beaman C, Tamayo P, Yu AL, Joshi S. BTK inhibition reverses MDSC-mediated immunosuppression and enhances response to anti-PDL1 therapy in neuroblastoma. Cancers (Basel) (2021) 13(4):817. doi: 10.3390/cancers13040817
71. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. Ebiomedicine (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
72. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70(8):3052–61. doi: 10.1158/0008-5472.CAN-09-3690
73. Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, et al. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother (2014) 63(2):175–83. doi: 10.1007/s00262-013-1502-y
74. Tang Y, Zhou C, Li QL, Cheng XJ, Huang TL, Li FL, et al. Targeting depletion of myeloid-derived suppressor cells potentiates PD-L1 blockade efficacy in gastric and colon cancers. Oncoimmunology (2022) 11(1):2131084. doi: 10.1080/2162402X.2022.2131084
75. Khaki Bakhtiarvand V, Ramezani-Ali Akbari K, Amir Jalali S, Hojjat-Farsangi M, Jeddi-Tehrani M, Shokri F, et al. Myeloid-derived suppressor cells (MDSCs) depletion by cabozantinib improves the efficacy of anti-HER2 antibody-based immunotherapy in a 4T1-HER2 murine breast cancer model. Int Immunopharmacol (2022) 113(Pt B):109470. doi: 10.1016/j.intimp.2022.109470
76. Choueiry F, Torok M, Shakya R, Agrawal K, Deems A, Benner B, et al. CD200 promotes immunosuppression in the pancreatic tumor microenvironment. J Immunother Cancer (2020) 8(1):e000189. doi: 10.1136/jitc-2019-000189
77. Su MT, Kumata S, Endo S, Okada Y, Takai T. LILRB4 promotes tumor metastasis by regulating MDSCs and inhibiting miR-1 family miRNAs. Oncoimmunology (2022) 11(1):2060907. doi: 10.1080/2162402X.2022.2060907
78. Thakkar D, Paliwal S, Dharmadhikari B, Guan S, Liu L, Kar S, et al. Rationally targeted anti-VISTA antibody that blockades the c-c' loop region can reverse VISTA immune suppression and remodel the immune microenvironment to potently inhibit tumor growth in an fc independent manner. J Immunother Cancer (2022) 10(2):e003382. doi: 10.1136/jitc-2021-003382
79. de Lera AR, Bourguet W, Altucci L, Gronemeyer H. Design of selective nuclear receptor modulators: RAR and RXR as a case study. Nat Rev Drug Discovery (2007) 6(10):811–20. doi: 10.1038/nrd2398
80. Weston AD, Blumberg B, Underhill TM. Active repression by unliganded retinoid receptors in development: less is sometimes more. J Cell Biol (2003) 161(2):223–8. doi: 10.1083/jcb.200211117
81. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67(22):11021–8. doi: 10.1158/0008-5472.CAN-07-2593
82. Wu CT, Huang YC, Chen WC, Chen MF. Effect of 1alpha,25-dihydroxyvitamin D3 on the radiation response in prostate cancer: Association with IL-6 signaling. Front Oncol (2021) 11:619365. doi: 10.3389/fonc.2021.619365
83. Hughes PJ, Marcinkowska E, Gocek E, Studzinski GP, Brown G. Vitamin D3-driven signals for myeloid cell differentiation–implications for differentiation therapy. Leuk Res (2010) 34(5):553–65. doi: 10.1016/j.leukres.2009.09.010
84. Hu XT, Zuckerman KS. Role of cell cycle regulatory molecules in retinoic acid- and vitamin D3-induced differentiation of acute myeloid leukaemia cells. Cell Prolif (2014) 47(3):200–10. doi: 10.1111/cpr.12100
85. Adorini L, Penna G. Dendritic cell tolerogenicity: a key mechanism in immunomodulation by vitamin d receptor agonists. Hum Immunol (2009) 70(5):345–52. doi: 10.1016/j.humimm.2009.01.016
86. Penna G, Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol (2000) 164(5):2405–11. doi: 10.4049/jimmunol.164.5.2405
87. Szeles L, Keresztes G, Torocsik D, Balajthy Z, Krenacs L, Poliska S, et al. 1,25-dihydroxyvitamin d-3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J Immunol (2009) 182(4):2074–83. doi: 10.4049/jimmunol.0803345
88. Gauzzi MC, Purificato C, Donato K, Jin YX, Wang L, Daniel KC, et al. Suppressive effect of 1 alpha,25-dihydroxyvitamin d-3 on type IIFN-mediated monocyte differentiation into dendritic cells: Impairment of functional activities and chemotaxis. J Immunol (2005) 174(1):270–6. doi: 10.4049/jimmunol.174.1.270
89. Pedersen AE, Gad M, Walter MR, Claesson MH. Induction of regulatory dendritic cells by dexamethasone and 1alpha,25-dihydroxyvitamin D(3). Immunol Lett (2004) 91(1):63–9. doi: 10.1016/j.imlet.2003.11.004
90. Suuring M, Moreau A. Regulatory macrophages and tolerogenic dendritic cells in myeloid regulatory cell-based therapies. Int J Mol Sci (2021) 22(15):7970. doi: 10.3390/ijms22157970
91. Chen PT, Hsieh CC, Chen MF. Role of vitamin D3 in tumor aggressiveness and radiation response for hepatocellular carcinoma. Mol Carcinog (2022) 61(8):787–96. doi: 10.1002/mc.23421
92. Rebe C, Vegran F, Berger H, Ghiringhelli F. STAT3 activation: A key factor in tumor immunoescape. JAKSTAT (2013) 2(1):e23010. doi: 10.4161/jkst.23010
93. Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res (2007) 67(23):11438–46. doi: 10.1158/0008-5472.CAN-07-1882
94. Ko JS, Bukowski RM, Fincke JH. Myeloid-derived suppressor cells: a novel therapeutic target. Curr Oncol Rep (2009) 11(2):87–93. doi: 10.1007/s11912-009-0014-6
95. Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res (2008) 68(14):5501–4. doi: 10.1158/0008-5472.CAN-08-0925
96. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69(4):1553–60. doi: 10.1158/0008-5472.CAN-08-1921
97. Schweizer MT, Carducci MA. From bevacizumab to tasquinimod: angiogenesis as a therapeutic target in prostate cancer. Cancer J (2013) 19(1):99–106. doi: 10.1097/PPO.0b013e31827e0b86
98. Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, et al. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res (2010) 70(9):3526–36. doi: 10.1158/0008-5472.CAN-09-3278
99. Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett (2007) 252(1):86–92. doi: 10.1016/j.canlet.2006.12.012
100. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res (2012) 72(4):876–86. doi: 10.1158/0008-5472.CAN-11-1792
101. Kirk PS, Koreckij T, Nguyen HM, Brown LG, Snyder LA, Vessella RL, et al. Inhibition of CCL2 signaling in combination with docetaxel treatment has profound inhibitory effects on prostate cancer growth in bone. Int J Mol Sci (2013) 14(5):10483–96. doi: 10.3390/ijms140510483
102. Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5(+) myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res (2018) 78(1):157–67. doi: 10.1158/0008-5472.CAN-17-0348
103. Tang Q, Jiang J, Liu J. CCR5 blockade suppresses melanoma development through inhibition of IL-6-Stat3 pathway via upregulation of SOCS3. Inflammation (2015) 38(6):2049–56. doi: 10.1007/s10753-015-0186-1
104. Chi N, Tan Z, Ma K, Bao L, Yun Z. Increased circulating myeloid-derived suppressor cells correlate with cancer stages, interleukin-8 and -6 in prostate cancer. Int J Clin Exp Med (2014) 7(10):3181–92.
105. David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R axis: A double agent in tumor immune resistance. Vaccines (Basel) (2016) 4(3):22. doi: 10.3390/vaccines4030022
106. Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev (2001) 12(4):375–91. doi: 10.1016/S1359-6101(01)00016-8
107. Asfaha S, Dubeykovskiy AN, Tomita H, Yang X, Stokes S, Shibata W, et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology (2013) 144(1):155–66. doi: 10.1053/j.gastro.2012.09.057
108. Citro A, Cantarelli E, Maffi P, Nano R, Melzi R, Mercalli A, et al. CXCR1/2 inhibition enhances pancreatic islet survival after transplantation. J Clin Invest (2012) 122(10):3647–51. doi: 10.1172/JCI63089
109. Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res (2016) 22(15):3924–36. doi: 10.1158/1078-0432.CCR-15-2463
110. Citro A, Valle A, Cantarelli E, Mercalli A, Pellegrini S, Liberati D, et al. CXCR1/2 inhibition blocks and reverses type 1 diabetes in mice. Diabetes (2015) 64(4):1329–40. doi: 10.2337/db14-0443
111. Dominguez C, McCampbell KK, David JM, Palena C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight (2017) 2(21):e94296. doi: 10.1172/jci.insight.94296
112. Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res (2010) 70(3):1063–71. doi: 10.1158/0008-5472.CAN-09-3965
113. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
114. Ohl K, Tenbrock K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
115. Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res (2010) 16(6):1812–23. doi: 10.1158/1078-0432.CCR-09-3272
116. Li B, Luo Y, Zhou Y, Wu J, Fang Z, Li Y. Role of sanguinarine in regulating immunosuppression in a Lewis lung cancer mouse model. Int Immunopharmacol (2022) 110:108964. doi: 10.1016/j.intimp.2022.108964
117. Alicea-Torres K, Sanseviero E, Gui J, Chen J, Veglia F, Yu Q, et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun (2021) 12(1):1717. doi: 10.1038/s41467-021-22033-2
118. Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev (2016) 31:1–15. doi: 10.1016/j.cytogfr.2016.05.001
119. Cohen PA, Ko JS, Storkus WJ, Spencer CD, Bradley JM, Gorman JE, et al. Myeloid-derived suppressor cells adhere to physiologic STAT3- vs STAT5-dependent hematopoietic programming, establishing diverse tumor-mediated mechanisms of immunologic escape. Immunol Invest (2012) 41(6-7):680–710. doi: 10.3109/08820139.2012.703745
120. Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest (2013) 123(10):4464–78. doi: 10.1172/JCI68189
121. Trillo-Tinoco J, Sierra RA, Mohamed E, Cao Y, de Mingo-Pulido A, Gilvary DL, et al. AMPK alpha-1 intrinsically regulates the function and differentiation of tumor myeloid-derived suppressor cells. Cancer Res (2019) 79(19):5034–47. doi: 10.1158/0008-5472.CAN-19-0880
122. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature (2022) 612(7939):338–46. doi: 10.1038/s41586-022-05443-0
123. Liu M, Zhou J, Liu X, Feng Y, Yang W, Wu F, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut (2020) 69(2):365–79. doi: 10.1136/gutjnl-2018-317257
124. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature (2019) 569(7754):73–8. doi: 10.1038/s41586-019-1118-2
125. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic gr-1(+)/CD11b(+) myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res (2005) 11(18):6713–21. doi: 10.1158/1078-0432.CCR-05-0883
126. Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M, et al. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol (2013) 190(5):2464–71. doi: 10.4049/jimmunol.1202781
127. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGF beta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Trans Med (2016) 14:282. doi: 10.1186/s12967-016-1037-z.
128. Van Wigcheren GF, De Haas N, Mulder TA, Horrevorts SK, Bloemendal M, Hins-Debree S, et al. Cisplatin inhibits frequency and suppressive activity of monocytic myeloid-derived suppressor cells in cancer patients. Oncoimmunology (2021) 10(1):1935557. doi: 10.1080/2162402X.2021.1935557
129. Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res (2020) 8(11):1440–51. doi: 10.1158/2326-6066.CIR-20-0111
130. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(11):749. doi: 10.1038/nrc.2016.71
131. Yang L, Venneti S, Nagrath D. Glutaminolysis: A hallmark of cancer metabolism. Annu Rev BioMed Eng (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
132. Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest (2020) 130(7):3865–84. doi: 10.1172/JCI131859
133. Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell (2018) 172(4):825–40.e18. doi: 10.1016/j.cell.2017.12.026
134. Tesi RJ. MDSC; the most important cell you have never heard of. Trends Pharmacol Sci (2019) 40(1):4–7. doi: 10.1016/j.tips.2018.10.008
135. Yu SJ, Ma C, Heinrich B, Brown ZJ, Sandhu M, Zhang Q, et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J Hepatol (2019) 70(3):449–57. doi: 10.1016/j.jhep.2018.10.040
136. Patra T, Cunningham DM, Meyer K, Toth K, Ray RB, Heczey A, et al. Targeting Lin28 axis enhances glypican-3-CAR T cell efficacy against hepatic tumor initiating cell population. Mol Ther (2023) 31(3):715–28. doi: 10.1016/j.ymthe.2023.01.002
137. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
138. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. New Engl J Med (2015) 372(4):320–30. doi: 10.1056/NEJMoa1412082
139. Ueno M, Chung HC, Nagrial A, Marabelle A, Kelley RK, Xu L, et al. Pembrolizumab for advanced biliary adenocarcinoma: Results from the multicohort, phase II KEYNOTE-158 study. Ann Oncol (2018) 29:210. doi: 10.1093/annonc/mdy282.009
140. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
141. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
142. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother (2014) 63(3):247–57. doi: 10.1007/s00262-013-1508-5
143. Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai YL, Tawbi H, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PloS One (2014) 9(2):e87705. doi: 10.1371/journal.pone.0087705
144. Weber J, Gibney G, Kudchadkar R, Yu B, Cheng PY, Martinez AJ, et al. Phase I/II study of metastatic melanoma patients treated with nivolumab who had progressed after ipilimumab. Cancer Immunol Res (2016) 4(4):345–53. doi: 10.1158/2326-6066.CIR-15-0193
145. Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun (2017) 8:14572. doi: 10.1038/ncomms14572
146. Lin H, Wei S, Hurt EM, Green MD, Zhao LL, Vatan L, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest (2018) 128(4):1708. doi: 10.1172/JCI120803
147. Tang HD, Liang Y, Anders RA, Taube JM, Qiu XY, Mulgaonkar A, et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest (2018) 128(2):580–8. doi: 10.1172/JCI96061
148. Wu Y, Chen W, Xu ZP, Gu W. PD-L1 distribution and perspective for cancer immunotherapy-blockade, knockdown, or inhibition. Front Immunol (2019) 10:2022. doi: 10.3389/fimmu.2019.02022
149. Ying HG, Zhang XZ, Duan Y, Lao MY, Xu J, Yang HS, et al. Non-cytomembrane PD-L1: An atypical target for cancer. Pharmacol Res (2021) 170:105741. doi: 10.1016/j.phrs.2021.105741
150. Loeuillard E, Yang JC, Buckarma E, Wang J, Liu YH, Conboy C, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest (2020) 130(10):5380–96. doi: 10.1172/JCI137110
151. Kim K, Skora AD, Li ZB, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. P Natl Acad Sci USA (2014) 111(32):11774–9. doi: 10.1073/pnas.1410626111
152. Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, et al. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol (2021) 359:104254. doi: 10.1016/j.cellimm.2020.104254
153. Jin K, Pandey NB, Popel AS. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res (2018) 20(1):54. doi: 10.1186/s13058-018-0981-3
154. Sumida K, Wakita D, Narita Y, Masuko K, Terada S, Watanabe K, et al. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol (2012) 42(8):2060–72. doi: 10.1002/eji.201142335
155. Liu H, Shen J, Lu K. IL-6 and PD-L1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem Biophys Res Commun (2017) 486(2):239–44. doi: 10.1016/j.bbrc.2017.02.128
156. Li J, Xu J, Yan X, Jin K, Li W, Zhang R. Targeting interleukin-6 (IL-6) sensitizes anti-PD-L1 treatment in a colorectal cancer preclinical model. Med Sci Monit (2018) 24:5501–8. doi: 10.12659/MSM.907439
157. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut (2018) 67(2):320–32. doi: 10.1136/gutjnl-2016-311585
158. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U.S.A. (2020) 117(33):20159–70. doi: 10.1073/pnas.1918986117
159. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with ipilimumab. Int Immunopharmacol (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
160. Zannikou M, Duffy JT, Levine RN, Seblani M, Liu Q, Presser A, et al. IL15 modification enables CAR T cells to act as a dual targeting agent against tumor cells and myeloid-derived suppressor cells in GBM. J Immunother Cancer (2023) 11(2):e006239. doi: 10.1136/jitc-2022-006239
161. Johnson LR, Lee DY, Eacret JS, Ye D, June CH, Minn AJ. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell (2021) 184(19):4981–95.e14. doi: 10.1016/j.cell.2021.08.004
162. Wang J, Liu X, Ji J, Luo J, Zhao Y, Zhou X, et al. Orthotopic and heterotopic murine models of pancreatic cancer exhibit different immunological microenvironments and different responses to immunotherapy. Front Immunol (2022) 13:863346. doi: 10.3389/fimmu.2022.863346
163. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res (2019) 7(3):363–75. doi: 10.1158/2326-6066.CIR-18-0572
164. Tumino N, Weber G, Besi F, Del Bufalo F, Bertaina V, Paci P, et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J Hematol Oncol (2021) 14(1):191. doi: 10.1186/s13045-021-01193-0
165. Yuti P, Wutti-In Y, Sawasdee N, Kongkhla K, Phanthaphol N, Choomee K, et al. Anti-CD19 chimeric antigen receptor T cells secreting anti-PD-L1 single-chain variable fragment attenuate PD-L1 mediated T cell inhibition. Int Immunopharmacol (2022) 113(Pt B):109442. doi: 10.1016/j.intimp.2022.109442
166. Ma Q, He X, Zhang B, Guo F, Ou X, Yang Q, et al. A PD-L1-targeting chimeric switch receptor enhances efficacy of CAR-T cell for pleural and peritoneal metastasis. Signal Transduct Target Ther (2022) 7(1):380. doi: 10.1038/s41392-022-01198-2
167. Wang Y, Fang X, Li M, Ye J, Zhao S, Yu L, et al. Mesothelin CAR-T cells secreting PD-L1 blocking scFv for pancreatic cancer treatment. Cancer Genet (2022) 268-269:103–10. doi: 10.1016/j.cancergen.2022.10.003
168. Ribechini E, Eckert I, Beilhack A, Du Plessis N, Walzl G, Schleicher U, et al. Heat-killed mycobacterium tuberculosis prime-boost vaccination induces myeloid-derived suppressor cells with spleen dendritic cell-killing capability. JCI Insight (2019) 5(13):e128664. doi: 10.1172/jci.insight.128664
169. Lurje I, Werner W, Mohr R, Roderburg C, Tacke F, Hammerich L. In situ vaccination as a strategy to modulate the immune microenvironment of hepatocellular carcinoma. Front Immunol (2021) 12:650486. doi: 10.3389/fimmu.2021.650486
170. Liechtenstein T, Perez-Janices N, Blanco-Luquin I, Goyvaerts C, Schwarze J, Dufait I, et al. Anti-melanoma vaccines engineered to simultaneously modulate cytokine priming and silence PD-L1 characterized using ex vivo myeloid-derived suppressor cells as a readout of therapeutic efficacy. Oncoimmunology (2014) 3(7):e945378. doi: 10.4161/21624011.2014.945378
171. Meng S, Whitt AG, Stamp BF, Eaton JW, Li C, Yaddanapudi K. Exosome-based cancer vaccine for prevention of lung cancer. Stem Cell Investig (2023) 10:2. doi: 10.21037/sci-2022-030
172. Mougel A, Mejean F, Tran T, Adimi Y, Galy-Fauroux I, Kabore C, et al. Synergistic effect of combining sunitinib with a peptide-based vaccine in cancer treatment after microenvironment remodeling. Oncoimmunology (2022) 11(1):2110218. doi: 10.1080/2162402X.2022.2110218
173. Gordy JT, Sandhu AK, Fessler K, Luo K, Kapoor AR, Ayeh SK, et al. IFNalpha and 5-Aza-2'-deoxycytidine combined with a dendritic-cell targeting DNA vaccine alter tumor immune cell infiltration in the B16F10 melanoma model. Front Immunol (2022) 13:1074644. doi: 10.3389/fimmu.2022.1074644
174. Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
175. Peng DJ, Tanikawa T, Li W, Zhao LL, Vatan L, Szeliga W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res (2016) 76(11):3156–65. doi: 10.1158/0008-5472.CAN-15-2528
176. Sainz B, Martin B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res (2014) 74(24):7309–20. doi: 10.1158/0008-5472.CAN-14-1354
177. Wan SS, Zhao ED, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147(6):1393–404. doi: 10.1053/j.gastro.2014.08.039
178. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004) 6(4):409–21. doi: 10.1016/j.ccr.2004.08.031
179. Wang PF, Song SY, Wang TJ, Ji WJ, Li SW, Liu N, et al. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology (2018) 7(10):e1494113. doi: 10.1080/2162402X.2018.1494113
180. Mao YM, Eissler N, Le Blanc K, Johnsen JI, Kogner P, Kiessling R. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res (2016) 22(15):3849–59. doi: 10.1158/1078-0432.CCR-15-1912
181. Liu J, Blake SJ, Smyth MJ, Teng MW. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin Transl Immunol (2014) 3(8):e22. doi: 10.1038/cti.2014.18
Keywords: myeloid-derived suppressor cells, tumor immunotherapy, tumor immune microenvironment, combinatorial strategies, cell therapy
Citation: Zhao Y, Du J and Shen X (2023) Targeting myeloid-derived suppressor cells in tumor immunotherapy: Current, future and beyond. Front. Immunol. 14:1157537. doi: 10.3389/fimmu.2023.1157537
Received: 02 February 2023; Accepted: 06 March 2023;
Published: 17 March 2023.
Edited by:
Mary Poupot-Marsan, INSERM U1037 Centre de Recherche en Cancérologie de Toulouse, FranceReviewed by:
Eva Sahakian, Moffitt Cancer Center, United StatesCopyright © 2023 Zhao, Du and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junfeng Du, ZHVqZjY2QDEyNi5jb20=; Xiaofei Shen, ZGcxNTM1MDU4QHNtYWlsLm5qdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.