 Yingying Liu
Yingying Liu Yu Shi
Yu Shi Ruiqin Han
Ruiqin Han Chaoge Liu5,6
Chaoge Liu5,6 Pengfei Li
Pengfei Li Renjun Gu
Renjun Gu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 18 April 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1139589
This article is part of the Research TopicNew immunotherapy strategies and related therapeutic targets for gastrointestinal malignanciesView all 14 articles
Gastric cancer is one of the top causes of cancer-related death globally. Although novel treatment strategies have been developed, attempts to eradicate gastric cancer have been proven insufficient. Oxidative stress is continually produced and continually present in the human body. Increasing evidences show that oxidative stress contributes significantly to the development of gastric cancer, either through initiation, promotion, and progression of cancer cells or causing cell death. As a result, the purpose of this article is to review the role of oxidative stress response and the subsequent signaling pathways as well as potential oxidative stress-related therapeutic targets in gastric cancer. Understanding the pathophysiology of gastric cancer and developing new therapies for gastric cancer depends on more researches focusing on the potential contributors to oxidative stress and gastric carcinogenesis.
Gastric cancer is the third most frequent cause of cancer-related death, and the fifth most diagnosed malignancy around the world (1). Gastric cancer is the major burden in male, accounting for 20% globally, only to lung and liver cancers (2). Anatomically, true gastric adenocarcinomas (non-cardia gastric tumors) and gastro-oesophageal junction adenocarcinomas (cardia gastric cancers) are two types of gastric cancer (3). The early stages of gastric cancer are frequently clinically unconscious, and patients are typically diagnosed at an advanced stage. The prognosis is poor once the neoplastic cells invade the muscularis propria, with the 5-year survival is almost 25% in Europe and US (4–6). With the development of economy and living standards, the prevalence of Helicobacter pylori (H. pylori) which is the key risk factor of non-cardia gastric cancer has decreased (7). Despite a consistent decrease in the rates of morbidity and mortality, more cases of gastric cancer can be seen in the future because of ageing populations (8). The disease’s late diagnosis and high mortality rate reveal a lack of knowledge regarding its etiology and pathology, as well as the absence of efficient treatments. Generally, gastric cancer is a consequence of the multifactorial interplay between host genetics, microbial factors, nutrition, and environmental milieu (9), where it is thought that oxidative stress plays a crucial role in the occurrence and development of gastric cancer.
Oxidative stress is the result of an imbalance of reactive oxygen species (ROS) production and natural antioxidant defenses, which can damage biological molecules and cells, with possible effects on the entire organism (10). Numerous studies demonstrate the tight relationship between ROS and cancer, indicating that cancer cells produced more ROS than healthy cells did (11). Increased ROS levels are thought to have an oncogenic effect, inducing DNA damage and chromosomal instability to activate proto-oncogenes and inactivate tumor suppressor genes (12, 13). Additionally, ROS also serve as signaling molecules in cancer, which affect receptor and oncogene activity, as well as alter several signaling pathways or oxidizing enzymes, facilitating tumorigenesis, angiogenesis, cellular proliferation, invasiveness, and metastasis (14). However, excessive intracellular levels of ROS may promote cell death by damaging proteins, lipid bilayers, and chromosomes. Therefore, cancer cells must fight against high level of ROS to strive for progression and develop resistance to apoptosis through antioxidant defense systems, especially at early stages (15). For this reason, both eliminating and elevating ROS production are potentially effective cancer therapies despite the fact that it is a challenging notion.
According to studies, increased levels of oxidative stress are found in individuals with gastric cancer, and this contributes to the development of gastric cancer (16). The significance of the link between oxidative stress and gastric cancer is becoming increasingly clear. This article reviews the current knowledge on the roles of oxidative stress in gastric cancer and the potential therapeutic applications of manipulating related signaling pathways in oxidative stress.
The human body continuously produces ROS which are oxygen-containing oxidants with reactive properties, represented as oxygen radicals including superoxide anions ( ), hydroxyl (HO·), alkoxyl (RO·), peroxyl (RO2·), and certain nonradicals either oxidizing agents and/or easily converted to radical including hydrogen peroxide (H2O2), hypochlorous acid (HOCl), singlet oxygen (1O2) and ozone (O3) (17). Reactive nitrogen species (RNS) are nitrogen-containing chemical species, which can damage cells via nitrosative stress. Reactive nitrogen species (RNS) include nitric oxide (·NO), nonradical compounds, peroxynitrite (ONOO–), nitrogen dioxide (·NO2) and dinitrogen trioxide (N2O3) (18) (Table 1). Most of these molecules are produced from oxygen in numerous metabolic processes occurring throughout the body, which primarily take place in the mitochondria, endoplasmic reticulum (ER) and peroxisomes. Approximately 2% of the oxygen consumed by the mitochondria is converted into superoxide, making it one of the largest sources of endogenous ROS (19). Peroxisomes mediate the production of ROS via β-oxidation of fatty acid and flavin oxidase reaction and degrading ROS via catalase-mediated breakdown of H2O2 (20). The ER provides an oxidizing environment, which promotes the protein folding and acts as a source of ROS (21).
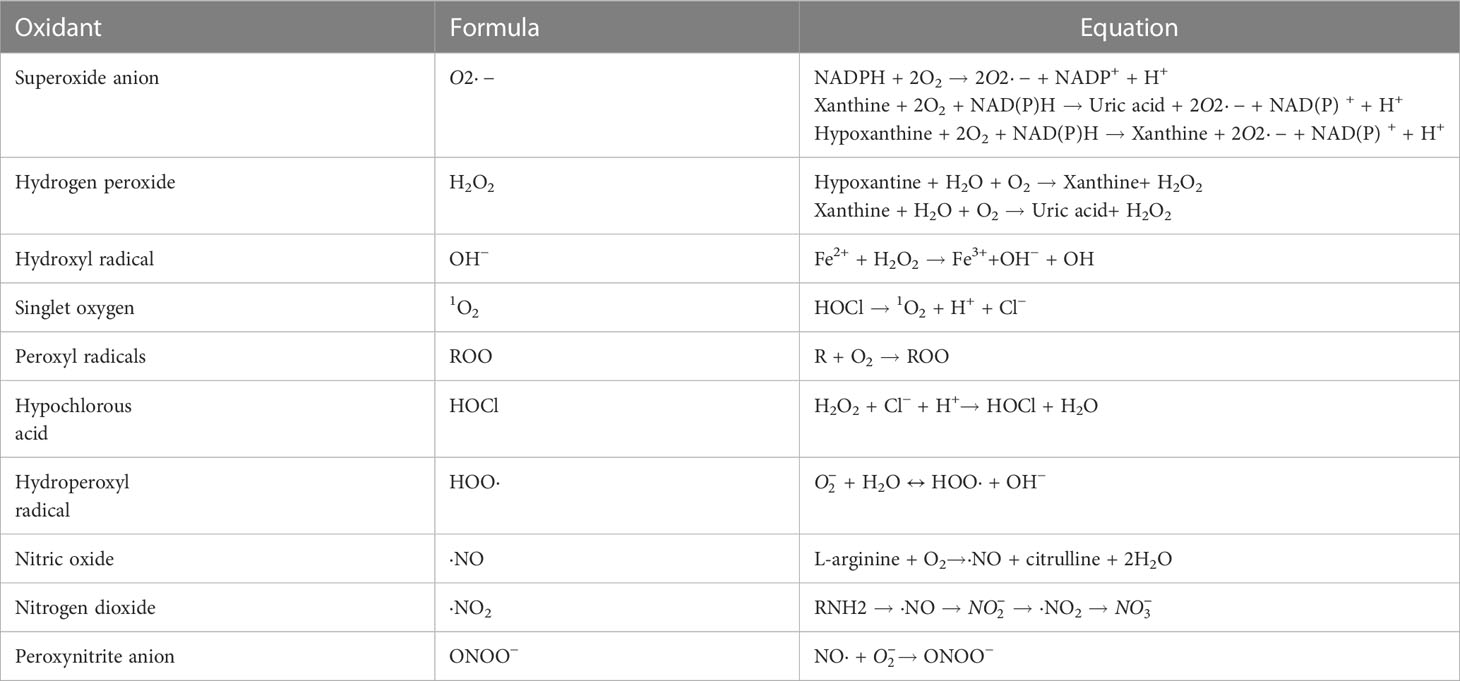
Table 1 Formation of major oxidants.
Enzymatic and non-enzymatic reactions are both necessary for ROS and RNS production. The main enzymes involved in enzymatic reactions are uncoupled endothelial nitric oxide synthase (eNOS), NADPH oxidase (NOX), xanthine oxidase (XO), arachidonic acid (ARA), peroxidase, and metabolic enzymes such the cytochrome P450 system, cyclooxygenase, and lipoxygenase. The major source of ROS comes from non-enzymatic processes in the mitochondrial respiratory chain (22). Generally, ROS are by-products of biological metabolism in healthy organisms, though at lower amounts, which activate different signaling pathways to promote survival, proliferation, or resistance to oxidative stress (15). However, numerous factors, including hypoxia, ER stress, infection, inflammation, environmental toxins, nutrition, and mitochondrial respiration, all participate in the excessive ROS generation in cells.
Everything has two sides, and it is crucial for cell to regulate ROS levels to avoid oxidative stress. Cells have developed antioxidant defense mechanisms to scavenge ROS in maintaining homeostasis. A number of nonenzymatic and enzymatic antioxidant defense mechanisms are responsible for neutralizing ROS. The nonenzymatic defense system includes glutathione (GSH), flavonoids, dietary antioxidants such as vitamins A, C, and E, selenium and β−carotene (23). The enzymatic antioxidant system includes superoxide dismutase (SOD), glutathione peroxidase (GPX), catalase (CAT), peroxiredoxin (PRX), glutathione S-transferases (GST), glutathione reductase (GSR) and thioredoxin reductase (TRX) (24–26). It is important for cells to use these antioxidant defense mechanisms to regulate ROS levels to avoid oxidative stress. However, oxidative stress happens when the antioxidant defense system of body is overwhelmed by the production of ROS (Figure 1). Oxidative stress is involved in numerous human diseases, such as neurodegenerative disease, cancer, cardiovascular disease, inflammatory disease, immune system dysfunctions, allergy, diabetes, aging. For instance, inflammatory cells release chemical mediators of inflammation, particularly ROS. Due to their high reactivity, ROS typically oxidize targets with or adjacent to the intracellular compartment where they are produced, affecting surrounding neighboring cells.
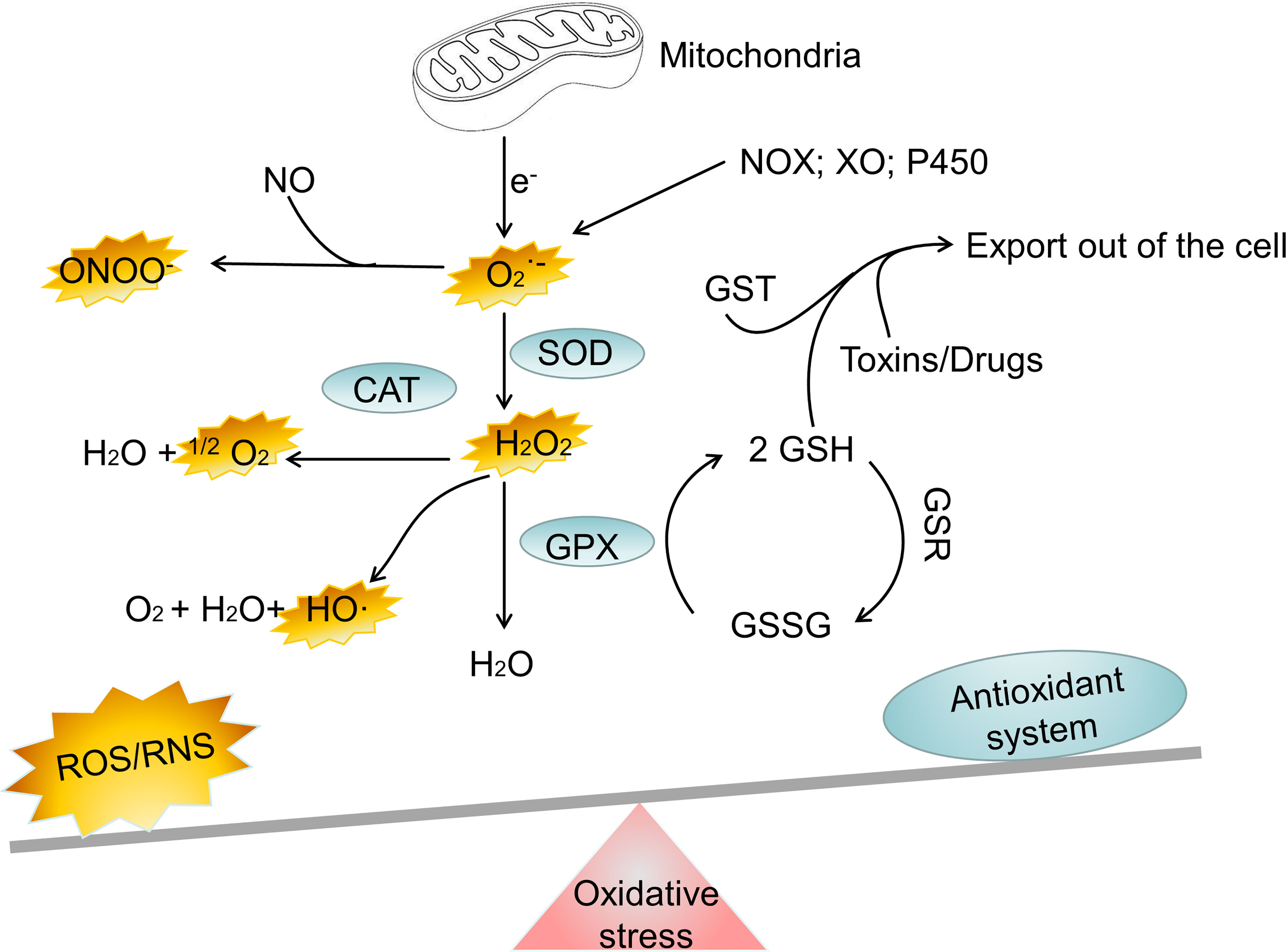
Figure 1 The major oxidant and antioxidant systems. NOX, NADPH oxidase; XO, xanthine oxidase; SOD, superoxide dismutase; CAT, catalase; GPX, glutathione peroxidase; GSH, glutathione; GSSG, reduced glutathione; GST, glutathione S-transferase; H2O2, hydrogen peroxide; ONOO−, peroxynitrite; HO·, hydroxyl radical; , superoxide; 1O2, singlet oxygen; Fe2+, Iron (II); Fe3+, Iron(III); ROS, reactive oxygen species; RNS, reactive nitrogen species.
A gram-negative, microaerophilic bacteria called H. pylori infects over 4.4 billion (or 59% of) people worldwide (7). The human gastric mucosa is selectively colonized by H. pylori, which can cause gastroduodenal diseases including chronic gastritis, mucosa-associated lymphoid tissue (MALT) lymphoma, peptic ulcers, and gastric adenocarcinoma (27). Sinus gastritis affects 10%-15% of H. pylori-infected patients and may potentially be connected to their own concurrent hypergastrinemia (28). Potential long-term complications for the patients include duodenal ulcers, intestinal metaplasia with dysplasia, gastric adenocarcinoma (non-cardia intestinal-type), and spontaneous diffuse gastric cancer (29). H. pylori can cause gastric lymphoma adenocarcinoma or gastric MALT lymphoma when it clings to the underlying epithelium (30, 31).
The principal producers of ROS and RNS in the body are neutrophils, macrophages and gastric epithelial cells (32) (Figure 2). In order to kill bacteria, NOX on the neutrophil membrane catalyzes the production of ROS (33). In an effort to eradicate the infection, phagocytic cells flood the area where H. pylori is present. In an effort to eliminate the bacteria, both neutrophils and macrophages phagocytose produce ROS. Additionally, the inducible nitric oxide synthase (iNOS), a crucial enzyme producing Nitric oxide, is expressed in the host neutrophils and epithelial cells (34). Despite the fact that H. pylori activates a strong innate and adaptive response, the human immune system is typically unable to completely eliminate the infection (35). DNA damage, oxidative stress, and chronic inflammation are all directly caused by this inadequate immune response (36). Patients with H. pylori infections exhibit higher amounts of ROS and NO-derived metabolites, which show that iNOS has been activated (37). Compared with phagocytic cells, the epithelial cells produce ROS at a much lower, which are involved in redox-sensitive signaling pathways but may not directly eradicate H. pylori (38). It is also known that the dual oxidases on the gastric epithelial cells produce H2O2 in response to infection, which likewise increases the levels of ROS (39). The environment of oxidative stress is available by the interaction of ROS generated by phagocytic and epithelial cell, which result in the growth of gastric cancer. On the one hand, one of the main causes of gastric cancer is oxidative stress by H. pylori infection.

Figure 2 The Various pathways of ROS production and DNA damage by the epithelial and immune cells. CagA, cytotoxin-associated gene A; SMO, spermine oxidase; H2O2, hydrogen peroxide; VacA, vacuolating cytotoxin A; HO-1, heme oxygenase 1; ROS, reactive oxygen species.
The main cause is oxidative stress by H. pylori infection in gastric cancer. Tumor forms by H. pylori water extract via ROS production (40). Reactive oxygen metabolites are terminated by H. pylori treatment to eliminate the infections (41). It was feasible to ascertain the impact of bacterial eradication on oxidative stress of mucosal by contrasting the levels of nitrotyrosine and 8-hydroxy-2’-deoxyguanosine (8-oxo-dG) in antral biopsies from patients with peptic ulcer and chronic atrophic gastritis before and after eradication. Human gastric mucosa experiences less oxidative stress when H. pylori is removed (42). The infection of H. pylori can be cured by prescribed vitamins E and C with antibody therapy (43). According to recent studies, H. pylori-infected gastric epithelial cells produce more ROS than healthy cells do. This increased ROS production may contribute to the infection-related apoptosis (44). Furthermore, numerous virulence factors in H. pylori strains may lead to oxidative stress in the host. There is a higher risk of gastric carcinogenesis in patients infected with CagA-positive compared to CagA-negative strains (45). Elevated hydrogen peroxide levels and oxidative DNA damage are shown in CagA positive strains (46). IL8 and tumor necrosis factor (TNF), markers of oxidative stress and inflammation, are also increasing (47). Despite the fact that the exact mechanism by which CagA causes carcinogenesis is still unknown, it is evident that these actions can contribute to raising the chance of developing gastric cancer (48). On the other hand, gastric cells can protect themselves against oxidative stress by producing scavenger molecules.
Gastric cells can protect themselves against oxidative stress by producing scavenger molecules. Metallothioneins are important components in preventing H. pylori-induced gastric erosive lesions in the animal model (49). Other antioxidant systems include those that control energy metabolism globally, such as AMP-activated protein kinase (AMPK) (50) and the cytoprotective activity of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (51). At the same time, H. Pylori has also developed oxidative stress defense mechanisms that might encourage the acquisition of potentially cancerous traits and accelerate the development of the condition into gastric cancer (52). For example, NO levels and superoxide dismutase activity were found to have a relevant and reverse association in gastric juice of patients suffering from H. pylori (53). Isogenic mutants deficient in the activities of thioredoxin (54), catalase (KatA) (55), NADPH quinone reductase (56), and superoxide dismutase (46) are sensitive to host colonization and susceptible to oxidative damage. Besides, it is interesting to note that the bacteria also produce ROS (32).
Tobacco smoke from tar and gas phases maintain a variety of compounds, including unstable free radicals and ROS, which can harm organism through oxidative stress. The burning of tobacco produces ROS in the gas phase inhaled by smokers, as part of the mainstream smoke (57). Several rather stable free radicals in the tar phase are included in the tarry matrix, such as the quinone/hydroquinone (Q/QH2) complex (58). This Q/QH2 polymer may act as the redox system by converting pulmonary O2 to or additional free radicals like H2O2 and ·OH (59). Another crucial point is that, when an individual’s antioxidant defense system is weak or saturated, inhaling additional ROS or other reactive metabolites produced by the biotransformation of chemicals in tobacco smoke can increase the amount of oxidative stress caused by the gas-phase and tar-phase derived ROS (60). In addition, tar builds up in the lungs from cigarette smoke particles and processes, producing an aqueous solution that goes through redox cycling to produce different reactive species, causing damage subsequently (61).
Increasing data indicate that the release of ROS from smoking and the subsequent oxidative stress have a substantial impact on inflammation and carcinogenesis. Estimates suggest that tobacco use causes about 80,000 cases of gastric cancer annually (11% of all estimated cases) (62). Despite the decline among population-attributable fractions, smoking remained the main risk factor for men’s gastric cancer in 2012, where the incidence is substantially higher in 2020 (63). Healthy smokers may be more susceptible to oxidant-mediated tissue damage and gastric cancer because of their poor antioxidant level. The levels of thiobarbituric acid reactive substances (TBARS) are higher in smokers than in non-smokers with gastric cancer, and smokers have lower levels of SOD, CAT, GPX, GST, GSR and decreased vitamins A, E, and C (64). Low-density lipoprotein cholesterol, high-density lipoprotein cholesterol and total cholesterol all dramatically rise in non-smokers while falling in smokers, whereas these reduced in smokers (65). It has been discovered that antioxidant-rich diet significantly influenced smokers’ cellular stress protection (66). Plasma levels of malondialdehyde (MDA) were substantially higher and melatonin levels were substantially lower in smokers than non-smokers, which appears that melatonin can lessen the respiratory system damage caused by free radicals brought on by cigarette smoke (67).
Under oxidative stress, increased ROS in cells may harm tissues and trigger carcinogenesis, especially in the gastrointestinal system (Figure 3). ROS are initiating factor in gastric carcinogenesis in both humans and mice. Serum and tissue samples from the human gastrointestinal have dysregulated ROS levels (41). In mice gastric cancer models induced by H. pylori and N-methyl-N’nitro-N’nitrosoguanidine (MNNG), the downstream pathways P53, Wnt, Ras, and mTOR are activated by ROS (70, 71). Proviral insertion in murine lymphomas 2 (PIM2) is reported to act as an oncogene in gastric cancer, controlling apoptosis via ROS-triggered ER stress, and promoting the development of gastric cancer (72). 13 biomarkers including β-catenin, C-MYC, GATA-4, CXCL13, DAPK1, TIMP3, DC-SIGN, EGFR, PIM2, GRIN2B, SLC5A8, VCAM-1 and CDH1 are related to the development of gastric cancer, and six of them including β-catenin, DC-SIGN, C-MYC, EGFR, CXCL13 and PIM2 have been reported overexpressed in gastric tissue from infected children and gastric cancer patients (73). Moreover, it has been shown that stomach cancer is more likely to develop as a result of the oxidative stress brought on by CagA-positive bacteria (74), in which H pylori CagA produces cells with oxidative DNA damage by inducing spermine oxidase (SMO), and a portion of these cells are apoptosis-resistant and therefore highly susceptible to developing cancer (75). Oxidative stress can cause DNA damage caused by H pylori infection. In vitro investigations have demonstrated that cells infected with H pylori that have defective DNA repair systems experience increased oxidative stress and DNA damage (76). In vivo studies using mice lacking a component of the base excision repair process revealed significant stomach lesions after H pylori infection (46). H. pylori’s propensity to generate DNA strand breaks undoubtedly contributes to genomic instability and may aid in carcinogenesis (77). NO can block 8-oxoguanine glycosylase from removing DNA mutations. Research has revealed that H. pylori infection increases phosphohistone H2AX, a marker of repair for double-strand DNA breaks (46). It has been reported that 8-hydroxy-2’deoxyguanosine buildup causes DNA damage. The loss of a base following damage would create an abasic site, which could result in a single-strand break in the DNA. Inadequate repair or continuous damage may cause double-strand breaks in the DNA, though DNA strands can be produced in various ways (46). If a cell does not heal enough fractures, it may die.
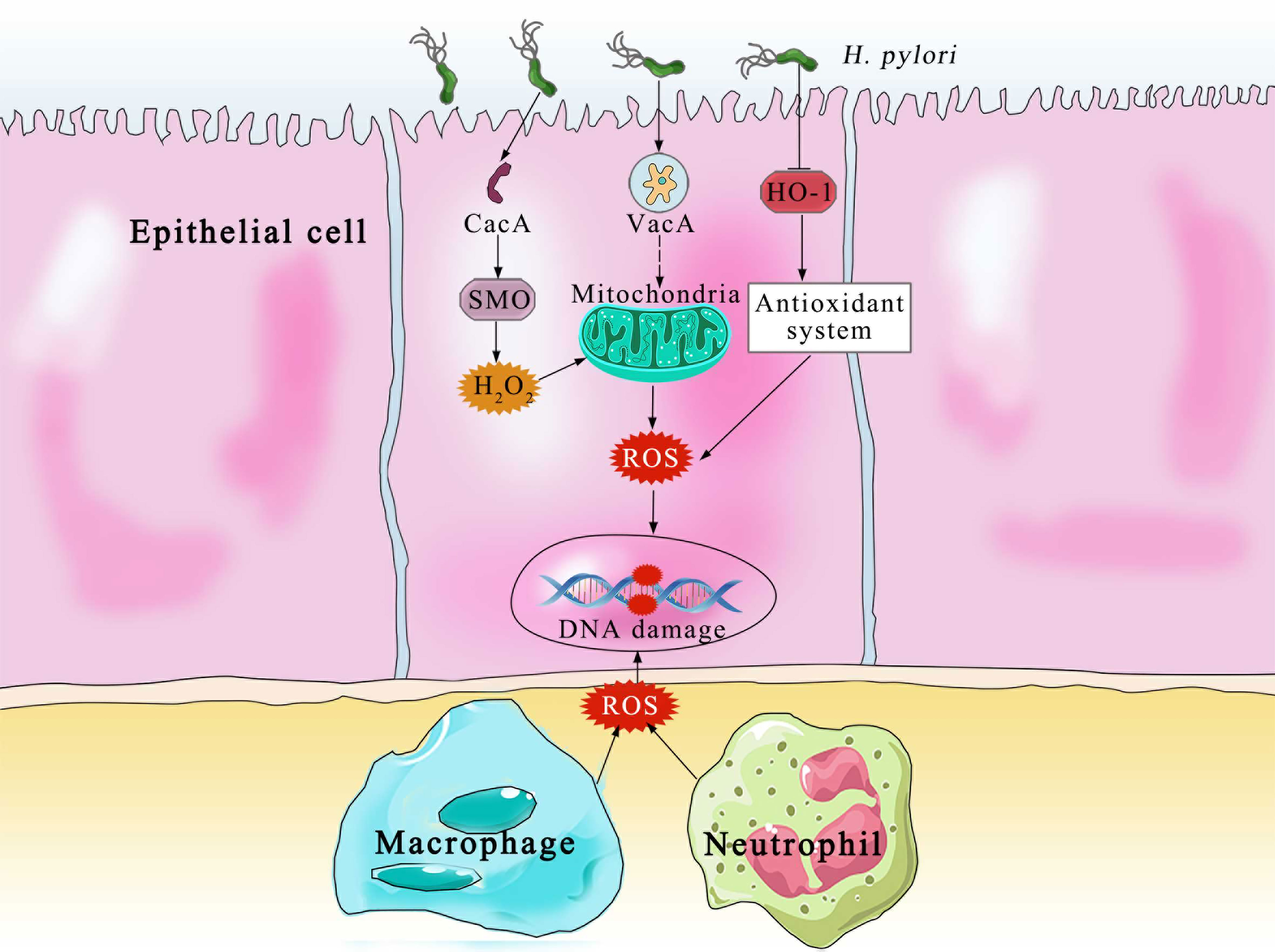
Figure 3 ROS and its pathophysiological effects in gastric carcinogenesis. At low to moderate concentrations, ROS function as signaling molecules that support cellular differentiation and proliferation and activate survival pathways in response to stress. Excessive ROS harms lipids, proteins, and DNA, causing mucosal injury and trigger carcinogenesis. Reactive aldehydes include 4-hydroxynonenal and other aldehydes (68). The mutator phenotype is shown by the self-directed arrow at mutation (69).
Tumor hypoxia is well recognized in oncology as a major cause of therapy resistance and poor prognosis. Hypoxia promotes the production of several gene products implicated in tumor development, invasion, and metastasis formation of gastric cancer. Hypoxia causes the production of ROS, which inhibit the degradation of the hypoxia-inducible factor 1 (HIF-1) (78). Subsequently, HIF-1α influences the expression of numerous genes that are crucial for gastric carcinogenesis. For instance, Angiogenesis is promoted by HIF-1 to stimulate the vascular endothelial growth factor (VEGF) pathway in gastric cancer (79). Caveolin-1 (Cav-1) is expressed less while induced by HIF-1, which regulates E-cadherin to cause the epithelial-mesenchymal transition (EMT) in gastric cancer (80). On the other hand, as a signaling molecule, ROS activates vital signaling pathways that are crucial to promote the onset and progression of gastric cancer. ROS, also as a second messenger, can activate tyrosine kinases and MAPK which promote cell development (81), and the protein kinase-B (Akt)/mTOR signaling pathway which promotes cell growth of gastric cancer (82). Additionally, ROS activates nuclear factor-B (NF-κB), facilitating invasion of gastric cancer (83).
Furthermore, H. pylori-colonized mucosal cells with deficient DNA repair systems are more vulnerable to oxidative stress and DNA damage (84). Spermine oxidase (SMOX) is activated in H. pylori in gastric epithelial cells, leading to oxidative stress (85). DNA damage promotes mutations of suppressor in tumor such as calcium/calmodulin dependent serine protein kinase (CASK), p53, as well as stimulation of the epidermal growth factor receptor (EFGR) signaling pathway, which are important early events in gastric carcinogenesis (86, 87). H. pylori colonization also negatively affects the expression of antioxidant proteins, along with epigenetic modifications and DNA methylation, such as GATA-4, GATA-5 and TWIST-1 (88), as well as miRNAs dysregulation, such as mir-21, mir-92a, mir-27a, mir-146a, mir-326, mir-155 and mir-663 (73, 89). It has been demonstrated that the expression of the purine-free/pyrimidine-free nucleic acid endonuclease 1 (APE1) is downregulated in gastric host cells infected with H. pylori, which ultimately reduces T-cell capacity for repair, increasing the likelihood of DNA carboxy-terminal genetic alterations. The oxidative stress defensive factors such as FOXO1, are known to be inhibited by miR-27a, which is recognized as an oncogenic miRNA in gastric cancer (90). miR-328 is downregulated in H. pylori -infected gastritis (90), and the low level of miR-328 activates CD44 to promote the differentiation of gastric stem cell (68). H. pylori increases the expression of miR-210 by controlling its methylation, which in turn suppressed dimethyl adenosine transferase 1 (DIMT1) and oncoprotein 18 or metablastic (STMN1), which promotes the proliferation of gastric epithelial cells (69). Due to the methylation of the gene promoter region by ROS, H. pylori infection may change the expression of miRNAs in oxidative stress, interfering with the methylation of miRNAs, which may contribute to the mechanism triggering the onset of gastric carcinogenesis.
The process of developing gastric cancer involves several stages, beginning with the change from normal mucosa to chronic superficial gastritis (non-atrophic gastritis). Atrophic gastritis, intestinal metaplasia, dysplasia and adenocarcinoma, among other conditions can be caused by gastritis (91). Gastritis caused by H. pylori is the only condition that always precedes diffuse gastric cancer. According to Correa’s idea, a series of events initiating with chronic superficial gastritis and progressing from atrophic gastritis, intestinal metaplasia, and dysplasia to gastric cancer (92). The especially high risk of cancer exists in people who have antibodies to the CagA protein, which is a marker for the more inflammatory and virulent strain of H. pylori that carries a pathogenicity island of genes. According to a meta-analysis of research, CagA-positive strains are two times more likely than CagA-negative strains to cause noncardia gastric cancer (93). The cag+ H. pylori strains have a stronger connection to gastric carcinogenesis than strains without cag (94). ROS or RNS production is substantially increased in vascular endothelium, gastric mucosa infected with H. pylori, and neutrophils aggregated in inflammatory mucosa (93). Following H. pylori infection, phagocytes that have gathered in the stomach mucosa produce O2·, HO·, and HOCl (95). Rat gastric mucosal cells have been shown to undergo apoptosis when exposed to NH2Cl (96).
Epstein-Barr virus (EBV) is recognized as a pathogen that causes stomach cancer. Nearly 10% of cases of gastric cancer are EBV-associated gastric cancer, which is the monoclonal proliferation of epithelial cells infected with EBV that only express a few EBV-latent genes (Latency I program) (97). The production of NH2Cl by infiltrating neutrophils can convert latent EBV into lytic EBV in the H. pylori-infected gastric, which may further contribute to gastric carcinogenesis (98). Although the function of the ROS generated by infected gastric epithelial cells is not fully known, it is thought that these ROS trigger signaling processes that control how H. pylori pathogenesis develops.
H. pylori infection directly causes oxidative stress in gastric epithelial cells by the production of ROS, and it also stimulates host responses that result in ROS and controls the production of proinflammatory cytokines, inflammation, and cell death (99). Continuous ROS production results in oncogene and tumor suppressor gene changes, as well as chromosomal abnormalities by oxidative genome damage, which includes the oxidation of guanine to form 8-OhdG and 8-oxo,7,8-dihydroguanosine (8-OHG) in RNA and DNA (100).
When compared to normal mucosa, gastric adenoma and H. pylori-infected or uninfected cancer tissues express ROS and APE1/Ref1 more mucosally (101). As a result of H. pylori infection, both the gastrointestinal lumen and gastric juice ascorbic acid content decrease. This antioxidant lessens the effects of carcinogens by lowering carcinogenic substances including nitrosamines and ROS. Depleting cellular antioxidants makes ROS more effective at killing cancer cells because this is the traditional treatment strategy for doing so. Perhaps, the disease can be regulated by blocking different antioxidant systems during neoadjuvant treatments.
Gastric MALT lymphomas are a slow-growing type of non-Hodgkins lymphoma, developed from the extranodal marginal zone of lymphoid follicles (102). Gastric MALT lymphoma is an illustration of the intimate pathogenetic relationship between chronic inflammation and tumor development. Approximately 92% of gastric MALT lymphomas have a tight connection to H. pylori infection which makes H. pylori easier to develop and diffuse (103). The H. pylori strains linked to gastric MALT lymphoma are less virulent than those linked to gastric adenocarcinoma. The latter strains may have the VacA m2 gene without the CagPAI, which could make H. pylori carriers easier to develop diffuse large B-cell lymphoma (104). H. pylori infection increased the incidence of low-grade gastric MALT lymphoma by an odds ratio of 2.8 times compared with H. pylori-negative individuals (105). Within gastric MALT lymphomas, T lymphocytes activated by H. pylori are responsible for B-cell proliferation (106).
Most individuals with early-stage H. pylori disease have been in durable remission for more than ten years after completing a single brief course of combination antibiotic therapy. A meta-analysis of more than 30 trials found that the overall remission rate of MALT lymphomas with a low histological grade that is restricted to the perigastric lymph nodes or the gastric wall (stage I or stage IIe1 illness) was 78% (107). Therefore, preventive removal of H. pylori is particularly helpful in reversing MALT lymphoma either in the early MALT stage or in the late bone marrow-involved stage. However, the recurring possibility of MALT lymphoma should not be ignored because it frequently returns several years following surgery, which may due to risk factors for gastric cancer have not been totally blocked.
Gastric MALT lymphoma is regarded as one of the greatest models for understanding how genetic events contribute to oncogenesis, influence tumor biology, govern clinical behavior, and represent feasible treatment targets. Genetic aberrations arise through the release of ROS, H. pylori-induced endonucleases, and other effects. Stronger oxidative stress is caused by H. pylori strains originating from gastric cancer in the host, and these strains may have suppressive effects on the host’s GSH-related defensive mechanisms (108). Surprisingly, the nucleotide-binding oligomerization domain protein 2 (NOD2) functions as a receptor for pattern recognition. H. pylori activates NF-κB signaling via NOD2. However, the NF-κB signaling is uncontrolled when the R702W gene is mutated, protecting the organism against the harm caused by oxidative stress induced by H. pylori (109). Thus, it is essential to consider how the gastric MALT lymphoma is influenced by the NOD2 gene (110) (Table 2).
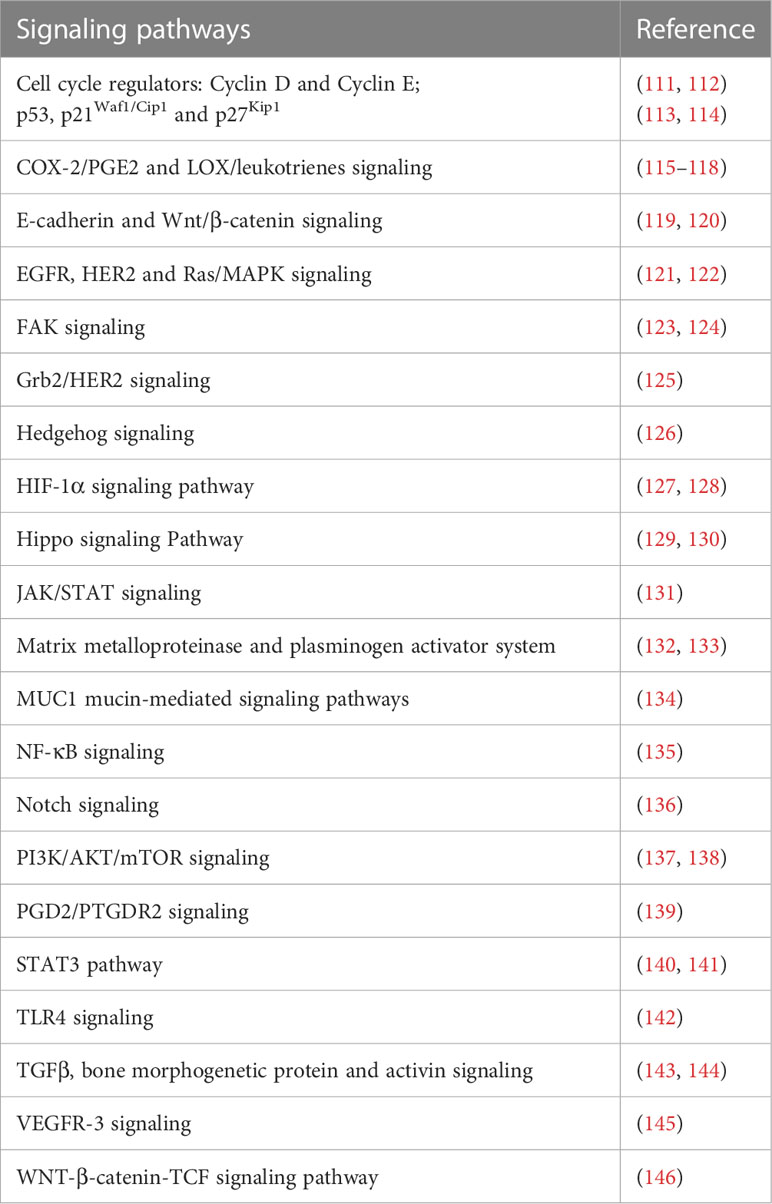
Table 2 A partial list of signaling pathways linked to oxidative stress in gastric cancer.
Regulation of redox homeostasis is crucial because increasing oxidative stress has a role in all stages of carcinogenesis either initiating/stimulating tumorigenesis and promoting cancer cells transformation/proliferation or leading to cell death. Enhancing antioxidant defense capability decreases ROS as a result of one strategy (Table 3). However, utilizing antioxidants has been shown to change the effectiveness of treatment and, in some cases, even speed up the development of tumors.
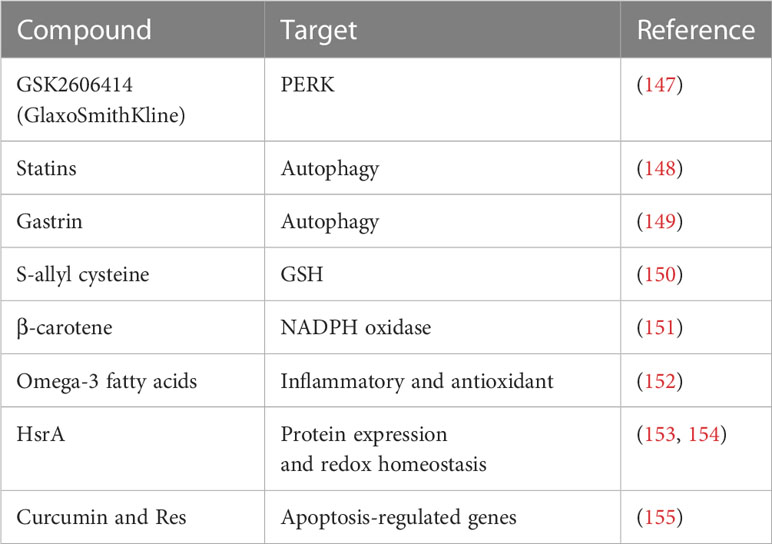
Table 3 Antioxidant therapy.
According to a recent study, the garlic compound S-allyl cysteine has anti-inflammatory and antioxidant properties, which greatly raises the GSH levels in the liver, gastric tissue, and serum of rat models of gastric cancer, and lowers the risk of developing gastric cancer (156). In experimental settings using AGS cells infected with H. pylori strains, GSH levels are lower in individuals with gastric cancer than in those with duodenal ulcers, indicating a more severe oxidative stress response to gastric cancer with H. pylori infection (157). The level of GSH and the ratio of GSSG/GSH significantly decline in patients of gastric cancer with H. pylori infection, and glutamine levels are also low. Additionally, the production of hydrogen peroxide is encouraged, aggravating the effects of oxidative stress. However, GSH therapy is proved successful in alleviating the high ROS buildup (158). In conclusion, intestinalization in the gastric host cells is caused by low GSH levels. Therefore, the risk of H. pylori-induced carcinogenesis of gastric mucosal may be ameliorated in rats by raising their GSH levels, which may also prevent oxidative stress damage (108).
Antioxidants, such as vitamin E and selenium, have been the subject of numerous research in this context. In 1993, the first large, randomized, double-blind, primary prevention trial to investigate the potential cancer prevention benefits of supplementing with vitamin E, selenium and β-carotene was conducted, and the cocktail has been found to dramatically lower mortality from gastric cancer (159). Interestingly, the protective effects of these antioxidants can still be noticeable ten years after the end of supplementation (160). Clinical studies have shown that consistent oral dose of β-carotene is advantageous for lowering bacterial colonization by 48% (151). It has been proposed that intake of diet rich in vitamin C, carotenoids, and alpha-lipoic acid (α-LA) may lessen the morbidity of gastric disease linked to H. pylori infection. α-LA, a naturally occurring dithiol with antioxidant and anti-inflammatory function, can decrease the interaction between Nrf2 and Keap1, inhibit the pro-inflammatory cytokine IL-8 production and minimize the infection via the Nrf2/HO-1 pathway in the AGS cells (161). It is reported that omega-3 fatty acids inhibit the oxidation of polyunsaturated long-chain fatty acids and boost the antioxidant and anti-inflammatory effects of other nutrients (162). However, omega-3 may result in oxidative stress, and the process is associated with the suppression of the production of antioxidant enzymes. Therefore, antibiotics such clarithromycin, metronidazole, quinolones, amoxicillin, and tetracycline to counteract the oxidative effects of omega-3 is recommended (74). The expression of SOD2 (Mn-SOD), superoxide anion scavenger, is elevated, but the expression of SOD1 (copper/zincSOD) is decreased while comparing gastric cancer tissues with their matching normal mucosa. In specifically, the Mn-SOD ratio (levels in normal and malignant tissue) is demonstrated as an independent predictive indicator in patients of gastric cancer, and it appears to be therapeutically relevant for the survival of patients, the higher the ratio, the poorer overall survival (163). MnSOD is elevated in primary tumors with lymph node metastases while comparing gastric cancer patients with and without metastasis, indicating that MnSOD and ROS are involved in metastasis (164).
More importantly, it is necessary to block oxidative stress completely sometimes. For instance, HsrA, the in vivo exclusive regulator for epsilon proteobacteria, is involved in altering redox homeostasis and protein expression. Consequently, it may serve as a potential therapeutic target to eradicate H. pylori (153, 165). The increased expression of apoptosis-regulated gene in the gastric host cells of patients with H. pylori infection, such as BID, ZMAT3, PMAIP1 and FAS, can also be successfully controlled by the combination of curcumin and Res, which causes apoptosis to decline (166, 167).
Gastric cancer is the third leading cause of cancer-related death worldwide. Free radicals and oxidative stress are continuously imposed upon cells in tissues and organs on a regular basis. More and more evidences show that ROS functions an essential role in the gastric cancer. Despite a number of mechanisms have been discussed in this review, most of the ROS-induced signaling targets are yet unknown. The elevated ROS production in gastric cancer can initiate genotoxic consequences, contributing to genetic instability, DNA damage, metabolic adaptation, drug resistance and occasional cell death. However, certain amounts of ROS can be advantageous because they trigger the antioxidant defense system and shield cells. There is an urgent need to find selective and readily available therapeutic therapies for gastric cancer and gastric cancer-predisposed patients. In order to treat and prevent ROS in gastric cancer, it may be crucial to focus on the enhancement of ROS by neutralizing antioxidants to induce cancer cell death, and the inhibition of ROS activity or increase of antioxidant capacity to regulate pro-tumorigenic signaling pathways. Nevertheless, considering that multiple studies have connected some dietary antioxidants with a rise in cancer incidence, it will be crucial to thoroughly investigate all biochemical reactions within cancer cells, including their precise targets and downstream effects while boosting antioxidant capacity. More researches are needed to put on the agenda to explore the function of elevated ROS and identify the exact ROS target pathways that will be most beneficial in treating gastric cancer.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Young Talent Project of Jiangsu Provincial Traditional Chinese Medicine Science and Technology Development Plan (QN202206).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YP declared a shared parent affiliation with the author YS to the handling editor at the time of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rawla P, Barsouk A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz Gastroenterol (2019) 14(1):26–38. doi: 10.5114/pg.2018.80001
2. Thrift AP, El-Serag HB. Burden of gastric cancer. Clin Gastroenterol Hepatol (2020) 18(3):534–42. doi: 10.1016/j.cgh.2019.07.045
3. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet (2016) 388(10060):2654–64. doi: 10.1016/S0140-6736(16)30354-3
4. Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: Descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev (2014) 23(5):700–13. doi: 10.1158/1055-9965.EPI-13-1057
5. Wang JP, Sun YH, Bertagnolli MM. Comparison of gastric cancer survival between Caucasian and Asian patients treated in the united states: Results from the surveillance epidemiology and end results (Seer) database. Ann Surg Oncol (2015) 22(9):2965–71. doi: 10.1245/s10434-015-4388-4
6. Bauer K, Schroeder M, Porzsolt F, Henne-Bruns D. Comparison of international guidelines on the accompanying therapy for advanced gastric cancer: Reasons for the differences. J Gastric Cancer (2015) 15(1):10–8. doi: 10.5230/jgc.2015.15.1.10
7. Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, et al. Global prevalence of helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology (2017) 153(2):420–9. doi: 10.1053/j.gastro.2017.04.022
8. Song Y, Liu X, Cheng W, Li H, Zhang D. The global, regional and national burden of stomach cancer and its attributable risk factors from 1990 to 2019. Sci Rep (2022) 12(1):11542. doi: 10.1038/s41598-022-15839-7
9. Jencks DS, Adam JD, Borum ML, Koh JM, Stephen S, Doman DB. Overview of current concepts in gastric intestinal metaplasia and gastric cancer. Gastroenterol Hepatol (N Y) (2018) 14(2):92–101.
10. Habtemariam S. Modulation of reactive oxygen species in health and disease. Antioxid (Basel Switzerland) (2019) 8(11):513. doi: 10.3390/antiox8110513
11. Sarmiento-Salinas FL, Perez-Gonzalez A, Acosta-Casique A, Ix-Ballote A, Diaz A, Trevino S, et al. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci (2021) 284:119942. doi: 10.1016/j.lfs.2021.119942
12. Barnes RP, Fouquerel E, Opresko PL. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech Ageing Dev (2019) 177:37–45. doi: 10.1016/j.mad.2018.03.013
13. Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discovery (2018) 8(5):537–55. doi: 10.1158/2159-8290.cd-17-1461
14. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. Ros in cancer therapy: The bright side of the moon. Exp Mol Med (2020) 52(2):192–203. doi: 10.1038/s12276-020-0384-2
15. Azmanova M, Pitto-Barry A. Oxidative stress in cancer therapy: Friend or enemy? Chembiochem (2022) 23(10):e202100641. doi: 10.1002/cbic.202100641
16. Braga-Neto MB, Costa DVS, Queiroz DMM, Maciel FS, de Oliveira MS, Viana-Junior AB, et al. Increased oxidative stress in gastric cancer patients and their first-degree relatives: A prospective study from northeastern Brazil. Oxid Med Cell Longev (2021) 2021:6657434. doi: 10.1155/2021/6657434
17. Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem (2015) 97:55–74. doi: 10.1016/j.ejmech.2015.04.040
18. Sharaf A, De Michele R, Sharma A, Fakhari S, Obornik M. Transcriptomic analysis reveals the roles of detoxification systems in response to mercury in chromera velia. Biomolecules (2019) 9(11):647. doi: 10.3390/biom9110647
19. Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med (2009) 47(4):333–43. doi: 10.1016/j.freeradbiomed.2009.05.004
20. Sandalio LM, Rodríguez-Serrano M, Romero-Puertas MC, del Río LA. Role of peroxisomes as a source of reactive oxygen species (Ros) signaling molecules. Sub-cellular Biochem (2013) 69:231–55. doi: 10.1007/978-94-007-6889-5_13
21. Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and er stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. BioMed Pharmacother (2019) 118:109249. doi: 10.1016/j.biopha.2019.109249
22. Zia A, Farkhondeh T, Pourbagher-Shahri AM, Samarghandian S. The roles of mitochondrial dysfunction and reactive oxygen species in aging and senescence. Curr Mol Med (2022) 22(1):37–49. doi: 10.2174/1566524021666210218112616
23. Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J (2015) 29(12):4766–71. doi: 10.1096/fj.15-275404
24. Veskoukis AS, Tsatsakis AM, Kouretas D. Dietary oxidative stress and antioxidant defense with an emphasis on plant extract administration. Cell Stress Chaperones (2012) 17(1):11–21. doi: 10.1007/s12192-011-0293-3
25. Benhar M. Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress. Free Radic Biol Med (2018) 127:160–4. doi: 10.1016/j.freeradbiomed.2018.01.028
26. Kirtonia A, Sethi G, Garg M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci (2020) 77(22):4459–83. doi: 10.1007/s00018-020-03536-5
27. Backert S, Neddermann M, Maubach G, Naumann M. Pathogenesis of helicobacter pylori infection. Helicobacter (2016) 21 Suppl 1:19–25. doi: 10.1111/hel.12335
28. Censini S, Stein M, Covacci A. Cellular responses induced after contact with helicobacter pylori. Curr Opin Microbiol (2001) 4(1):41–6. doi: 10.1016/s1369-5274(00)00162-4
29. Banks M, Graham D, Jansen M, Gotoda T, Coda S, di Pietro M, et al. British Society of gastroenterology guidelines on the diagnosis and management of patients at risk of gastric adenocarcinoma. Gut (2019) 68(9):1545–75. doi: 10.1136/gutjnl-2018-318126
30. Kalisperati P, Spanou E, Pateras IS, Korkolopoulou P, Varvarigou A, Karavokyros I, et al. Inflammation, DNA damage, helicobacter pylori and gastric tumorigenesis. Front Genet (2017) 8:20. doi: 10.3389/fgene.2017.00020
31. Zhang XY, Zhang PY, Aboul-Soud MA. From inflammation to gastric cancer: Role of helicobacter pylori. Oncol Lett (2017) 13(2):543–8. doi: 10.3892/ol.2016.5506
32. Handa O, Naito Y, Yoshikawa T. Helicobacter pylori: A ros-inducing bacterial species in the stomach. Inflammation Res (2010) 59(12):997–1003. doi: 10.1007/s00011-010-0245-x
33. Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med (2009) 47(9):1239–53. doi: 10.1016/j.freeradbiomed.2009.07.023
34. Saini R, Singh S. Inducible nitric oxide synthase: An asset to neutrophils. J Leukoc Biol (2019) 105(1):49–61. doi: 10.1002/JLB.4RU0418-161R
35. Karkhah A, Ebrahimpour S, Rostamtabar M, Koppolu V, Darvish S, Vasigala VKR, et al. Helicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases. Microbiol Res (2019) 218:49–57. doi: 10.1016/j.micres.2018.09.011
36. Souliotis VL, Vlachogiannis NI, Pappa M, Argyriou A, Ntouros PA, Sfikakis PP. DNA Damage response and oxidative stress in systemic autoimmunity. Int J Mol Sci (2019) 21(1):55. doi: 10.3390/ijms21010055
37. Lin TY, Lan WH, Chiu YF, Feng CL, Chiu CH, Kuo CJ, et al. Statins’ regulation of the virulence factors of helicobacter pylori and the production of ros may inhibit the development of gastric cancer. Antioxid (Basel Switzerland) (2021) 10(8):1293. doi: 10.3390/antiox10081293
38. Jain U, Saxena K, Chauhan N. Helicobacter pylori induced reactive oxygen species: A new and developing platform for detection. Helicobacter (2021) 26(3):e12796. doi: 10.1111/hel.12796
39. Grasberger H, El-Zaatari M, Dang DT, Merchant JL. Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent helicobacter felis infection and inflammation in mice. Gastroenterology (2013) 145(5):1045–54. doi: 10.1053/j.gastro.2013.07.011
40. Ishikawa T, Yoshida N, Tokuda H, Kokura S, Nakabe N, Kuchide M, et al. Role of oxygen-derived free radicals in helicobacter pylori water extract-induced mouse skin carcinogenesis. Biofactors (2006) 28(1):1–7. doi: 10.1002/biof.5520280101
41. Mashimo M, Nishikawa M, Higuchi K, Hirose M, Wei Q, Haque A, et al. Production of reactive oxygen species in peripheral blood is increased in individuals with helicobacter pylori infection and decreased after its eradication. Helicobacter (2006) 11(4):266–71. doi: 10.1111/j.1523-5378.2006.00410.x
42. Zou Y, Chen X, Sun Y, Li P, Xu M, Fang P, et al. Antibiotics-free nanoparticles eradicate helicobacter pylori biofilms and intracellular bacteria. J Control Release (2022) 348:370–85. doi: 10.1016/j.jconrel.2022.05.044
43. Sezikli M, Cetinkaya ZA, Sezikli H, Güzelbulut F, Tiftikçi A, Ince AT, et al. Oxidative stress in helicobacter pylori infection: Does supplementation with vitamins c and e increase the eradication rate? Helicobacter (2009) 14(4):280–5. doi: 10.1111/j.1523-5378.2009.00686.x
44. Ding SZ, Minohara Y, Fan XJ, Wang J, Reyes VE, Patel J, et al. Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect Immun (2007) 75(8):4030–9. doi: 10.1128/IAI.00172-07
45. Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K, et al. Reactive oxygen species-induced autophagic degradation of helicobacter pylori caga is specifically suppressed in cancer stem-like cells. Cell Host Microbe (2012) 12(6):764–77. doi: 10.1016/j.chom.2012.10.014
46. Butcher LD, den Hartog G, Ernst PB, Crowe SE. Oxidative stress resulting from helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol Gastroenterol Hepatol (2017) 3(3):316–22. doi: 10.1016/j.jcmgh.2017.02.002
47. O’Hara AM, Bhattacharyya A, Bai J, Mifflin RC, Ernst PB, Mitra S, et al. Tumor necrosis factor (Tnf)-Alpha-Induced il-8 expression in gastric epithelial cells: Role of reactive oxygen species and ap endonuclease-1/Redox factor (Ref)-1. Cytokine (2009) 46(3):359–69. doi: 10.1016/j.cyto.2009.03.010
48. Amieva M, Peek RM Jr. Pathobiology of helicobacter pylori-induced gastric cancer. Gastroenterology (2016) 150(1):64–78. doi: 10.1053/j.gastro.2015.09.004
49. Mita M, Satoh M, Shimada A, Okajima M, Azuma S, Suzuki JS, et al. Metallothionein is a crucial protective factor against helicobacter pylori-induced gastric erosive lesions in a mouse model. Am J Physiol Gastrointestinal liver Physiol (2008) 294(4):G877–84. doi: 10.1152/ajpgi.00251.2007
50. Zhao H, Zhu H, Lin Z, Lin G, Lv G. Compound 13, an Alpha1-selective small molecule activator of ampk, inhibits helicobacter pylori-induced oxidative stresses and gastric epithelial cell apoptosis. Biochem Biophys Res Commun (2015) 463(4):510–7. doi: 10.1016/j.bbrc.2015.05.059
51. Skowron MA, Niegisch G, Albrecht P, van Koeveringe G, Romano A, Albers P, et al. Various mechanisms involve the nuclear factor (Erythroid-derived 2)-like (Nrf2) to achieve cytoprotection in long-term cisplatin-treated urothelial carcinoma cell lines. Int J Mol Sci (2017) 18(8):1680. doi: 10.3390/ijms18081680
52. Wang G, Alamuri P, Maier RJ. The diverse antioxidant systems of helicobacter pylori. Mol Microbiol (2006) 61(4):847–60. doi: 10.1111/j.1365-2958.2006.05302.x
53. Ansari MR-N M, Dolatkhah H, Fattahi E, Aghazade AM, Mojtabaii-Motlag S. Comparison of levels of nitric oxide, superoxide dismutase and glutathione peroxidase of gastric juice in infected and non-infected patients with helicobacter Pylori. Acta Medica Iranica (2006) 44(3):159–66.
54. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med (2014) 66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036
55. Harris AG, Wilson JE, Danon SJ, Dixon MF, Donegan K, Hazell SL. Catalase (Kata) and kata-associated protein (Kapa) are essential to persistent colonization in the helicobacter pylori Ss1 mouse model. Microbiol (Reading England) (2003) 149(Pt 3):665–72. doi: 10.1099/mic.0.26012-0
56. Wang G, Maier RJ. An nadph quinone reductase of helicobacter pylori plays an important role in oxidative stress resistance and host colonization. Infect Immun (2004) 72(3):1391–6. doi: 10.1128/IAI.72.3.1391-1396.2004
57. Seyler RW Jr., Olson JW, Maier RJ. Superoxide dismutase-deficient mutants of helicobacter pylori are hypersensitive to oxidative stress and defective in host colonization. Infect Immun (2001) 69(6):4034–40. doi: 10.1128/iai.69.6.4034-4040.2001
58. Stone KK, Bermudez E, Pryor WA. Aqueous extracts of cigarette tar containing the tar free radical cause DNA nicks in mammalian cells. Environ Health Perspect (1994) 102 Suppl 10:173–8. doi: 10.1289/ehp.94102s10173
59. Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutat Res (2015) 763:212–45. doi: 10.1016/j.mrrev.2014.11.002
60. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J (2012) 5(1):9–19. doi: 10.1097/WOX.0b013e3182439613
61. Wang S, Gallimore PJ, Liu-Kang C, Yeung K, Campbell SJ, Utinger B, et al. Dynamic wood smoke aerosol toxicity during oxidative atmospheric aging. Environ Sci Technol (2023) 57(3):1246–56. doi: 10.1021/acs.est.2c05929
62. Lyons K, Le LC, Pham YT, Borron C, Park JY, Tran CTD, et al. Gastric cancer: Epidemiology, biology, and prevention: A mini review. Eur J Cancer Prev (2019) 28(5):397–412. doi: 10.1097/cej.0000000000000480
63. Peleteiro B, Castro C, Morais S, Ferro A, Lunet N. Worldwide burden of gastric cancer attributable to tobacco smoking in 2012 and predictions for 2020. Dig Dis Sci (2015) 60(8):2470–6. doi: 10.1007/s10620-015-3624-x
64. Pasupathi P, Chinnaswamy P, Saravanan G, Kumar US. Effect of chronic smoking on lipid peroxidation and antioxidant status in gastric carcinoma patients. Bangladesh Med Res Counc Bull (2009) 35(1):1–6. doi: 10.3329/bmrcb.v35i1.2132
65. Alvarez-Parrilla E, de la Rosa LA, Legarreta P, Saenz L, Rodrigo-Garcia J, Gonzalez-Aguilar GA. Daily consumption of apple, pear and orange juice differently affects plasma lipids and antioxidant capacity of smoking and non-smoking adults. Int J Food Sci Nutr (2010) 61(4):369–80. doi: 10.3109/09637480903514041
66. Bøhn SK, Myhrstad MC, Thoresen M, Holden M, Karlsen A, Tunheim SH, et al. Blood cell gene expression associated with cellular stress defense is modulated by antioxidant-rich food in a randomised controlled clinical trial of Male smokers. BMC Med (2010) 8:54. doi: 10.1186/1741-7015-8-54
67. Cizmarova B, Tomeckova V, Hubkova B, Hurajtova A, Ohlasova J, Birkova A. Salivary redox homeostasis in human health and disease. Int J Mol Sci (2022) 23(17):10076. doi: 10.3390/ijms231710076
68. Ishimoto T, Sugihara H, Watanabe M, Sawayama H, Iwatsuki M, Baba Y, et al. Macrophage-derived reactive oxygen species suppress mir-328 targeting Cd44 in cancer cells and promote redox adaptation. Carcinogenesis (2014) 35(5):1003–11. doi: 10.1093/carcin/bgt402
69. Kiga K, Mimuro H, Suzuki M, Shinozaki-Ushiku A, Kobayashi T, Sanada T, et al. Epigenetic silencing of mir-210 increases the proliferation of gastric epithelium during chronic helicobacter pylori infection. Nat Commun (2014) 5:4497. doi: 10.1038/ncomms5497
70. Candido MF, Medeiros M, Veronez LC, Bastos D, Oliveira KL, Pezuk JA, et al. Drugging hijacked kinase pathways in pediatric oncology: Opportunities and current scenario. Pharmaceutics (2023) 15(2):664. doi: 10.3390/pharmaceutics15020664
71. Tatsuta M, Iishi H, Baba M, Mikuni T, Narahara H, Uedo N, et al. Suppression by iron chelator phenanthroline of sodium chloride-enhanced gastric carcinogenesis induced by n-Methyl-N’-Nitro-N-Nitrosoguanidine in wistar rats. Cancer Lett (2003) 191(1):9–16. doi: 10.1016/s0304-3835(01)00797-2
72. Xin H, Deng Y, Cao J. Proviral insertion in murine lymphomas 2 promotes stomach cancer progression by regulating apoptosis Via reactive oxygen species-triggered endoplasmic reticulum stress. Biochem Biophys Res Commun (2018) 506(1):145–52. doi: 10.1016/j.bbrc.2018.09.062
73. George S, Lucero Y, Torres JP, Lagomarcino AJ, O’Ryan M. Gastric damage and cancer-associated biomarkers in helicobacter pylori-infected children. Front Microbiol (2020) 11:90. doi: 10.3389/fmicb.2020.00090
74. Han L, Shu X, Wang J. Helicobacter pylori-mediated oxidative stress and gastric diseases: A review. Front Microbiol (2022) 13:811258. doi: 10.3389/fmicb.2022.811258
75. Chaturvedi R, Asim M, Romero-Gallo J, Barry DP, Hoge S, de Sablet T, et al. Spermine oxidase mediates the gastric cancer risk associated with helicobacter pylori caga. Gastroenterology (2011) 141(5):1696–708 e1-2. doi: 10.1053/j.gastro.2011.07.045
76. Bagchi D, McGinn TR, Ye X, Bagchi M, Krohn RL, Chatterjee A, et al. Helicobacter pylori-induced oxidative stress and DNA damage in a primary culture of human gastric mucosal cells. Dig Dis Sci (2002) 47(6):1405–12. doi: 10.1023/a:1015399204069
77. Hanada K, Uchida T, Tsukamoto Y, Watada M, Yamaguchi N, Yamamoto K, et al. Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect Immun (2014) 82(10):4182–9. doi: 10.1128/IAI.02368-14
78. Pan Z, Ma G, Kong L, Du G. Hypoxia-inducible factor-1: Regulatory mechanisms and drug development in stroke. Pharmacol Res (2021) 170:105742. doi: 10.1016/j.phrs.2021.105742
79. Rath S, Das L, Kokate SB, Pratheek BM, Chattopadhyay S, Goswami C, et al. Regulation of noxa-mediated apoptosis in helicobacter pylori-infected gastric epithelial cells. FASEB J (2015) 29(3):796–806. doi: 10.1096/fj.14-257501
80. Kannan A, Krishnan A, Ali M, Subramaniam S, Halagowder D, Sivasithamparam ND. Caveolin-1 promotes gastric cancer progression by up-regulating epithelial to mesenchymal transition by crosstalk of signalling mechanisms under hypoxic condition. Eur J Cancer (2014) 50(1):204–15. doi: 10.1016/j.ejca.2013.08.016
81. Hao W, Yuan X, Yu L, Gao C, Sun X, Wang D, et al. Licochalcone a-induced human gastric cancer bgc-823 cells apoptosis by regulating ros-mediated mapks and Pi3k/Akt signaling pathways. Sci Rep (2015) 5:10336. doi: 10.1038/srep10336
82. Lin J, Chen Z, Huang Z, Chen F, Ye Z, Lin S, et al. Effect of T-cadherin on the Akt/Mtor signaling pathway, gastric cancer cell cycle, migration and invasion, and its association with patient survival rate. Exp Ther Med (2019) 17(5):3607–13. doi: 10.3892/etm.2019.7350
83. Chaithongyot S, Jantaree P, Sokolova O, Naumann M. Nf-Kb in gastric cancer development and therapy. Biomedicines (2021) 9(8):870. doi: 10.3390/biomedicines9080870
84. Kidane D, Murphy DL, Sweasy JB. Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during helicobacter pylori infection. Oncogenesis (2014) 3(11):e128. doi: 10.1038/oncsis.2014.42
85. Chaturvedi R, Asim M, Piazuelo MB, Yan F, Barry DP, Sierra JC, et al. Activation of egfr and Erbb2 by helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology (2014) 146(7):1739–51.e14. doi: 10.1053/j.gastro.2014.02.005
86. Zhou X, Xu G, Yin C, Jin W, Zhang G. Down-regulation of mir-203 induced by helicobacter pylori infection promotes the proliferation and invasion of gastric cancer by targeting cask. Oncotarget (2014) 5(22):11631–40. doi: 10.18632/oncotarget.2600
87. Su M, Zhang Z, Zhou L, Han C, Huang C, Nice EC. Proteomics, personalized medicine and cancer. Cancers (Basel) (2021) 13(11):2512. doi: 10.3390/cancers13112512
88. Crowe SE. Helicobacter pylori infection. N Engl J Med (2019) 380(12):1158–65. doi: 10.1056/NEJMcp1710945
89. Singh N, Baby D, Rajguru JP, Patil PB, Thakkannavar SS, Pujari VB. Inflammation and cancer. Ann Afr Med (2019) 18(3):121–6. doi: 10.4103/aam.aam_56_18
90. Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. Foxo forkhead transcription factors induce G(2)-m checkpoint in response to oxidative stress. J Biol Chem (2002) 277(30):26729–32. doi: 10.1074/jbc.C200256200
91. Piscione M, Mazzone M, Di Marcantonio MC, Muraro R, Mincione G. Eradication of helicobacter pylori and gastric cancer: A controversial relationship. Front Microbiol (2021) 12:630852. doi: 10.3389/fmicb.2021.630852
93. El Hafa F, Wang T, Ndifor VM, Jin G. Association between helicobacter pylori antibodies determined by multiplex serology and gastric cancer risk: A meta-analysis. Helicobacter (2022) 27(3):e12881. doi: 10.1111/hel.12881
94. Wroblewski LE, Choi E, Petersen C, Delgado AG, Piazuelo MB, Romero-Gallo J, et al. Targeted mobilization of Lrig1(+) gastric epithelial stem cell populations by a carcinogenic helicobacter pylori type iv secretion system. Proc Natl Acad Sci U.S.A. (2019) 116(39):19652–8. doi: 10.1073/pnas.1903798116
95. Toh JWT, Wilson RB. Pathways of gastric carcinogenesis, helicobacter pylori virulence and interactions with antioxidant systems, vitamin c and phytochemicals. Int J Mol Sci (2020) 21(17):6451. doi: 10.3390/ijms21176451
96. Naito Y, Yoshikawa T, Fujii T, Boku Y, Yagi N, Dao S, et al. Monochloramine-induced cell growth inhibition and apoptosis in a rat gastric mucosal cell line. J Clin Gastroenterol (1997) 25 Suppl 1:S179–85. doi: 10.1097/00004836-199700001-00029
97. Wallaschek N, Reuter S, Silkenat S, Wolf K, Niklas C, Kayisoglu O, et al. Ephrin receptor A2, the epithelial receptor for Epstein-Barr virus entry, is not available for efficient infection in human gastric organoids. PloS Pathog (2021) 17(2):e1009210. doi: 10.1371/journal.ppat.1009210
98. Akkus S, Gareayaghi N, Saribas S, Demiryas S, Ozbey D, Kepil N, et al. Co-Infection relationship with Epstein-Barr virus in gastroduodenal diseases with helicobacter pylori. quantitative pcr and ebna-1 gene-based approach. Acta Gastroenterol Belg (2022) 85(2):301–8. doi: 10.51821/85.2.9440
99. Yang H, Hu B. Immunological perspective: Helicobacter pylori infection and gastritis. Mediators Inflammation (2022) 2022:2944156. doi: 10.1155/2022/2944156
100. Saunders RM, Biddle M, Amrani Y, Brightling CE. Stressed out - the role of oxidative stress in airway smooth muscle dysfunction in asthma and copd. Free Radic Biol Med (2022) 185:97–119. doi: 10.1016/j.freeradbiomed.2022.04.011
101. Futagami S, Hiratsuka T, Shindo T, Horie A, Hamamoto T, Suzuki K, et al. Expression of Apurinic/Apyrimidinic endonuclease-1 (Ape-1) in h. pylori-associated gastritis, gastric adenoma, and gastric cancer. Helicobacter (2008) 13(3):209–18. doi: 10.1111/j.1523-5378.2008.00605.x
102. Du MQ, Atherton JC. Molecular subtyping of gastric malt lymphomas: Implications for prognosis and management. Gut (2006) 55(6):886–93. doi: 10.1136/gut.2004.061663
103. Gong EJ, Ahn JY, Jung HY, Park H, Ko YB, Na HK, et al. Helicobacter pylori eradication therapy is effective as the initial treatment for patients with h. pylori-negative and disseminated gastric mucosa-associated lymphoid tissue lymphoma. Gut liver (2016) 10(5):706–13. doi: 10.5009/gnl15510
104. Floch P, Megraud F, Lehours P. Helicobacter pylori strains and gastric malt lymphoma. Toxins (Basel) (2017) 9(4):132. doi: 10.3390/toxins9040132
105. Kim SS, Ruiz VE, Carroll JD, Moss SF. Helicobacter pylori in the pathogenesis of gastric cancer and gastric lymphoma. Cancer Lett (2011) 305(2):228–38. doi: 10.1016/j.canlet.2010.07.014
106. Della Bella C, Soluri MF, Puccio S, Benagiano M, Grassi A, Bitetti J, et al. The helicobacter pylori cagy protein drives gastric Th1 and Th17 inflammation and b cell proliferation in gastric malt lymphoma. Int J Mol Sci (2021) 22(17):9459. doi: 10.3390/ijms22179459
107. Zullo A, Hassan C, Cristofari F, Andriani A, De Francesco V, Ierardi E, et al. Effects of helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin Gastroenterol Hepatol (2010) 8(2):105–10. doi: 10.1016/j.cgh.2009.07.017
108. Matsuoka K, Nishiumi S, Yoshida M, Kodama Y. Effects of helicobacter pylori on the glutathione-related pathway in gastric epithelial cells. Biochem Biophys Res Commun (2020) 526(4):1118–24. doi: 10.1016/j.bbrc.2020.04.019
109. Poplawski T, Chojnacki C, Czubatka A, Klupinska G, Chojnacki J, Blasiak J. Helicobacter pylori infection and antioxidants can modulate the genotoxic effects of heterocyclic amines in gastric mucosa cells. Mol Biol Rep (2013) 40(8):5205–12. doi: 10.1007/s11033-013-2622-3
110. Deng R, Mo F, Chang B, Zhang Q, Ran H, Yang S, et al. Glucose-derived ages enhance human gastric cancer metastasis through Rage/Erk/Sp1/Mmp2 cascade. Oncotarget (2017) 8(61):104216–26. doi: 10.18632/oncotarget.22185
111. Ahn MJ, Kim BH, Jang SJ, Hong EK, Lee WM, Baik HK, et al. Expression of cyclin D1 and cyclin e in human gastric carcinoma and its clinicopathologic significance. J Korean Med Sci (1998) 13(5):513–8. doi: 10.3346/jkms.1998.13.5.513
112. Aoyagi K, Koufuji K, Yano S, Murakami N, Terasaki Y, Yamasaki Y, et al. Immunohistochemical study on the expression of cyclin D1 and e in gastric cancer. Kurume Med J (2000) 47(3):199–203. doi: 10.2739/kurumemedj.47.199
113. Arendt T, Rodel L, Gartner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor P16 in alzheimer’s disease. Neuroreport (1996) 7(18):3047–9. doi: 10.1097/00001756-199611250-00050
114. Gamboa-Dominguez A, Seidl S, Reyes-Gutierrez E, Hermannstädter C, Quintanilla-Martinez L, Busch R, et al. Prognostic significance of P21waf1/Cip1, P27kip1, P53 and e-cadherin expression in gastric cancer. J Clin Pathol (2007) 60(7):756–61. doi: 10.1136/jcp.2006.038976
115. Shin VY, Wu WK, Chu KM, Wong HP, Lam EK, Tai EK, et al. Nicotine induces cyclooxygenase-2 and vascular endothelial growth factor receptor-2 in association with tumor-associated invasion and angiogenesis in gastric cancer. Mol Cancer Res (2005) 3(11):607–15. doi: 10.1158/1541-7786.MCR-05-0106
116. Leung WK, To KF, Go MY, Chan KK, Chan FK, Ng EK, et al. Cyclooxygenase-2 upregulates vascular endothelial growth factor expression and angiogenesis in human gastric carcinoma. Int J Oncol (2003) 23(5):1317–22. doi: 10.3892/ijo.23.5.1317
117. Shimakura S, Boland CR. Eicosanoid production by the human gastric cancer cell line ags and its relation to cell growth. Cancer Res (1992) 52(7):1744–9.
118. Ji XK, Madhurapantula SV, He G, Wang KY, Song CH, Zhang JY, et al. Genetic variant of cyclooxygenase-2 in gastric cancer: More inflammation and susceptibility. World J Gastroenterol (2021) 27(28):4653–66. doi: 10.3748/wjg.v27.i28.4653
119. Li Y, Liu C, Zhang X, Huang X, Liang S, Xing F, et al. Cct5 induces epithelial-mesenchymal transition to promote gastric cancer lymph node metastasis by activating the Wnt/Beta-catenin signalling pathway. Br J Cancer (2022) 126(12):1684–94. doi: 10.1038/s41416-022-01747-0
120. Tian S, Peng P, Li J, Deng H, Zhan N, Zeng Z, et al. Serpinh1 regulates emt and gastric cancer metastasis Via the Wnt/Beta-catenin signaling pathway. Aging (Albany NY) (2020) 12(4):3574–93. doi: 10.18632/aging.102831
121. Lei ZN, Teng QX, Tian Q, Chen W, Xie Y, Wu K, et al. Signaling pathways and therapeutic interventions in gastric cancer. Signal Transduct Target Ther (2022) 7(1):358. doi: 10.1038/s41392-022-01190-w
122. Vitiello PP, Cardone C, Martini G, Ciardiello D, Belli V, Matrone N, et al. Receptor tyrosine kinase-dependent Pi3k activation is an escape mechanism to vertical suppression of the Egfr/Ras/Mapk pathway in kras-mutated human colorectal cancer cell lines. J Exp Clin Cancer Res (2019) 38(1):41. doi: 10.1186/s13046-019-1035-0
123. Yan H, Zheng C, Li Z, Bao B, Yang B, Hou K, et al. Nptx1 promotes metastasis Via Integrin/Fak signaling in gastric cancer. Cancer Manag Res (2019) 11:3237–51. doi: 10.2147/CMAR.S196509
124. Wang X, Zhou Q, Yu Z, Wu X, Chen X, Li J, et al. Cancer-associated fibroblast-derived lumican promotes gastric cancer progression Via the integrin B1-fak signaling pathway. Int J Cancer (2017) 141(5):998–1010. doi: 10.1002/ijc.30801
125. Yu GZ, Chen Y, Wang JJ. Overexpression of Grb2/Her2 signaling in Chinese gastric cancer: Their relationship with clinicopathological parameters and prognostic significance. J Cancer Res Clin Oncol (2009) 135(10):1331–9. doi: 10.1007/s00432-009-0574-8
126. Katoh Y, Katoh M. Hedgehog signaling pathway and gastric cancer. Cancer Biol Ther (2005) 4(10):1050–4. doi: 10.4161/cbt.4.10.2184
127. Li H, Jia Y, Wang Y. Targeting hif-1α signaling pathway for gastric cancer treatment. Die Pharmazie (2019) 74(1):3–7. doi: 10.1691/ph.2019.8674
128. Wang J, Ni Z, Duan Z, Wang G, Li F. Altered expression of hypoxia-inducible factor-1α (Hif-1α) and its regulatory genes in gastric cancer tissues. PloS One (2014) 9(6):e99835. doi: 10.1371/journal.pone.0099835
129. Zhou GX, Li XY, Zhang Q, Zhao K, Zhang CP, Xue CH, et al. Effects of the hippo signaling pathway in human gastric cancer. Asian Pac J Cancer Prev (2013) 14(9):5199–205. doi: 10.7314/apjcp.2013.14.9.5199
130. Kang W, Cheng AS, Yu J, To KF. Emerging role of hippo pathway in gastric and other gastrointestinal cancers. World J Gastroenterol (2016) 22(3):1279–88. doi: 10.3748/wjg.v22.i3.1279
131. Khanna P, Chua PJ, Bay BH, Baeg GH. The Jak/Stat signaling cascade in gastric carcinoma (Review). Int J Oncol (2015) 47(5):1617–26. doi: 10.3892/ijo.2015.3160
132. Ji F, Chen YL, Jin EY, Wang WL, Yang ZL, Li YM. Relationship between matrix metalloproteinase-2 mrna expression and clinicopathological and urokinase-type plasminogen activator system parameters and prognosis in human gastric cancer. World J Gastroenterol (2005) 11(21):3222–6. doi: 10.3748/wjg.v11.i21.3222
133. Lee KH, Choi EY, Kim MK, Kim KO, Jang BI, Kim SW, et al. Inhibition of histone deacetylase activity down-regulates urokinase plasminogen activator and matrix metalloproteinase-9 expression in gastric cancer. Mol Cell Biochem (2010) 343(1-2):163–71. doi: 10.1007/s11010-010-0510-x
134. Ganguly K, Rauth S, Marimuthu S, Kumar S, Batra SK. Unraveling mucin domains in cancer and metastasis: When protectors become predators. Cancer Metastasis Rev (2020) 39(3):647–59. doi: 10.1007/s10555-020-09896-5
135. Sokolova O, Naumann M. Nf-Kb signaling in gastric cancer. Toxins (Basel) (2017) 9(4):119. doi: 10.3390/toxins9040119
136. Brzozowa M, Mielanczyk L, Michalski M, Malinowski L, Kowalczyk-Ziomek G, Helewski K, et al. Role of notch signaling pathway in gastric cancer pathogenesis. Contemp Oncol (Pozn) (2013) 17(1):1–5. doi: 10.5114/wo.2013.33765
137. Matsuoka T, Yashiro M. The role of Pi3k/Akt/Mtor signaling in gastric carcinoma. Cancers (Basel) (2014) 6(3):1441–63. doi: 10.3390/cancers6031441
138. Tapia O, Riquelme I, Leal P, Sandoval A, Aedo S, Weber H, et al. The Pi3k/Akt/Mtor pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch (2014) 465(1):25–33. doi: 10.1007/s00428-014-1588-4
139. Zhang B, Bie Q, Wu P, Zhang J, You B, Shi H, et al. Pgd2/Ptgdr2 signaling restricts the self-renewal and tumorigenesis of gastric cancer. Stem Cells (2018) 36(7):990–1003. doi: 10.1002/stem.2821
140. Ashrafizadeh M, Zarrabi A, Orouei S, Zarrin V, Rahmani Moghadam E, Zabolian A, et al. Stat3 pathway in gastric cancer: Signaling, therapeutic targeting and future prospects. Biol (Basel) (2020) 9(6):126. doi: 10.3390/biology9060126
141. Giraud AS, Menheniott TR, Judd LM. Targeting Stat3 in gastric cancer. Expert Opin Ther Targets (2012) 16(9):889–901. doi: 10.1517/14728222.2012.709238
142. Yuan X, Zhou Y, Wang W, Li J, Xie G, Zhao Y, et al. Activation of Tlr4 signaling promotes gastric cancer progression by inducing mitochondrial ros production. Cell Death Dis (2013) 4:e794. doi: 10.1038/cddis.2013.334
143. Zhang J, Ge Y, Sun L, Cao J, Wu Q, Guo L, et al. Effect of bone morphogenetic protein-2 on proliferation and apoptosis of gastric cancer cells. Int J Med Sci (2012) 9(2):184–92. doi: 10.7150/ijms.3859
144. Shirai YT, Ehata S, Yashiro M, Yanagihara K, Hirakawa K, Miyazono K. Bone morphogenetic protein-2 and -4 play tumor suppressive roles in human diffuse-type gastric carcinoma. Am J Pathol (2011) 179(6):2920–30. doi: 10.1016/j.ajpath.2011.08.022
145. Hsieh HL, Tsai MM. Tumor progression-dependent angiogenesis in gastric cancer and its potential application. World J Gastrointest Oncol (2019) 11(9):686–704. doi: 10.4251/wjgo.v11.i9.686
146. Song L, Guo X, Zhao F, Wang W, Zhao Z, Jin L, et al. Ttc36 inactivation induce malignant properties Via wnt-Beta-Catenin pathway in gastric carcinoma. J Cancer (2021) 12(9):2598–609. doi: 10.7150/jca.47292
147. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-Methyl-5-(1-[3-(Trifluoromethyl)Phenyl]Acetyl-2,3-Dihydro-1h-Indol-5-Yl)-7h-Pyrrolo[2,3-D]Pyrimidin-4-Amine (Gsk2606414), a potent and selective first-in-Class inhibitor of protein kinase r (Pkr)-like endoplasmic reticulum kinase (Perk). J Med Chem (2012) 55(16):7193–207. doi: 10.1021/jm300713s
148. Liao WC, Huang MZ, Wang ML, Lin CJ, Lu TL, Lo HR, et al. Statin decreases helicobacter pylori burden in macrophages by promoting autophagy. Front Cell Infect Microbiol (2016) 6:203. doi: 10.3389/fcimb.2016.00203
149. Rao SV, Solum G, Niederdorfer B, Norsett KG, Bjorkoy G, Thommesen L. Gastrin activates autophagy and increases migration and survival of gastric adenocarcinoma cells. BMC Cancer (2017) 17(1):68. doi: 10.1186/s12885-017-3055-5
150. Velmurugan B, Bhuvaneswari V, Nagini S. Effect of s-allylcysteine on oxidant-antioxidant status during n-Methyl-N’-Nitro-N-Nitrosoguanidine and saturated sodium chloride-induced gastric carcinogenesis in wistar rats. Asia Pac J Clin Nutr (2003) 12(4):488–94.
151. Park Y, Lee H, Lim JW, Kim H. Inhibitory effect of B-carotene on helicobacter pylori-induced traf expression and hyper-proliferation in gastric epithelial cells. Antioxid (Basel Switzerland) (2019) 8(12):637. doi: 10.3390/antiox8120637
152. Sepidarkish M, Akbari-Fakhrabadi M, Daneshzad E, Yavari M, Rezaeinejad M, Morvaridzadeh M, et al. Effect of omega-3 fatty acid plus vitamin e Co-supplementation on oxidative stress parameters: A systematic review and meta-analysis. Clin Nutr (2020) 39(4):1019–25. doi: 10.1016/j.clnu.2019.05.004
153. Pelliciari S, Pinatel E, Vannini A, Peano C, Puccio S, De Bellis G, et al. Insight into the essential role of the helicobacter pylori Hp1043 orphan response regulator: Genome-wide identification and characterization of the DNA-binding sites. Sci Rep (2017) 7:41063. doi: 10.1038/srep41063
154. Gonzalez A, Salillas S, Velazquez-Campoy A, Espinosa Angarica V, Fillat MF, Sancho J, et al. Identifying potential novel drugs against helicobacter pylori by targeting the essential response regulator hsra. Sci Rep (2019) 9(1):11294. doi: 10.1038/s41598-019-47746-9
155. Xu XY, Meng X, Li S, Gan RY, Li Y, Li HB. Bioactivity, health benefits, and related molecular mechanisms of curcumin: Current progress, challenges, and perspectives. Nutrients (2018) 10(10):1553. doi: 10.3390/nu10101553
156. Velmurugan B, Subapriya R, Nagini S. Chemoprotection against n-Methyl-N’-Nitro-N-Nitrosoguanidine-Induced oxidative stress by s-allylcysteine, a garlic constituent, in wistar rats. Toxicol Mech Methods (2003) 13(2):83–7. doi: 10.1080/15376510309844
157. Chitcholtan K, Hampton MB, Keenan JI. Outer membrane vesicles enhance the carcinogenic potential of helicobacter pylori. Carcinogenesis (2008) 29(12):2400–5. doi: 10.1093/carcin/bgn218
158. Lee YM, Kim MJ, Kim Y, Kim H. Glutamine deprivation causes hydrogen peroxide-induced interleukin-8 expression Via Jak1/Stat3 activation in gastric epithelial ags cells. J Cancer Prev (2015) 20(3):179–84. doi: 10.15430/JCP.2015.20.3.179
159. Li JY, Taylor PR, Li B, Dawsey S, Wang GQ, Ershow AG, et al. Nutrition intervention trials in linxian, China: Multiple Vitamin/Mineral supplementation, cancer incidence, and disease-specific mortality among adults with esophageal dysplasia. J Natl Cancer Inst (1993) 85(18):1492–8. doi: 10.1093/jnci/85.18.1492
160. Qiao YL, Dawsey SM, Kamangar F, Fan JH, Abnet CC, Sun XD, et al. Total and cancer mortality after supplementation with vitamins and minerals: Follow-up of the linxian general population nutrition intervention trial. J Natl Cancer Inst (2009) 101(7):507–18. doi: 10.1093/jnci/djp037
161. Kyung S, Lim JW, Kim H. A-lipoic acid inhibits il-8 expression by activating Nrf2 signaling in helicobacter pylori-infected gastric epithelial cells. Nutrients (2019) 11(10):2524. doi: 10.3390/nu11102524
162. Sepidarkish M, Morvaridzadeh M, Akbari-Fakhrabadi M, Almasi-Hashiani A, Rezaeinejad M, Heshmati J. Effect of omega-3 fatty acid plus vitamin e Co-supplementation on lipid profile: A systematic review and meta-analysis. Diabetes Metab Syndr (2019) 13(2):1649–56. doi: 10.1016/j.dsx.2019.03.018
163. Janssen AM, Bosman CB, van Duijn W, Oostendorp-van de Ruit MM, Kubben FJ, Griffioen G, et al. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin Cancer Res (2000) 6(8):3183–92.
164. Jelic MD, Mandic AD, Maricic SM, Srdjenovic BU. Oxidative stress and its role in cancer. J Cancer Res Ther (2021) 17(1):22–8. doi: 10.4103/jcrt.JCRT_862_16
165. Gonzalez A, Casado J, Chueca E, Salillas S, Velazquez-Campoy A, Espinosa Angarica V, et al. Repurposing dihydropyridines for treatment of helicobacter pylori infection. Pharmaceutics (2019) 11(12):681. doi: 10.3390/pharmaceutics11120681
166. Xu H, Yu WB, Gao Y, Zhang MJ, Malhotra A, Yu WH. Modulatory potential of curcumin and resveratrol on P53 post-translational modifications during gastric cancer. J Environ Pathol Toxicol Oncol (2018) 37(2):93–101. doi: 10.1615/JEnvironPatholToxicolOncol.2018025547
Keywords: gastric cancer, oxidative stress, signaling pathways, pathophysiology, therapeutic targets
Citation: Liu Y, Shi Y, Han R, Liu C, Qin X, Li P and Gu R (2023) Signaling pathways of oxidative stress response: the potential therapeutic targets in gastric cancer. Front. Immunol. 14:1139589. doi: 10.3389/fimmu.2023.1139589
Received: 07 January 2023; Accepted: 20 March 2023;
Published: 18 April 2023.
Edited by:
Qun Zhao, Fourth Hospital of Hebei Medical University, ChinaReviewed by:
Laiping Zhong, Shanghai Jiao Tong University, ChinaCopyright © 2023 Liu, Shi, Han, Liu, Qin, Li and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renjun Gu, cmVuanVuZ3VAaG90bWFpbC5jb20=; Pengfei Li, bGlwZW5nZmVpQG5qdWNtLmVkdS5jbg==; Xiaogang Qin, NTAyNjkzNjgwQHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.